
Tunable regiodivergent phosphine-catalyzed [3 + 2] cycloaddition of alkynones and trifluoroacetyl phenylamides†
Yao-Liang
Sun
a,
Yin
Wei
 *b and
Min
Shi
*abc
*b and
Min
Shi
*abc
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
bState Key Laboratory of Organometallic Chemistry, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@sioc.ac.cn; mshi@sioc.ac.cn
cState Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 29th August 2017
Abstract
Alkynones can be activated by phosphine as a nucleophilic catalyst, and then trapped by a series of trifluoroacetyl phenylamides to afford cycloaddition products. Through subtly adjusting the substituent of trifluoroacetyl phenylamides, the addition of water and changing the reaction temperature, two kinds of highly regioselective cycloaddition products were obtained in moderate to excellent yields. Plausible mechanisms were proposed and supported by the deuterium-labeling experiments and DFT calculations. DFT calculations demonstrate that the currently accepted intramolecular proton transfer processes involved in these reactions are impossible, and these proton transfer processes can proceed with the assistance of substrates containing an acidic moiety or by the addition of water. Our mechanistic studies provide reasonable explanations for the regioselectivity affected by the protic additive H2O, and the reaction temperature.
Introduction
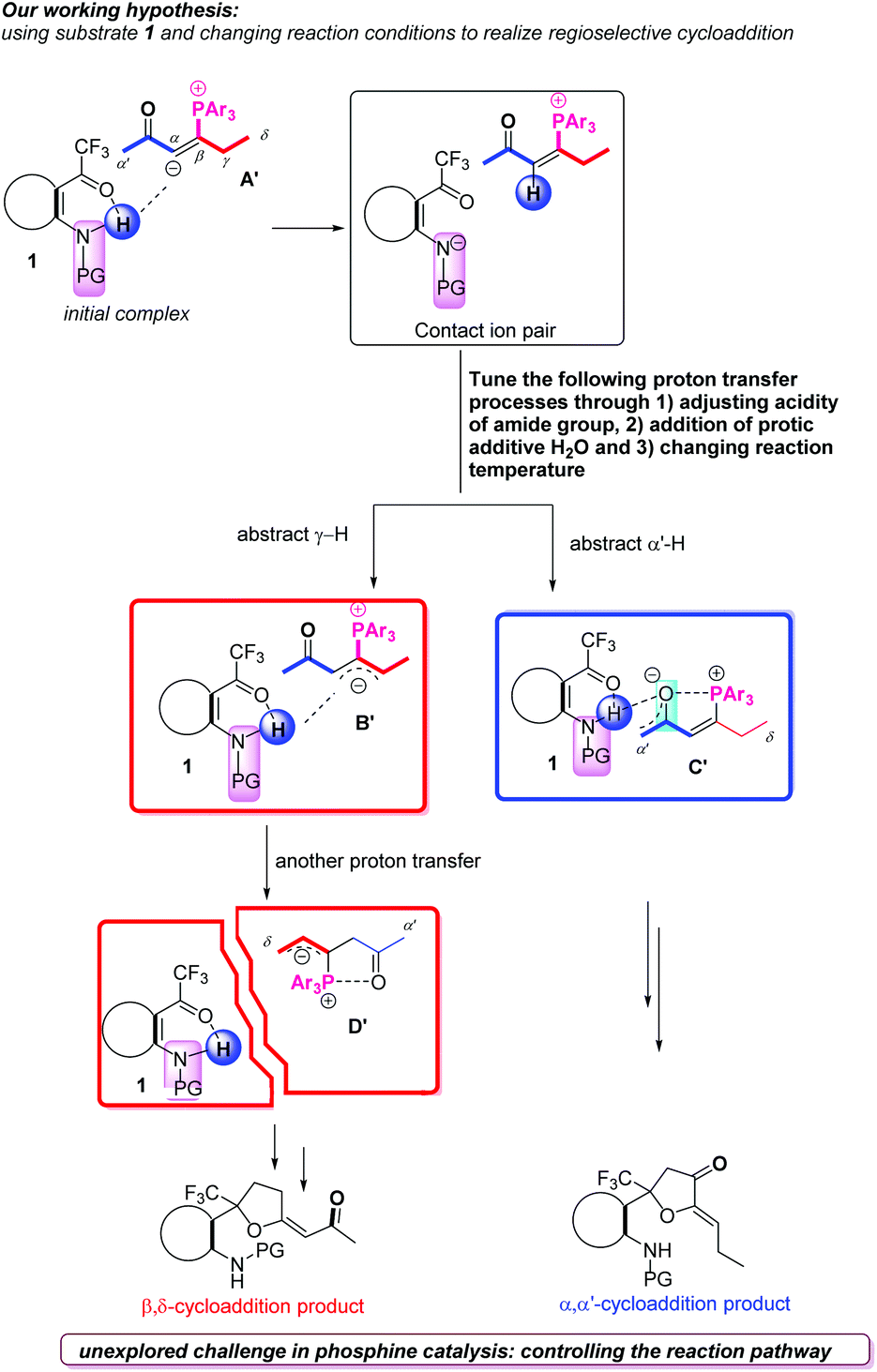
In recent years, the nucleophilic phosphine-catalyzed reactions1 have been recognized as reliable methods which could combine olefins,2 allenes,3 alkynes,4 or Morita–Baylis–Hillman adducts (MBHADs)5 with other electrophilic or nucleophilic blocks to afford highly functionalized products; the related isomerization, addition and cycloaddition reactions have been explored, expanded and applied. Alkynones as activated π system substrates have been frequently utilized in a large number of nucleophilic phosphine-catalyzed reactions, such as addition,6 umpolung addition,4b,7 and cycloaddition4a,8a,b reactions. Based on the commonly accepted mechanisms for the nucleophilic phosphine-catalyzed reactions of electrophilic π systems,9 the first step is nucleophilic addition of phosphine to electrophilic π systems. In this case, zwitterionic intermediate A is first formed by the nucleophilic addition of tertiary phosphine to the alkynones, which subsequently undergo α-addition,10 β-addition11 or di-addition12 with a variety of nucleophiles to afford highly functionalized products (Scheme 1). On the other hand, the zwitterionic intermediate A can be transformed to a zwitterionic intermediate Bviaa 1,3-proton shift (γ–α shift), which subsequently undergoes γ-umpolung addition4b,13 with nucleophiles or cycloaddition reactions with electrophilic π systems (Scheme 1).14 Alternatively, the zwitterionic intermediate A can be transformed to another zwitterionic intermediate Cvia 1,3-proton shift (α′–α shift), which can conduct α′,α-cycloadditions with a range of electrophilic π systems (Scheme 1).15 Very recently, we first reported the phosphine-catalyzed δ-carbon addition of alkynones with electron-deficient carbonyl groups to generate 1,3-diene derivatives (Scheme 1);16 a novel zwitterionic intermediate D, which was probably converted from zwitterionic intermediate A′via two proton shift processes, was identified as the key intermediate and reacted with trifluoroacetyl compounds to afford δ-addition products. On the basis of previous reports, the related four zwitterionic intermediates A, B, C and D were critical to direct the subsequent transformations. These zwitterionic intermediates A, B, C and D can be converted reversibly, and their versatile reactivities can be tuned by adjusting substituents in the substrates.17 However, there are challenges to deliberately control their conversions, and further achieve highly regioselective phosphine-catalyzed reactions of alkynones with other substrates.18 | ||
| Scheme 1 The reaction modes of phosphine-catalyzed reactions of alkynones with other substrates. | ||
On the basis of previous reports by our group and others, we realized that these zwitterionic intermediates A–D shown in Scheme 1 are critical intermediates to direct the following transformations and envisaged that adjusting the substituents in the substrates or changing the reaction conditions can further achieve highly regioselective phosphine-catalyzed reactions of alkynones with a range of electrophilic π systems. Considering the amide group,19 widely utilized as an important moiety in organocatalysts,20 bifunctional catalysts,1j ligands21 and directing groups22 in substrates to direct transformations, we designed substrate 1 having an amide group, and hypothesized that it could assist the proton transfer processes23 due to the acidity of the NHAc moiety, which makes the zwitterionic intermediate A′ be transformed into zwitterionic intermediates B′, C′ or D′ (Scheme 2). In addition, the addition of a protic additive, such as H2O, and changing the reaction temperature may tune these proton transfer processes to afford different regioselective cycloaddition products (Scheme 2). Based on the working hypothesis, we systematically investigated the phosphine-catalyzed cycloaddition reactions of alkynones with a range of trifluoroacetyl phenylamides, producing cycloadducts containing a CF3 group, which may have further synthetic applications since in a variety of pharmaceutical compounds the incorporation of a CF3 group is often found to give special chemical properties, metabolic stability, lipophilicity and solubility.24 Herein, we would like to report the regiodivergent25 phosphine-catalyzed [3 + 2] cycloaddition of alkynones with a series of trifluoroacetyl phenylamides, affording two kinds of cycloaddition products in moderate to excellent yields; we also conducted mechanistic studies to reveal the detailed mechanisms through deuterium-labeling experiments and DFT calculations.
 | ||
| Scheme 2 Tunable regioselective cycloaddition. | ||
Results and discussion
Experimental investigations
We initially tested the reaction outcomes in the presence of P(4-FC6H4)3 and optimized the reaction conditions employing 2-(2,2,2-trifluoroacetyl)phenylamide 1a and alkynone 2a as substrates. The results are shown in Table 1. It was found that α′,α-cycloaddition product 4a was obtained in 74% yield with 3.0 equiv. of 2a in toluene at room temperature for 72 h (entry 1). The solvent effect was next investigated, and it was identified that toluene was still better than other solvents, such as THF, dioxane, and DCM (entries 2–4). Decreasing the reaction temperature to 10 °C improved the yield to 88% (entry 5). However, the reaction did not take place when a 4 Å molecular sieve was added as an additive (entry 6), suggesting that a trace of water might be a co-catalyst in this cycloaddition reaction at low temperature. Interestingly, the β,δ-cycloaddition product 3a was mainly accessed at 65 °C with the same substrates 1a and 2a in 67% yield and hydration product 3a′ was obtained in 11% yield as a by-product (entry 7). Enhancing the concentration of the reaction mixture, the yield of β,δ-cycloaddition product 3a decreased to 52%; however, the yield of hydration product 3a′ simultaneously increased to 24% (entry 8). Adding a 4 Å molecular sieve into the reaction system did not influence the reaction proceeding (entry 9), but the addition of phenol would impede the reaction proceeding, only affording the desired β,δ-cycloaddition product 3a in 23% yield (entry 10).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Additive (mg) | Solvent | Temp (°C) | Time (h) | Yield (%)b | ||
| 3a | 3a′ | 4a | |||||
| a The reaction was carried out using 1a (0.2 mmol), 2a (3.0 equiv.), cat. (0.04 mmol), in the indicated solvent (1.0 mL) in a Schlenk tube at the indicated temperature. b Determined by 19F NMR analysis of the crude reaction mixture by using 4-Bromobenzotrifluoride as the internal standard. | |||||||
| 1 | — | Toluene | r.t | 72 | — | — | 74 |
| 2 | — | THF | r.t | 72 | — | — | 13 |
| 3 | — | Dioxane | r.t | 72 | — | — | 36 |
| 4 | — | DCM | r.t | 54 | — | — | 73 |

|

|

|
|
|

|

|

|
| 6 | 4Å MS (100) | toluene | 10 | 54 | — | — | — |

|

|

|

|

|

|

|

|
| 8 | — | Toluene (0.5 mL) | 65 | 48 | 52 | 24 | — |
| 9 | 4Å MS (100) | Toluene | 65 | 48 | 57 | 12 | — |
| 10 | PhOH | Toluene | 65 | 48 | 23 | 17 | — |

|
|||||||

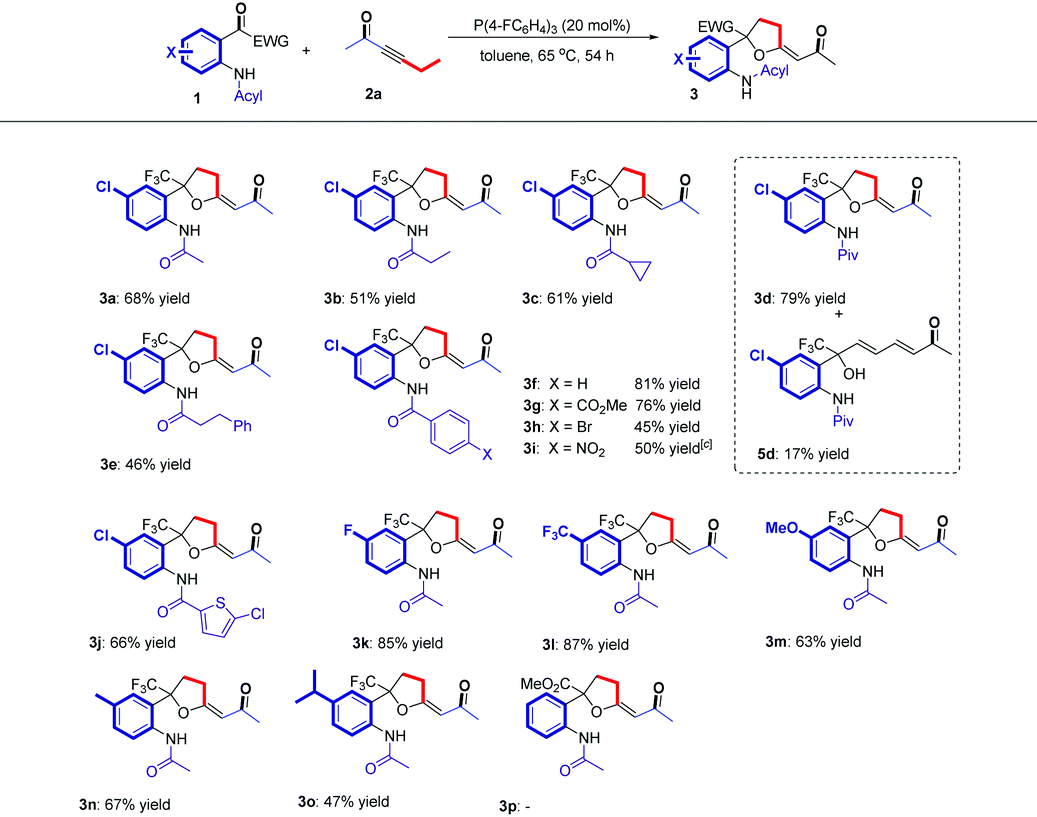
Having established the optimal reaction conditions, we next surveyed the substrate scope of the reaction by using a range of 2-(2,2,2-trifluoroacetyl)phenylamides 1 with different substituents (Table 2). First, we tested 2-(2,2,2-trifluoroacetyl)phenylamides 1 with different N-acyl groups. It was found that acetyl, propionyl, cyclopropanecarbonyl, pivaloyl and 3-phenylpropanoyl substituted 2-(2,2,2-trifluoroacetyl)phenylamides all afforded the desired β,δ-cycloaddition products 3a–3e in 46–79% yields. We isolated diene product 5d in 17% yield, which indicated that increasing the steric hindrance of the amide group would impede the cycloaddition process. Using the benzoyl substituted 2-(2,2,2-trifluoroacetyl)phenylamide 1f as the substrate also gave the corresponding product 3f in 81% yield. Changing the substituents (CO2Me, Br, NO2) on the benzene ring afforded the corresponding products 3g–3i in yields ranging from 45% to 76%, but the NO2 substituted product 3i was a mixture of cis- and trans-isomers (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E = 1:1). Substrate 1j bearing the 5-chlorothiophene-2-carbonyl group was also tolerated, affording the corresponding product 3j in 66% yield. Next, the substituents on the aniline ring were examined with substrates 1k–1o. Substrates 1k and 1l, containing electron-withdrawing group (F, CF3), afforded the corresponding products in higher yields (85% yield and 87% yield) than those of substrates 1m–1o, containing electron-donating groups such as OMe, Me and i-Pr (67% yield to 47% yield). These results indicate that an electron-withdrawing group on the aniline ring probably facilitates the addition of zwitterionic intermediate D′ to the substrate 1. However, using substrate 1p having a CO2Me moiety instead of the CF3 moiety failed to give the desired product 3p; probably, 1p was unstable at high temperature and the reaction only give a complex mixture.
:E = 1:1). Substrate 1j bearing the 5-chlorothiophene-2-carbonyl group was also tolerated, affording the corresponding product 3j in 66% yield. Next, the substituents on the aniline ring were examined with substrates 1k–1o. Substrates 1k and 1l, containing electron-withdrawing group (F, CF3), afforded the corresponding products in higher yields (85% yield and 87% yield) than those of substrates 1m–1o, containing electron-donating groups such as OMe, Me and i-Pr (67% yield to 47% yield). These results indicate that an electron-withdrawing group on the aniline ring probably facilitates the addition of zwitterionic intermediate D′ to the substrate 1. However, using substrate 1p having a CO2Me moiety instead of the CF3 moiety failed to give the desired product 3p; probably, 1p was unstable at high temperature and the reaction only give a complex mixture.
Subsequently, we examined the phosphine-catalyzed cycloadditions of substrates 1a–1p at 10 °C. As shown in Table 3, changing the acyl group on the aniline with propionyl and cyclopropanecarbonyl groups could improve the product yields, giving the desired products 4b in 96% yield and 4c in 91% yield, respectively. Increasing the steric hindrance of the carbonyl group or extending the carbon chain of the N-protecting group gave the desired products 4d and 4e in moderate yields. Unlike the reactions carried out at high temperature, the replacement of the aliphatic carbonyl group with a benzoyl group on substrate 1 did not give the corresponding product 4f; however, introducing substituents such as CO2Me, Br, or NO2 on the benzoyl group could afford the corresponding products 4g–4i in good to excellent yields ranging from 81% to 93%, presumably due to the influence of the electronic properties of the benzoyl group. Substrate 1j bearing the 5-chlorothiophene-2-carbonyl group afforded the corresponding product 4j in 85% yield. Furthermore, the substituents on the aniline ring were also examined at 10 °C. Substrates 1k and 1l, containing an electron-withdrawing group, such as F and CF3, gave the corresponding products 4k and 4l in moderate yields (69% yield and 73% yield); and substrates containing electron-donating groups, such as OMe, Me or i-Pr, impeded the reaction proceeding, producing the desired products 4m–4o in low yields. Similarly, using substrate 1p having a CO2Me moiety instead of a CF3 moiety could not give the desired product 4p; most of 1p was recovered, indicating the low reactivity of a substrate having an ester substituted carbonyl group.

The examination of substrate scope indicated that the amide group at the ortho position of the trifluoroacetyl group played a decisive role to affect the product selectivity. We hypothesized that varying the acidity of the NHAcyl moiety of substrate 1 probably affected the product regioselectivity. Thus, we synthesized a series of substrates 1q–1s containing different N-sulfonyl groups, which are more acidic than those substrates containing the N-acyl group,26 which were tested in these phosphine-catalyzed cycloaddition reactions. In addition, substrates 1t–1v containing the phenolic hydroxyl group were also investigated. We found that the α′,α-cycloaddition product 4q was the only isolated product in 89% yield at 10 °C or in 31% yield at 65 °C. Products 4r and 4s were obtained at 10 °C in 85% yield and 69% yield, respectively. Similarly, 4t was the only isolated product at 10 °C in 84% yield or at 65 °C in 51% yield, as in a preliminary exploration in our previous work.16 Products 4u and 4v were obtained at 10 °C in 85% yield and 73% yield, respectively. When the amide group was replaced by N-sulfonyl groups or the hydroxyl group, the obtained products could not be switched to β,δ-cycloaddition products 3 by increasing the reaction temperature and only complex reaction mixtures were obtained at 65 °C (Scheme 3a).
 | ||
| Scheme 3 Substrate containing substituents with different acidities. | ||
It was interesting that the reaction of 1t containing an OH moiety with 2a proceeded smoothly under the standard reaction conditions with a 4 Å molecular sieve to remove water, affording the α′,α-cycloaddition product 4t at 10 °C, which is contrary to earlier results that a trace of water is an indispensable co-catalyst for the reaction of 1a having an NHAc moiety (Scheme 3b); the preliminary calculation results show that the abstraction of α′-H is more favored over the abstraction of γ-H, which leads to the α′,α-cycloaddition product and may account for the experimental observation (for details, see ESI†). In addition, (E)-hexa-3,5-dien-2-one was detected as a byproduct simultaneously (Scheme 3b). These observations showed that varying the acidity of the NHAcyl moiety indeed affected the product regioselectivity, exclusively giving the corresponding α′,α-cycloaddition product without the assistance of water. Reducing the pKa of substrate 1via varying the NHAcyl moiety probably makes the α′,α-H shift process more favorable, leading to an α′,α-cycloaddition product without the assistance of water. On the other hand, substrates having a pyrrole or indole moiety, which are less acidic than those substrates containing an N-acyl group, were also tested. The β,δ-cycloaddition products 3w and 3x were obtained at 65 °C in 43% yield and 61% yield, respectively. When the reaction was carried out at 10 °C, only the δ-addition products 5w and 5x were isolated in 67% yield and 74% yield, respectively. The addition of 2-bromo-4-fluorophenol as an additive could not switch the product's regioselectivity, only generating the δ-addition products 5w in a 38% yield (Scheme 3c). Upon moving the trifluoroacetyl group from the C2 position of indole to the C3 position, the reaction only gave a complex mixture at 65 °C. Substrate 1z without the N-acyl group could not afford the cycloaddition product at 10 °C or at 65 °C, and only the δ addition product 5z was detected in 63% yield, as in our previous report. When 6-phenylhex-3-yn-2-one 2b was replaced to hex-3-yn-2-one 2a, steric hindrance probably influenced the reaction pathway, leading to the α′,α-cycloaddition product 4aa, obtained at 65 °C in 59% yield, and the α′ addition product 5aa accessed at 10 °C in 34% yield (Scheme 3d). A phenyl group substituted in the δ position prevented the δ addition occurring and benefited the addition at the α′ position.
Mechanistic studies
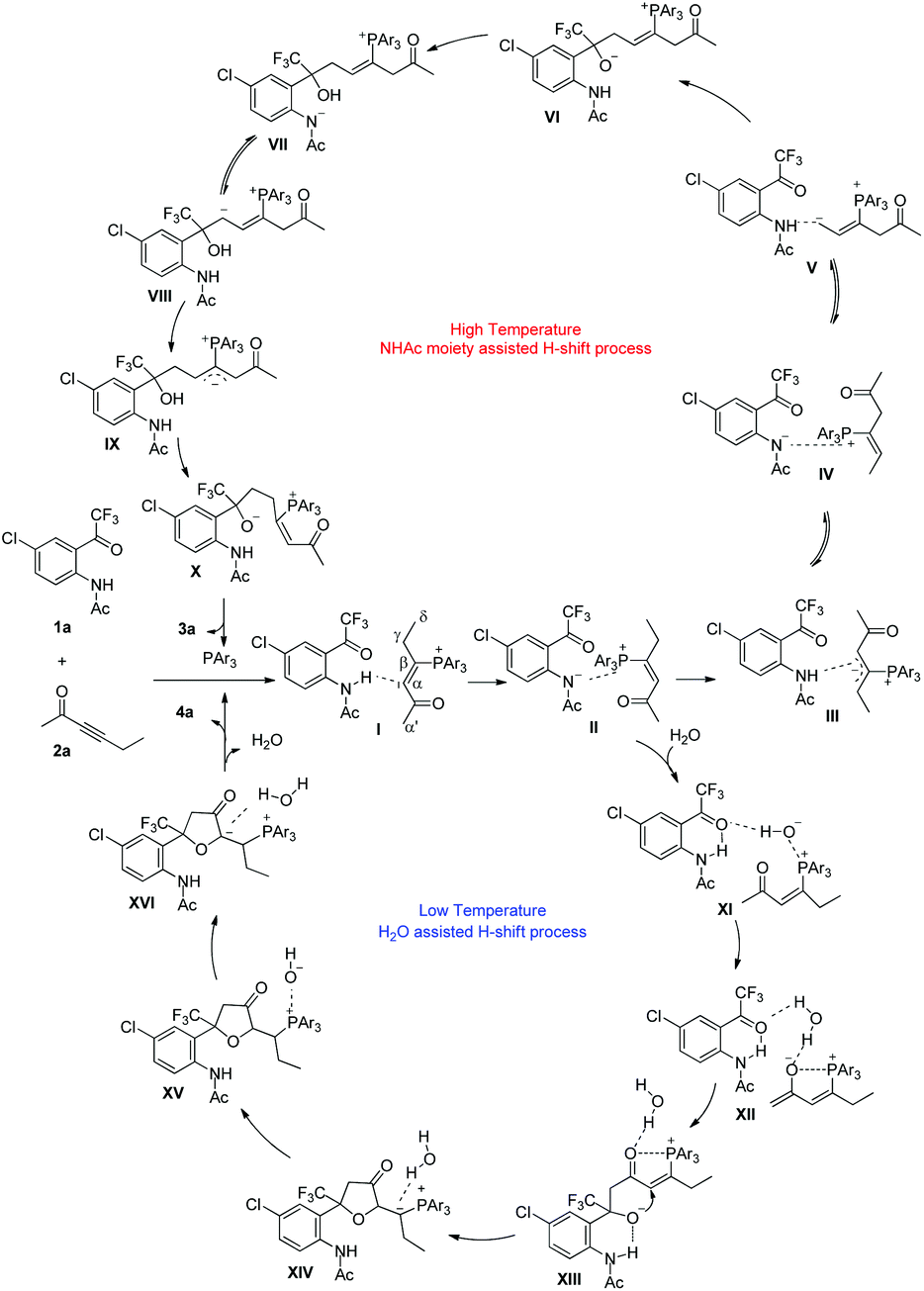 | ||
| Scheme 4 A plausible reaction mechanism. | ||
If the protic additive H2O is added to the reaction system, the nitrogen anion moiety in II probably first abstracts proton from H2O to give intermediate XI. Subsequently, the OH moiety in XI abstracts one α′-H to give a stable complex XII through hydrogen-bonding interaction. A subsequent addition takes place to give intermediate XIII, which undergoes cyclization to form intermediate XIV. With the assistance of water, 1,2-H shift processes proceed via intermediate XV to afford XVI, which is similar to Yu's studies on Lu's [3 + 2] cycloaddition reaction.27 Elimination of water and the tertiary phosphine generates the α′,α-cycloaddition product 4. Our proposed mechanism involves a series of H-shift processes, and thus deuterium-labeling experiments have been undertaken to understand and give evidences to the suggested reaction mechanism. DFT calculations were also carried out to rationalize the regioselectivity affected by the substrates, the additives and the reaction temperatures, and to understand the detailed reaction mechanism.
Deuterium-labeling experimental investigations
For the optimal conditions, trace water in the solvent was an indispensable co-catalyst to afford α′,α-cycloaddition products at 10 °C; in contrast, water was no longer necessary during the β,δ-cycloaddition process (Table 1). To gain some insights into the reaction mechanism, several deuterium-labeling experiments were performed at different temperatures. The reaction of 1a with 2a was first carried out in the presence of D2O (20 equiv.) at 65 °C under the standard reaction conditions. The reaction proceeded smoothly to give the corresponding partially deuterated addition product [D]-3a in 58% yield along with the deuterium incorporation at the C1–C6 positions (Scheme 5a). The addition of D2O did not improve the product yield significantly, indicating that water did not play a significant role in the reaction at high temperature; however, the reaction involving several H-shift processes led to the deuterium incorporation at D1–D6 positions. Under the standard reaction conditions with the addition of a 4 Å molecular sieve, using 80% N–H deuterated [D]-1a as the substrate produced the corresponding deuterated product [D]-3a′ in 61% yield with 18% and 20% D contents incorporated at the D1 and D5 positions, respectively (Scheme 5b). The deuterium incorporated at the D5 position was probably due to the deuterium being transferred from the NDAc moiety to the β-carbon, as in the suggested process from I to II shown in Scheme 4. The deuterium incorporated at the D1 position was in a syn position with respect to the CF3 substituent. This unexpected and stereospecific deuterium incorporation revealed that intramolecular proton exchange processes probably existed, as illustrated in Scheme 5c. The oxygen anion moiety of intermediate VI can abstract the proton in the D1 position to afford intermediate VIII, and then the deuterium of N–D is transferred to C1, generating intermediate VII; the nitrogen anion moiety can abstract the proton of the hydroxy group to give intermediate VI again (Scheme 5c). During the proton exchange cycle, the electrostatic interaction between phosphonium and the oxygen anion maintained the configuration, leading to stereospecific deuterium incorporation. Moreover, the acetyl group made the proton exchange reversible and stabilized the oxygen anion, which benefited the next cycloaddition step. Without the assistance of the acetyl group, zwitterion VI-3 probably prefers to undergo isomerization to give another intermediate VI-4, which subsequently produces zwitterion VI-5 through a classic proton transfer, finally affording the diene product 3′ (Scheme 5d). Using deuterated hex-3-yn-2-one [D]-2a with a 98% D content as the substrate produced the corresponding deuterated product [D]-3a′′ in 63% yield, along with very low deuterium content at the D1 and D5 positions, suggesting that the hydrogen atoms at the α′ position of 2a did not involve several proton transfer processes in the reaction at 65 °C (Scheme 5e). Using deuterated hex-3-yn-2-one [D]-2a′ with a 98% D content as the substrate, the reaction did not take place efficiently, and only gave trace amounts of the desired product (Scheme 5f). | ||
| Scheme 5 Deuterium-labeling experiment at 65 °C and result discussion. | ||
Next, the reaction of 1a with 2a was investigated in the presence of D2O (20 equiv.) at 10 °C under the standard reaction conditions, giving the corresponding partially deuterated addition product [D]-4a in 74% yield along with the deuterium incorporation only at the D1–D3 positions (Scheme 6a). The use of 1.0 equiv. of D2O significantly decreased the deuterium incorporation ratio at the D1–D3 positions (Scheme 6a). This result suggested that water assisted the proton transfer processes shown in Scheme 4 and the hydrogen atoms at the γ-position and δ-position of 2a did not involve the proton transfer processes. Under the standard reaction conditions, using 80% N–H deuterated [D]-1a as the substrate produced the corresponding deuterated product [D]-4a′′ in 71% yield, in which the deuterium was only transferred from the N atom to the D2 position with a 35% D content (Scheme 6b). As proposed in Scheme 4, the deuterium is transferred from the NDAc moiety to the β-carbon and then, through a water-assisted 1,2-H shift, arrives at the D2 position; the deuterium content is lost, probably due to the water involved in these proton transfer processes. Under the standard reaction conditions, using deuterated hex-3-yn-2-one [D]-2a with a 98% D content as the substrate produced the corresponding deuterated product [D]-4a′′′ in 72% yield, in which the deuterium incorporation at C1 totally decreased, probably due to the exchange of H2O with the zwitterionic intermediates in solvent (Scheme 6c).
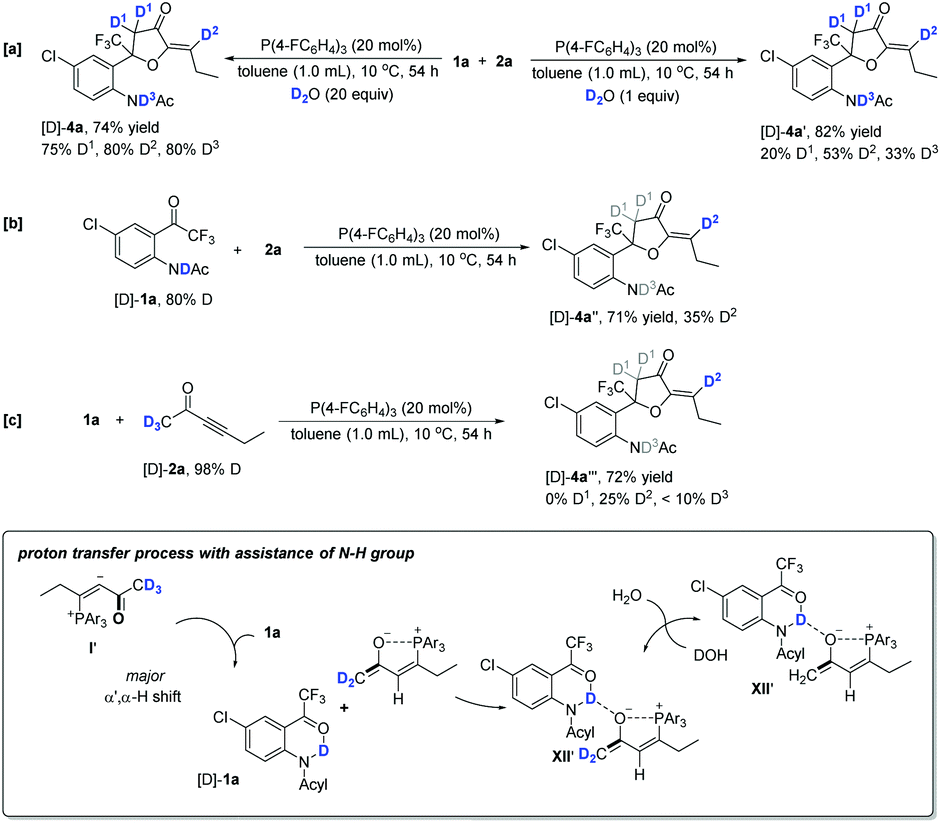 | ||
| Scheme 6 Deuterium-labeling experiment at 10 °C and result discussion. | ||
As aforementioned, the reaction of 1t containing an OH moiety with 2a conducted smoothly without a trace of water. Thus, the deuterium-labeling experiments of 1t with 2a were also carried out in the absence of water. Under the standard reaction conditions, using deuterated hex-3-yn-2-one [D]-2a (2.0 equiv.) with a 98% D content as the substrate produced the corresponding deuterated product [D]-4t in 89% yield, in which the deuterium incorporation at D1 was totally lost (Scheme 7a). However, reducing the amount of [D]-2a to 1.0 equiv. provided [D]-4t′ in 78% yield with a 30% D content at D1 (Scheme 7b). Considering the obtained byproduct (E)-hexa-3,5-dien-2-one, shown in Scheme 3b, we deduced that the deuterium at D1 probably exchanged with the hydrogen atoms in other zwitterionic intermediates if the concentration of 2a was high. Without substrate 1t, the [D]-2a itself could not afford the (E)-hexa-3,5-dien-2-one under the standard conditions, indicating that the substrate 1t assisted the generation of (E)-hexa-3,5-dien-2-one (Scheme 7c). Based on crude 31P NMR spectrum analysis (see ESI† for the details) and the proposed mechanism, we considered that the OH group of substrate 1t also assists the α′,α-H shift process via intermediates II′ and XII′ to afford product 4t; however, these steps, involving proton transfer processes, are reversible, which allows intermediate I′ to be converted into byproduct (E)-hexa-3,5-dien-2-one. Interestingly, using 50% O–H deuterated [D]-1t as the substrate, the product was obtained without any deuterium incorporation. The deuterium of the OH group was probably easy to lose under this reaction condition, which failed to transfer to the product (Scheme 7d).
 | ||
| Scheme 7 Deuterium-labeling experiment using 1t at 10 °C and result discussion. | ||
 | ||
| Scheme 8 Solvation Gibbs free energy profiles for the generation of key zwitterionic intermediates and the formation of product 3a. | ||
We next investigated the suggested reaction pathway for the PPh3 catalyzed reaction of 1a and 2a to produce 4a, and the solvation Gibbs free energy profiles in toluene are shown in Scheme 9. Since a trace amount of H2O was necessary for the reaction at 10 °C, we speculated that the H2O plays a role in assisting the H-shift processes. A similar H2O assisted [1,2] proton shift process in Lu's [3 + 2] cycloaddition reaction has been investigated theoretically by Yu's group.27a In this case, a contact ion pair INT3 is also generated at the initial stage; however, the nitrogen anion in INT3 does not directly abstract γ-H or abstract α′-H in the presence of water. The addition of H2O leads to the formation of a stable complex INT13. Subsequently, the nitrogen anion moiety in INT13 abstracts a proton from H2O to give complex INT14via transition state TS11, which only requires 3.3 kcal mol−1. The hydroxyl anion in INT14 is highly attracted by the PPh3 moiety, which can abstract α′-H to give complex INT15 or γ-H abstract to give complex INT15-1. In this case, the abstraction of α′-H by the hydroxyl anion with an activation free energy of 17.3 kcal mol−1via transition state TS12 gives complex INT15; however, it requires 28.0 kcal mol−1 to overcome transition state TS12-1 to form complex INT15-1. Thus, the abstraction of α′-H is more favorable, indicating that the H2O assisted α′,α-H shift process is preferred in the presence of water, which is probably due to the stronger basicity of the hydroxyl anion than the nitrogen anion. In addition, complex INT15 is probably stabilized by hydrogen bonding between water, substrate 1a and the zwitterion, which probably makes the following addition step proceed easily. Passing through transition state TS13 with an activation free energy of 9.1 kcal mol−1, INT15 is converted into INT16, which is exergonic by 10.7 kcal mol−1. Undergoing cyclization, the intermediate INT17 is generated via transition state TS14 with an energy barrier of 6.7 kcal mol−1. The [1,2] proton transfer from INT17 to INT19 also requires water's assistance; the energy barrier without water's assistance is 47.6 kcal mol−1 (for the whole reaction pathway without water, see ESI†). The proton transfer from water to the carbon atom connected with the phosphorus atom via transition state TS15 requires an activation free energy of 20.9 kcal mol−1, generating intermediate INT18. Similarly, the abstraction of a proton by the hydroxyl anion with an activation free energy of 16.6 kcal mol−1via transition state TS16 gives rise to another complex, INT19. Passing through transition state TS17, the elimination of PPh3 produces the product complex INT20. Cleavage of complex INT20 to yield the separate components 4a, H2O and PPh3 is exothermic by 17.9 kcal mol−1. In this case, the highest activation free energy is only 20 kcal mol−1 or so, and the reaction can proceed at low reaction temperature. Our calculation results agree with the results in deuterium-labeling experiments, which found deuterium incorporation ratios at D1–D3 position (see Scheme 6a). However, the thermodynamically controlled β,δ-cycloaddition product is still preferred at higher temperatures under the water-assisted pathway. Thus, with or without water-assistance, the β,δ-cycloaddition product is always obtained at a higher temperature, which agrees with the experimental results.
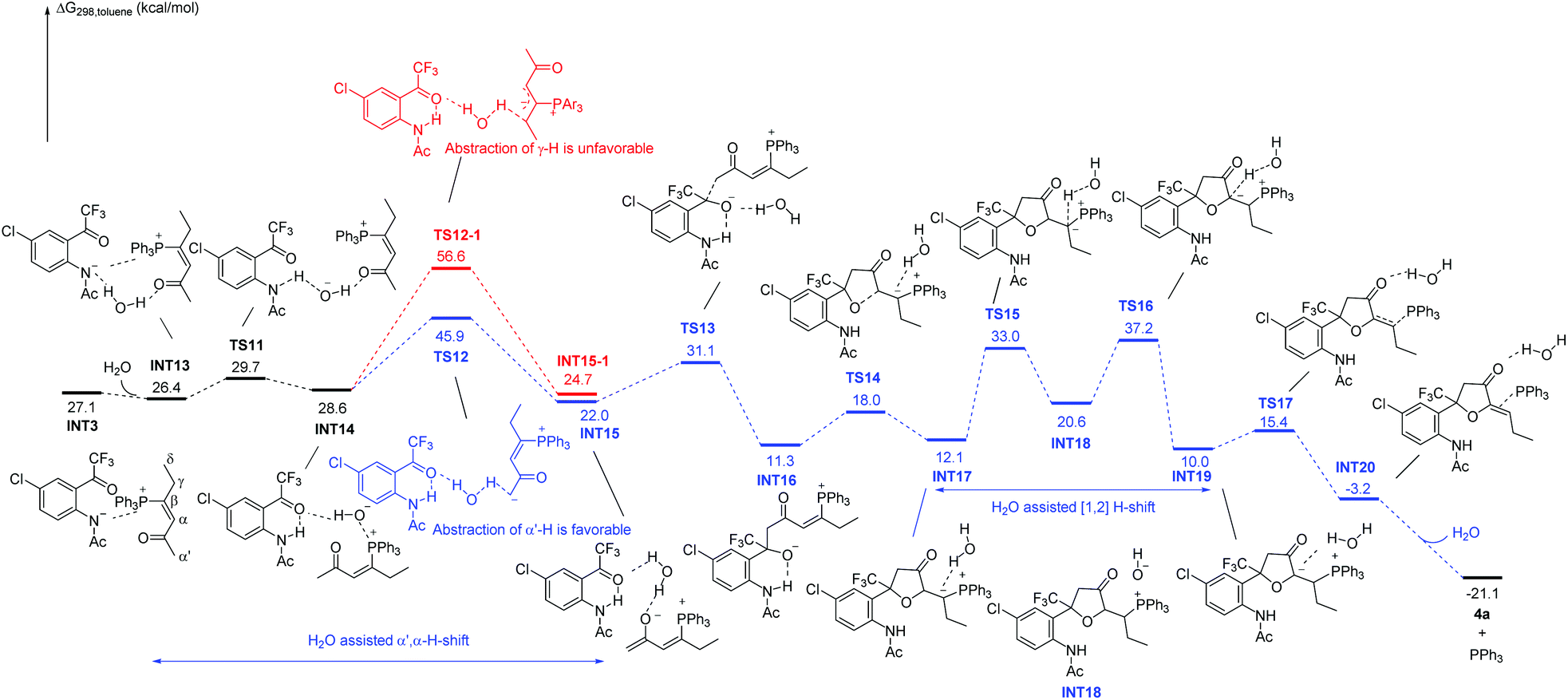 | ||
| Scheme 9 Solvation Gibbs free energy profiles for the generation of key zwitterionic intermediates and the formation of product 4a. | ||
Conclusions
We have disclosed the regiodivergent phosphine-catalyzed [3 + 2] cycloaddition of 2-(2,2,2-trifluoroacetyl)phenylamides and alkynones, affording a series of β,δ-cycloaddition products 3 or α′,α-cycloaddition products 4 in moderate to good yields. The experimental results demonstrated that product regioselectivity could be switched through adjusting the acidities of substrates via varying the NHAcyl moiety in substrate 1, changing the reaction temperature and the addition of the protic additive H2O. Based on deuterium labeling experiments and systematic theoretical studies, we have disclosed a detailed reaction mechanism for the phosphine-catalyzed cycloaddition reactions of alkynones with trifluoroacetyl phenylamides. The calculation results ruled out the intramolecular proton transfer processes frequently proposed in previous reports; our theoretical studies also provide reasonable explanations for why the regioselectivities of products can be controlled by the protic additive H2O and the reaction temperature. In the absence of water, the NHAc moiety of substrate 1 plays an important role in assisting the proton transfer process, leading to the key zwitterionic intermediate which directly forms the β,δ-cycloaddition products 3; the high reaction temperature is required to overcome the high activation energy barriers for some proton transfer processes. For the addition of protic additive H2O, the important proton transfer processes are assisted by H2O, affording the key zwitterionic intermediate which directly generates α′,α-cycloaddition products 4; in this case, the high reaction temperature is not necessary. Further applications of the phosphine-catalyzed cycloaddition reactions are underway in our lab.Computational methods
All DFT calculations were performed with the Gaussian 09 program.28 The geometries of all the minima and transition states have been optimized at the M06-2X/6-31G(d) level of theory.29 The subsequent frequency calculations on the stationary points were carried out at the same level of theory to ascertain the nature of the stationary points as minima or first-order saddle points on the respective potential energy surfaces. All the transition states were characterized by one and only one imaginary frequency pertaining to the desired reaction coordinate. The intrinsic reaction coordinate (IRC) calculations were carried out at the same level of theory to further authenticate the transition states. The conformational space of the flexible systems was first searched manually. Thermochemical corrections to 298.15 K were calculated for all minima from unscaled vibrational frequencies obtained at this same level. The solvent effect was estimated by the IEFPCM method with radii and nonelectrostatic terms for the SMD solvation model30 in toluene (ε = 2.3741). Solution-phase single point energy calculations were performed at the M06-2X/6-311+G(d,p) level based on the gas phase optimized structures.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank financial support from the National Basic Research Program of China (973)-2015CB856603, the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB20000000, and the National Natural Science Foundation of China (21372250, 20472096, 21372241, 21361140350, 20672127, 21421091, 21121062, 21302203, 20732008, 21572052, 21772226 and 21772037), and the Fundamental Research Funds for the Central Universities 222201717003. We are grateful for the facility support from the Shanghai Supercomputer Center.References
- (a) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC; (b) A. Marinetti and A. Voituriez, Synlett, 2010, 2010, 174 CrossRef; (c) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (d) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC; (e) C. Gomez, J.-F. Betzer, A. Voituriez and A. Marinetti, ChemCatChem, 2013, 5, 1055 CrossRef CAS; (f) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (g) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (h) Y. Wei and M. Shi, Chem. – Asian J., 2014, 9, 2720 CrossRef CAS PubMed; (i) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657 RSC; (j) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed.
- (a) M. Yang, T. Wang, S. Cao and Z. He, Chem. Commun., 2014, 50, 13506 RSC; (b) X.-N. Zhang, G.-Q. Chen, X.-Y. Tang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 10768 CrossRef CAS PubMed; (c) J. Chen, J. Li, J. Wang, H. Li, W. Wang and Y. Guo, Org. Lett., 2015, 17, 2214 CrossRef CAS PubMed; (d) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621 CrossRef CAS PubMed; (e) Y. Li, X. Su, W. Zhou, W. Li and J. Zhang, Chem. – Eur. J., 2015, 21, 4224 CrossRef CAS PubMed; (f) X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874 CrossRef CAS PubMed.
- (a) V. R. Gandi and Y. Lu, Chem. Commun., 2015, 51, 16188 RSC; (b) Y. Gu, P. Hu, C. Ni and X. Tong, J. Am. Chem. Soc., 2015, 137, 6400 CrossRef CAS PubMed; (c) S. Y. Lee, Y. Fujiwara, A. Nishiguchi, M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 4587 CrossRef CAS PubMed; (d) X. Han, W.-L. Chan, W. Yao, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2016, 55, 6492 CrossRef CAS PubMed; (e) E. Li, H. Jin, P. Jia, X. Dong and Y. Huang, Angew. Chem., Int. Ed., 2016, 55, 11591 CrossRef CAS PubMed; (f) H. Liu, Y. Liu, C. Yuan, G.-P. Wang, S.-F. Zhu, Y. Wu, B. Wang, Z. Sun, Y. Xiao, Q.-L. Zhou and H. Guo, Org. Lett., 2016, 18, 1302 CrossRef CAS PubMed.
- (a) C. T. Mbofana and S. J. Miller, ACS Catal., 2014, 4, 3671 CrossRef CAS; (b) D. T. Ziegler and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 12069 CrossRef CAS PubMed.
- (a) F. Zhong, G.-Y. Chen, X. Han, W. Yao and Y. Lu, Org. Lett., 2012, 14, 3764 CrossRef CAS PubMed; (b) L. Zhang, H. Liu, G. Qiao, Z. Hou, Y. Liu, Y. Xiao and H. Guo, J. Am. Chem. Soc., 2015, 137, 4316 CrossRef CAS PubMed.
- H. Murayama, K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2016, 18, 1706 CrossRef CAS PubMed.
- D. Lecercle, M. Sawicki and F. Taran, Org. Lett., 2006, 8, 4283 CrossRef CAS PubMed.
- (a) M. Sampath and T.-P. Loh, Chem. Sci., 2010, 1, 739 RSC; (b) C.-Z. Zhu, Y.-L. Sun, Y. Wei and M. Shi, Adv. Synth. Catal., 2017, 359, 1263–1270 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed.
- (a) I. Junji, B. Yoshiyasu and H. Takeshi, Chem. Lett., 1993, 22, 241 CrossRef; (b) R. B. Grossman, S. Comesse, R. M. Rasne, K. Hattori and M. N. Delong, J. Org. Chem., 2003, 68, 871 CrossRef CAS PubMed.
- (a) B. M. Trost and G. R. Dake, J. Am. Chem. Soc., 1997, 119, 7595 CrossRef CAS; (b) L. Zhu, Y. Xiong and C. Li, J. Org. Chem., 2015, 80, 628 CrossRef CAS PubMed.
- (a) C. Lu and X. Lu, Org. Lett., 2002, 4, 4677 CrossRef CAS PubMed; (b) V. Sriramurthy, G. A. Barcan and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12928 CrossRef CAS PubMed; (c) V. Sriramurthy and O. Kwon, Org. Lett., 2010, 12, 1084 CrossRef CAS PubMed.
- (a) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 3167 CrossRef CAS; (b) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 10819 CrossRef CAS.
- (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS; (b) J.-C. Wang and M. J. Krische, Angew. Chem., Int. Ed., 2003, 42, 5855 CrossRef CAS PubMed; (c) J.-C. Wang, S.-S. Ng and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 3682 CrossRef CAS PubMed.
- (a) H. Kuroda, I. Tomita and T. Endo, Org. Lett., 2003, 5, 129 CrossRef CAS PubMed; (b) J. E. Wilson, J. Sun and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 161 CrossRef CAS PubMed; (c) Z. Lian and M. Shi, Eur. J. Org. Chem., 2012, 2012, 581 CrossRef CAS; (d) Z. Lian and M. Shi, Org. Biomol. Chem., 2012, 10, 8048 RSC; (e) L. Yang, P. Xie, E. Li, X. Li, Y. Huang and R. Chen, Org. Biomol. Chem., 2012, 10, 7628 RSC; (f) L. Liang, E. Li, P. Xie and Y. Huang, Chem. – Asian J., 2014, 9, 1270 CrossRef CAS PubMed; (g) L. Liang and Y. Huang, Org. Lett., 2016, 18, 2604 CrossRef CAS PubMed.
- Y.-L. Sun, X.-N. Zhang, Y. Wei and M. Shi, ChemCatChem, 2016, 8, 3112 CrossRef CAS.
- W. Yao, X. Dou, S. Wen, J. E. Wu, J. J. Vittal and Y. Lu, Nat. Commun., 2016, 7, 13024 CrossRef PubMed.
- (a) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed; (b) D. Wang, G.-P. Wang, Y.-L. Sun, S.-F. Zhu, Y. Wei, Q.-L. Zhou and M. Shi, Chem. Sci., 2015, 6, 7319 RSC; (c) T. Wang, Z. Yu, D. L. Hoon, K.-W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912 RSC; (d) T. Wang, Z. Yu, D. L. Hoon, C. Y. Phee, Y. Lan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 265 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- (a) P. García-García, F. Lay, P. García-García, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2009, 48, 4363 CrossRef PubMed; (b) L. Ratjen, P. García-García, F. Lay, M. E. Beck and B. List, Angew. Chem., Int. Ed., 2011, 50, 754 CrossRef CAS PubMed; (c) Q. Wang, M. Leutzsch, M. van Gemmeren and B. List, J. Am. Chem. Soc., 2013, 135, 15334 CrossRef CAS PubMed.
- K. Ishihara, M. Fushimi and M. Akakura, Acc. Chem. Res., 2007, 40, 1049 CrossRef CAS PubMed.
- (a) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS PubMed; (b) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef CAS PubMed; (c) Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed.
- (a) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2015, 137, 4134 CrossRef CAS PubMed; (b) Y. Wang, P.-J. Cai and Z.-X. Yu, J. Org. Chem., 2017, 82, 4604 CrossRef CAS PubMed; (c) Z.-X. Yu and Y. Wang, Org. Biomol. Chem., 2017 10.1039/c7ob01628j.
- (a) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (b) M. Schlosser, Angew. Chem., Int. Ed., 2006, 45, 5432 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545 CrossRef CAS PubMed.
- (a) B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS PubMed; (b) L. C. Miller and R. Sarpong, Chem. Soc. Rev., 2011, 40, 4550 RSC; (c) N. Funken, Y.-Q. Zhang and A. Gansaeuer, Chem. – Eur. J., 2017, 23, 19 CrossRef CAS PubMed.
- In general, sulfoamides, phenols have smaller pKa values than amides. See: Bordwell pKa values on http://www.chem.wisc.edu/areas/reich/pkatable/index.htm and references therein.
- (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (b) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem. – Eur. J., 2008, 14, 4361 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed. See ESI† for full citation.
- (a) W. J. Hehre, R. Ditchfield and J. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 1500323, 1521105 and 1451585. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00512a |
| This journal is © the Partner Organisations 2017 |