
Functional nanostructures for enzyme based biosensors: properties, fabrication and applications
Ali
Othman
,
Anahita
Karimi
and
Silvana
Andreescu
*
Department of Chemistry and Biomolecular Science, Clarkson University, Potsdam, New York 13699-5810, USA. E-mail: eandrees@clarkson.edu
First published on 22nd September 2016
Abstract
With the increased use of nanobiotechnology-enabled solutions for diagnostics, therapeutics, bioelectronics, environmental, and energy-related applications, there exists a great demand for developing manufacturing processes that can reliably and reproducibly generate functional nanostructures with bioactive properties at low cost and in large quantities for implementation in practical devices. Interest in the development of field portable monitoring devices has increased throughout the past decade. Herein we describe fabrication, characterization and performance of portable and printable enzyme biosensors based on functional enzyme–nanoparticle conjugates. We review specific physicochemical and surface properties of nanoparticles used as carriers and sensing components for the design of portable biosensors and describe the role of these parameters in enhancing the performance, stability and field operability of these devices. Assembly of enzyme–nanoparticle conjugates is also discussed with an overview of current and emerging techniques enabling large scale roll-to-roll fabrication and miniaturization, including screen-printing, inkjet and 3D printing methods and their integration in flexible, wearable and inexpensive point-of-use devices. Current status and implementation challenges are provided with examples of applications in the clinical, environmental, public safety and food monitoring fields.
1. Introduction
There is a growing demand for the development of inexpensive measurement technology that can provide field monitoring and real time data for home diagnostics, point-of-care testing, food and environmental monitoring. Enzyme-based bioanalytical systems and devices continue to be of interest due to their high selectivity, potential for reusability, ease of preparation and broad range of applications. Development of such devices requires methods enabling large scale fabrication and assembly of biofunctionalized surfaces that have appropriate detection sensitivity and selectivity. Efforts during the past decade that combine the selective biorecognition capabilities of enzymes with the opto-electronic, catalytic and mechanical properties of nanomaterials enabled the rapid development of a broad array of diagnostic devices1,2 with significantly enhanced sensitivity and increased shelf life.3 In addition to the conventional enzyme based bioacatalytic systems, the development of wearable and flexible devices,4 driven by an increased market demand, has seen tremendous advancement in recent years. These devices show high potential for applications in personalized medicine, computerized and connected bioelectronic devices, and smart and responsive surfaces. Current efforts in the fabrication of these devices focus on miniaturization, large scale roll-to-roll manufacturing and the rational design of material systems and interfaces to improve field detection capabilities, maintain enzyme activity and enhance sensitivity and stability for measurements in ‘real world’ environments. A key requirement in the production of enzyme-based nanostructures for portable low cost sensing is to achieve uniform deposition of both the enzyme and nanostructure elements to ensure the required detection sensitivity and preserve the biological activity of the enzyme.Advanced manufacturing methods can make it possible to economically fabricate enzyme based devices on low cost surfaces such as paper, plastic, thread or metal, and generate large numbers of identically fabricated devices. Controlling the assembly and conserving the catalytic properties of the enzyme during manufacturing are critical parameters for achieving reproducible fabrication and maintaining biorecognition properties. Meeting these challenges is key to creating commercially viable devices with the required detection capabilities. Fig. 1 illustrates a general strategy for designing portable enzyme based biosensors. The use of nano-manufacturing techniques, microarray printing, nanolithography, and 2D and 3D printing facilitates miniaturization, mass production and multiplexing, extending their capabilities for real-time and point-of-care testing.2,5
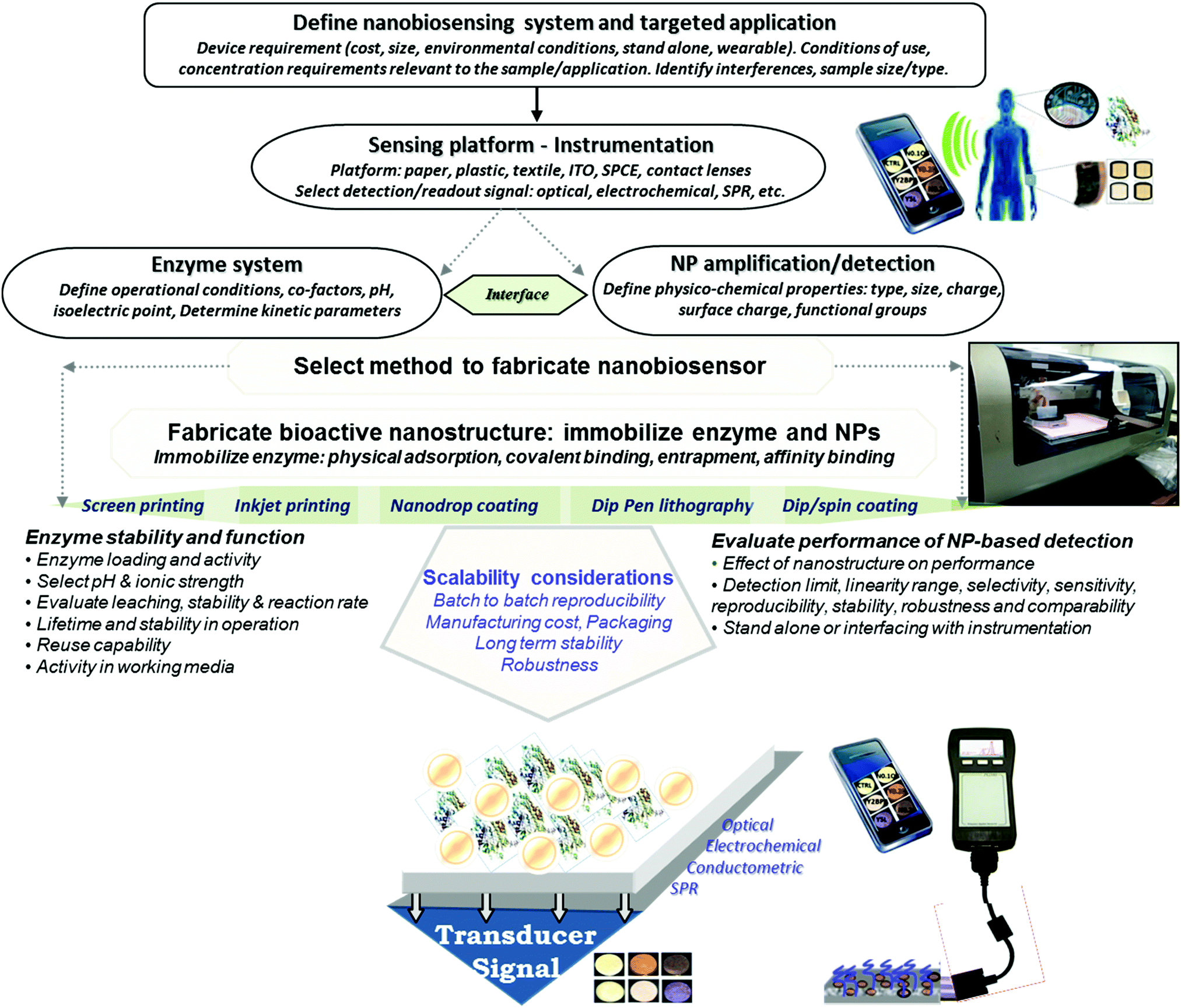 | ||
| Fig. 1 General strategy for designing portable enzyme based nanobiosensors. | ||
The increased performance has created opportunities for the development of inexpensive, portable and easy-to-use devices with on-site detection capabilities, as alternatives to conventional instrumentation for field analysis.7,16 These devices can meet the criteria set by The World Health Organization (WHO) of being Affordable, Sensitive, Specific, User-friendly, Rapid and Robust, Equipment free and Deliverable to end-users, termed ‘ASSURED’6 to improve health care, the environment and the safety of food, and be used in resource-limited communities and remote regions. Their applications include point-of-care medical diagnosis, environmental monitoring and food quality control.7–10 Such devices can be miniaturized, are disposable and can be used for on-site analysis, as wearable devices, home diagnostic tools and portable screening units.7,16 Recently, these devices have been developed on paper, thread or plastic7,16 and have been integrated with easy to use colorimetric11,12 and electrochemical13–15 signal transduction systems.
This review provides an overview of the recent advancements in the use, properties and assembly of nanostructured materials for the production of functional enzyme–NP conjugates and their integration into portable biosensors. Selection criteria for various types of nanoparticles (NPs) and their assembly in functional portable devices are discussed with examples of applications in the environment, clinical, public safety and food monitoring fields. Special emphasis is placed on the large scale, roll-to-roll fabrication of such devices for achieving field measurement capabilities. The first part of the review describes the main classes of NPs, the methods of assembly and their recognition and sensing properties that make them suitable for the design of portable enzyme-based assays. The second part provides examples of nanostructured enzyme based platforms fabricated on low cost substrates and highlights their applications. The third part discusses recent trends towards miniaturization, multiplexing, automatization and methods for large scale fabrication including: lab-on-a-chip devices, microfluidic based arrays, inkjet and 3D based enzyme printing, and wearable enzyme sensors, with examples of applications. Challenges for implementation in practical applications and future trends are also discussed.
2. Enzyme–NP conjugates: properties, bio-immobilization and assembly
The unique physicochemical properties of NPs (i.e., catalysis, surface plasmon, magnetism, luminescence, conductivity, etc.) associated with their small size and tailored surface chemistry used in conjunction with the high selectivity of enzymes can generate powerful enzyme–NP conjugate systems that can serve as main components in portable biosensing devices. In a biosensing system, the NPs can be used for multiple purposes; e.g. to increase sensitivity, stabilize the enzyme, increase surface area, enhance conductivity or provide a spectroscopic read-out in optical sensing schemes (e.g. colorimetric, fluorescent). A large diversity of NPs of characteristic sizes, surface properties and chemical composition with electronic, optical or catalytic functions are now available for sensing applications.17 The selection of the NP system and parameters such as pH, temperature, viscosity, and salt concentrations is critical for maintaining enzyme function.18–20 The NP chemistry, the structure, surface charge, and composition are critical for the binding, biological activity, orientation, structure and functionality of enzymes.21,22 The configuration, orientation, density and activity of the immobilized enzyme as well as the NP characteristics such as size and surface chemistry all have an impact on the sensing performance. These characteristics can be tailored through different strategies such as covalent (e.g. through glutaraldehyde or carbodiimide chemistry) or affinity (e.g. thiol) attachment or enzyme entrapment within composite materials like polyelectrolytes or conducting polymers.23–25 The main challenge is maintaining stability, activity, and function of the enzyme as close as possible to its native state.3 The use of immobilized enzymes is preferred due to their ease of handling, prolonged availability and robustness, increased resistance to environmental changes and reusability.26 Ensuring enzyme stability during manufacturing and use is essential for the development of practical biosensors. Enzymes immobilized within polymeric composite layers can undergo conformational changes of the native protein27 and can have slow diffusion rates of the substrate.28 Enzymes immobilized on NPs, nanostructured materials and NP-based composites have demonstrated increased stability over free enzymes23 and improved catalytic properties as compared to micro-size particles.29 NPs can be used as enzyme carriers30 to increase enzyme loading and stability,31,32 to electrically connect enzymes to electrode surfaces33 or impart detection functions (spectral, imaging, electrical readout). Developing biofunctional enzyme–NP assemblies offers broad opportunities for biosensing,34,35 biocatalysis,36 tissue engineering,37 disease diagnosis,37 biofuel cells,38 and drug delivery applications.39Fig. 2 shows a summary of various NP systems with an overview on their properties and applications in enzyme based biosensors. In the following section, we discuss the factors influencing enzyme–NP interactions with a summary of the enzyme immobilization principles and the main structural considerations affecting the assembly of NP–enzyme conjugates.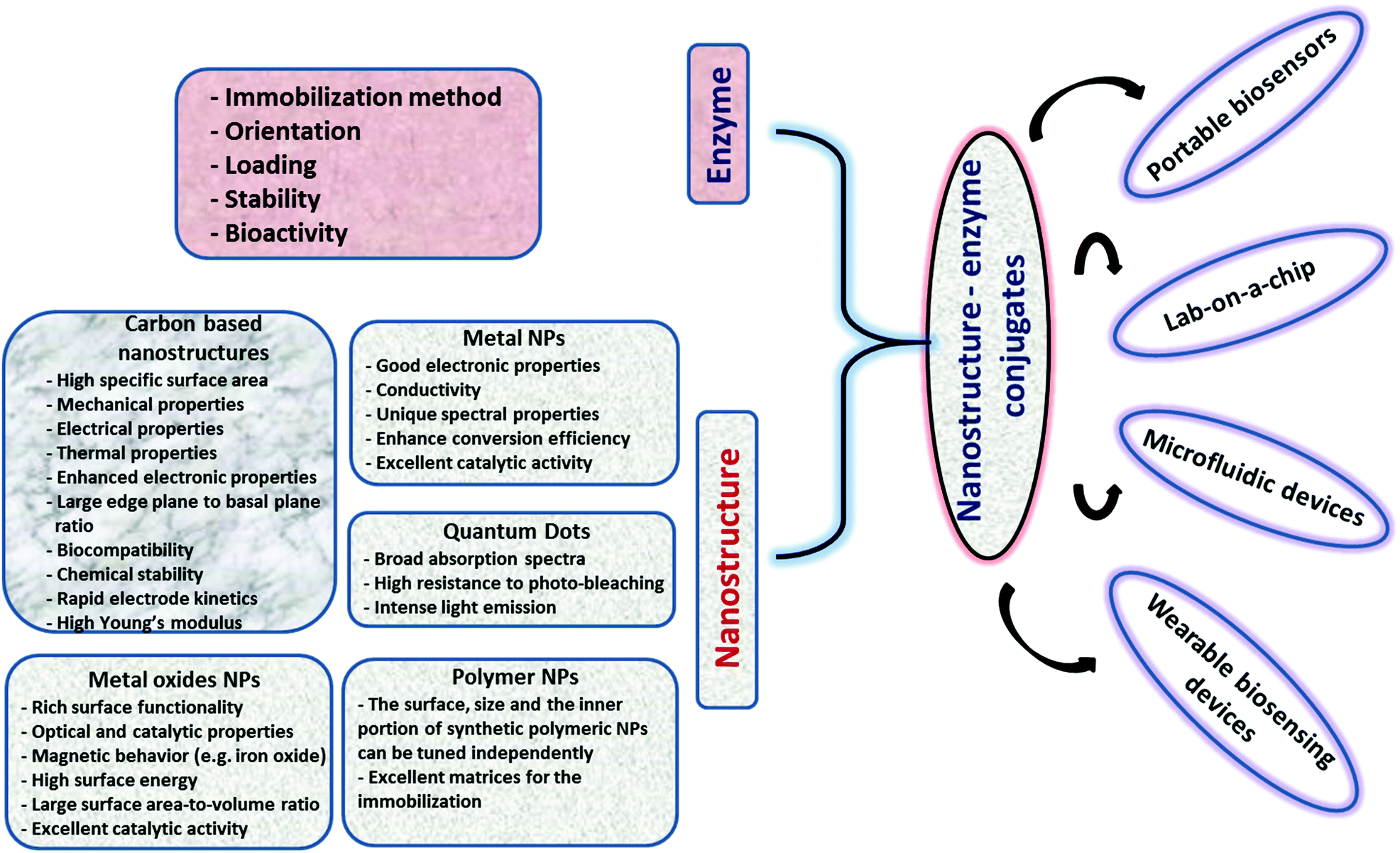 | ||
| Fig. 2 Overview of various nanostructures, their properties, and examples of applications in enzyme based biosensors. | ||
2.1 Factors influencing enzyme–NP interactions
The surface characteristics of NPs dictate the interaction and performance of NP–enzyme conjugates. The physicochemical properties of the NP system can affect enzyme activity and binding characteristics.40 The properties and biocompatibility of the additives/surface coatings that were used to stabilize the NPs should also be taken into account. Most NPs used in biosensors contain a surface coating to increase stability and dispersibility. These coatings facilitate the immobilization of enzymes via conjugation with the functional groups of the surface ligands. Examples include organic ligands, polymeric stabilizers, inorganic metals or metal oxide surfaces. Both covalent and non-covalent interactions (i.e., electrostatic interaction, van der Waals forces, and π–π stacking) can be used to stabilize enzymes. The surface coating can affect enzyme activity in various ways. For example, Fischer et al.41 showed that a carboxylic NP coating can inhibit the activity of α-chymotrypsin. On the other hand, a poly(ethylene glycol) (PEG) coating improved its biocompatibility.42 Other studies reported superior characteristics for enzyme immobilization of NPs functionalized with chitosan43 and poly(aminomethylenephosphonic) acid.44The size of NPs is also a key factor in modulating protein structure and function. In a study of lysozyme adsorption on silica NPs of various diameters (4, 20 and 100 nm)45 it was found that the particle size influences the structure and activity of the adsorbed lysozyme. A study reporting protein binding on different metal oxides (TiO2, SiO2, and ZnO) of different sizes46 showed that despite the fact that these NPs have a similar surface charge (i.e., zeta potential) in buffer, they bound different plasma proteins.47 Aggregation of NPs in water was observed with all NPs. The shape of TiO2 was also found to play an important role in protein binding. In general, it was suggested that protein–NP interactions are complex and that each system should be examined on a case-by-case basis.40Fig. 3 shows the binding behavior of different proteins to various sizes of poly(acrylic acid)/Au NPs (PAAGNPs). The binding of the protein to the NPs can result in changes in protein conformation and protein unfolding, a process that is dependent on the size, surface area, charge, and concentration of the NPs.48 Studies showed that subtle changes in NP size can influence protein binding both with the surface of the NP and within the protein corona.49
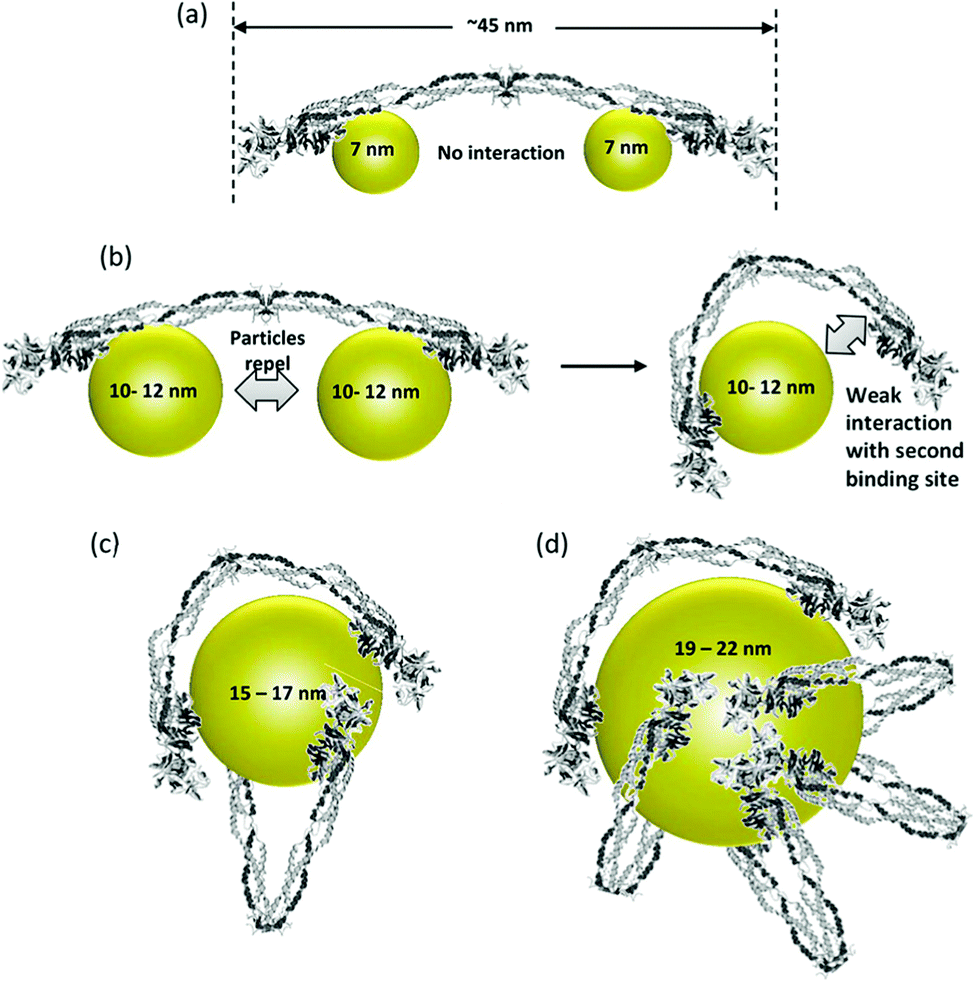 | ||
| Fig. 3 (a) Binding of small (7 nm) PAA–GNPs to fibrinogen showing two NPs bound to each protein molecule. (b) Larger size (10–12 nm) NPs inhibit the binding of two particles to each fibrinogen. As the diameter of the NP increases, the second binding site can come into contact with the NP due to flexibility at the E-binding domain of the protein. (c and d) Larger NPs can accommodate multiple fibrinogen molecules due to the larger surface area. Figure adapted with permission from ref. 49. | ||
2.2. Assembly of enzyme–NP conjugates
The immobilization of the enzyme and the assembly of enzyme–NP conjugates are critical steps in the fabrication of enzyme-based biosensors. Fig. 4 summarizes enzyme immobilization principles and the main structural considerations affecting the assembly of NP–enzyme conjugates. Enzyme stabilization is driven by parameters like enzyme orientation, enzyme density (loading), NP surface, morphology (shape) and size. This interaction is complex and the fundamental parameters affecting enzyme stability and function and the interfacial forces at the molecular level are not completely understood. Some studies indicate the formation of surface adsorbed NP–enzyme complexes.50,51 The specific characteristics of enzyme–NP conjugates influence enzyme activity and device performance.52–54 To facilitate assembly and design compact biosensors, the conjugates can be deposited in conjunction with other materials such as conducting polymers, biopolymers or silica sol gel materials. A comprehensive review describing the interplay between enzyme and NPs and the analysis of nanomaterial bioconjugates can be found in ref. 55.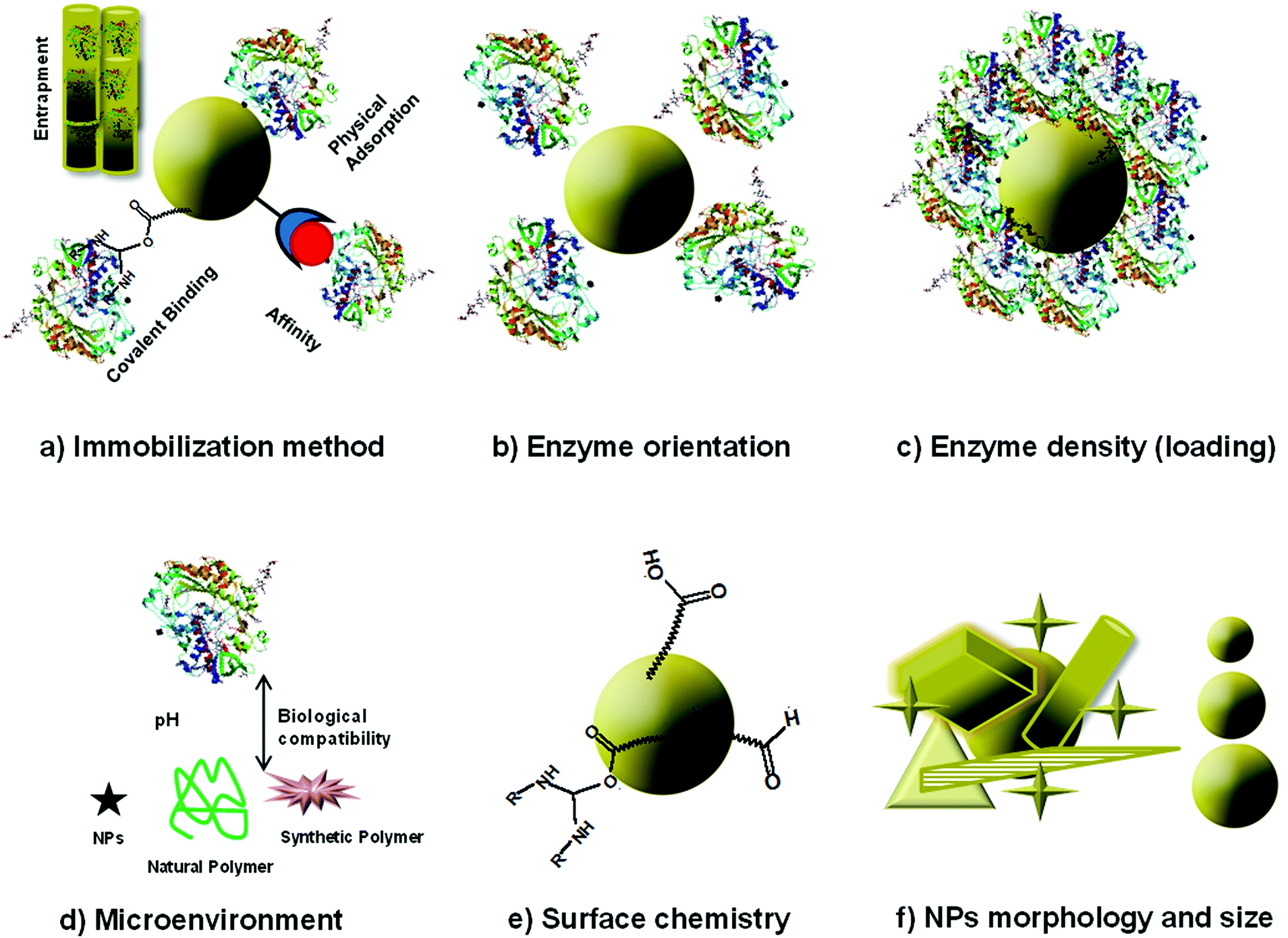 | ||
| Fig. 4 Schematic of immobilization methods and parameters determining the activity of enzyme–NP conjugates: (a) immobilization method, (b) enzyme orientation, (c) surface loading, (d) microenvironment, (e) surface chemistry, (f) morphology and size. | ||
2.3. Nanostructure materials for enzyme based biosensors
Magnetic NPs are widely used in biological research due to their magnetic behavior, high surface energy and large surface area-to-volume ratio.108,109 The immobilization of enzymes on the surface of these particles enables easy enzyme recovery using a magnetic field and enhances mass-transfer processes associated with recognition of substrates by enzymes.46,54,109 Iron oxide (Fe3O4) NPs are most commonly used because of their good biocompatibility, strong magnetic properties, low toxicity and easy preparation.110 Silanization is the most widely used surface modification technique to introduce surface functional groups on bare magnetic NPs to enable further enzyme binding.111 Sinigaglia et al.112 developed a superparamagnetic surface-active maghemite nanoparticles (SAMNs, γ-Fe2O3) with a dimension of ∼10 nm modified with bovine serum amine oxidase which used a rhodamine B isothiocyanate (RITC) adduct as a fluorescent spacer arm. This design provided an inexpensive and simple amine oxidase-based magnetic nanocatalyst tagged with a fluorescent probe.
Several types of enzyme-based magnetic NPs have been employed to fabricate enzyme biosensors. Iron core–shell Fe3O4@poly(dopamine) magnetic NPs were also developed and used as solid supports for the covalent immobilization of horseradish peroxidase (HRP) and to construct a biosensor for H2O2 (Fig. 5). This biosensor showed a sensitivity of 442.14 mA M−1 cm−2 and stability for 1 month.113 Xu et al. fabricated NTA modified magnetic NPs and used these conjugates as a substrate, a carrier and an anchor for the binding of His-tagged proteins via a transition metal terminated affinity ligand NTA group.114 Ni-IDA functionalized magnetic NPs were also used to site specifically immobilize genetically engineered acetylcholinesterase (AChE) for the highly sensitive detection of chlorpyriphos and chlorfenvinphos insecticides.115 Gan et al.116 developed a disposable organophosphorus pesticide (OP) enzyme biosensor based on magnetic composite NPs deposited on a screen printed carbon electrode (SPCE). The fabrication procedure involves the preparation of a AChE-coated Fe3O4/Au (GMP) magnetic nanoparticulate system (GMP-AChE) and then, absorption of GMP-AChE on the surface of a SPCE modified by a carbon nanotube (CNT)/nano-ZrO2/prussian blue (PB)/Nafion (Nf) composite membrane by an external magnetic field.
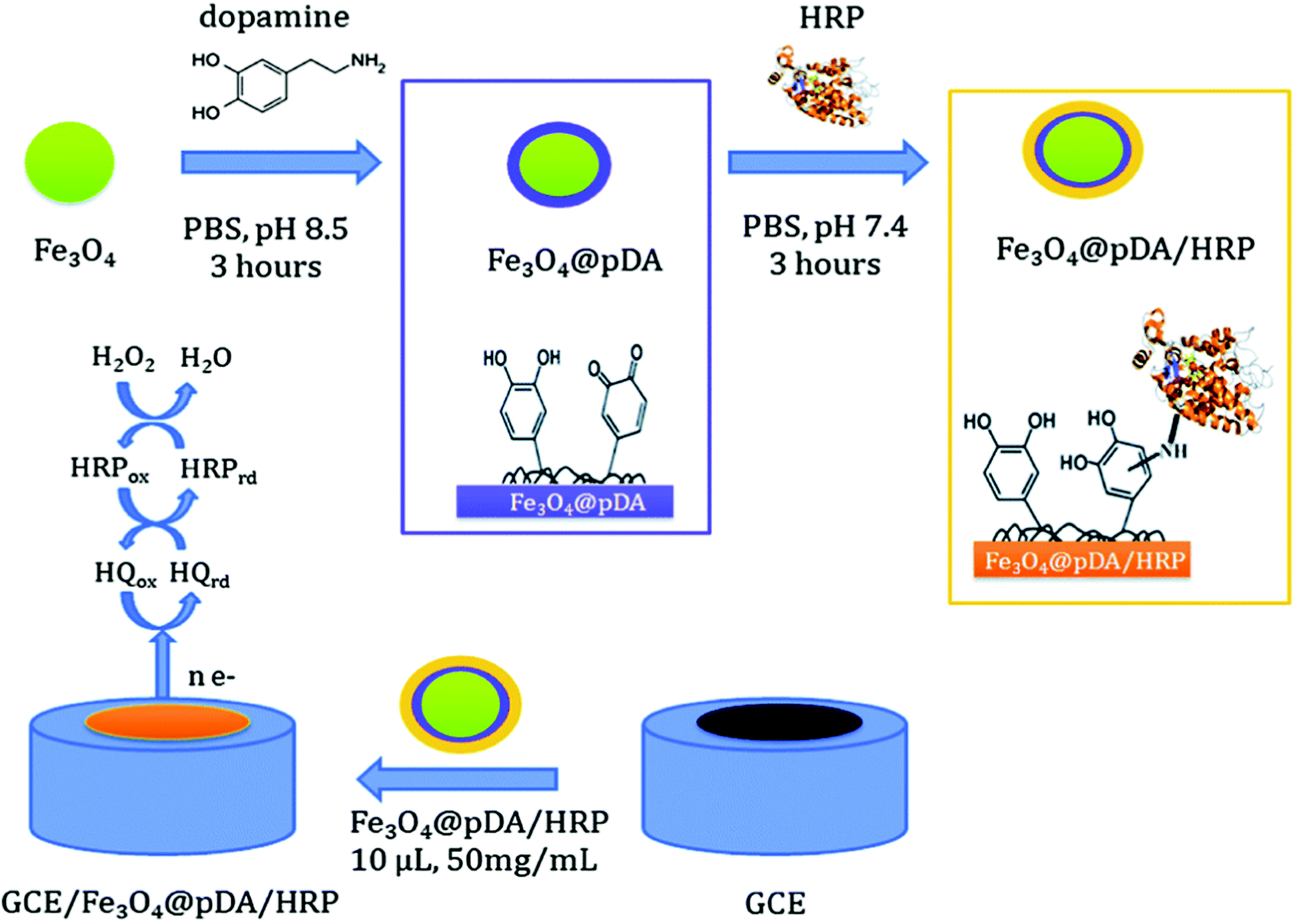 | ||
| Fig. 5 Schematic showing the synthesis of core–shell Fe3O4@pDA/HRP NPs and the construction of a Fe3O4@pDA/HRP-modified GC electrode. Figure adapted with permission from ref. 113. | ||
MnO2 is another material that has shown great interest in enzymatic biosensing applications. Hierarchical MnO2 spheres were synthesized and used as new electrode materials for the mediatorless detection of glucose.117 A glucose sensitive enzyme field-effect transistor (ENFET) was fabricated by co-immobilizing GOx and MnO2 NPs on the gate of an ion-sensitive field-effect transistor (ISFET).118 The proposed ENFET exhibits a linear response for glucose in the range of 0.025–1.90 mM and a dynamic upper limit of 3.5 mM glucose. A glassy carbon electrode modified with β-MnO2 nanowires and GOx enabled the amperometric detection of glucose with a sensitivity of 38.2 μA mM−1 cm−2 and a response time of <5 s.119 TiO2 nanorods, nanowires and nanotubes have also found broad biosensing applications. Sensitive detection of glucose was demonstrated using a tetragonal columnar-shaped TiO2 nanorod ((TCS-TiO2)/chitosan) modified electrode.120 The biosensor was characterized by a linear range from 5.0 μM to 1.32 mM with a sensitivity and a detection limit of 23.2 mA M−1 cm−2 and 2.0 μM, respectively. An electrochemical biosensor (HRP/Au NWs/TiO2 NPs/Au electrode) enabled the determination of H2O2121 from 2.3 μM to 2.4 mM with a detection limit of 0.7 μM.
A promising class of oxide NPs is the lanthanide oxide series that have unique luminescence, optical and electronic properties122–124 in addition to some therapeutic properties (e.g. antioxidants to inactivate reactive oxygen species) making them interesting candidates as labels for detection, imaging agents and NP theranostic probes. Cerium oxide (CeO2, ceria) NPs have been extensively investigated for such applications in recent years. The unique properties of CeO2 NPs is derived from their ability to act both as oxidation and reduction catalysts due to the presence of cerium in dual oxidation states (Ce3+/Ce4+).125 In addition to their redox reactivity, these NPs have high catalytic activity, oxygen rich surface and low toxicity126 which make them an excellent enzyme immobilization material for a variety of enzymes.127,128 CeO2 NPs have high oxygen mobility at their surface129,130 and a large oxygen diffusion coefficient, which facilitate the conversion between valance states Ce3+/Ce4+ and allow oxygen to be released or stored in its crystalline structure.131–134 This surface oxygen enables the functionality of oxidase enzymes under hypoxic conditions by providing an oxygen supply to the enzyme through a continuous release and recycling to/from the NP structure.135,136
For portable biosensing design, CeO2 NPs can be used as a colorimetric indicator to replace commonly used soluble dyes in oxidase enzyme assays. This approach takes advantage of the color changes resulting from alterations in the redox state and composition of these NPs in response to the product of the enzymatic reaction. Research by our group has demonstrated the possibility of incorporating CeO2 NPs and enzymes into a paper-based colorimetric format for the detection of H2O2 and of oxidase enzyme substrates, using glucose and glucose oxidase as a model example.127 The sensor was constructed by co-immobilization of CeO2 NPs and glucose oxidase onto filter paper using a silanization procedure. In the presence of glucose, the enzymatically generated H2O2 induces a visual color change of the CeO2 NPs immobilized onto the bioactive sensing paper, from white-yellowish to dark orange, in a concentration-dependent manner. The general principle of the sensor is shown in Fig. 6.
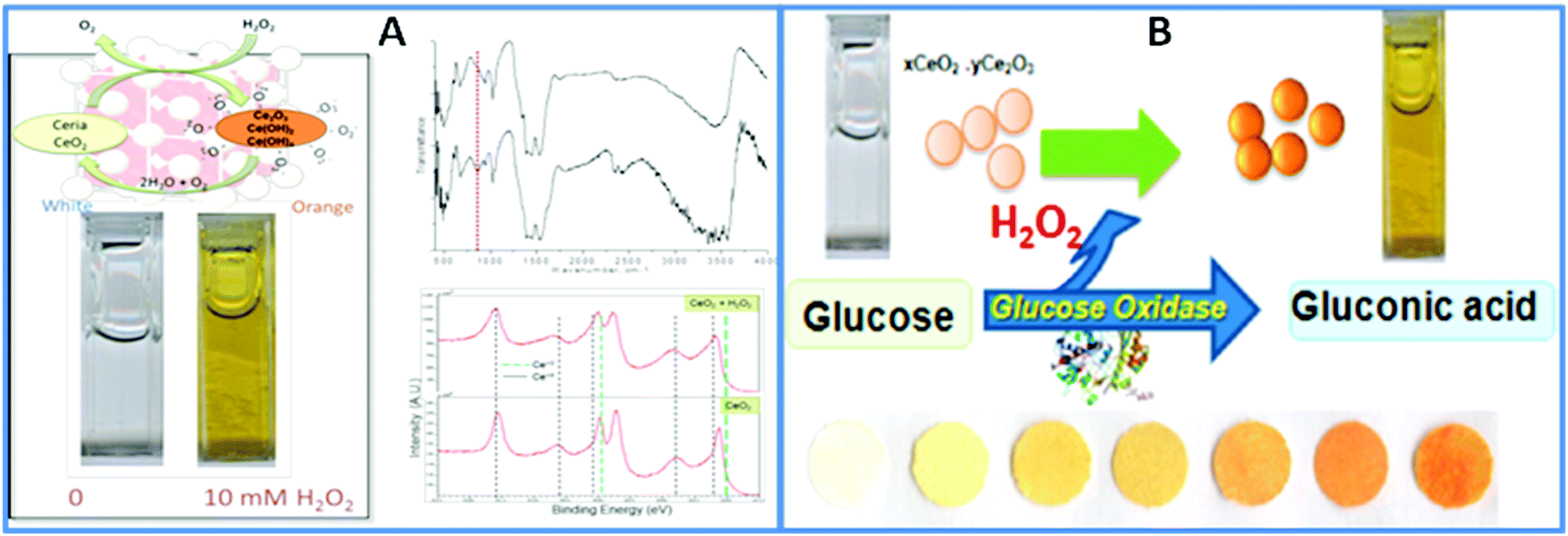 | ||
| Fig. 6 (A) Surface changes of CeO2 NPs in reaction with H2O2. The color change indicates the formation of surface adsorbed O2−, confirmed by FTIR and XPS. (B) Working principle of the portable colorimetric CeO2 sensor for the detection of glucose. This principle has been demonstrated on paper surfaces with both the GOX enzyme and the CeO2 NPs immobilized on paper to create a reagentless assay (with permission from ref. 127). | ||
The changes in the physicochemical and optical properties of CeO2 NPs in response to the analyte, measured visually as a colorimetric readout without additional instrumentation enabled a detection limit of 0.5 mM glucose with a linear range up to 100 mM and a reproducibility of 4.3%. Moreover, the bioassay can be stored for at least 79 days at room temperature while maintaining the same analytical performance. Both the particles and the enzyme can be inkjet-printed within a multilayered structure of biocompatible polymers such as chitosan and alginate.137,138 An extension of this assay in a portable and multiplexed format was demonstrated for the detection of food antioxidants by monitoring changes in the physicochemical properties of NPs.139,140
Another unique characteristic of CeO2 NPs is their oxygen storage and release behavior. The catalytic and oxygen storage/release capacity of CeO2 NPs was used to eliminate or minimize problems associated with low oxygen levels for enzymes that require oxygen as a co-substrate and fabricate ‘oxygen rich’ bioelectrodes.141 Enzymes, glutamate oxidase (GmOx), GOx and lactate oxidase (LOx) were co-immobilized with CeO2 NPs onto a microelectrode surface in a chitosan matrix. The use of CeO2 NPs in implantable sensors has advantages, which include biocompatibility and enhanced catalytic activity, acting as ‘oxidase enzyme mimetics’142–144 and as internal oxygen reservoirs, which expands the range of low oxygen environments in which these biosensors can be used reliably. Co-immobilization of oxidase enzymes with CeO2 NPs greatly improved the performance of in vivo biosensors under hypoxic conditions for real time measurements of dopamine,145 lactate,135 and glutamate141 using the enzymes tyrosinase, GOx, GmOx and LOx. Enhanced reactivity can be achieved by addition of dopants such as Pt, Au and Eu that create higher oxygen vacancies for oxygen transfer reactions while also imparting additional capabilities (e.g. conductivity, fluorescence). For example, Pt-doped CeO2136 enhanced detection sensitivity and enabled the measurement of physiologically relevant concentrations of glucose and lactate under oxygen limited conditions, with a detection limit of 37.4 μM and 3.3 μM when interfaced with GOx and LOx, respectively. When the strategy was applied to a Pt microelectrode, the biosensor enabled lactate monitoring during in vivo hypoxia (Fig. 7), demonstrating the applicability and versatility of this material for use in miniaturized implantable enzyme bioelectrodes.135,136 The oxidase enzyme–CeO2 conjugates can be particularly useful in increasing the detection sensitivity of biosensing devices under conditions where a low detection limit is needed. Other applications including implantable enzymatic biofuel cells and wearable patches for lactate monitoring can be designed based on this principle.
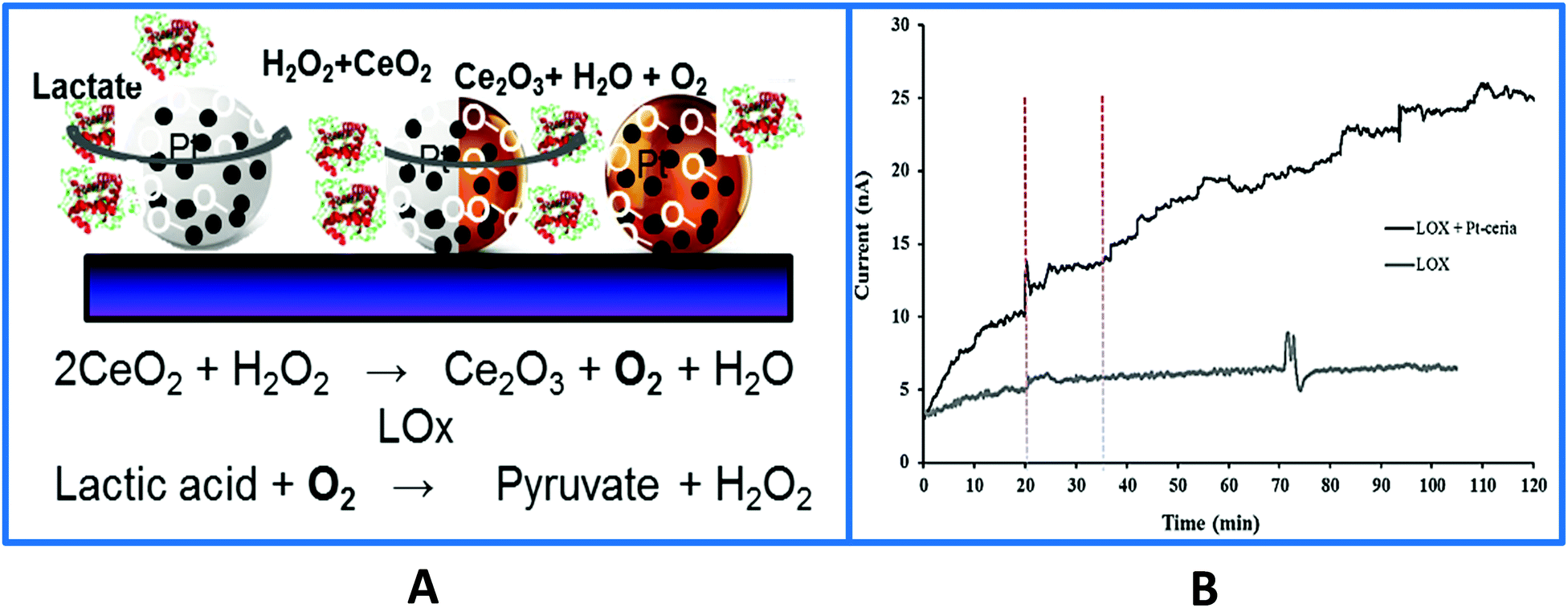 | ||
| Fig. 7 (A) Surface design of a Pt-doped-CeO2 biosensor used to monitor in vivo lactate levels during ischemia and reperfusion. The sensor was fabricated by co-immobilizing the particles and the enzyme on the surface of a Pt microelectrode. In the absence of particles the LOx biosensor enables real-time monitoring of in vivo lactate during hypoxia which was attributed to the local oxygen provided by the particles and their enhanced catalytic properties enhancing H2O2 (the product of the enzyme reaction) oxidation. (B) Typical in vivo current–time responses of the biosensor with and without Pt–CeO2 particles showing continuous monitoring of lactate in rat brain hippocampus at an applied biosensor potential of +0.55 V vs. Ag/AgCl. The vertical dotted lines indicate the onset of ischemia at 20 min and reperfusion at 35 min (with permission from ref. 135). | ||
These particles can be used in the development of fluorescence based biosensors, lab-on-a-chip and optoelectronic devices.150 Kim et al.151 reported a fluorescent cholesterol sensor by using enzyme immobilized CdSe/ZnS QDs. The particles were synthesized using trioctylphosphine oxide (TOPO) templates. For biological applications, TOPO was replaced with mercaptoacetic acid (MAA), which was used to immobilize the cholesterol oxidase (COx) enzyme via its carboxylic groups. In another design CdSe/ZnS core–shell QDs-organophosphorus hydrolase (OPH) bioconjugate enabled the detection of paraoxon.152 The OPH was coupled to CdSe/ZnS core–shell QDs through electrostatic interaction between the negatively charged QD surface and the positively charged protein side chain ending groups (–NH2). Despite their exciting optical properties, the application of these particles in practical assays has raised several concerns due to their potential toxicity associated with the release of highly toxic metals from their composition such as Cd.153–155
Conducting polymers such as polyaniline (PANI) and poly(3,4-ethylenedioxythiophene) (PEDOT) have been extensively investigated due to their electrochemical and optical properties. These polymers have been widely used as electrode materials owing to their high-electron affinities.159 Stočes et al.160 described the fabrication and characterization of a PANI–GOx biosensor for glucose detection. GOx was immobilized in a carbon paste with electro-polymerized aniline. In another work, urease was immobilized on indium tin oxide (ITO) electrodes that were previously modified with electroactive nanostructured membranes made with PANI and Ag NPs stabilized in polyvinyl alcohol (PANI/PVA–Ag NP). The environment promoted the efficient catalytic conversion of urea into ammonium and bicarbonate ions.161 Glucose biosensors were also fabricated by using electrochemically deposited PdNPs on nanofibrous PEDOT.162 The sensor displayed excellent performance for glucose with a sensitivity of 1.6 mA M−1 cm−2 and a linear concentration range from 0.5 to 30 mM. A modified electrode with a conducting film containing PEDOT and poly(methylene blue) on a glassy carbon electrode (GCE) enables the sensitive detection of ascorbate and glucose163 with a linear range from 0.02 to 1.40 mM, a sensitivity of 31.4 ± 1.9 μA cm−2 mM−1 and a limit of detection of 7.20 ± 0.35 μM for glucose. While improving the performance of the sensor by maximizing the exposed area using high surface area nanomaterials is important, controlling the geometry is also important for enhancing the sensor response and sensitivity. For example, the use of thick film deposition can suffer from enzyme–substrate accessibility issues. In addition, films of randomly oriented polymer nanotube/nanowire/nanofiber structures in a collapsed state may suffer from slow electron transport.164,165 Gokhale et al.166 evaluated the performance of a PEDOT/nitrate reductase nanowire sensor and demonstrated the superior performance of the nanowire design as compared to a flat 2D PEDOT/nitrate reductase film grown using similar electropolymerization conditions.
3. Scalable fabrication of enzyme based nanoassemblies for portable biosensing
Scalable fabrication of bioactive nanostructures is essential for the construction of inexpensive, lightweight, disposable, flexible and wearable devices. A requirement for the incorporation of nanomaterials in practical devices is their ability to mass produce the nanocomponents at low cost and with high reproducibility. While manufacturing individual nanostructures, NPs, carbon nanotubes, etc. has been widely demonstrated, there is significant less work on the scalable production of nanoassemblies supporting biomolecules.Portable stand-alone biosensors require the integration of the NPs and enzymes into compact functional devices. A key requirement is to achieve uniform deposition of biofunctional NP–enzyme conjugates to ensure the required detection sensitivity provided by the nanomaterial and preserve the biological activity of the enzyme. The use of automatic deposition procedures such as inkjet and 3D printing enables scalable production with increased reproducibility.167,168 More recently, substrates such as paper have been introduced as inexpensive platforms for enzyme immobilization and fabrication of low cost biosensors. Automatically fabricated lab-on-a-chip and microfluidic systems are also gaining increasing acceptance in recent years.
An important challenge to produce functional bioactive nanostructures for portable biosensing devices is the need to create conditions for patterning both the biological reagent and the nanomaterial and to precisely control and quantify the properties of these hybrid structures. Although a variety of processes have been developed for the deposition of NPs on surfaces, these methods are not directly applicable to nanobiomolecular assemblies. Many techniques used for the deposition of inorganic nanomaterials are not directly applicable to bioreagents due to the requirement of biocompatibility. On the other hand, procedures amenable to immobilize biomolecules are not directly applicable to NPs due to their potential in induced aggregation and loss of colloidal dispersity, which would result in reduction of their surface area and subsequent loss of their unique optoelectronic properties. Conventional fabrication methods that involve the use of harsh chemicals, organic solvents and/or high temperatures for the sintering of the NPs are not compatible with biomolecules. In many biosensing systems, NPs are embedded in polymeric films and are prone to aggregation losing their nanoscale features. As a result there is little control over the assembly, and both NPs and biomolecules are randomly distributed within the nanostructure, which strongly affect the sensitivity and reproducibility of sensing devices. In general, deposition of biomolecules requires the use of mild reagents and fabrication conditions that do not affect bioactivity and stability. Many enzyme biosensors reported in the literature involve complicated multistep procedures and expensive substrates and reagents, some of which are carried out manually, and require special conditions to conserve bioactivity. In many cases reagents needed for detection or signal amplification (including bio-reagents, colorimetric dyes and nanoelements) are added post-fabrication, adding additional cost and inconvenience to analysis.
Screen-printing, inkjet and 3D printing167,168 and advanced nanomanufacturing techniques such as dip-pen lithography169,170 are possible methods to generate nanobiomolecular assemblies in a controlled manner with stabilized NPs and biomolecules. These methods enable roll-to-roll fabrication and patterning with precise dimensional control and replication. Most printing methods require the development of specific pastes with appropriate composition (e.g. additives, biocompatible linkers) and viscosity. Advantages of printing technologies for biofabrication include: (i) simple and inexpensive hardware, (ii) mild deposition conditions favorable for handling biomaterials and maintaining bioactivity, (iii) precise placement of small pico-to-nanoliter quantities of material onto predetermined computer-generated spots without prior patterning and with minimal waste, (iv) the possibility of obtaining rapid sequential deposition of several layers on multiple predetermined locations within seconds enabling rapid and scalable production, and (v) fully automated, computer controlled operation.171 Most work to date on biosensor manufacturing was done using screen printing, followed by a few examples illustrating inkjet printing of biomolecules. In the following section we discuss various fabrication procedures that are amenable to scale up the manufacturing of nanoassemblies containing both NPs and enzymes for automatic fabrication of portable biosensing devices. The current status and performance characteristics of sensors based on screen, ink-jet, and 3D printing technologies, bioactive paper, lab-on-a-chip and microfluidic devices are reviewed with examples of applications. Integration of biomolecular recognition, detection and signal transduction capabilities into a stand-alone measuring device with minimal reagent use and power output, which can be used as a wearable device, has also been discussed.
3.1. Screen printed enzyme–NP based biosensors
Screen printing technology is a widely used method for the fabrication of screen printed electrodes (SPEs). Portable electrochemical biosensors that use SPEs are successfully commercialized and widely used. The ‘glucose pen’ that integrates enzyme functionalized SPE electrodes with a low cost electrochemical analyzer is a well-known example of a portable cost effective enzyme biosensor with good sensitivity and selectivity of real time measurements with minimal space, power requirement and instrumentation.172 Classical configurations of SPE biosensors include a three electrode configuration (working, counter and reference electrodes) printed on various types of plastic or ceramic substrates, modified with a wide variety of commercial or custom-made inks. A silver ink is typically used as a conducting track. A carbon paste is used on working electrodes which are then functionalized with active sensing materials to achieve the required selectivity and sensitivity.173 These include graphite or polymeric inks containing the enzyme and can incorporate nanoscale materials such as the ones described in previous sections. A gold paste for the generation of self-assembled monolayers (SAMs) through strong Au–S bonds174 was also employed. Differences in ink composition e.g. type, size, viscosity or loading in the printing and curing conditions can strongly affect the electron transfer, conductivity and electrochemical properties of the NPs, as well as the stability of the enzyme, and affect the overall analytical performance of the resulting sensors.175,176In the last ten years there has been significant interest in the use of NPs and nanostructures to enhance detection capabilities of SPE. Examples include Au, Ag, Pt, and Pd NPs, CNTs and graphene. These materials have been included in the composition of printing inks in conjunction with polymeric linkers or other surface modifiers, or deposited on the surface of the working electrode. The use of these materials has enhanced the immobilization efficiency of enzymes and has increased the conductivity and the electron transfer rate at the electrode surface. The electrocatalytic behavior is dependent on the nature of the nanostructured material and the surface modifier.177 Depending on the composition and the properties of the NPs, different electroanalytical signals can be obtained. For example, a tyrosinase based SPE for the detection of bisphenol A modified with three different types of NPs (Ni, Au, and Fe3O4) showed that Ni provided comparable or better characteristics in terms of detection limit and sensitivity than Fe3O4 and Au NPs.104 The detection limits for bisphenol A for the three types of biosensors were 7.1 × 10−9, 8.3 × 10−9, and 1 × 10−8 mol L−1 for Ni, Fe3O4 and Au NPs respectively. A low cost and small sample volume assay has been developed for the in situ detection of glucose in human whole blood by using a disposable Au NP-modified SPE coupled with a paper disk. The electrode was modified with a graphene/polyaniline/Au NPs/GOx biocomposite and then covered by a paper disk impregnated with the sample. The use of this composite material provided a favorable microenvironment for facilitating direct electron transfer between the GOx and the electrode.178 The fabrication and detection procedures are illustrated in Fig. 8. Table 1 provides a summary of varying SPE biosensor based enzyme-nanostructure configurations.
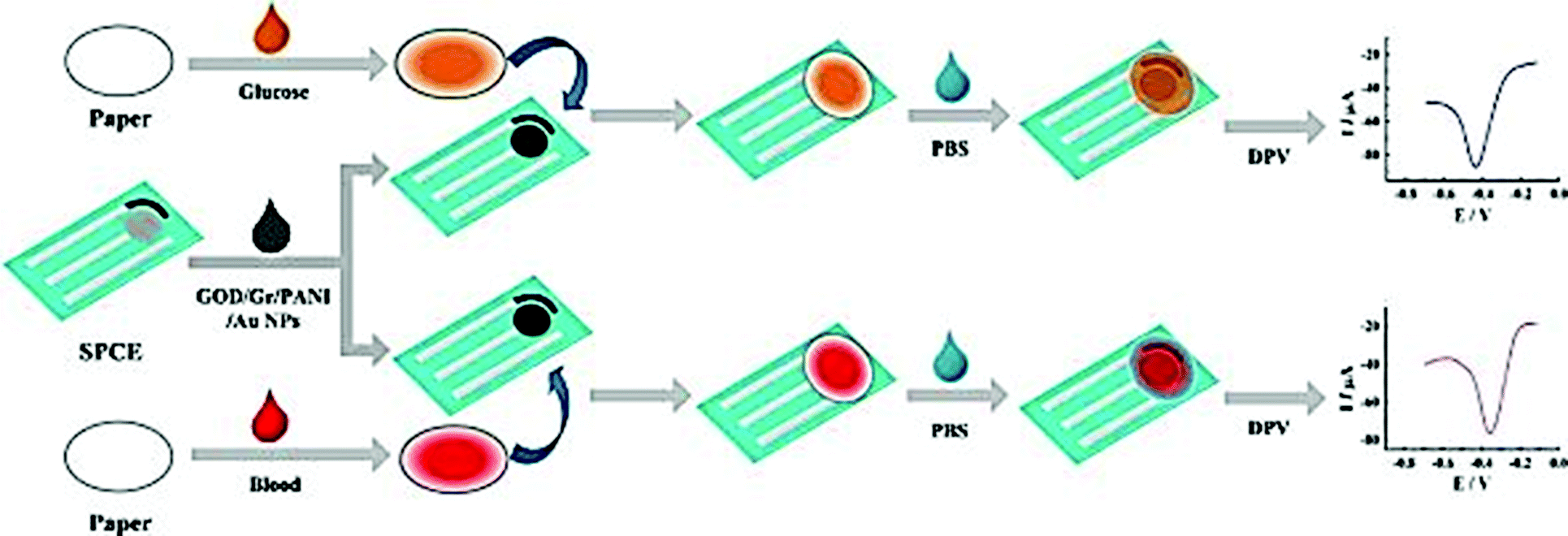 | ||
| Fig. 8 Schematic representation of the fabrication and assay procedure of an Au NP/graphene/polyaniline SPE biosensor for the detection of glucose. Figure adapted with permission from ref. 178. | ||
| Nanostructure | Enzyme | Analyte | Specific properties of the nanostructure | Technique | Sensitivity/LOD | Ref. |
|---|---|---|---|---|---|---|
| Au NP/graphene (GR)/polyaniline | GOx | Blood glucose determination | A favorable microenvironment for facilitating direct electron transfer | Amperometric | 20.32 μA mM−1 cm−2/1.0 × 10−4 M | 178 |
| Au NPs anchored on reduced graphene oxide (RGO) | L-lactate dehydrogenase (LDH) | L-lactate tumor biomarker | Electrocatalytic properties/facilitates fast electron transfer processes. | Amperometric | 154 μA mM−1 cm−2/0.13 μM | 179 |
| Nanocomposites of magnetic NPs functionalized with iridium oxide NPs (IrOx NPs) | Tyrosinase | Methimazole (drug) | Stability and catalytic properties/the use of magnetic NP carriers for immobilization of tyrosinase | Amperometric lab-on-a-chip (LOC) microsystem with an integrated SPE | 20.00 μA M−1/0.004 μM | 180 |
| Graphitized carbon nanofiber–Pt NP hybrids | Lactate oxidase | Lactate | Electrocatalytic activity towards H2O2 oxidation | Amperometric | 36.8 mA M−1 cm−2/11 μM | 181 |
| Au or ZnO NPs/carbon nanotube SPEs | Sarcosine oxidase | Sarcosine | Increasing enzyme stability and biosensor sensitivity | Amperometric | 0.034 μA mM−1/16 nM | 182 |
| Au NPs | Alkaline phosphatase | Vanadium | Excellent electrocatalytic response/fast response time/long term stability and reproducibility | Amperometric | 4.6 × 10−3 A M−1/0.39 ± 0.06 μM | 183 |
| Au NPs | Acetylcholinesterase | Aluminum(III) | Electrocatalytic activity/high analytical response | Amperometric | 0.0325 A M−1/2.1 ± 0.1 μM | 184 |
| Au NPs | Monoamine oxidase | Putrescine and cadaverine | Increasing surface area/catalytic properties | Amperometric | 1.16 nA μM−1/9.9 μM for Put 1.05 nA μM−1/19.9 ± 0.9 μM for Cad | 185 |
| Au NPs | Tyrosinase | Phenol | Electrical properties/large active surface areas for enzyme immobilization/catalytic activities and biocompatibility/robust immobilization of enzymes via the combined use of alkanethiol self-assembly and cross-linking chemistry | Cyclic voltammetry and square wave voltammetry | 15.7 μA ppm−1/47 ppb | 186 |
| Prussian Blue (PB) NPs | Glucose-6-phosphate dehydrogenase, and glutathione reductase | Glucose-6-phosphate | Enhance the electrochemical response of the bienzymatic reaction by accelerating the electron transfer between the electrodes and the enzymes | Amperometric | 63.3 μA mM−1/2.3 μM | 187 |
| Pt NPs–RGO | Laccase | Determination of total polyphenolic content | Synergetic effects by deposition of metallic NPs onto graphene which can reduce the overpotential of the electrochemical reactions/increased electroactive surface area and better electron transfer | Amperometric | 2147.38 nA μM−1 forCaffeic acid and 99.78 nA μM−1 for Rosmarinic acid/0.09 μM | 188 |
| Fe3O4/Au NPs | Alkaline phosphatases | Mutant alleles/BRAF V600E mutations | Carriers for loading enzymes | Electrochemical impedance spectroscopy and cyclic voltammetry | NA | 189 |
| Magnetic nanoparticle/[Fe(tris(3,5-dimethyl-pyrazolyl)borate)2]+[FeCl4]− | Glucose oxidase | Blood glucose | High enzyme loadings | Amperometric flow system | NA/0.08 mM | 190 |
| Bismuth NPs | Tyrosinase | Phenolic compounds | Larger specific surface area and biocompatibility | Amperometric | 44.05 nA μM−1/26 nM for catechol and 14.25 nA μM−1/62 nM for phenol | 191 |
| Snowflake-like Pt–Pd bimetallic nanoclusters (Pt–PdBNC)/Au nanofilm electrode (SPGFE) | GOx | H2O2 and glucose | Electrochemical stability and catalytic activity/Pt–Pd composites enhance its catalytic performance for H2O2 reduction/3D framework offers sufficient electron transfer pathways | Amperometric and cyclic voltammetry | 804 mA M−1 cm−2/0.87 μM | 192 |
| Au nanocubes | Tyrosinase | Catechol | Catalytic properties | Amperometric | 13.72 A M−1/0.4 nM | 193 |
| Graphene oxide(GO)/Au NPs | Tyrosinase | Catechol | Microenvironment for enzyme immobilization and enhanced electron transfer | Amperometric | 160 mA M−1/2.4 × 10−8 M | 194 |
| Au NPs | Trimethylamine dehydrogenase | Trimethylamine | Higher electrochemical response than that of a planar gold electrode | Voltammetric | NA/1 μM | 195 |
| GO–Au NPs | Tyrosinase | Phenolic compounds | Integration of GO–Au the hybrid material with good biocompatibility and catalytic properties | Amperometric | ||
| Nanoscale Pt particles (nPt) and GR | Cholesterol oxidase | H2O2 and Cholesterol | Catalyzes the electrochemical oxidation of H2O2/facilitated electron transfer | Amperometric | 8.07 μA μM−1 and 0.5 nM for H2O2/2.07 ± 0.1 μA μM−1 cm−2 and 0.2 μM for Cholesterol | 196 |
| Fe3O4 NP core and a mesoporous silica shell (Fe3O4/m-silica) | Horseradish peroxidase (HRP) | H2O2 | Matrix for immobilization | Amperometric | of 84.4 μA mM−1 cm−2/4.3 × 10−7 M | 31 |
| Ferrocene-capped Au NPs | Cholesterol esterase | Cholesterol | Increasing surface area/biocompatibility and catalytic properties | Amperometric | 0.18 μA μM−1/12 μM | 197 |
| Ni NPs | Tyrosinase | Bisphenol A | Enzyme immobilization platform and electrode material | Amperometric | 758.1 μA mM−1/7.1 × 10−9 M | 104 |
| RGO-carboxymethylcellulose layered with Pt/PAMAM dendrimer/magnetic NP hybrids (MNP–PAMAM–Pt NP) | Xanthine oxidase | Xanthine | Pt NPs-decorated layer-by-layer polyelectrolytic nanoassemblies | Amperometric | 40 mA M−1 cm−2/13 nM | 198 |
3.2. Ink-jet printing
In recent years, there has been increased interest in using ink-jet printing as a non-impact technique to fabricate biosensors. Ink-jet sputtering of printing inks reduces the risk of damaging reagents due to the direct contact between the printing head and the biosensor support. This method shows great potential for large scale fabrication of sensors due to its speed, low cost and suitability towards automation.199,200 The printer can transfer any computer-generated pattern onto substrates by dispensing a volume of 1–1000 pL per drop of deposition.Inkjet printing is an established technology201 for printing NP dispersions. Printing inks consist of preformed particles and a dispersing agent or a surfactant to prevent agglomeration. An example of inkjet printing is the deposition of Ag NP based tracks from a Ag NP dispersion.202 Most NP inks, especially those designed for creating electrically conductive layers require sintering using procedures like laser microwave, pulsed light, IR or by high temperature oven sintering,203 which are not compatible with biomolecules. Several successful applications have been reported for bioprinting of proteins or genes.171,204 This method can be used for multilayer deposition of enzymes, stabilizing agents and biocompatible polymeric layers. In bioanalysis, inkjet printing has been used to create electrically conductive tracks on planar electrodes. An ink containing dodecane thiol protected Au NPs dispersed in toluene was used to create conductive Au arrays on plastic. The platform was used for immunodetection of a cancer biomarker205 but the application of the antibody was performed manually and involved multiple functionalization steps. Few printing methods have been reported to create biomolecular assemblies in conjunction with NPs. Due to the requirement for biocompatibility, parameters used for printing bioreagents are very different from those used for printing inorganic nanomaterials. Another challenge is obtaining and controlling the homogeneity of the layer and preventing clogging of the cartridges. Moreover, the physicochemical properties of the printed layers need to be carefully controlled to maintain the nanoscale properties and ensure sensitivity and selectivity for biodetection applications.
Ink-jet printing of enzymes within sequential layers of chitosan and alginate biopolymers has been demonstrated on paper for fabrication of a paper based tyrosinase biosensor for the detection of phenolic compounds.137,138 Weng et al.206 have reported inkjet printable polypyrrole (PPy) NPs to fabricate an electrochemical sensor chip. The enzymes HRP and GOx were blended with PPy NPs and the resultant inks were printed onto SPCEs. The biosensors showed stable and continuous responses to H2O2 or glucose over a relatively broad range (H2O2: 10−5–10−2 M, glucose: 1 to 5 mM). Sensor sensitivity varied with the loading of the materials. Stability of more than 90 and ∼70% for PPy/HRP and PPy/GOx biosensors was demonstrated after 30 days of vacuum dry storage at 4 °C. Creran et al.207 used inkjet printing to co-pattern β-galactosidase (β-gal) and Au NPs on a paper-based test strip for rapid detection of bacteria (Fig. 9). A colorimetric response is generated on a paper substrate that allows for visual detection of contamination without the need for expensive instrumentation.
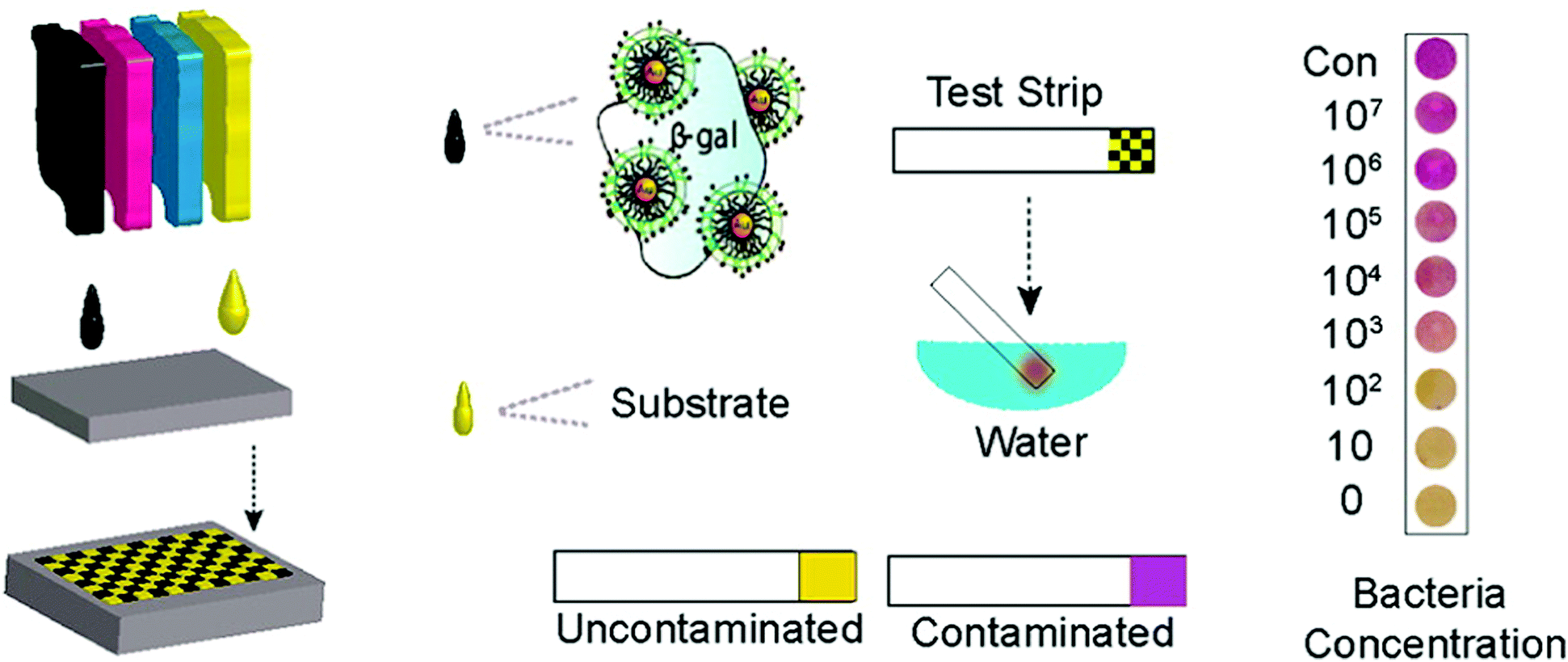 | ||
| Fig. 9 Inkjet printing of test strips used for rapid detection of bacteria. Figure adapted with permission from ref. 207. | ||
3.3. 3D printing technology
3D printing has been introduced recently as an emerging technique for bioprinting (e.g. tissue and organ printing).208,209 3D printing allows modular design of 3D structures with precise dimensional control and replication enabling integration with commercially available components.168 The method has received significantly less attention from the biosensing community. The first inkjet printing of biomolecules such as proteins, enzymes, antibodies and DNA was achieved in the 1980s. However, the first use on 3D bioprinting was reported in 2009 with commercially available 3D printers becoming widely available in 2012.The development of 3D technology of biomolecules (i.e. 3D bioprinting) offers the advantages of mechanical flexibility, thinness, and flexible design. This method is expected to become an important fabrication tool for novel commercial sensing technologies in the future.206 According to IDTechEx research, the fully printed sensors and 3D bioprinting market will be over $6 billion by 2024.210,211 Taking advantages of the 3D biomimetic structure in enrichment, separation and detection, Gou et al.212 developed a bioinspired 3D detoxification device by installing polydiacetylene (PDA) NPs. The PDA NPs can attract, capture and sense pore-forming toxins (PFTs) (Fig. 10). The 3D printed device is designed to efficiently collect and sense PFTs for future detoxification applications.
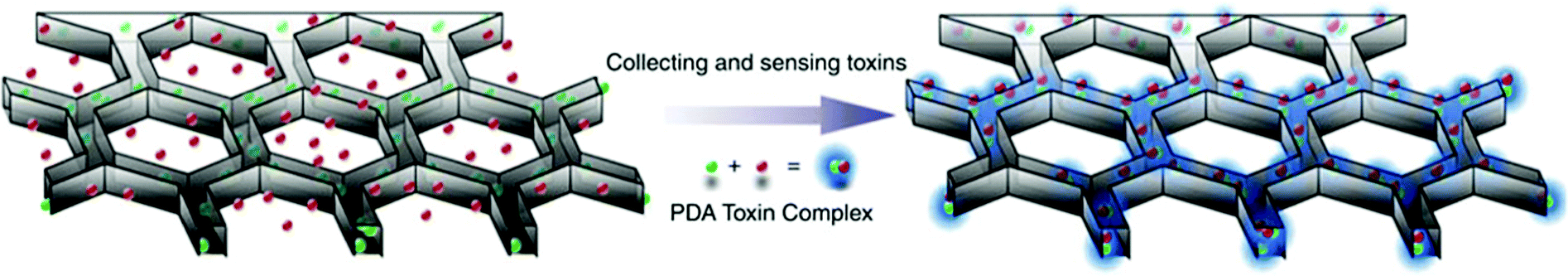 | ||
| Fig. 10 Bio-inspired 3D detoxification device. PDA NPs (green) are installed in the PEGDA hydrogel matrix (grey) with a liver-mimetic 3D structure fabricated by 3D printing. The NPs attract, capture and sense toxins (red), while the 3D matrix with a modified liver lobule structure allows toxins to be trapped efficiently. This biomimetic 3D detoxifier has promising clinical applications for detoxification by collecting and removing toxins. Figure adapted with permission from ref. 212. | ||
3.4. Nanolithography and nano-dispersing techniques
Nanoarraying and nanopatterning of biomolecules are important for the fabrication of protein microarrays that can be used as high-throughput screening tools in applications such as cell biology, diagnostics and toxicity screening applications. Nanoscale fabrication of protein nanostructures can be achieved by dip-pen nanolithography (DPN) involving the use of chemically modified AFM tips. DPN is a well-established technology for the deposition and assembly of hierarchical nanostructures on surfaces in a flexible manner.213 Patterning is achieved by the delivery of an ink coating the AFM tip upon scanning and contacting with surfaces. Fabrication of protein nanostructures formed via direct-write DPN was demonstrated by the pioneering work of Mirkin.169 Nanolithographic patterning of metallic, semiconductor, and metal oxide nanostructures on surfaces, their formation mechanism and applications have been reviewed in ref. 214. The nanoscale resolution and preservation of the biological activity can be achieved by controlling deposition and avoiding exposure of the patterning surface to harsh conditions. DPN was used as a patterning method to fabricate Au NP-modified enzymes that act as “biocatalytic inks” for the generation of metallic (Au and Ag) nanowires on Si surfaces215 and generate nanoarrays of NP functionalized-rabbit IgG proteins.216 Preservation of the biological activity of a fluorescent protein adsorbed onto the porous layer by layer (LbL) DPN tip was demonstrated.217 More recently, development of a surface chemical method to control the surface architecture was demonstrated. This method enabled site-specific immobilization of histidine-tagged proteins to aminosiloxane films with photoremovable protein-resistant protecting groups in a controlled manner.218Direct patterning and localized deposition of NPs and biomolecules can also be achieved by liquid nanodispensing (NADIS),170 a technique consisting of depositing droplets of a solution through a nanochannel drilled at the apex of an AFM tip. Fabrication of large arrays of single (or pairs of) NPs and deposition of fluorescent proteins without loss of protein function was demonstrated by this method, as an alternative to other scanning probe lithography techniques.
Another method, NanoPen, enabled real time light actuating patterning of single or multiple NPs and one-dimensional nanostructures for the fabrication of surface enhanced Raman spectroscopy hot-spots.219 This method could be extended to patterning proteins and enzymes, in conjunction with NPs to produce opto and nanoelectronic biosensing devices in the future. The advantages and disadvantages, and limitations and challenges of these methods are summarized in Table 2.
| Method | Advantages | Disadvantages, limitations and challenges |
|---|---|---|
| Screen printed enzyme–NP based biosensors |
– Portable and disposable
– Cost effective – Easy-to-use – Good sensitivity and selectivity with minimal space, power requirement and instrumentation – Ability to change the composition of the printing inks |
– Unsuitable for very long runs
– Potential dissolution of inks |
| Ink-jet printing |
– Reduced risk of damaging reagents
– Low cost and suitability towards automation – Can transfer any computer-generated pattern onto substrates – Can be used for multilayer deposition of enzymes |
– Controlling the homogeneity of the layer, preventing clogging of the nozzle and irregular droplet size
– Printing action may affect bioactivity. – The physicochemical properties of the printed layer need to be carefully controlled – Probability of contamination of the ink supply system due to frequent refilling of bio-inks |
| 3D printing technology |
– Mechanical flexibility
– Flexible design – Ability to dispense high viscous and dense biomaterials – Capable of parallel patterning on various substrates |
– The high dispensing pressure and mechanical stress applied may damage biomolecules
– Requires the development of printable formulations |
| Nanolithography and nano-dispersing techniques |
– High nanoscale resolution
– Capable of patterning bimolecular substrates on various substrates |
– Expensive
– Risk of denaturing from thermal bending |
4. Biosensing applications
4.1. Printed paper-based nanostructured biosensors
Paper-based analytical devices (PADs) have emerged as powerful analytical devices for portable and low cost detection applications. PADs first introduced by the Whitesides group in 2007 use porous cellulose (i.e., common filter paper) as a sensing support. Paper bioassays have been patterned and fabricated by photolithography for the detection of glucose and bovine serum albumin,7 sol–gel bio-inks for the detection of neurotoxins,134 and aptamer-NP based lateral flow devices for the detection of DNA sequences.220 The paper can store reagents including colorimetric dyes, cofactors and immobilized enzymes. Reagents can also be added on hydrophobic channels on modified paper and migrate to the reaction/sensing spot via capillary action.221–224 Quantification can be obtained by peripheral technologies such as digital scanners, cameras, cell phones or other optical techniques.9 These devices are relatively cheap, portable, and easy to use. However, traditional performance parameters, such as stability, detection limits, etc., must be balanced with the need for speed, cost, and simplicity. Several NP-based detection schemes have been adapted to paper to increase their sensitivity and detection capabilities. Detection of glucose in urine samples using colloidal Au NPs functionalized with GOx225 and glucose and phosphatase detection in serum using CeO2 NPs127 were reported. Table 3 summarizes recent examples of portable paper-based assays based on nanostructures and enzymes with colorimetric and electrochemical detection, and their performance. The use of NPs provided catalytic enhancement and/or increased enzyme loading leading to improved detection sensitivity. An example of an electrochemical paper-based device based on Ag NP-modified boron-doped diamond (BDD) on paper with immobilized cholesterol oxidase was reported for the detection of cholesterol.226 The use of electrodeposited Ag NPs onto the BDD electrode increased the specific surface area and provided electrocatalytic activity for H2O2 reduction. Fig. 11 shows the fabrication and assay procedure of this sensor and the electrochemical measurement for the detection of cholesterol. To accelerate adoption of this technology, there is a need to develop procedures for the large scale manufacturing of these devices with low cost and high yield fabrication. Various printing techniques, such as screen printing227 and inkjet printing228 have been utilized to mass produce these biosensors. A novel technique based on flexographic printing has been reported for the fabrication of Au NP-based devices, enabling printing of multiple layers consecutively. The main advantage of this method is that it does not suffer from clogging of the printing heads as observed in the inkjet printing technique.227| Nanostructure | Enzyme | Analyte | Which characteristic of nanostructure? | Method | Sensitivity/LOD | Ref. |
|---|---|---|---|---|---|---|
| Tungsten disulfide (WS2) nanosheets | GOx | Blood glucose | Catalyze the peroxidase substrate (TMB) to produce a colored reaction | Colorimetric method and portable test kit | NA/2.9 μM | 229 |
| Au NPs | HRP | Nucleic acid | Increasing enzyme loading/optical properties of Au NPs | Lateral flow strip biosensor | NA/0.3 pM | 230 |
| MFe2O4 (M = Mg, Ni, Cu) magnetic NPs | GOx | Glucose in the urine sample | Catalytic activities similar to those of biological enzymes such as catalase and peroxidase | Colorimetric biosensing | NA/4.5 × 10−7 M | 231 |
| Carbon nanotubes (CNTs)–SPE | Acetylcholinesterase (AChE) | Organophosphorus pesticides and nerve agents | Signal is further enhanced by using carbon nanotubes (CNTs) | Integrated lateral flow test strip with an electrochemical sensor | 1.366 μA nM−1/0.02 nM | 232 |
| Colloidal Au NPs | HRP | H2O2 | The matrix for the immobilization of enzymes which retain the macromolecules' bioactivity | With flow injection amperometric analysis | NA/0.005 mM | 233 |
| Ag NPs | Cholesterol oxidase | Cholesterol | Increase in specific surface area and furthermore for electrocatalytic activity of H2O2 reduction | Amperometric detection | 49.61 μA mM−1 cm−2/0.25 mg dL−1 | 226 |
| Au NPs | Glucose dehydrogenase | Dihydronicotinamide adenine dinucleotide (NADH) | Investigating the dissolution of Au NPs with NADH followed by a color change | Colorimetric readout | NA/12.5 μM | 234 |
| CeO2 NPs | GOx | Hydrogen peroxide | Color changes resulting from alterations in the redox state and composition of these NPs in response to the analyte | Colorimetric readout | 31.989 mM−1/0.5 mM | 127 |
| ZnO NWs | GOx | D-(+)-Glucose/PBS | Hydrothermal growth of ZnO NWs on the carbon WE | Electrochemical (CV) | 8.24 μA mM−1 cm−2/59.5 μM | 235 |
| Au NPs | AChE | Solution containing acetylthiocholine (ATCh) and Au(III) salt | Biocatalyzed hydrolysis of ATCh leads to the formation of thiocholine, which in turn reduces Au(III), producing a concomitant increase in color intensity | A bioactive paper-based colorimetric ‘‘dipstick” | NA/0.5 μM | 236 |
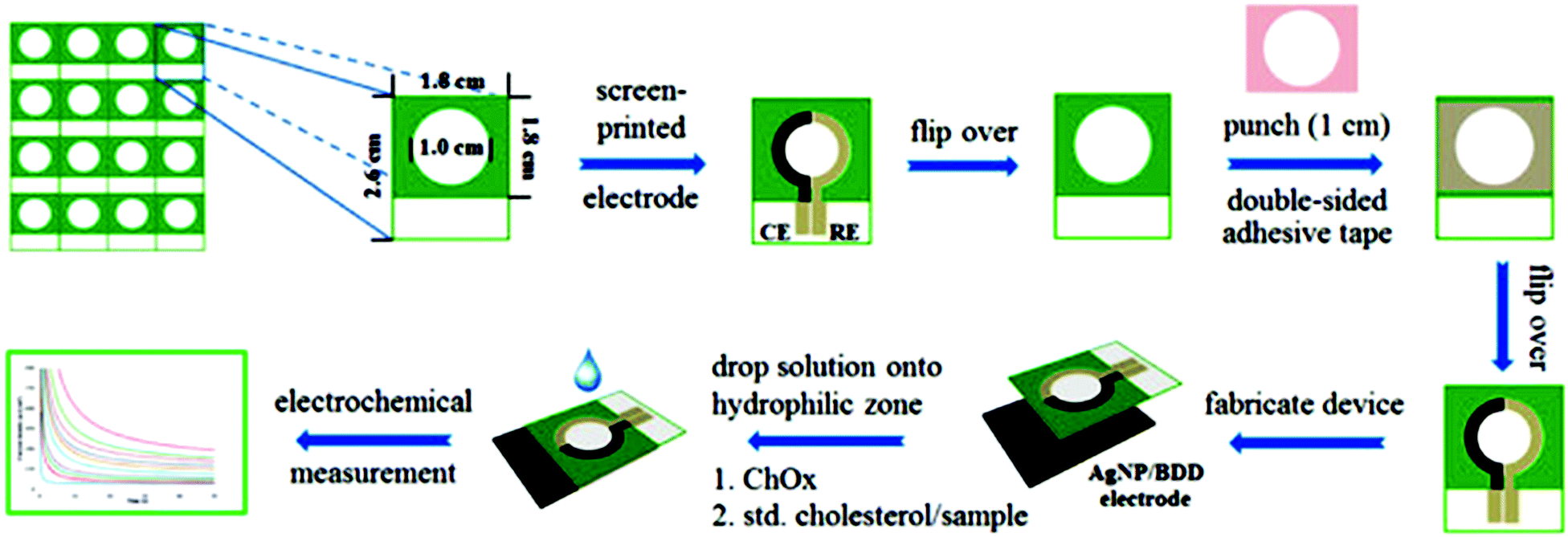 | ||
| Fig. 11 Fabrication and analytical measurement procedure of a cholesterol sensor based on the coupling of the Ag NP/BDD electrode with PAD. Figure adapted with permission from ref. 226. | ||
4.2. Lab-on-a-chip and microfluidic devices
As the field of biosensing continues to evolve, new types of electrodes and bioanalytical devices for point-of-care testing have emerged. Lab-on-a-chip biosensing devices with integrated microfluidics have been developed to provide capabilities for automatic sample analysis of low amounts of sample in a portable format (typical dimensions of 10–100 mm) with minimum reagent and power requirements. By appropriate integration of fluidic reservoirs and pumps, it is possible to design systems that can simultaneously perform multiple functions including sample and reagents handling, preparation (e.g., separation, concentration, extraction, mixing, incubation) and detection.237 The ultimate aim is to achieve selective and sensitive detection of a large number of samples without manual sample preparation or user input. Several types of nanostructures such as single-walled, metallic and semiconductor carbon nanotubes have been integrated on a chip and investigated for the detection of biomolecules,238 mostly in an immunoassay format.239 A recent example for the detection of trace radioactive elements such as uranium has been reported.240In addition to lab-on-a-chip devices, molecular nanoscale electronic devices that measure the response at a molecular level have been developed as inexpensive low power systems241,242 for medical care.243 The use of nanoscale electrodes and nanobiosensors and their potential in medical diagnosis have been reviewed by H. Shi et al.244 and H. Shi and Yeh.245 Self-assembly, the independent organization of different components into patterns or structures, is one of the most common methods of creating molecular electronic based nanosensors.246 One of the biggest challenges in the development of molecular electronics is the sensitivity of the measurement at the molecular level.246,247 Nanoelectrodes can provide molecular interfaces for accessing and stabilizing the redox center of enzymes and establishing biomolecular recognition through electrical communication. Blum and colleagues248 designed electronic nanosensors by using hierarchical self-assembly of the individual components built on a viral capsid scaffold. They showed that these nanosensors can detect proteins through changes in the molecular network conductance and can differentiate among analytes.
4.3. Wearable biosensing devices
One of the most exciting areas of growth in the biosensing field is the development and integration of low cost sensors with smart and wearable devices such as smartphones, tablets, smart-watches, flexible and low power bioelectronics and wearable health-monitoring devices.249–251 These devices are expected to bring significant improvement in the healthcare sector, and enable a variety of applications. According to a Transparency Market Research (TMR) research report on the global wearable sensor market, the global wearable sensor market is expected to expand at a 45.20% CAGR by 2020. At present, commercially available wearable sensors are only capable of tracking an individual's physical activities (such as body movement) and vital signs (such as heart rate), and fail to provide insight into the user's health state (e.g. monitoring disease biomarkers).252,253 Wearable devices are attractive, because they require no medical procedure and travel with the patient. Ideally, these sensors should be cheap, operate without or with a minimum power output, be easy to use, provide a fast response and be non-invasive. This section discusses the current status and development challenges of enzymatic biosensors as flexible and wearable devices.Development of wearable biosensors for continuous and non-invasive monitoring of glucose continues to be of interest. A flexible, noninvasive, and wearable amperometric biosensor to monitor the level of glucose in tears, with a linear concentration range of 0.025–1.475 mM, was reported by Iguchi et al.254 Another glucose sensor fabricated using microelectromechanical system (MEMS) techniques was constructed by immobilizing GOx on a flexible H2O2 electrode (Pt working electrode and Ag/AgCl counter/reference electrode).255 The output current of the H2O2 electrode was linearly related to the H2O2 concentration in a range of 0.20–2.50 mM, while that of glucose was over a range of 0.06–2.00 mM, which includes the reported concentration of tear glucose in a normal human subject (0.14 mM). Yao et al.256 reported a design of a contact lens with an integrated amperometric glucose sensor, proposing the possibility of in situ human health monitoring simply by wearing a contact lens. The glucose sensor was constructed by creating microstructures with immobilized GOx deposited within a titania sol–gel film on a polymer substrate. The sensor exhibited a fast response (20 s), a high sensitivity (240 μA cm−2 mM−1), with good linearity covering the typical range of glucose concentrations in tears (0.1–0.6 mM), and good reproducibility. The minimum detectable concentration was 0.01 mM. Wearable biosensors on flexible or wearable materials for physical exercise have also seen rapid expansion. Wearable tattoo-based biosensors have been designed for the continuous assessment of lactate levels in human perspiration257 and for alcohol monitoring in induced sweat using the alcohol-oxidase enzyme and a Prussian Blue modified electrode as an electrochemical transducer (Fig. 12A).258 Wearable sensing devices on textiles have also attracted attention due to the possibility of achieving conformal contact between the sensor and the body and hence the ability to operate under extreme mechanical tensions without compromising performance. Parrilla et al. reported a stretchable and printable textile-based potentiometric sensor array for simultaneous multi-ion analysis in sweat using a variety of fabric materials.259 Chuang et al.260 examined the influence of mechanical bending and stress upon the performance of an amperometric biosensor of GOx–Nafion-coated SPEs. Their findings indicate that extreme bending of flexible GOx/Nafion-coated SPE biosensors (including a 180° pinch) does not cause a sensor failure but actually leads to a substantial enhancement in sensitivity and improved sensor performance. Table 4 provides examples of wearable biosensors reported in the literature with their characteristics.
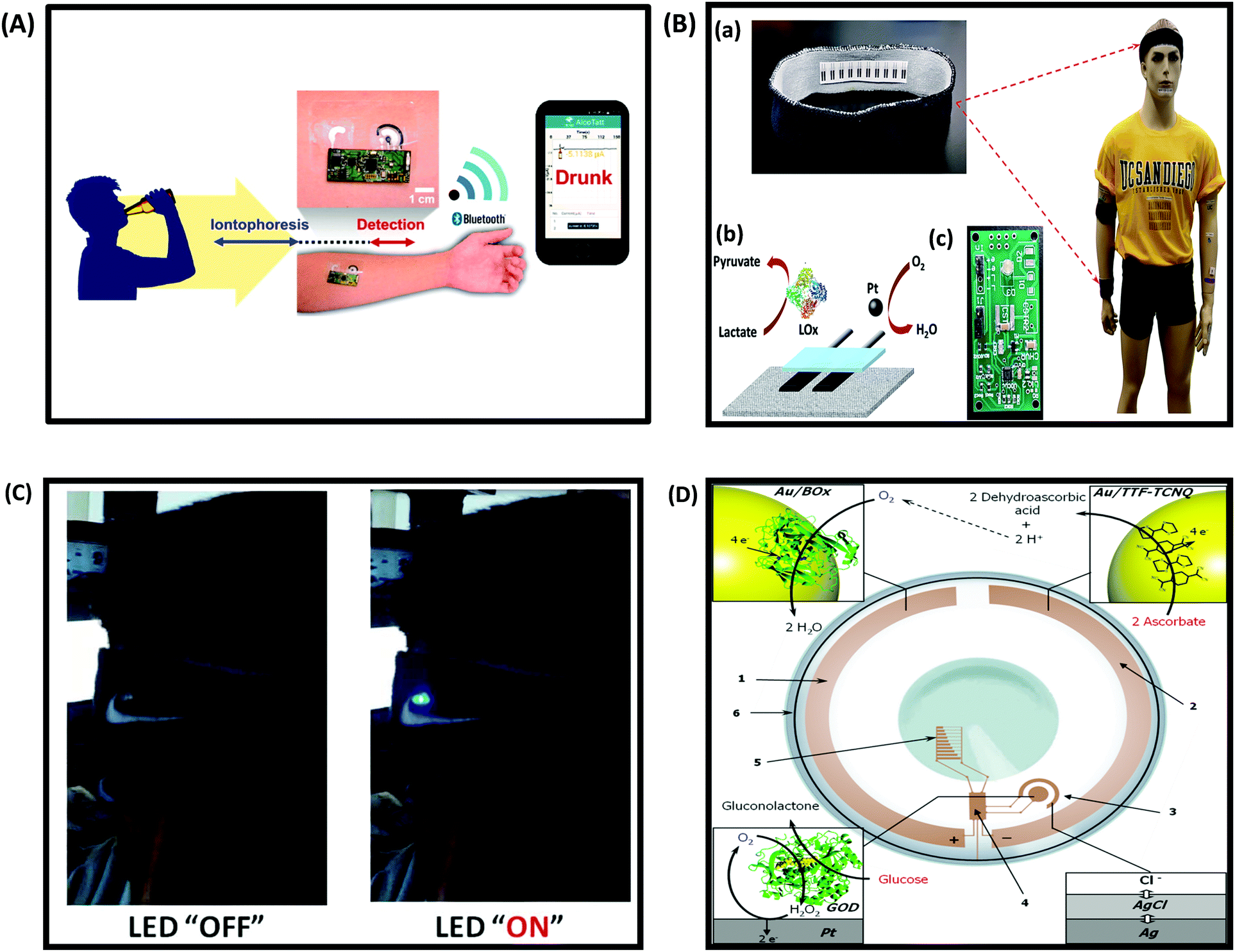 | ||
| Fig. 12 (A) Schematic diagram of a wireless operation of the iontophoretic-sensing tattoo device for transdermal alcohol sensing. In the diagrams of the tattoo-based device, blue and red highlights show the active zones during iontophoresis and amperometric detection, respectively. Figure adapted with permission from ref. 263, (B) wearable textile BFCs. (a) Textile BFCs integrated into various garments such as headbands or wristbands, (b) a scheme of a textile BFC including a bioanode for lactate oxidation and cathode for oxygen reduction, (c) a customized printed circuit board prototype for the conversion, conditioning and temporary storage of extracted energy. Figure adapted with permission from ref. 69, (C) photographs of LED operation powered by four parallel BFCs, integrated into a headband, before and after a stationary bicycle exercise. Figure adapted with permission from ref. 69, (D) conceptual scheme of a bionic contact lens consisting of (1) a biocathode, modified with Au NPs and BOx, (2) an anode, modified with Au NPs and a TTF–TCNQ complex, (3) a glucose biosensor, (4) an interface chip, (5) a simple display, and (6) an antenna. The BFC supplies power to the other electronic components by converting the chemical energy existing in tears into electrical energy, where ascorbate is oxidized to dehydroascorbic acid and oxygen is reduced to water, thereby creating a glucose-sensing electronic contact lens. Figure adapted with permission from ref. 264. | ||
| Type of wearable sensor | Electrochemical detection | Analyte | Analytical performance | Ref. |
|---|---|---|---|---|
| Contact lens | Amperometry | Tear glucose | Fast response (20 s), a high sensitivity (240 A cm−2 mM−1)/detection of less than 0.01 mM, linearity (0.1–0.6 mM) | 271 |
| Contact lens | Amperometry | Tear glucose | Linear relationship between glucose concentration and output current in a range of 0.025–1.475 mmol l−1, with a sensitivity of 0.491 μM mM−1 | 254 |
| Wearable sensor based on functional polymers with Soft-MEMS techniques | Amperometry | Tear glucose | The current output of the glucose sensor related to the glucose level over a range of 0.06–2.00 mmol l−1, with a correlation coefficient of 0.997, and sensitivity of 0.183 nA mM−1 | 255 |
| Wearable patch/graphene based | Impedance, amperometry, potentiometry, tremor sensor (strain gauge) | Sweat-based diabetes monitoring and feedback therapy | The patch consists of a heater, temperature, humidity, glucose and pH sensors and polymeric microneedles that can be thermally activated to deliver drugs transcutaneously | 272 |
| Highly stretchable and printable textile-based sensor array | Potentiometry | Simultaneous multi-ion sweat analysis | Sensor array exhibited a Nernstian response of 59.4 mV/log[Na+], linear range from 10−4 M up to 10−1 M and a limit of detection (LOD) of 10−4.9 M for sodium and a Nernstian response of 56.5 mV/log[K+] from 10−4 M up to 10−1 M and a LOD of 10−4.9 M for the potassium | 259 |
| Wearable sensor based flexible polyimide | Impedance | Glucuronide (EtG) in human sweat | EtG detection over a dose concentration of 0.001–100 μg L−1 with a detection sensitivity of 0.001 μg L−1. The wearable sensor has the ability to detect alcohol consumption of up to 11 standard drinks in the US over a period of 4 to 9 hours | 273 |
| Headband embedded with the fully integrated flexible PET substrate sensing system | Potentiometry | Monitoring of Ca2+ and pH | The Ca2+ sensor shows a near-Nernstian response with an average of 32.7 mV per decade in two complete cycles. The senor shows fast response to changes in Ca2+ levels with a 3.0% relative standard deviation of sensitivity | 251 |
| Flexible tattoo based CNT sensors fabricated via conventional screen printing methods | Amperometry | Lactate | The tattoo sensor linear response throughout the 1 mM to 20 mM range. The linear dynamic range is characterized with high sensitivity (slope, 644.2 nA mM−1 or 10.31 μA mM−1 cm2; correlation coefficient, 0.996) | 257 |
| Wearable flexible screen-printed on-body sensors | Amperometry | Glucose | Influence of mechanical bending upon the sensing performance of the flexible Nafion/GOx/carbon SPEs was studied. Bending the SPEs over a wide range of curvatures does not cause any adverse effect on the current response, but actually leads to an enhanced sensitivity | 260 |
| Smart wristband | Amperometry, potentiometry | Sweat metabolites, skin temperature | Sensitivities of 2.35 nA μM−1 for glucose sensors and 220 nA mM−1 for lactate sensors were reported. Both ion-selective sensors show a near-Nernstian behavior with sensitivities of 64.2 mV and 61.3 mV per decade of concentration for Na+ and K+ sensors, respectively | 252 |
| Graphene-based silk film | Field effect transistor | Glucose | The biosensor showed a large linear detection range of 0.1–10 mM. The detection limit was approximately 0.1 mM (S/N = 3) with excellent selectivity, and the average sensitivity was 2.5 μA mM−1 measured at Vds = 100 mV and Vg = 0 V | 269 |
| Wearable stacked metal/metal-oxide (gold/zinc oxide) thin films within porous polyamide substrates | Non-Faradaic electrochemical impedance spectroscopy | Glucose and cortisol in human sweat | The detection of glucose over a concentration range from 0.01 to 200 mg dL−1 spiked in synthetic and human sweat. Correlation of the sensor response with that of a commercial glucose meter TRUEresult™ sensor (LOD of 20 mg dL−1 in blood) was found to be 0.9 | 274 |
| Wearable sensor patch integrated with a biofuel cell as a power source | Amperometry | Lactate | Linear current response with increasing lactate concentrations with limits of detection ranging from 5 to 100 mM lactate and a sensitivity of 0.2 μA mM−1. The biofuel cell showed excellent performance, achieving over 80 mA at 0.4 V (16 mW) in a footprint of 3.5 × 3.5 × 0.7 cm | 265 |
Another area of extensive development is to create portable and independently operated biofuel cells (BFCs) to power wearable electronics. However, most of the reported on-body BFCs are implemented within the wearer and requires invasive and sophisticated procedures, because it relies on blood glucose as fuel.69,261,262 Thus, utilization of such devices as external energy sources is currently limited. Development of easy-to-wear non-invasive BFCs that utilize metabolites present on the epidermis to generate usable energy and power on-body devices is of great interest for future development.
Jia et al.69 reported the fabrication of a wearable BFC printed directly onto textile substrates and demonstrated the system functionality under a realistic environment (Fig. 12B and C). The textile based BFC utilizes physiologically produced sweat lactate as the fuel to generate electrical energy, generating up to 100 μW cm−2 at 0.34 V during in vitro experimentation, even after repeated bending stress. A wearable NAD+-dependent enzymatic sensor integrated with a BFC power source was designed for non-invasive monitoring of lactate in sweat.265 The overall system is comprised of a lactate biosensor, a glucose oxidase BFC power source, an energy harvester and a micropotentiostat. Integration of a BFC into the bionic contact lens, where enzyme catalysts were employed to convert the chemical energy from biofuel (glucose) and biooxidant (oxygen) available in tear fluid into electrical energy, was proposed264 (Fig. 12D). Human perspiration was also found to be able to generate substantial levels of electrical power in a noninvasive and continuous fashion through the use of epidermal BFCs based on temporary transfer tattoos (tBFCs).266 Integration of nanomaterials such as CNTs267 and graphene268–270 within enzyme-based approaches for wearable applications is under development and is expected to generate more powerful characteristics in the coming years. Table 5 provides some examples of wearable biofuel cells and their performance.
| Wearable BFC characteristics | Fuel/oxidant | Analytical performance of a biofuel cell | Ref. |
|---|---|---|---|
| Biscrolled multi-walled carbon nanotube (MWCNT) yarn BFCs woven into textiles | Glucose/oxygen | Open-circuit voltage of 0.70 V, and a maximum areal power density of 2.18 mW cm−2 | 275 |
| Wearable headband and wristband textile based BFCs | Sweat lactate/dissolved oxygen | Producing up to 100 mW cm2 of power density at 0.34 V during in vitro experimentation | 276 |
| Epidermal BFCs based on temporary transfer tattoos | Lactate/oxygen | Power density can be correlated with the fitness level and aerobic capacity of an individual. It ranged from 5 to 70 mW cm2 | 277 |
| Miniature BFC based Au nanostructured as a power source for glucose sensing contact lenses | Ascorbate/oxygen naturally present in tears | An open circuit voltage of 0.54 V, maximal power densities of 3.1 μW cm−2 at 0.25 V and 0.72 μW cm−2 at 0.4 V, with a stable current density output of over 0.55 μA cm−2 at 0.4 V during 6 h of continuous operation | 264 |
Although the number of biosensing systems that have been reported in the literature is very large, their adaptation to a wearable, lightweight and economical sensing unit is not trivial. A variety of challenges may be encountered when designing a wearable, robust, simple to use and independently operated device. Two main parts are involved in this technology, the enzyme system and the sensing/transduction component, both of which must be integrated to ensure connectivity and functionality in “real world” conditions. The design of wearable devices should also take into consideration the environments in which they will be used. Challenges and limitations related to the enzymatic system are related to the inherent instability of enzymes, the success of the immobilization process with preservation of activity, sensitivity to vibration, mechanical stretch, pH and temperature. Here the key is in the design of the immobilization strategy and the selection of the sensing materials to preserve enzyme conformation and ensure long term stability and functionality. In addition to field stability, other issues such as self-calibration and functionality under variable conditions of changing temperature and humidity need to be resolved. Miniaturizing and connecting the individual sensing units, including the biorecognition unit, the signal transducer and processing components into a portable and compact system, and developing a user friendly interface are still areas that need improvements in the future. Manufacturing these units at a large scale and in a reproducible and economical way is also of interest. The recent advancements in nanopatterning and printing techniques including nano-drop deposition, 2D and 3D printing are expected to generate innovative designs of portable and inexpensive components. Most likely, these will incorporate nanostructured materials and will be interfaced with biomolecules. Of special interest will be the fabrication of the entire biosensing unit, together with signal recognition, transduction and amplification on existing portable platforms such as contact lenses, plastic, textiles, etc. Miniaturization and integration with widespread communication devices such as smart phones and tablets have been proposed. This area of integration is growing and can facilitate data storage, transmission and centralization of information in a computerized form. As these devices become available, ensuring privacy and security of the growing amount of information will be a challenge. Further development of BFCs is also anticipated and will include the fabrication of BFCs incorporated into flexible materials, integration of microelectronics, new materials and mechanisms to improve self-powered efficiency and utilization of alternate fuels to further increase the operation time.
5. Conclusion
Significant efforts are currently underway to fabricate bioactive nanostructures with defined and controllable properties for implementation in portable biosensors. In order to ensure commercial viability, the development of technology for low cost, high yield and scalable fabrication of these devices is also of interest. Nanoarchitectural materials have shown significant promise for enhancing enzyme stability and function and have made a significant impact in biotechnology and biosensing. This paper provided an overview of the main classes of NPs and enzyme-based nanoassembly methods used for the fabrication of portable biosensors. Different strategies used to functionalize nanostructures with enzymes, development and characterization of functional nanoassemblies, evaluation of biological activity and generation of response and sensing function have been discussed. Methods enabling roll-to-roll fabrication have been reviewed, with focus on emerging technologies such as paper based, flexible and wearable devices. While several flexible and wearable biosensing systems have been reported in the literature, their implementation into routine functional devices still remains a challenge. With the advancement of nanofabrication, 2D, 3D and microarray printing, and the large investment and promotion of this technology in the commercial sector, we can expect that an increased activity can be seen in this field in the future. Improvements in nanoassembly and device fabrication are expected to provide better dimensional control and stability of enzymes within nanostructures. In addition to novel prototypes, connectivity and integration of biosensing units with smartphones and wireless communication devices are also expected. These future efforts can lead to improved measurement tools and portable devices for clinical diagnosis, environmental monitoring, food quality control, aerospace and defense, safety and security applications. These devices are particularly appealing for field analysis as low cost screening devices, where specialized equipment is not available.Acknowledgements
This work was funded by the National Science Foundation under Grants No. 1561491 and 1200180. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.Notes and references
- G. S. Wilson and Y. Hu, Chem. Rev., 2000, 100, 2693–2704 CrossRef CAS PubMed.
- C. R. Ispas, G. Crivat and S. Andreescu, Anal. Lett., 2012, 45, 168–186 CrossRef CAS.
- S. Andreescu, J. Njagi and C. Ispas, New Frontiers of Organic and Composite Nanotechnology, 2008, pp. 355–394 DOI:10.1016/B978-008045052-0.50009-9.
- Y. Khan, A. E. Ostfeld, C. M. Lochner, A. Pierre and A. C. Arias, Adv. Mater., 2016, 28, 4373–4395 CrossRef CAS PubMed.
- M. Gabig-Ciminska, Microb. Cell Fact., 2006, 5, 9 CrossRef PubMed.
- D. Mabey, R. W. Peeling, A. Ustianowski and M. D. Perkins, Nat. Rev. Microbiol., 2004, 2, 231–240 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- W. A. Zhao, M. M. Ali, S. D. Aguirre, M. A. Brook and Y. F. Li, Anal. Chem., 2008, 80, 8431–8437 CrossRef CAS PubMed.
- R. Pelton, TrAC, Trends Anal. Chem., 2009, 28, 925–942 CrossRef CAS.
- A. Martinez, S. Phillips, M. Butte and G. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chim. Acta, 2010, 674, 227–233 CrossRef CAS PubMed.
- M. Reches, K. A. Mirica, R. Dasgupta, M. D. Dickey, M. J. Butte and G. M. Whitesides, ACS Appl. Mater. Interfaces, 2010, 2, 1722–1728 CAS.
- Z. H. Nie, C. A. Nijhuis, J. L. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab Chip, 2010, 10, 477–483 RSC.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chem., 2009, 81, 5821–5826 CrossRef CAS PubMed.
- J. P. Metters, S. M. Houssein, D. K. Kampouris and C. E. Banks, Anal. Methods, 2013, 5, 103–110 RSC.
- R. Baron, B. Willner and I. Willner, Chem. Commun., 2007, 323–332 RSC.
- A. Popat, S. B. Hartono, F. Stahr, J. Liu, S. Z. Qiao and G. Qing Lu, Nanoscale, 2011, 3, 2801–2818 RSC.
- J. Kim, J. W. Grate and P. Wang, Chem. Eng. Sci., 2006, 61, 1017–1026 CrossRef CAS.
- D. H. Jo, J. H. Kim, T. G. Lee and J. H. Kim, Nanomed. Nanotech. Biol. Med., 2015, 11, 1603–1611 CrossRef CAS PubMed.
- C. Czeslik and R. Winter, Phys. Chem. Chem. Phys., 2001, 3, 235–239 RSC.
- H. Jia, G. Zhu and P. Wang, Biotechnol. Bioeng., 2003, 84, 406–414 CrossRef CAS PubMed.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef PubMed.
- R. Ahmad and M. Sardar, Biochem. Anal. Biochem., 2015, 4.2, 1–8 Search PubMed.
- A. Karimi, A. Othman, A. Uzunoglu, L. Stanciu and S. Andreescu, Nanoscale, 2015, 7, 6909–6923 RSC.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, Biotechnology, 2013, 3, 1–9 Search PubMed.
- J. Bryjak and B. N. Kolarz, Process Biochem., 1998, 33, 409–417 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- J. P. Lata, L. Gao, C. Mukai, R. Cohen, J. L. Nelson, L. Anguish, S. Coonrod and A. J. Travis, Bioconjugate Chem., 2015, 26, 1931–1938 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- Y.-H. Won, D. Aboagye, H. S. Jang, A. Jitianu and L. A. Stanciu, J. Mater. Chem., 2010, 20, 5030–5034 RSC.
- J. Njagi and S. Andreescu, Biosens. Bioelectron., 2007, 23, 168–175 CrossRef CAS PubMed.
- A. Heller, Acc. Chem. Res., 1990, 23, 128–134 CrossRef CAS.
- S. Demin and E. A. H. Hall, Bioelectrochemistry, 2009, 76, 19–27 CrossRef CAS PubMed.
- J. M. Pingarrón, P. Yáñez-Sedeño and A. González-Cortés, Electrochim. Acta, 2008, 53, 5848–5866 CrossRef.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- D. L. Arruda, W. C. Wilson, C. Nguyen, Q. W. Yao, R. J. Caiazzo, I. Talpasanu, D. E. Dow and B. C. S. Liu, Expert Rev. Mol. Diagn., 2009, 9, 749–755 CrossRef CAS PubMed.
- J. Naruse, L. Q. Hoa, Y. Sugano, T. Ikeuchi, H. Yoshikawa, M. Saito and E. Tamiya, Biosens. Bioelectron., 2011, 30, 204–210 CrossRef CAS PubMed.
- H. Ai, S. Jones and Y. Lvov, Cell Biochem. Biophys., 2003, 39, 23–43 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5018–5023 CrossRef CAS PubMed.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed.
- M. J. Chaichi and M. Ehsani, Sens. Actuators, B, 2016, 223, 713–722 CrossRef CAS.
- C. Monteil, N. Bar, R. Retoux, J. Henry, B. Bernay and D. Villemin, Sens. Actuators, B, 2014, 192, 269–274 CrossRef CAS.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- Z. J. Deng, G. Mortimer, T. Schiller, A. Musumeci, D. Martin and R. F. Minchin, Nanotechnology, 2009, 20, 455101 CrossRef PubMed.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CAS.
- Z. J. Deng, M. T. Liang, I. Toth, M. J. Monteiro and R. F. Minchin, ACS Nano, 2012, 6, 8962–8969 CrossRef CAS PubMed.
- E. Sharpe, T. Frasco, D. Andreescu and S. Andreescu, Analyst, 2013, 138, 249–262 RSC.
- W. Putzbach and N. Ronkainen, Sensors, 2013, 13, 4811 CrossRef CAS PubMed.
- J. C. Claussen, A. Malanoski, J. C. Breger, E. Oh, S. A. Walper, K. Susumu, R. Goswami, J. R. Deschamps and I. L. Medintz, J. Phys. Chem. C, 2015, 119, 2208–2221 CAS.
- B. J. Johnson, W. Russ Algar, A. P. Malanoski, M. G. Ancona and I. L. Medintz, Nano Today, 2014, 9, 102–131 CrossRef CAS.
- P.-H. Chou, S.-H. Chen, H.-K. Liao, P.-C. Lin, G.-R. Her, A. C.-Y. Lai, J.-H. Chen, C.-C. Lin and Y.-J. Chen, Anal. Chem., 2005, 77, 5990–5997 CrossRef CAS PubMed.
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83, 4453–4488 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- E. H. Alsharaeh and A. A. Othman, Polym. Compos., 2014, 35, 2318–2323 CrossRef CAS.
- H. Wang, M. Chhowalla, N. Sano, S. Jia and G. Amaratunga, Nanotechnology, 2004, 15, 546–550 CrossRef CAS.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS PubMed.
- J. Lu, I. Do, L. T. Drzal, R. M. Worden and I. Lee, ACS Nano, 2008, 2, 1825–1832 CrossRef CAS PubMed.
- K. Balasubramanian and M. Burghard, Anal. Bioanal. Chem., 2006, 385, 452–468 CrossRef CAS PubMed.
- B. S. Sherigara, W. Kutner and F. D'Souza, Electroanalysis, 2003, 15, 753–772 CrossRef CAS.
- M. Shim, N. W. Shi Kam, R. J. Chen, Y. Li and H. Dai, Nano Lett., 2002, 2, 285–288 CrossRef CAS.
- J. Zhang, F. Zhang, H. Yang, X. Huang, H. Liu, J. Zhang and S. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- A. A. Sehat, A. A. Khodadadi, F. Shemirani and Y. Mortazavi, Int. J. Electrochem. Sci., 2015, 10, 272–286 Search PubMed.
- M. Patila, A. Kouloumpis, D. Gournis, P. Rudolf and H. Stamatis, Sensors, 2016, 16, 287 CrossRef PubMed.
- S. Hermanová, M. Zarevúcká, D. Bouša, M. Pumera and Z. Sofer, Nanoscale, 2015, 7, 5852–5858 RSC.
- W. Jia, X. Wang, S. Imani, A. J. Bandodkar, J. Ramírez, P. P. Mercier and J. Wang, J. Mater. Chem. A, 2014, 2, 18184–18189 CAS.
- Y. Zhang, J. Zhang, X. Huang, X. Zhou, H. Wu and S. Guo, Small, 2012, 8, 154–159 CrossRef CAS PubMed.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901–905 CrossRef CAS PubMed.
- S. K. Tuteja, M. Kukkar, C. R. Suri, A. K. Paul and A. Deep, Biosens. Bioelectron., 2015, 66, 129–135 CrossRef CAS PubMed.
- M. Zhang, C. Liao, C. H. Mak, P. You, C. L. Mak and F. Yan, Sci. Rep., 2015, 5, 8311 CrossRef CAS PubMed.
- Y. Song, C. Chen and C. Wang, Nanoscale, 2015, 7, 7084–7090 RSC.
- V. Mani, R. Devasenathipathy, S.-M. Chen, B. Subramani and M. Govindasamy, Int. J. Electrochem. Sci., 2015, 10, 691–700 Search PubMed.
- K. ul Hasan, M. H. Asif, O. Nur and M. Willander, J. Biosens. Bioelectron., 2012, 3, 114 CAS.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 26, 6158–6160 CrossRef CAS PubMed.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Biosens. Bioelectron., 2010, 25, 1070–1074 CrossRef CAS PubMed.
- S. G. Wang, Q. Zhang, R. Wang, S. F. Yoon, J. Ahn, D. J. Yang, J. Z. Tian, J. Q. Li and Q. Zhou, Electrochem. Commun., 2003, 5, 800–803 CrossRef CAS.
- S. Palanisamy, S. Cheemalapati and S.-M. Chen, Int. J. Electrochem. Sci., 2012, 7, 8394–8407 CAS.
- J. C. Claussen, A. D. Franklin, A. ul Haque, D. M. Porterfield and T. S. Fisher, ACS Nano, 2009, 3, 37–44 CrossRef CAS PubMed.
- A. P. Periasamy, Y.-J. Chang and S.-M. Chen, Bioelectrochemistry, 2011, 80, 114–120 CrossRef CAS PubMed.
- G. Li, J. Liao, G. Hu, N. Ma and P. Wu, Biosens. Bioelectron., 2005, 20, 2140–2144 CrossRef CAS PubMed.
- K. Besteman, J.-O. Lee, F. G. M. Wiertz, H. A. Heering and C. Dekker, Nano Lett., 2003, 3, 727–730 CrossRef CAS.
- T. Liu, J. Zhong, X. Gan, C. Fan, G. Li and N. Matsuda, ChemPhysChem, 2003, 4, 1364–1366 CrossRef CAS PubMed.
- C. Ren, Y. Song, Z. Li and G. Zhu, Anal. Bioanal. Chem., 2005, 381, 1179–1185 CrossRef CAS PubMed.
- D. Du, S. Chen, J. Cai and A. Zhang, Biosens. Bioelectron., 2007, 23, 130–134 CrossRef CAS PubMed.
- G. A. Petkova, K. Záruba, P. Žvátora and V. Král, Nanoscale Res. Lett., 2012, 7, 287 CrossRef PubMed.
- C.-L. Wu, Y.-P. Chen, J.-C. Yang, H.-F. Lo and L.-L. Lin, J. Mol. Catal. B: Enzym., 2008, 54, 83–89 CrossRef CAS.
- D. Lan, B. Li and Z. Zhang, Biosens. Bioelectron., 2008, 24, 934–938 CrossRef CAS PubMed.
- X. Fu, X. Feng, K. Xu and R. Huang, Anal. Methods, 2014, 6, 2233–2238 RSC.
- J. D. Keighron and C. D. Keating, Langmuir, 2010, 26, 18992–19000 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- M. Ganesana, G. Istarnboulie, J. L. Marty, T. Noguer and S. Andreescu, Biosens. Bioelectron., 2011, 30, 43–48 CrossRef CAS PubMed.
- K. B. Lee, S. Park and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 3048–3050 CrossRef CAS PubMed.
- A. A. Awang, J. Lee, G. Jenikova, A. Mulchandani, N. V. Myung and W. Chen, Nanotechnology, 2006, 17, 3375–3379 CrossRef PubMed.
- J. M. Abad, S. F. L. Mertens, M. Pita, V. M. Fernandez and D. J. Schiffrin, J. Am. Chem. Soc., 2005, 127, 5689–5694 CrossRef CAS PubMed.
- L. Wang and E. Wang, Electrochem. Commun., 2004, 6, 49–54 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- P. Raghu, T. Madhusudana Reddy, K. Reddaiah, L. R. Jaidev and G. Narasimha, Enzyme Microb. Technol., 2013, 52, 377–385 CrossRef CAS PubMed.
- H. Yin, S. Ai, W. Shi and L. Zhu, Sens. Actuators, B, 2009, 137, 747–753 CrossRef CAS.
- K. Murata, M. Suzuki, K. Kajiya, N. Nakamura and H. Ohno, Electrochem. Commun., 2009, 11, 668–671 CrossRef CAS.
- R. S. J. Alkasir, M. Ganesana, Y.-H. Won, L. Stanciu and S. Andreescu, Biosens. Bioelectron., 2010, 26, 43–49 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- E. Hutter and D. Maysinger, Trends Pharmacol. Sci., 2013, 34, 497–507 CrossRef CAS PubMed.
- M. Willander, K. Khun and Z. Ibupoto, Sensors, 2014, 14, 8605 CrossRef PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- Y. Xu and E. Wang, Electrochim. Acta, 2012, 84, 62–73 CrossRef CAS.
- J. Xu, J. Sun, Y. Wang, J. Sheng, F. Wang and M. Sun, Molecules, 2014, 19, 11465–11486 CrossRef PubMed.
- G. Sinigaglia, M. Magro, G. Miotto, S. Cardillo, E. Agostinelli, R. Zboril, E. Bidollari and F. Vianello, Int. J. Nanomed., 2012, 7, 2249–2259 CAS.
- M. Martin, P. Salazar, R. Villalonga, S. Campuzano, J. M. Pingarron and J. L. Gonzalez-Mora, J. Mater. Chem. B, 2014, 2, 739–746 RSC.
- C. J. Xu, K. M. Xu, H. W. Gu, X. F. Zhong, Z. H. Guo, R. K. Zheng, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 3392–3393 CrossRef CAS PubMed.
- T. Noguer, G. Istarnboulie, S. Andreescu and J. L. Marty, Biosens. Bioelectron., 2007, 23, 506–512 CrossRef PubMed.
- N. Gan, X. Yang, D. Xie, Y. Wu and W. Wen, Sensors, 2010, 10, 625–638 CrossRef CAS PubMed.
- P. Si, P. Chen and D.-H. Kim, J. Mater. Chem. B, 2013, 1, 2696–2700 RSC.
- X.-L. Luo, J.-J. Xu, W. Zhao and H.-Y. Chen, Biosens. Bioelectron., 2004, 19, 1295–1300 CrossRef CAS PubMed.
- L. Zhang, S.-M. Yuan, L.-m. Yang, Z. Fang and G.-C. Zhao, Microchim. Acta, 2013, 180, 627–633 CrossRef CAS.
- Z. Yang, Y. Tang, J. Li, Y. Zhang and X. Hu, Biosens. Bioelectron., 2014, 54, 528–533 CrossRef CAS PubMed.
- H. Zhong, R. Yuan, Y. Chai, W. Li, Y. Zhang and C. Wang, Bioprocess Biosys. Eng., 2011, 34, 923–930 CrossRef CAS PubMed.
- G.-Y. Adachi and N. Imanaka, Chem. Rev., 1998, 98, 1479–1514 CrossRef CAS.
- R. Li, Z. Ji, C. H. Chang, D. R. Dunphy, X. Cai, H. Meng, H. Zhang, B. Sun, X. Wang, J. Dong, S. Lin, M. Wang, Y.-P. Liao, C. J. Brinker, A. Nel and T. Xia, ACS Nano, 2014, 8, 1771–1783 CrossRef CAS PubMed.
- M. Nichkova, D. Dosev, S. J. Gee, B. D. Hammock and I. M. Kennedy, Anal. Chem., 2005, 77, 6864–6873 CrossRef CAS PubMed.
- T. Pirmohamed, J. M. Dowding, S. Singh, B. Wasserman, E. Heckert, A. S. Karakoti, J. E. S. King, S. Seal and W. T. Self, Chem. Commun., 2010, 46, 2736–2738 RSC.
- M. Das, S. Patil, N. Bhargava, J.-F. Kang, L. M. Riedel, S. Seal and J. J. Hickman, Biomaterials, 2007, 28, 1918–1925 CrossRef CAS PubMed.
- M. Ornatska, E. Sharpe, D. Andreescu and S. Andreescu, Anal. Chem., 2011, 83, 4273–4280 CrossRef CAS PubMed.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, J. Biotechnol., 2009, 142, 179–184 CrossRef CAS PubMed.
- G. Preda, A. Migani, K. M. Neyman, S. T. Bromley, F. Illas and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 5817–5822 CAS.
- M. Zhang, H. Wang, X. Wang and W. Li, Mater. Des., 2006, 27, 489–493 CrossRef CAS.
- D. Wang, Y. Kang, V. Doan-Nguyen, J. Chen, R. Küngas, N. L. Wieder, K. Bakhmutsky, R. J. Gorte and C. B. Murray, Angew. Chem., Int. Ed., 2011, 50, 4378–4381 CrossRef CAS PubMed.
- J. Xu, J. Harmer, G. Li, T. Chapman, P. Collier, S. Longworth and S. C. Tsang, Chem. Commun., 2010, 46, 1887–1889 RSC.
- P. Dutta, S. Pal, M. S. Seehra, Y. Shi, E. M. Eyring and R. D. Ernst, Chem. Mater., 2006, 18, 5144–5146 CrossRef CAS.
- S. M. Z. Hossain, R. E. Luckham, A. M. Smith, J. M. Lebert, L. M. Davies, R. H. Pelton, C. D. M. Filipe and J. D. Brennan, Anal. Chem., 2009, 81, 5474–5483 CrossRef CAS PubMed.
- N. P. Sardesai, M. Ganesana, A. Karimi, J. C. Leiter and S. Andreescu, Anal. Chem., 2015, 87, 2996–3003 CrossRef CAS PubMed.
- N. P. Sardesai, A. Karimi and S. Andreescu, ChemElectroChem, 2014, 1, 2082–2088 CrossRef CAS.
- R. S. J. Alkasir, M. Ornatska and S. Andreescu, Anal. Chem., 2012, 84, 9729–9737 CrossRef CAS PubMed.
- R. S. J. Alkasir, A. Rossner and S. Andreescu, Environ. Sci. Technol., 2015, 49, 9889–9897 CrossRef CAS PubMed.
- E. Sharpe, R. Bradley, T. Frasco, D. Jayathilaka, A. Marsh and S. Andreescu, Sens. Actuators, B, 2014, 193, 552–562 CrossRef CAS PubMed.
- A. Vasilescu, E. Sharpe and S. Andreescu, Curr. Anal. Chem., 2012, 8, 495–505 CrossRef CAS.
- R. E. Ozel, C. Ispas, M. Ganesana, J. C. Leiter and S. Andreescu, Biosens. Bioelectron., 2014, 52, 397–402 CrossRef CAS PubMed.
- A. K. Sinha and K. Suzuki, J. Phys. Chem. B, 2005, 109, 1708–1714 CrossRef CAS PubMed.
- C. Ispas, J. Njagi, M. Cates and S. Andreescu, J. Electrochem. Soc., 2008, 155, F169–F176 CrossRef CAS.
- R. Khan and M. Dhayal, Electrochem. Commun., 2008, 10, 263–267 CrossRef CAS.
- J. Njagi, M. M. Chernov, J. C. Leiter and S. Andreescu, Anal. Chem., 2010, 82, 989–996 CrossRef CAS PubMed.
- P. Reiss, M. Protiere and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Delivery Rev., 2012, 64, 967–978 CrossRef CAS PubMed.
- E. Petryayeva and U. J. Krull, Langmuir, 2012, 28, 13943–13951 CrossRef CAS PubMed.
- K.-E. Kim, T. Kim and Y.-M. Sung, J. Nanopart. Res., 2012, 14, 1–9 Search PubMed.
- X. Ji, J. Zheng, J. Xu, V. K. Rastogi, T.-C. Cheng, J. J. DeFrank and R. M. Leblanc, J. Phys. Chem. B, 2005, 109, 3793–3799 CrossRef CAS PubMed.
- S. George, T. Xia, R. Rallo, Y. Zhao, Z. Ji, S. Lin, X. Wang, H. Zhang, B. France and D. Schoenfeld, ACS Nano, 2011, 5, 1805–1817 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- W. Chen, Y. Inoue and K. Ishihara, Colloids Surf., B, 2015, 135, 365–370 CrossRef CAS PubMed.
- K. Welser, M. D. A. Perera, J. W. Aylott and W. C. Chan, Chem. Commun., 2009, 6601–6603, 10.1039/b914358k.
- L.-M. Zhao, L.-E. Shi, Z.-L. Zhang, J.-M. Chen, D.-D. Shi, J. Yang and Z.-X. Tang, Braz. J. Chem. Eng., 2011, 28, 353–362 CrossRef CAS.
- D. Ateh, H. Navsaria and P. Vadgama, J. R. Soc., Interface, 2006, 3, 741–752 CrossRef CAS PubMed.
- M. Stočes, K. Kalcher, I. Švancara and K. Vytřas, Int. J. Electrochem. Sci., 2011, 6, 1917–1926 Search PubMed.
- F. N. Crespilho, R. M. Iost, S. A. Travain, O. N. Oliveira and V. Zucolotto, Biosens. Bioelectron., 2009, 24, 3073–3077 CrossRef CAS PubMed.
- P. Santhosh, K. M. Manesh, S. Uthayakumar, S. Komathi, A. I. Gopalan and K. P. Lee, Bioelectrochemistry, 2009, 75, 61–66 CrossRef CAS PubMed.
- S. Kakhki, M. M. Barsan, E. Shams and C. Brett, Electroanalysis, 2013, 25, 77–84 CrossRef CAS.
- H. Liu, J. Kameoka, D. A. Czaplewski and H. Craighead, Nano Lett., 2004, 4, 671–675 CrossRef CAS.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS PubMed.
- A. A. Gokhale, J. Lu, R. R. Weerasiri, J. Yu and I. Lee, Electroanalysis, 2015, 27, 1127–1137 CrossRef CAS.
- K. Abe, K. Suzuki and D. Citterio, Anal. Chem., 2008, 80, 6928–6934 CrossRef CAS PubMed.
- S. A. Gowers, V. F. Curto, C. A. Seneci, C. Wang, S. Anastasova, P. Vadgama, G. Z. Yang and M. G. Boutelle, Anal. Chem., 2015, 87, 7763–7770 CrossRef CAS PubMed.
- K. B. Lee, J. H. Lim and C. A. Mirkin, J. Am. Chem. Soc., 2003, 125, 5588–5589 CrossRef CAS PubMed.
- L. Fabie, P. Agostini, M. Stopel, C. Blum, B. Lassagne, V. Subramaniam and T. Ondarcuhu, Nanoscale, 2015, 7, 4497–4504 RSC.
- J. T. Delaney, P. J. Smith and U. S. Schubert, Soft Matter, 2009, 5, 4866–4877 RSC.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS PubMed.
- S. Andreescu, L. Barthelmebs and J. L. Marty, Anal. Chim. Acta, 2002, 464, 171–180 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- J. Wang, B. Tian, V. B. Nascimento and L. Angnes, Electrochim. Acta, 1998, 43, 3459–3465 CrossRef CAS.
- P. Fanjul-Bolado, D. Hernández-Santos, P. J. Lamas-Ardisana, A. Martín-Pernía and A. Costa-García, Electrochim. Acta, 2008, 53, 3635–3642 CrossRef CAS.
- S. Cinti, F. Arduini, M. Carbone, L. Sansone, I. Cacciotti, D. Moscone and G. Palleschi, Electroanalysis, 2015, 27, 2230–2238 CrossRef CAS.
- F.-Y. Kong, S.-X. Gu, W.-W. Li, T.-T. Chen, Q. Xu and W. Wang, Biosens. Bioelectron., 2014, 56, 77–82 CrossRef CAS PubMed.
- S. Azzouzi, L. Rotariu, A. M. Benito, W. K. Maser, M. Ben Ali and C. Bala, Biosens. Bioelectron., 2015, 69, 280–286 CrossRef CAS PubMed.
- S. Kurbanoglu, C. C. Mayorga-Martinez, M. Medina-Sánchez, L. Rivas, S. A. Ozkan and A. Merkoçi, Biosens. Bioelectron., 2015, 67, 670–676 CrossRef CAS PubMed.
- P. J. Lamas-Ardisana, O. A. Loaiza, L. Añorga, E. Jubete, M. Borghei, V. Ruiz, E. Ochoteco, G. Cabañero and H. J. Grande, Biosens. Bioelectron., 2014, 56, 345–351 CrossRef CAS PubMed.
- T. S. C. R. Rebelo, C. M. Pereira, M. G. F. Sales, J. P. Noronha, J. Costa-Rodrigues, F. Silva and M. H. Fernandes, Anal. Chim. Acta, 2014, 850, 26–32 CrossRef CAS PubMed.
- A. L. Alvarado-Gámez, M. A. Alonso-Lomillo, O. Domínguez-Renedo and M. J. Arcos-Martínez, Sensors, 2014, 14, 3756–3767 CrossRef PubMed.
- M. Barquero-Quirós, O. Domínguez-Renedo, M. A. Alonso-Lomillo and M. J. Arcos-Martínez, Sensors, 2014, 14, 8203–8216 CrossRef PubMed.
- W. Henao-Escobar, O. Domínguez-Renedo, M. Asunción Alonso-Lomillo and M. Julia Arcos-Martínez, Talanta, 2013, 117, 405–411 CrossRef CAS PubMed.
- M. Nurul Karim and H. J. Lee, Talanta, 2013, 116, 991–996 CrossRef CAS PubMed.
- S. Banerjee, P. Sarkar and A. P. F. Turner, Anal. Biochem., 2013, 439, 194–200 CrossRef CAS PubMed.
- S. A. V. Eremia, I. Vasilescu, A. Radoi, S. C. Litescu and G. L. Radu, Talanta, 2013, 110, 164–170 CrossRef CAS PubMed.
- B. Situ, N. Cao, B. Li, Q. Liu, L. Lin, Z. Dai, X. Zou, Z. Cai, Q. Wang, X. Yan and L. Zheng, Biosens. Bioelectron., 2013, 43, 257–263 CrossRef CAS PubMed.
- P. Hernandez, J. A. Rodriguez, C. A. Galan, Y. Castrillejo and E. Barrado, Biosens. Bioelectron., 2013, 41, 244–248 CrossRef CAS PubMed.
- C. C. Mayorga-Martinez, M. Cadevall, M. Guix, J. Ros and A. Merkoçi, Biosens. Bioelectron., 2013, 40, 57–62 CrossRef CAS PubMed.
- X. Niu, C. Chen, H. Zhao, Y. Chai and M. Lan, Biosens. Bioelectron., 2012, 36, 262–266 CrossRef CAS PubMed.
- M. N. Karim, J. E. Lee and H. J. Lee, Biosens. Bioelectron., 2014, 61, 147–151 CrossRef CAS PubMed.
- W. Song, D.-W. Li, Y.-T. Li, Y. Li and Y.-T. Long, Biosens. Bioelectron., 2011, 26, 3181–3186 CrossRef CAS PubMed.
- Y. B. Choi, H. G. Kim, G. H. Han, H. H. Kim and S. W. Kim, Biotechnol. Bioprocess Eng., 2011, 16, 631–637 CrossRef CAS.
- R. S. Dey and C. R. Raj, J. Phys. Chem. C, 2010, 114, 21427–21433 CAS.
- B. Feng and Y. N. Liu, Int. J. Electrochem. Sci., 2015, 10, 4770–4778 CAS.
- B. Borisova, A. Sánchez, S. Jiménez-Falcao, M. Martín, P. Salazar, C. Parrado, J. M. Pingarrón and R. Villalonga, Sens. Actuators, B, 2016, 232, 84–90 CrossRef CAS.
- Y. Zhang, F. Lyu, J. Ge and Z. Liu, Chem. Commun., 2014, 50, 12919–12922 RSC.
- Y. H. Yun, B. K. Lee, J. S. Choi, S. Kim, B. Yoo, Y. S. Kim, K. Park and Y. W. Cho, Anal. Sci., 2011, 27, 375 CrossRef CAS PubMed.
- P. Calvert, Chem. Mater., 2001, 13, 3299–3305 CrossRef CAS.
- T. T. Nge, M. Nogi and K. Suganuma, J. Mater. Chem. C, 2013, 1, 5235–5243 RSC.
- E. Halonen, T. Viiru, K. Ostman, A. L. Cabezas and M. Mantysalo, IEEE Trans. Compon., Packag., Manuf. Technol., 2013, 3, 350–356 CrossRef CAS.
- N. Komuro, S. Takaki, K. Suzuki and D. Citterio, Anal. Bioanal. Chem., 2013, 405, 5785–5805 CrossRef CAS PubMed.
- G. C. Jensen, C. E. Krause, G. A. Sotzing and J. F. Rusling, Phys. Chem. Chem. Phys., 2011, 13, 4888–4894 RSC.
- B. Weng, A. Morrin, R. Shepherd, K. Crowley, A. J. Killard, P. C. Innis and G. G. Wallace, J. Mater. Chem. B, 2014, 2, 793–799 RSC.
- B. Creran, X. Li, B. Duncan, C. S. Kim, D. F. Moyano and V. M. Rotello, ACS Appl. Mater. Interfaces, 2014, 6, 19525–19530 CAS.
- F. Pati, D. H. Ha, J. Jang, H. H. Han, J. W. Rhie and D. W. Cho, Biomaterials, 2015, 62, 164–175 CrossRef CAS PubMed.
- K. Markstedt, A. Mantas, I. Tournier, H. M. Avila, D. Hagg and P. Gatenholm, Biomacromolecules, 2015, 16, 1489–1496 CrossRef CAS PubMed.
- G. Chansin, IDTechEx, 2015.
- J. Harrop, IDTechEx research, 2014.
- M. Gou, X. Qu, W. Zhu, M. Xiang, J. Yang, K. Zhang, Y. Wei and S. Chen, Nat. Commun., 2014, 5, 3774 CAS.
- D. Wouters and U. S. Schubert, Angew. Chem., Int. Ed., 2004, 43, 2480–2495 CrossRef CAS PubMed.
- B. Basnar and I. Willner, Small, 2009, 5, 28–44 CrossRef CAS PubMed.
- B. Basnar, Y. Weizmann, Z. Cheglakov and I. Willner, Adv. Mater., 2006, 18, 713–718 CrossRef CAS.
- Z. Hua, L. Ki-Bum, L. Zhi and A. M. Chad, Nanotechnology, 2003, 14, 1113 CrossRef.
- C.-C. Wu, H. Xu, C. Otto, D. N. Reinhoudt, R. G. Lammertink, J. Huskens, V. Subramaniam and A. H. Velders, J. Am. Chem. Soc., 2009, 131, 7526–7527 CrossRef CAS PubMed.
- S. J. Xia, M. Cartron, J. Morby, D. A. Bryant, C. N. Hunter and G. J. Leggett, Langmuir, 2016, 32, 1818–1827 CrossRef CAS PubMed.
- A. Jamshidi, S. L. Neale, K. Yu, P. J. Pauzauskie, P. J. Schuck, J. K. Valley, H. Y. Hsu, A. T. Ohta and M. C. Wu, Nano Lett., 2009, 9, 2921–2925 CrossRef CAS PubMed.
- J. W. Liu, D. Mazumdar and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 7955–7959 CrossRef CAS PubMed.
- R. Pelton, TrAC, Trends Anal. Chem., 2009, 28, 925–942 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- D. M. Cate, W. Dungchai, J. C. Cunningham, J. Volckens and C. S. Henry, Lab Chip, 2013, 13, 2397–2404 RSC.
- A. W. Martinez, S. T. Phillips, E. Carrilho, S. W. Thomas Iii, H. Sindi and G. M. Whitesides, Anal. Chem., 2008, 80, 3699–3707 CrossRef CAS PubMed.
- C. S. K. Radhakumary, Anal. Chem., 2011, 83, 2829–2833 CrossRef CAS PubMed.
- S. Nantaphol, O. Chailapakul and W. Siangproh, Anal. Chim. Acta, 2015, 891, 136–143 CrossRef CAS PubMed.
- J. Benson, C. M. Fung, J. S. Lloyd, D. Deganello, N. A. Smith and K. S. Teng, Nanoscale Res. Lett., 2015, 10, 1–8 CrossRef CAS PubMed.
- L. Setti, A. Fraleoni-Morgera, B. Ballarin, A. Filippini, D. Frascaro and C. Piana, Biosens. Bioelectron., 2005, 20, 2019–2026 CrossRef CAS PubMed.
- T. Lin, L. Zhong, Z. Song, L. Guo, H. Wu, Q. Guo, Y. Chen, F. Fu and G. Chen, Biosens. Bioelectron., 2014, 62, 302–307 CrossRef CAS PubMed.
- Y. He, S. Zhang, X. Zhang, M. Baloda, A. S. Gurung, H. Xu, X. Zhang and G. Liu, Biosens. Bioelectron., 2011, 26, 2018–2024 CrossRef CAS PubMed.
- L. Su, W. Qin, H. Zhang, Z. U. Rahman, C. Ren, S. Ma and X. Chen, Biosens. Bioelectron., 2015, 63, 384–391 CrossRef CAS PubMed.
- D. Du, J. Wang, L. Wang, D. Lu and Y. Lin, Anal. Chem., 2012, 84, 1380–1385 CrossRef CAS PubMed.
- J. Lin, W. Qu and S. Zhang, Anal. Biochem., 2007, 360, 288–293 CrossRef CAS PubMed.
- P. Liang, H. Yu, B. Guntupalli and Y. Xiao, ACS Appl. Mater. Interfaces, 2015, 7, 15023–15030 CAS.
- X. Li, C. Zhao and X. Liu, Microsystems & Nanoengineering, 2015, 1, 15014 Search PubMed.
- R. E. Luckham and J. D. Brennan, Analyst, 2010, 135, 2028–2035 RSC.
- C. R. Ispas, G. Crivat and S. Andreescu, Anal. Lett., 2012, 45, 168–186 CrossRef CAS.
- K. Balasubramanian, Biosens. Bioelectron., 2010, 26, 1195–1204 CrossRef CAS PubMed.
- D. Tang, J. Tang, B. Su, J. Ren and G. Chen, Biosens. Bioelectron., 2010, 25, 1658–1662 CrossRef CAS PubMed.
- Y. Huang, L. Fang, Z. Zhu, Y. Ma, L. Zhou, X. Chen, D. Xu and C. Yang, Biosens. Bioelectron., 2016, 85, 496–502 CrossRef CAS PubMed.
- A. K. Feldman, M. L. Steigerwald, X. Guo and C. Nuckolls, Acc. Chem. Res., 2008, 41, 1731–1741 CrossRef CAS PubMed.
- M. E. Roberts, A. N. Sokolov and Z. Bao, J. Mater. Chem., 2009, 19, 3351–3363 RSC.
- D. J. Macaya, M. Nikolou, S. Takamatsu, J. T. Mabeck, R. M. Owens and G. G. Malliaras, Sens. Actuators, B, 2007, 123, 374–378 CrossRef CAS.
- H. Shi, T. Xia, A. E. Nel and J. I. Yeh, Nanomedicine, 2007, 2, 599–614 CrossRef CAS PubMed.
- H. Shi and J. I. Yeh, Nanomedicine, 2007, 2, 587–598 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- N. C. Seeman and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6451–6455 CrossRef CAS PubMed.
- A. S. Blum, C. M. Soto, K. E. Sapsford, C. D. Wilson, M. H. Moore and B. R. Ratna, Biosens. Bioelectron., 2011, 26, 2852–2857 CrossRef CAS PubMed.
- I. Ortac, S. C. Esener, I. G. Yayla and B. Messmer, Enzyme-encapsulated nanoparticle platform, US Pat., WO/2015/042204, 2015 Search PubMed.
- X. Huang, Y. Liu, K. Chen, W. J. Shin, C. J. Lu, G. W. Kong, D. Patnaik, S. H. Lee, J. F. Cortes and J. A. Rogers, Small, 2014, 10, 3083–3090 CrossRef CAS PubMed.
- H. Y. Y. Nyein, W. Gao, Z. Shahpar, S. Emaminejad, S. Challa, K. Chen, H. M. Fahad, L.-C. Tai, H. Ota, R. W. Davis and A. Javey, ACS Nano, 2016, 10, 7216–7224 CrossRef CAS PubMed.
- W. Gao, S. Emaminejad, H. Y. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki, D. Kiriya, D.-H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nature, 2016, 529, 509–514 CrossRef CAS PubMed.
- S. Coyle, K.-T. Lau, N. Moyna, D. O'Gorman, D. Diamond, F. Di Francesco, D. Costanzo, P. Salvo, M. G. Trivella and D. E. De Rossi, IEEE Trans. Inf. Technol. Biomed., 2010, 14, 364–370 CrossRef PubMed.
- S. Iguchi, H. Kudo, T. Saito, M. Ogawa, H. Saito, K. Otsuka, A. Funakubo and K. Mitsubayashi, Biomed. Microdevices, 2007, 9, 603–609 CrossRef CAS PubMed.
- H. Kudo, T. Sawada, E. Kazawa, H. Yoshida, Y. Iwasaki and K. Mitsubayashi, Biosens. Bioelectron., 2006, 22, 558–562 CrossRef CAS PubMed.
- H. Yao, A. J. Shum, M. Cowan, I. Lähdesmäki and B. A. Parviz, Biosens. Bioelectron., 2011, 26, 3290–3296 CrossRef CAS PubMed.
- W. Jia, A. J. Bandodkar, G. Valdés-Ramírez, J. R. Windmiller, Z. Yang, J. Ramírez, G. Chan and J. Wang, Anal. Chem., 2013, 85, 6553–6560 CrossRef CAS PubMed.
- J. Kim, I. Jeerapan, S. Imani, T. N. Cho, A. Bandodkar, S. Cinti, P. P. Mercier and J. Wang, ACS Sens., 2016, 1, 1011–1019 CrossRef CAS.
- M. Parrilla, R. Cánovas, I. Jeerapan, F. J. Andrade and J. Wang, Adv. Healthcare Mater., 2016, 5, 996–1001 CrossRef CAS PubMed.
- M.-C. Chuang, Y.-L. Yang, T.-F. Tseng, T. Chou, S.-L. Lou and J. Wang, Talanta, 2010, 81, 15–19 CrossRef CAS PubMed.
- K. MacVittie, J. Halamek, L. Halamkova, M. Southcott, W. D. Jemison, R. Lobel and E. Katz, Energy Environ. Sci., 2013, 6, 81–86 CAS.
- C. Agnes, M. Holzinger, A. Le Goff, B. Reuillard, K. Elouarzaki, S. Tingry and S. Cosnier, Energy Environ. Sci., 2014, 7, 1884–1888 CAS.
- J. Kim, I. Jeerapan, S. Imani, T. N. Cho, A. Bandodkar, S. Cinti, P. P. Mercier and J. Wang, ACS Sens., 2016, 1, 1011–1019 CrossRef CAS.
- M. Falk, V. Andoralov, M. Silow, M. D. Toscano and S. Shleev, Anal. Chem., 2013, 85, 6342–6348 CrossRef CAS PubMed.
- S. O. Garcia, Y. V. Ulyanova, R. Figueroa-Teran, K. H. Bhatt, S. Singhal and P. Atanassov, ECS J. Solid State Sci. Technol., 2016, 5, M3075–M3081 CrossRef CAS PubMed.
- W. Jia, G. Valdés-Ramírez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236 CrossRef CAS PubMed.
- A. A. Arbab, K. C. Sun, I. A. Sahito, M. B. Qadir and S. H. Jeong, Phys. Chem. Chem. Phys., 2015, 17, 12957–12969 RSC.
- H. Lee, T. K. Choi, Y. B. Lee, H. R. Cho, R. Ghaffari, L. Wang, H. J. Choi, T. D. Chung, N. Lu and T. Hyeon, Nat. Nanotechnol., 2016, 11, 566–572 CrossRef CAS PubMed.
- X. You and J. J. Pak, Sens. Actuators, B, 2014, 202, 1357–1365 CrossRef CAS.
- Y. H. Kwak, D. S. Choi, Y. N. Kim, H. Kim, D. H. Yoon, S.-S. Ahn, J.-W. Yang, W. S. Yang and S. Seo, Biosens. Bioelectron., 2012, 37, 82–87 CrossRef CAS PubMed.
- H. Yao, A. J. Shum, M. Cowan, I. Lähdesmäki and B. A. Parviz, Biosens. Bioelectron., 2011, 26, 3290–3296 CrossRef CAS PubMed.
- H. Lee, T. K. Choi, Y. B. Lee, H. R. Cho, R. Ghaffari, L. Wang, H. J. Choi, T. D. Chung, N. Lu, T. Hyeon, S. H. Choi and D.-H. Kim, Nat. Nanotechnol., 2016, 11, 566–572 CrossRef CAS PubMed.
- A. Panneer Selvam, S. Muthukumar, V. Kamakoti and S. Prasad, Sci. Rep., 2016, 6, 23111 CrossRef CAS PubMed.
- R. D. Munje, S. Muthukumar and S. Prasad, Sens. Actuators, B, 2017, 238, 482–490 CrossRef CAS.
- C. H. Kwon, S.-H. Lee, Y.-B. Choi, J. A. Lee, S. H. Kim, H.-H. Kim, G. M. Spinks, G. G. Wallace, M. D. Lima, M. E. Kozlov, R. H. Baughman and S. J. Kim, Nat. Commun., 2014, 5, 3928 CAS.
- W. Jia, X. Wang, S. Imani, A. J. Bandodkar, J. Ramirez, P. P. Mercier and J. Wang, J. Mater. Chem. A, 2014, 2, 18184–18189 CAS.
- W. Jia, G. Valdés-Ramírez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |