
Control of the size and density of ZnO-nanorods grown onto graphene nanoplatelets in aqueous suspensions†
C. R.
Chandraiahgari
ab,
G.
De Bellis
ab,
S. K.
Balijepalli
c,
S.
Kaciulis
c,
P.
Ballirano
bd,
A.
Migliori
e,
V.
Morandi
e,
L.
Caneve
f,
F.
Sarto
f and
M. S.
Sarto
*ab
aDepartment of Astronautics, Electrical and Energetics Engineering, Sapienza University of Rome, via Eudossiana 18, Rome 00184, Italy. E-mail: c.chandraiahgari@uniroma1.it; mariasabrina.sarto@uniroma1.it
bResearch Center for Nanotechnology Applied to Engineering of Sapienza (CNIS), SNNLab, Sapienza University of Rome, Piazzale Aldo Moro, 5, Rome 00185, Italy
cCNR – ISMN, P.O. Box 10, 00015 Monterotondo Stazione, Rome, Italy
dDepartment of Earth Sciences, Sapienza University of Rome, Piazzale Aldo Moro 5, Rome 00185, Italy
eCNR – IMM Section of Bologna, via Gobetti 101, Bologna 40129, Italy
fENEA, Centro Ricerche Frascati, Via Enrico Fermi 45, Frascati 00044, Italy
First published on 22nd August 2016
Abstract
Zinc oxide nanorods (ZnO-NRs) with high density and chemical purity were grown onto unsupported graphene nanoplatelets (GNPs) in aqueous suspensions, using two different growth approaches namely: a hydrothermal method and ultrasonic probe sonication. The size and density of the ZnO-nanorods grown onto graphene nanoplatelets were controlled through seed layer deposition and through the proper setting of the process parameters, in particular through the control of the fluidodynamics of the colloidal suspension during the growth. The highest growth density of the ZnO nanorods having a diameter of ∼45 nm was obtained onto GNPs seeded by the probe sonication method and through the hydrothermal method in dynamic conditions. XRD and XPS investigations confirmed that all produced ZnO-GNP composites are characterized by high crystallinity and purity, although solution dynamics affected their UV luminescence. The proposed approaches enable the controlled high-density growth of crystalline ZnO-NRs onto GNPs in an aqueous suspension, at a low cost, and are suitable for large scale production.
1. Introduction
Graphene, owing to its exceptional electrical, thermal, and mechanical properties, has been widely explored in a variety of fields, such as electronics, sensors, catalysis, drug delivery, solar cells, energy harvesting and storage systems.1,2 Decoration or functionalization of pristine graphene with inorganic nanostructures is a way to bring additional functionality to it.3 Recently, hybrid systems made of graphene-based materials and ZnO-nanostructures have been investigated for their potentiality as a new class of multifunctional nanomaterials exploiting the salient features of graphene and ZnO.4–8Hybrid nanostructures or composites made of ZnO and graphene have resulted in multifunctional and enhanced properties such as high UV sensing capabilities, excellent field emission, ultrafast nonlinear optical switching, gas sensing, improved photocatalytic activity, and piezoelectricity.5–10 Such enhanced properties mainly arise from the combination of the superior electrical properties of the carbon-based materials with the optical properties and polarity of ZnO.11,12 Several studies demonstrated that shape, size, concentration and distribution of the nanostructures in a composite are greatly affecting the functional properties of the final material.13–15 Therefore, the control of shape, size and morphology through the proper setting of the growth conditions plays a crucial role in tuning the functional properties.
ZnO crystals have a noncentral symmetric wurtzite structure and are composed of closely packed O2− and Zn2+ layers piled alternatively along the c-axis, producing positively charged Zn-terminated [0001] polar surfaces and negatively charged O-terminated [0001] polar surfaces.16 ZnO-nanocrystals with varying surface configurations and polarities display different optical and electronic properties. However, synthesis of ZnO-nanostructures (i.e. c-axis oriented nanowires or a-axis oriented nanobelts17) is very sensitive to the growth conditions18–20 and, consequently, special care must be paid in order to assure reproducibility and controllability.
Recently, interesting progresses have been made towards the fabrication of ZnO–graphene hybrid nanostructures by using different techniques, such as: chemical vapor deposition (CVD) of ZnO nanostructures on CVD-grown graphene; synthesis in liquid solution of ZnO-NRs onto graphene flakes transferred via scotch-tape; synthesis in liquid solution of ZnO-NRs on CVD-grown graphene; synthesis in liquid solution of ZnO-NRs onto chemically-reduced graphene oxide (RGO).10,21–23 In these studies, the functional properties of the hybrid nanostructures were controlled through the synthesis methods. Various process parameters such as growth solution concentration, pH and temperature, tilt position and seed deposition affect the controlled growth of nanostructures.23 J. Ding et al. (2015),21 Y. Kim et al. (2011)23 and M. Khenfouch et al. (2014)24 have grown ZnO-NRs through hydrothermal method onto supported few-layer-graphene flakes and unsupported RGO without the use of a seed layer: this approach resulted in low density and poor homogeneity of the growth. Differently, R. Chung et al. (2013)22 and D. Shin et al. (2014)5 deposited a seed layer either through electron-gun evaporation or through spin-coating onto supported CVD-grown graphene, and achieved a high-density growth, thus demonstrating the importance of seed layer. J. Fan et al. (2015)25 deposited the seed layer by spin-coating onto supported monolayer graphene, and obtained high-density growth of twinned structures and microrod arrays, although clearly lacking in homogeneity and in morphology control.
It should be highlighted that in most of these studies, ZnO nanostructures were grown onto graphene supported on a substrate, so that decoration or functionalization was typically achieved only over the exposed surface of graphene. Moreover, it has not been shown, how different hydrodynamic conditions of the colloidal suspension during the growth enable the control of the density and shape of the ZnO nanostructures.
In this paper, we report the synthesis of ZnO-nanostructures with controlled shape and density onto unsupported multilayer-graphene flakes (also known as graphene nanoplatelets – GNPs2), using a seed layer, through aqueous solution-based low-temperature approaches.26,27 In particular, we investigate the role that the hydrodynamic conditions of the growth suspension have on the morphology, density, crystallinity and photoluminescence properties of ZnO-NR decorated GNPs (ZNGs). General models for particle nucleation and growth by hydrothermal method suggest that atoms fluctuating in liquid phase may collide and stick to existing seeds and/or already formed agglomerates. The so-formed clusters will remain stable if the energy gain associated to solid formation is higher than the energy cost of building the solid/liquid interface.28 Changing the hydrodynamic growth conditions from static to dynamic regime leads to an increase of the energy gain associated to solid formation, due to the increased concentration of the (precipitating) ions in the supersaturated phase, which may favour smaller nanostructures to become stable.
Therefore, in this study, we demonstrate that the proper setting of the seed-layer deposition process and of the hydrodynamic conditions during the ZnO-nanostructure growth lead to the production of ZNGs with controlled morphology, density, crystallinity and PL properties. To this purpose, FE-SEM, HR-TEM, XRD, XPS, and PL techniques have been used to probe the properties of the produced ZNGs.
2. Experimental
2.1. Materials
All the chemicals were of reagent-grade and were used as received, without any further purification: graphite intercalation compound (GIC), zinc acetate dihydrate (Zn(CH3COO)2·2H2O, Sigma-Aldrich, ACS reagent, ≥98%), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Acros Organics, 98%), hexamethylenetetramine (C6H12N4, Fisher Scientific, ≥99%), isopropanol ((CH3)2CHOH, Sigma-Aldrich, ACS reagent, ≥99%) and ethanol (CH3CH2OH, Fluka, ACS reagent, ≥99%). The water, used in all the experiments, was deionized (DI) and had a resistivity of 18 MΩ cm.2.2. Methods
The deposition of SL by probe sonication method was also performed using a very similar procedure, as described above, but replacing the step of magnetic stirring with a probe sonication. Probe sonication was performed for 15 min with constant ultrasound amplitude set at 45% of its maximum level. Finally, we obtained a suspension of GNPs coated with quasi spherical seed particles of ZnO, having an average diameter of ∼15 nm.
The influence of the continuous stirring of the seeded-GNP suspension during the ZnO-NR growth reaction was assessed by implementing the aforementioned hydrothermal synthesis process keeping the suspension in a static condition (i.e. without the action of the mechanical stirring) at the same temperature of 60 °C and for the same reaction time of 4.5 h. These resulted in GNPs decorated with ZnO microrods (ZnO-MRs) as detailed in the following. This procedure, hereinafter referred to as hydrothermal method in static condition, is denoted as HT2.
2.3. Characterizations
Samples for field emission scanning electron microscope (FE-SEM), high resolution transmission electron microscopy (HR-TEM), X-ray Photoelectron Spectroscopy (XPS), Auger Electron Spectroscopy (AES), Ultraviolet Photoelectron Spectroscopy (UPS) and photoluminescence (PL) analyses were prepared from the suspensions produced through either hydrothermal or probe sonication method. To this purpose, a concentrated suspension of hybrid nanostructures obtained upon centrifugation (for 30 min at 3095g) was drop-casted onto cleaned silicon substrates (holey carbon grid coated on a 200 mesh Cu grids for HR-TEM), with subsequent drying in oven at 120 °C for 30 min. X-ray diffraction (XRD) was performed on ZNG powders obtained by centrifugation of the ZNG suspension and subsequent drying in oven at 120 °C for 30 min.The morphology, homogeneity and density of the produced hybrid nanostructures were observed using a FE-SEM (Zeiss Auriga). Samples were imaged without any metal coating, with an accelerating voltage varying between 3 and 5 keV.
The crystalline analysis was carried out using a HR-TEM (FEI TECNAI F20) equipped with a Schottky emitter and operated at 200 kV, by means of high resolution imaging and electron diffraction.
The structural characterization and phase identification was performed by XRD using a Bruker AXS D8-Advance diffractometer, operating in θ–θ transmission geometry, equipped with focusing Göbel mirrors along the incident beam, Soller slits on both incident and (radial) diffracted beams, and a PSD VANTEC-1 detector. Samples for XRD were prepared loading the powders into 0.3 mm diameter borosilicate capillaries that were aligned onto a standard goniometer head. XRD data were collected at room temperature, using Cu Kα radiation (λ = 1.5418 Å, 40 kV at 40 mA), in a 2θ angular range ranging from 20° to 145° with a step size of 0.022° and 1 s of counting time. Structural refinements were performed by the Rietveld method using TOPAS v.4.2 (Bruker AXS 2009) software. Starting structural parameters of hexagonal ZnO (zincite) were taken from Sowa and Ahsbahs.30
The chemical composition analysis was performed by XPS using a monochromatized spectrometer Escalab 250Xi (Thermo Fisher Scientific, UK) equipped with a six-channeltron detection system for spectroscopy and a multichannel plate for chemical imaging. The measurements were carried out on the powders deposited onto silicon substrates and placed in an ultra-high vacuum (UHV) chamber, using the Al Kα X-ray source set to the diameter of 900 µm, the analyser – at constant pass energy of 20 eV and standard mode of electromagnetic input lenses (about 1 mm in diameter). Acquired spectroscopic data were processed by the Avantage v.5 software (Thermo Fisher Scientific, UK). UPS measurements were carried out using ultraviolet light source of He I and II. A constant pass energy of 5 eV and 20 eV were used for He I and He II sources, respectively. Chemical images were acquired using 500 µm X-ray source, electromagnetic mode of input lenses, 100 eV pass energy of the analyzer and multichannel plate detector.
The crystalline defect analysis was studied through room temperature PL using as excitation source the 266 nm line of a Nd:YAG pulsed laser (Thomson DIVA), with 10 ns pulse length and 20 Hz repetition rate. Energy dispersion of the PL signal was obtained by using a 3648-element linear silicon CCD array, operating in the 200–1100 nm wavelength range. The PL spectra were repeated 16 times on each sample at two different levels of the laser mean power (i.e. 700 and 1300 ± 100 W m−2) and the average values were reported versus the emission energy.
3. Results and discussions
3.1. Morphology analysis by FE-SEM
All the produced samples, with their main morphological characteristics, are listed in Table 1.| Sample | Description | Production method | Morphological characteristics (average values) | |
|---|---|---|---|---|
| G | GNP | Thermochemical exfoliation of GIC | Thickness | 6 nm |
| Lateral dimensions | 5.5 µm | |||
| G1 | GNP + SL | SL deposition through stirring method | SL nanoparticle size | 20 nm |
| G2 | GNP + SL | SL deposition through probe sonication method | SL nanoparticle size | 15 nm |
| G1-HT1 | ZNG | SL deposition through stirring method | ZnO-NRs diameter | 45 nm |
| ZnO-NR decoration through hydrothermal method in dynamic condition (HT1) | ZnO-NRs length | 405 nm | ||
| G1-HT2 | ZNG | SL deposition through stirring method | ZnO-NRs diameter | 180 nm |
| ZnO-NR decoration through hydrothermal method in static condition (HT2) | ZnO-NRs length | 1.2 µm | ||
| G2-PS | ZNG | SL deposition through probe sonication | ZnO-NRs diameter | 50 nm |
| ZnO-NR decoration by probe sonication method (PS) | ZnO-NRs length | 170 nm | ||
| G2-HT1 | ZNG | SL deposition through probe sonication | ZnO-NRs diameter | 34 nm |
| ZnO-NR decoration through hydrothermal method in dynamic condition (HT1) | ZnO-NRs length | 418 nm | ||
| G-HT2 | ZNG | No SL | ZnO-NRs diameter | 119 nm |
| ZnO-NR decoration through hydrothermal method in static condition (HT2) | ZnO-NRs length | 493 nm | ||
| G-PS | ZNG | No SL | ZnO-NRs diameter | 130 nm |
| ZnO growth assisted by probe sonication method (PS) | ZnO-NRs length | 304 nm | ||
The surface morphology observed by FE-SEM of seeded GNPs, produced either by magnetic stirring (sample G1) or by probe sonication (sample G2) of the GNPs suspension in the seed solution is shown in Fig. 1. In both cases, the seed ZnO-nanoparticles are uniformly deposited onto the GNP surfaces, they are quasi spherical in shape and anchored very well to the GNP surfaces. The diameter of the seed nanoparticles in sample G1 (Fig. 1A) is in the range of 10–30 nm, whereas in sample G2 (Fig. 1B) they are slightly smaller, being in the range of 10–20 nm, but with a higher coverage density. Hence, the seed layer deposition assisted by probe sonication, with respect to the one assisted by magnetic stirring, resulted in more densely decorated GNPs with smaller seed nanoparticles.
 | ||
| Fig. 1 Electron microscopy images of GNPs with seed-layer: (A) seeded GNPs produced with magnetic stirring of the seed solution (sample G1 in Table 1) and (B) seeded GNPs produced with probe-sonication of the seed solution (sample G2 in Table 1). | ||
Fig. 2 shows the morphology of the different samples of Table 1. ZnO-NRs with the highest aspect ratio (i.e. length-to-diameter ratio) are obtained either using seeded GNPs produced with magnetic stirring (Fig. 2A and B) or probe sonication (Fig. 2G and H), through hydrothermal method under dynamic condition (method HT1). In both cases, the seed particles are acting as nucleation sites of the ZnO nanostructures, which growth in a flower-like configuration.
 | ||
| Fig. 2 Electron microscopy images of ZNGs at different magnifications produced starting from (A–D) seeded GNPs obtained through mechanical stirring (sample G1) or (E–H) probe sonication of the seed solution (sample G2): (A and B) sample G1-HT1 obtained by hydrothermal growth in dynamic condition of seeded graphene G1; (C and D) sample G1-HT2 obtained by hydrothermal growth in static condition of seeded graphene G1; (E and F) sample G2-PS obtained by probe sonication of seeded graphene G2; (G and H) sample G2-HT1 obtained by hydrothermal growth in dynamic condition of seeded graphene G2. | ||
Due to the high-density of the growth, ZnO-NRs appear mostly vertically oriented with respect to the GNP surface, uniform in size, with a diameter ranging from 20 to 50 nm and length ranging from 300 to 500 nm.
Sample G2-HT1 shown in Fig. 2G and H is characterized by a higher density of ZnO-NRs with respect to sample G1-HT1 of Fig. 2A and B: this is mainly due to the better coverage of seed nanoparticles produced by the seed-layer deposition assisted by probe-sonication.
Hydrothermal growth onto seeded GNPs carried out in static condition (method HT2) produces ZnO microrods (ZnO-MRs) having diameter ranging from 100 to 300 nm and length ranging from 600 nm to 1.50 µm (as shown in Fig. 2C and D), with a lower coverage density. In this case, the flower-like nucleation of ZnO-MRs from the seed nanoparticles is more evident.
The use of PS method during both seed deposition and nanostructure growth originates flower like bunches of ZnO-NRs with a low aspect ratio. These ZnO-NRs have diameter ranging from 40 to 60 nm and length ranging from 150 to 200 nm (Fig. 2E and F). It is observed that in this case nanostructure coverage over GNPs surface is not very uniform. The main advantage of this technique is that nanorods growth takes place in few minutes, whereas the hydrothermal method needs several hours.
The role of the SL deposition in the high density growth of ZnO-NRs onto GNPs was assessed by performing the growth in the absence of such layer. The obtained FE-SEM results are shown in the ESI (page 2, Fig. S2†). It is evident that in this case it is not possible to achieve a uniform and dense distribution of ZnO-nanostructures over the GNP exposed surfaces. In fact, the growth without SL can be mainly ascribed to the presence of surface defects on the unseeded GNPs, which might act like nucleation centers for the growth.31 This confirms the importance of the deposition of the SL onto unsupported GNPs.
Table 2 summarizes the difference of the effects on the size and density of the ZnO-NRs grown onto the graphene nanoplatelets, between hydrothermal method and ultrasonic probe sonication method.
| Process | Hydrothermal method | Probe sonication method |
|---|---|---|
| Seed layer (SL) depostion over GNPs | Small seed particles | Very small seed particles |
| Good coverage and good density | Best coverage and highest density | |
| SL coating reaction (30 min) | Faster SL coating reaction (15 min) | |
| Growth of ZnO nanostructures | Nanorods by dynamic condition | Flower like bunches of nanorods |
| Microrods by HT2 | ||
| Vertical orientation for nanorods | Vertical orientation | |
| Random orientation for microrods | ||
| Narrow size range for nanorods | Narrow size range | |
| Wide size range for microrords | ||
| High aspect ratio | Low aspect ratio | |
| Higher coverage density by HT1 | Poor coverage density | |
| Lower coverage density by HT2 | ||
| Slower growth rate (growth reaction for 4.5 h) | Faster growth rate (growth reaction for 15 min) |
3.2. Crystalline and structural analysis by HR-TEM and XRD
The interaction between ZnO-seed particles and GNPs was investigated by HR-TEM analysis. To this purpose, sample G1-HT1 of Table 1 was considered, since it is quite representative of all the other samples described. Fig. 3A and B shows HR-TEM images of the ZNGs produced by hydrothermal method in dynamic condition, starting from seeded GNPs produced by SL deposition assisted by magnetic stirring (sample G1-HT1). Fig. 3A clearly demonstrates that the planar and transparent graphene sheets are decorated with rod-like ZnO nanostructures. Fig. 3B highlights the intimate physical interaction between graphene and ZnO-seed particle. It can also be seen from Fig. 3B that the ZnO-NRs are radially evolved from these seed particles, which are anchored over the unsupported graphene sheets. Thereby, these seed particles play an important role in the growth mechanism of ZnO-NRs over the GNPs with high density. Selected area electron diffraction (SAED) pattern shown in the inset of Fig. 3B, with highlighted the (100) and (110) reflections of ZnO as well as the (002) of graphene, confirms that the crystalline ZnO nanorods lie on the graphene sheets. | ||
| Fig. 3 (A) HR-TEM images of ZNGs produced through hydrothermal growth in dynamic condition starting from seeded GNP obtained by seed-layer deposition assisted by magnetic stirring, and (B) high magnification image showing the intergration of seed particle and GNP. In the inset the SAED pattern shows the presence of ZnO crystals and graphene (Gr). | ||
Fig. 4 shows the diffraction patterns of the produced ZNGs, compared with one of pristine GNPs. The sharp and intense peaks of the patterns clearly indicate the highly crystalline nature of the ZNGs. Moreover, peaks positions and intensities agree with those published in the Joint Committee on Powder Diffraction Standards (JCPDS) card 036-1451 for the hexagonal polymorph (zincite) of ZnO.
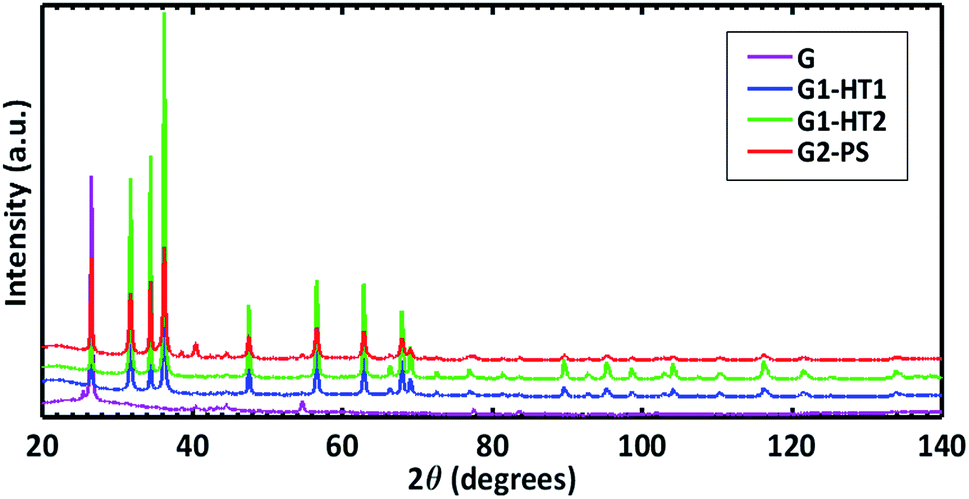 | ||
| Fig. 4 XRD patterns of pristine GNPs (sample G of Table 1) and of the different types of ZNGs obtained in different growth conditions experienced in this study: samples G1-HT1, G1-HT2, and G2-PS. | ||
Detailed structural information on the analyzed samples is reported in the ESI (page 4, Table S1†). The cell parameters and 〈Zn–O〉 bond distance of 1.978–1.979 Å obtained for all the samples is in perfect agreement with the findings of Sowa and Ahsbahs.30
3.3. Chemical purity analysis by XPS
The surface chemical composition of the produced ZNG samples was analyzed by XPS in order to detect traces of any impurities and to assess the homogeneity of the surface composition. For comparison, the same analysis was carried out on the reference sample containing pristine GNPs. All measurements were carried out at three different points on each sample. These three points on each sample were found to be very similar to each other, which indicate that ZnO is homogeneously distributed on the surface of the investigated samples.In all cases, zinc is always present in the form of ZnO, i.e. in Zn+2 chemical state, which is confirmed by the binding energy (BE) value of Zn2p3/2 peak at BE ≈ 1022.5 eV and by the position of Auger Zn LMM peak at kinetic energy KE ≈ 987.3 eV.32,33 In addition, the value of modified Auger parameter (α ≈ 2009.8 eV) corresponds to the Zn (+2) chemical state.33 The spectra of Zn2p and Zn LMM regions for the ZNG samples are shown in Fig. 5A and B respectively.
 | ||
| Fig. 5 (A) Zn2p3/2 and (B) Zn LMM regions of the ZNGs produced in the different growth conditions experienced in this study: samples G1-HT1, G1-HT2, and G2-PS. | ||
The O1s spectra, including the peak fitting, of the hybrid ZNG samples and of the reference sample are presented in Fig. 6.
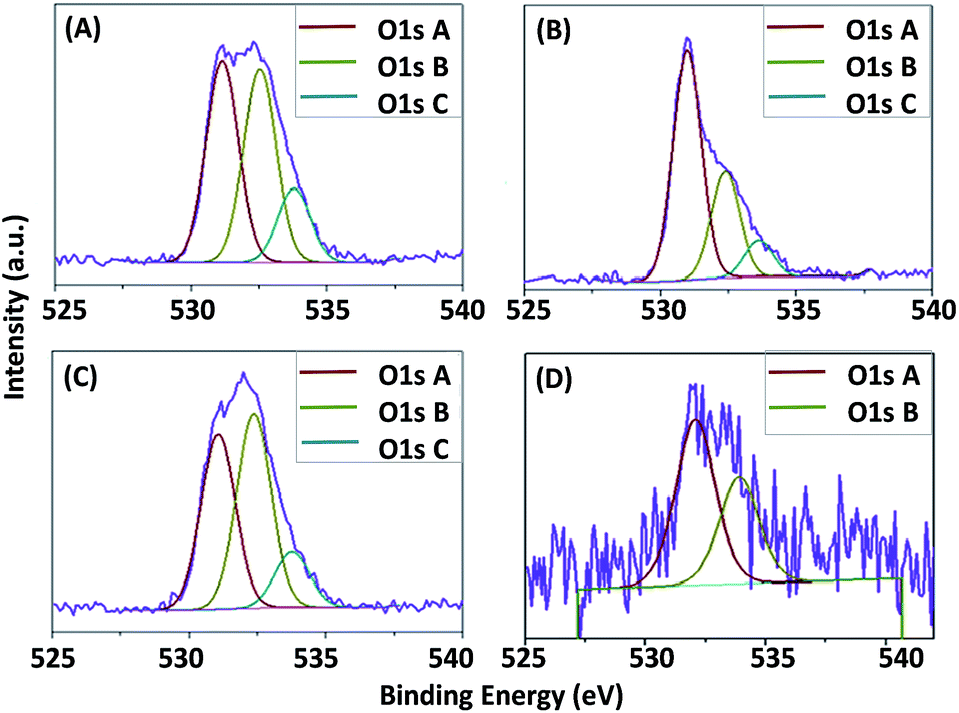 | ||
| Fig. 6 O1s spectra of the ZNGs produced in the different growth conditions experienced in this study: (A) G1-HT1, (B) G1-HT2, (C) G2-PS, and (D) reference pristine GNPs. | ||
The O1s spectra of all the ZNG samples consist of three components indicated as A, B and C. The first component A is attributed to O2− ions in the wurtzite structure of ZnO,47 while the second one is attributed to the hydroxyl groups OH− and/or CO3 radicals and the third one is assigned to adsorbed H2O molecules.32,34
It should be noted that in the reference sample the first component (oxide) is completely absent, while the other two components (OH− and H2O) are very low. Whereas in all other ZNGs samples noticeable variation in the atomic percent of O1s components is observed, ascribed to the respective variance in the growth conditions. Although samples G1-HT1 and G1-HT2 are prepared by same method but the growth solution dynamics are different, resulting in high variation in the atomic percent of OH− groups and their effect on crystalline quality is assessed by PL and discussed in Section 3.5.
For all produced ZNG samples, the values of asymmetric C1s peak at BE ≈ 284.4 eV and the valence band (VB) spectra, starting at 2.5 eV above Fermi level (typical for graphene34), indicate the presence of graphene. In the reference sample, carbon is present in the form of graphite, because even if the C1s peak remains at BE ≈ 284.4 eV, the VB spectrum starts at the Fermi level, i.e. at BE = 0 eV (typical for graphite). The C1s and VB spectra of the samples are reported in Fig. 7A and B respectively. A high peak immediately after the valence band at BE ≈ 11 eV is attributed to Zn3d core level and is absent in the reference sample without ZnO (see Fig. 7B). The main difference between the samples is the variation in the atomic concentrations of C and Zn. The relative reduction of carbon amount corresponds to the higher coverage of ZnO on the surface. In the order of higher C content, the samples can be listed as: G2-PS > G1-HT2 > G1-HT1. This indicates that the sample G1-HT1 contains the best coverage of ZnO, which can be listed in the following order: G1-HT1 > G1-HT2 > G2-PS. These results indicate that the preparation methods ensured the production of pure oxide (ZnO) using all three methods G1-HT1, G1-HT2 and G2-PS, as the Zn and O were the only elements detected on the surface of graphene. The chemical composition and chemical states of elements for all the samples are reported in the ESI (page 4, Table S2†).
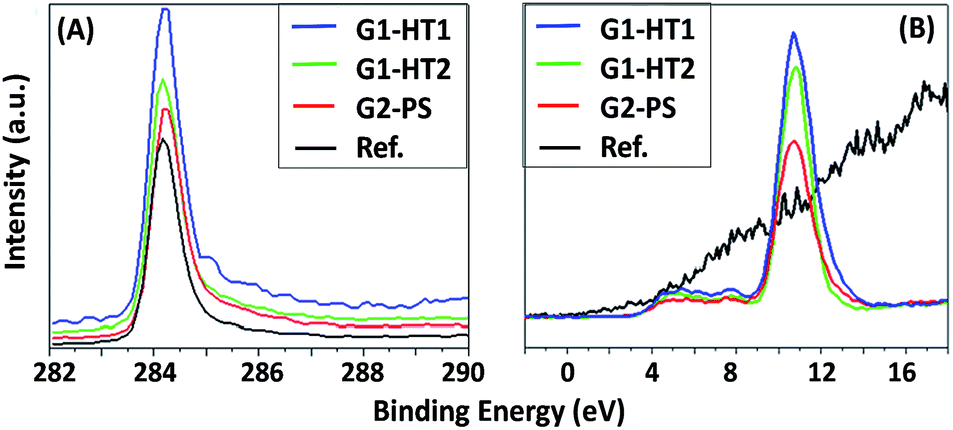 | ||
| Fig. 7 (A) C1s and (B) XPS valence band spectra for the produced ZNGs (samples G1-HT1, G1-HT2 and G2-PS) and pristine GNPs (sample G). | ||
Furthermore, the XPS chemical images were acquired over the area of 100 × 100 µm2 on all the samples and demonstrated that the Zn2p signal is homogeneous and densely distributed all over the surface as shown in the ESI (page 5, Fig. S3†).
3.4. Valence band analysis by UPS
The work function (WF) values of the hybrid ZNG samples were analyzed by UPS in order to assess the doping and homogeneity of the composition. Fig. 8A shows the valence band spectra acquired by He I source to calculate the WF values. WF of the samples was calculated by subtracting the spectra cut-off values from the energy of the He I source which is equal to 21.2 eV. The values of WF were the following: for G1-HT1 sample 21.2–16.2 = 5 eV; for G1-HT2 sample 21.2–16.2 = 5 eV; for G2-PS sample 21.2–16.2 = 5 eV; for reference sample 21.2–16 = 5.2 eV. All the ZNG samples have similar values of WF equal to 5 eV, which corresponds to the value of ZnO from the literature.35–37 The reference sample has the value of the WF of 5.2 eV, which is in agreement to the value of graphite from the literature.33–35 This confirms doping and that the state of zinc is ZnO. | ||
| Fig. 8 UPS valence band spectra obtained by using; (A) He I and (B) He II sources. Work function was calculated from the cut-off in He I spectra. | ||
Valence band (VB) spectra were also acquired by using He II source with a higher energy of 40 eV, as shown in Fig. 8B. These spectra confirm the conclusions derived from VB spectra acquired by using X-ray source as shown in Fig. 7. In the samples G1-HT1, G1-HT2 and G2-PS, the signal starts approximately at BE = 2.5 eV, which is a typical behavior for graphene. In the reference sample, the spectrum started at 0 eV, which is typical for graphite. In all three ZNG samples, the core-level peak of Zn3d is identified at approximately BE = 11 eV, which confirms that in them GNPs are being doped successfully with ZnO. In sample G1-HT1, the intensity of Zn3d peak is higher than in G1-HT2 and G2-PS samples, i.e. is confirmed our earlier conclusion that G1-HT1 sample has more homogeneous distribution and higher amount of ZnO than other samples.
3.5. Crystalline defects analysis by photoluminescence
The effect on the ZNG luminescence properties of the static- or dynamic-growth condition was studied via PL spectroscopy. The obtained results were compared with the ones of the XPS analysis in order to assess the effect of observed differences in the atomic percent of OH− groups of samples produced applying method HT1 or HT2 (Fig. 6A and B).Fig. 9 shows the PL spectra of samples G1-HT1 and G1-HT2, produced by dynamic and static methods, respectively. Both samples exhibited PL spectra which are typical of ZnO nanostructures, consisting of a strong UV excitonic emission at ∼3.26 eV and a weak and broad VIS emission with its maximum at about 2.13 eV. A similar spectrum is obtained when low lasing power is used and it is shown in ESI (page 5, Fig. S4†). The UV/VIS peak intensity ratio is used to evaluate the crystal quality, i.e. the absence of electronic defects which may act as recombination centers for charge carriers and quench the excitonic peak.38,39 As evident from Fig. 9, the intensity of UV emission is much higher for sample G1-HT2 than G1-HT1, despite there is not any difference in the weak VIS emission. This suggests that the concentration of defects is lower in samples grown in static hydrodynamic conditions. This observation is consistent with the evidenced differences in the O1s components of XPS spectra (Fig. 6A and B), since a more intense O1s B line is known to be associated to the presence of various defects, responsible for damping the UV PL emission.40,41
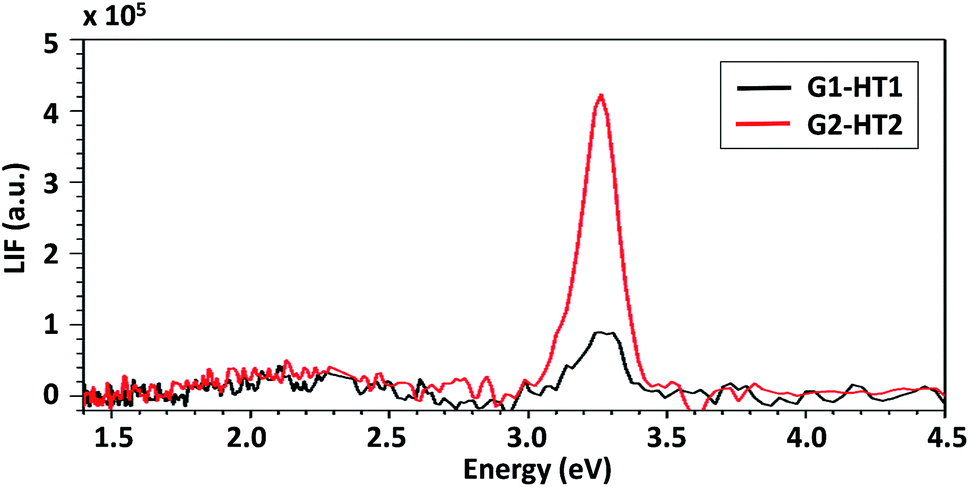 | ||
| Fig. 9 PL spectra of samples G1-HT1 and G1-HT2; incident laser power = 1300 W m−2. | ||
Erki Kärber et al. (2011)42 reported the various factors affecting the PL of ZnO nanorods deposited by chemical spray pyrolysis including the spray rate. They observed that by increasing the spray rate from 1.2 ml min−1 to 6.2 ml min−1 the intensity of UV excitonic peak is diminished and slightly shifted, due to the increase of the growth rate (which is consistent with what we observed, i.e. growth rate of sample G1-HT2 is lower than of G1-HT1). Hatim Alnoor et al. (2015)40 reported the influence of varying the duration of the precursor solution stirring (prior to the growth) on the concentration and spatial distributions of deep level defects in ZnO-NRs, attributed to variation in the concentration of the oxygen-deficiency. T. Dai et al. (2014)41 reported on the enhancement of PL emission of ZnO sol–gel nanoparticles by thermal annealing, resulting from the elimination of surface defects associated to adsorbed OH bonds. However, to the best of our knowledge, no particular study was performed to investigate the dynamics of nutrient solution during the growth and their effect on PL emission.
Actually during the solution growth, the presence of OH− which further reacts with Zn+2 and later (during the formation of ZnO nanostructures) promotes the growth mechanism, is higher for G1-HT1 than in G1-HT2. This favors the formation of smaller nanostructures (as explained by DLA theory43), as well as the presence of surface adsorbed OH− groups (i.e. ZnO(OH), due to the excess solvent capped on the surface of ZnO41). Both these effects may be responsible for a higher number of oxygen-related surface defects, either due to a larger mass specific area, either due to higher concentration of surface adsorbed OH− groups.
XRD analysis already demonstrated that all the samples are perfectly crystalline. Therefore, the relatively lower PL emission evinced in sample G1-HT1 can be more likely ascribed to the presence of surface defects than to structural (bulk) ones. Although a significant variation in the oxygen-component is observed for static and dynamic conditions which affect the optical quality, the ZNG nanostructures produced are of perfectly crystalline and chemically pure.
3.6. Potential applications of ZNGs with controlled size and density of ZnO-NRs
Over the last decade, vertically aligned ZnO-NRs has been intensively studied due to their remarkable piezoelectric properties and potential for various applications. The improvement and control in the density and size of ZnO-NRs grown onto graphene sheet has a direct beneficial effect on the heterostructured nanogenerator performance.5 The facile fabrication process developed in this study suggests that ZnO–graphene heterostructures with tailored morphology would improve the performance of electrical and optoelectrical device, such as touch pad, pressure sensor, biosensor and dye-sensitized solar cells.5,22,23In a recent study,44 it has been shown how the control of size and morphology of ZnO-NRs grown onto graphene oxide sheets contributes to an enhancement of the photocatalytic properties of these material with respect to UV radiation. The comparison of the photocatalytic performances of different types of graphene–ZnO hybrids in photodegradation of methylene blue was also reported.44
A new promising field of application of the ZNGs developed in this study is related to their antibacterial and antibiofilm properties. In previous studies, the authors have demonstrated the stronger potential of ZnO-NRs as antimicrobial agent with respect to other types of ZnO nanostructures (like nanoparticles or microrods), due to their particular shape and size. In fact, ZnO-NRs, having diameter in the range 40–50 nm behave as nano-needles against pathogens,8 although they are characterized by very low cytotoxicity as shown through in vitro studies.45 At the same time, GNPs have demonstrated relevant antimicrobial effect against both Gram-negative46 and Gram-positive47 bacteria, due to the direct mechanical damage induced on the cell wall through their sharp edges and the wrapping effect. However, the use of GNPs as antibacterial material has some limitations in those applications in which their characteristic grey color is a problem, like in dental use or in cultural heritage. Weak antimicrobial efficacy of GNPs was also observed against spherical-shaped bacteria with relatively small size, like S. aureus or S. mutans,47 offering a limited interaction surface with the 2D-nanostructures, having lateral dimensions from 500 nm up to a few microns. ZNGs, with high-density of ZnO-NRs less than 50 nm in diameter, allow to combine the antimicrobial and antibiofilm properties of both ZnO-NRs and GNPs and, at the same time they enable to overcome the color limitation of GNPs thanks to the optical properties of ZnO.
4. Conclusions
Facile versatile aqueous methods to decorate unsupported GNPs with shape-controlled ZnO-NRs have been demonstrated. Different hydrodynamic conditions during the ZNG synthesis are experimented, on the basis of two distinct growth methods (i.e. the hydrothermal method and probe sonication), starting from seeded GNPs suspended in aqueous solution.Hydrothermal growth performed in dynamic condition resulted in a dense growth of mostly vertically oriented crystalline wurtzite ZnO-NRs having diameter of ∼45 nm and length of ∼405 nm. On the contrary, HT synthesis in static condition resulted mainly in the growth of flower-like bunches of ZnO-MRs with a lower coverage density, average diameter of ∼180 nm and length up to 1–1.5 µm.
Probe sonication growth method is demonstrated to be a faster process producing flower-like bunches of ZnO-NRs with low aspect ratio (i.e. average diameter of ∼50 nm and length of ∼170 nm).
In conclusions, the highest growth density with utmost homogeneity of nanorods is obtained onto GNPs seeded by probe sonication method and growth carried out by hydrothermal method in dynamic condition. This type of ZNGs has very promising applications as building block in piezoelectric nanogenerator or as antimicrobial/antibiofilm device.
Although solution dynamics affected the UV luminescence, XRD and XPS investigations confirmed that all the produced ZNGs are characterized by high crystallinity and purity. UPS work function analysis suggested a possible doping effect of ZnO-NRs grown over GNPs.
Acknowledgements
This study was performed under the project PON03PE_00214_1.References
- X. Huang, X. Qi, F. Boeyab and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- L. Paliotta, G. De Bellis, A. Tamburrano, F. Marra, A. Rinaldi, S. K. Balijepalli, S. Kaciulis and M. S. Sarto, Carbon, 2015, 89, 260 CrossRef CAS.
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483 CAS.
- C. Hsieh, C. Lin, Y. Chen and J. Lin, Electrochim. Acta, 2013, 111, 359 CrossRef CAS.
- D. M. Shin, E. L. Tsege, S. H. Kang, W. Seung, S. W. Kim, H. K. Kim, S. W. Hong and Y. H. Hwang, Nano Energy, 2015, 12, 268–277 CrossRef CAS.
- S. Xu and Z. L. Wang, Nano Res., 2011, 4, 1013 CrossRef CAS.
- I. Rago, C. R. Chandraiahgari, M. P. Bracciale, G. De Bellis, E. Zanni, M. C. Guidi, D. Sali, A. Broggi, C. Palleschi, M. S. Sarto and D. Uccelletti, RSC Adv., 2014, 4, 56031 RSC.
- H. Chang, Z. Sun, K. Y. F. Ho, X. Tao, F. Yan, W. M. Kwok and Z. Zheng, Nanoscale, 2011, 3, 258 RSC.
- R. Zou, G. He, K. Xu, Q. Liu, Z. Zhang and J. Hu, J. Mater. Chem. A, 2013, 1, 8445 CAS.
- X. Dong, Y. Cao, J. Wang, M. B. Chan-Park, L. Wang, W. Huanga and P. Chen, RSC Adv., 2012, 2, 4364 RSC.
- T. Song, J. W. Choung, J. G. Park, W. I. Park, J. A. Rogers and U. Paik, Adv. Mater., 2008, 20, 4464 CrossRef CAS.
- Z. Chen, N. Zhang and Y. Xu, CrystEngComm, 2013, 15, 3022 RSC.
- L. Zhang, X. Liu, Z. Lian, X. Wang, G. Shen, D. Shen and Q. Yan, J. Mater. Chem. C, 2014, 2, 3965 RSC.
- A. G. D'Aloia, F. Marra, A. Tamburrano, G. De Bellis and M. S. Sarto, Carbon, 2014, 73, 175–184 CrossRef.
- G. De Bellis, A. Tamburrano, A. Dinescu, M. L. Santarelli and M. S. Sarto, Carbon, 2011, 49, 4291–4300 CrossRef CAS.
- C. M. Lieber, Sci. Am., 2001, 285, 50 CrossRef.
- Y. Ding, P. X. Gao and Z. L. Wang, J. Am. Chem. Soc., 2004, 126, 2066 CrossRef CAS PubMed.
- P. Yang, H. Yan, S. Mao, R. Russo, J. Johnson, R. Saykally, N. Morris, J. Pham, R. He and H.-J. Choi, Adv. Funct. Mater., 2002, 12, 323 CrossRef CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 4430 CrossRef CAS PubMed.
- C. R. Chandraiahgari, G. De Bellis, P. Ballirano, S. K. Balijepalli, S. Kaciulis, L. Caneve, F. Sarto and M. S. Sarto, RSC Adv., 2015, 5, 49861 RSC.
- J. Ding, S. Zhu, T. Zhu, W. Sun, Q. Li, G. Wei and Z. Su, RSC Adv., 2015, 5, 22935 RSC.
- R. Chung, Z. Lin, P. K. Yang, K. Lai, S. F. Jen and P. W. Chiu, Nanoscale Res. Lett., 2013, 8, 350 CrossRef PubMed.
- Y. J. Kim, Hadiyawarman, A. Yoon, M. Kim, G. C. Yi and C. Liu, Nanotechnology, 2011, 22, 245603 CrossRef PubMed.
- M. Khenfouch, M. Baïtoul and M. Maaza, Opt. Mater., 2014, 36, 27 CrossRef.
- J. Fan, T. Li, H. Heng, B. Markovic and I. Djerdj, Acta Chim. Slov., 2015, 62, 902 CrossRef CAS PubMed.
- L. Vayssieres, Adv. Mater., 2003, 15, 465 CrossRef.
- T. O. Okyay, R. K. Bala, H. N. Nguyen, R. Atalay, Y. Bayam and D. F. Rodrigues, RSC Adv., 2015, 5, 2568 RSC.
- G. Cao, Nanostructures and Nanomaterials: Synthesis, Properties and Applications, Imperial College Press, London, UK, 2004 Search PubMed.
- L. E. Greene, M. Law, H. D. Tan, M. Montano, J. Goldberger, G. Somorjai and P. Yang, Nano Lett., 2005, 5, 1231 CrossRef CAS PubMed.
- H. Sowa and H. Ahsbahs, J. Appl. Crystallogr., 2006, 39, 169 CAS.
- J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. Il Park, J. Phys. Chem. C, 2009, 113, 19134 CAS.
- S. Kaciulis, L. Pandolfi, E. Comini, G. Faglia, M. Ferroni, G. Sberveglieri, S. Kandasamy, M. Shafiei and W. Wlodarski, Surf. Interface Anal., 2008, 40, 575 CrossRef CAS.
- S. Kaciulis, G. Mattogno, A. Galdikas, A. Mironas and A. Setkus, J. Vac. Sci. Technol., A, 1996, 14, 3164 CAS.
- S. Kaciulis, A. Mezzi, P. Calvani and D. M. Trucchi, Surf. Interface Anal., 2014, 46, 966 CrossRef CAS.
- Q. Zhao, X. Y. Xu, X. F. Song, X. Z. Zhang, D. P. Yu, C. P. Li and L. Guo, Appl. Phys. Lett., 2006, 88, 033102 CrossRef.
- J. O. Hwang, D. H. Lee, J. Y. Kim, T. H. Han, B. H. Kim, M. Park, K. No and S. O. Kim, J. Mater. Chem., 2011, 21, 3432 RSC.
- W. Song, S. Y. Kwon, S. Myung, M. W. Jung, S. J. Kim, B. K. Min, M. A Kang, S. H. Kim, J. Lim and K. Seok An, Sci. Rep., 2014, 4, 4064 Search PubMed.
- L. Wu, Y. Wu, X. Pan and F. Kong, Opt. Mater., 2006, 28, 418 CrossRef CAS.
- A. B. Djurisic, Y. H. Leung, K. H. Tam, L. Ding, W. K. Ge, H. Y. Chen and S. Gwo, Appl. Phys. Lett., 2006, 88, 103107 CrossRef.
- H. Alnoor, C. O. Chey, G. Pozina, X. Liu, V. Khranovskyy, M. Willander and O. Nur, AIP Adv., 2015, 5, 087180 CrossRef.
- T. Dai, W. Hsu and H. Hsu, Opt. Express, 2014, 22, 27170 Search PubMed.
- E. Kärber, T. Raadik, T. Dedova, J. Krustok, A. Mere, V. Mikli and M. Krunks, Nanoscale Res. Lett., 2011, 6, 359 CrossRef PubMed.
- G. Cao and Y. Wang, Nanostructures and Nanomaterials: Synthesis, Properties and Applications, Imperial College Press, London, UK, 2004 Search PubMed.
- E. Rokhsat and O. Akhavan, Appl. Surf. Sci., 2016, 371, 590 CrossRef CAS.
- E. Zanni, S. De Palma, C. R. Chandraiahgari, G. De Bellis, S. Cialfi, C. Talora, C. Palleschi, M. S. Sarto, D. Uccelletti and P. Mancini, Mater. Lett., 2016, 179, 90 CrossRef CAS.
- E. Zanni, G. De Bellis, M. P. Bracciale, A. Broggi, M. L. Santarelli, M. S. Sarto, C. Palleschi and D. Uccelletti, Nano Lett., 2012, 12, 2740 CrossRef CAS PubMed.
- I. Rago, A. Bregnocchi, E. Zanni, A. G. D'Aloia, F. De Angelis, M. Bossu, G. De Bellis, A. Polimeni, D. Uccelletti and M. S. Sarto, 2015 IEEE 15th International Conference on Nanotechnology (IEEE-NANO), Rome, 2015, pp. 9–12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18317d |
| This journal is © The Royal Society of Chemistry 2016 |