
Exploring potential cooperative effects in dicopper(I)-di-mesoionic carbene complexes: applications in click catalysis†
Stephan
Hohloch‡
,
Lisa
Suntrup‡
and
Biprajit
Sarkar
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195, Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
First published on 2nd December 2015
Abstract
Triazolylidenes are an emerging class of mesoionic carbenes with potential as ligands in homogeneous catalysis. In this contribution we present well-defined dicopper(I) complexes with di-mesoionic carbenes where the copper centers are held at a distance of about 2.8 Å. All complexes display excellent activity as pre-catalysts for the azide–alkyne cycloaddition (click) reaction. Comparisons with analogous mononuclear complexes show the dinuclear pre-catalysts to be twice as active for the same amount of copper used. These results thus point to potentially strong cooperative effects in these dinuclear complexes, and further support the recently established dinuclear reaction pathway for the click reaction. The results presented here provide a synthetic route for generating dinuclear complexes with di-mesoionic carbenes with relatively short metal–metal distances, and opens a general platform for investigating potential cooperative effects in catalysis.
 Biprajit Sarkar | Biprajit grew up in Assam, studied in Delhi and Mumbai before moving to Stuttgart to pursue his doctoral studies. After spending a post-doctoral year in Strasbourg, Biprajit moved back to Stuttgart to work on his Habilitation, which he completed in 2011. At the same time, he was offered a Professor's position at the Free University in Berlin where he is stationed now. His group uses a molecular synthetic approach for designing catalysts and switchable molecular materials with a strong focus on redox-active and mesoionic carbene ligands. The group also specializes in electrochemical and spectroelectrochemical methods. Recently, Biprajit was selected as one of the rising stars at the ICCC-41, was a visiting Professor at IIT Bombay, and was awarded the Zasshikai Lectureship at the University of Tokyo. |
Introduction
The copper(I) catalyzed azide–alkyne cycloaddition reaction (CuAAC), the best click reaction known, is one of the most utilized reactions in contemporary chemistry.1 1,2,3-Triazoles and 1,2,3-triazol-5-ylidenes derived through this method are excellent ligands in coordination and organometallic chemistry.2 Despite finding wide use in all branches of chemistry, the mechanism of the CuAAC reaction has been a topic of intense debate. Recent theoretical and experimental work points to the involvement of a dicopper site as the active catalyst.3 Even though the in situ generation of the active copper(I) catalyst has been the most widely used form for the CuAAC reaction,1 the potential for using well-defined copper(I) complexes was recognized earlier. Obvious advantages of this route is the ease of mechanistic studies with well-defined copper(I) complexes.4Copper(I) complexes of N-heterocyclic carbene (NHC) ligands of the imidazolylidene type have been one of the most widely investigated copper(I) catalysts for this reaction (Fig. 1).5 The copper(I)–NHC combination has not only been shown to deliver highly active catalysts with a large functional group tolerance, but has also intensely contributed to the mechanistic understanding of the CuAAC reaction.3a,d,4a,5 We and others have pioneered the use of copper(I) complexes with mesoionic carbenes (MIC) of the triazolylidene type as catalysts for the CuAAC reaction (Fig. 2).6 Comparative studies have shown the Cu–MIC complexes to be more active than their Cu–NHC counterparts under certain conditions.6c Considering the importance of a dinuclear active catalytic center for the CuAAC reaction, it is surprising that only a few reports exist on the use of well-defined dicopper(I) complexes as catalysts for that reaction.3b,c,d Among those reports, to the best of our knowledge, there is just one instance where a direct comparison of the activity of the related monocopper(I) complex has been made with its dicopper(I) counterpart (Fig. 1).3d However, even in that case, the NHC ligands used for the mononuclear case was of the imidazolylidene type, while a 1,2,4-triazolylidene derived ligand was used for the dinuclear copper complex.
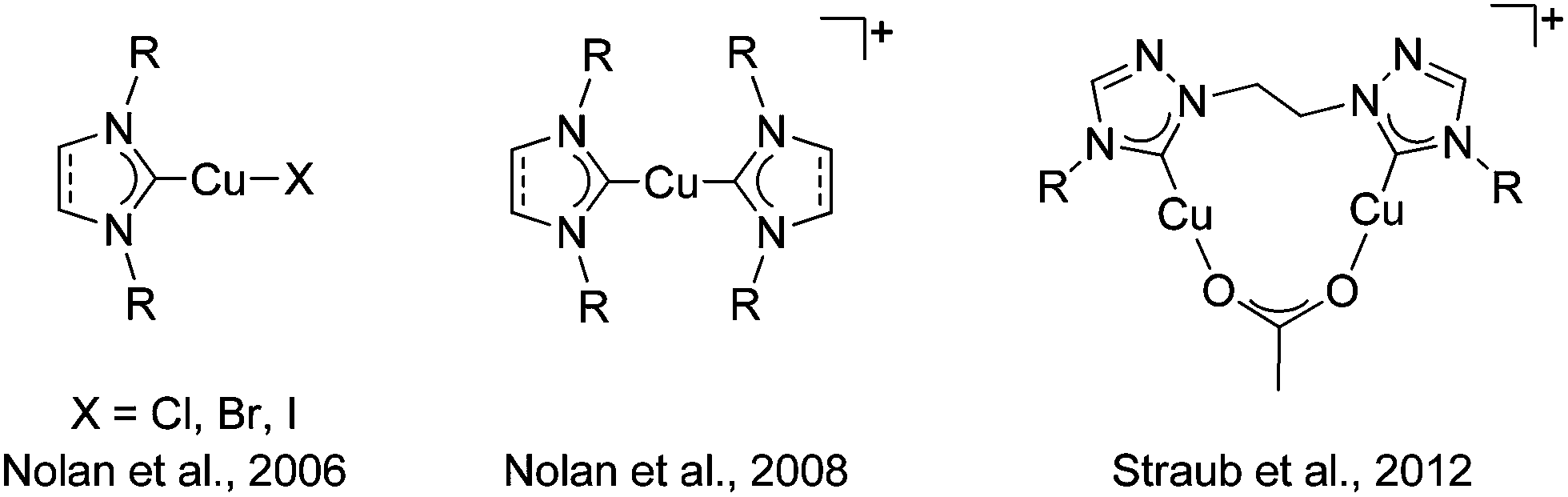 | ||
| Fig. 1 Cu–NHC catalysts for the CuAAC reaction reported in the literature. | ||
 | ||
| Fig. 2 Mononuclear Cu–MIC complexes reported previously (left) and the new dinuclear Cu–MIC complexes (right). | ||
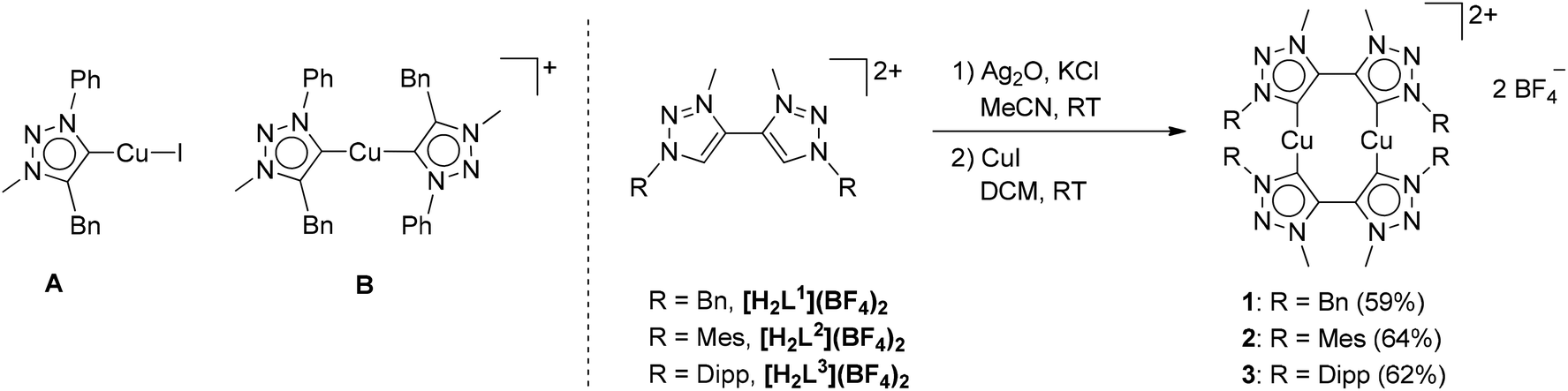
Having established the excellent activity of monocopper(I)-MIC complexes in the CuAAC reaction in the past (Fig. 2, left),6 we turned our attention to the generation of dicopper(I)-MIC complexes as catalysts, keeping in mind the recent proposal of the involvement of a dicopper(I) active site for this reaction.3a In the following work, we present the synthesis and characterization of three dicopper(I) complexes with di-mesoionic carbenes (Fig. 2). The small Cu–Cu distance in these complexes makes them ideal for the investigation of potential cooperative effects. We compare the activity of these complexes with their mononuclear counterparts with a similar ligand backbone. Finally, we show the substrate scope and comment on the reaction order of these reactions with respect to the nuclearity of the complexes.
Results and discussion
Methylation of di-triazoles7 using Meerweins's salt in dichloromethane afforded the desired di-triazolium salts [H2Lx](BF4)2 (x = 1, 2 or 3) in reasonable yields (Fig. 2).7c The identity and purity of the triazoles and the triazolium salts were established using 1H and 13C NMR spectroscopy, mass spectrometry and elemental analysis. The transmetallation route using basic silver oxide in the presence of a halide donor turned out to be the method of choice for the synthesis of the complexes.6a,7c The silver–carbene complex that was formed under the exclusion of light was further reacted with CuI or [Cu(CH3CN)4]BF4. Recrystallization of the crude mixture provided the desired complexes 1–3 (Fig. 2). Even though similar dinuclear complexes have been reported earlier for other metal centers, the distance between the MIC donors in the present cases is smaller, thus making the metal–metal distance shorter than what has been observed for related reported systems (2.796–2.882 Å, see below).2m–pFormation of the triazolylidene complexes was proven by their 1H NMR spectra, which showed the disappearance of the characteristic low field triazolium-5H signal of the ligand precursors. Additionally, the 1H NMR spectra already gave indications that the molecule probably has a complex dinuclear structure. For example, the 1H NMR spectra of 1 showed two doublets for the benzylic CH2-protons, with a relatively large coupling constant of 14 Hz pointing to germinal coupling. On the other hand for 3, four doublets for the CH3 protons of the iso-propyl groups are observed (Fig. S5 and S9†). In the case of 2, all signals are broadened indicating some hindered rotation within this molecule (Fig. S7†). Further indications of the formation of a triazolylidene complex were obtained by 13C NMR spectroscopy. Signals at ppm values of 167.8, 170.7 and 170.2 for 1, 2 and 3, respectively, unambiguously proved the existence of a triazolylidene species in solution. Finally, we were able to obtain single crystals of 1, 2 and 3 suitable for X-ray diffraction analysis, and these results confirmed the structure of the complexes.
For the triazolium salt [H2L2](BF4)2 single crystals were obtained by slow diffusion of diethyl ether into a concentrated solution in dichloromethane. The molecule exhibits an inversion center along the C3–C3 axis (Fig. S34†). The bond lengths and angles are consistent with similar compounds reported in the literature (Table S2†).6 Additionally, single crystals of 1, 2 and 3 were obtained for X-ray diffraction measurements by layering a concentrated solution in dichloromethane with hexane. Unfortunately, in the case of 2 huge accessible voids are present in the crystal structure, which leads the molecule to crystallize with 14 partly disordered molecules of dichloromethane making a reasonable solution of the structure nearly impossible. Crystallization of 2 from other solvents always led to the formation of amorphous white powders. However, the connectivity of 2 was concluded to be the same as observed for 1 and 3 (Fig. 3 and S36†). The triazolylidene rings within the di-triazolylidene ligands are twisted with respect to each other and this twist is different for complexes 1 and 3 (43–48°, Fig. S35†). The Cu–C distances are in the range of the reported values (1.86–1.90 Å).6d
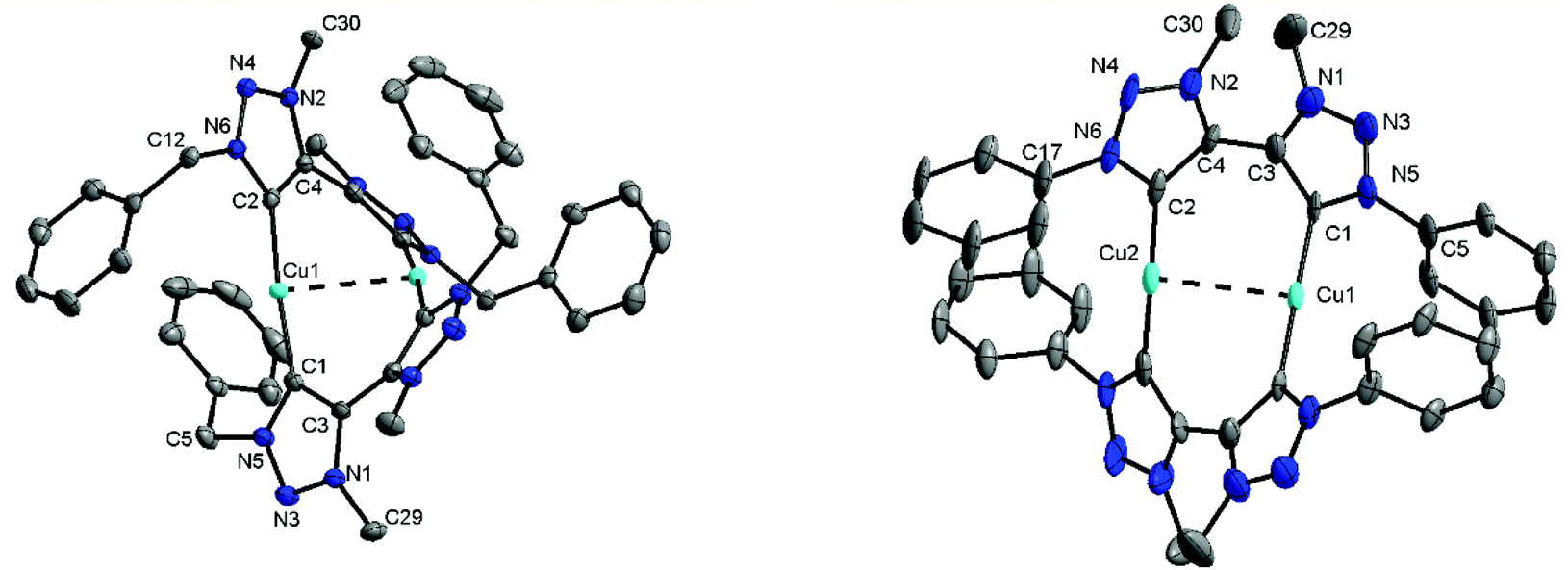 | ||
| Fig. 3 ORTEP-plot of 1 (left) and 3 (right). Hydrogen atoms, solvent molecules and counter ions are omitted for clarity. For 3 the iPr groups are omitted for clarity. Ellipsoids are shown at a probability level of 50%. | ||
Interestingly, both molecules exhibit a somewhat short Cu–Cu distance being 2.796(1) Å in 1 and 2.882(2) Å in 3.
Recent investigations have shown that the active form of the catalyst in the CuAAC reaction is a dinuclear species.3 These results were obtained using Cu–NHC complexes. The dinuclear complexes with short Cu–Cu distances presented here thus provide an ideal platform for investigating the nature of the catalytically active species using well-defined dinuclear copper(I) complexes. Such an investigation is particularly interesting in view of the comparable mononuclear complexes with similar ligands that have been recently reported by our group (Fig. 2).6d To investigate the catalytic performances of such systems and the mechanism, we first started to identify the most active complex of the new series 1–3. The reaction between phenyl azide and phenyl acetylene (Scheme 1) is a good benchmark test for this purpose, since it is a fast reaction but still slow enough to monitor6d (see Fig. 4).
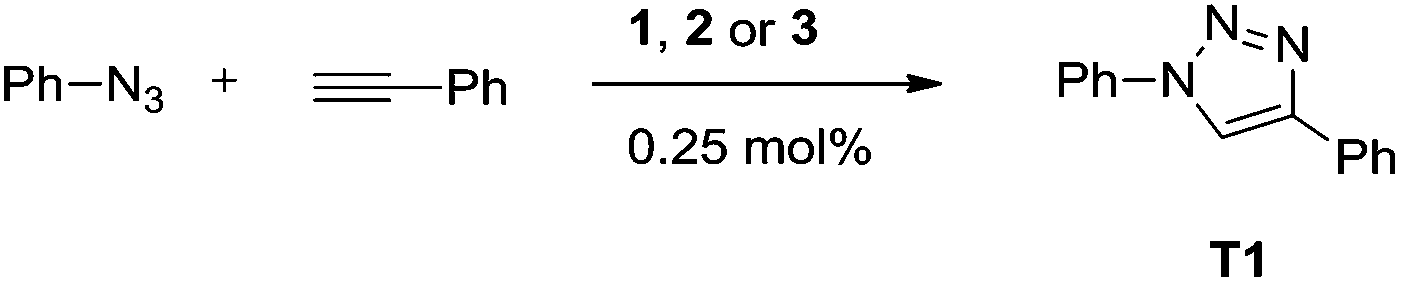 | ||
| Scheme 1 Benchmark reaction to investigate the catalytic performance of Cu–MIC complexes (under neat conditions). | ||
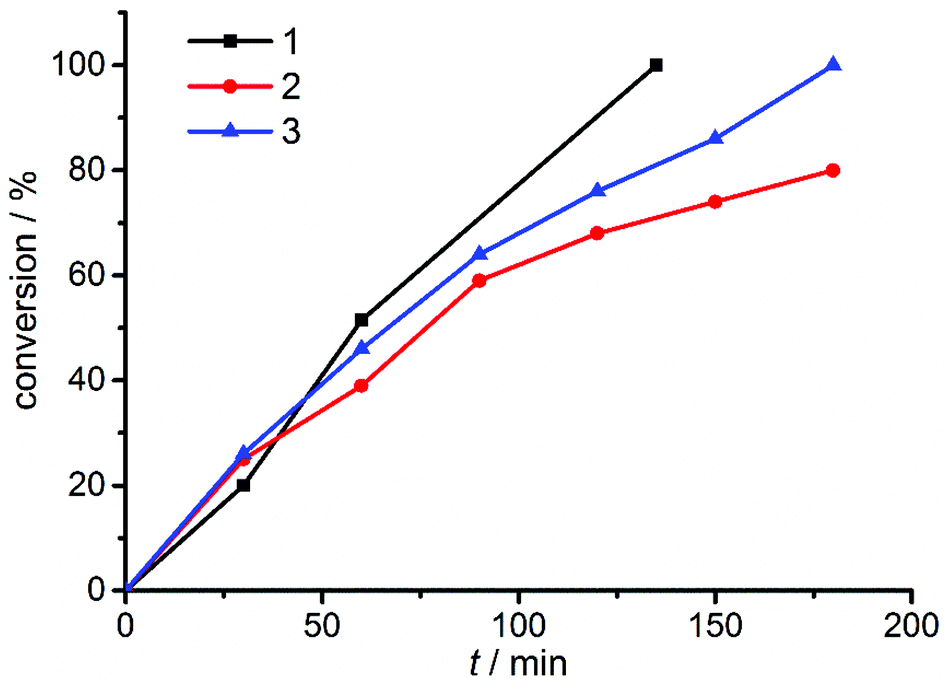 | ||
| Fig. 4 Conversion-time diagram of the benchmark reaction using different binuclear copper complexes 1–3. | ||
As can be seen from Fig. 4 and Table 1, the benzyl substituted (pre)catalyst 1 is the most effective catalyst in this new series of complexes. This is probably related to steric crowding of the Mes and Dipp substituents on the triazolylidene moieties in 2 and 3. Having established the most active catalyst, we then compared this catalyst to our previously reported systems. The reactions were carried out with an overall loading of 0.5 mol% of copper atoms. The results are summarized in Table 2.
| (Pre)catalyst | Time [min] | Conversion [%] |
|---|---|---|
| 1 | 135 | 100 |
| 2 | 180 | 80 |
| 3 | 180 | 100 |
| (Pre)catalyst | Time [min] | Conversion [%] | TOF [h−1] |
|---|---|---|---|
| A | 135 | 28 | 25 |
| B | 135 | 73 | 64 |
| 1 | 135 | >99 | 89 |
Table 2 clearly shows a trend we had previously observed for the mononuclear complexes:6d the neutral mononuclear (MIC)–Cu halide complex A (see Fig. 2) is the least active (pre-)catalyst in our series of triazolylidene copper complexes, converting the starting materials only to 28% after 135 min and 72% after 24 hours, respectively. Stirring the reaction for another 24 hours did not result in higher conversions. In comparison, the most active mononuclear complex with a similar substitution pattern reported by us to date, complex B (Fig. 2), shows conversions of 73% after 135 min6d and full conversion was reached after 420 minutes. In contrast to these results, the new dicationic, dinuclear complex 1 shows full conversion of the starting materials already after 135 minutes. The calculated turnover frequencies (TOF, after 135 min) are significantly higher for complex 1 (89 h−1) than for the mononuclear complexes A (25 h−1) and B (64 h−1). These results already point to a potentially strong cooperative effect in 1. In view of previous reports,5c,6d we believe, that the active species in catalysis is created by breaking a copper–MIC bond and one MIC-ligand acts as an internal base to deprotonate the alkyne which then coordinates to the copper center forming an acetylide species, which reacts further. Fig. 5 shows five possible activation pathways that can occur during catalysis.
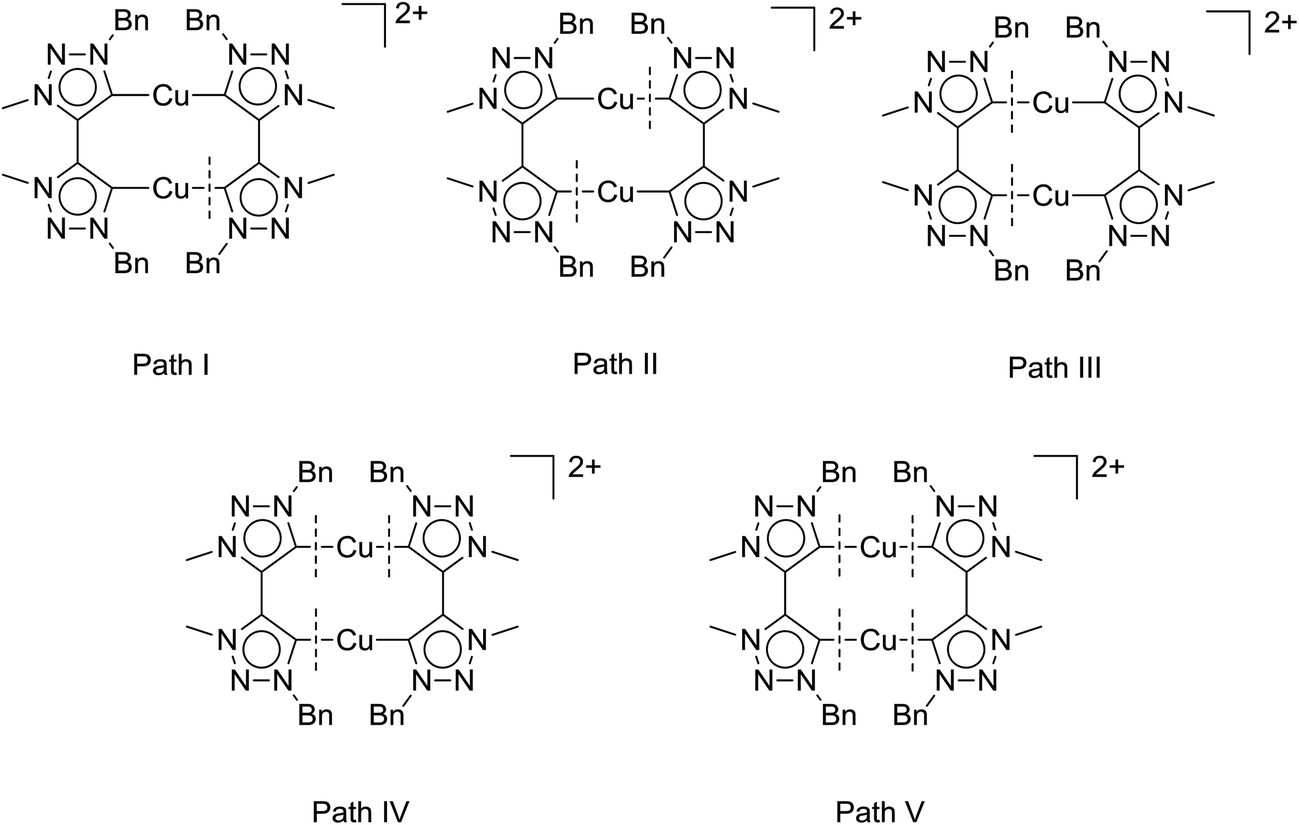 | ||
| Fig. 5 Possible activation/breaking pathways for dinuclear complex 1. | ||
To obtain strong cooperative effects between the metal centers, it is most likely that the active species in solution is of a dinuclear nature. The bond breaking pathway II would result in two mononuclear active species whereas pathway IV would deliver one mononuclear active species and a “naked” copper atom. Pathway V would deliver two “naked” copper species which should not be that active as a catalyst as can be judged from the catalytic performance of [Cu(MeCN)4][BF4] for example6d (conversion after 135 min: 19% with 1,2-dibromoethane as an internal standard, see Fig S38†). Furthermore, the “naked” copper species would also be likely highly susceptible to oxidation reactions. Therefore, bond breaking in these systems is likely to occur in accordance with either Path I or Path III. By determining the reaction order with respect to the catalyst one can usually distinguish between the different pathways.3c In case of Path I and III monomolecular, dinuclear complexes form the active species in the catalytic process, and thus these pathways should result in a reaction order of 1 with respect to the catalyst.
Kinetic NMR studies were carried out with our benchmark reaction between phenyl acetylene and phenyl azide in CD3CN to determine the reaction order of the catalyst (Fig. 6). The slope of the line from the logarithm of the starting reaction rate plotted against the logarithm of the used catalyst concentrations gives the reaction order with respect to the catalyst as 1. Vint was calculated from the slope of the curves between 20 and 35 minutes. Observing a reaction order of 1 thus supports the hypothesis that either pathway I or III delivers the active catalyst. Additionally, we found by NMR spectroscopy, the formation of the free ligand [H2L1]2+ during catalysis, which is consistent with previous reports for the activation of cationic triazolylidene complexes in the CuAAC.5c,6d Furthermore, the addition of a stoichiometric amount of phenyl acetylene to a solution of 2 (2 was used because of the ease of assignment of the signals in the aromatic region as compared to 1) led to the progressive decrease in the intensity of the acetylene C–H protons (Fig. S39 and S40†). The addition of a stoichiometric amount of benzyl azide to the same solution also led to the formation of the substituted triazole. We were also able to observe signals for the free ligand [H2L2]2+ from the reaction mixture containing 2 and phenyl acetylene. All these observations likely point to the breaking of the C–H bond of the phenyl acetylene as well as the release of one of the ligands attached to the copper centers as important steps in these reactions.
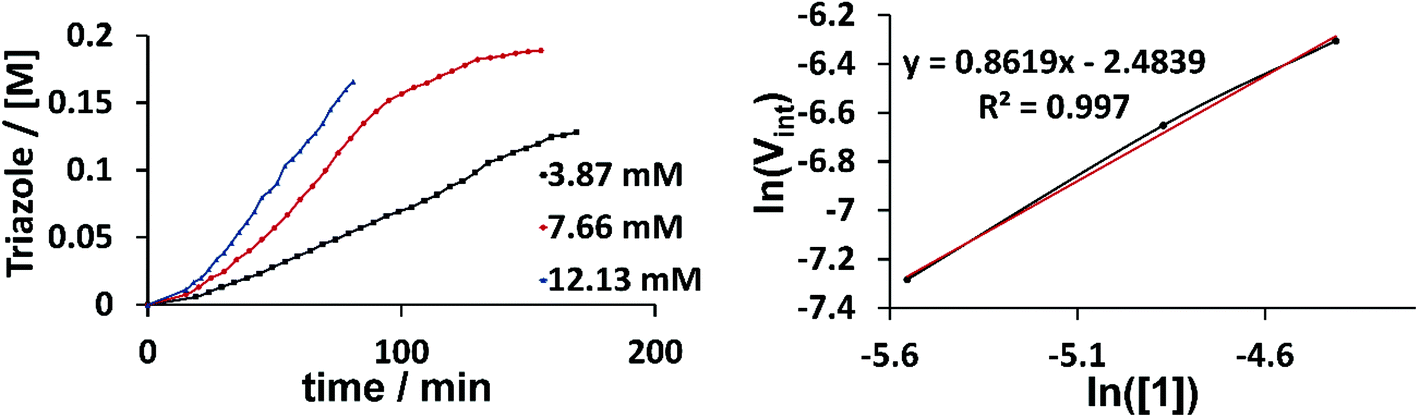 | ||
| Fig. 6 Conversion-time profile (top) and reaction order plot (bottom) for the reaction between phenyl azide and phenyl acetylene using 1 as the catalyst. | ||
Taken together, these results support pathway III as the activation pathway for the formation of the active catalyst, and also point to the dinuclear nature of this species. A similar behavior with dinuclear copper catalysts based on silicotungstates has been reported previously.3b While the occurrence of other possible reaction pathways cannot be completely ruled out, from the data that we have in hand up to now, it appears that it is pathway III (Fig. 5) that delivers the active catalytic species.
We next investigated the catalytic scope of the new catalytic system. Chart 1 gives an overview of the 1,2,3-triazoles that have been synthesized using 1 as a (pre)catalyst at catalyst loadings of 0.25 mol%. As the reactions were run either neat or in dichloromethane, yields were determined directly from the reaction mixture by 1H NMR spectroscopy (either directly or after removing dichloromethane, see the Experimental section and ESI Fig. S11–S32†). To prove the integrity of the method, for selected examples the conversions were also determined by using an internal standard or by determination of the isolated yields.
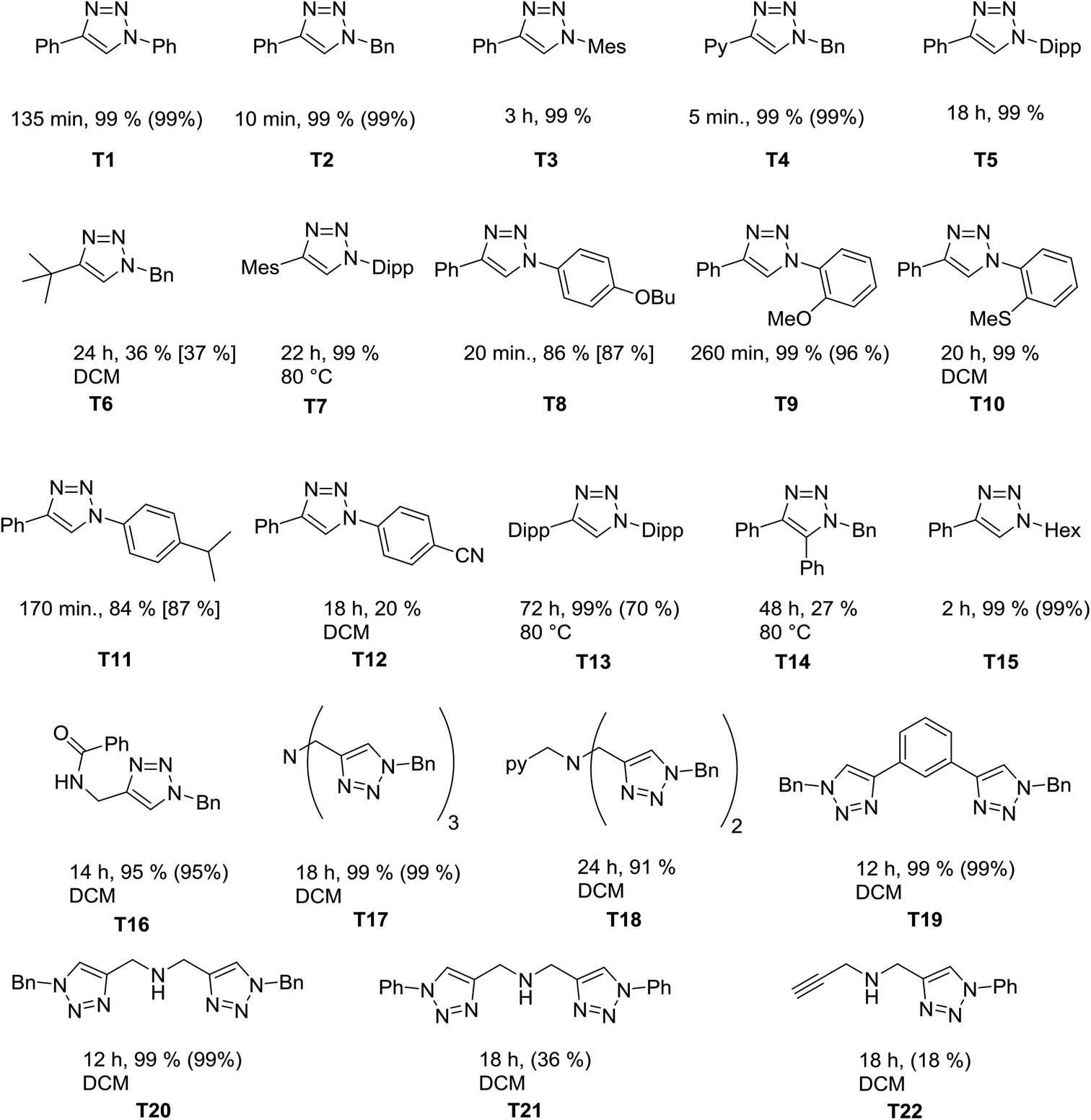 | ||
| Chart 1 Substituted triazoles obtained with the dinuclear complex 1 as a (pre)catalyst. Triazoles T21 and T22 were obtained from the same reaction. Conversion was determined via1H NMR spectroscopy. Numbers in brackets are conversions determined with the use of 1,2-dibromoethane (0.25 mmol) as an internal standard. Numbers in parenthesis refer to isolated yields. For the compounds where DCM is mentioned under the structure, reactions were run in 2 mL of dichloromethane. For all other cases they were run under neat conditions. | ||
Of particular interest here are the triazoles with sterically demanding substituents (T7 and T13). These are difficult products for the CuAAC, and 1 is able to deliver these products in good yields. The tripodal triazole tbta, T17, is another triazole that is challenging to synthesize. T17 is a useful molecule in various branches of chemistry.8 The usual route for synthesizing this molecule uses a mixture of CuSO4 and sodium ascorbate with heating over 2 days at 50 °C. This route uses a high catalyst loading and most of the times column chromatography is necessary to remove incompletely “clicked” side products or remaining copper(II) salts. In contrast to this, we have been able to perform the reaction on a 530 mg (1 mmol) product scale without the observation of any side products. The triazole T18 is an even harder substrate for the click reaction, since it can coordinate to the copper centers and make them highly sensitive towards oxygen.12k With the method developed here, we were able to achieve conversions of 91% within 24h for T18 under ambient conditions (moisture and air). It should be mentioned that this triazole has previously been synthesized under the exclusion of moisture and air with higher catalyst loadings in lower yields over 3 days.8c This once again points out the high activity of our catalysts in this reaction.
Overall the new (pre)-catalyst 1 shows a high tolerance towards different functional groups (thio-ethers, ethers, amines, amides and pyridines) and is successful in catalyzing [3 + 2] cycloaddition reaction of electron-rich aryl azides (4-substitutions and 2-substiutions patterns) or alkyl azides and the reactions can be performed smoothly also in solution if needed. However, in the case of electron poor aryl azides (e.g. nitrile substituted azides, T12) and sterically demanding alkynes, such as in T6 and T14, the catalytic performance is still not optimum, and can likely be improved further.
Conclusions
In conclusion, we have presented dicopper(I) complexes of di-MIC ligands with Cu–Cu distances of about 2.8 Å. This dicopper(I) platform is an extremely efficient catalyst for the CuAAC reaction. Kinetic measurements have established the reaction order as one with respect to the catalyst, thus indirectly pointing to the involvement of a dinuclear species in catalysis. Such dinuclear complexes are thus drastically more active as catalysts than their mononuclear counterparts, making them leading candidates in the Cu–carbene family for the CuAAC reaction. Such well-defined, highly active catalysts could possibly find use in bio-conjugation studies where low copper loading is a pre-requisite. It is also the first instance where a well-defined mono-copper(I) complex has been compared to its dicopper(I) counterpart (with virtually the same ligands) with respect to catalytic activity in the CuAAC reaction. The new catalysts display a wide substrate scope and are capable of catalyzing reactions between demanding substrates in good yields. Such di-MIC ligands are expected to be useful in generating possible cooperative effects9 with other metal centers as well, thus opening up new doors to investigate cooperative catalysis with MIC ligands.Experimental section
Materials and physical methods
Copper iodide and trimethyloxonium tetrafluoroborate (Meerwein's salt) was purchased from Sigma-Aldrich. All the reagents were used as supplied. [Cu(MeCN)4]BF4 was synthesized following literature known procedures.10 The solvents used for metal complex synthesis and methylations via Meerwein salt were dried and distilled under argon and degassed by common techniques prior to use. DCM-d2 for NMR experiments was dried over molecular sieves (4 Å) prior to degassing. All ditriazoles and the ditriazolium salts [H2L3(BF4)2] are literature known.7b,7c,11 The 1H and 13C NMR spectra were recorded on a Jeol ECS 400 spectrometer. Chemical shifts are reported in ppm relative to TMS. Spectra are referenced to the residual solvent peak. Multiplicities are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), sept (septet), m (multiplet), br (broad) or combinations thereof. Elemental analyses were performed on a Vario Elementar EL III. Mass spectrometry was performed on an Agilent ESI-TOF instrument.Synthesis of triazolium salts
The corresponding triazole (1.00 equiv., 3.00 mmol) was dissolved in dry dichloromethane and Meerwein salt (2.50 equiv., 7.50 mmol) was added at room temperature. The mixture was closed and stirred under an inert gas atmosphere for 3 days. The reaction was quenched by addition of 1 mL of methanol and poured into diethyl ether (300 mL), resulting in precipitation of the triazolium salts. The white solids were collected by filtration, washed with diethyl ether and dried to give the desired triazolium salts in good yields of 68% and higher.Synthesis of the copper complexes
Purification method A
The compounds were isolated by dissolving the solids in dichloromethane (10 mL) and washing them with ammonia/EDTA solution twice (2 × 10 mL), followed by one washing with deionized water (10 mL). The aqueous washings were re-extracted with dichloromethane (15 mL) and the combined organic layers were dried over sodium sulfate, filtered and evaporated to give the corresponding triazoles as pure, white to off-white solids in excellent yields.Purification method B
Similar to Method A. Additionally, the solids were recrystallized from hot hexanes to give the corresponding triazoles in moderate yields.Purification method C
Similar to Method A. Additionally, the solids were subjected to silica gel column chromatography using dichloromethane and methanol (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 80:20 v/v) as the eluent.
:5 to 80:20 v/v) as the eluent.
The respective NMR spectra can be found in the ESI (Fig. S11–S33†). Triazoles T1–T20 have all been reported previously in the literature.2i,6a,b,8c,12
Triazoles T21 and T22 were obtained from the same reaction using dipropargylamine (1.00 equiv., 1.00 mmol, 93 mg) and phenyl azide (2.00 equiv., 2.00 mmol, 238 mg). The reaction was stirred at room temperature for 18 h. Crude NMR showed the formation of 2 different triazole species and Method C was applied for the workup of the reaction to give T21 and T22.
Kinetic experiments
Stock solutions of phenylacetylene and phenylazide (each 0.2 M) were prepared in 5 mL of MeCN-d3. For kinetic experiments, the desired amount of catalyst was weighed in a 5 mL vial. Then the corresponding azide stock solution was added and the catalyst was completely dissolved. Afterwards, the phenylacetylene stock solution was added and the mixture was transferred into an NMR tube which was sealed with a parafilm. 1H NMR spectra were taken and analyzed every 3–5 minutes. Yields were determined by comparison of the integrals from the acetylene-H and the triazole-5H peak of the corresponding product (compare ESI, Fig. S37†). Vint was determined from the slope of the reaction–conversion plot between the times from 20 to 35 minutes and was plotted as the lg against the lg of the corresponding catalyst concentration to give the reaction order of the catalyst.NMR experiments
Catalyst 2 (0.01 mmol, 9.8 mg) was dissolved in dry and degassed, deuterated dichloromethane (0.4 mL) in an NMR tube with a J. Young valve before 0.1 mL stock solution containing phenylacetylene (0.1 mmol, 10.9 μL, 0.1 M) and 1,2-dibromoethane (25.0 μmol, 2.4 μL, 25.0 mM) in DCM-d2 was added. The reaction was then monitored via1H NMR. After 12 h, a stock solution of benzyl azide in DCM-d2 was added and the mixture was again monitored via1H NMR.X-ray crystallography
X-ray quality crystals of [H2L2](BF4)2 were obtained by slow diffusion of ether into a concentrated solution of dichloromethane at 8 °C. X-ray quality crystals of 1, 2 and 3 were obtained by slow diffusion of n-hexane into a concentrated solution of these complexes in dichloromethane at 8 °C. Single crystal X-ray structural studies were performed on a Bruker smart AXS diffractometer. Data were collected at 100(2) K using graphite-monochromated Mo Kα radiation (λα = 0.71073 Å). The strategy for the data collection was evaluated by using Smart software. The data were collected by the standard ‘omega-scan’ techniques, and were scaled and reduced using Saint+ software. The structures were solved by direct methods using SHELXS-97 and refined by full matrix least-squares with SHELXL-97, refining on F2.13 CCDC 954946, 954947 and 954948 contain the CIF-Files for L,3b1 and 3 respectively.Acknowledgements
We are grateful to the Fonds der Chemischen Industrie (FCI) and the Freie Universität Berlin for financial support for this work.References
- (a) V. V. Rostostev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef; (b) C. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed.
- For selected reviews and examples see: (a) H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC; (b) D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nachr. Chem., 2011, 59, 937 CrossRef CAS; (c) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC; (d) K. F. Donnelly, A. Petnonilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC; (e) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS; (f) J. D. Crowley and D. McMorran, in Topics in Heterocyclic Chemistry, ed. J. Kosmrlj, Springer, Berlin/Heidelberg, 2012, vol. 22, p. 31 Search PubMed; (g) J. M. Aizpurua, R. M. Fratila, Z. Monasterio, N. Perez-Esnaola, E. Andreieff, A. Irastorza and M. Sagartzazu-Aizpurua, New J. Chem., 2014, 38, 474 RSC; (h) J. D. Crowley, A. Lee and K. J. Kiplin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS; (i) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (j) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164 CrossRef CAS; (k) S. Hohloch, W. Frey, C.-Y. Su and B. Sarkar, Dalton Trans., 2013, 42, 11355 RSC; (l) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (m) L. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 456 CrossRef; (n) J. Cai, X. Yang, K. Arumugam, C. W. Bielawski and J. L. Sessler, Organometallics, 2011, 30, 5033 CrossRef CAS PubMed; (o) M. T. Zamora, M. J. Ferguson and M. Cowie, Organometallics, 2012, 31, 5384 CrossRef CAS; (p) K. J. Kiplin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328 RSC; (q) A. Petronilho, J. A. Woods, S. Bernhard and M. Albrecht, Eur. J. Inorg. Chem., 2014, 708 CrossRef CAS; (r) J. M. Aizpurua, M. Sagartzazu-Aizpurua, Z. Monasterio, I. Azcune, C. Mendicute, J. I. Miranda, C. Garcia-Lecina, A. Altube and R. M. Fratila, Org. Lett., 2012, 14, 1866 CrossRef CAS PubMed; (s) R. Maity, M. van der Meer and B. Sarkar, Dalton Trans., 2015, 44, 46 RSC; (t) R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem. – Eur. J., 2014, 20, 9952 CrossRef CAS PubMed; (u) R. Maity, M. van der Meer, S. Hohloch and B. Sarkar, Organometallics, 2015, 34, 3090 CrossRef CAS; (v) L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949 RSC.
- For selected examples see: (a) B. T. Worell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef PubMed; (b) K. Kamata, Y. Nakagawa, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15304 CrossRef CAS PubMed; (c) G.-C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984 CrossRef CAS PubMed; (d) R. Berg, J. Straub, E. Schreiner, S. Mader, F. Rominger and B. F. Straub, Adv. Synth. Catal., 2012, 354, 3445 CrossRef CAS; (e) V. O. Rodiniv, S. I. Presolski, D. Díaz Díaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef PubMed; (f) V. O. Rodinov, S. I. Presolski, S. Gardinier, Y.-H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696 CrossRef PubMed; (g) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 Search PubMed.
- (a) C. Nolte, P. Mayer and B. F. Straub, Angew. Chem., Int. Ed., 2007, 46, 2101 CrossRef CAS PubMed; (b) P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459 RSC; (c) R. Berg and B. F. Straub, Beilstein J. Org. Chem., 2013, 9, 2715 CrossRef PubMed; (d) S. Lal, H. S. Rzepa and S. Diez-Gonzalez, ACS Catal., 2014, 4, 2274 CrossRef CAS; (e) C. Shao, G. Cheng, D. Su, J. Xu, X. Wang and Y. Hu, Adv. Synth. Catal., 2010, 352, 1587 CrossRef CAS.
- For selected examples see: (a) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2006, 12, 7558 CrossRef PubMed; (b) S. Díez-González, E. D. Stevens and S. P. Nolan, Chem. Commun., 2008, 4747 RSC; (c) S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881 CrossRef PubMed; (d) C. Gibard, D. Avignant, F. Cisnetti and A. Gautier, Organometallics, 2012, 31, 7902 CrossRef CAS; (e) M.-L. Teyssot, A. Chevry, M. Traikia, M. El-Ghozzi, D. Avignaut and A. Gautier, Chem. – Eur. J., 2009, 15, 6322 CrossRef CAS PubMed; (f) B. Liu, C. Chen, Y. Zhang, X. Liu and W. Chen, Organometallics, 2013, 32, 5451 CrossRef CAS; (g) F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969 CrossRef CAS; (h) S. C. Sau, S. R. Roy, T. K. Sen, D. Mullangi and S. K. Mandal, Adv. Synth. Catal., 2013, 355, 2982 CrossRef CAS; (i) L. R. Collins, T. M. Rookes, M. F. Mahon, I. M. Riddlestone and M. K. Whittlesey, Organometallics, 2014, 33, 5882 CrossRef CAS.
- (a) T. Nakamura, T. Terashima, K. Ogata and S. Fukuzawa, Org. Lett., 2011, 13, 620 CrossRef CAS PubMed; (b) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067 CrossRef CAS; (c) S. Hohloch, B. Sarkar, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808 CrossRef CAS; (d) S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 3956 CrossRef CAS.
- (a) U. Monkowius, S. Ritter, B. König, M. Zabel and H. Yersin, Eur. J. Inorg. Chem., 2007, 4597 CrossRef CAS; (b) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Organometallics, 2011, 30, 6017 CrossRef CAS PubMed; (c) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376 CrossRef CAS; (d) J. T. Flechter, B. J. Bumgarner, N. D. Engels and D. A. Skoglund, Organometallics, 2008, 27, 5430 CrossRef; (e) S. Hohloch, S. Kaiser, F. L. Dücker, A. Bolje, R. Maity, J. Kosmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686 RSC.
- (a) D. Schweinfurth, F. Weisser, D. Bubrin, L. Bogani and B. Sarkar, Inorg. Chem., 2011, 50, 6114 CrossRef CAS PubMed; (b) D. Schweinfurth, J. Klein, S. Hohloch, S. Dechert, S. Demeshko, F. Meyer and B. Sarkar, Dalton Trans., 2013, 42, 6944 RSC; (c) F. Weisser, S. Hohloch, S. Plebst, D. Schweinfurth and B. Sarkar, Chem. – Eur. J., 2014, 20, 781 CrossRef CAS PubMed; (d) D. Schweinfurth, S. Demeshko, M. M. Khusniyarov, S. Dechert, V. Gurram, M. R. Buchmeiser, F. Meyer and B. Sarkar, Inorg. Chem., 2012, 51, 7592 CrossRef CAS PubMed; (e) D. Schweinfurth, J. Krzystek, I. Schapiro, S. Demeshko, J. Klein, J. Telser, A. Ozarowski, C.-Y. Su, M. Atanasov, F. Neese and B. Sarkar, Inorg. Chem., 2013, 52, 6880 CrossRef CAS PubMed; (f) D. Schweinfurth, S. Demeshko, S. Hohloch, M. Steinmetz, J. G. Brandenburg, S. Dechert, F. Meyer, S. Grimme and B. Sarkar, Inorg. Chem., 2014, 53, 8203 CrossRef CAS PubMed; (g) D. Schweinfurth, M. G. Sommer, M. Atanasov, S. Demeshko, S. Hohloch, F. Meyer, F. Neese and B. Sarkar, J. Am. Chem. Soc., 2015, 137, 1993 CrossRef CAS PubMed; (h) F. Weisser, S. Plebst, S. Hohloch, M. van der Meer, S. Manck, F. Führer, V. Radtke, D. Leichnitz and B. Sarkar, Inorg. Chem., 2015, 54, 4621 CrossRef CAS PubMed; (i) F. Weisser, H. Stevens, J. Klein, M. van der Meer, S. Hohloch and B. Sarkar, Chem. – Eur. J., 2015, 21, 8926 CrossRef CAS PubMed; (j) T. U. Connell, C. Schieber, I. P. Silvestri, J. M. White, S. J. Williams and P. S. Donnelly, Inorg. Chem., 2014, 53, 6503 CrossRef CAS PubMed.
- M. Weiss and R. Peters, in Bimetallic Catalysis: Cooperation of Carbophilic Metal Centers in: Cooperative Catalysis – Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) D. Fortin, M. Drouin, M. Turcotte and P. D. Harvey, J. Am. Chem. Soc., 1997, 119, 531 CrossRef CAS; (b) G. J. Kubas, Inorg. Synth., 1979, 19, 90 CrossRef CAS.
- C. E. Welby, L. Gilmartin, R. R. Marriott, A. Zahid, C. R. Rice, E. A. Gibson and P. I. P. Elliott, Dalton Trans., 2013, 42, 13527 RSC.
- (a) K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809 CrossRef CAS PubMed; (b) R. Thorwirth, A. Stolle, B. Ondruschka, A. Wild and U. S. Schubert, Chem. Commun., 2011, 47, 4370 RSC; (c) J. E. M. N. Klein, M. S. Holzwarth, S. Hohloch, B. Sarkar and B. Plietker, Eur. J. Org. Chem., 2013, 6310 CrossRef CAS; (d) X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 5409 CrossRef CAS; (e) Z.-J. Cai, X.-M. Lu, Y. Zi, L.-J. Shen, J. Li, S.-Y. Wang and S. J. Ji, Org. Lett., 2014, 16, 5108 CrossRef CAS PubMed; (f) J. Deng, Y. M. Wu and Q. Y. Chen, Synthesis, 2005, 2730 CAS; (g) P. Li, L. Wang and Y. Zhang, Tetrahedron, 2008, 64, 10825 CrossRef CAS; (h) N. Candelon, D. Lastécouères, A. K. Diallo, J. R. Aranzaes, D. Astruc and J.-M. Vincent, Chem. Commun., 2008, 741 RSC; (i) S. L. Lee, M. Yi, S. Y. Chu, J. Y. Lee, H. R. Kwon, K. R. Lee, D. Kang, W. S. Kim, H. B. Lim, J. Lee, H.-J. Youn, D. Y. Chi and N. H. Hur, Chem. Commun., 2010, 46, 3935 RSC; (j) P. Fabrizzi, S. Cicchi, A. Brandi, E. Sperotto and G. van Koten, Eur. J. Org. Chem., 2009, 5423 CrossRef; (k) S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570 CrossRef CAS PubMed; (l) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS PubMed; (m) M. Liu and O. Reiser, Org. Lett., 2011, 13, 1102 CrossRef CAS PubMed.
- SHELXS-97, G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, ORTEP plots, tables with crystallographic details. CCDC 954946–954948. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00163c |
| ‡ Both the authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |