
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in synthetic methods for benzo[b]heteroles
Bin
Wu
and
Naohiko
Yoshikai
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: nyoshikai@ntu.edu.sg
First published on 12th February 2016
Abstract
Benzo[b]heteroles containing heteroatoms other than nitrogen and oxygen have received considerable attention for their potential applications in materials science. This poses an increasing demand for efficient, selective, and broad-scope methods for their synthesis. This review article summarizes the recent developments in synthetic methods and approaches to access representative members of the benzoheterole family.
 Bin Wu | Bin Wu was born in Jiangsu Province, China in 1986. He received his B.Sc. degree in 2009 and M.Sc. degree in 2012 from Soochow University, China. He subsequently moved to Singapore and enrolled as a Ph.D. student in Prof. Naohiko Yoshikai's group at Nanyang Technological University. His current research is focused on the development of novel synthetic approaches to heteroatom-bridged π-conjugated systems such as benzoheteroles and dibenzoheteroles. |
 Naohiko Yoshikai | Naohiko Yoshikai was born in 1978 and raised in Tokyo, Japan. He received his B.Sc. (2000), M.Sc. (2002), and Ph.D. (2005) degrees from the University of Tokyo under the guidance of Prof. Eiichi Nakamura, and then served as an Assistant Professor at the same institute (2005–2009). In 2009, he moved to Singapore to join the faculty of Nanyang Technological University as an Assistant Professor and a Research Fellow of the Singapore National Research Foundation. His research interests are focused on the development and mechanistic study of novel transition metal-catalyzed reactions and their synthetic applications. |
1. Introduction
Indole and benzofuran represent the most ubiquitous benzo-fused five-membered heterocycles in natural products.1 They are also common structural elements in man-made drugs and other functional molecules. Consequently, numerous methods have been developed for their synthesis over the years.2 On the other hand, analogous heterocycles containing other Group 13–16 heteroatom elements (called benzoheteroles hereafter) have also received increasing interest in recent years. Besides the presence of benzothiophene moieties in pharmaceutically relevant compounds, a major reason for this increasing interest revolves around unique optoelectronic properties of benzoheteroles. Benzoheteroles can be viewed as heteroatom-bridged styrenes, whose electronic properties (e.g., HOMO/LUMO levels and gaps) should be substantially influenced by the heteroatom moiety as well as by the peripheral substituents.3 As such, benzoheteroles with different heteroatoms such as silicon, phosphorus, sulfur and selenium, along with their non-benzo-fused and dibenzo-fused analogues (i.e., heteroles and dibenzoheteroles), may exhibit attractive optical and electronic properties for applications as materials for organic light-emitting devices (OLED), photovoltaics (OPV), and field-effect transistors (OFET) as well as fluorescent probes for biological studies (Fig. 1).4–7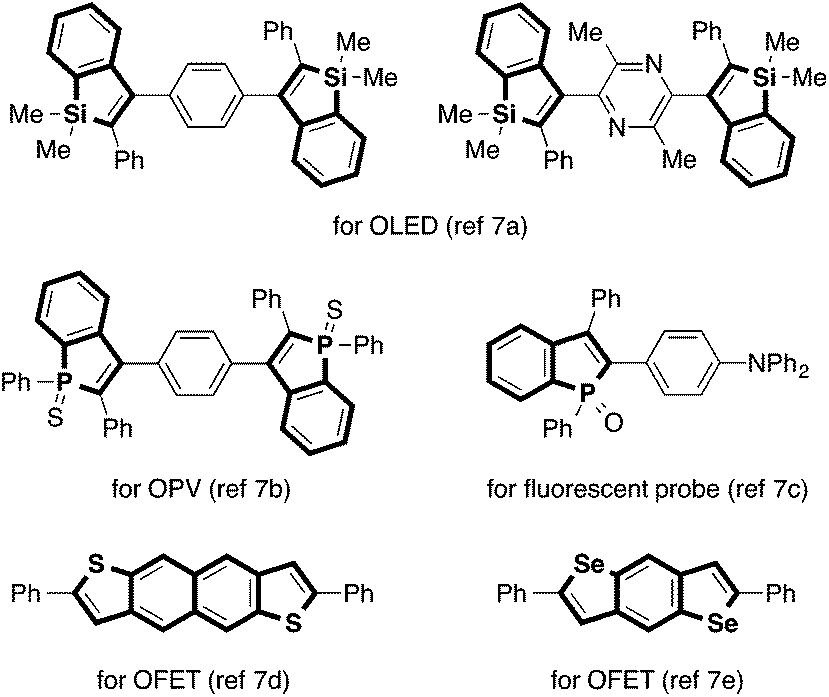 | ||
| Fig. 1 Examples of benzoheterole-containing functional molecules. | ||
From an application perspective, benzoheteroles may be discussed together with heteroles and dibenzoheteroles. On the other hand, from a synthetic standpoint, it may be worthwhile to focus the discussion on benzoheteroles while leaving the rest. This is because many of the synthetic approaches to benzoheteroles are based on intra- or intermolecular reactions between functionalized arenes and alkynes (Scheme 1), which are not always relevant to the synthesis of heteroles and dibenzoheteroles. One of the common approaches employs a heteroatom-functionalized arene bearing an ortho-alkynyl group (called type A precursor), which is cyclized to the corresponding benzoheterole with the aid of a reagent or a catalyst that activates either the heteroatom or the alkyne moiety (Scheme 1a). The Castro indole synthesis represents a prototypical example of this approach.8 In another common approach, a heteroatom-functionalized arene bearing an ortho-halogen atom and an alkyne (type B precursors) are annulated under transition metal catalysis or otherwise appropriate conditions (Scheme 1b). In some cases, the halogen atom may be replaced by another functional group or even a hydrogen atom. The Larock indole synthesis using ortho-haloaniline derivatives is a prototypical example of this approach.9
 | ||
| Scheme 1 Two common approaches to benzoheteroles (E and EY denote a heteroatom and a heteroatom-containing group). | ||
The purpose of this review article is to provide an overview of the recent progress in synthetic methods for representative members of the benzoheterole family (except indole and benzofuran) based on the above-discussed and alternative approaches.10 The following sections are organized according to the central heteroatom element, starting from Group 14 (benzosilole and benzogermole) to Group 15 (benzophosphole) and then to Group 16 (benzothiophene and benzoselenophene). In each section, methods employing type A or type B precursors are discussed first, followed by other notable methods. The article focuses on methods developed in the last decade, while earlier reports are also discussed where considered necessary. Note that synthesis of benzoheteroles fused with extra aromatic moieties at the 2,3-positions (e.g., bis-heteroatom-bridged stilbenes) will not be covered in this article.11
2. Benzosiloles and benzogermoles
A series of intramolecular cyclization reactions of type A substrates have been developed for the synthesis of benzosiloles. Simple cyclization of ortho-alkynylarylsilanes into 2-substituted benzosiloles through (formal) trans-hydrosilylation has been achieved using a Lewis acidic AlCl3 catalyst by Yamamoto,12 a cationic Ru catalyst by Murakami,13 or KH as a promoter by Tsuji and Nakamura (Scheme 2).14 The latter KH-promoted protocol was demonstrated to show a good scope with respect to the alkyne substituent.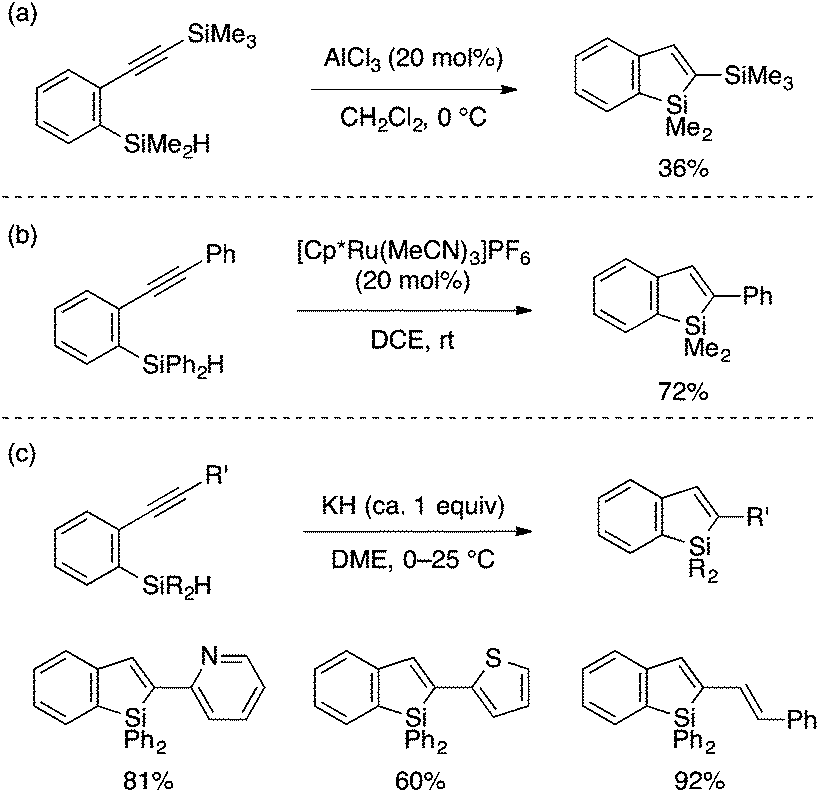 | ||
| Scheme 2 Cyclization of ortho-alkynylarylsilanes to 2-substituted benzosiloles. | ||
Transformation of ortho-alkynylarylsilanes into 2,3-disubstituted benzosiloles is also possible with careful choice of substrates and reaction conditions. Yamaguchi pioneered a reductive cyclization approach for such transformation, which features an alkenyl 1,2-dianion as a putative reactive intermediate (Scheme 3a).15 Cyclization of this anion through Si–H bond cleavage leads to a benzosilol-3-yl anion, which can be intercepted by an electrophile to afford a 2,3-disubstituted benzosilole. Interestingly, the reductive cyclization proceeds more efficiently with π-extended starting materials, and multi-fold cyclization of such substrates offers novel synthetic routes to ladder-shaped Si,C-bridged oligo(p-phenylenevinylene)s (Scheme 3b).16
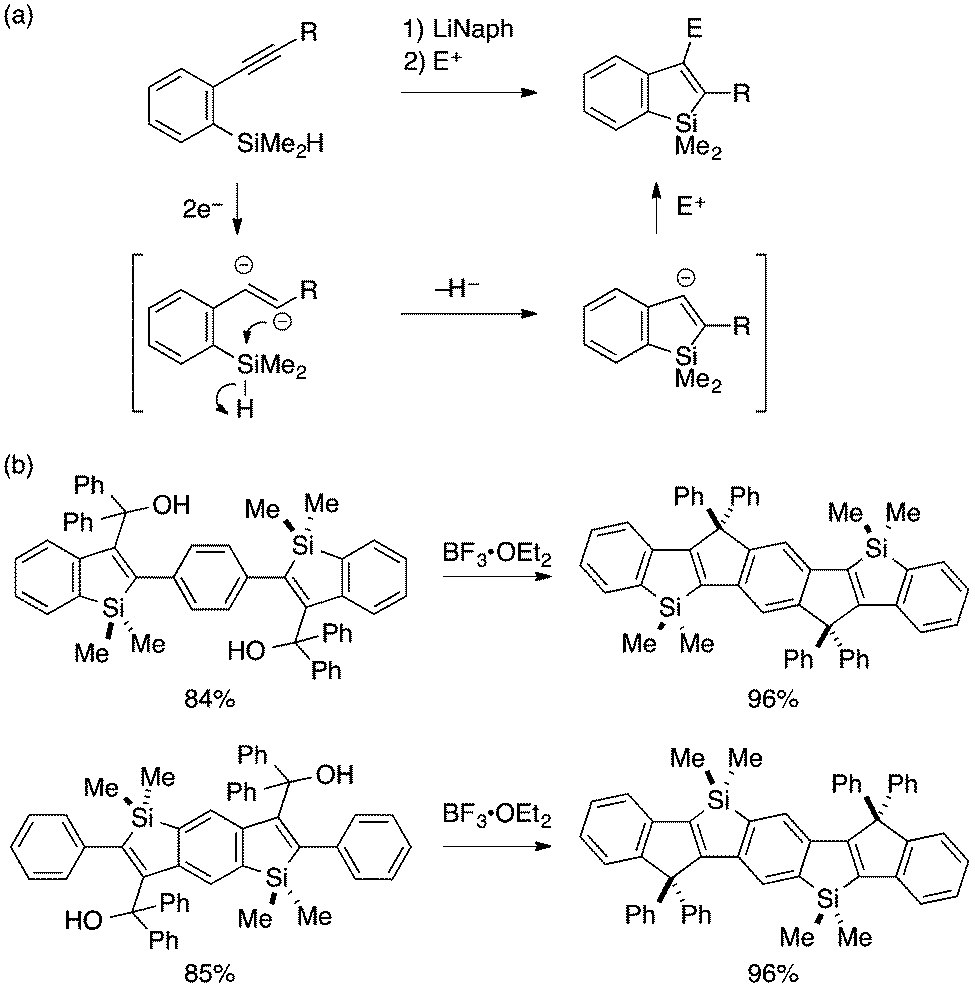 | ||
| Scheme 3 (a) Reductive cyclization–electrophilic trapping approach to 2,3-disubstituted benzosiloles and (b) its application to Si,C-bridged oligo(p-phenylenevinylene)s. | ||
Tsuji, Nakamura and coworkers developed a novel stannyllithium-mediated cyclization reaction of an ortho-alkynylarylsilane to afford a 2-phenyl-3-stannylbenzosilole in high yield (Scheme 4a).17 The reaction is proposed to proceed through trans-stannyllithiation of the alkynyl moiety followed by intramolecular displacement of the Si–H bond with the alkenyl anion. Importantly, 3-stannylated benzosilole may be used directly as a nucleophile for Stille coupling or converted to the corresponding iodide for use in Negishi coupling. These protocols allow for divergent synthesis of benzosilole-containing extended π-conjugated molecules (Scheme 4b), some of which have proved to exhibit high electron drift mobility and thus serve as hole-blocking materials for OLED.7a
 | ||
| Scheme 4 (a) Stannylative cyclization-cross-coupling approach to 2,3-disubstituted benzosiloles and (b) the thus-synthesized π-extended molecules containing benzosilole units. | ||
Murakami and coworkers developed a gold(I)-catalyzed cyclization reaction of ortho-alkynylaryl(allyl)silanes to afford 3-allylbenzosiloles through trans-allylsilylation of the alkyne moiety (Scheme 5).18 The reaction tolerates a variety of substituents on the alkynyl group, including alkyl, aryl, and ester groups as well as Cl and Br atoms. The proposed mechanism involves nucleophilic attack of the allylic terminus on the gold-activated alkynyl group, ring-opening of the resulting seven-membered cycle via Si–C cleavage, and Si–C bond formation between the alkenylgold and silyl cation moieties. Note that an analogous germanium-containing substrate is also amenable to the gold catalysis, affording a benzogermole.
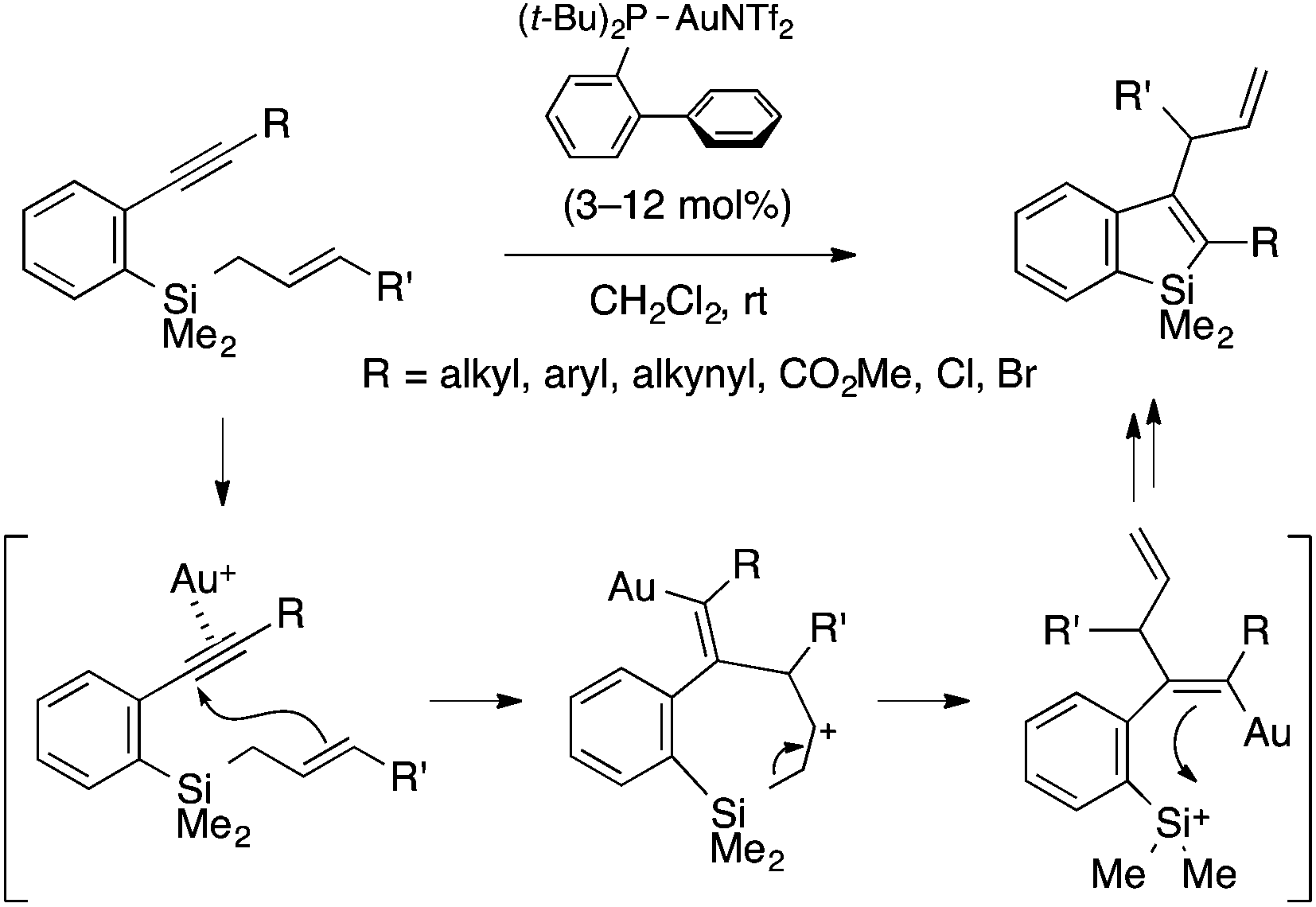 | ||
| Scheme 5 Gold(I)-catalyzed intramolecular trans-allylsilylation of alkyne leading to 2,3-disubstituted benzosiloles. | ||
Matsuda, Murakami and coworkers extended their study on the gold(I) catalysis to find other interesting benzosilole-forming cyclization modes using ortho-ethynylarylsilanes bearing an alkenyl or an aryl group on the silicon atom (Scheme 6).19 A substrate bearing an isobutenyl–Si moiety underwent intramolecular trans-alkenylsilylation of the alkyne moiety to afford a 3-isobutenylbenzosilole derivative (Scheme 6a). The reaction is likely initiated by 6-endo addition of the alkenyl group to the gold-activated alkyne, followed by a sequence of skeletal rearrangements of carbocation species. In contrast, a substrate bearing a phenyl–Si moiety gave a 2-phenylbenzosilole through 1,1-phenylsilylation (Scheme 6b).
 | ||
| Scheme 6 Gold-catalyzed trans-alkenylsilylation and 1,1-arylsilylation routes to benzosiloles. | ||
Matsuda and coworkers reported a rhodium-catalyzed cyclization reaction of ortho-alkynylaryldisilanes to afford 3-silylbenzosiloles through trans-bis-silylation of the alkyne moiety (Scheme 7).20 The reaction is likely initiated by oxidative addition of the Si–Si bond to Rh(I) followed by intramolecular insertion of the alkyne into the Rh–Si bond. While not fully supported experimentally, the resulting vinylrhodium intermediate is proposed to undergo an interesting rearrangement process involving a η2-vinylrhodium (1-rhodacyclopropene) species.
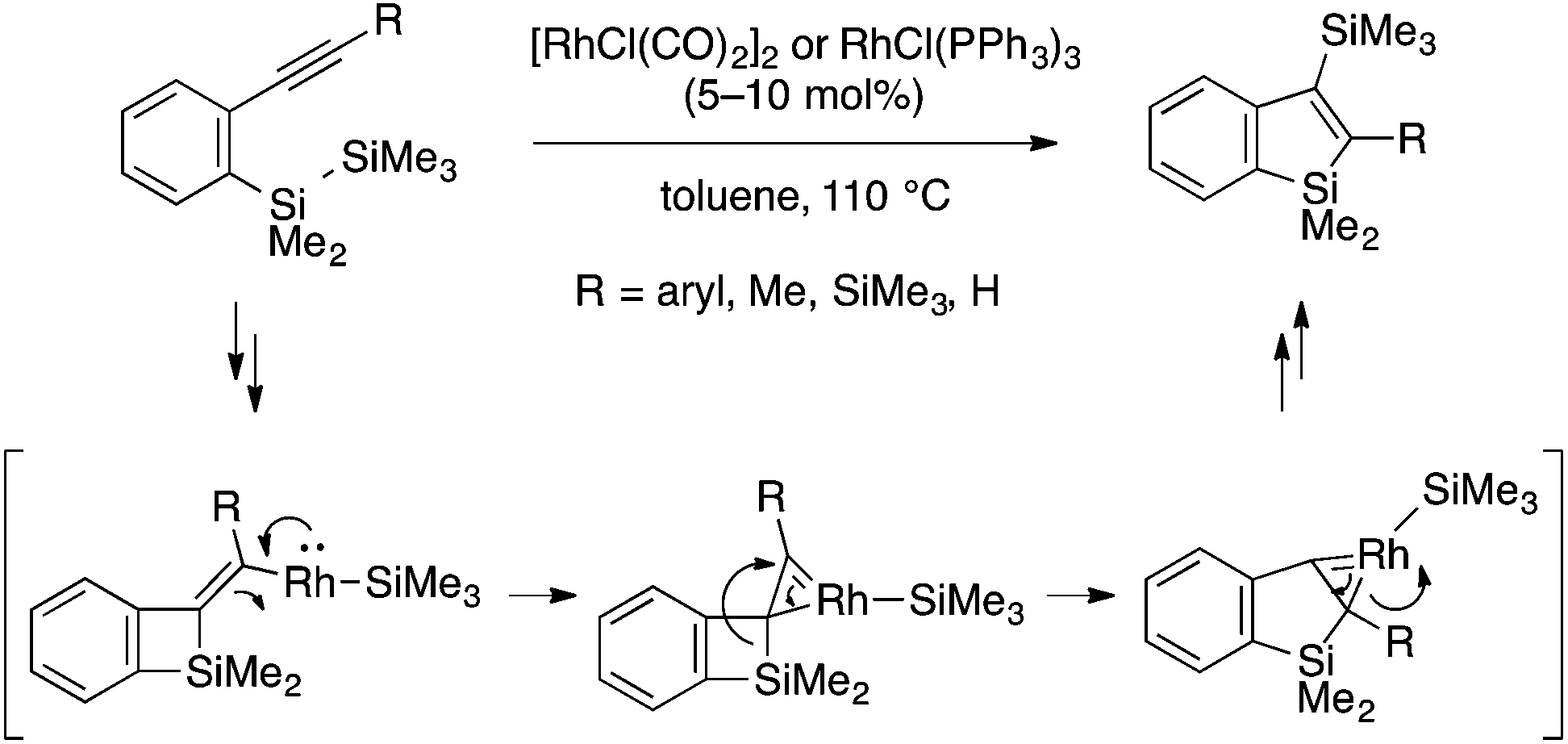 | ||
| Scheme 7 Rhodium-catalyzed trans-bis-silylation of alkyne leading to 3-silylated benzosiloles. | ||
Chatani and coworkers reported the first successful example of the annulation approach to benzosiloles. The reaction of an ortho-trimethylsilylphenylboronic acid (or ester) and an internal alkyne in the presence of a rhodium(I) catalyst proceeds smoothly to afford a 2,3-disubstituted benzosilole derivative (Scheme 8a).21 The reaction is considered to involve insertion of the alkyne into an arylrhodium species to form an alkenylrhodium intermediate. The key Si–C bond-forming cyclization may proceed through a σ-bond metathesis mechanism with concomitant elimination of a Rh–Me species, which can undergo protodemetalation and then transmetalation with the arylboron reagent to regenerate the arylrhodium species. The reaction employing an arylboronic ester bearing a SiMe2i-Pr group and a chiral diphosphine–Rh catalyst occurs with chemo- and enantioselective activation of the Si–Me bond to afford a benzosilole with Si-centered chirality (Scheme 8b). Notably, the Rh(I) catalytic system is also effective for the annulation of an ortho-trimethylgermylphenylboronic ester and an alkyne, which occurs at a much lower temperature to afford a benzogermole (Scheme 8c).22
 | ||
| Scheme 8 (a) Rhodium-catalyzed annulation of ortho-silylarylboronic acids/esters and internal alkynes leading to 2,3-disubstituted benzosiloles, (b) its enantioselective variant, and (c) its germanium variant. | ||
Xi and coworkers developed a coupling reaction of an ortho-trimethylsilylaryl bromide and an internal alkyne as an alternative annulation method for benzosilole synthesis (Scheme 9).23 The reaction is achieved by a Pd(0) catalytic system with a carefully optimized ligand, a base, and other reaction conditions depending on the type of the alkyne reaction partner. Interestingly, the addition of 4-nitrobenzaldehyde is crucial to achieve high yield using arylalkynes. As is the case with the rhodium-catalyzed annulation reaction, the reaction likely involves insertion of the alkyne into an arylpalladium species. While the exact mechanism is not clear, the resulting alkenylpalladium species would formally undergo metathesis of the Si–Me and Pd–C bonds to afford the benzosilole product and a MePdBr species, the latter being eventually reduced back to Pd(0). Xi and coworkers further expanded the substrate scope of this annulation reaction to ortho-trimethylsilylaryl triflates.24
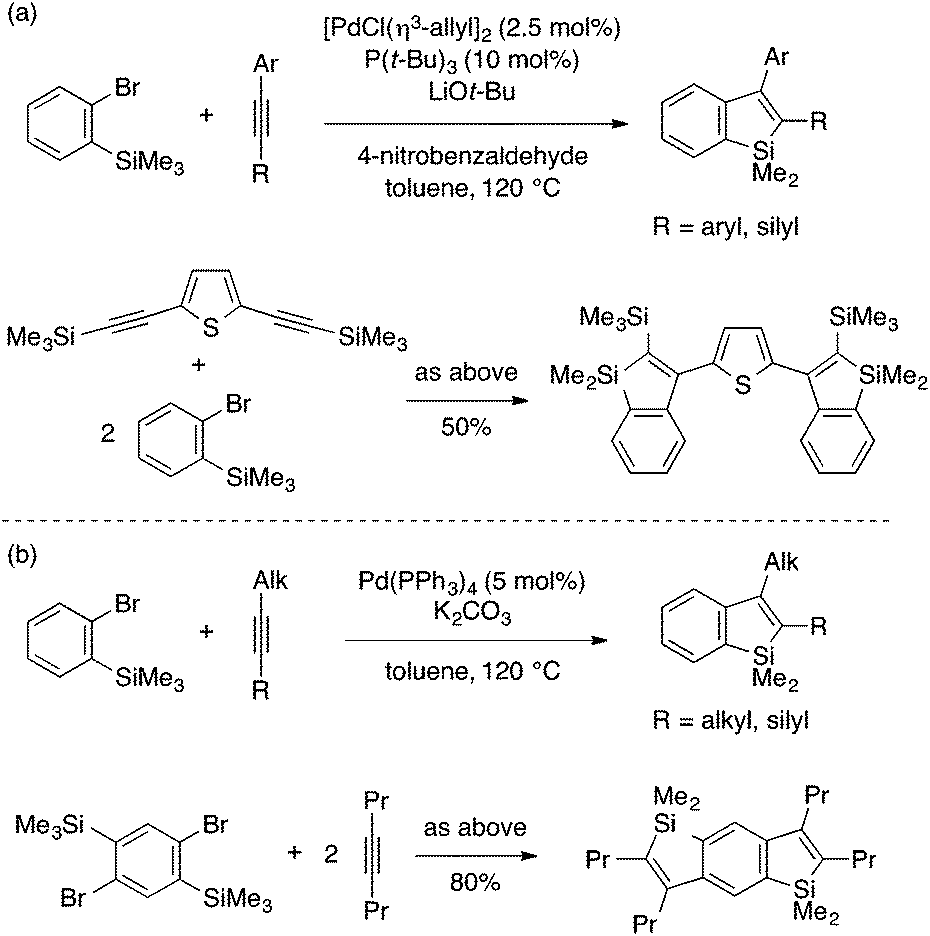 | ||
| Scheme 9 Pd-catalyzed annulation of ortho-trimethylsilylaryl bromides with arylalkynes (a) or alkylalkynes (b) leading to 2,3-disubstituted benzosiloles. | ||
Shirakawa, Hayashi and coworkers reported an annulative benzosilole synthesis employing iron-catalyzed aryllithiation as a key step (Scheme 10).25 Thus, an ortho-trimethylsilylaryllithium and an internal alkyne are coupled in the presence of catalytic Fe(acac)3 to give a 2,3-disubstituted silole. The reaction most likely involves iron-catalyzed aryllithiation of the alkyne and subsequent intramolecular nucleophilic attack of the alkenyllithium moiety to the SiMe3 group with concomitant loss of MeLi.
 | ||
| Scheme 10 Iron-catalyzed addition of ortho-trimethylsilylaryllithium to alkynes leading to 2,3-disubstituted benzosiloles. | ||
Recently, Li and coworkers reported an annulative benzosilole synthesis under transition metal-free oxidative conditions, which likely involves a silyl radical as a key reactive intermediate (Scheme 11).26 Thus, the reaction of a tertiary arylsilane without an ortho-functional group and an internal alkyne proceeds in the presence of di-tert-butyl peroxide (DTBP), affording a 2,3-disubstituted benzosilole through Si–H and C–H cleavage. The reaction is proposed to involve hydrogen abstraction of the Si–H bond with a t-BuO radical, addition of the silyl radical to the alkyne, and cyclization of the resulting alkenyl radical.
 | ||
| Scheme 11 Peroxide-mediated oxidative annulation of arylsilanes and alkynes leading to 2,3-disubstituted benzosiloles (Li et al., 201526). | ||
Besides ortho-alkynylarylsilanes, a few other types of cyclization precursors have been utilized for benzosilole synthesis. Matsuda, Murakami and coworkers demonstrated synthesis of 2-substituted benzosilole by ring-closing metathesis (RCM) of an arene precursor bearing vinyl and alkenylsilyl groups on the ortho positions.27 The RCM approach is also effective for benzogermole synthesis. Furthermore, the RCM approach can be integrated with Ru-catalyzed alkyne hydrosilylation, thus enabling streamlined preparation of 2-substituted benzosiloles (Scheme 12).28
 | ||
| Scheme 12 Intermolecular hydrosilylation/ring-closing metathesis approach to benzosilole. | ||
In another example, Xi and coworkers demonstrated benzosilole synthesis by intramolecular Mizoroki–Heck reaction (Scheme 13).29 Thus, 5-endo-type cyclization of an aryl bromide bearing an ortho-vinylsilyl moiety takes place in the presence of an appropriate Pd(0) catalyst to afford a 2,3-unsubstituted benzosilole derivative.
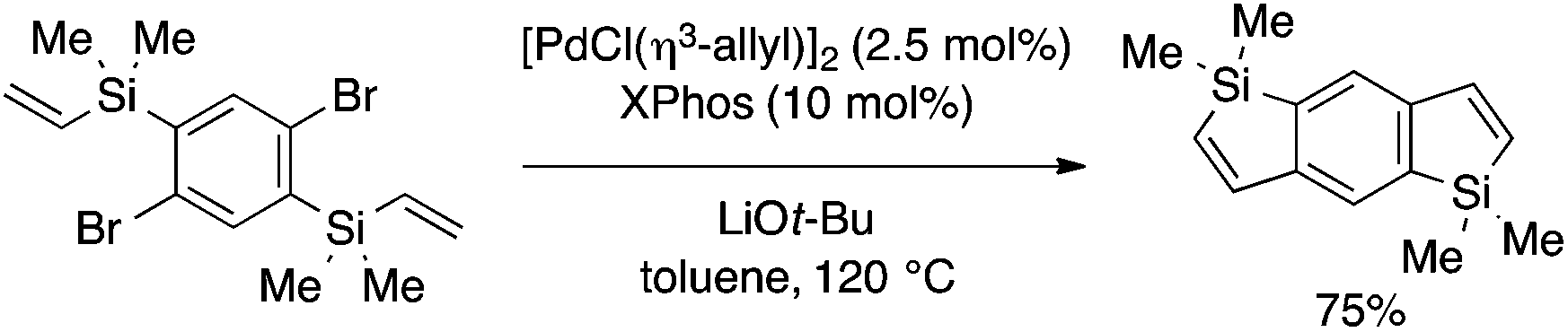 | ||
| Scheme 13 Intramolecular Mizoroki–Heck approach to benzosilole. | ||
While all the above synthetic methods start with substrates with preformed aryl–Si bonds, a few approaches involving aryl–Si bond formation as the cyclization step have been reported. Kuznetsov and Gevorgyan developed a one-pot protocol for the synthesis of dihydrobenzosiloles starting from vinylarenes and diphenylsilane by combining Ni-catalyzed linear-selective hydrosilylation and Ir-catalyzed dehydrogenative cyclization reactions (Scheme 14),30 of which the latter was developed earlier by Hartwig.31 The products can be readily converted to the corresponding 2,3-unsubstituted benzosiloles by oxidation using DDQ.
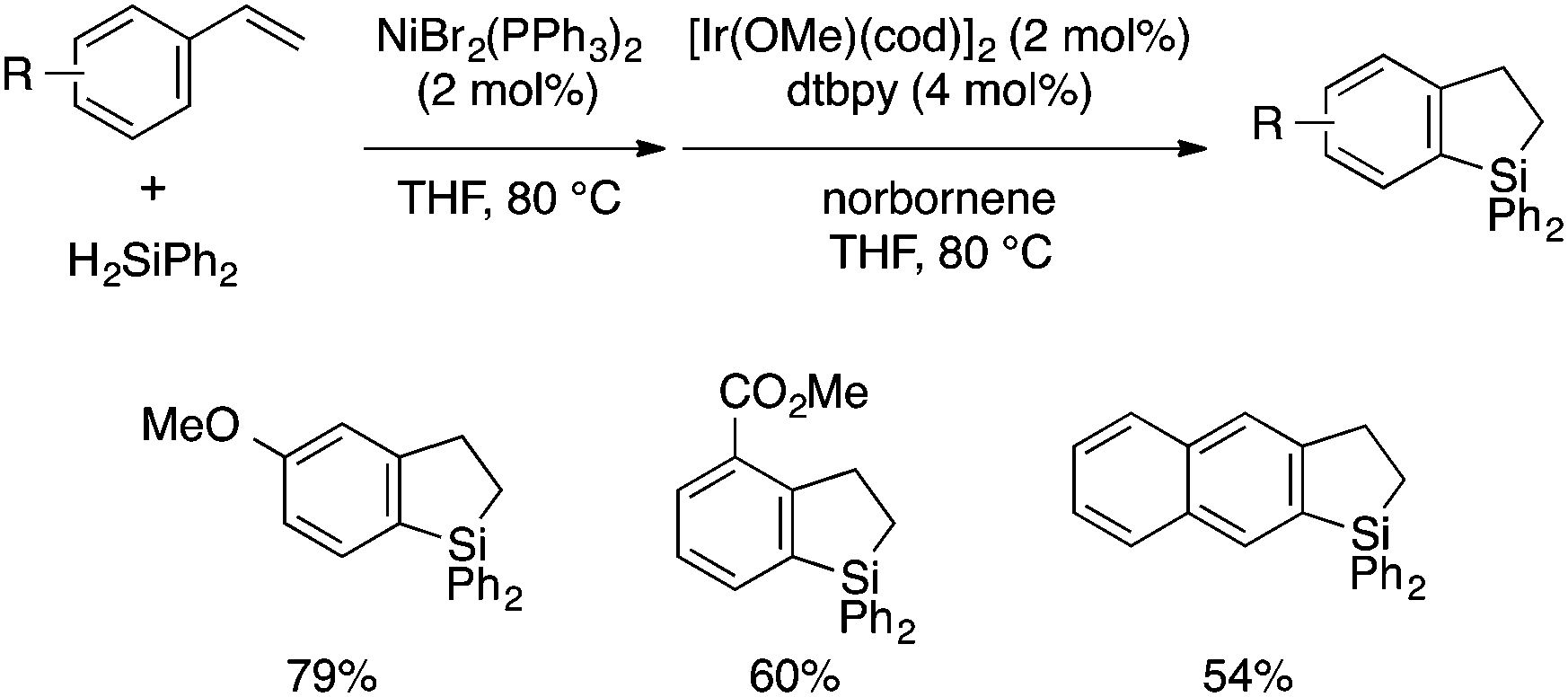 | ||
| Scheme 14 A one-pot approach to dihydrobenzosiloles through Ni-catalyzed hydrosilylation and Ir-catalyzed dehydrogenative cyclization. | ||
Curless and Ingleson reported that B(C6F5)3-catalyzed trans-hydrosilylation of 1-phenyl-1-propyne with Ph2SiH2 produces an alkenylsilane intermediate, which, upon addition of 2,6-dichloropyridine, undergoes sila-Friedel–Crafts cyclization to afford a benzosilole product in one pot (Scheme 15).32,33 This approach, however, is rather limited because of a narrow scope of the hydrosilylation.
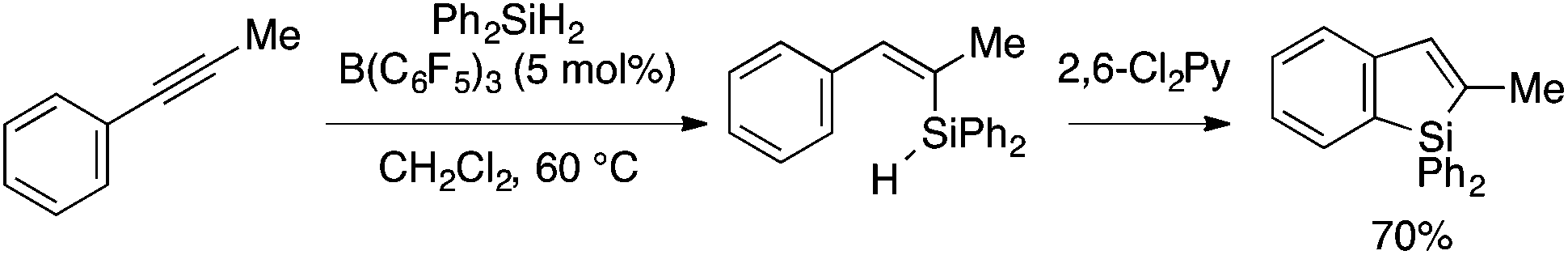 | ||
| Scheme 15 B(C6F5)3-catalyzed hydrosilylation/sila-Friedel–Crafts route to benzosilole. | ||
3. Benzophospholes
Several cyclization reactions of type A precursors leading to benzophospholes have been developed in the last several years. In 2008, Tsuji, Nakamura and coworkers reported on a BuLi-mediated cyclization of a secondary phosphine bearing ortho-alkynylaryl and bulky Mes* (2,4,6-tri-tert-butylphenyl) groups (Scheme 16).34 The resulting 3-lithiated benzophosphole can be utilized for a variety of electrophilic trapping reactions including Pd-catalyzed Negishi coupling (upon transmetalation with ZnCl2), allowing the modular preparation of 2,3-disubstituted benzophospholes. The Mes* group can be removed under reductive conditions and transformed into different substituents, and some of the thus-synthesized benzophosphole derivatives proved to serve as semiconductor materials for OLED and OPV applications.7b,35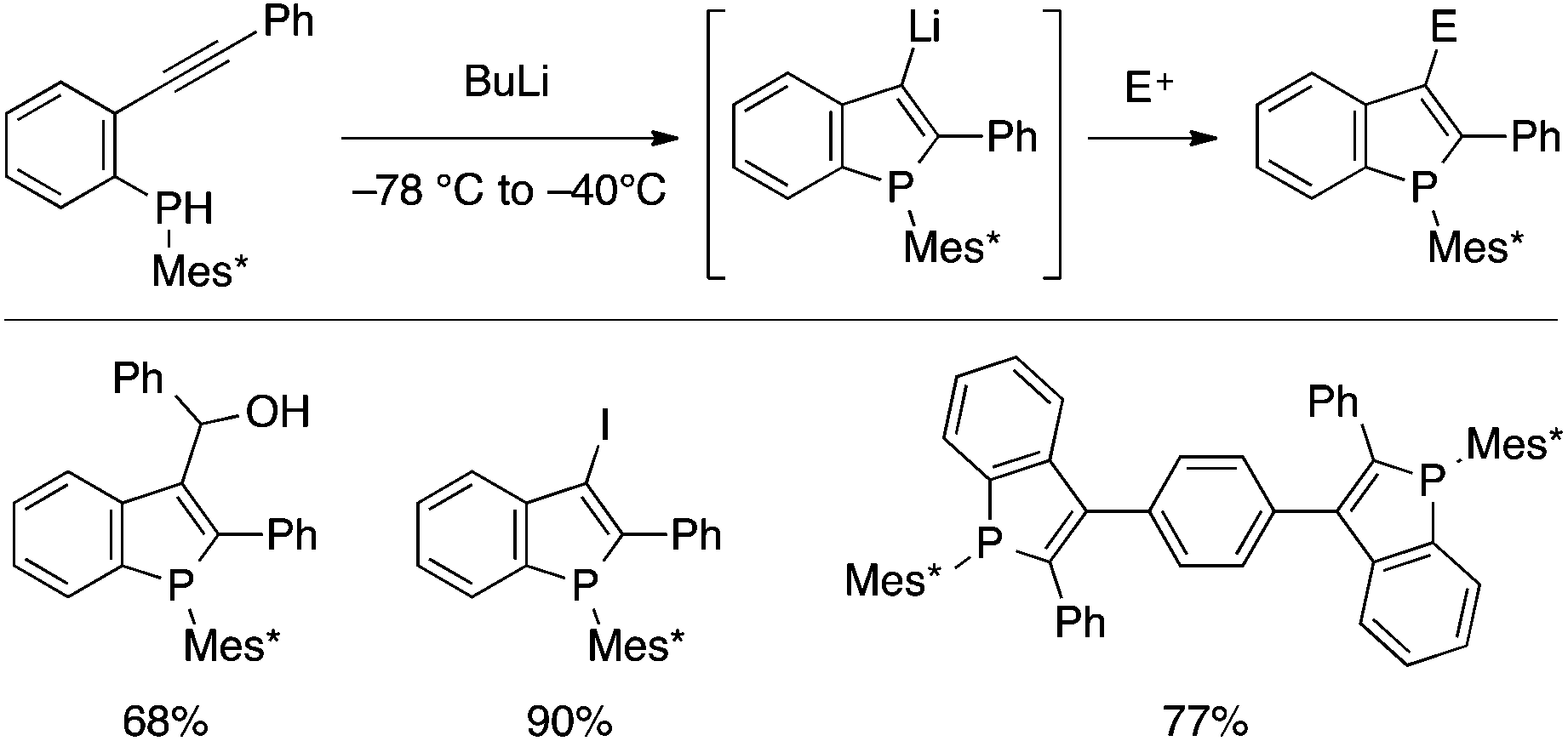 | ||
| Scheme 16 BuLi-mediated cyclization of ortho-alkynylarylphosphine leading to 3-lithiobenzophosphole (Mes* = 2,4,6-tri-tert-butylphenyl). | ||
Sanji and Tanaka developed a t-BuOK-catalyzed cyclization reaction of an ortho-alkynylphenyl(aryl)phosphine oxide to afford a benzophosphole oxide (Scheme 17).36 This convenient protocol allowed them to perform a systematic study on the photophysical properties of a series of 2-substituted benzophospholes, which generally exhibited intense blue-green fluorescence.
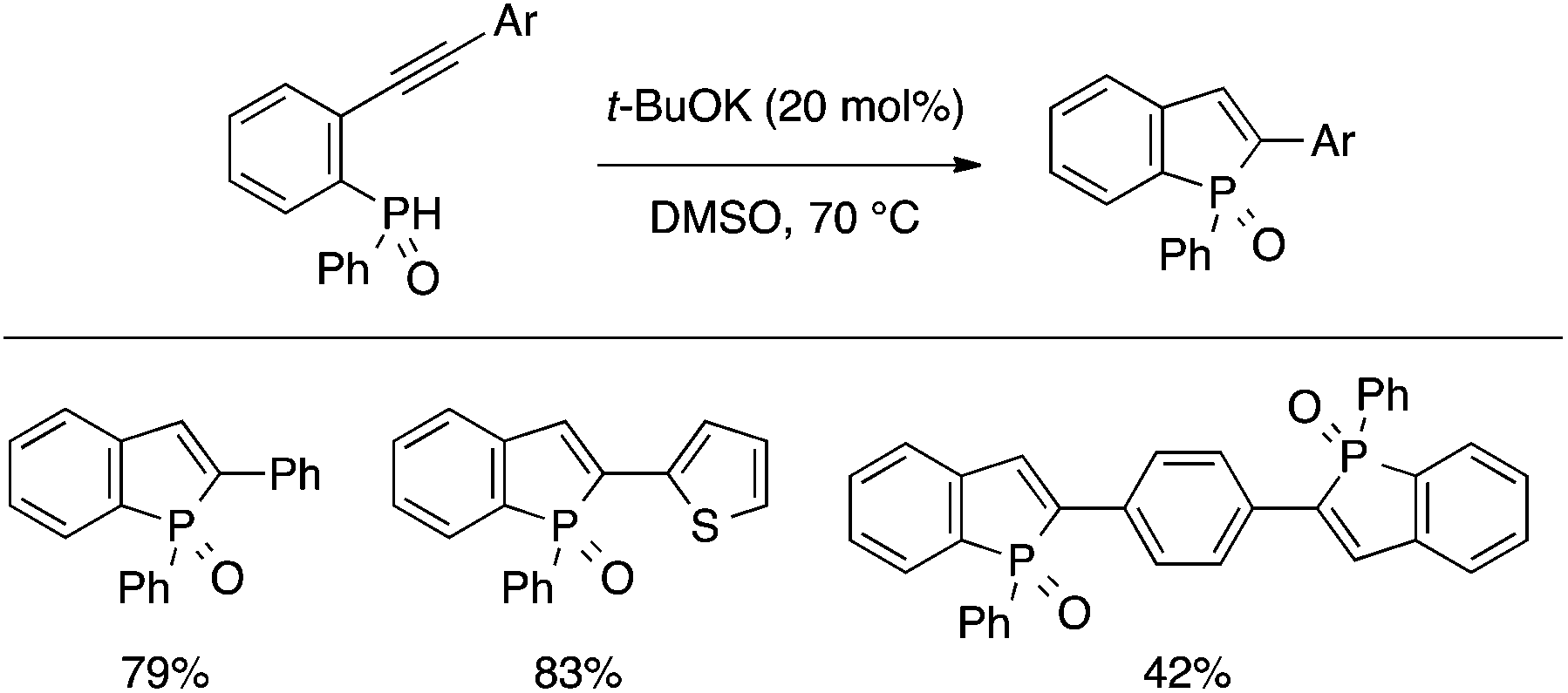 | ||
| Scheme 17 t-BuOK-catalyzed cyclization of ortho-alkynylarylphosphine oxide leading to benzophosphole oxide. | ||
Fukazawa and Yamaguchi achieved a one-pot synthesis of 3-halogenated benzophosphole oxides based on intramolecular trans-halophosphanylation (Scheme 18).37 Treatment of a 2-(aminophosphanyl)phenylacetylene intermediate, generated in situ from a brominated precursor through Br/Li exchange and electrophilic trapping, with PBr3 affords, upon oxidation with H2O2, a 3-bromobenzophosphole oxide derivative. A 3-iodo analogue can also be obtained by performing the cyclization step in the presence of LiI. The 3-bromo substituent allows subsequent transformations such as Br/Li exchange and Suzuki–Miyaura coupling to access structurally diverse benzophosphole derivatives, some of which have proved to serve as environment-sensitive fluorescent probes.7c
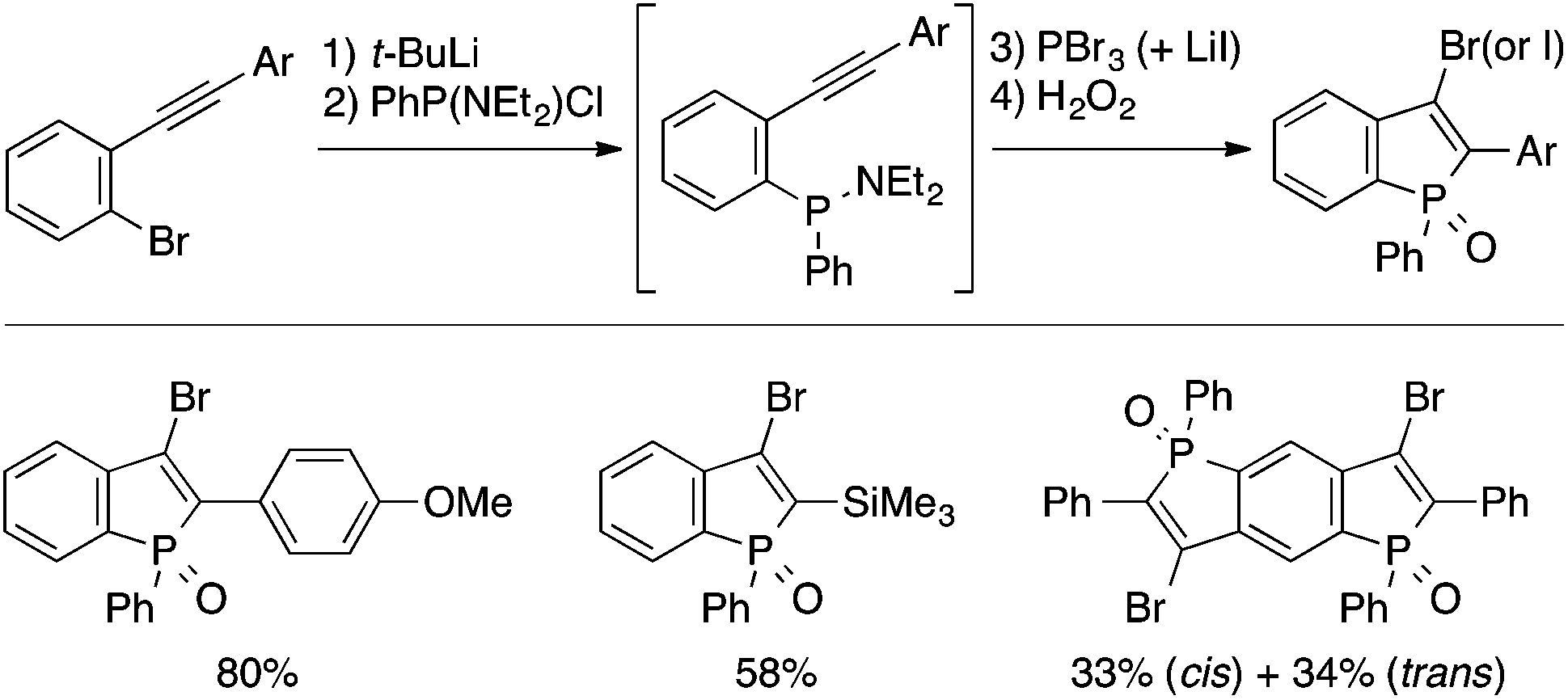 | ||
| Scheme 18 Intramolecular trans-halophosphanylation approach to 3-halogenated benzophospholes. | ||
A diarylphosphino group can also be used as a viable phosphorus group for intramolecular cyclization leading to benzophospholes. Duan and Mathey demonstrated that treatment of an ortho-diphenylphosphinophenylalkyne with excess Li metal leads to the formation of a benzophospholide anion presumably through reduction of the first P–Ph bond, 5-endo cyclization of the resulting phosphide, and reduction of the second P–Ph bond (Scheme 19a).38 Trapping of the benzophospholide with an electrophile such as MeI followed by oxidation with H2O2 leads to a 2-substituted benzophosphole oxide. Duan and Mathey also developed a novel Pd/Cu-cocatalyzed reaction of the same substrate to afford a 2,3-disubstituted benzophosphole, whose 3-substituent originates from the phosphorus atom (Scheme 19b).39 The reaction is proposed to involve oxidative addition of the P–Ph bond to Pd, intramolecular phospha-palladation of the alkyne, and reductive elimination, while the role of the copper catalyst remains unclear.
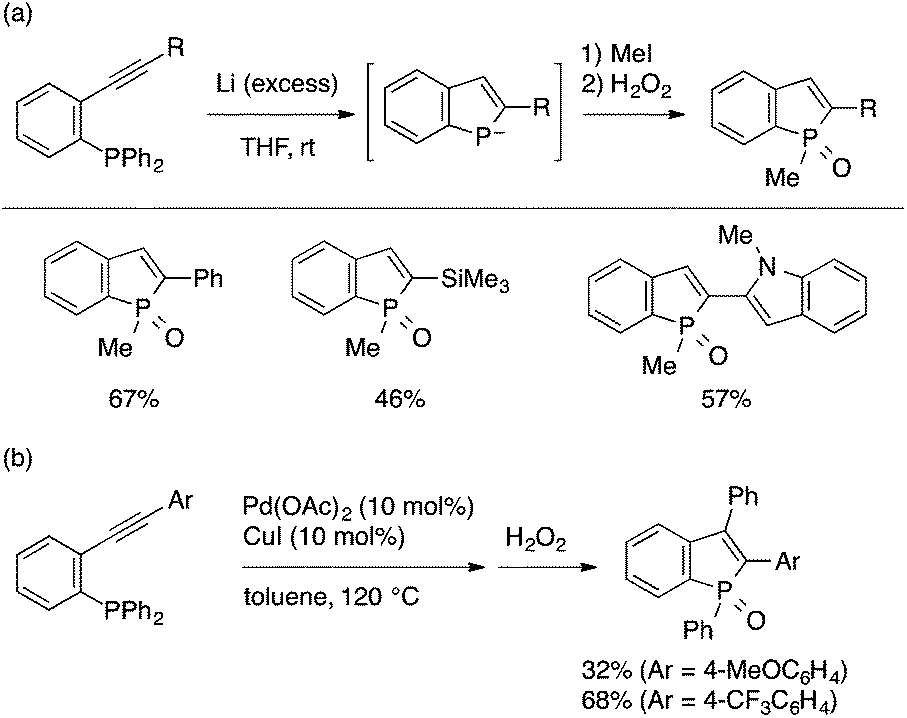 | ||
| Scheme 19 Intramolecular cyclization reactions of ortho-alkynylphenyl(diphenyl)phosphines to benzophospholes. | ||
The groups of Satoh/Miura and Duan independently developed an annulation approach to benzophosphole based on dehydrogenative coupling of a secondary arylphosphine oxide and an internal alkyne (Scheme 20).40 This atom-economical reaction takes place smoothly using a metallic oxidant such as AgOAc, Ag2O, and Mn(OAc)3, and exhibits excellent functional group compatibility. At the same time, the reaction has a few intrinsic limitations with respect to regioselectivity. For example, the reaction of 4-substituted arylphosphine oxide ends up in a mixture of the expected 5-substituted benzophosphole oxide and its 6-substituted isomer (Scheme 20b). This regiochemical outcome is explained by a radical-based mechanism. The addition of a P-centered radical generated by oxidation of the P–H bond with Ag(I) to the alkyne forms an alkenyl radical, which then undergoes cyclization to form either a 5-membered fused bicyclic intermediate or a 4-membered spirocyclic intermediate. The former intermediate may undergo further one-electron oxidation and loss of proton to form the 5-substituted benzophosphole. While the latter intermediate may rearrange into the former, it may also rearrange into another fused bicycle via P–C cleavage, which leads to the 6-substituted benzophosphole. It should also be noted that a diarylphosphine oxide bearing different aryl groups undergoes annulation at either of the aryl groups with low regioselectivity. Besides the reports of Satoh/Miura and Duan, the groups of Ackermann and Gao also disclosed similar Ag-mediated annulation reactions to form benzophospholes.41 Furthermore, Tang and coworkers reported that a copper-based catalytic system employing TBHP as a stoichiometric oxidant is also capable of promoting the annulation reaction.42
 | ||
| Scheme 20 Ag-mediated oxidative annulation of secondary arylphosphine oxides and alkynes leading to benzophospholes. | ||
Recently, Gao and coworkers reported another radical-based approach to benzophospholes, which involves a cyclization step similar to the one in the above reaction while employing different starting materials and reaction conditions (Scheme 21).43 Thus, heating a toluene solution of diaryl(arylethynyl)phosphine oxide in the presence of Bu4NI catalyst and DTBP oxidant results in 2-benzyl-3-arylbenzophosphole oxide. The reaction is considered to involve the addition of a benzyl radical to the alkynyl moiety followed by cyclization of the resulting alkenyl radical.
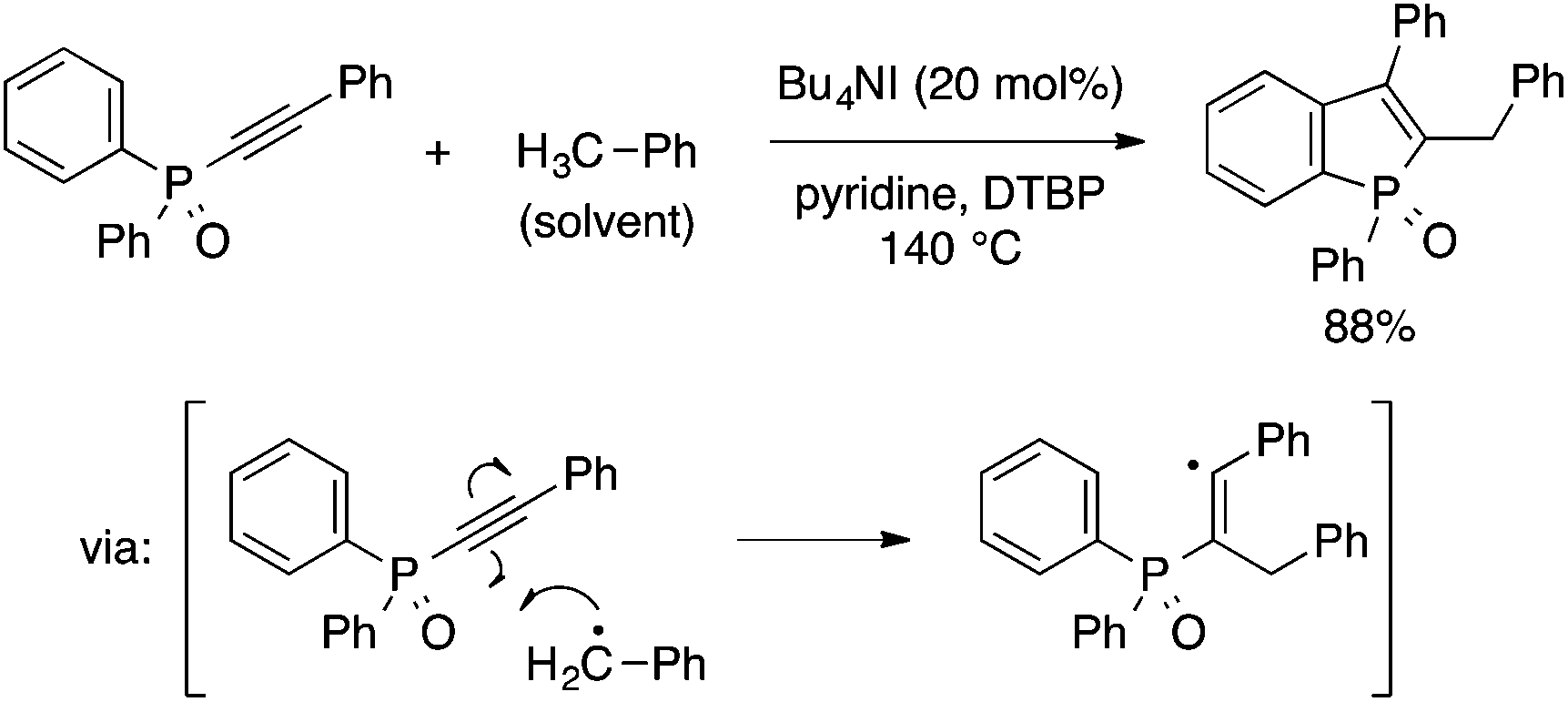 | ||
| Scheme 21 Oxidative coupling of diaryl(arylethynyl)phosphine oxide and toluene leading to 2-benzyl-3-arylbenzophosphole. | ||
Electrophilic trapping of organometallic intermediates with phosphorus electrophiles has also been utilized as a key step in benzophosphole synthesis. Matano and coworkers have demonstrated the synthetic utility of 2-bromobenzophosphole derivatives, which are prepared in a few steps from linear precursors via dilithio species as key intermediates, by establishing protocols for their transformations using Stille, Suzuki–Miyaura, Heck, and Sonogashira couplings (Scheme 22).44 These methods allowed them to explore photophysical properties of a variety of benzophosphole derivatives depending on the core structural and the peripheral substituents.
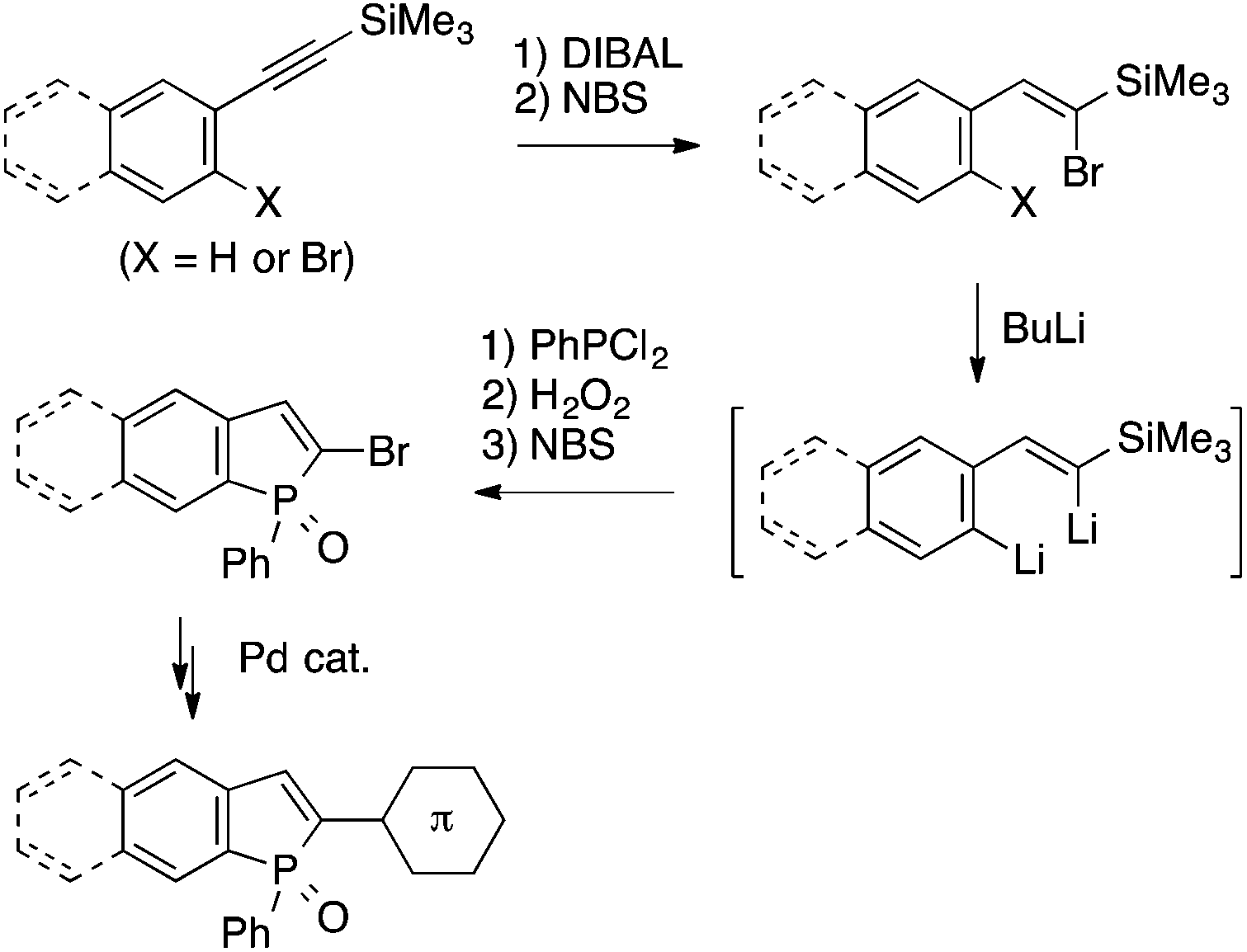 | ||
| Scheme 22 Benzophosphole synthesis utilizing dilithio intermediates. | ||
Ogasawara and Takahashi demonstrated benzophosphole synthesis through electrophilic trapping of a zirconaindene species, generated from a zirconium–benzyne complex and an alkyne, with PhPCl2 (Scheme 23).45,46 The thus-synthesized benzophospholes were further transformed into novel benzophosphaferrocenes.
 | ||
| Scheme 23 Benzophosphole synthesis utilizing zirconaindene intermediates. | ||
A major limitation common to the above-discussed synthetic approaches concerns the difficulty in diversification of substituents on the benzo moiety as well as the P atom of benzophosphole. This is due to the requirement for multistep preparation of starting materials for the cyclization approach, and due to the low regioselectivity for the annulation approach. Addressing this problem, Yoshikai and coworkers developed two one-pot multicomponent approaches for the construction of benzophospholes from three simple reactants, that is, an arylmetal reagent, an internal alkyne, and a phosphorus electrophile (Scheme 24). One approach employs cobalt-catalyzed migratory arylzincation of the alkyne47 and subsequent copper-catalyzed trapping of the resulting ortho-alkenylarylzinc species with PhPCl2 (Scheme 24a).48 The zinc species may also be treated with PCl3 and then with a Grignard reagent, which allows variation of the P-substituent. The limitation of the alkyne scope in this approach can be complemented by another approach based on alkyne arylmagnesiation. Thus, an alkenylmagnesium species generated by Ni-catalyzed alkyne arylmagnesiation49 can be converted to a benzophosphole derivative through electrophilic trapping with dichloroorganophosphine (Scheme 24b).50 These two approaches likely involve phospha-Friedel–Crafts-type P–C bond formation as the cyclization step.
 | ||
| Scheme 24 One-pot benzophosphole synthesis from arylmetal reagents, alkynes, and phosphorus through alkyne (migratory) arylmetalation, electrophilic trapping, and phospha-Friedel–Crafts-type cyclization. | ||
4. Benzothiophenes and benzoselenophenes
In the early 2000s, Larock and coworkers developed a prototypical method for the synthesis of benzothiophenes from type A precursors (Scheme 25).51,52 Thus, an ortho-alkynylaryl methyl sulfide is cyclized with the aid of various electrophiles such as I2, Br2, N-bromosuccinimide (NBS), and PhSeCl, which ends up in a benzothiophene skeleton with the incorporation of the electrophile into the C3 position. Later, other reagents such as copper(II) halide (CuCl2 and CuBr2)53 and a combination of trifluoromethanesulfanylamide (PhNHSCF3) and BiCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 were also demonstrated to serve as viable electrophiles for this cyclization, thus allowing the preparation of benzothiophenes bearing 3-halogen and -trifluoromethylthio groups, respectively. Larock demonstrated that an analogous cyclization of an ortho-alkynylaryl methyl selenide is also feasible, thus affording 2,3-disubstituted benzoselenophenes.55
54 were also demonstrated to serve as viable electrophiles for this cyclization, thus allowing the preparation of benzothiophenes bearing 3-halogen and -trifluoromethylthio groups, respectively. Larock demonstrated that an analogous cyclization of an ortho-alkynylaryl methyl selenide is also feasible, thus affording 2,3-disubstituted benzoselenophenes.55
 | ||
| Scheme 25 Benzothiophene and benzoselenophene synthesis through electrophile-mediated 5-endo cyclization. | ||
Alkynophilic transition metal catalysts also allow for cyclization of type A precursors to benzothiophenes and benzoselenophenes. In 2006, Nakamura and coworkers reported a gold(I)-catalyzed transformation of an ortho-alkynylaryl thioether bearing an α-alkoxyalkyl group into a 2,3-disubstituted benzothiophene, whose C3-substituent originates from the S atom (Scheme 26a).56 The reaction was proposed to proceed through nucleophilic attack of the sulfur atom on the gold-activated alkyne moiety and subsequent migration of the α-alkoxyalkyl group to the gold-bonded carbon atom. Besides α-alkoxyalkyl groups, 4-methoxybenzyl, allyl, and 1-phenethyl groups also serve as viable migrating groups for this reaction, suggesting that their ability to form stabilized carbocations is crucial.57 Nakamura also extended this conceptual approach to benzoselenophene synthesis using PtCl2 as a catalyst (Scheme 26b).58
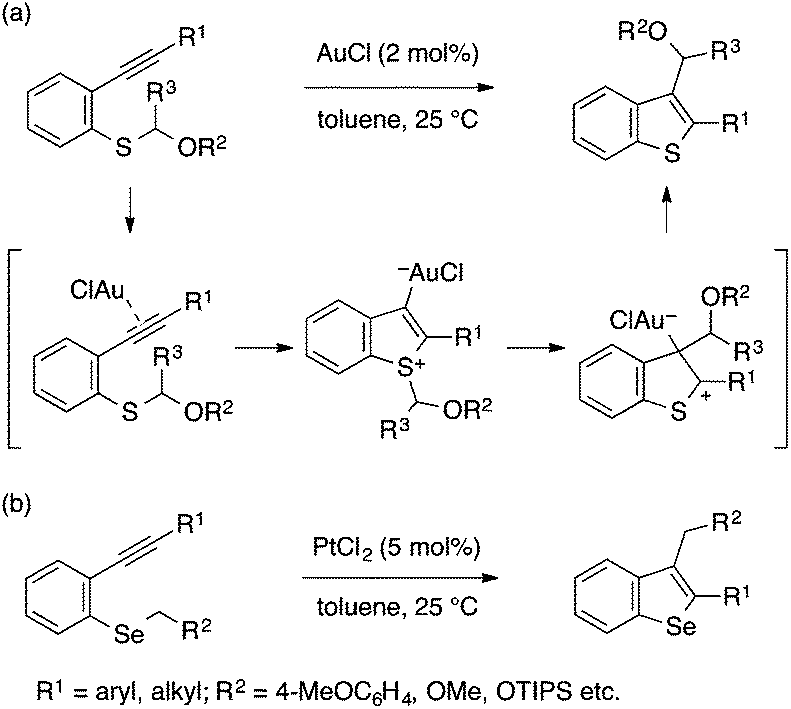 | ||
| Scheme 26 Gold- or platinum-catalyzed cyclization leading to benzothiophenes and benzoselenophenes. | ||
Besides the preformed ortho-alkynylaryl sulfides and selenides used in the above reactions, ortho-alkynylaryl bromides have also been frequently used as precursors for the synthesis of benzothiophenes and benzoselenophenes through sequential aryl–chalcogen bond formation and intramolecular cyclization. An early report of such transformations was made by Sashida and coworkers, employing Br/Li exchange and electrophilic trapping with elemental chalcogens.59 Takimiya later utilized this reaction in the synthesis of benzodichalcogenophene derivatives and demonstrated their promising performances as OFET materials (Scheme 27).7e,60
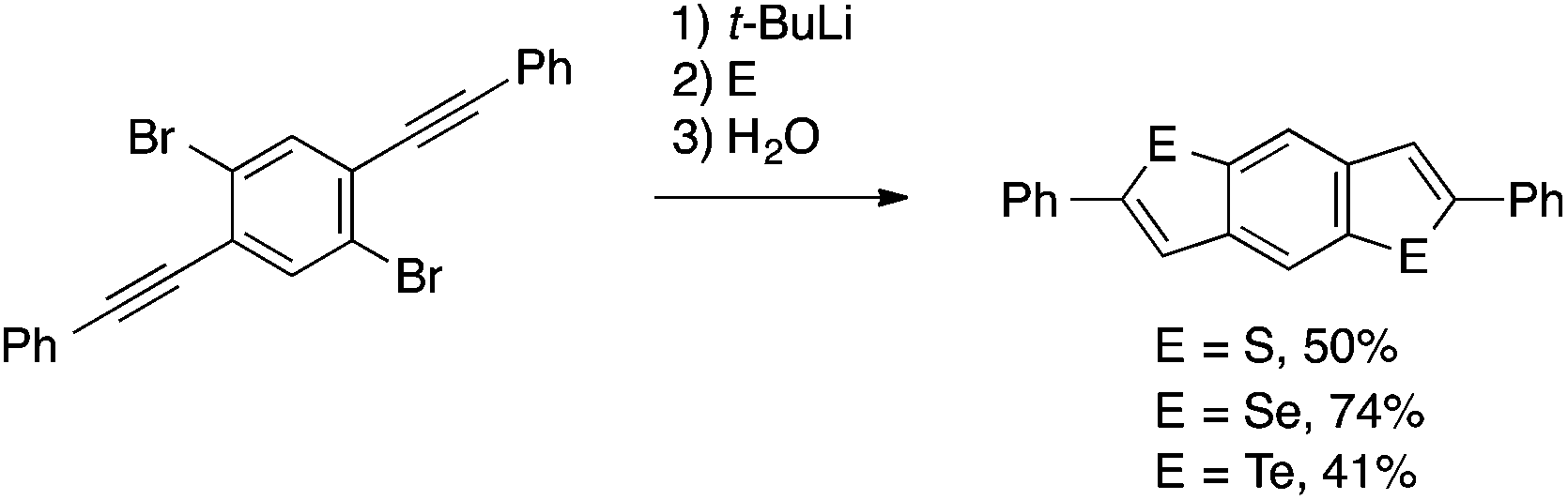 | ||
| Scheme 27 Chalcogenative cyclization of ortho-alkynylaryl bromide leading to 2,6-diphenylbenzo[1,2-b:4,5-b‘]dichalcogenophenes. | ||
Takimiya developed a more convenient method for the conversion of the above and related starting materials into benzothiophene and benzoselenophene derivatives. Thus, treatment of ortho-alkynylaryl bromides or chlorides with sodium sulfide or -selenide at a high temperature (180–190 °C) in NMP effects chalcogenative cyclization to afford the corresponding benzochalcogenophenes (Scheme 28).61 Later, Zhang and coworkers demonstrated that the reaction of ortho-alkynylaryl bromides with Na2S may be performed using a CuI/TMEDA catalyst at a lower temperature (80 °C).62
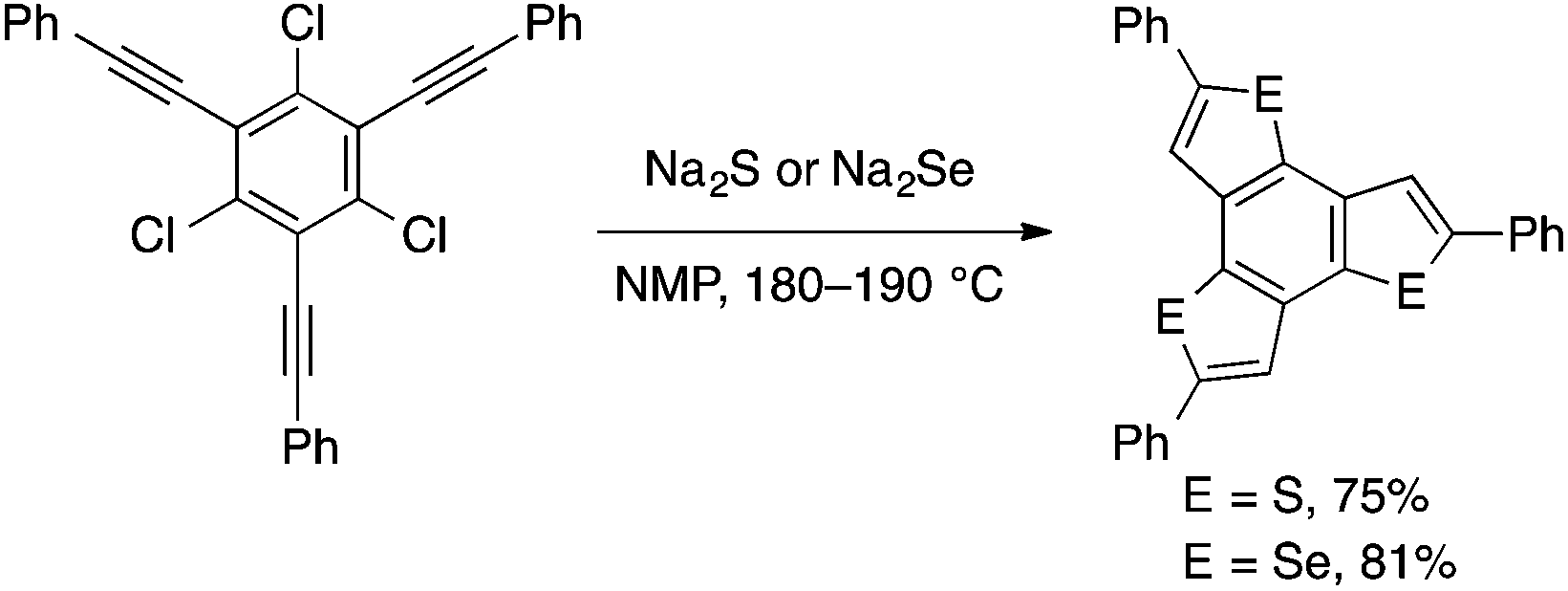 | ||
| Scheme 28 Chalcogenative cyclization using Na2E (E = S or Se) reagents. | ||
A few other methods for the transformation of ortho-alkynylaryl bromides to benzothiophenes using different sulfur sources have been reported. Paradies and coworkers demonstrated that thiourea serves as a hydrogen sulfide surrogate for Pd-catalyzed reaction of ortho-alkynylaryl bromides, thus affording benzothiophenes in one-pot operation (Scheme 29a).63 Sanz and coworkers developed two Pd-catalyzed protocols for one-pot stepwise construction of benzothiophenes (Scheme 29b).64 In one protocol, triisopropylsilanethiol is used as a sulfur source for C–S coupling, followed by fluoride-mediated deprotection and intramolecular cyclization. Another protocol involves C–S coupling using potassium thioacetate and subsequent deacetylation/cyclization using Cs2CO3.
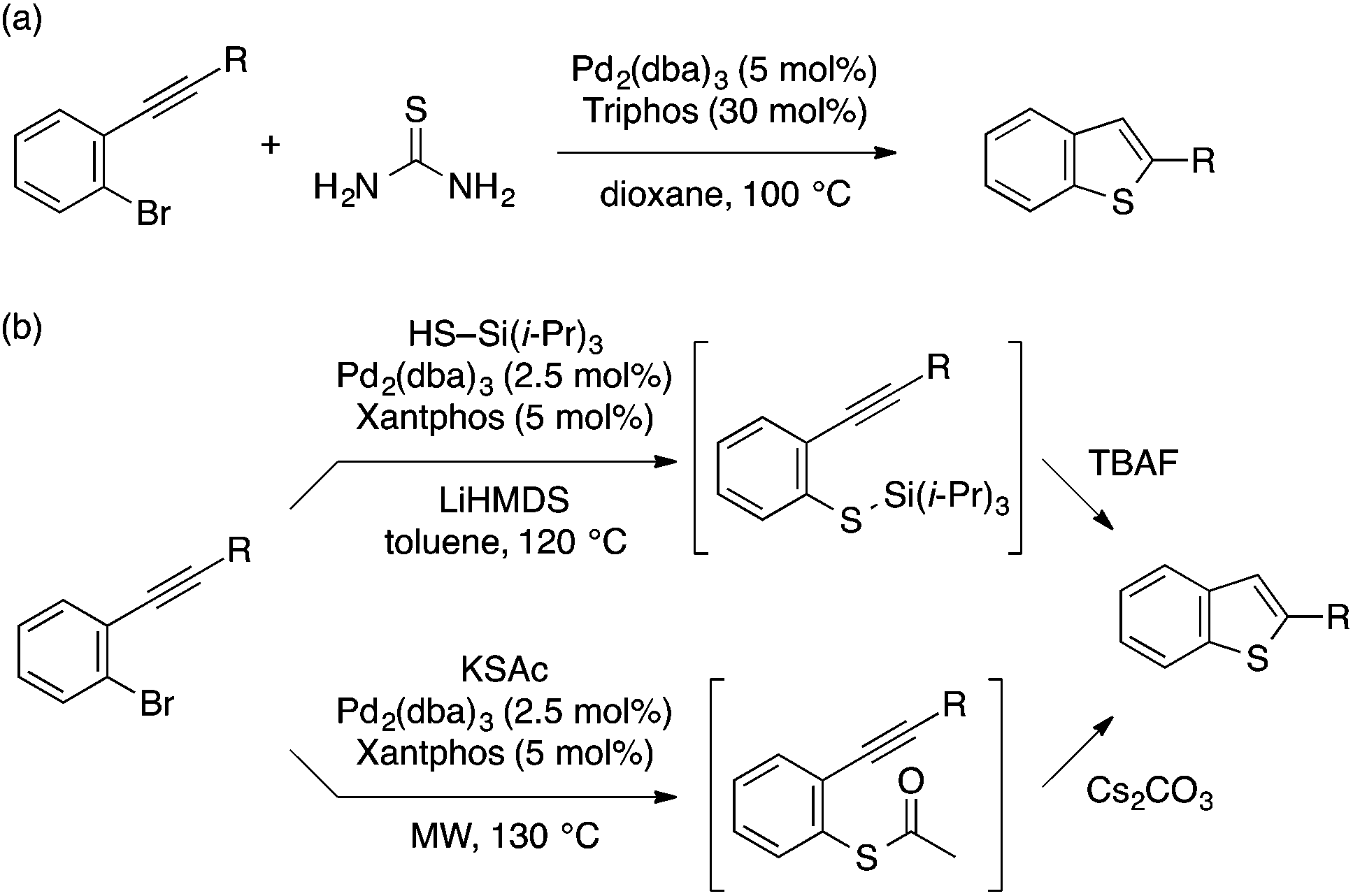 | ||
| Scheme 29 Pd-catalyzed thiolative cyclization of ortho-alkynylaryl bromide using different sulfur sources. | ||
A few examples of benzothiophene and benzoselenophene synthesis starting from type B precursors have been reported in recent years. König and coworkers developed an annulation reaction of ortho-methylthioaryldiazonium salts and alkynes under visible-light photocatalytic conditions using eosin Y (Scheme 30a).65 The reaction is applicable to terminal arylacetylenes, propiolates, and dimethyl acetylenedicarboxylate (DMAD) derivatives. The proposed mechanism involves initiation by single-electron transfer from the excited state of the photocatalyst to the diazonium salt, which generates an aryl radical as a reactive species. Schiesser and coworkers reported an analogous annulation reaction using ortho-methylselenoaryldiazonium salt in the presence of FeSO4, while the yields remained only modest (Scheme 30b).66
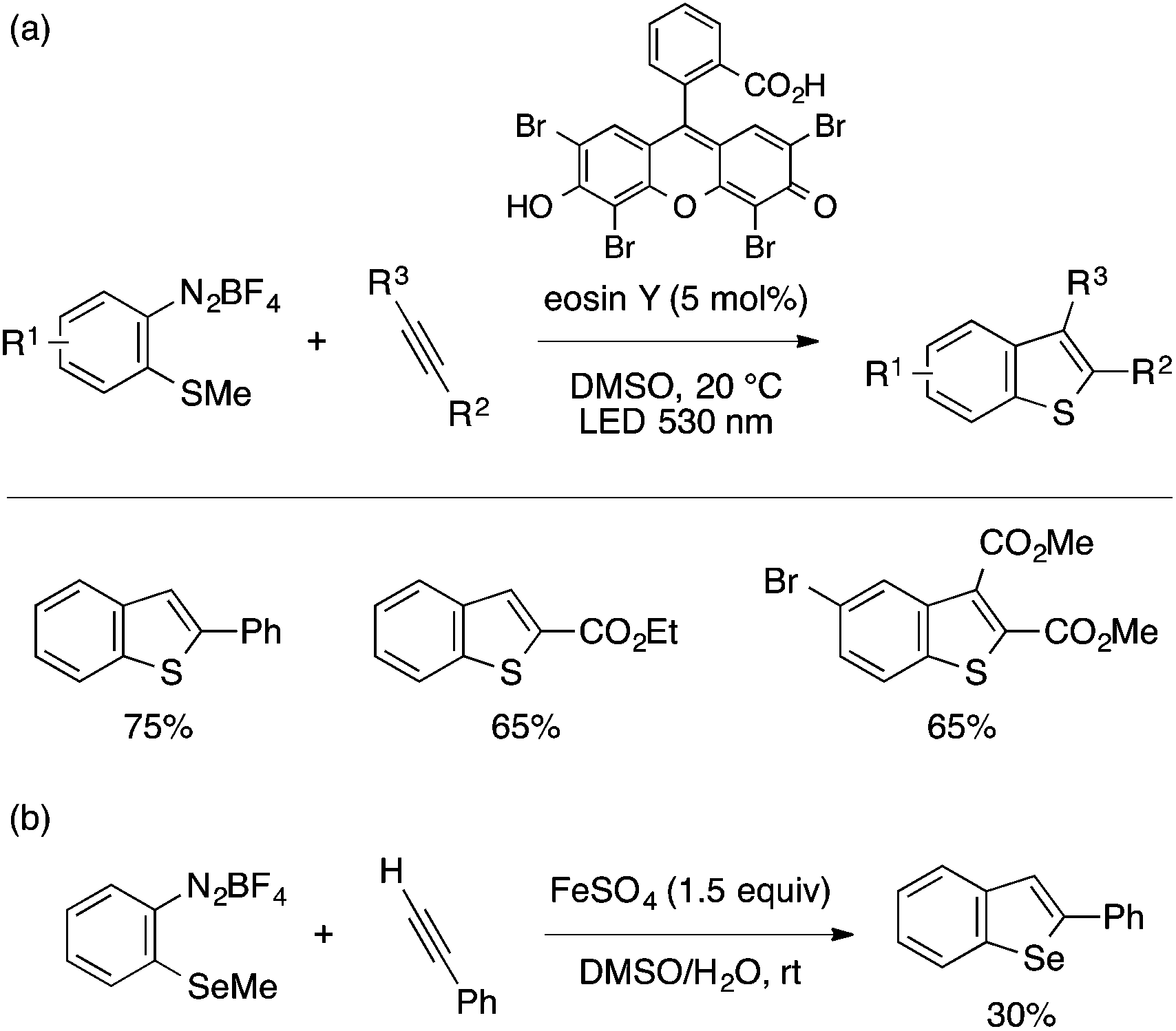 | ||
| Scheme 30 Annulation of ortho-methylthio- and methylselenoaryldiazonium salts with alkynes. | ||
Li and coworkers achieved annulation of simple thiophenols with DMAD and related activated internal alkynes to form 2,3-disubstituted benzothiophenes using Mn(OAc)3 as a catalyst under aerobic conditions (Scheme 31).67 While the reaction proceeds with Mn(OAc)3 alone, the addition of benzoic acid further improves the efficiency. The reaction appears to be initiated by O2-triggered hydrogen abstraction from the thiol to form a thiyl radical as a reactive species, which undergoes addition to the C–C triple bond. The manganese catalyst likely plays a role to activate O2 and/or stabilize the hydroperoxy radical.
 | ||
| Scheme 31 Oxidative annulation of thiophenols and activated internal alkynes under manganese catalysis. | ||
Wang and coworkers reported another example of direct annulation of thiophenols and activated internal alkynes for benzothiophene synthesis (Scheme 32).68 Thus, a metal-free catalytic system comprising I2 and DTBP promotes the annulation reaction using DMAD and phenyl propiolate derivatives to afford the corresponding 2,3-disubstituted benzothiophenes in good yields. The proposed mechanism involves the initial oxidation of thiophenol into diphenyldisulfide, which reacts with I2 to form PhSI as an electrophilic reactive species.
 | ||
| Scheme 32 Annulation of thiophenols and activated internal alkynes under iodine catalysis. | ||
Besides type A precursors, many other ortho-difunctionalized arenes have been exploited for intramolecular cyclization to benzothiophenes. In contrast to Larock's electrophile-assisted 5-endo cyclization (Scheme 25), Flynn utilized 5-exo cyclization using a substrate bearing a propargylic alcohol moiety (Scheme 33).69 For this example, 5-exo cyclization is followed by rearrangement and loss of HI to afford a 2-acylbenzothiophene derivative. Gabriele and coworkers utilized similar substrates featuring a free SH group to achieve palladium-catalyzed and radical-mediated 5-exo-cyclizations leading to benzothiophenes.70
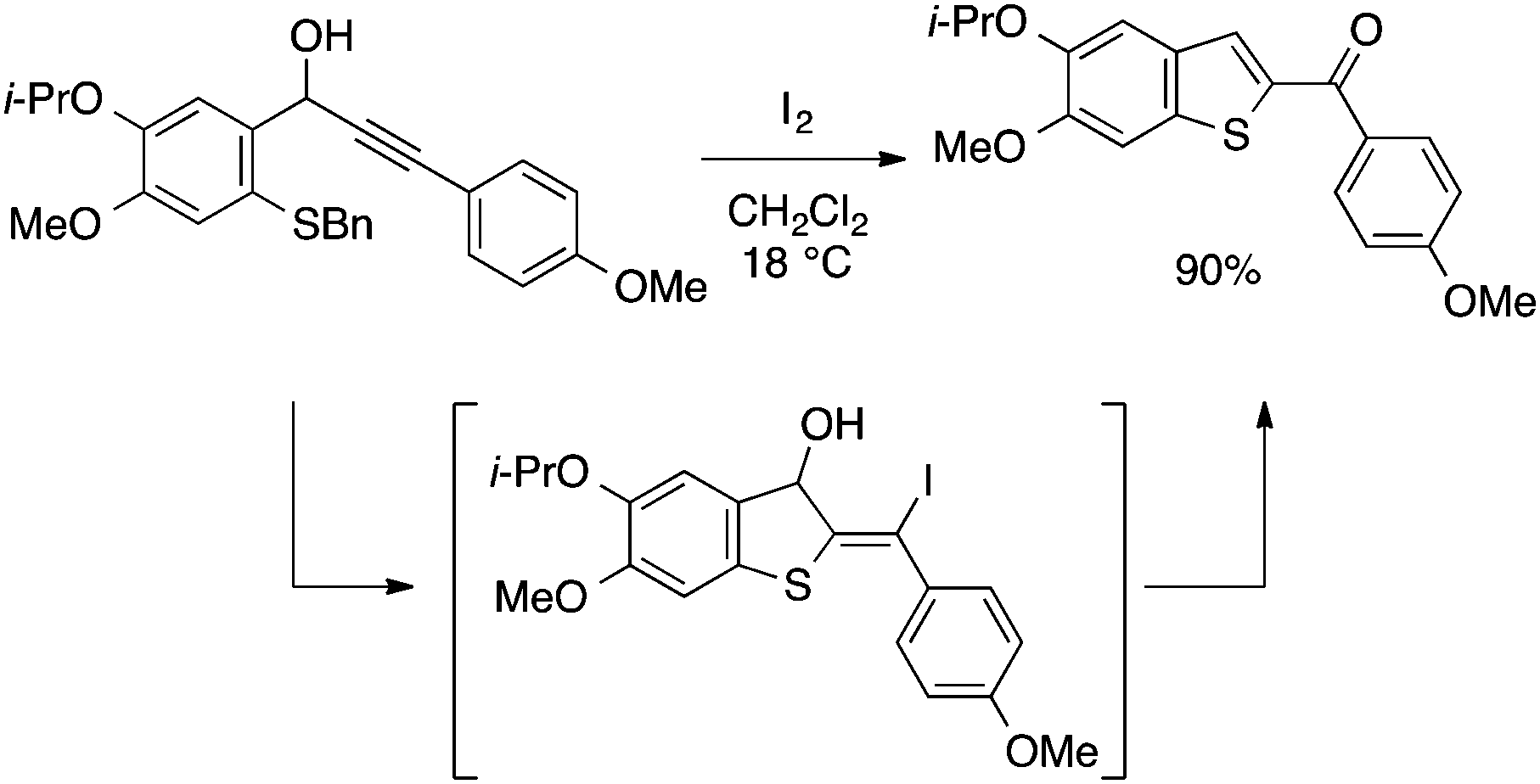 | ||
| Scheme 33 Benzothiophene synthesis through I2-mediated 5-exo cyclization. | ||
Recently, Procter and coworkers have significantly expanded the scope and utility of the 5-exo cyclization approach to benzothiophenes (Scheme 34a).71 Importantly, they utilized sulfoxide-directed deoxygenative ortho-propargylation of aryl sulfoxide to prepare cyclization precursors, i.e., ortho-propargyl aryl thioethers,72 thus streamlining the entire synthetic route to benzothiophenes. Furthermore, they established three different conditions for 5-exo cyclization to achieve chemoselective synthesis of 2-acyl, 2-alkyl, and 2-alkenylbenzothiophenes. The utility of this approach was demonstrated by the preparation of benzothiophene-containing polyaromatics including naphthodithiophenes. Satoh and Miura also demonstrated the utility of aryl sulfoxides as precursors for stepwise synthesis of benzothiophenes (Scheme 34b).73 Thus, Rh(III)-catalyzed ortho-alkenylation via sulfoxide-directed C–H activation can be followed by TfOH-induced interrupted Pummerer cyclization to afford 2,3-disubstituted benzothiophenes.
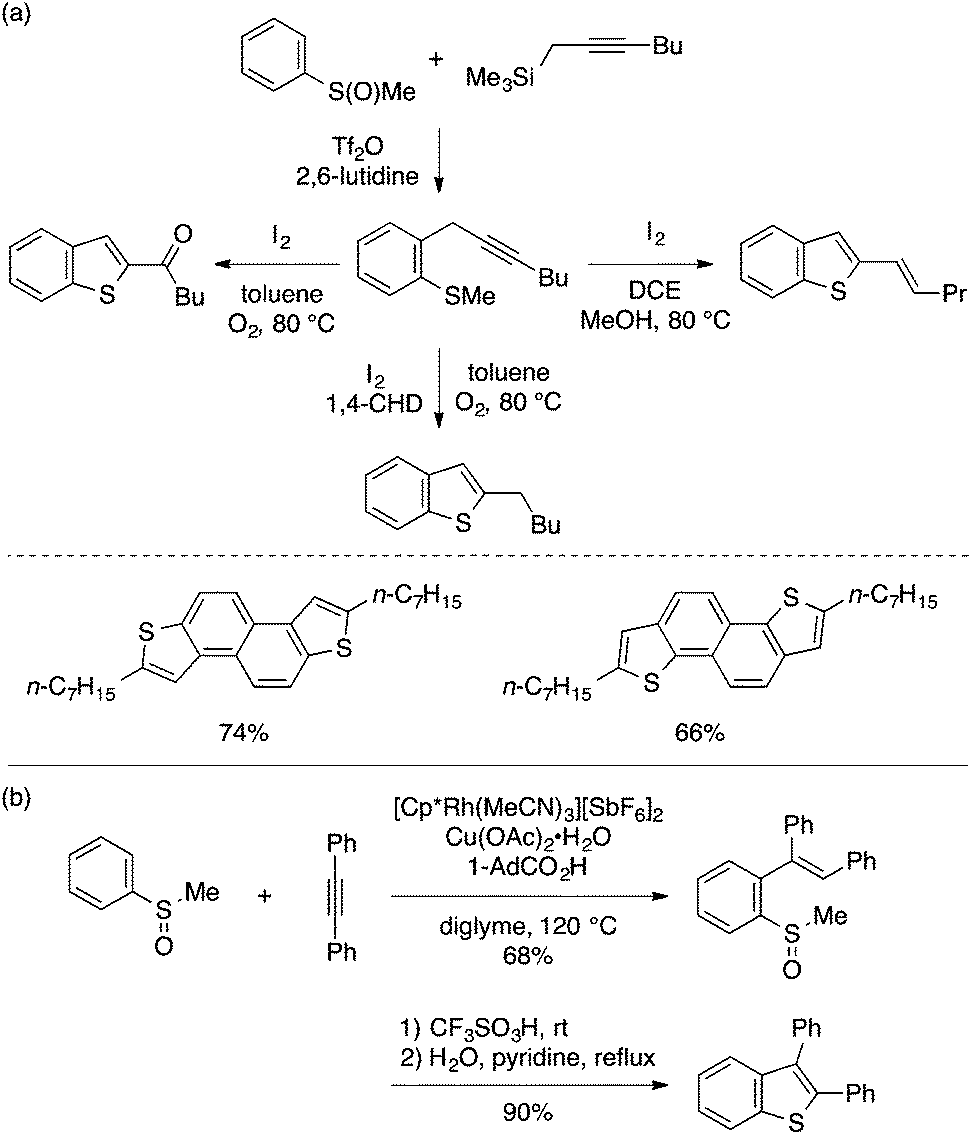 | ||
| Scheme 34 Aryl sulfoxide as a precursor for stepwise synthesis of benzothiophenes. | ||
Lautens and coworkers demonstrated that a thiophenol bearing a gem-dibromoethenyl group serves as a versatile precursor to benzothiophenes through transition metal-catalyzed C–S coupling (Scheme 35). While intramolecular Ullmann reaction using a copper catalyst affords 2-bromobenzothiophenes (Scheme 35a),74 the use of a palladium catalyst and an appropriate coupling partner enables pairing of the intramolecular C–S bond formation and various C–C couplings such as Suzuki–Miyaura (Scheme 35b), Sonogashira, and Heck reactions, affording a variety of 2-substituted benzothiophenes in an expedient manner.75 The former transformation was also achieved using TBAF76 or a trace copper catalyst.77 The scope of coupling partners for the latter tandem approach was later extended to CO + nucleophiles (e.g., alcohol, amine),78 azoles,79 K4Fe(CN)6,80 and organosilanes.81
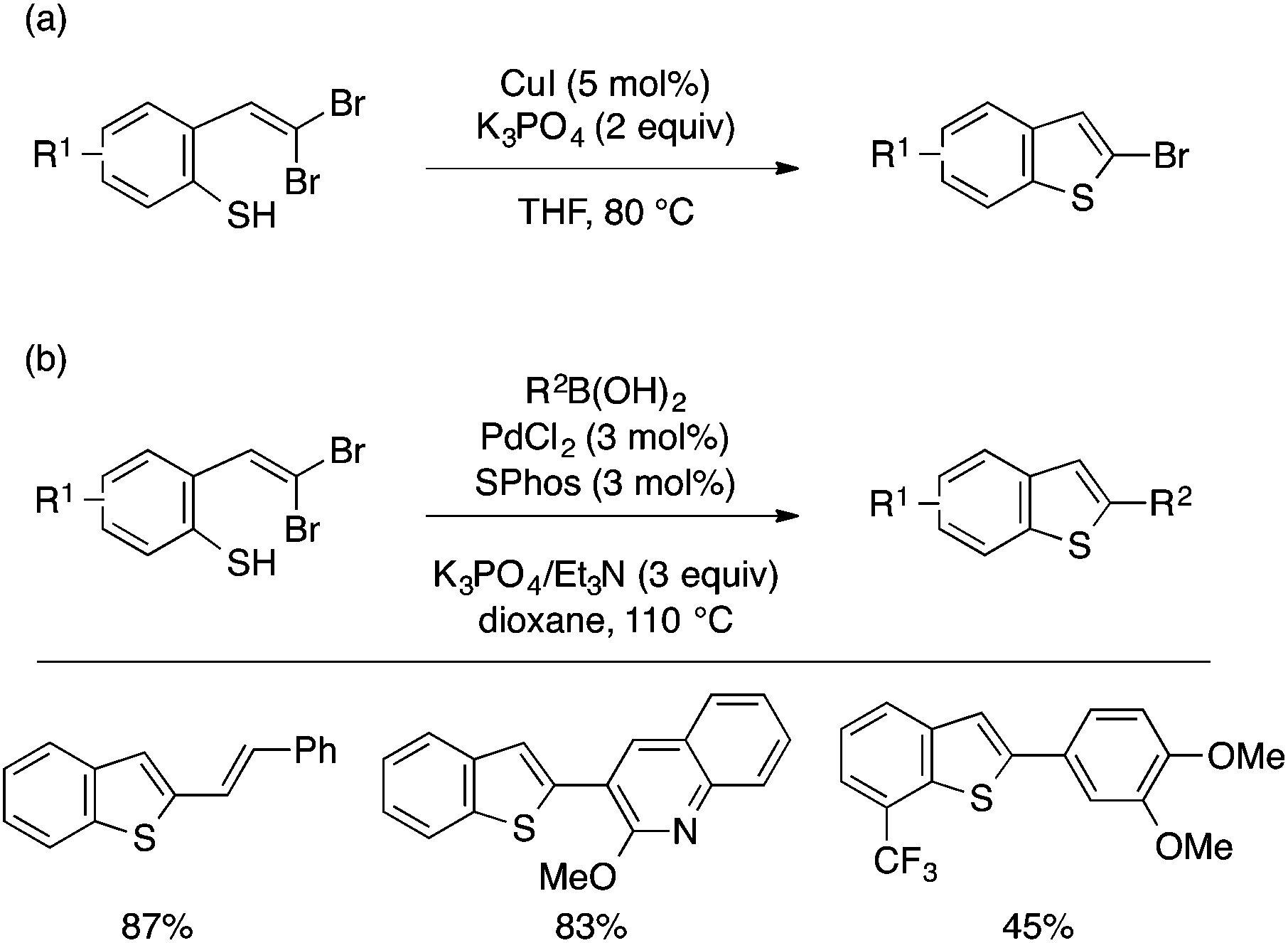 | ||
| Scheme 35 ortho-(gem-Dibromoethenyl)thiophenol as a versatile precursor to benzothiophenes under Cu or Pd catalysis. | ||
ortho-Functionalized aryl halides other than ortho-alkynyl derivatives (Schemes 27–29) have also been utilized as substrates for benzothiophene synthesis through domino aryl–S bond formation/cyclization. Such examples include Cu-catalyzed double Ullmann coupling leading to 2-trifluoromethylbenzothiophene (Scheme 36a),82 Pd-catalyzed aryl–S formation/thiolate addition to nitrile leading to 2-aminobenzothiophene (Scheme 36b),83 and Cu-catalyzed aryl–S formation using a thiocarboxylic acid and intramolecular Wittig reaction to give 2-substituted benzothiophene (Scheme 36c).84 Conversion of styrene or stilbene-type substrates bearing ortho-fluorine substituents into 2-arylbenzothiophenes was also achieved under transition metal-free conditions using K2S (Scheme 36d).85
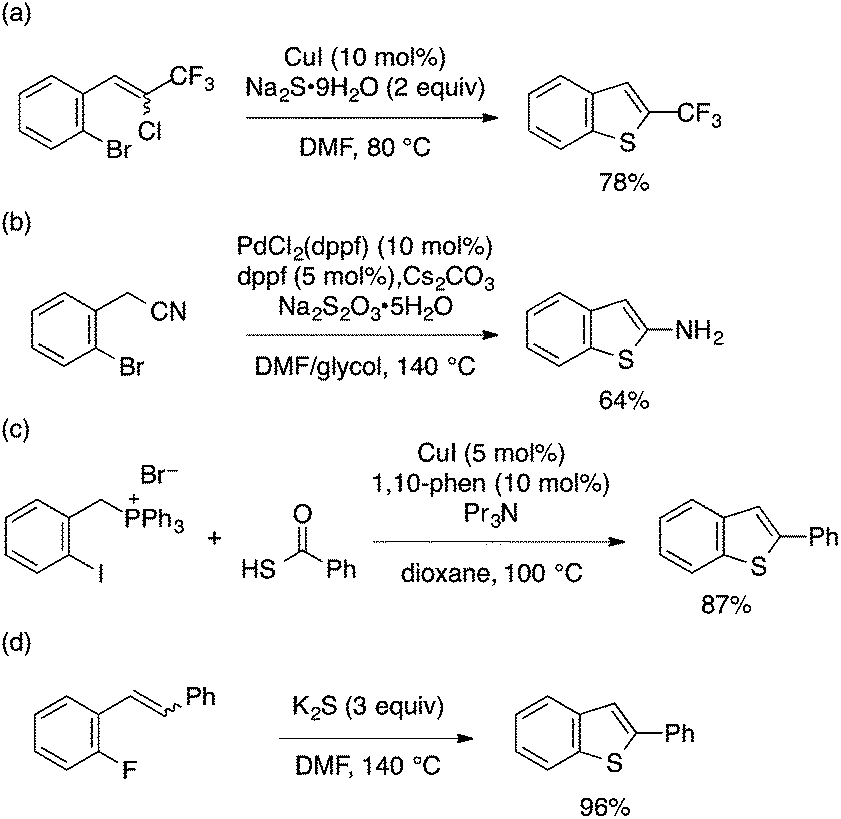 | ||
| Scheme 36 Transformation of ortho-functionalized aryl halides into benzothiophenes through domino aryl–S bond formation/cyclization. | ||
Wu and Yoshikai developed a modular approach to benzothiophenes and benzoselenophenes capitalizing on the cobalt-catalyzed migratory arylzincation reaction of an alkyne, which affords an ortho-alkenylarylzinc species as a versatile precursor (Scheme 37; see also Scheme 24).86 This zinc species may be directly converted to a benzothiophene derivative by the reaction with elemental sulfur in the presence of CuI. Alternatively, the zinc species may be first converted to the corresponding iodide, which is then subjected to the copper-catalyzed chalcogenative cyclization reaction with elemental sulfur or selenium. Owing to the wide availability of arylzinc reagents, these methods allow for expedient preparation of multisubstituted benzothiophenes and benzoselenophenes, which are particularly rich in diversity of the substituents on the benzo moiety.
 | ||
| Scheme 37 Synthesis of benzothiophenes and benzoselenophenes based on cobalt-catalyzed migratory arylzincation. | ||
Methods for benzothiophene synthesis through intramolecular aryl–S bond-forming cyclization have also been reported. Yorimitsu, Oshima and coworkers developed a unique cyclization reaction of arylketene dithioacetal monoxide using trifluoromethanesulfonic anhydride, which affords a 2-methylthiobenzothiophene derivative (Scheme 38).87 The reaction is considered to proceed through triflation of the sulfoxide moiety and subsequent S–O cleavage to form a stabilized dication, which undergoes intramolecular C–S cyclization.
 | ||
| Scheme 38 Benzothiophene synthesis through cyclization of arylketene dithioacetal monooxides under Pummerer-like conditions. | ||
Ila and coworkers reported a benzothiophene synthesis through cyclization of a bifunctional substrate bearing aryl–Br and alkenyl–SMe moieties using catalytic Bu3SnCl and AIBN along with NaCNBH3 (Scheme 39).88 The reaction most likely involves the formation of an aryl radical and its attack on the sulfur atom, and has proved effective for the preparation of 3-cyanobenzothiophene derivatives bearing various 2-substituents.
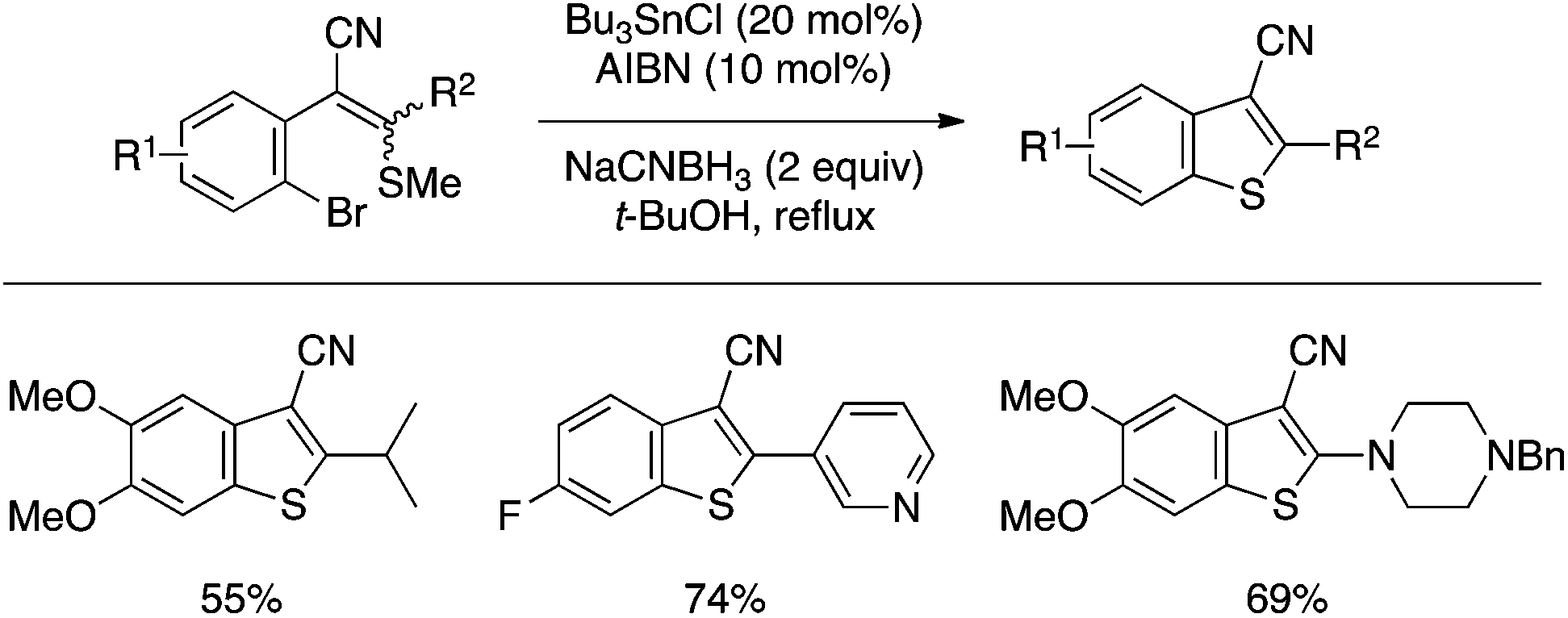 | ||
| Scheme 39 Benzothiophene synthesis through aryl–S bond-forming cyclization under radical conditions. | ||
Inamoto and Doi achieved direct synthesis of 2,3-diarylbenzothiophenes through intramolecular aryl–S cyclization of thioenol precursors using a palladium catalyst (Scheme 40a).89 The proposed mechanism of this reaction involves the initial oxidation of the thioenol to a disulfide, oxidative addition of the disulfide to Pd, and intramolecular C–H activation. Ila and coworkers developed two methods for benzothiophene synthesis through intramolecular aryl–S bond formation of a thioenolate moiety generated by base-mediated condensation of arylacetonitrile or related substrate with dithioester (Scheme 40b). One relies on Ullmann-type chemistry using a copper catalyst and a brominated substrate,90 while another capitalizes on the above Inamoto/Doi-type direct aryl–S bond formation using a modified Pd catalytic system.91 In particular, the latter method allows rapid preparation of a broad range of polyfunctionalized benzothiophenes.
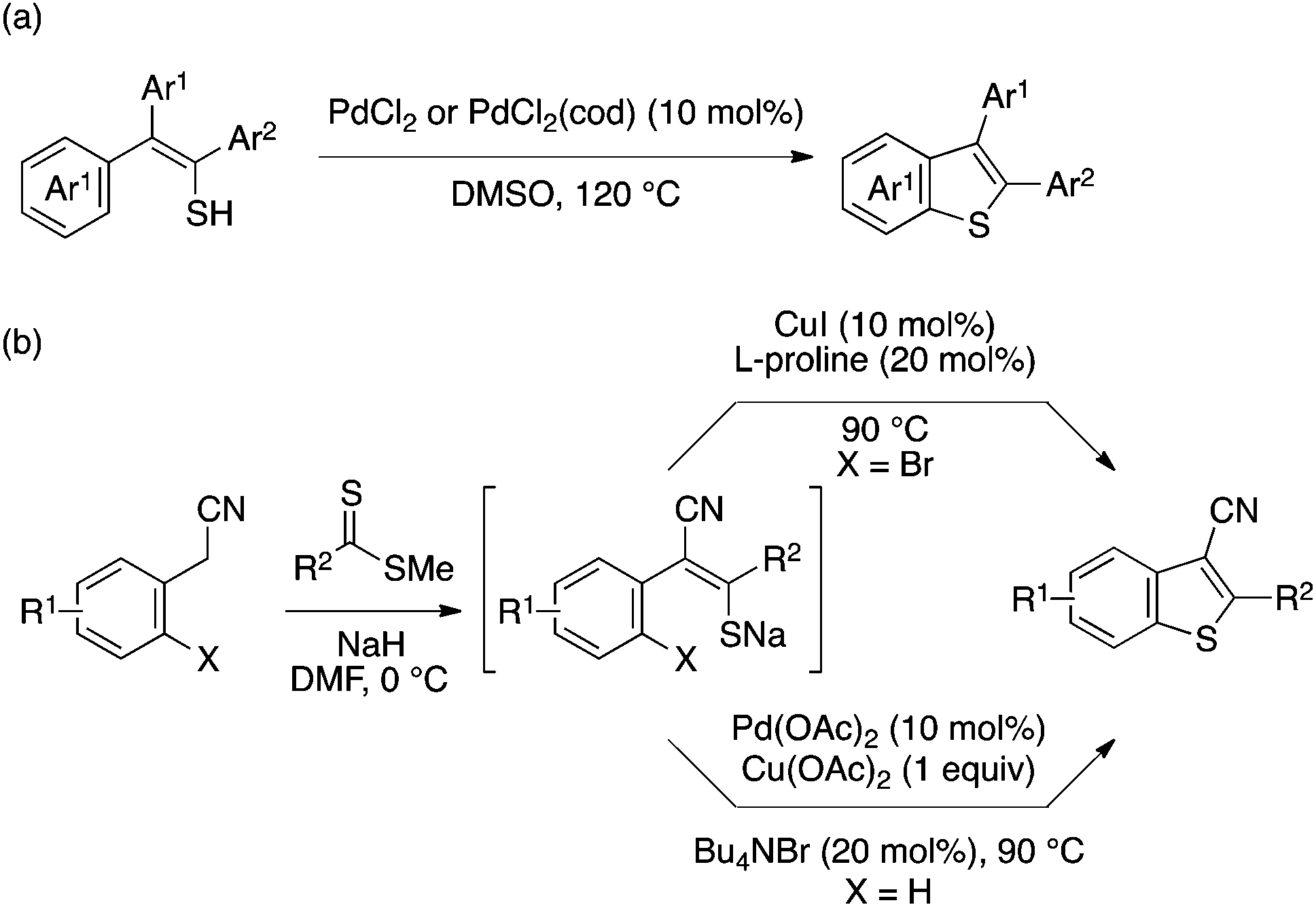 | ||
| Scheme 40 Benzothiophene synthesis through Pd- or Cu-catalyzed aryl–S bond-forming cyclization. | ||
Benzoselenophene synthesis involving aryl–Se bond formation as the cyclization step has also been reported. Arsenyan and coworkers developed a reaction of arylalkynes with SeBr4 generated in situ from SeO2 and HBr to afford 3-bromobenzoselenophenes under mild conditions (Scheme 41).92 The reaction is proposed to occur through trans-selenobromination of the alkyne followed by the intramolecular SEAr reaction.93
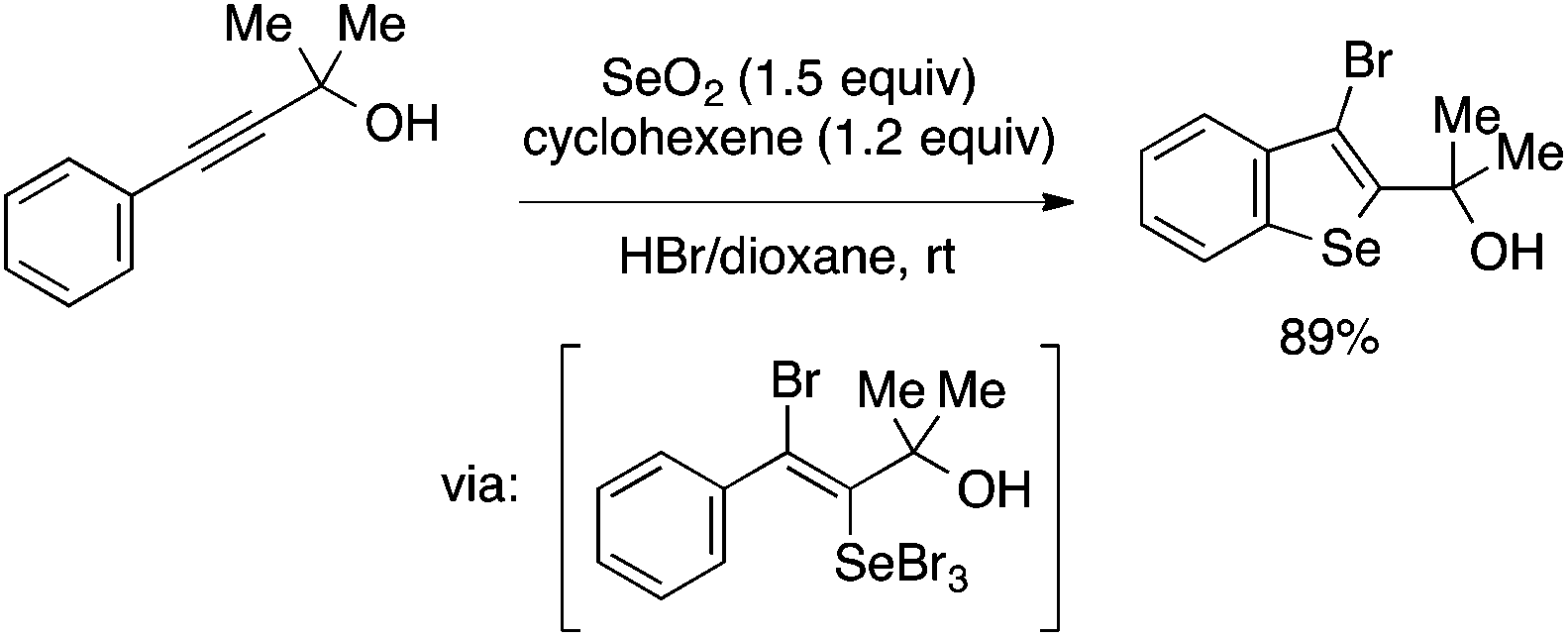 | ||
| Scheme 41 Benzoselenophene synthesis through alkyne selenobromination/intramolecular SEAr reaction. | ||
While many of the intramolecular cyclization approaches to benzothiophenes features C–S bond formation as the key step, Knochel and coworkers developed a unique intramolecular C–C bond-forming cyclization approach to benzothiophenes (Scheme 42).94 Thus, an ortho-alkynylthioaryl bromide is transformed into a benzothiophen-2-ylmagnesium species through a sequence of halogen–metal exchange and copper-catalyzed intramolecular carbomagnesiation. The organomagnesium species can be diverted into a variety of 2,3-disubstituted benzothiophenes by electrophilic trapping.
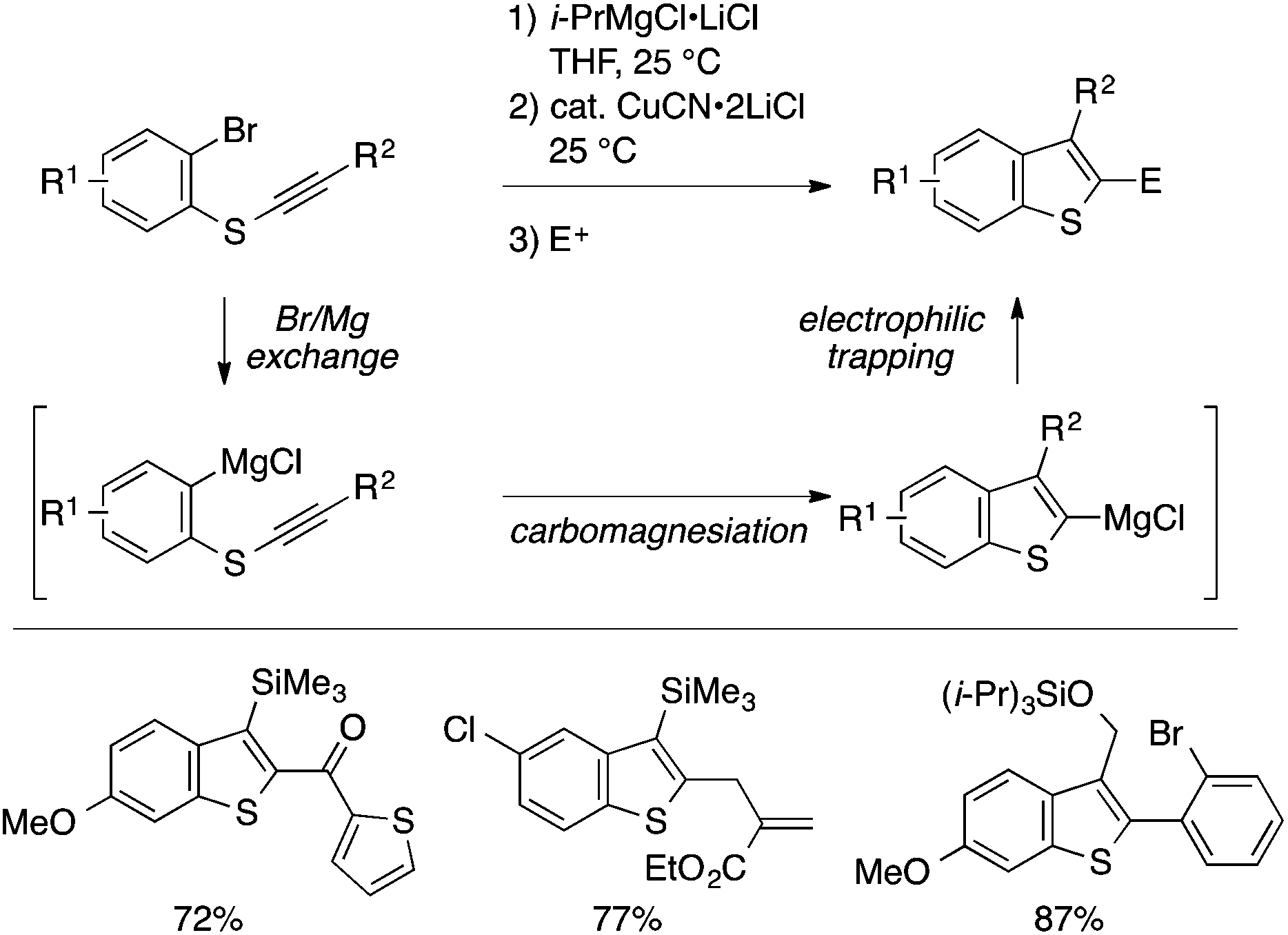 | ||
| Scheme 42 Benzothiophene synthesis through intramolecular carbomagnesiation of thioalkyne. | ||
5. Conclusions
The last several years have witnessed significant new developments in synthetic methodology for benzoheteroles containing various heteroatom elements across the periodic table. Such new synthetic methods have enabled facile variation of peripheral substituents on the benzo moiety, the C2- and C3-positions, and also the heteroatom itself. This has therefore triggered the emergence of new benzoheterole-based materials elaborately designed and synthesized for optoelectronic applications. That said, there remains plenty of room for further improvement of existing synthetic methods as well as for the development of new methods. From a fundamental point of view, synthetic methods for other members of the benzoheterole family, benzoborole and benzotellurophene for example, remain scarce regardless of their intriguing properties,95 and thus warranting further development. From an application perspective, more flexible, robust, and broad-scope methods are desired for the synthesis of benzoheteroles of established or promising utility including benzosiloles, benzophospholes, and benzothiophenes.Acknowledgements
We thank the Singapore National Research Foundation (NRF-RF-2009-05) and Nanyang Technological University for the financial support of our works discussed in this article.Notes and references
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed.
- (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC.
- S. Yamaguchi and K. Tamao, Chem. Lett., 2005, 34, 2 CrossRef CAS.
- For selected reviews on siloles and their benzo-fused analogues, see: (a) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS; (b) W. W. H. Wong, J. F. Hooper and A. B. Holmes, Aust. J. Chem., 2009, 62, 393 CAS; (c) H. Fu and Y. Cheng, Curr. Org. Chem., 2012, 16, 1423 CrossRef CAS.
- For selected reviews on phospholes and their benzo-fused analogues, see: (a) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681 CrossRef CAS PubMed; (b) Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258 RSC; (c) M. Stolar and T. Baumgartner, Chem. – Asian J., 2014, 9, 1212 CrossRef CAS PubMed.
- For selected reviews on chalcogenophenes and their benzo-fused analogues, see: (a) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347 CrossRef CAS PubMed; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- For specific examples shown in Fig. 1, see: (a) L. Ilies, Y. Sato, C. Mitsui, H. Tsuji and E. Nakamura, Chem. – Asian J., 2010, 5, 1376 CAS; (b) H. Tsuji, K. Sato, Y. Sato and E. Nakamura, Chem. – Asian J., 2010, 5, 1294 CAS; (c) E. Yamaguchi, C. G. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539 CrossRef CAS PubMed; (d) S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024 CrossRef CAS PubMed; (e) K. Takimiya, Y. Kunugi, Y. Konda, N. Niihara and T. Otsubo, J. Am. Chem. Soc., 2004, 126, 5084 CrossRef CAS PubMed.
- C. E. Castro, E. J. Gaughan and D. C. Owsley, J. Org. Chem., 1966, 31, 4071 CrossRef CAS.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CrossRef CAS.
- For comprehensive review articles on this topic, see: Comprehensive Heterocyclic Chemistry III, ed. G. Jones and C. A. Ramsden, Elsevier, Oxford, 2008, vol. 3 Search PubMed.
- A. Fukazawa and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1386 CrossRef CAS PubMed.
- T. Sudo, N. Asao and Y. Yamamoto, J. Org. Chem., 2000, 65, 8919 CrossRef CAS PubMed.
- T. Matsuda, S. Kadowaki and M. Murakami, Chem. Commun., 2007, 2627 RSC.
- L. Ilies, H. Tsuji and E. Nakamura, Org. Lett., 2009, 11, 3966 CrossRef CAS PubMed.
- C. Xu, A. Wakamiya and S. Yamaguchi, Org. Lett., 2004, 6, 3707 CrossRef CAS PubMed.
- C. Xu, A. Wakamiya and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 1638 CrossRef CAS PubMed.
- L. Ilies, H. Tsuji, Y. Sato and B. Nakamura, J. Am. Chem. Soc., 2008, 130, 4240 CrossRef CAS PubMed.
- T. Matsuda, S. Kadowaki, Y. Yamaguchi and M. Murakami, Chem. Commun., 2008, 2744 RSC.
- T. Matsuda, Y. Yamaguchi, M. Shigeno, S. Sato and M. Murakami, Chem. Commun., 2011, 47, 8697 RSC.
- T. Matsuda and Y. Ichioka, Org. Biomol. Chem., 2012, 10, 3175 CAS.
- (a) M. Tobisu, M. Onoe, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 7506 CrossRef CAS PubMed; (b) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477 CrossRef CAS PubMed.
- M. Tobisu, K. Baba and N. Chatani, Org. Lett., 2011, 13, 3282 CrossRef CAS PubMed.
- Y. Liang, W. Geng, J. Wei and Z. Xi, Angew. Chem., Int. Ed., 2012, 51, 1934 CrossRef CAS PubMed.
- T. Meng, K. Ouyang and Z. Xi, RSC Adv., 2013, 3, 14273 RSC.
- E. Shirakawa, S. Masui, R. Narui, R. Watabe, D. Ikeda and T. Hayashi, Chem. Commun., 2011, 47, 9714 RSC.
- L. Xu, S. Zhang and P. Li, Org. Chem. Front., 2015, 2, 459 RSC.
- T. Matsuda, Y. Yamaguchi and M. Murakami, Synlett, 2008, 561 CAS.
- T. Matsuda, Y. Yamaguchi, N. Ishida and M. Murakami, Synlett, 2010, 2743 CrossRef CAS.
- K. Ouyang, Y. Liang and Z. Xi, Org. Lett., 2012, 14, 4572 CrossRef CAS PubMed.
- A. Kuznetsov and V. Gevorgyan, Org. Lett., 2012, 14, 914 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092 CrossRef CAS PubMed.
- L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241 CrossRef CAS.
- S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192 CrossRef CAS PubMed.
- H. Tsuji, K. Sato, L. Ilies, Y. Itoh, Y. Sato and E. Nakamura, Org. Lett., 2008, 10, 2263 CrossRef CAS PubMed.
- H. Tsuji, K. Sato, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 3364 RSC.
- T. Sanji, K. Shiraishi, T. Kashiwabara and M. Tanaka, Org. Lett., 2008, 10, 2689 CrossRef CAS PubMed.
- A. Fukazawa, Y. Ichihashi, Y. Kosaka and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1729 CrossRef CAS PubMed.
- Y. Xu, Z. Wang, Z. Gan, Q. Xi, Z. Duan and F. Mathey, Org. Lett., 2015, 17, 1732 CrossRef CAS PubMed.
- Y. Zhou, Z. Gan, B. Su, J. Li, Z. Duan and F. Mathey, Org. Lett., 2015, 17, 5722 CrossRef CAS PubMed.
- (a) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed; (b) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed.
- (a) W. Ma and L. Ackermann, Synthesis, 2014, 2297 CAS; (b) G. Hu, Y. Zhang, J. Su, Z. Li, Y. Gao and Y. Zhao, Org. Biomol. Chem., 2015, 13, 8221 RSC.
- P. Zhang, Y. Gao, L. Zhang, Z. Li, Y. Liu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2016, 358, 138 CrossRef CAS.
- Y. Zhang, G. Hu, D. Ma, P. Xu, Y. Gao and Y. Zhao, Chem. Commun., 2016, 52, 2815 RSC.
- (a) Y. Hayashi, Y. Matano, K. Suda, Y. Kimura, Y. Nakao and H. Imahori, Chem. – Eur. J., 2012, 18, 15972 CrossRef CAS PubMed; (b) Y. Matano, Y. Hayashi, K. Suda, Y. Kimura and H. Imahori, Org. Lett., 2013, 15, 4458 CrossRef CAS PubMed; (c) Y. Matano, Y. Motegi, S. Kawatsu and Y. Kimura, J. Org. Chem., 2015, 80, 5944 CrossRef CAS PubMed.
- M. Ogasawara, S. Arae, S. Watanabe, V. Subbarayan, H. Sato and T. Takahashi, Organometallics, 2013, 32, 4997 CrossRef CAS.
- X. Yan and C. Xi, Acc. Chem. Res., 2015, 48, 935 CrossRef CAS PubMed.
- B.-H. Tan, J. Dong and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 9610 CrossRef CAS PubMed.
- B. Wu, M. Santra and N. Yoshikai, Angew. Chem., Int. Ed., 2014, 53, 7543 CrossRef CAS PubMed.
- F. Xue, J. Zhao and T. S. A. Hor, Chem. Commun., 2013, 49, 10121 RSC.
- B. Wu, R. Chopra and N. Yoshikai, Org. Lett., 2015, 17, 5666 CrossRef CAS PubMed.
- (a) R. C. Larock and D. Yue, Tetrahedron Lett., 2001, 42, 6011 CrossRef CAS; (b) D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905 CrossRef CAS PubMed.
- For a review on electrophilic cyclizations, see: B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937 CrossRef CAS PubMed.
- W.-D. Lu and M.-J. Wu, Tetrahedron, 2007, 63, 356 CrossRef CAS.
- J. Sheng, C. Fan and J. Wu, Chem. Commun., 2014, 50, 5494 RSC.
- T. Kesharwani, S. A. Worlikar and R. C. Larock, J. Org. Chem., 2006, 71, 2307 CrossRef CAS PubMed.
- I. Nakamura, T. Sato and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 4473 CrossRef CAS PubMed.
- I. Nakamura, T. Sato, M. Terada and Y. Yamamoto, Org. Lett., 2007, 9, 4081 CrossRef CAS PubMed.
- T. Sato, I. Nakamura and M. Terada, Eur. J. Org. Chem., 2009, 5509 CrossRef CAS.
- H. Sashida, K. Sadamori and T. Tsuchiya, Synth. Commun., 1998, 28, 713 CrossRef CAS.
- K. Takimiya, Y. Konda, H. Ebata, N. Niihara and T. Otsubo, J. Org. Chem., 2005, 70, 10569 CrossRef CAS PubMed.
- T. Kashiki, S. Shinamura, M. Kohara, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Org. Lett., 2009, 11, 2473 CrossRef CAS PubMed.
- L. L. Sun, C. L. Deng, R. Y. Tang and X. G. Zhang, J. Org. Chem., 2011, 76, 7546 CrossRef CAS PubMed.
- M. Kuhn, F. C. Falk and J. Paradies, Org. Lett., 2011, 13, 4100 CrossRef CAS PubMed.
- V. Guilarte, M. A. Fernandez-Rodriguez, P. Garcia-Garcia, E. Hernando and R. Sanz, Org. Lett., 2011, 13, 5100 CrossRef CAS PubMed.
- D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334 CrossRef CAS PubMed.
- M. K. Staples, R. L. Grange, J. A. Angus, J. Ziogas, N. P. H. Tan, M. K. Taylor and C. H. Schiesser, Org. Biomol. Chem., 2011, 9, 473 CAS.
- K. Liu, F. Jia, H. Xi, Y. Li, X. Zheng, Q. Quo, B. Shen and Z. Li, Org. Lett., 2013, 15, 2026 CrossRef CAS PubMed.
- K. Yan, D. Yang, M. Zhang, W. Wei, Y. Liu, L. Tian and H. Wang, Synlett, 2015, 1890 CAS.
- K. O. Hessian and B. L. Flynn, Org. Lett., 2003, 5, 4377 CrossRef CAS PubMed.
- B. Gabriele, R. Mancuso, E. Lupinacci, L. Veltri, G. Salerno and C. Carfagna, J. Org. Chem., 2011, 76, 8277 CrossRef CAS PubMed.
- A. J. Eberhart, H. Shrives, Y. Zhang, A. Carrër, A. V. S. Parry, D. J. Tate, M. L. Turner and D. J. Procter, Chem. Sci., 2016, 7, 1281 RSC.
- A. J. Eberhart and D. J. Procter, Angew. Chem., Int. Ed., 2013, 52, 4008 CrossRef CAS PubMed.
- K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed.
- S. G. Newman, V. Aureggi, C. S. Bryan and M. Lautens, Chem. Commun., 2009, 5236 RSC.
- C. S. Bryan, J. A. Braunger and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 7064 CrossRef CAS PubMed.
- W. Chen, Y. Zhang, L. Zhang, M. Wang and L. Wang, Chem. Commun., 2011, 47, 10476 RSC.
- Y. Ji, P. Li, X. Zhang and L. Wang, Org. Biomol. Chem., 2013, 11, 4095 CAS.
- F. Zeng and H. Alper, Org. Lett., 2011, 13, 2868 CrossRef CAS PubMed.
- X. Qin, X. Cong, D. Zhao, J. You and J. Lan, Chem. Commun., 2011, 47, 5611 RSC.
- W. Zhou, W. Chen and L. Wang, Org. Biomol. Chem., 2012, 10, 4172 CAS.
- J. Liu, W. Chen, Y. Ji and L. Wang, Adv. Synth. Catal., 2012, 354, 1585 CrossRef CAS.
- C.-L. Li, X.-G. Zhang, R.-Y. Fang, P. Zhong and J.-H. Li, J. Org. Chem., 2010, 75, 7037 CrossRef CAS PubMed.
- C. Hou, Q. He and C. Yang, Org. Lett., 2014, 16, 5040 CrossRef CAS PubMed.
- H. Yu, M. Zhang and Y. Li, J. Org. Chem., 2013, 78, 8898 CrossRef CAS PubMed.
- X. Zhang, W. Zeng, Y. Yang, H. Huang and Y. Liang, Synlett, 2013, 1687 CAS.
- B. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 10496 CrossRef CAS PubMed.
- S. Yoshida, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 5573 CrossRef CAS PubMed.
- P. P. Singh, A. K. Yadav, H. Ila and H. Junjappa, J. Org. Chem., 2009, 74, 5496 CrossRef CAS PubMed.
- K. Inamoto, Y. Arai, K. Hiroya and T. Doi, Chem. Commun., 2008, 5529 RSC.
- A. Acharya, S. V. Kumar, B. Saraiah and H. Ila, J. Org. Chem., 2015, 80, 2884 CrossRef CAS PubMed.
- A. Acharya, S. V. Kumar and H. Ila, Chem. – Eur. J., 2015, 21, 17116 CrossRef CAS PubMed.
- E. Paegle, S. Belyakov and P. Arsenyan, Eur. J. Org. Chem., 2014, 3831 CrossRef CAS.
- For other miscellaneous approaches to benzoselenophenes, see: C. R. B. Rhoden and G. Zeni, Org. Biomol. Chem., 2011, 9, 1301 CAS.
- T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 1958 CrossRef CAS PubMed.
- For example, see: (a) A. Fukazawa, H. Yamada and S. Yamaguchi, Angew. Chem., Int. Ed., 2008, 47, 5582 CrossRef CAS PubMed; (b) G. He, B. D. Wiltshire, P. Choi, A. Savin, S. Sun, A. Mohammadpour, M. J. Ferguson, R. McDonald, S. Farsinezhad, A. Brown, K. Shankar and E. Rivard, Chem. Commun., 2015, 51, 5444 RSC.
| This journal is © The Royal Society of Chemistry 2016 |