
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocompatibility assessment of haemodialysis membrane materials by proteomic investigations†
Luisa
Pieroni
ab,
Stefano
Levi Mortera
ab,
Viviana
Greco
ab,
Vittorio
Sirolli
c,
Maurizio
Ronci
bde,
Paolo
Felaco
c,
Giorgio
Fucci
a,
Silvia
De Fulviis
c,
Renato
Massoud
a,
Stefano
Condò
f,
Ambrogio
Capria
f,
Nicola
Di Daniele
f,
Sergio
Bernardini
a,
Andrea
Urbani‡
ab and
Mario
Bonomini‡§
*c
aDepartment of Experimental Medicine and Surgery, University “Tor Vergata”, Rome, Italy
bIRCCS-Santa Lucia Foundation, Rome, Italy
cNephrology and Dialysis Institute, Department of Medicine, “G. d'Annunzio” University, Chieti-Pescara, Italy
dDepartment of Medical, Oral and Biotechnological Sciences, University G. D'Annunzio of Chieti-Pescara, Chieti, Italy
eMawson Institute, University of South Australia, Mawson Lakes, SA, 5095 Australia
fDepartment of System Medicine, University “Tor Vergata”, Rome, Italy
First published on 26th March 2015
Abstract
The exposure of blood to an artificial surface such as the haemodialysis membrane results in the nearly instantaneous deposition of a layer of plasma proteins. The composition of the protein layer profoundly influences all subsequent events, and to a large extent determines the biocompatibility of the biomaterial. In the present study, we examine the protein adsorption capacity and coagulation profiles of the polysulfone-based helixone material in comparison to cellulose triacetate. A differential profiling investigation using shotgun proteomics data-independent analysis was applied to eluates obtained with each membrane after a dialysis session, in order to assess the function of desorbed proteins. Functional classification and network analysis performed using bioinformatics tools shed light on the involvement of adsorbed proteins into important molecular processes, such as lipid transport and metabolism, cell growth differentiation and communication, and the coagulation cascade. The collected evidence was further validated by targeted mass spectrometry using selected reaction monitoring on proteotypic transitions of key protein effectors, confirming the different panels of adsorbed protein on each membrane. The coagulation profile during haemodialysis of patients under polysulfone-based helixone filter cartridges was also assessed showing a slightly higher platelet activation profile after the dialysis session. The overall collected evidence highlights a modulation of the coagulation biological pathway during haemodialysis, which is largely influenced by the biomaterial used.
Introduction
The membrane packed into the haemodialyser represents the ultimate determinant of the quality and success of haemodialysis therapy, by far the most commonly used modality of renal replacement treatment for end-stage renal disease (ESRD) patients. The removal of excess water and unwanted solutes via the membrane device lessens the uremic intoxication, thereby allowing patient survival and rehabilitation, though the non-selectivity of the removal processes may also cause useful substances to be lost. In addition, the bio(in)compatibility of a haemodialysis membrane, which in a broad sense refers to all the potentially harmful consequences of blood contact with the membrane material, is an established contributing factor to several complications frequently encountered in patients on chronic haemodialysis.1The membrane properties for protein adsorption represent one important feature in membrane performance. Adsorption of plasma proteins onto a biomaterial surface affects the removal capacity of a membrane material and is vital to its biocompatibility in particular. The interaction of surface-adsorbed activated proteins may indeed trigger almost every cellular and plasmatic component of the blood,2–6 with potentially pathophysiological consequences.
Activation of the coagulation cascade via blood–membrane contact, together with changes in the rheological conditions,7 triggers clotting of the extracorporeal circuit during haemodialysis. To avoid the risk of premature clotting/premature termination of the dialysis session, intradialytic anticoagulation is necessary, usually by unfractionated heparin or its low-molecular weight derivatives. Optimal anticoagulation of the extracorporeal circuit remains, however, a controversial issue for standard practice,8 and even sophisticated and expensive tests assessing coagulation cannot adequately predict the risk of clotting.9,10 As a result, the patient may be exposed not only to the risk of clotting in the extracorporeal circuit, but also to the risk of bleeding.
While there are ample data on the systemic effects of the interaction between blood and artificial membrane material, the molecular substrates remain largely obscure. A better understanding of the mechanism(s) responsible for foreign pattern recognition and the subsequent reaction cascade during haemodialysis could help to prevent adverse sequelae.1 This may be accomplished in an unbiased manner by elution of dialysers and subsequent proteomic analyses. In recent studies, proteomics has proved its usefulness in investigating proteins adsorbed onto the dialysis membrane.1,11–15 In the present study, we conducted a shotgun proteomics investigation by data-independent differential profiling in order to assess the protein functional ontologies associated with membrane retention onto different materials (cellulose triacetate and helixone) during in vivo haemodialysis. The evidence collected was further validated by targeted mass spectrometry using selected reaction monitoring on proteotypical transitions of key protein effectors. Our results highlight a modulation of the coagulation system in particular that associated with activation of the collagen pathway of platelet activation, which is largely dependent on the biomaterial used.
Results
In this work, we analysed the biocompatibility of cellulose triacetate (CTA) and polysulfone-based helixone (HE) materials by means of a differential proteomics profiling experiment by data-independent analysis based on a shotgun discovery proteomics method.16,17 We have previously found comparable quantitative protein adsorption properties of the two materials.14 The present experimental approach was designed to compare the protein repertoire adsorbed on different membranes after a dialysis session.The analysis was performed on proteins adsorbed from the different membranes after two independent dialysis sessions of 3 chronic patients (from now on indicated as cases 1, 2 and 3), routinely treated by haemodialysis therapy. For each patient, four replicate runs were analysed upon treatment with each membrane; we found an average of 65 proteins (respectively 69 in case 1, 66 in case 2 and 60 in case 3) differentially adsorbed on the two membranes (Table S1, ESI†), and an average of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 525 EMRT, exact mass retention time clusters (respectively 57024 in case 1, 60339 in case 2 and 58212 in case 3) (Fig. S1, ESI†). Quality control measures16 were performed on the replicates to determine the analytical reproducibility of the analysis (Fig. S2, ESI†).
525 EMRT, exact mass retention time clusters (respectively 57024 in case 1, 60339 in case 2 and 58212 in case 3) (Fig. S1, ESI†). Quality control measures16 were performed on the replicates to determine the analytical reproducibility of the analysis (Fig. S2, ESI†).
A slightly higher percentage of proteins adsorbed onto the CTA membrane was observed than onto HE (Fig. 1A). To obtain a more comprehensive picture of the class of proteins retained by the two membranes, we pooled the results relative to the different adsorption onto membrane profiles obtained for the 3 patients; then, using those pooled datasets, we performed an unsupervised bioinformatics analysis. To this end, panther classification tool18,19 allowed us to obtain a functional classification of the differentially adsorbed proteins.
 | ||
| Fig. 1 Characterization of proteins adsorbed by CTA or HE membranes. (A) Percentage of proteins adsorbed onto the two membranes in each patient. (B) Representation of the Gene Ontology functional classification of the proteins adsorbed by the two membranes restricted to the ontology cellular component. | ||
As already shown in our previous studies,14,15 most of the proteins retained by CTA belong to the extracellular matrix and extracellular region component, being represented by a high abundance of plasma proteins such as the immunoglobulin chain, albumin, apolipoproteins or complement factors, while HE membrane mostly retains a low abundance of plasma proteins belonging to macromolecular complexes, organelles, cell parts and a low level of proteins belonging to the extracellular matrix or extracellular region (Fig. 1B). The extension of the previously collected data allowed us to perform a systematic bioinformatics analysis to highlight the key ontological functional classes associated with the retained protein repertoire on the different filter materials.
The complete datasets were also uploaded onto QIAGEN's Ingenuity Pathway Analysis (IPA®, QIAGEN Redwood City, http://www.qiagen.com/ingenuity) system software, for comprehensive pathway and network analysis. Fig. 2A shows the first two top networks and the molecules involved, generated by IPA with the pooled datasets of proteins preferentially adsorbed by the CTA membrane, suggesting an involvement of those molecules (green coloured symbols) in networks related to lipid metabolism, molecular transport, small molecule biochemistry, cellular assembly and organization, cell death and survival, and cardiovascular disease. Moreover, the merged network design (Fig. 2B) confirms, as stated above and in our previous work,15 that most of these molecules are secreted in the extracellular region.
 | ||
| Fig. 2 Ingenuity Pathway Analysis (IPA) of CTA retained proteins pool of dataset. (A) Network table of the two top ranking networks and (B) merge of the first two top ranking networks generated. Network focus molecules are mostly extracellular (secreted) proteins involved in: lipid metabolism, molecular transport, small molecule biochemistry, cellular assembly and organization, cell death and survival, and cardiovascular disease. | ||
Fig. 3A and B report the corresponding analysis performed on the HE-membrane adsorbed proteins, showing their involvement in hematological disease, hereditary disorder, developmental disorder, organism injury and abnormalities, renal and urological disease, and molecular transport. Fig. 3B shows a merger of these networks whereby the focus molecules (red symbols) are both secreted and cellular proteins.
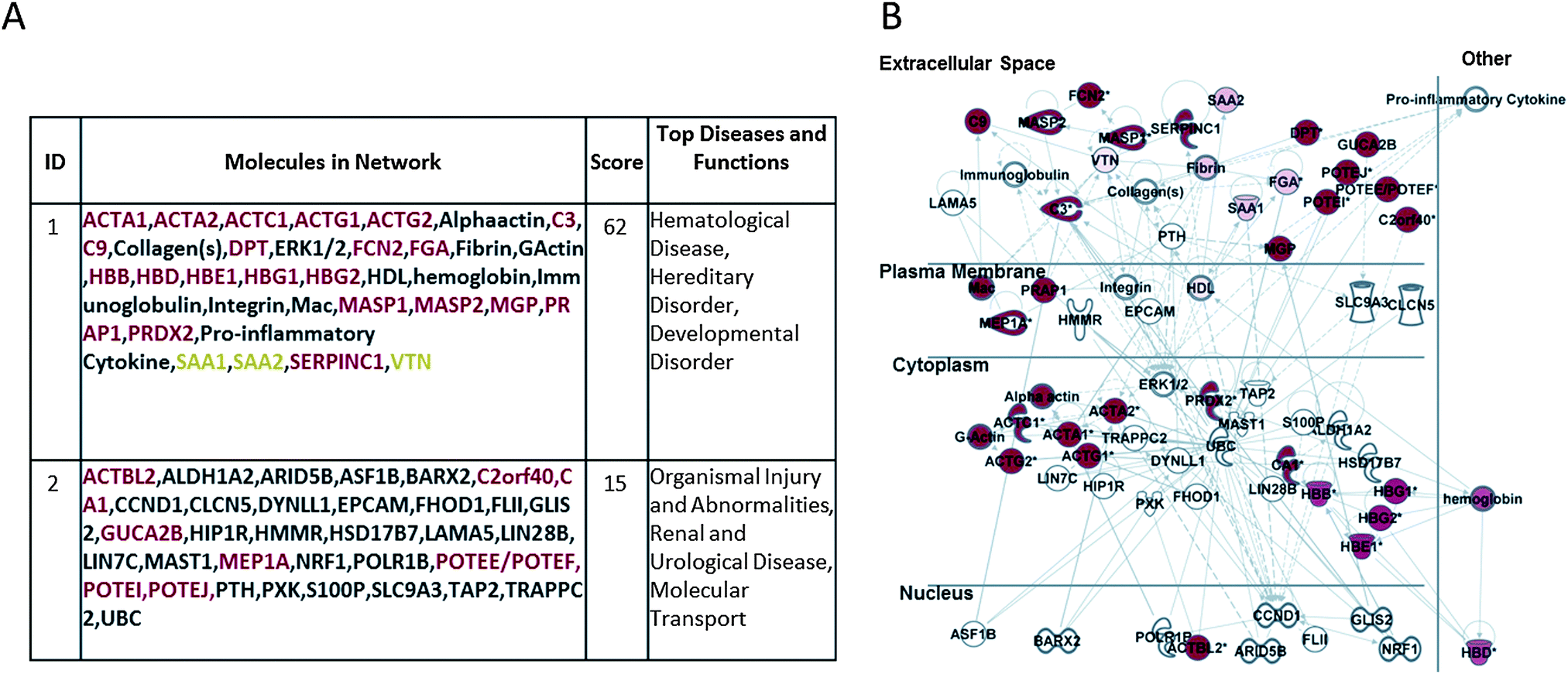 | ||
| Fig. 3 Ingenuity Pathway Analysis (IPA) of HE retained proteins pool of dataset. (A) Network table of the two top ranking networks and (B) merger of the first two top ranking networks generated. Network focus molecules are both extracellular (secreted) and cytoplasmic proteins involved in: hematological disease, hereditary disorder, developmental disorder, organism injury and abnormalities, renal and urological disease, and molecular transport. | ||
Within the core analysis performed by IPA, we further investigated the involvement of those proteins in canonical pathways. As shown in Fig. S3 (ESI†), the proteins mostly retained by CTA material are involved in the activation of the canonical pathway related to the activation of LXR/RXR, the retinoid X nuclear receptors (RXRs) that mediate the biological effects of retinoids via their involvement in retinoic acid-mediated gene activation. On the other hand, the proteins mostly retained by HE filters include a number of actin cytoskeleton components which are involved in the canonical pathways of VEGF signalling and integrin signalling (Fig. S4, ESI†).
Coagulation is one of the key functions which is kept under strict control in patients undergoing haemodialysis; for this reason one purpose of this work was to understand which molecular mechanism(s) may be activated upon protein–membrane interaction, with particular regard to the coagulation cascade. Taking advantage of the IPA knowledge base, we were able to confirm that several proteins, retained preferentially by either CTA or HE membranes, match with molecular targets included in the canonical pathway of the coagulation system included in the IPA database (Fig. 4).
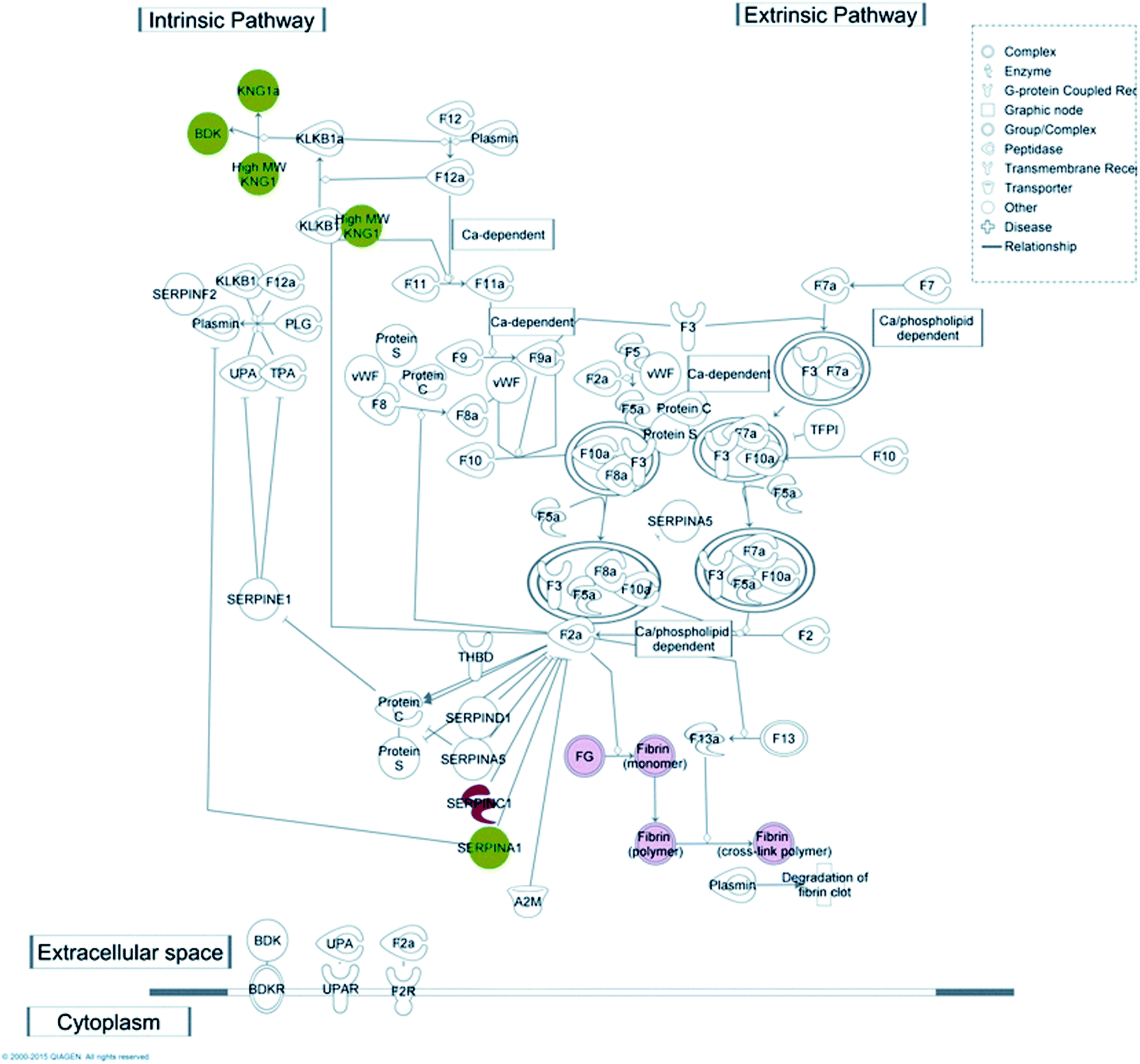 | ||
| Fig. 4 Coagulation system canonical pathway matches with CTA and HE retained molecules. Several proteins from our dataset, either retained from HE (red symbols) or CTA dialysers (green symbols) are involved in the coagulation system pathway, both intrinsic and extrinsic. | ||
On the basis of label-free quantitation experiments and subsequent IPA analysis, a number of proteins were selected for both technical and functional validation of the evidence reported. To this end we performed a targeted validation analysis using selective proteotypical MS/MS transition on a restricted number of proteins. This experiment was run in two phases.
A preliminary shotgun approach through mudPIT chromatography was designed to confirm previous data and to widen the number of proteins identified allowing the inclusion of other targets in the list of peptides for MRM. This analysis, consisting of a total of four gradient runs, was fundamental if we were to know the elution time of each targeted peptide and develop a method where MS and MS/MS transitions might be monitored in different time windows. The acquisition method was used to analyse a maximum of ten transitions in each segment, minimising the number of concurrent MRMs and trying to obtain the highest sensitivity possible, ensuring a higher duty cycle for each peptide. Chromatographic reproducibility was assessed (Fig. S5, ESI†). The transitions selected are listed in Table S2 (ESI†).
In the second phase, MRM analysis was performed by means of mono-dimensional chromatographic runs, where targeted peptides may co-elute with a higher amount of molecules than in the mudPIT step and ion-suppression is likely to occur with less abundant species. Although it was not possible to follow up all the selected transitions or integrate peak areas for all their respective Extracted Ion Chromatograms (EIC), so that we could not provide a quantitative estimate for all the targeted proteins, MRM analysis showed a good agreement with the label-free approach and with previously published shotgun experiments,15,20–24 providing a wider protein coverage and a further tool to quantitate proteins of interest. In Fig. 5 we plotted the natural log of the ratios of averaged peak areas for each protein identified. These values are indicative of the relative amount of each protein differentially retained by the CTA or HE membrane. For fibrinogen chains, serum amyloid proteins, insulin-like growth factor, and inhibin chains we observed a positive log ratio confirming that those proteins are more retained by the HE membrane, as well as for antithrombin and integrin which had not been detected before. A negative value was found for WISP2 (WNT1 inducible signalling pathway protein 2) and RET4 (retinol-binding protein 4), which were more abundant in samples from CTA filters (Fig. 5). These data confirmed the retention on polysulfone-based HE membranes of proteins associated with the coagulation cascade, possibly suggesting an active role by this material in modulating this molecular mechanism.
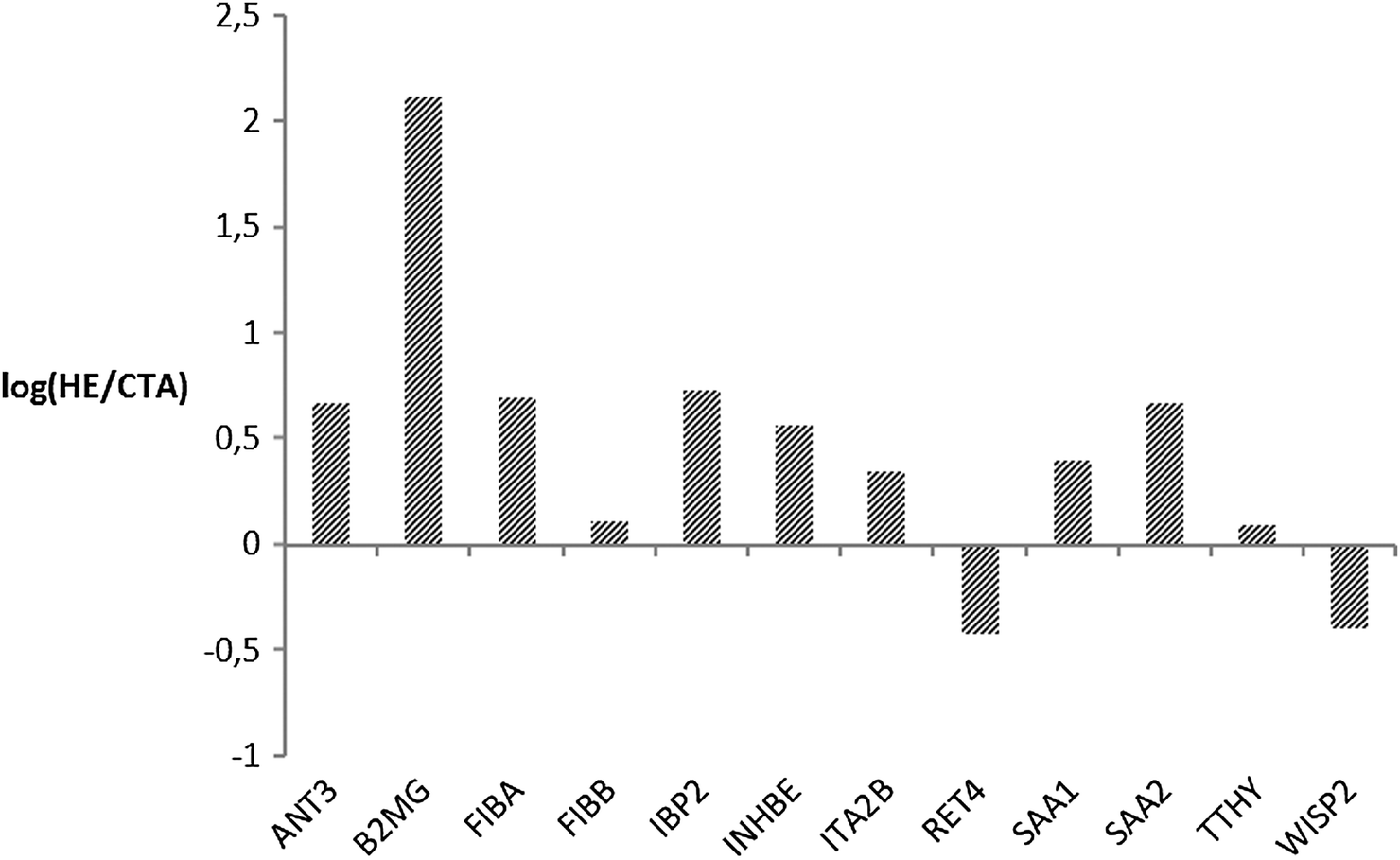 | ||
| Fig. 5 Graphical representation of the fold change of some targeted proteins in the MRM experiment. Bars correspond to the logarithm of the ratio of the averaged peak areas measured in the EIC of monitored transitions. Proteins associated with positive values are more retained on HE membrane, while a negative value indicates that peptides are more retained by the CTA membrane. | ||
The present data are in agreement with and extend our observation described in previous reports.14,15 Interestingly, while most of the proteins that we found adsorbed to CTA and HE membranes by means of two-dimensional electrophoresis analysis have been confirmed (e.g. apolipoprotein family, serum albumin and retinol binding protein 4 bound to CTA, and fibrinogen or ficolin 2 bound to HE), we now further increase the number of identified proteins by using the most potent analysis provided by nUPLC-MSe supported by a targeted MRM analysis (e.g. WNT1-inducible-signalling pathway protein 2 and transthyretin bound to CTA, or mannan-binding lectin serine protease 2 and serum amyloid A-1 protein retained by HE).
In order to assess the coagulation profile during the haemodialysis procedure with both materials, four key routine test parameters were measured: prothrombin time, partial thromboplastin time, international normalized ratio (INR), and fibrinogen.
The coagulation profile of patients undergoing dialysis with the CTA membrane (Fig. 6) did not show any significant difference in the pre- vs. post-session investigation. Moreover, the correlation between fibrinogen and PTT level was not altered by the dialytic session.
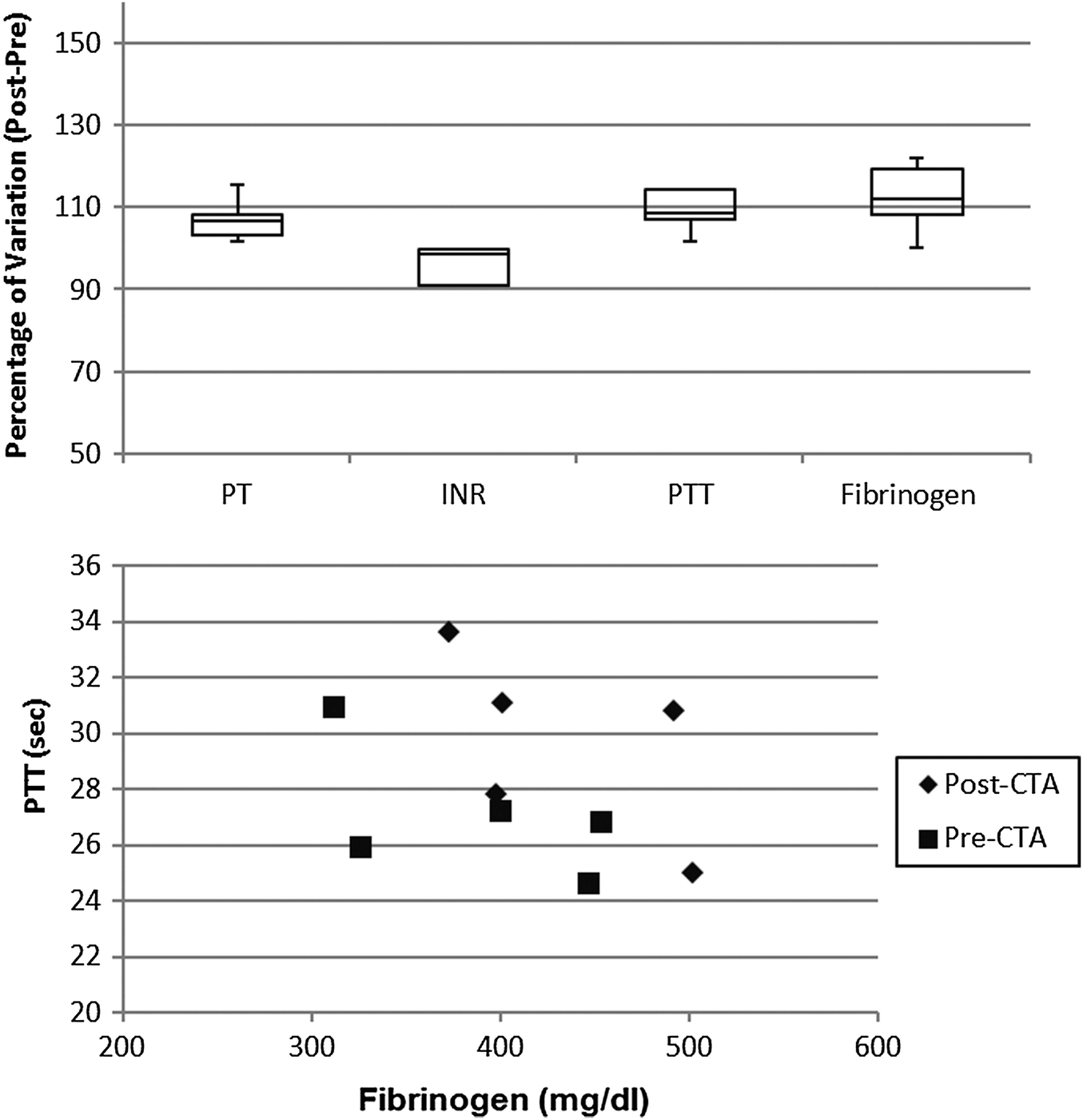 | ||
| Fig. 6 Percentage variation after-before dialysis session of coagulation parameters (upper panel) and correlations between fibrinogen concentration and partial thromboplastin time (lower panel) in patients undergoing haemodialysis with the CTA membrane. | ||
For the polysulfone-based HE membrane there were no major significant differences in the single parameters when statistically analysed but a small difference in fibrinogen concentration as already reported,14 yet the distribution of levels proved to be quite heterogeneous within the HE population. Such a broad distribution was also observable in the PTT (partial thromboplastin time) of patients undergoing haemodialysis with such a filter material (Fig. 7). These two coagulation parameters are strictly linked in the predialysis condition (Fig. 7) and show a strong correlation coefficient on a Pearson test. The value of R is −0.8298, suggestive of a strong negative correlation which is physiologically expected in a condition where a high fibrinogen value is associated with low PTT scores (and vice versa). Such correlation however is lost after the patients undergo the haemodialysis session, reflecting a potential rearrangement of the intrinsic pathway of the coagulation cascade. That pathway is well known to be activated upon blood contact with negatively charged surfaces or with the collagen interface on the intravasal surface. In order to test the intrinsic pathway activation hypothesis, we examined the platelet aggregation capacity from ESRD patients dialysed with the HE membrane (Fig. 8) using a system for the quantitative in vitro determination of platelet function. Two specific tests were assayed, collagen and a Trap-6 based analysis, in order to evaluate the collagen-induced aggregation pathways versus a reference pathway of platelet stimulation via the thrombin receptor. The collagen activation pathway test highlighted a small but significant (p < 0.03) increase in the platelet activation profile after patients had undergone treatment with the HE material. In order to test the specificity of this pathway, we included a TRAP receptor based analysis in the platelet aggregation test. This was not affected by the haemodialysis treatment, all the reported values being within the standard reference intervals, thus showing that patient stabilization had been established.
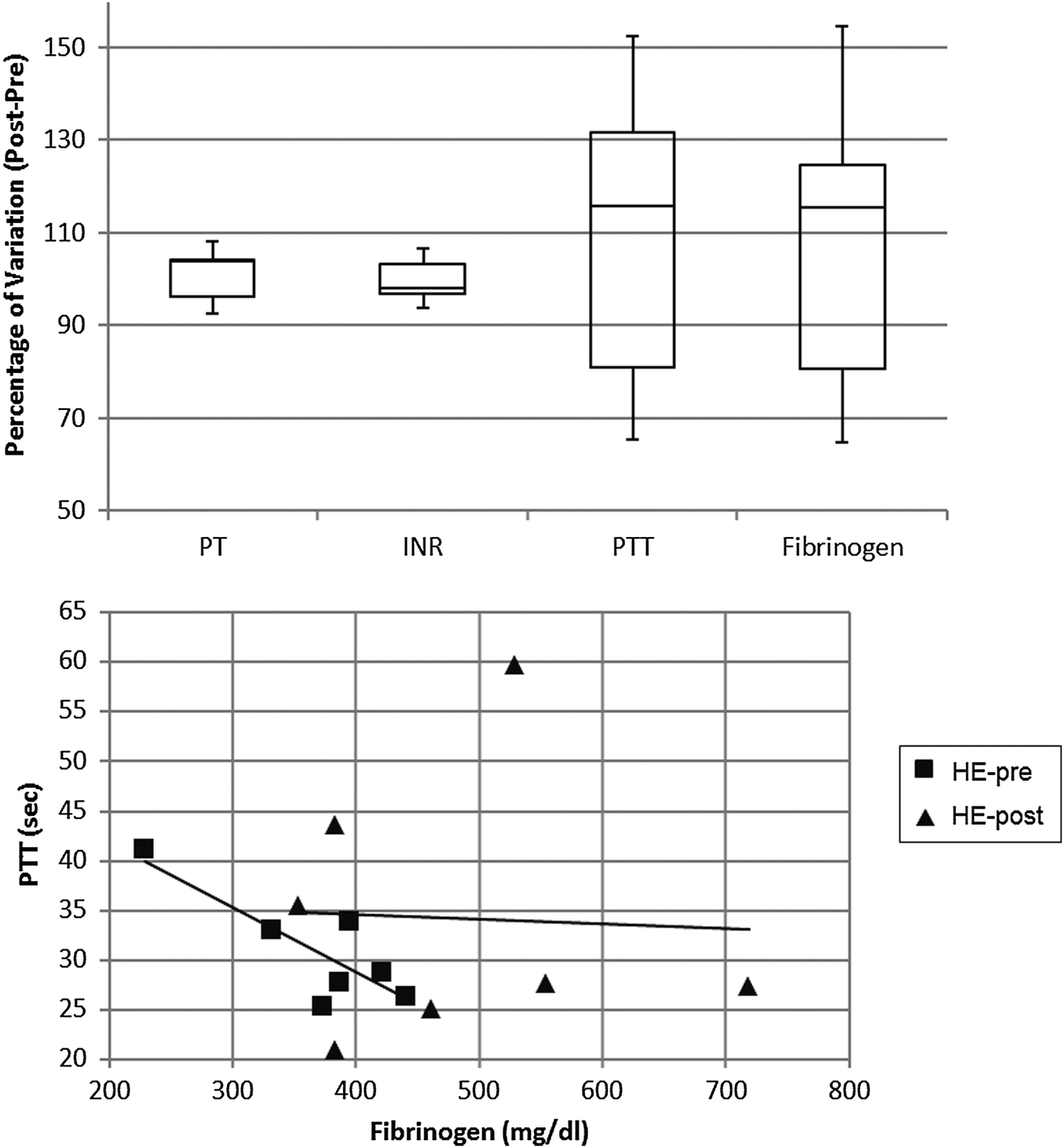 | ||
| Fig. 7 Percentage variation after-before dialysis session of coagulation parameters (upper panel) and correlations between fibrinogen concentration and partial thromboplastin time (lower panel) in patients undergoing haemodialysis with the HE membrane. | ||
 | ||
| Fig. 8 Platelet activation test on a multiplate reader. Samples from three patients were tested before (pre) and after (post) dialysis session for the collagen (COL) and TRAP activation pathway. The COL pathway returns a significant increase under post-dialysis conditions (*p < 0.03). | ||
Discussion
Adsorption of proteins onto artificial surfaces such as HD membranes occurs rapidly within seconds to minutes upon blood-contact. Membrane protein adsorption onto biomaterials is a complex process, which may be affected by a series of factors including the actual blood composition and surface characteristics such as hydrophobicity, roughness, pore structure, charge, and so on.25,26 It has been proposed as a two-step process. The first is characterized by preferential competitive adsorption on the membrane surface of high MW proteins (albumin, fibrinogen, fibronectin, etc.), a phenomenon which has limited capacity and limited selectivity due to the saturation of the surface with albumin and clotting factors. In the second step, adsorption of low and medium MW proteins occurs in the body of the membrane. This latter phenomenon is slower, depends on membrane characteristics such as thickness and structure, is limited by membrane permselectivity, and is dynamic being characterized by continuing enzymatic reactions and substrate replacement.27 With regard to biocompatibility, an important feature of adsorbed proteins is the potential variation of the protein conformation caused by a foreign surface.26The deposition of heterogeneous protein films on foreign surfaces is of critical importance for subsequent events such as the activation of blood cells, plasma proteins, and the enzymatic cascades of the coagulation system and the complement system. Since surface-adsorbed proteins play an important role in the events occurring at the blood/dialyzer membrane interface, the identification of adsorbed proteins is of considerable interest when assessing the in vivo biocompatibility of a biomaterial. Proteomic applications might help in defining the molecular substrates which are involved in the interaction between blood and the membrane material, causing plasma protein adsorption and the following biological reactions during HD procedure.1
Our data support the concept that each dialysis membrane material has many different physiochemical properties which may contribute to interactions with blood components, and hence acquires a unique and characteristic protein layer. The two membrane materials examined here during clinical hemodialysis, CTA and HE, displayed both similarities and significant differences in their protein-adsorption capacity, a difference that seems to be not patient- or protein -specific but rather related to membrane properties. The polysulfone-based HE material is hydrophobic, whereas CTA has a more balanced hydrophilic/hydrophobic profile. Protein adsorption to a hydrophobic surface is stronger and usually irreversible;28 by contrast, the adsorption of proteins onto a hydrophilic surface is weaker, more sensitive to electrostatic interactions and has better reversibility.29,30 Differences between membranes in terms of charge, roughness, and thickness might also have played a role in the differences displayed in their protein adsorption patterns.
Identification of proteins surface-adsorbed onto the dialysis membrane can provide insights into the reactions which may occur during the haemodialysis procedure.
The experimental approach we used in the present study allows us to not only confirm our previous observations about CTA membrane adsorption of soluble proteins and HE membrane attraction of fibrinogen or complement components, which may be involved in haematological diseases or inflammatory disorders, but also widen the protein repertoire detectable and to reinforce the data by MRM measurements. Moreover, by means of a bioinformatics pathway analysis tool we show that CTA retained proteins, including the RTB4 protein, also assessed by targeted analysis, are involved in the pathway related to activation of LXR/RXR, confirming their involvement in mechanisms related to lipid metabolism, molecular transport, and small molecule biochemistry. Furthermore, proteins retained from the HE membrane are involved in the canonical pathways of VEGF signalling and integrin signalling: the first is a very important pathway in the growth and differentiation of vascular as well as lymphatic endothelial cells, while the second, being integrins involved in cell–cell and cell–extracellular matrix (ECM) interactions, includes important interactions responsible for mechanisms such as cell migration and anchorage, cell growth, and cell differentiation.
The retention on polysulfone-based HE membrane of proteins associated with the coagulation cascade suggests an active role by the material in modulating this biological process, in keeping with recent observations of a significant coagulation activation during haemodialysis with PS-based materials.14,15,31–33 The interaction of plasma proteins with artificial surfaces can initiate the activation of intrinsic as well as extrinsic blood coagulation, the kinin, complement, and fibrinolytic systems,34 followed by the adhesion of blood cells such as leukocytes and platelets to physically adsorbed proteins, and their subsequent activation. At the end of the dialysis session, as compared to predialysis, we observed that the HE membrane was associated with the retention of proteins due to the coagulation cascade and platelet activation. An increase in specific coagulation proteins was further highlighted by a highly specific targeted differential SRM analysis on the protein remnants of both membrane materials. The reported presence of fibrinogen fragments is also indicative of proteolytic activity.15 Interestingly, platelet reactivity does not seem to be a generalized process but is restricted to some pathways such as the collagen pathway, which is suggestive of a mechanism associated with the physico–chemical properties of the HE material, especially the negatively charged surface. This evidence is also supported by platelet GpIIb/IIIa expression after haemodialysis with the polysulfone membrane.31 The increase in collagen-induced platelet activation may be explained by blood cell contact that activates platelets, although other reasons cannot be unequivocally ruled out. The platelet thrombin-based activation test was unmodified by haemodialysis, suggestive of a non-generalized activation mechanism.
Because activation of blood coagulation is a necessary component of artificial surface-induced thrombus formation, its inhibition is a key issue in preventing thrombotic occlusion of the extracorporeal circuit. Inhibition of the coagulation cascade within the extracorporeal circuit during haemodialysis requires the judicious use of anticoagulants, possibly tailored to the specific needs or bleeding risk of the individual patient, including age-related factors, treatment with oral anticoagulants for cardiac arrhythmias, and the presence of atherosclerotic vascular disease with the risk of plaque rupture. Our results suggest that the membrane material might also be taken into account in the anticoagulant prescription for haemodialysis, in order to provide each patient with adequate, safe and personalized anticoagulation, although it is clearly too early to say so based on the current results.
Proteomics has progressively emerged as a powerful tool for research in renal replacement therapy.35,36 The results of the present study confirm the successful use of proteomics for assessing the biocompatibility of a biomaterial.
The biocompatibility of a haemodialysis membrane, even though it remains somewhat nebulous in some aspects, has gained a general clinical acceptance over the last few years and represents the most important challenge to be overcome for improving dialysis quality.27 Bioincompatibility is held (at least partially) responsible for complications found in haemodialysis patients including malnutrition, accelerated atherosclerosis, and thrombotic diathesis.37–39 Our data indicate that, in terms of the tendency to activate the coagulation cascade, CTA seems to be more biocompatible than the polysulfone-based HE material. Whether this translates into a favourable outcome requires a longitudinal clinical study, which is currently being planned.
Conclusions
Protein adsorption is a key process bound up with the biocompatibility of materials used in medical applications such as haemodialysis therapy. Interfacial phenomena taking place on or in the material (adsorption and alteration of proteins) are crucial for biocompatibility and function since they can initiate complex cascade processes in the biological system.26 Identification of proteins surface-adsorbed onto the dialysis membrane material can thus provide important insights into reactions occurring during the haemodialysis procedure. We show here that the use of proteomic techniques may give valuable molecular and submolecular information on interfacial structures. Improved knowledge about the impact of biomaterial surface parameters upon interaction with plasma patients may hopefully lead to the development of more biocompatible polymers for the potential benefit of the uraemic patient.Methods
Study design
The present work examined and compared the blood protein adsorption patterns and coagulation profiles of two different membrane materials used for clinical haemodialysis in ESRD patients: CTA, a chemically engineered modified cellulosic membrane (Sureflux 210 E, Nipro Corporation, Osaka, Japan, gamma ray sterilized), and the fully synthetic polymer polysulfone-based HE, a high-flux membrane characterized by nanoscale modulation of the innermost surface structures (FX 80, Fresenius Medical Care, Bad Homburg, Germany, gamma ray sterilized).The protein adsorption pattern on the test membranes was analyzed in eluates which had been obtained from a previous prospective cross-over study.15 The current proteomics analysis was carried out on eluates from 3 male non-diabetic haemodialysis patients (aged 73.6. ± 4 years) routinely treated at the Dialysis Centre of Chieti University Hospital. All patients had been on regular dialysis treatment for at least six months, were on a stable intradialytic anticoagulation regimen (unfractionated heparin), and had a native arterio-venous fistula allowing a blood flow of 300 mL min−1 or more. Exclusion criteria included a history of thrombotic complications or known coagulation disorder, anticoagulation therapy other than during the dialysis session, active inflammation or infection, malignancy, or liver disease.
A clinical observational validation study was likewise carried out in 10 non-diabetic ESRD patients (6 males and 4 females, aged 69.4 ± 16 years) who were being routinely treated at the Dialysis Centre of Chieti University Hospital and of Rome Tor Vergata University Hospital, had been on regular dialysis treatment for at least six months and were on a stable intradialytic anticoagulation regimen. Coagulation parameters (prothrombin time, partial thromboplastin time, fibrinogen) were measured before and after the haemodialysis session using a nephelometric method; the INR was calculated as the ratio of a patient's prothrombin time to that of a normal (control) sample, raised to the power of the ISI value for the analytical system being used. Platelet aggregation testing was performed by a multiplate reader (Roche) using the TRAPtest, platelet stimulation via the thrombin receptor (TRAP-6) cascade, and the COLtest, collagen-induced aggregation. GpIIb/IIIa antagonist reagent was applied as a negative quality control.
The work was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans, and informed written consent was obtained from each subject prior to participation.
Proteomic analysis
Qualitative identification of proteins was obtained using the embedded ion accounting algorithm of the software PLGS, sought in the human database UniProt KB/Swiss-Prot Protein Knowledgebase (UniProt KB/Swiss-Prot Protein Knowledgebase release 2014_03 of 19-March-14 containing 542782 sequence entries, taxonomical restrictions: Homo Sapiens, 20263 sequence entries) to which data from S. cerevisiae enolase were appended (UniProtKB/Swiss-Prot AC: P00924). The search parameters included: automatic tolerance for precursor ions and for product ions, a minimum of 3 fragment ions matched per peptide, a minimum of 7 fragment ions matched per protein, a minimum of 2 peptides matched per protein, 1 missed cleavage, carbamidomethylation of cysteine as the fixed modification and oxidation of methionine as the variable modification.16,17
Statistical analyses were performed following the hypothesis that each dialyser (CTA or HE) is an independent variable. Expression analysis was performed considering the 4 technical replicates available for each experimental condition (e.g. 1 exp. condition × 3 biological replicates × 4 technical replicates). For differential analysis, the identified proteins were normalized against P00924 entry (enolase S. cerevisiae) in the quantitative analysis.16,17,40
The datasets, including the gene identifier and relative expression values (fold change HE/CTA) of each protein, were uploaded for core analysis application which delivers a rapid assessment of the signalling and metabolic pathways, molecular networks, and biological processes that are most significantly perturbed in the dataset of interest.
Within the core analysis, we performed a canonical pathway analysis enabling us to identify the most relevant metabolic and signaling pathways among those present in the dataset.41,42
000 and a maximum acquisition time of 50 ms in the range between 330 and 1400 m/z. Source: capillary voltage 4500 V, end plate offset −500 V, Nebulizer 8.7 psi, dry gas 5.0 L min−1, dry T 220 °C. AutoMSn (DDA) acquisitions were executed selecting 10 precursors for every survey scan during the whole run time, leaving the active exclusion option enabled for 30 seconds after 1 spectrum. MS/MS fragmentation amplitude was set to 0.80 V. Raw data were processed by compass data analysis 1.3 (Bruker Daltonics) while protein identification and 2D fraction data combining were performed by compass proteinscape 2.1 (Bruker Daltonics).
The SRM experiments were performed using the same instruments in a mono-dimensional HPLC step. The SCX precolumn was removed and gradient elution was carried out using the chromatographic method previously employed. In this case the three samples for each condition were run individually in two replicates, injecting 3 μg (6 μL) of protein tryptic digest. The mass spectrometer source and analyser settings were the same as those described previously. The MS method was adjusted to analyse all 56 transitions in a 220 min run time, dividing it into 11 segments. In the first and last segments the instrument was acquired in MS mode, while in the nine segments between 20 and 180 min, the analyser parameters were set to monitor not more than 10 transitions (5 peptides) together in MS3. Data were processed by compass data analysis 1.3 (Bruker Daltonics) integrating the peak area of the Extracted Ion Chromatogram (EIC) of each transition after smoothing.
Acknowledgements
The study was partially supported by unconditional grants from Nipro Corporation, Japan.References
- J. Mares, V. Thongboonkerd, Z. Tuma, J. Moravec and M. Matejovic, Kidney Int., 2009, 76, 404–413 CrossRef CAS PubMed.
- J. M. Anderson, T. L. Bonfield and N. P. Ziats, Int. J. Artif. Organs, 1990, 13, 375–382 CAS.
- A. Sagripanti, A. Cupisti, U. Baicchi, M. Ferdeghini, E. Morelli and G. Barsotti, Nephron, 1993, 63, 273–278 CrossRef CAS PubMed.
- R. Johnson, Nephrol., Dial., Transplant., 1994, 9, 36–45 Search PubMed.
- R. D. Franck, J. Weber, H. Dresbach, H. Thelen, C. Weiss and J. Floege, Kidney Int., 2001, 60, 1972–1981 CrossRef PubMed.
- W. H. Horl, J. Am. Soc. Nephrol., 2002, 13(suppl. 1), S62–S71 CAS.
- L. Lucchi, G. Ligabue, M. Marietta, A. Delnevo, M. Malagoli, S. Perrone, L. Stipo, F. Grandi and A. Albertazzi, Artif. Organs, 2006, 30, 106–110 CrossRef CAS PubMed.
- R. Ouseph and R. A. Ward, Semin. Dial., 2000, 13, 181–187 CrossRef CAS.
- J. Mulvihill, T. Crost, J. L. Renaux and J. P. Cazenave, Nephrol., Dial., Transplant., 1997, 12, 1968–1973 CrossRef CAS PubMed.
- S. Sagedal, A. Hartmann, K. Sundstrom, S. Bjørnsen and F. Brosstad, Nephrol., Dial., Transplant., 2001, 16, 987–993 CrossRef CAS PubMed.
- M. Bonomini, B. Pavone, V. Sirolli, F. Del Buono, M. Di Cesare, P. Del Boccio, L. Amoroso, C. Di Ilio, P. Sacchetta, G. Federici and A. Urbani, J. Proteome Res., 2006, 5, 2666–2674 CrossRef CAS PubMed.
- I. Aoike, Nephrol., Dial., Transplant., 2007, 22(suppl. 5), 13–19 CrossRef PubMed.
- B. Pavone, V. Sirolli, S. Bucci, F. Libardi, P. Felaco, L. Amoroso, P. Sacchetta, A. Urbani and M. Bonomini, J. Blood Transfus., 2010, 8(suppl. 3), S113–S119 Search PubMed.
- A. Urbani, S. Lupisella, V. Sirolli, S. Bucci, L. Amoroso, B. Pavone, L. Pieroni, P. Sacchetta and M. Bonomini, Mol. BioSyst., 2012, 8, 1029–1039 RSC.
- A. Urbani, V. Sirolli, S. Lupisella, S. Levi-Mortera, B. Pavone, L. Pieroni, L. Amoroso, R. Di Vito, S. Bucci, S. Bernardini, P. Sacchetta and M. Bonomini, J. Blood Transfus., 2012, 10(suppl. 2), S97–108 Search PubMed.
- L. Pieroni, F. Finamore, M. Ronci, D. Mattoscio, V. Marzano, S. Levi-Mortera, S. Quattrucci, G. Federici, M. Romano and A. Urbani, Mol. BioSyst., 2011, 7, 630–639 RSC.
- T. Alberio, H. Bondi, F. Colombo, I. Alloggio, L. Pieroni, A. Urbani and M. Fasano, Mol. BioSyst., 2014, 10(suppl. 6), 1332–1344 RSC.
- H. Mi, A. Muruganujan and P. D. Thomas, Nucleic Acids Res., 2012, 41, 377–386 CrossRef PubMed.
- H. Mi, A. Muruganujan, J. T. Casagrande and P. D. Thomas, Nat. Protoc., 2013, 8, 1551–1566 CrossRef PubMed.
- I. Pešić, G. A. Müller, C. Baumann, G. H. Dihazi, M. J. Koziolek, M. Eltoweissy, C. Bramlage, A. R. Asif and H. Dihazi, Biochim. Biophys. Acta, 2013, 1834, 754–762 CrossRef PubMed.
- S. W. Holman, P. F. Sims and C. E. Eyers, The use of selected reaction monitoring in quantitative proteomics, Bioanalysis, 2012, 4(14), 1763–1786 CrossRef CAS PubMed.
- P. Picotti and R. Aebersold, Selected reaction monitoring-based proteomics: workflows, potential, pitfalls, and future directions, Nat. Methods, 2012, 9(6), 555–566 CrossRef CAS PubMed.
- B. Domon, Considerations on selected reaction monitoring experiments: implications for the selectivity and accuracy of measurements, Proteomics: Clin. Appl., 2012, 6(11–12), 609–614 CrossRef CAS PubMed.
- A. Maiolica, M. A. Junger, I. Ezkundica and R. Aebersold, Targeted proteome investigation via selected reaction monitoring mass spectrometry, J. Proteomics, 2012, 75(12), 3495–3513 CrossRef CAS PubMed.
- S. Sun, Y. Yue, X. Huang and D. Meng, J. Membr. Sci., 2003, 222, 3–18 CrossRef CAS.
- C. Wermer and H.-J. Jacobash, Int. J. Artif. Organs, 1999, 22, 160–176 Search PubMed. ,
- J. Chanard, S. Lavaud, C. Randoux and P. Rieu, Nephrol., Dial., Transplant., 2003, 18, 252–257 CrossRef CAS PubMed.
- J. D. Andrade, V. L. Hlady and R. A. Van Wagenen, Pure Appl. Chem., 1984, 56, 1345–1350 CrossRef CAS.
- G. A. Bomzin and I. F. Miller, J. Colloid Interface Sci., 1982, 86, 539–541 CrossRef.
- U. Jonsson, B. Ivarson, I. Lundstrom and L. Bergheim, J. Colloid Interface Sci., 1982, 90, 148–155 CrossRef.
- T. Kuragano, T. Kuno, I. Takahashi, C. Yamamoto, Y. Nagura, S. Takahashi and K. Kanmatsuse, Blood Purif., 2003, 21, 176–182 CrossRef CAS PubMed.
- M. Salame, S. Uerheye, R. More and S. B. King III, Int. J. Cardiol., 1999, 69, 231–236 CrossRef CAS.
- F. Olafiranye, W. Kyaw and O. Olafiranye, Case Rep. Med., 2011, 2011, 134295 Search PubMed.
- R. G. Mason, H. Y. K. Chuang, S. F. Mohammad and H. I. Saba, in The Thromboembolic Disorders, ed. J. Van der Loo, C. R. M. Prentice and F. K. Beller, Schattauer Verlag, New York, 1983, p. 533 Search PubMed.
- M. Bonomini, V. Sirolli, F. Magni and A. Urbani, J. Nephrol., 2012, 25, 865–871 CrossRef CAS PubMed.
- V. Thongboonkerd, J. Proteomics, 2010, 73, 521–526 CrossRef CAS PubMed.
- G. A. Kaysen, J. Am. Soc. Nephrol., 2001, 12, 1549–1557 CAS.
- W. H. Horl, J. Am. Soc. Nephrol., 2002, 13(suppl. 2), 562–571 Search PubMed.
- F. Locatelli, B. Canaud and K. U. Eckardt, Nephrol., Dial., Transplant., 2003, 18, 1272–1280 CrossRef CAS PubMed.
- V. Marzano, S. Santini, C. Rossi, M. Zucchelli, A. D'Alessandro, C. Marchetti, M. Mingardi, V. Stagni, D. Barilà and A. Urbani, J. Proteomics, 2012, 75(suppl.15), 4632–4646 CrossRef CAS PubMed.
- D. Ciavardelli, M. D'Orazio, L. Pieroni, A. Consalvo, C. Rossi, P. Sacchetta, C. Di Ilio, A. Battistoni and A. Urbani, Mol. BioSyst., 2013, 9(suppl. 6), 1117–1126 RSC.
- S. Marinelli, F. Nazio, A. Tinari, L. Ciarlo, M. D'Amelio, L. Pieroni, V. Vacca, A. Urbani, F. Cecconi, W. Malorni and F. Pavone, Pain, 2014, 155(suppl. 1), 93–107 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00058k |
| ‡ Both co-authors acted as senior investigators. |
| § Nephrology and Dialysis Institute, SS Annunziata Hospital, Via dei Vestini, 66013, Chieti, Italy. E-mail: m.bonomini@nephro.unich.it; Fax: +39-0871-574736. |
| This journal is © The Royal Society of Chemistry 2015 |