
Insights into dioxygen binding on metal centers: an ab initio multireference electronic structure analysis†

Abstract
Why does binding of dioxygen (O2) to metal centers, the initial step of O2 storage, transportation, and activation, almost inevitably induce metal-to-O2 single-electron transfer and generate superoxo (O2−˙) species, instead of genuine O02 adducts? To address this question, this study describes highly correlated wavefunction-based ab initio calculations using CASSCF/NEVPT2 (CASSCF = complete active space self-consistent field, and NEVPT2 = N-electron valence state second-order perturbation theory) approaches to explore the electronic-structure evolution of O2 association on Fe(II)(BDPP) (H2BDPP = 2,6-bis((2-(S)-diphenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine) and Co(II)(BDPP) to produce S = 3 Fe(III)(BDPP)(O2−˙) (1) and 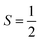 Co(III)(BDPP)(O2−˙) (2). CASSCF/NEVPT2 calculations suggest that the processes furnishing 1 and 2 feature an avoided crossing resulting from interactions of two diabatic curves, of which one is characterized as Co(II) and Fe(II) centers interacting with a triplet O2 ligand and the other as Co(III) and Fe(III) centers bound to a superoxo ligand. In both cases, the avoided crossing induces a one-electron transfer from the divalent metal center to the incoming O2 ligand and leads to formation of trivalent metal–O2−˙ complexes. To facilitate the interpretation of complicated multireference wavefunctions, we formulated two-fragment spin eigenfunctions utilizing Clebsch–Gordan coefficients (CGCs) to rationalize computed spin populations on the metal centers and the O2 ligand and compared these results with usual valence bonding (VB) analyses. It turns out that both methods give the same results and are complementary to each other. Finally, the limitation of DFT approaches in describing complex electronic structures involving metal–ligand magnetic couplings is delineated.
Co(III)(BDPP)(O2−˙) (2). CASSCF/NEVPT2 calculations suggest that the processes furnishing 1 and 2 feature an avoided crossing resulting from interactions of two diabatic curves, of which one is characterized as Co(II) and Fe(II) centers interacting with a triplet O2 ligand and the other as Co(III) and Fe(III) centers bound to a superoxo ligand. In both cases, the avoided crossing induces a one-electron transfer from the divalent metal center to the incoming O2 ligand and leads to formation of trivalent metal–O2−˙ complexes. To facilitate the interpretation of complicated multireference wavefunctions, we formulated two-fragment spin eigenfunctions utilizing Clebsch–Gordan coefficients (CGCs) to rationalize computed spin populations on the metal centers and the O2 ligand and compared these results with usual valence bonding (VB) analyses. It turns out that both methods give the same results and are complementary to each other. Finally, the limitation of DFT approaches in describing complex electronic structures involving metal–ligand magnetic couplings is delineated.

- This article is part of the themed collection: Quantum Bio-Inorganic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

