
Deciphering the origin of million-fold reactivity observed for the open core diiron [HO–FeIII–O–FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation†
O]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation†

Abstract
High-valent metal–oxo species have been characterised as key intermediates in both heme and non-heme enzymes that are found to perform efficient aliphatic hydroxylation, epoxidation, halogenation, and dehydrogenation reactions. Several biomimetic model complexes have been synthesised over the years to mimic both the structure and function of metalloenzymes. The diamond-core [Fe2(μ-O)2] is one of the celebrated models in this context as this has been proposed as the catalytically active species in soluble methane monooxygenase enzymes (sMMO), which perform the challenging chemical conversion of methane to methanol at ease. In this context, a report of open core [HO(L)FeIII–O–FeIV(O)(L)]2+ (1) gains attention as this activates C–H bonds a million-fold faster compared to the diamond-core structure and has the dual catalytic ability to perform hydroxylation as well as desaturation with organic substrates. In this study, we have employed density functional methods to probe the origin of the very high reactivity observed for this complex and also to shed light on how this complex performs efficient hydroxylation and desaturation of alkanes. By modelling fifteen possible spin-states for 1 that could potentially participate in the reaction mechanism, our calculations reveal a doublet ground state for 1 arising from antiferromagnetic coupling between the quartet FeIV centre and the sextet FeIII centre, which regulates the reactivity of this species. The unusual stabilisation of the high-spin ground state for FeIVO is due to the strong overlap of 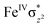 with the
with the 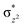 orbital, reducing the antibonding interactions via spin-cooperation. The electronic structure features computed for 1 are consistent with experiments offering confidence in the methodology chosen. Further, we have probed various mechanistic pathways for the C–H bond activation as well as –OH rebound/desaturation of alkanes. An extremely small barrier height computed for the first hydrogen atom abstraction by the terminal FeIVO unit was found to be responsible for the million-fold activation observed in the experiments. The barrier height computed for –OH rebound by the FeIII–OH unit is also smaller suggesting a facile hydroxylation of organic substrates by 1. A strong spin-cooperation between the two iron centres also reduces the barrier for second hydrogen atom abstraction, thus making the desaturation pathway competitive. Both the spin-state as well as spin-coupling between the two metal centres play a crucial role in dictating the reactivity for species 1. By exploring various mechanistic pathways, our study unveils the fact that the bridged μ-oxo group is a poor electrophile for both C–H activation as well for –OH rebound. As more and more evidence is gathered in recent years for the open core geometry of sMMO enzymes, the idea of enhancing the reactivity via an open-core motif has far-reaching consequences.
orbital, reducing the antibonding interactions via spin-cooperation. The electronic structure features computed for 1 are consistent with experiments offering confidence in the methodology chosen. Further, we have probed various mechanistic pathways for the C–H bond activation as well as –OH rebound/desaturation of alkanes. An extremely small barrier height computed for the first hydrogen atom abstraction by the terminal FeIVO unit was found to be responsible for the million-fold activation observed in the experiments. The barrier height computed for –OH rebound by the FeIII–OH unit is also smaller suggesting a facile hydroxylation of organic substrates by 1. A strong spin-cooperation between the two iron centres also reduces the barrier for second hydrogen atom abstraction, thus making the desaturation pathway competitive. Both the spin-state as well as spin-coupling between the two metal centres play a crucial role in dictating the reactivity for species 1. By exploring various mechanistic pathways, our study unveils the fact that the bridged μ-oxo group is a poor electrophile for both C–H activation as well for –OH rebound. As more and more evidence is gathered in recent years for the open core geometry of sMMO enzymes, the idea of enhancing the reactivity via an open-core motif has far-reaching consequences.
![Graphical abstract: Deciphering the origin of million-fold reactivity observed for the open core diiron [HO–FeIII–O–FeIV [[double bond, length as m-dash]] O]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation](/ko-kr/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=D0SC02624G&imageInfo.ImageIdentifier.Year=2020)
- This article is part of the themed collection: Celebrating 10 years of Chemical Science
 Please wait while we load your content...
Please wait while we load your content...
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation
O]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation

