
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinacridone dyes: versatile molecules and materials for photo- and photoelectrochemical processes
Elena
Rossin†
 a,
Yunshuo
Yang†
a,
Martina
Chirico
a,
Greta
Rossi
a,
Pierluca
Galloni
*b and
Andrea
Sartorel
*a
a,
Yunshuo
Yang†
a,
Martina
Chirico
a,
Greta
Rossi
a,
Pierluca
Galloni
*b and
Andrea
Sartorel
*a
aDept. Chemical Sciences, University of Padova, via Marzolo 1, 35131, Padova, Italy. E-mail: andrea.sartorel@unipd.it
bDepartment of Chemical Science and Technologies, University of Rome “Tor Vergata” via della Ricerca Scientifica snc, 00133 Rome, Italy. E-mail: galloni@scienze.uniroma2.it
First published on 2nd July 2024
Abstract
The renaissance of photochemistry and the explosion of photo- and photoelectro-catalysis open new opportunities in organic photocatalyst design and applications towards solar fuels and sustainable organic reactivity. In this perspective, we discuss the relevant case of quinacridone (QA) dyes: these have long been known to the scientific community, but their application in photocatalysis is recent and still explored in a limited way. This is somehow surprising given that QA is a cheap and readily available organic pigment, and in front of the appealing properties of QA derivatives, including intense absorption in the visible region, balanced redox properties making them suitable for both oxidative and reductive photochemistry, and versatility to several operative conditions. We will discuss recent examples of photo- and photoelectrochemical processes taking advantage of QA dyes, from solution photocatalysis to photoactive materials and devices (nanoparticles, covalent organic frameworks, photoelectrodes); the target applications include water splitting, carbon dioxide reduction, and organic transformations. We aim to show the potential of organic photocatalyst design and implementation, and to inspire the readers with new opportunities in this field.
 From left to right: Elena Rossin, Yunshuo Yang, Martina Chirico and Greta Rossi | E. R. is a third year PhD student in Molecular Sciences at the University of Padova working on photosynthetic systems based on proton-coupled electron transfer (supervisor A. S.). Y. Y. is a third year PhD student in Materials Science and Technology at the University of Padova working on hybrid organic/inorganic materials for photocatalysis (supervisor A. S.). M. C. is a first year PhD student in Materials Science and Technology at the University of Padova with a project on reductive electrophotocatalysis (supervisor A. S.). G. R. completed her master course in Chemistry at the University of Padova with a thesis on oxidative photocatalysis in water (supervisor A. S.). |
 Andrea Sartorel (left) and Pierluca Galloni (right) | Pierluca Galloni (on the right in the picture) is Associate Professor at the Department of Chemical Science and Technologies, University of Rome Tor Vergata. His research activity is dedicated to the synthesis and study of the photo and electrochemical properties of organic molecules to be used as dyes in photoelectrochemical and photocatalytic applications and in studying their properties in solution and in catalytic systems using DFT calculations. Andrea Sartorel (on the left in the picture) is professor at the Department of Chemical Sciences, University of Padova. His research interests are focused on photosynthetic processes for small molecule activation and for sustainable organic synthesis. |
Introduction
In 1912, ahead of his time, the day-dreamer Italian photochemist Giacomo Ciamician envisioned solar light as an inexhaustible and clean source of energy to fuel the world, in contrast to the exploitation of fossil fuels.1 Nowadays, considering the energy crisis and the climate change, this concept is more current than ever.This led to the renaissance of photochemistry applications and to an explosion of reports dealing with photocatalysis starting from the beginning of XXI Century.2–8 Photocatalysis is often associated to redox chemistry for the production of solar fuels, with relevant examples dealing with water splitting9,10 and carbon dioxide reduction,11–13 and for the development of sustainable organic synthesis.14–16 The progress in new photocatalyst families design and the expansion of novel photochemical processes are therefore high-target objectives for the scientific community.
Among the necessary features of future investigations, we identify three important aspects.
First, photocatalysts should be affordable and cheap, avoiding the use of noble, rare, or expensive elements. Second, the photocatalyst should be modular through a chemical design, to afford the required photophysical and redox properties.4,17
Third, the development of photocatalytic materials and devices should be considered.18 As possible technologies for water splitting, Harry B. Gray identified photovoltaic modules coupled to electrolysers, photoelectrochemical cells or mixed photocatalytic colloids,19 with this latter being the less developed but more scalable solution. Indeed, this include the recently assessed wireless, particulate materials based on aluminum-doped strontium titanate in a 100 m2 prototype composed of an array of photoreactors, enabling hydrogen production for one year.20 A record solar-to-hydrogen value of 9.2% has been achieved using a particulate photocatalysts based on indium gallium nitride.21 Other examples of photocatalytic particulate materials include dye-sensitised nanoparticles22 and covalent–organic frameworks, with some cases discussed in the following paragraphs of this perspective.
In order to respond to the requirements above, the use of organic photocatalysts is a promising road to follow.17 Organic photocatalysts can be either commercially available or synthesisable in large scale with few steps; many of them have low or null toxicity and are exploited in food or cosmetic industry. The molecular structure allows to reach tuneable chemical, photophysical and redox properties, upon suitable design and/or functionalisation of the parent scaffold.4 Finally, they can be implemented in heterogeneous systems exploiting chemical bonds or physisorption to surfaces.23,24
In this perspective, we will discuss these aspects dealing with quinacridone (QA) dyes as a selected example (Scheme 1). Quinacridone (pigment violet 19) belongs to industrially relevant pigments, it is non-toxic, commercially available and cheap, and is exploited in inks and paints.25 Its pentacyclic aromatic structure provides an extended absorption in the visible region of the electromagnetic spectrum. The N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups induce the formation of an extended hydrogen bonding network, providing features as a semiconducting pigment.26 Among its applications, it was considered in field-effect transistors27 and in light emitting diodes,28 while more recently it was the subject of several investigations towards photocatalysis.
O groups induce the formation of an extended hydrogen bonding network, providing features as a semiconducting pigment.26 Among its applications, it was considered in field-effect transistors27 and in light emitting diodes,28 while more recently it was the subject of several investigations towards photocatalysis.
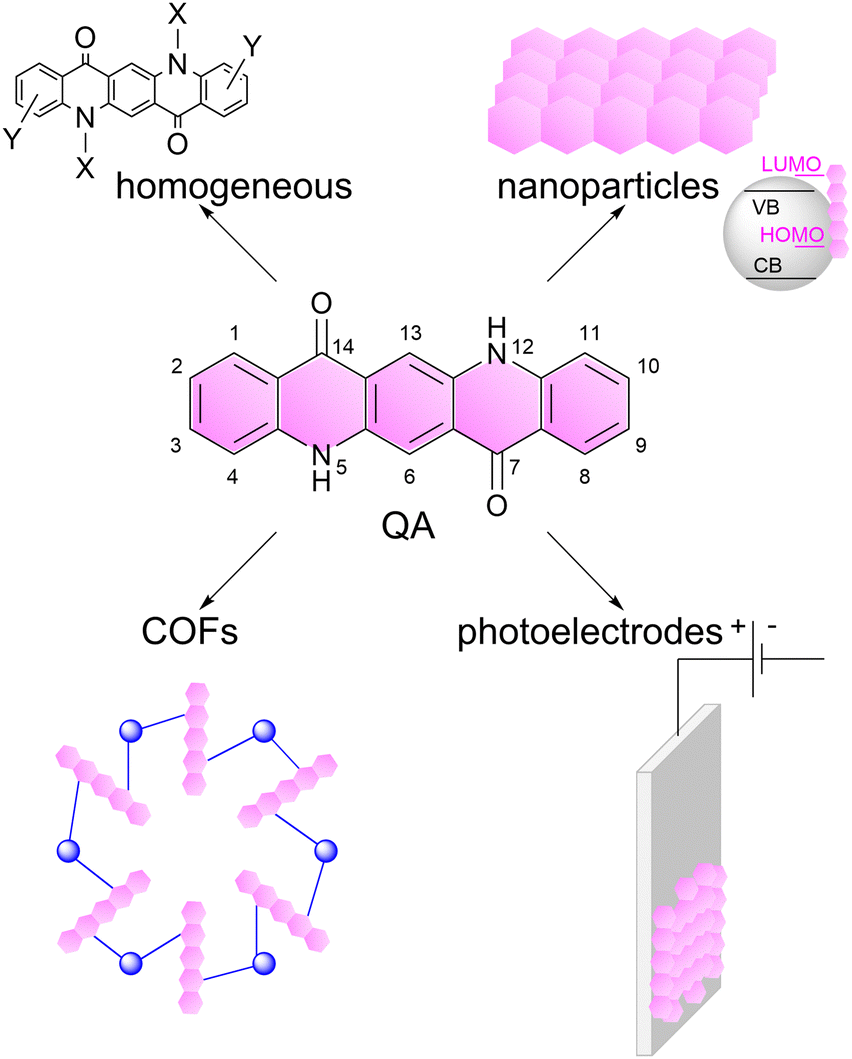 | ||
| Scheme 1 Representation of the linear trans-isomer of quinacridone (QA), and of its versatility for application in photo- and photo-electrocatalysis, which is the subject of this perspective article. The numeration of the atoms in QA scaffold is also reported.25 | ||
Herein, we will focus on photo- and photoelectrochemical processes based on QA derivatives: we will discuss the versatility of this molecule towards several photocatalytic processes (including water splitting, carbon dioxide reduction, organic functionalisation) and concerning its application in several operative conditions including homogeneous systems, nanoparticles, covalent organic frameworks and photoelectrodes, Scheme 1. Our aim is to show the potential of organic photocatalyst design and implementation, inspiring the readers with new opportunities in this field.
Photophysical and redox properties of QA derivatives
Photophysical properties
The absorption properties of QA depend on the degree of aggregation of the molecules through intramolecular hydrogen bonding; it appears yellowish in diluted solutions, while it is intense red in aggregated forms and in the solid state.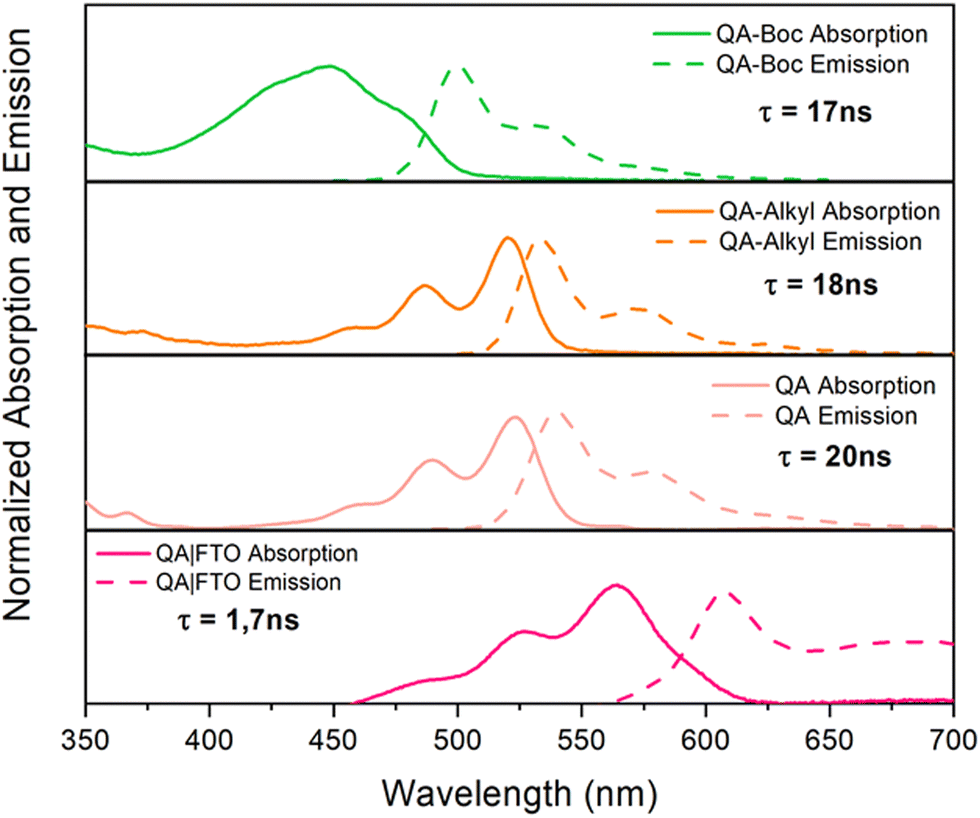 | ||
| Fig. 1 Normalised absorption and emission properties of QA derivatives. QA-Boc, QA-alkyl and QA spectra are reported in DMSO solution; spectra of QA films prepared by vacuum sublimation over FTO are registered under air atmosphere. For the emission spectra, the major lifetime component is also indicated. | ||
Redox properties
The QA scaffold allows the occurrence of both oxidative and reductive redox processes. Due to its poor solubility, electrochemical characterisation of parent QA is typically conducted in heterogeneous conditions,31,39 and in the presence of protic medium the redox processes are proton-coupled involving the CO and N–H moieties (vide infra). N-functionalised QA are instead amenable of solution analysis.32,35 For instance, in DCM solution N,N′-dioctyl quinacridone (DOQA) shows reversible one-electron anodic waves at +0.68 and +1.15 V, and two reversible one-electron cathodic processes at −1.82 and −2.18 V (all potentials reported vs. Fc+/Fc), as shown in the differential pulse voltammograms in Fig. 2.32,35 Notably, although the reversibility of the processes may depend on the solvent, the bis-oxidised and bis-reduced species are characterised by fully conjugated systems with an even number of electrons (Scheme 2). For QA-Boc, the waves are shifted to more positive potentials by 300/400 mV. More importantly, the redox properties of QA derivatives are tuneable and balanced,40 allowing their application in both oxidative and reductive photocatalytic processes, as it will be presented in the next sections.
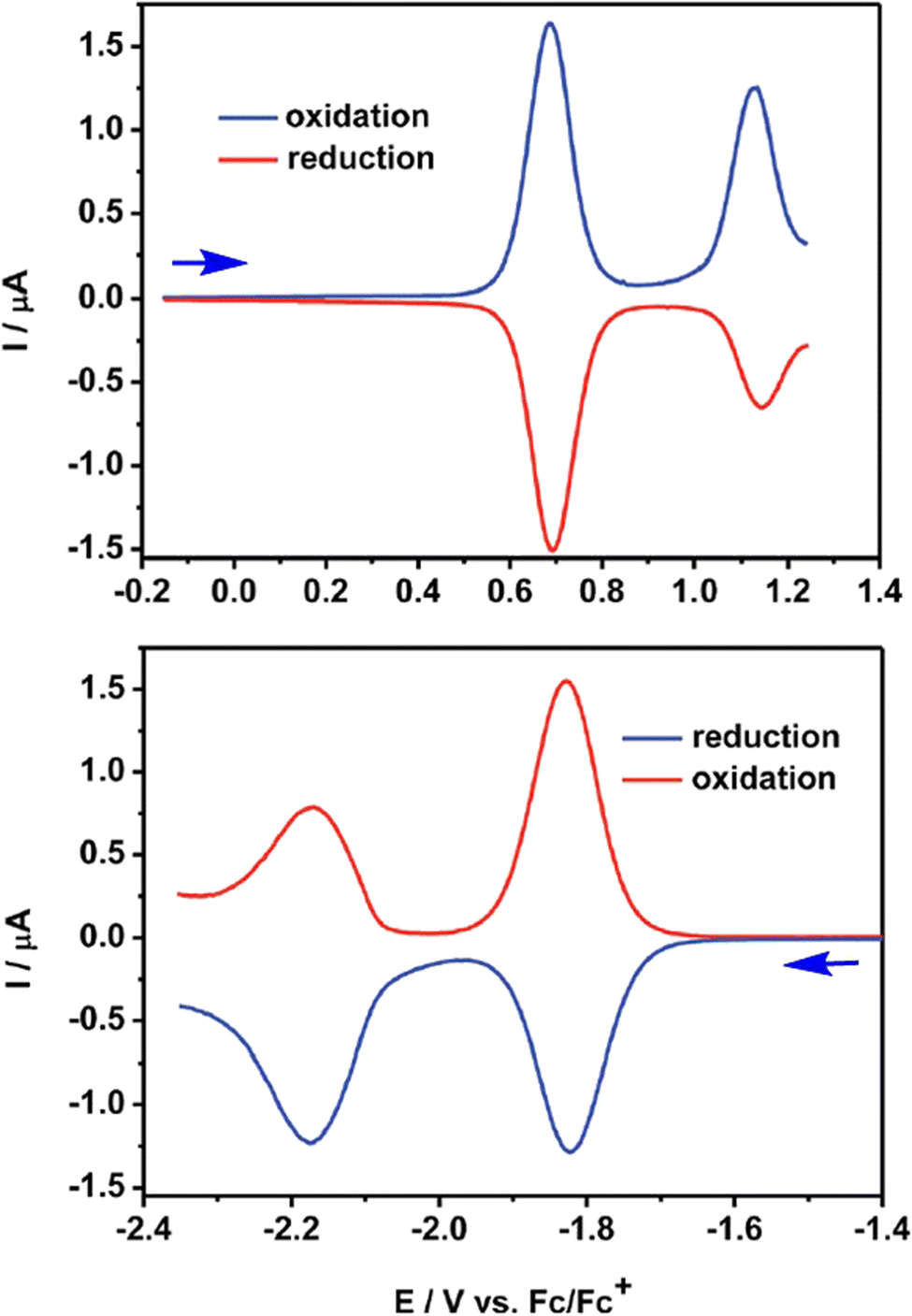 | ||
| Fig. 2 Differential pulse voltammograms registered for N,N′-dioctyl QA (DOQA) in DCM. Reproduced from ref. 32 with permission from the Royal Society of Chemistry, copyright 2017. | ||
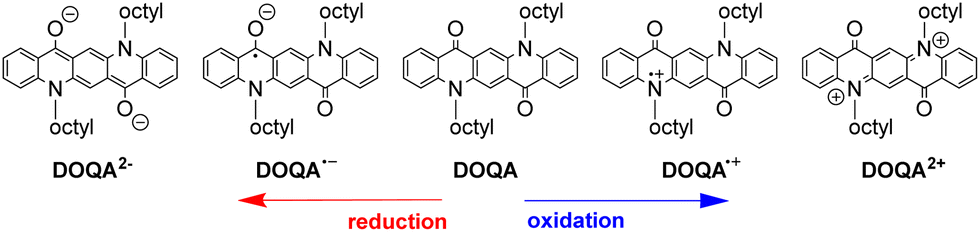 | ||
| Scheme 2 Representation of oxidised and reduced forms of DOQA. | ||
Application of QA in photo- and photoelectrocatalysis
Homogeneous photocatalysis
Functionalized QAs (on the ring or alkylating the N) show higher solubility in common organic solvents, such as DMF or chlorinated ones. The solubility is even higher in fluorinated alcohols, in particular hexafluoroisopropanol (HFIP). This feature allows to use QA derivatives as photocatalysts in homogeneous conditions.Moreover, considering the spectroscopic and electrochemical properties of QA derivatives, these compounds can act as photocatalysts both in reducing and oxidation processes. In fact, the potentials of the excited state, calculated combining the absorption spectra and the redox potential of the ground state,41,42 show suitable values in order to promote photoinduced oxidative or reductive quenching pathways (Scheme 3).
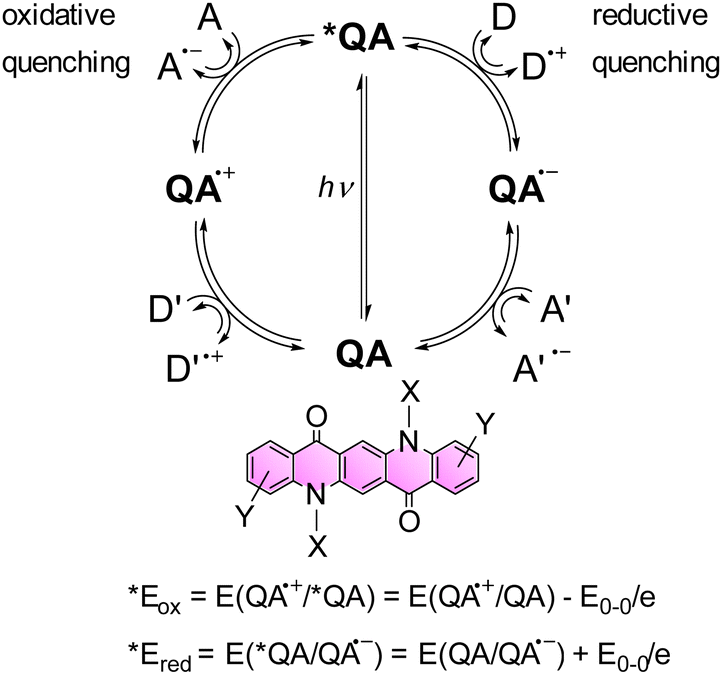 | ||
| Scheme 3 Representation of possible reaction pathways of the *QA excited state through oxidative and reductive quenching, leading to the generation of oxidised QA˙+ and reduced QA˙−, respectively. A and A′ represent electron acceptors, while D and D′ represent electron donors. The two equations enable the estimation of *Eox and *Ered of the *QA excited state.41,42 | ||
N,N′-disubstituted QAs were probed as efficient photocatalysts in polymerization of methyl (MMA) or butyl methacrylate (BMA). In this system QA is functionalized at the nitrogen sites with Boc or a long alkyl chain (Fig. 3). The resulting catalysts are soluble in DMF and toluene, giving interesting results in polymerization reactions. In particular, the QA-Boc derivative showed the best results in term of polymer yield. The mechanism involves a halogenated initiator that is reduced by the photoexcited QA photocatalyst obtaining a radical initiator and a halide X−, together with the oxidised photocatalyst (oxidative quenching, with the oxidised PC˙+ and X− forming an ion pair). The radical initiator starts the chain reaction with the monomer (MMA or BMA) propagating the polymer, while the PC˙+/X− ion pair is responsible for halogenation of radical intermediates, that re-enter the catalytic cycle (Fig. 3).
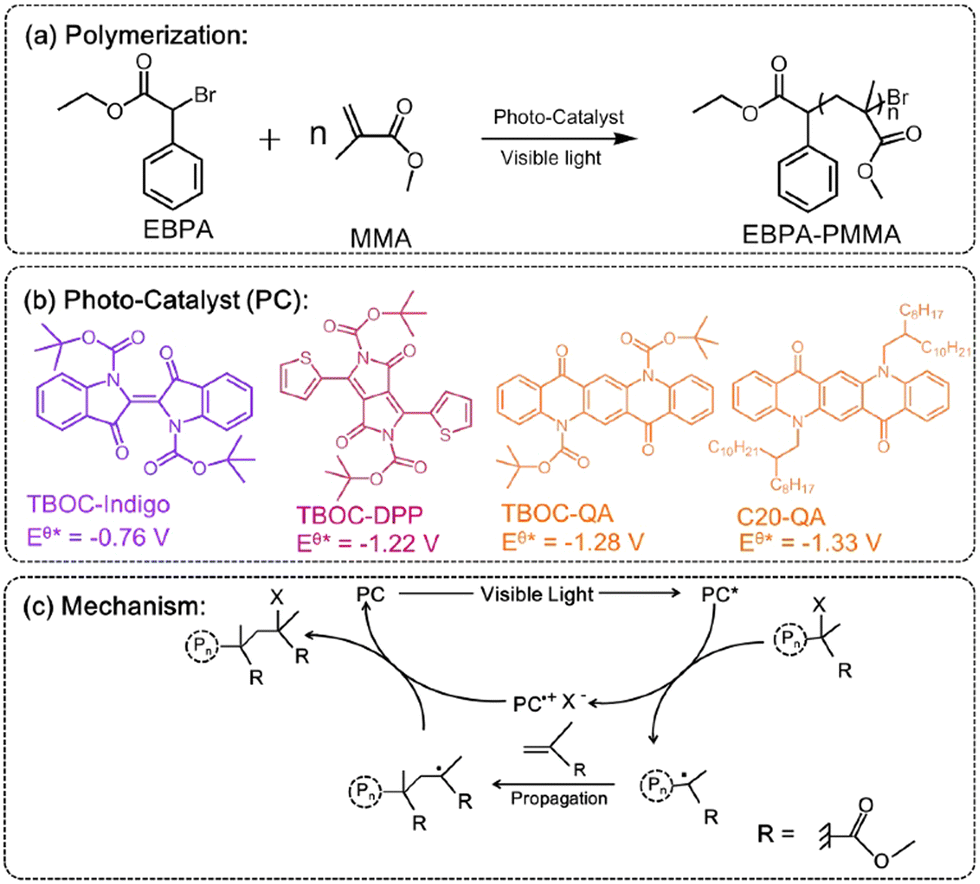 | ||
| Fig. 3 (a–b) Photocatalysed polymerisation reaction of MMA with QA based photocatalysts, and (c) proposed mechanism (adapted from ref. 34 with permission from Elsevier, copyright 2019). | ||
Recently, N-functionalized QA compounds have been used as photocatalysts in thioethers oxidation in homogenous reactions.43 The N,N′ di-butyl quinacridone derivative (DBQA) was obtained from the reaction of deprotonated QA and n-butyl bromide in a 65% yield, and was characterized in apolar DCM and polar, protic HFIP, showing large difference in terms of maximum absorption spectra, oxidation and reduction potentials, and consequently on the redox energy of the excited state.
In the reaction involving thioethers, the excited state of DBQA can oxidise directly the substrate through a reductive quenching pathway, with E(DBQA*/DBQA˙−) estimated +1.29 V vs. SCE in HFIP and with reduced DBQA˙− that is then oxidised by O2. Alternatively, the excited state of DBQA can be oxidised by molecular oxygen through an oxidative quenching, with E(DBQA˙+/DBQA*) estimated −1.28 V vs. SCE in HFIP; oxidised DBQA˙+ further reacts with the thioether, Fig. 4. The overall result is a very efficient catalytic reaction, obtaining selectively the desired sulphoxide in excellent yields and very short reaction time. DBQA was also effective in the selective oxidation of thianthrene to thianthrene-5-oxide, which is a valuable reagent in organo-catalysis.43
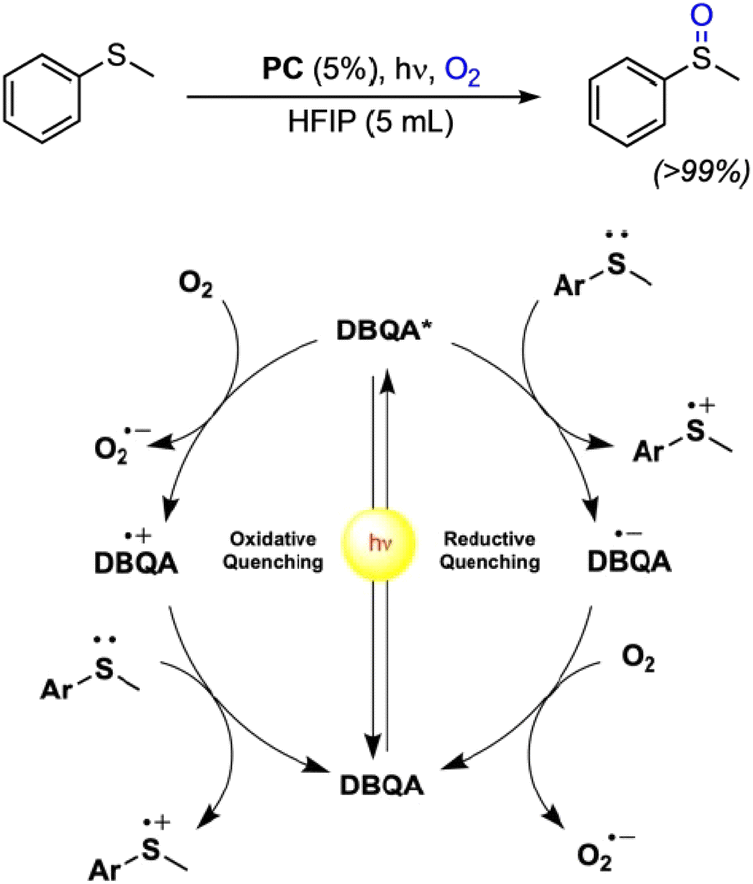 | ||
| Fig. 4 Photocatalytic oxidation of thioethers with QA derivatives, and proposed reaction mechanism on the basis of scavenging experiments. Reproduced from ref. 43 with permission from Wiley Inc., copyright 2023. | ||
The possibility of QA to react with oxygen through the oxidative quenching pathway may be exploited for the generation of reactive oxygen species (ROS), that can be competent agents towards the degradation of organic contaminants. An example deals with a photocatalytic system exploiting a Ni-QA complex for the aerobic oxidation of acridine orange (AO) in water, Fig. 5.44 The complex Ni-QA can be suspended in a water solution of AO, and after irradiation with a Xe lamp the AO visible absorption is completely suppressed in 3 hours, whereas using a heterogeneous solution QA the degradation of AO is only around 50%. Although this system is actually heterogeneous, we report it in this section given the similar mechanism discussed previously for the oxidation of thioethers. Indeed, the Ni-QA complex acts as reducing agent of molecular oxygen (oxidative quenching) forming the superoxide radical anion that degrades the AO.
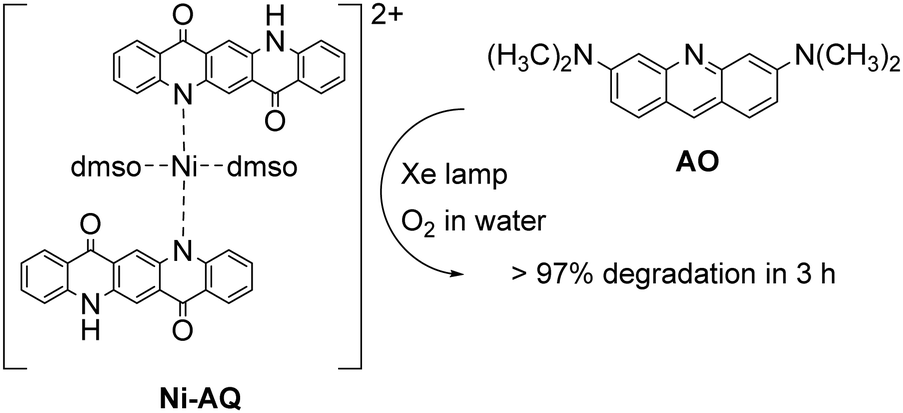 | ||
| Fig. 5 Ni–AQ complex (obtained as the nitrate salt) for the aerobic photodegradation of acridine orange (AO) in water with visible light.44 | ||
The photoinduced reactivity of organic dyes towards oxygen can be exploited for the photochemical synthesis of hydrogen peroxide.45 H2O2 is considered a green oxidant (47% oxygen efficiency, with water as the sole by-product), while it is industrially synthesized through the anthraquinone process, that requires harsh and inconvenient reaction conditions (high temperatures, noble metal catalysts, high pressure of H2, use of organic solvents). In the case of QA, photosynthesis of H2O2 was achieved with a water soluble 2,9-disulfonate derivative; in aqueous medium, when coupled with electron donors such as formate, glucose or oxalate, the disulfonate QA was able to photogenerate H2O2 in a mM concentration range, although with a limited turnover number of ca. 5 (Fig. 6).46 The one electron oxygen reduction to superoxide radical anion is the first key step, with O2˙− further converting to H2O2. We will discuss an example of H2O2 photosynthesis also in the photoelectrochemical cells section.
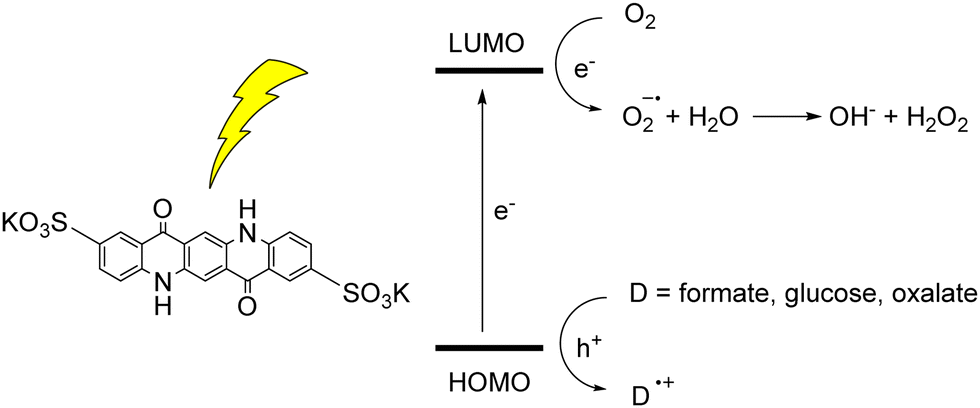 | ||
| Fig. 6 Water soluble QA disulfonate derivative, and photochemical hydrogen peroxide production coupled to oxidation of donors D. Adapted from ref. 46 with permission from Wiley Inc., copyright 2019. | ||
QA sensitised nanoparticles, covalent organic frameworks, nanostructures
The investigation of QA sensitised nanoparticles for photocatalysis applications was pioneered by Shan et al.,47 who reported the sol–gel deposition of a TiO2/QA layer onto the inner pore wall of mesoporous SBA-15 silica. The resulting material was employed in photocatalytic decomposition of indigo carmine with visible light irradiation, showing enhanced activity with respect to anatase TiO2 and Ti-modified SBA-15 materials.47Following a similar approach, Lin et al. prepared TiO2/SBA-15 particles sensitised with 2,9-dichloro QA by impregnation method. The material was exploited for photocatalytic carbon dioxide reduction to methanol in aqueous solution, achieving a production of 0.44 μmol h−1 per gram of photocatalyst, significantly higher than the productivity of P25 TiO2 sensitised particles of 0.16 μmol h−1 per gram of photocatalyst, ascribed to a higher quinacridone loading on the material.48
A more recent report using QA composites as photocatalyst is related to a photochemical reduction of CO2 using QA deposited onto reduced graphene oxide (rGO), Fig. 7.49 The hybrid photocatalyst was prepared by irradiation with a Xe lamp of a DMSO solution of QA and suspended GO, with the resulting QA/rGO composite that precipitates and can be recovered by filtration. As a reference material, the authors also prepared QA nanoparticles, and they showed that the heterogeneous QA/rGO photocatalyst gave the best result in term of CO2 reduction, exploiting a synergic effect of the QA and rGO components. In particular, excited QA can reduce directly the CO2 or reduce the rGO that then transfer the electron to the carbon dioxide. Using a sacrificial electron donor such as triethanolamine (TEOA) allowed to obtain photocatalytic production of CO and CH4 with rates of 450 and 275 μmol h−1 per gram of catalyst.49
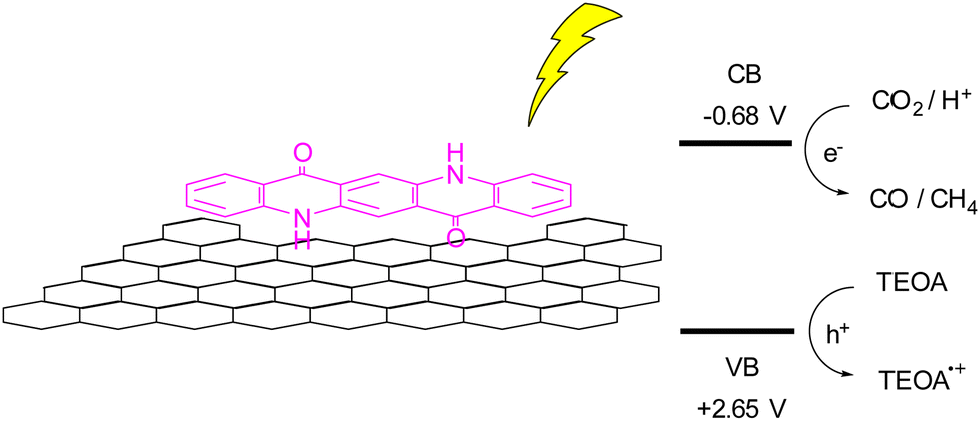 | ||
| Fig. 7 Reduction of CO2 over QA/rGO photocatalyst, and schematic representation of CO2 reduction to CO and CH4, coupled with oxidation of triethanolamine (TEOA); the potential of the conduction and valence bands (CB and VB, respectively) are reported versus the reversible hydrogen electrode.49 | ||
More recently, photocatalytic reduction of CO2 to CO was achieved over a Z-scheme inspired Quinacridone/BiVO4 nanocomposites.50 The QA/nBiVO4 hybrid materials were prepared in different compositions (n = 0.2–20, n is mass ratio of BiVO4 to QA) by hydrogen-bonding self-assembly of QNC over hydroxylated BiVO4 nanosheets.
The QA/nBiVO4 materials were proven to photocatalyse the reduction of CO2 to CO as the main product (traces of methane and H2 were also detected), when immobilised onto a glass sheet (5 mg of material onto a 9 cm2 surface), immerged and irradiated in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:TEOA solution, saturated with CO2. The optimal material was found to be QA/10BiVO4, achieving 407 μmol h−1 production of CO per gram of photocatalyst, 24 times higher than QA alone, while BiVO4 nanosheets almost showed no activity (Fig. 8; indeed, the conduction band of BiVO4 does not reach the thermodynamic potential to drive reduction of CO2). Interestingly, the proposed mechanism foresees a Z-scheme, with excitation of both QA and BiVO4, and electron transfer from the conduction band of BiVO4 to the previous HOMO of QA. The holes on BiVO4 are exploited for oxidation of TEOA, while reducing equivalents conveyed to QA drive the CO2 reduction. It is worth to mention that similarly to quinones,51 electrogenerated reduced forms of QA are capable of reversible carbon dioxide binding, leading to the formation of a QA˙ carbonate anion.52 The formation of such intermediates could be responsible for the observed photocatalytic activity of QA/nBiVO4 composites.
:1 water:TEOA solution, saturated with CO2. The optimal material was found to be QA/10BiVO4, achieving 407 μmol h−1 production of CO per gram of photocatalyst, 24 times higher than QA alone, while BiVO4 nanosheets almost showed no activity (Fig. 8; indeed, the conduction band of BiVO4 does not reach the thermodynamic potential to drive reduction of CO2). Interestingly, the proposed mechanism foresees a Z-scheme, with excitation of both QA and BiVO4, and electron transfer from the conduction band of BiVO4 to the previous HOMO of QA. The holes on BiVO4 are exploited for oxidation of TEOA, while reducing equivalents conveyed to QA drive the CO2 reduction. It is worth to mention that similarly to quinones,51 electrogenerated reduced forms of QA are capable of reversible carbon dioxide binding, leading to the formation of a QA˙ carbonate anion.52 The formation of such intermediates could be responsible for the observed photocatalytic activity of QA/nBiVO4 composites.
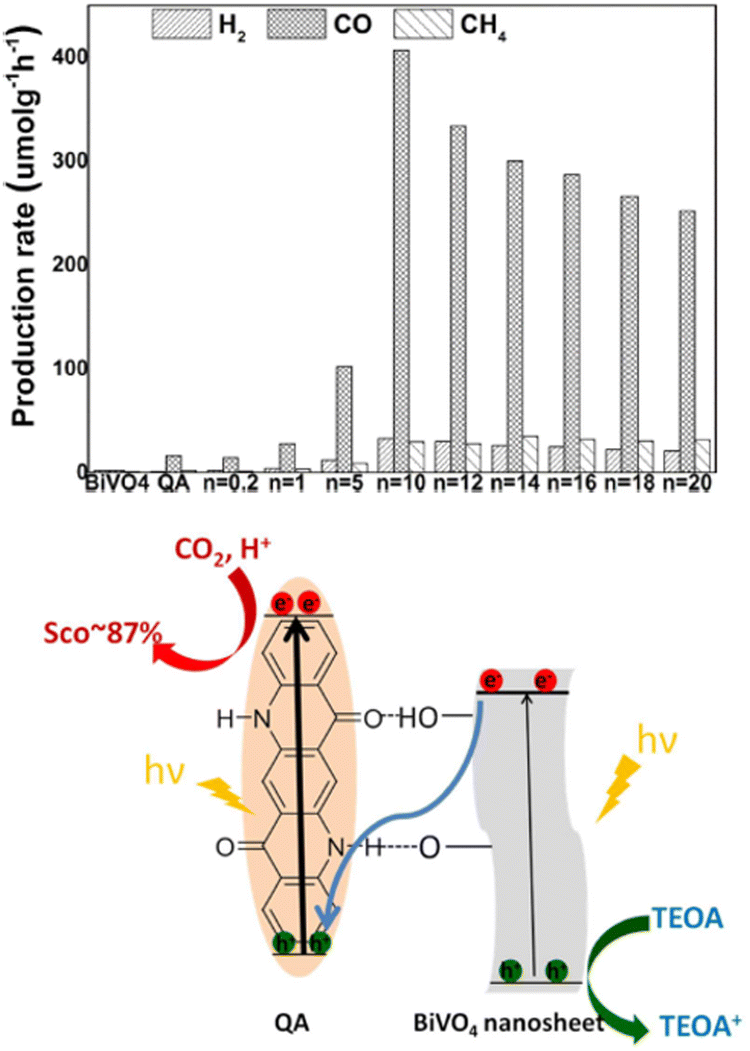 | ||
| Fig. 8 Top panel: Production rates from CO2 photoreduction (average over 10 h) on BiVO4, QA, and QA/nBiVO4 for 10 h. n is the mass ratio between BiVO4 and QA, impacting the photocatalytic performance. Bottom panel: Schematic representation of photochemical CO2 reduction on QA/BiVO4 composites. Reproduced from ref. 50 with permission from Wiley Inc., copyright 2020. | ||
The planar structure of QA makes it suitable for its incorporation in 2D covalent organic frameworks, as in the case of the report by Zhang, Chen and co-workers, Fig. 9.53 QA-TAPT-COF and QA-TPB-COF were prepared by solvothermal synthesis starting from the molecular components, necessary to form the imine linkers. The resulting COF are characterised by large pore size (>4.7 nm).
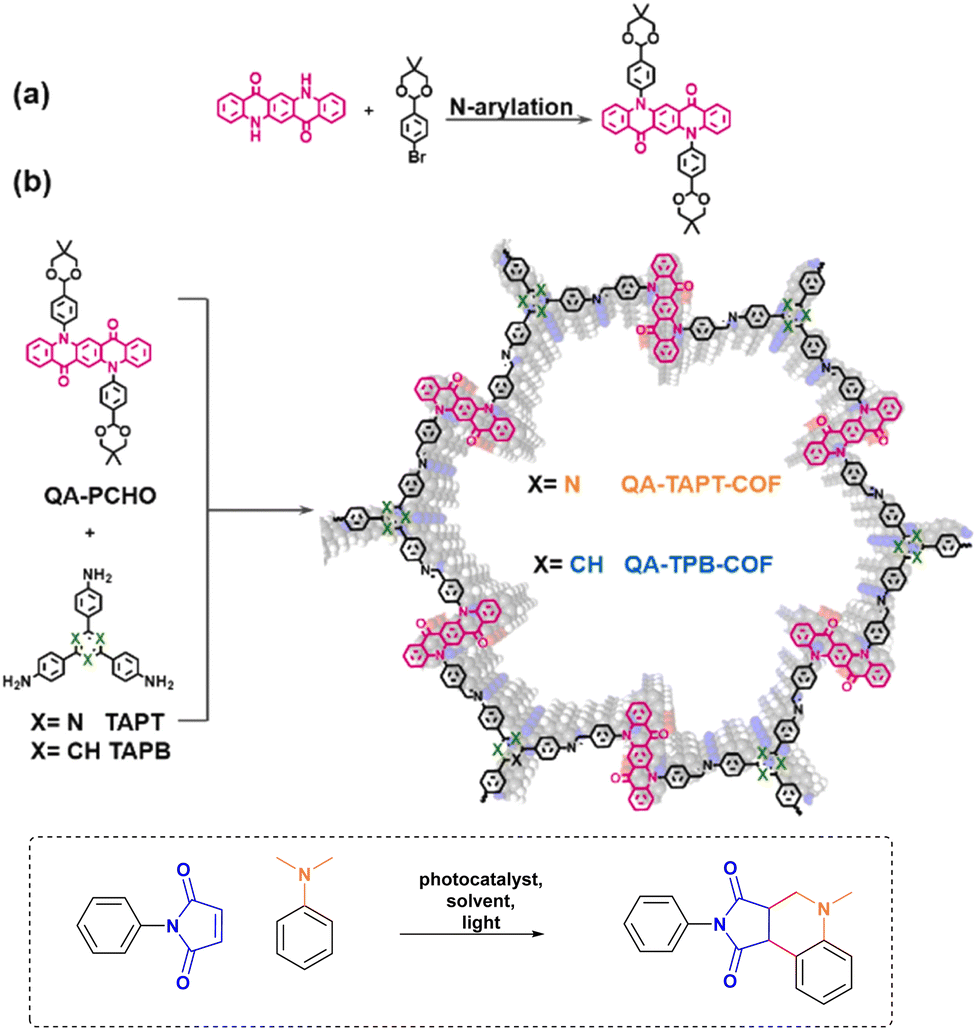 | ||
| Fig. 9 Incorporation of QA derivatives in 2D covalent organic frameworks via (a) synthesis of QA-PCHO component and (b) solvothermal synthesis of QA-TAPT-COF and QA-TPB-COF. Photochemical application towards the Povarov reaction. Reproduced from ref. 53 with permission from Elsevier, copyright 2022. | ||
The two COFs show a redshifted absorption with respect to the molecular QA monomers, due to the extended conjugated structure of the materials; the band gaps of 2.03 and 2.09 eV for QA-TAPT-COF and QA-TPB-COF, respectively, are in the range expected for aggregate states of QA (vide supra).
The QA-based 2D COFs were applied as photocatalysts for the Povarov reaction, targeting the condensation of maleimide and N,N-dimethylaniline derivatives under aerobic atmosphere to produce tetrahydroquinolines (Fig. 9). Both COFs were effective in photocatalysing the reaction (quantitative yields were achieved in ca. 40 minutes irradiation in DMF as the solvent), with a superior reactivity of QA-TAPT-COF with respect to QA-TPB-COF; QA-TAPT-COF was also more active than soluble QA derivatives alkylated at the nitrogen sites, and could also be recycled up to 10 times. Concerning the mechanism, the first step of the Povarov reaction typically involves oxidation by single electron transfer of the arylamine by the excited photocatalyst, with formation of the arylamine radical cation and of the reduced photocatalyst (reductive quenching), that is then re-oxidised by oxygen to form O2˙−, this latter being involved in an oxidation step leading to the final product.40 As a mechanistic proof, the authors were able to support O2˙− formation by trapping it with 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO) and characterising the adduct by electron paramagnetic resonance.53
As a final selected example of QA based particulate materials for photocatalysis application, we report the development of supramolecular nanobelts.54 In particular, these were designed through alkylation of both nitrogen atoms of QA with n-butyl or n-octyl chains, and by insertion of hydrophilic pyridine-2,6-dicarboxylic acid moieties at positions 3 and 10 of the pentacyclic core (Fig. 10). The molecular system is characterised by donor–acceptor interactions between the electron rich QA donor and the electron-deficient pyridine ring acceptor. The monomers self-assemble in water through hydrogen bonding and π–π stacking interactions, forming nanobelts with a width of ca. 50 nm and a length of hundreds of nm for SQNCP-C4, and a width of 100 nm and a length up to micrometers for SQNCP-C8. As expected and similarly to the cases discussed above, the formation of supramolecular aggregated systems leads to a substantial redshift of the nanobelts with respect to the isolated monomers, and the band gap of SQNCP-C4 and SQNCP-C8 was measured to be 1.76 eV and 1.94 eV, respectively. Both of them show emission peaks around 650 nm, with the emission intensity that was significantly lower in the case of SQNCP-C4; this was interpreted as a proof of intramolecular charge separation within the donor/acceptor system.
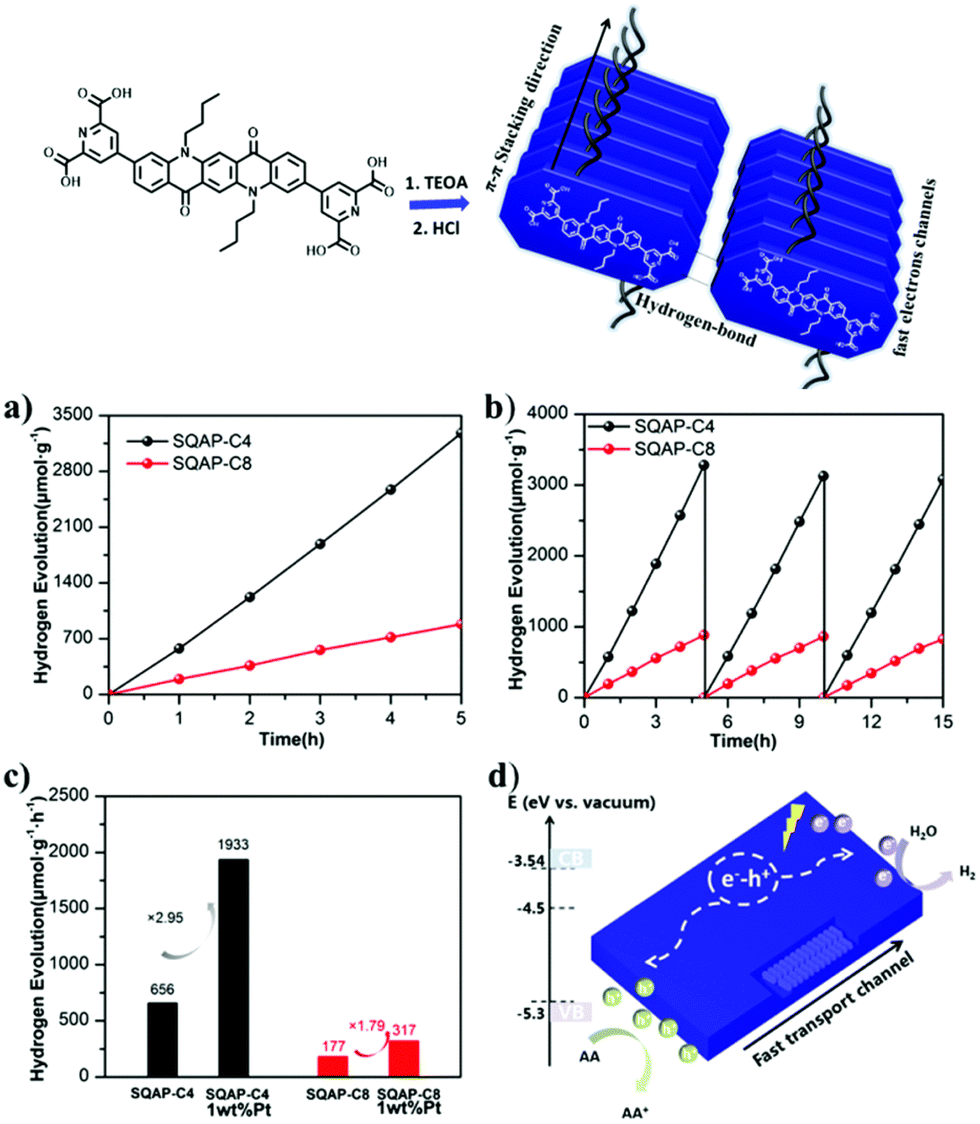 | ||
| Fig. 10 Formation of QA based nanobelts, and application in photocatalytic water reduction to hydrogen: (a) photocatalytic hydrogen evolution over SQAP-C4 and SQAP-C8; (b) recycle tests; (c) hydrogen evolution rate comparison of SQAP-C4, SQAP-C8, Pt-loaded SQAP-C4 and Pt-loaded SQAP-C8; (d) proposed mechanism. Reproduced from ref. 54 with permission from the Royal Society of Chemistry, copyright 2020. | ||
The materials were tested towards photocatalytic reduction of water to hydrogen, in aqueous solution in the presence of ascorbate as electron donor. The SQNCP-C4 material exhibited the highest activity of ca. 656 μmol h−1 per gram of photocatalyst; the H2 production rate was increased by ca. 3 times in the presence of 1% w/w Pt co-catalyst. The material maintained the activity over three runs, for a total of 15 h irradiation (Fig. 10).
Photoelectrochemical cells
Photoelectrochemical cells are considered a viable technology to conduct redox transformations; they are composed by an anode and a cathode, where at least one of them is activated by light. In photoelectrochemical cell, upon light absorption the electrons are thus conveyed through an external circuit, and thus it is not necessary to use sacrificial electron donors or acceptors, as it is often employed in solution photocatalysis. The first example of a photoelectrochemical cell was provided by Fujishima and Honda, that developed a TiO2 based photoanode coupled to a Pt cathode for overall water splitting into hydrogen and oxygen.55 Since then, many efforts have been conducted to design more efficient photoelectrodes, with one strategy being the integration of molecular components for more efficient light absorption (photosensitizers) and for accelerating the redox reactions (catalysts).56,57Exploiting the well-balanced redox properties of QA, its derivatives have been recently employed at both the photoanode and photocathode compartments of a photoelectrochemical cell for water splitting, capable to operate in the absence of external applied bias (Fig. 11).58 In particular, the QA was functionalised at 2 and 9 carbon positions with pyridine dicarboxylic acid pendants to anchor the dye to TiO2 (n-type semiconductor) and to NiO (p-type semiconductor), to develop the photoanode and photocathode, respectively. QA was also functionalized at both nitrogen sites with long alkyl chain substituents (C4–C16), to prevent excessive adhesion of the dye to the hydrophilic surface of titania (likely mitigating unproductive charge recombination events).
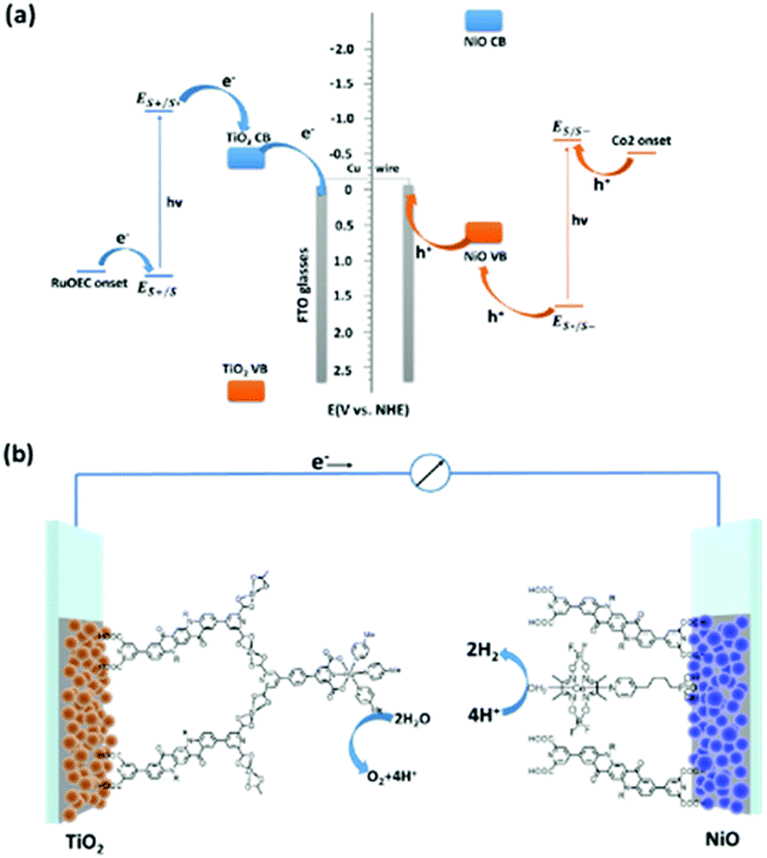 | ||
| Fig. 11 QA based photoelectrochemical cell for water splitting into hydrogen and oxygen: representation of the energy levels (a) and molecular assembly of the cell (b). Reproduced from ref. 58 with permission from the Royal Society of Chemistry, copyright 2022. | ||
The water oxidation catalyst was a single site Ru coordination complex, still bearing the pyridine dicarboxylic acid ligand; anchoring of the catalyst to the QA dye was achieved through Zr(IV) bridges, as pioneered by T. J. Meyer and co-workers.59 The water reduction catalyst was instead a Cobalt cobaloxime,60 directly anchored to the NiO through phosphonate pendants. When the two photoelectrodes were combined in a photoelectrochemical cell and both irradiated with visible light in aqueous solution (pH 7), water splitting was achieved in the absence of applied bias, reaching a photocurrent density of ca. 110 μA cm−2, a faradaic efficiency for hydrogen evolution of 89% and a solar to hydrogen efficiency of 0.11%.
An operative advantage of photoelectrochemical cells development is that the photoelectrodes can be designed and optimized independently, by coupling them with a suitable counter-electrode. Moreover, other processes with respect to water splitting can be targeted. As two final case studies, we will discuss the development of QA photocathodes for photoelectrochemical synthesis of hydrogen peroxide and of QA based photoanodes for photoelectrochemical oxidative C–H activation.
Epindolidione (EPI) and QA based photocathodes for hydrogen peroxide production were developed by Głowacki and co-workers, and constituted the first example of an organic semiconductor photocathode for H2O2 photosynthesis (Fig. 12).39 Polycrystalline films of QA (80 nm thickness) were prepared on gold electrode surface by vacuum sublimation of the pigment; this films behave as organic semiconductors, displaying charge mobility in the range 0.1–1 cm2 V−1 s−1.39 A cathodic photocurrent response up to 10 μA cm−2 was observed in aqueous electrolyte in a wide pH range from 1 to 12 and attributed to oxygen reduction (indeed, the photocurrent response was 10–20 times lower when measured in N2 degassed solutions). At pH 1, the photocurrent of the QA/Au photocathode was associated to H2O2 production with a 80 ± 5% faradaic yield; under these conditions and under illumination the photocathode was operative for 4 days, with photocurrent response in the range 1–4 μA cm−2. The initial photocathodic current drop was attributed to oxygen consumption in the cell, while the subsequent photocurrent increase was ascribed to a recrystallization of the QA dye, as shown in the scanning electron microscopy images taken before and after 20 hours of electrolysis, showing the conversion of rod-like crystallites into rectangular platelets (Fig. 12). As a final remark, key features of the QA films for photoelectrochemically reducing oxygen to H2O2 are the high reducing level of the conduction band (or of the LUMO), and the two electron reductive chemistry couple to proton transfer in aqueous solution, similarly to quinones.61,62
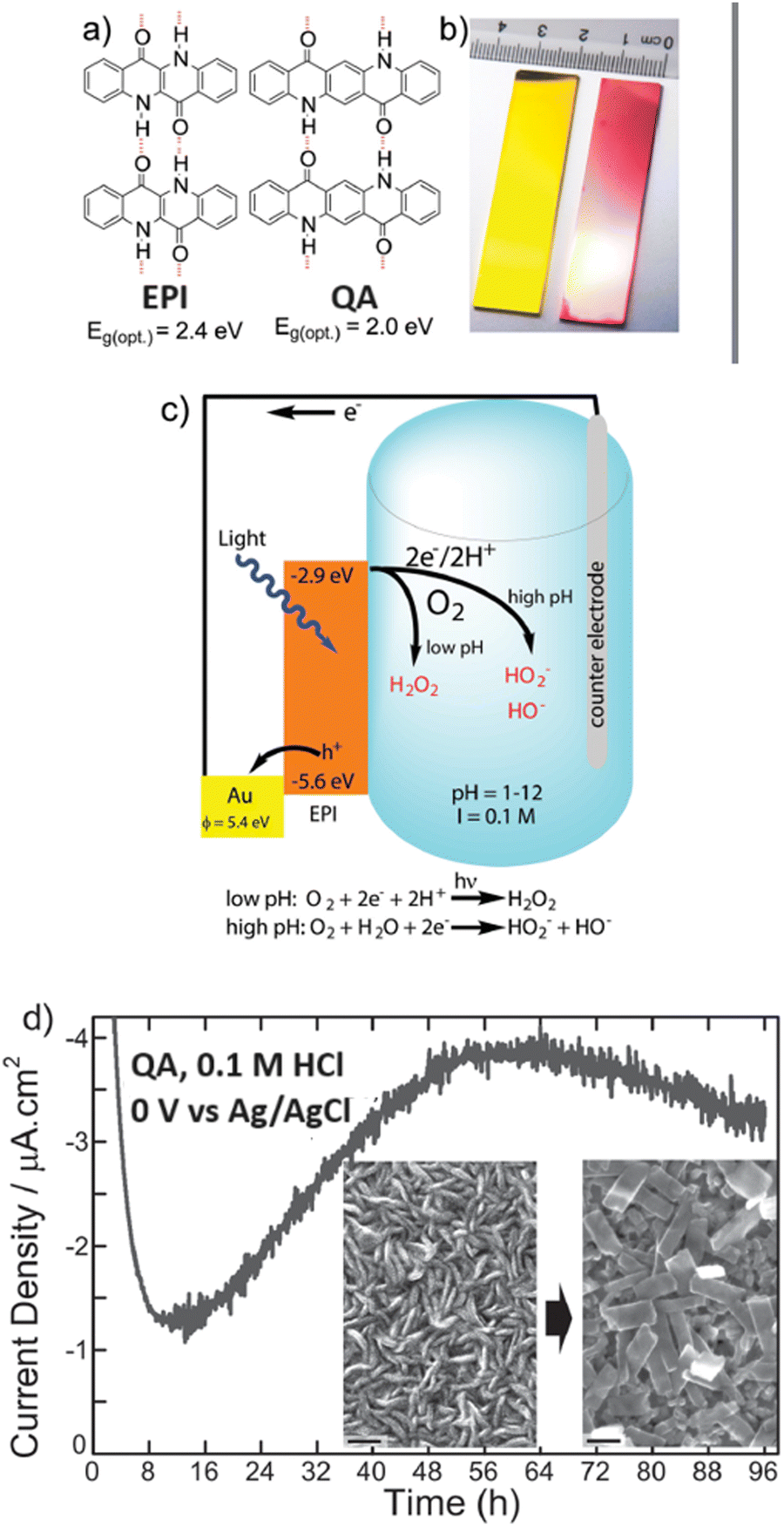 | ||
| Fig. 12 (a) and (b) Epindolidione (EPI) and QA photocathodes for photoelectrochemical synthesis of H2O2 upon reduction of oxygen. Panel (c) represents the operating mechanism and the energy levels involved. Panel (d) shoes the morphological evolution of QA films along the photoelectrocatalysis by scanning electron microscopy. Reproduced from ref. 39 with permission from Wiley Inc., copyright 2016. | ||
The proton coupled electron transfer (PCET) reactivity of QA can be achieved also along its oxidative chemistry, and is the subject of the last example discussed in this perspective. Indeed, cyclic voltammetries of QA/carbon inks casted on glassy carbon registered in aqueous solution (pH between 4.5 and 8.3) show that the QA dye is characterized by an anodic wave with a potential shifting with the pH close to 58 mV pH−1 unit, typical of a Nernstian behavior associated to a PCET event. The oxidation potential can thus be conveniently reported as 1.23 V vs. the reversible hydrogen electrode (RHE), and is associated to the oxidation of the QA core with release of H+ from the nitrogen sites to the aqueous solution, and formation of a nitrogen centered radical (Fig. 13).31 The oxidative PCET reactivity of QA was then exploited in photoanodes, sensitizing TiO2 or SnO2 mesoporous semiconductors through vacuum sublimation of QA; the photoelectrodes were characterized by an absorption typical of QA aggregate films (40 nm thickness) with an absorbance of 0.2 at 564 nm. Ultrafast spectroscopy confirmed electron injection from the QA dye to the semiconductor surface in a ps timescale (oxidative quenching of the QA dye), likely accompanied by proton transfer from the N–H group of the dye to the metal–oxide surface. This process is expected to generate a nitrogen centered radical on the QA dye, for which a bond dissociation free energy (BDFE) of 80.5 ± 2.3 kcal mol−1 can be estimated. Consistently, photoelectrochemical reactivity through hydrogen atom abstraction towards C–H bonds was observed. Indeed, photoelectrochemical oxidation of γ-terpinene to p-cymene was observed, occurring through the initial hydrogen atom transfer followed by a radical chain mechanism involving dissolved oxygen.
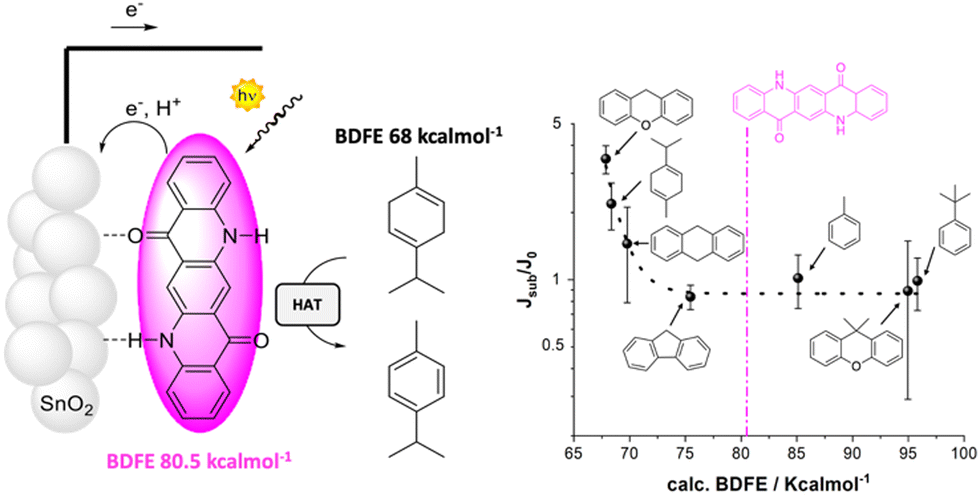 | ||
| Fig. 13 QA based photoanode for oxidative C–H activation. The right figure shows the photoelectrochemical response (given by the ratio of the photocurrent in the presence and in the absence of the organic substrate) versus the BDFE of the C–H bond of the substrate. Reproduced from ref. 31 with permission from Wiley Inc., copyright 2023. | ||
Expanding the scope to other C–H moieties, a photocurrent response was observed for substrates containing allylic or benzylic C–H bonds with a BDFE lower than the threshold of 80.5 kcal mol−1 for the N–H bond in QA. As expected, more challenging substrates (containing C–H bonds with BDFE > 80.5 kcal mol−1) provided a null photocurrent enhancement since they could not be oxidized by hydrogen atom abstraction.
It is worth to mention that oxidative C–H bond activation is considered a Holy Grail in organic chemistry, while this is the first report where this reaction is conducted photoelectrochemically with a dye-sensitized electrode. Therefore, this proof-of-concept can open a novel way of conducting this challenging transformation.
Conclusions and perspectives
In this perspective article, we have discussed examples of photocatalytic and photoelectrocatalytic processes taking advantage of QA derivatives. QA provides an example of a cheap and readily available organic pigment, showing intense absorption in the visible region of the electromagnetic spectrum and whose properties can be modulated by chemical design. Moreover, its versatility allows its use in several operative conditions, spanning from homogeneous photocatalysis to photoactive materials and devices. Finally, its balanced redox properties allow its application in both oxidative and reductive routines, embracing water splitting, carbon dioxide reduction, organic photocatalysis, as discussed in the previous sections.We believe that QA represents an appropriate example (although investigated only in a limited manner) of the potential of organic photocatalyst design. This research field is in rapid evolution, and examples of emerging research lines in the recent literature landscape include reactivity in flow,63,64 multiphoton excitation,65,66 or the combination of light with electrochemistry to promote photochemistry of oxidised and reduced photocatalysts.67 Targets for the future include the development of new processes and an improved sustainability of existing ones, together with a fundamental understanding of the reaction mechanism and identification of active species.68
Abbreviations
| AO | Acridine orange |
| BDFE | Bond dissociation free energy |
| BMA | Butyl methacrylate |
| Boc | tert-Butyloxycarbonyl |
| COF | Covalent organic framework |
| DBQA | N,N′ Di-butyl quinacridone |
| DCM | Dichloromethane |
| DMF | N,N′-Dimethylformamide |
| DMPO | 5,5-Dimethyl-1-pyrrolidine N-oxide |
| DMSO | Dimethylsulfoxide |
| DOQA | N,N′-Dioctyl quinacridone |
| EPI | Epindolidione |
| FTO | Fluorine doped tin oxide |
| HFIP | Hexafluoroisopropanol |
| MMA | Methyl methacrylate |
| PCET | Proton coupled electron transfer |
| QA | Quinacridone |
| RHE | Reversible hydrogen electrode |
| rGO | Reduced graphene oxide |
| ROS | Reactive oxygen species |
| TEOA | Triethanolamine |
Author contributions
All authors discussed the concepts and contributed to the first draft of the manuscript. P. G. and A. S. supervised and edited the writing, and were responsible for funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. acknowledges Dr Eric Daniel Glowacki (Brno University of Technology) for having inspired his interest in QA pigments and for fruitful discussions. Financial support from the Italian MUR (PRIN 2022KPK8WM PROMETEO, to A. S. and P. G.), and by the European Union – Next Generation UE (PRIN2022 PNRR PHOTOCORE, P2022ZSPWF to A. S.) is gratefully acknowledged.Notes and references
- G. Ciamician, Science, 1912, 36, 386–399 CrossRef PubMed.
- S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell’Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 8–11 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- C. Bie, L. Wang and J. Yu, Chem, 2022, 8, 1567–1574 CAS.
- B.-J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S.-P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- W. Zhang, A. R. Mohamed and W. J. Ong, Angew. Chem., Int. Ed., 2020, 59, 22894–22915 CrossRef CAS PubMed.
- V. Nikolaou, C. Govind, E. Balanikas, J. Bharti, S. Diring, E. Vauthey, M. Robert and F. Odobel, Angew. Chem., Int. Ed., 2024, 63, e202318299 CrossRef CAS PubMed.
- C. Bizzarri, Eur. J. Org. Chem., 2022, e202200185 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- T. Bortolato, S. Cuadros, G. Simionato and L. Dell’Amico, Chem. Commun., 2022, 58, 1263–1283 RSC.
- D. Cappelletti, M. Barbieri, A. Aliprandi, M. Maggini and L. Đorđević, Nanoscale, 2024, 16, 9153–9168 RSC.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- D. A. Kader and S. J. Mohammed, RSC Adv., 2023, 13, 26484–26508 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS PubMed.
- T. Gobbato, F. Rigodanza, E. Benazzi, P. Costa, M. Garrido, A. Sartorel, M. Prato and M. Bonchio, J. Am. Chem. Soc., 2022, 144, 14021–14025 CrossRef CAS PubMed.
- S. S. Labana and L. L. Labana, Chem. Rev., 1967, 67, 1–18 CrossRef CAS.
- M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gasiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, M. Piero, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 CrossRef PubMed.
- E. D. Głowacki, R. R. Tangorra, H. Coskun, D. Farka, A. Operamolla, Y. Kanbur, F. Milano, L. Giotta, G. M. Farinola and N. S. Sariciftci, J. Mater. Chem. C, 2015, 3, 6554–6564 RSC.
- J. Mizuguchi and T. Senju, J. Phys. Chem. B, 2006, 110, 19154–19161 CrossRef CAS PubMed.
- Y. Miyashita, H. Yokoyama, M. Tanabe, H. Kasai, H. Nakanishi and T. Miyashita, J. Photochem. Photobiol., A, 2009, 201, 208–213 CrossRef CAS.
- Y. Yang, G. A. Volpato, E. Rossin, N. Peruffo, F. Tumbarello, C. Nicoletti, R. Bonetto, L. Paoloni, P. Umari, E. Colusso, L. Dell’Amico, S. Berardi, E. Collini, S. Caramori, S. Agnoli and A. Sartorel, ChemSusChem, 2023, e202201980 CrossRef CAS PubMed.
- P. Kurzep, Ł. Skórka, M. Zagorska, P. A. Guńka, M. Banasiewicz, B. Kozankiewicz and I. Kulszewicz-Bajer, RSC Adv., 2017, 7, 8627–8632 RSC.
- M. Sytnyk, E. D. Głowacki, S. Yakunin, G. Voss, W. Schöfberger, D. Kriegner, J. Stangl, R. Trotta, C. Gollner, S. Tollabimazraehno, G. Romanazzi, Z. Bozkurt, M. Havlicek, N. S. Sariciftci and W. Heiss, J. Am. Chem. Soc., 2014, 136, 16522–16532 CrossRef CAS PubMed.
- T. Jia, S. Huang, H. Bohra and M. Wang, Dyes Pigm., 2019, 165, 223–230 CrossRef CAS.
- J. J. A. Chen, T. L. Chen, B. Kim, D. A. Poulsen, J. L. Mynar, J. M. J. Fréchet and B. Ma, ACS Appl. Mater. Interfaces, 2010, 2, 2679–2686 CrossRef CAS PubMed.
- J. Jia, D. Feng, Y. Sha, C. Zhou, G. Liang and Y. She, Tetrahedron, 2020, 76, 131057 CrossRef CAS.
- G. D. Potts, W. Jones, J. F. Bullock, S. J. Andrews and S. J. Maginn, J. Chem. Soc., Chem. Commun., 1994, 2565–2566 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- J. Mateos, F. Rigodanza, A. Vega-Peñaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyó, M. Bonchio and L. Dell’Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS PubMed.
- F. Puntoriero, S. Serroni, G. La Ganga, A. Santoro, M. Galletta, F. Nastasi, E. La Mazza, A. M. Cancelliere and S. Campagna, Eur. J. Inorg. Chem., 2018, 3887–3899 CrossRef CAS.
- V. Balzani, G. Bergamini and P. Ceroni, Angew. Chem., Int. Ed., 2015, 54, 11320–11337 CrossRef CAS PubMed.
- M. Forchetta, V. Baia, F. Sabuzi, V. Conte and P. Galloni, Eur. J. Org. Chem., 2023, e202300523 CrossRef CAS.
- N. Mousavi, H. Hadadzadeh, A. A. Ensafi, H. Farrokhpour and K. Z. Mousaabadi, Mater. Chem. Phys., 2023, 303, 127823 CrossRef CAS.
- M. Gryszel, T. Schlossarek, F. Wurthner, M. Natali and E. D. Glowacki, ChemPhotoChem, 2023, 7, e202300070 CrossRef CAS.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- H. Ding, H. Sun and Y. Shan, J. Photochem. Photobiol., A, 2005, 169, 101–107 CrossRef CAS.
- C.-J. Lin, Y.-H. Liou, S.-Y. Chen and M.-C. Tsai, Sustainable Environ. Res., 2012, 22, 167–172 CAS.
- P. Yang, S. Guo, X. Yu, F. Zhang, B. Yu, H. Zhang, Y. Zhao and Z. Liu, Ind. Eng. Chem. Res., 2019, 58, 9636–9643 CrossRef CAS.
- X. Yu, F. Wen, F. Zhang, P. Yang, Y. Zhao, Y. Wu, Y. Wang and Z. Liu, ChemSusChem, 2020, 13, 5565–5570 CrossRef CAS PubMed.
- K. M. Diederichsen, R. Sharifian, J. S. Kang, Y. Liu, S. Kim, B. M. Gallant, D. Vermaas and T. A. Hatton, Nat. Rev. Methods Prim., 2022, 2, 68 CrossRef CAS.
- D. H. Apaydin, E. D. Głowacki, E. Portenkirchner and N. S. Sariciftci, Angew. Chem., Int. Ed., 2014, 53, 6819–6822 CrossRef CAS PubMed.
- Z. Liu, X. Yang, Z. Yang, X. Su, Z. Xie, W. Chen, W. Zhang and L. Chen, Appl. Catal., B, 2022, 312, 121406 CrossRef CAS.
- M. Xu, K. Kong, H. Ding, Y. Chu, S. Zhang, F. Yu, H. Ye, Y. Hu and J. Hua, J. Mater. Chem. C, 2020, 8, 930–934 RSC.
- K. Fujishima and A. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- L. Fei, L. Lei, T. J. Meyer and D. Wang, Acc. Mater. Res., 2024, 5, 124–135 CrossRef CAS.
- T. Gobbato, G. A. Volpato, A. Sartorel and M. Bonchio, Chem. Sci., 2023, 14, 12402–12429 RSC.
- L. Shen, S. Zhang, H. Ding, F. Niu, Y. Chu, W. Wu, Y. Hu, K. Hu and J. Hua, Chem. Commun., 2021, 57, 5634–5637 RSC.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS PubMed.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed.
- E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC.
- G. Laudadio, Y. Deng, K. van der Walls, D. Ravelli, M. Nunõ, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- J. Schütte, D. Corsi, W. Haumer, S. Schmid, J. Žurauskas and J. P. Barham, Green Chem., 2024, 26, 6446–6453 RSC.
- G. Han, G. Li, J. Huang, C. Han, C. Turro and Y. Sun, Nat. Commun., 2022, 13, 2288 CrossRef CAS PubMed.
- B. Pfund, D. Gejsnæs-schaad, O. S. Wenger and B. Lazarevski, Nat. Commun., 2024, 15, 4738 CrossRef CAS PubMed.
- J. Žurauskas, S. Boháčová, S. Wu, V. Butera, S. Schmid, M. Domański, T. Slanina and J. P. Barham, Angew. Chem., Int. Ed., 2023, 62, e202307550 CrossRef PubMed.
- C. Wang, H. Li, T. H. Bürgin and O. S. Wenger, Nat. Chem., 2024 DOI:10.1038/s41557-024-01482-4.
Footnote |
| † E. R. and Y. Y. equally contributed. |
| This journal is © The Royal Society of Chemistry 2024 |