
Synergistic dual-interface engineering with self-organizing Li-ion/electric fields for enhanced lithium metal anode stability†
Zhiqiang
Li‡
 *a,
Kemeng
Liao‡
b,
Lihong
Yin
b,
Zongrun
Li
b,
Yingzhi
Li
b,
Hongzhi
Wang
b,
Ning
Qin
b,
Shuai
Gu
b,
Jingjing
Chen
b,
Weihua
Wan
*c and
Zhouguang
Lu
*b
*a,
Kemeng
Liao‡
b,
Lihong
Yin
b,
Zongrun
Li
b,
Yingzhi
Li
b,
Hongzhi
Wang
b,
Ning
Qin
b,
Shuai
Gu
b,
Jingjing
Chen
b,
Weihua
Wan
*c and
Zhouguang
Lu
*b
aDepartment of Electric Power Engineering, Hebei Key Laboratory of Green and Efficient New Electrical Materials and Equipment, North China Electric Power University, Baoding 071003, P. R. China. E-mail: lizq3@ncepu.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, P. R. China. E-mail: zglu@sustech.edu.cn
cGui Zhou Mei Ling Power Sources Co. Ltd, State Key Laboratory of Advanced Chemical Power Source, Zunyi, Guizhou 563003, China. E-mail: wanwei006@126.com
First published on 11th September 2024
Abstract
Lithium metal is promising anode material for next-generation ultra-high energy batteries due to its unparalleled theoretical capacity. Nonetheless, its practical application is largely hindered by interfacial instability. Herein, we propose an interfacial engineering strategy employing a sandwich-structured interface comprising a nano-silver (Ag) inner layer and a lithium chitosan sulfonate (LCS) outer layer. The lithophilic nano-silver layer, with its uniformly distributed three-dimensional structure, ensures a consistent interfacial electric field and robustly anchors the LCS, mitigating delamination or decoupling from the Li metal surface during Li plating/stripping. Simultaneously, the LCS coating, characterized by its polysaccharide glycosidic structure, not only delivers exceptional elasticity and mechanical strength but also serves as a robust artificial solid-electrolyte interphase (SEI) layer, preserving the interface's structural integrity. Additionally, the LCS's sulfonic acid groups (–SO3Li) further promote uniform Li-ion flux and maintain high Li+ ionic conductivity. These synergistic effects significantly improve the specific discharge capacity and cycling stability of a C–AgLi‖LiCoO2 full cell, achieving a capacity retention of 83.8% after 350 cycles. These findings elucidate a pathway towards the practical utilization of Li metal anodes by enhancing Li-ion flux, electric field uniformity, and interface adhesion, thus effectively inhibiting Li dendrites.
 Zhiqiang Li | Zhiqiang Li is an assistant professor at the Key Laboratory of Green and Efficient Electrical Materials and Devices, Department of Electrical Engineering, North China Electric Power University (NCEPU). He received his Bachelor's degree from Sichuan University in 2014 and his Master's degree from Central South University in 2017, both located in China. In 2023, he obtained his PhD at Southern University of Science and Technology. He joined NCEPU in September 2023, he currently serves as an Assistant professor. His research interests include Li-ion batteries, solid-state batteries, flowing batteries, and Li metal batteries. |
Introduction
In advancing energy storage systems, key goals for lithium-ion batteries include higher energy density, longer lifespan, improved safety, and cost reduction.1,2 Lithium metal batteries, with an ultra-high theoretical capacity (3860 mA h g−1) and low reduction potential (−3.04 V vs. SHE), are promising for next-generation high-energy-density applications like smart grids, electric vehicles, and advanced electronics.3–6 These batteries use a plating/stripping mechanism, unlike the intercalation chemistry of graphite anodes, offering superior performance.7,8 However, challenges such as poor cycling stability, low coulombic efficiency (CE), and safety issues hinder their widespread use. Li metal anodes are prone to spontaneous reduction and decomposition of organic electrolytes, resulting in the formation of a fragile, inhomogeneously distributed solid electrolyte interphase (SEI) that promotes uneven Li+ flux and interface instability.9,10 Additionally, significant volume expansion during cycling damages the SEI, exacerbating Li dendrite formation and electrolyte depletion, which diminishes battery efficiency and lifespan.11,12 The detachment of Li dendrites due to volume changes and polarization further contributes to battery degradation.13,14 These issues have been seriously impeding the application and development of Li metal batteries, therefore, it is indispensable to develop superior modified artificial interfaces to implement high-performance Li metal anodes.15Recent efforts to mitigate lithium metal battery (LMB) challenges include interface engineering strategies such as artificial solid electrolyte interphase (SEI) layers,16,17 3D current collectors,18,19 and lithiophilic nucleating sites.20,21 Among these, functionalized artificial SEI layers offer significant benefits by reducing side reactions, facilitating fast Li-ion transport, and ensuring uniform ion flux.22 These layers are generally composed of flexible organic components that, despite their pliability, cannot resist dendrite penetration due to the uneven lithium surface, and robust inorganic components that prevent dendrite growth but fail to accommodate the anode's volumetric expansion during cycling.23 Furthermore, despite advancements, issues like dendrite formation due to inhomogeneous nucleation persist, especially with repeated lithium plating/stripping.24 A combined approach using artificial SEI, lithiophilic nucleation, and 3D structures provides a comprehensive solution to enhance and stabilize the anode interface.25,26 However, challenges remain with lithiophilic materials in achieving homogeneous distribution within 3D structures, affecting dense lithium deposition and reducing battery lifespan.27 Additionally, repeated lithium deposition and stripping can alter the anode interface configuration, significantly affecting the SEI's physicochemical properties. The mismatch in affinity between the SEI and both lithium metal and lithiophilic substrates, coupled with the stress from volumetric changes during cycling, often leads to delamination at the SEI-anode interface.28 Therefore, developing an atomic-scale adjustable interface protective layer that guides lithium nucleation, suppresses dendrite growth, maintains SEI integrity, and achieves prolonged interface stability is crucial.
Herein, we report a two-pronged strategy to guide uniform lithium nucleation and mitigate dendrite growth through the construction of a 3D lithiophilic Ag-nanoparticles layer and a uniformly adherent LCS protective layer. Initially, the 3D Ag-nanoparticles layer is synthesized via a spontaneous in situ substitution reaction between Li atoms and Ag+ ions at room temperature, effectively reducing the nucleation energy barrier and facilitating uniform Li deposition. Simultaneously, the LCS layer is in situ lithiated and coordinated with the 3D Ag-nanoparticles via a simple solution immersion process, optimizing Li-ion flux and providing rapid ion transport channels while isolating the Li electrode surface from the electrolyte. This ingeniously integrated LCS–AgLi anode, featuring a 3D nano-Ag layer coupled with an LCS film, exhibits a dendrite-free morphology, high CE, low overpotential, and superior cycle stability. The full cells in combination with LiCoO2 cathode exhibited stable cycling life over 350 cycles, underlining the efficacy of our engineered interface in advancing the practical application of lithium metal anodes.
Results and discussion
To achieve a dual-protective artificial interface with a lithiophilic substrate and robust film of high Li+ ion channel for stable lithium metal anodes, LCS–Ag layer has been chosen. The different electrochemical deposition behaviors of Li metal surfaces with and without the dual-modified interface comprising nano-Ag inner layer and a LCS outer layer are proposed in Fig. 1. The nano-Ag and LCS-modified anode showcases a uniform interfacial electric field, promoting homogeneous Li-ion flux and stable Li deposition (Fig. 1a). Conversely, the bare Li anode demonstrates significant irregularities in Li-ion flux, leading to uneven Li deposition characterized by the formation of dendrites and dead Li (Fig. 1b). This arrangement significantly mitigates dendritic growth and enhances the longevity and safety of the Li anode. | ||
| Fig. 1 Schematic illustration of Li deposition behaviors on (a) LCS–Ag–Li substrate and (b) bare Li substrate. | ||
To assess the viability of this dual-protective artificial interface for Li metal anode, a nano-Ag and LCS-modified anode was prepared by a sample solution immersion reaction. Initially, LCS molecules—characterized by numerous hydrogen-bonded sulfonate and amino groups, as well as flexible glycosyl rings—were synthesized through a simple sulfonation reaction.29 The successful synthesis of LCS was confirmed by Fourier-transform infrared spectroscopy (FTIR) spectra and X-ray photoelectron spectroscopy (XPS) analyses (Fig. 2a and b). The FTIR spectra revealed absorption peaks corresponding to the functional groups (–SO3Li), characterized by distinctive bands associated with sulfur–oxygen (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and S–O) and carbon–oxygen (C–O–S) linkages. Notably, the typical absorption peaks for SO at 1203 cm−1 and –SO3Li at 1620 cm−1 in LCS (Fig. 2a, green line) and on the surface of LCS-coated Li foil (Fig. 2a, orange line) displayed a slightly red shift, indicating successful integration of the LCS film with Li foils.30 Nano-Ag, possessing charge distribution capabilities and lithium-ion flux regulation, actively forms a lithium–silver alloy instead of serving as a passive layer. Consequently, it has been selected to construct the inner lithiophilic nucleation layer of the dual protective artificial interface, which ensures uniform lithium deposition and inhibits dendrite formation on the surface of the lithium metal anode.31
O and S–O) and carbon–oxygen (C–O–S) linkages. Notably, the typical absorption peaks for SO at 1203 cm−1 and –SO3Li at 1620 cm−1 in LCS (Fig. 2a, green line) and on the surface of LCS-coated Li foil (Fig. 2a, orange line) displayed a slightly red shift, indicating successful integration of the LCS film with Li foils.30 Nano-Ag, possessing charge distribution capabilities and lithium-ion flux regulation, actively forms a lithium–silver alloy instead of serving as a passive layer. Consequently, it has been selected to construct the inner lithiophilic nucleation layer of the dual protective artificial interface, which ensures uniform lithium deposition and inhibits dendrite formation on the surface of the lithium metal anode.31
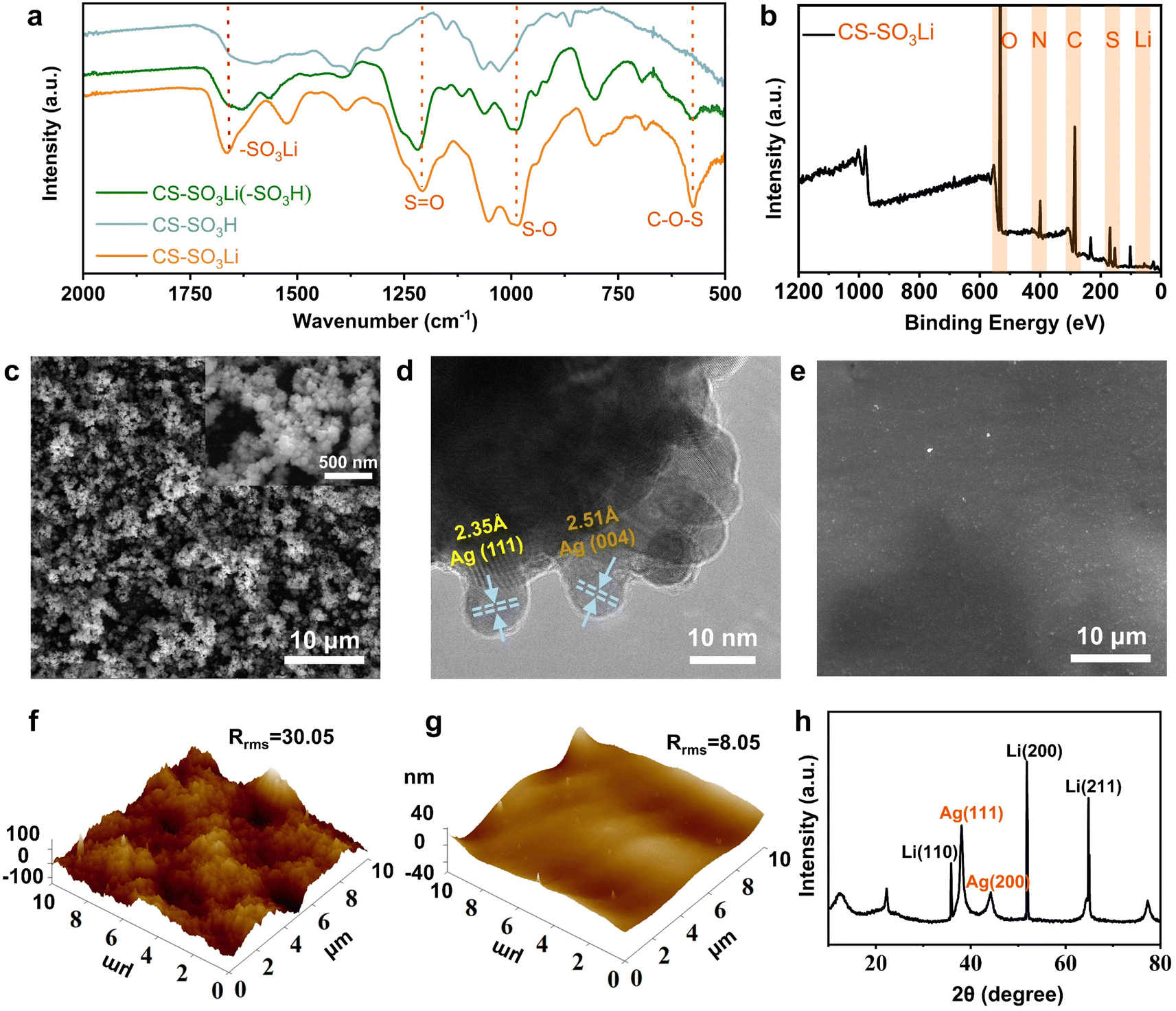 | ||
| Fig. 2 Chemical and morphological analysis of LCS and nano-Ag modifications: (a) FTIR spectra of CS–SO3H (blue line), CS–SO3Li (green line) and AgLi–CS–SO3Li (orange line) samples; (b) XPS spectra of LCS; (c) SEM and (d) TEM micrographs of nano-Ag modified lithium anodes; (e) SEM images of LCS–Ag coated anodes; AFM topographies with (f) nano-Ag and (g) LCS–Ag coatings; (h) XRD patterns of LCS–Ag modified anode. | ||
To further obtain the optimal composite interface layer, the nano-Ag layer was grown in situ on one side of a Li foil through a solution immersion reaction with AgNO3 as the silver source. Subsequently, the LCS powder are introduced into this solution to coordinate with or interact with the AgLi layer, forming a dual-protective SEI. The synthesis procedures are shown in Fig. S1a (ESI†). The morphology of the nano-Ag modified Li (AgLi) anode and LCS–Ag coated Li (C–AgLi) anode was characterized by scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM) respectively. The SEM images of the nano-Ag modified Li anode revealed a dense and uniformly distributed Ag nanoparticles formation over the anode surface (Fig. 2c). Further, the HR-TEM images and its Ag elemental mapping detail the crystalline structure of the nano-Ag with the lattice spacings of 2.35 Å and 2.51 Å corresponding to the Ag (111) and Ag (004) planes, respectively (Fig. 2d and S2†). This microstructure is similar to the lattice parameters of lithium metal which facilitates the formation of lithium–silver alloy, reducing the active sites for uneven Li deposition and promoting interaction between the nano-Ag and the Li ions.32 The SEM images and its EDS elemental mapping of the LCS–Ag coating on the Li anodes as showed in Fig. 2e, displaying the smooth and homogenous surface, which is advantageous for diminishing surface irregularities, thereby mitigating uneven Li deposition (Fig. S3†). Cross-sectional SEM images in Fig. S4 (ESI†) of the C–Li and C–AgLi anode also confirm the integration of the LCS film with AgLi anode. Atomic force microscopy (AFM) results further revealed a notably smooth surface, which contrasts sharply with the typical rough morphology of uncoated Li anodes (Fig. 2f, g and S5†). X-ray diffraction (XRD) patterns are showed in Fig. 2h, indicating distinct peaks associated with both the metallic Ag and Li phases, which suggest successful integration of the nano-Ag into the Li matrix without disrupting the inherent Li crystal structure. The flexibility of LCS was test by AFM's force–displacement curves and the Young's modulus of LCS was conducted to 281 MPa (Fig. S6†), which is flexible enough to resist stress and buffer the volume change during the Li plating/stripping process. According to the linear elasticity theory proposed by Newman and Monroe, the interfacial stability in lithium/polymer systems was highly dependent on the bulk mechanical characteristics of the polymers exerting on electrodes.33 In terms of the elasticity and affinity of LCS for AgLi anode, this dual-protective artificial interface was competent for LMBs. The ionic conductivity of the LCS film was determined to evaluate the practicality of the artificial SEI for Li metal anode. The ionic conductivity of the LCS film, reached to 7.81 × 10−5 S cm−1 (Fig. S7†), surpasses that of traditional SEI materials like LiF (10−31 S cm−1) and Li2CO3 (10−8 S cm−1).34 This enhancement highlights the LCS–Ag layer's ability to maintain uniform Li-ion flux, significantly improving upon standard SEI components.
To demonstrate the dendrite-free, uniform lithium deposition facilitated by the LCS–Ag coating, SEM images and electrochemical characterizations were performed on various anodes. The lithium deposition process on various substrates depicted in Fig. 3a and b. For bare lithium foil, an uneven distribution of ion flux and random lithium nucleation lead to irregular SEI formation and subsequent growth of lithium dendrites, which contribute to interfacial cracking and the development of inactive (‘dead’) lithium (Fig. 3a). In contrast, the C–AgLi substrate modulates Li+ flux and ensures uniform nucleation sites, resulting in homogeneous and dendrite-free lithium deposition (Fig. 3b). These phenomena are initially confirmed through SEM analysis of the Li plating/stripping behaviors on the different substrates after a plating capacity of 2 mA h cm−2. Notably, the bare Li foil exhibited a mass of twisted and tangled moss-like dendrites (Fig. 3c), while the 3D nano-Ag modified Li foil displayed no discernible dendritic structures, featuring relatively compact lithium depositions (Fig. 3d). Lithium deposition was constrained under the LCS film coating due to its notable mechanical elasticity on the C–Li substrate (Fig. 3e). However, star-shaped pores and uneven wrinkles emerged on the surface, attributed to internal stress. This suggests that a single LCS layer can only partially mitigate lithium dendrite growth. Conversely, due to the synergistic effects of the lithiophilic 3D nano-Ag structure and a robust LCS coating layer, the formation of lithium dendrites has been effectively and completely suppressed on the C–AgLi substrate (Fig. 3f). To further support this conclusion, we have provided additional in situ optical microscopy using a visualization cuvette-type optical cell, detailed further in ESI Videos 1 and 2.† The imagery captured at various time intervals shows that moss-like dendrites progressively form on the bare Li electrode, leading to reduced Li utilization and the risk of irreversible electrode damage (Fig. S8†). In contrast, the C–AgLi anode, tested under identical conditions, demonstrates that lithium deposition was constrained under the LCS film coating. The results consistently demonstrate that the presence of the mechanically elastic LCS film leads to constrained and more controlled lithium deposition.
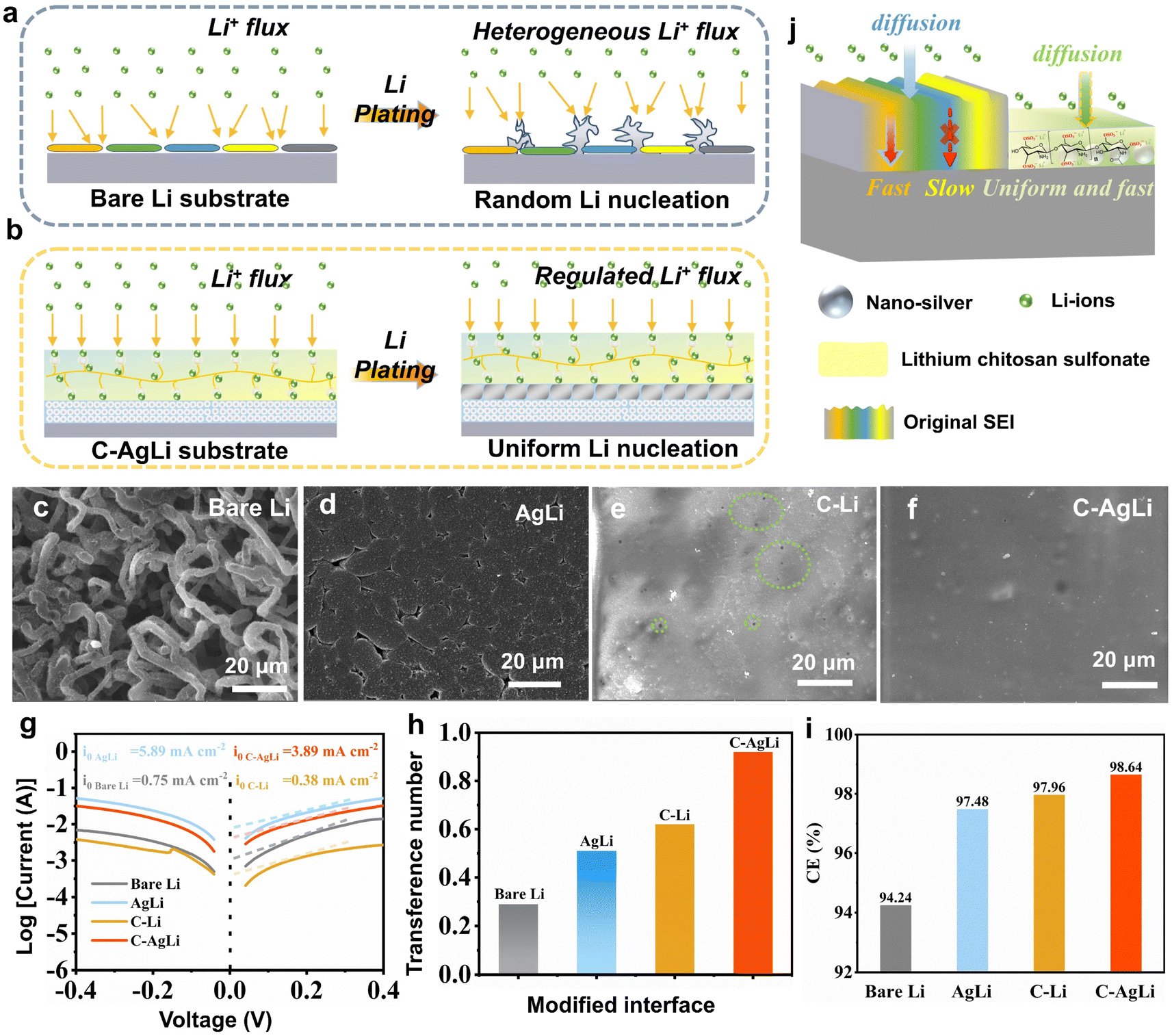 | ||
| Fig. 3 Comparison for lithium deposition behaviors on (a) bare lithium substrate and (b) C–AgLi substrate. SEM morphologies of lithium metal deposition on (c) bare Li, (d) AgLi, (e) C–Li, (f) C–AgLi substrates, respectively. Electrochemical characterizations: (g) Tafel curves, (h) ion transference number and (i) CE measurements for various modified interfaces. (j) Schematic diagram depicting the interfacial ion transport mechanism. | ||
To investigate aforementioned mechanisms of Li plating/stripping, comprehensive electrochemical tests were performed on four types of anodes, including assessments of exchange current density, Li-ion transference number, and CE. Initially, Tafel plots derived from Li‖Li cell tests were analyzed to calculate the exchange current density (I0), as illustrated in Fig. 3g. The I0 value for the cell assembled with C–AgLi reached 3.89 mA cm−2, significantly higher than that of bare Li by over fivefold (0.75 mA cm−2) and C–Li by over tenfold (0.38 mA cm−2). The AgLi modified anode exhibited a substantial increase in exchange current density, indicating that a uniform interfacial electric field enhances surface charge transfer. Conversely, the exchange current density on the C–AgLi modified anode decreased slightly, attributed to the abundant sulfonate groups on the polymer surface that bind with lithium ions, thereby impeding their transfer. These results corroborate the enhanced Li+ transfer kinetics within the SEI formed with LCS–Ag, an enhancement facilitated by the synergistic effects arising from the interaction of nano-Ag with uniform ion and electric fields. Additionally, the FTIR absorption peaks corresponding to the –SO3Li groups shifted from at SO (1223 cm−1) to 1118 cm−1, a slightly red shift indicative of coordination and intermolecular interactions with nano-silver (Fig. S9†). This shift further confirms that these interactions, which influence the arrangement of the sulfonate groups, play a crucial role in ion conduction and interfacial stability. Moreover, the capability of Li-ion transfer through the protective layer is crucial for achieving uniform Li metal deposition. However, most artificial SEIs have low Li-ion transference numbers, leading to uneven transfer and large concentration gradients near the Li anode, resulting in non-uniform Li metal deposition. The Li ion transference number (t+) is calculated based on the Bruce–Vincent formula by steady-state current tests to determine the Li-ion conducting ability of the various modified interface layer (Fig. S10†). A comparative analysis of t+ across different Li metal anodes is summarized in Fig. 3h. In symmetric cells employing bare Li, a low t+ value of 0.29 is recorded, indicative of the rapid migration of anions relative to solvated Li ions in the liquid electrolyte. The introduction of a nano-Ag-modified layer significantly enhances the t+ to 0.55. For LCS coated Li (C–Li) metal substrate, there is a slight improvement in t+, reaching 0.63. By integrating nano-Ag with an LCS coating, the C–AgLi substrate achieves a t+ of 0.92, which is indicative of a single Li-ion conductor. This configuration not only promotes homogeneous Li-ion transfer but also enhances ionic conductivity and reduces concentration polarization.
High CE and long cycle life typically indicate stable Li plating/stripping behaviors with fewer parasitic reactions. A modified Aurbach method, as described in previous reports, was employed to investigate the CE of various electrodes.35 The CEs of bare Li, AgLi, and C–Li electrodes displayed minimal differences, registering at 94.24%, 97.49%, and 97.96%, respectively (Fig. S11†). In contrast, the C–AgLi cells demonstrated an exceptional CE of 98.64%. These results further underscore the effectiveness of the dual-regulation of Li-ion/electric field in enhancing the CE.
To demonstrate the enduring protection offered by the modified layer of Li metal during prolonged cycling, it is crucial for the artificial SEI to effectively regulate Li deposition morphology, reduce interface impedance, and mitigate side reactions. Surface compositional changes offer a direct measure of interface stability throughout the cycling process. To elucidate the evolution of different electrodes, SEM imaging and ex situ XPS analyses were conducted on Li metal anodes based on different substrates after 100 cycles: bare Li, AgLi, C–Li, and C–AgLi substrates. As depicted in Fig. 4a, following 100 cycles, the surface of the bare Li anode displays pronounced corrosion and a notably rough morphology. This degradation is attributed to ongoing reactions between the unprotected metallic lithium and the electrolyte during lithium deposition, leading to substantial accumulation of ‘dead lithium’ at the interface. In contrast, the AgLi anode, though exhibiting a rough surface, shows a relatively lower quantity of ‘dead lithium’, but the SEI on the AgLi anode is more uniform (Fig. 4b). This uniformity is facilitated by the consistent Li+ ion flux near the electrode surface, which is conducive to the formation of a structurally coherent SEI, thereby mitigating interface degradation. On the other hand, the lithium metal anode modified solely with LCS displays numerous interface cracks and a small amount of dead lithium after 100 cycles, as illustrated in Fig. 4c. In stark contrast, the C–AgLi anode, benefiting from the dual protection afforded by the LCS and a nano-Ag layer, exhibits a completely smooth and intact surface morphology, as evidenced in Fig. 4d. This finding underscores the significant role of controlling lithium nucleation and regulating the deposition flux of lithium ions in enhancing interface stability during the electrochemical cycling process.
 | ||
| Fig. 4 Comparison for morphologies, components, and EIS of lithium metal anodes based on different substrates after 50 cycles. Detailed SEM images of (a) bare Li, (b) AgLi, (c) C–Li, and (d) C–AgLi electrode after Li deposition; XPS spectra for the (e, f, g and h) C 1s and (i, j, k and l) Li 1s of the (e and i) bare Li, (f and j) AgLi, (g and k) C–Li, and (h and l) C–AgLi electrode, respectively. EIS spectra of symmetric cells with different substrates as working electrode after 1st, 10th and 50th cycles: (m) bare Li, (n) AgLi, (o) C–Li, and (p) C–AgLi. The capacities of Li deposition are 2 mA h cm−2. | ||
High-resolution XPS analysis after 50 cycles assessed interface stability across electrodes. The bare Li anode showed peaks for interfacial reaction byproducts (Fig. 4e and i), and its Li 1s spectrum indicated side reactions with peaks for Li2O, LiF, and Li–CO2− (53.4, 56.3, and 54.95 eV, respectively). Similarly, the AgLi anode's SEI closely resembled that of the bare Li, underscoring the necessity for effective electrolyte interaction prevention (Fig. 4f and j). The C–Li anode still exhibited polymeric side products, including oxidized Li states (Fig. 4g and k). In stark contrast, the C–AgLi anode demonstrated minimal electrolyte decomposition and a predominantly stable SEI, evidenced by its C 1s spectrum showing only native polymer peaks and negligible byproduct peaks (Fig. 4h), with its Li 1s spectrum confirming superior interfacial stability (Fig. 4l). Moreover, a notable shift in the C–S bond for the C–AgLi samples compared to the C–Li samples (Fig. 4g and h), evident from the XPS spectra, is attributed to coordination interactions between the sulfonic groups and –NH– functionalities on the LCS film with Ag. This coordination is further supported by a red shift in the –SO3Li bands observed in the infrared spectra (Fig. S9†), confirming the modification in the electronic structure. Extended XPS depth profiling was employed to assess the chemical stability of the LCS–AgLi layer after prolonged cycling, with the findings illustrated in Fig. S12.† The XPS analyses reveal that the chemical states of carbon (C) and sulfur (S) within the LCS–AgLi layer exhibit negligible changes, even following extensive depth. This remarkable chemical stability underscores the layer's resistance to common degradation mechanisms, including electrolyte decomposition and the deterioration of active materials, thereby affirming the efficacy of the LCS–AgLi layer in maintaining its structural and chemical integrity under rigorous operational conditions. Fig. 4m–p presents the EIS results of symmetric cells employing various substrates over the 1st, 10th, and 50th cycles, detailing changes in interface impedance and ion diffusion through the SEI. Each plot displays a semicircle at high to mid frequencies, indicative of the charge transfer resistance (Rct), and a sloping line at low frequencies, representing the Warburg impedance due to ion diffusion through the SEI. The EIS plots reveal that modifications with nano-Ag, LCS, and particularly C–AgLi (Fig. 4p) significantly reduce the impedance values, suggesting enhanced ion transport and reduced interface resistance, which contribute to improved battery performance and cycle stability.
Electrochemical cycling stability was assessed using Li‖Li symmetric cells with various electrodes. Under conditions of 1.0 mA cm−2 and 1.0 mA h cm−2, the Li‖Li cell exhibited increasing overpotentials, reaching 121 mV by 800 hours before failure, as shown in Fig. 5a. In contrast, the C–AgLi‖C–AgLi cell maintained a low overpotential of 14 mV even after 1000 hours. The C–Li‖C–Li and AgLi‖AgLi cells, although stable, had higher overpotentials than the C–AgLi cell. Further tests at increased current densities and deposition capacities revealed the C–AgLi anode's superior stability. While the bare Li anode faced significant overpotentials of ∼200 mV after 200 hours, the C–AgLi cell remained stable with only 130 mV overpotential after 400 hours (Fig. 5c). Rate cycling tests to assess interfacial charge and mass transfer showed that the C–AgLi anode significantly outperformed other electrodes in reducing overpotentials. As current density increased from 0.5 mA cm−2 to 5.0 mA cm−2, the C–AgLi anode reduced overpotentials by 65.72% compared to bare Li, 60.33% to C–Li, and 57.21% to AgLi (Fig. 5b and d). This further demonstrates significant performance improvement after modification with the ion-conducting polymer LCS and lithiophilic nano-silver.
 | ||
| Fig. 5 (a) Cycling performance of Li‖Li symmetric cells with four different Li anodes at 1.0 mA cm−2 with a capacity of 1.0 mA h cm−2. (b) Rate performance of Li‖Li symmetric cells under different current densities (0.5, 1.0, 2.0, 3.0, 4.0, and 5 mA cm−2) with a fixed plating/stripping capacity of 1.0 mA h cm−2. (c) Voltage–time curves of Li‖Li symmetric cells at 5.0 mA cm−2 with a capacity of 2.0 mA h cm−2. (d) Overpotential measurements at varying current densities. (e) Cycling performance and (f) rate performance of Li‖LCO full cells. | ||
The cycling performance and rate capability of the Li‖LCO and Li‖LFP full cells further demonstrate the practical viability of the C–AgLi substrate in complete battery systems, respectively. Full cells with C–AgLi and bare Li anodes were evaluated for their application potential. Initially, both C–AgLi‖LiCoO2 and Li‖LiCoO2 cells showed identical discharge capacities of 142 mA h g−1. However, after 350 cycles, the C–AgLi‖LiCoO2 cell maintained a capacity of 119 mA h g−1, with an 83.8% retention rate, significantly outperforming the Li‖LiCoO2 cell's 42.9% retention at 61 mA h g−1, highlighting superior interface stability and capacity retention (Fig. 5e). Furthermore, LFP full cells featuring C–AgLi and bare Li anodes exhibited cycling performance consistent with that observed in LCO cells (Fig. S13†). Moreover, the Li‖LiCoO2 cells maintained high capacity retention under conditions of elevated areal capacities and rapid cycling rates, highlighting the interface's robust ability to accommodate swift changes in charge and discharge cycles while sustaining minimal losses in capacity or efficiency (Fig. S14†). Rate charge–discharge tests revealed better kinetic performance for the C–AgLi anode; at current densities from 1.0C to 10C, capacities were 142, 135, 123, and 93 mA h g−1, respectively, compared to the Li‖LiCoO2 cell's 126, 119, 108, and 86 mA h g−1 (Fig. 5f). This enhanced performance is attributed to the nano-silver layer in the C–AgLi anode, which facilitates uniform lithium ion deposition and inhibits dendrite growth, enhanced by the ionic polymer's protective effects for stable, long-term performance.
The initial capacity of LDS–Cu–Li∥LFP was 154 mA h g−1, and after 300 cycles, the capacity was 142 mA h g−1, corresponding to capacity retention of more than 92% (Fig. S13†). However, the capacity of the PVDF–Cu–Li∥LFP rapidly decreased to 16 mA h g−1 after 300 cycles, with a capacity retaining rate of less 10%. As a result, the significantly improved rate capability and cycle performance unambiguously demonstrated that LDS can effectively prevent the growth of Li dendrites and the production of dead Li, as well as stabilize the interface.
Conclusions
In summary, we developed a self-organizing, dual-modified interface for lithium metal anodes, incorporating lithiophilic nano-Ag and LCS film. This interface significantly enhanced electric field uniformity and ionic conductivity, leading to uniform lithium deposition and improved electroplating/stripping performance. Symmetric cells equipped with the C–AgLi electrode demonstrated over 1000 h of stability at 1 mA cm−2. At increased current densities of 5 mA cm−2, the lifespan of C–AgLi cells was doubled compared to those with bare lithium anodes. In full Li‖LiCoO2 cells, C–AgLi maintained 83.8% capacity retention after 350 cycles, substantially improving safety and energy density, crucial for the practical application of lithium metal batteries.Experimental section/methods
Experimental details are available from ESI.†Data availability
All datasets generated and analyzed during the current study are available in the Baidu Cloud, which can be accessed at https://1drv.ms/f/s!AiblYpjI7x0Ha5fCyEWkdik3e6k?e=AIKb2q. The datasets include all experimental results and characterization data relevant to the development and analysis of lithium battery technologies discussed in this manuscript. Additional ESI,† such as detailed experimental procedures and supplementary analyses, is included in the ESI† files accompanying this manuscript. If there are specific data that are not available in the public repository due to ethical or legal restrictions (such as proprietary data or sensitive information), these data can be made available from the corresponding author upon reasonable request, subject to necessary confidentiality agreements.Author contributions
Zhiqiang Li: conceptualization, methodology, investigation, data curation, formal analysis, visualization, writing-original draft, writing-review. Kemeng Liao: formal analysis, writing-review, data curation. Lihong Yin: investigation and data curation. Zongrun Li: investigation and data curation. Yingzhi Li: data curation and writing-review. Hongzhi Wang: investigation and data curation. Ning Qin: writing-review. Shuai Gu: formal analysis, visualization. Yingzhi Li: writing-review. Jingjing Chen: data curation. Weihua Wan: writing – review, and editing. Zhouguang Lu: conceptualization, supervision, resources, funding acquisition, project administration, writing – review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Basic Research Project of the Science and Technology Innovation Commission of Shenzhen (No. JCYJ 20220818100418040), the Southern University of Science and Technology Teaching Enhancement and Innovation Project (Y01251846), the Fundamental Research Funds for the Central Universities (2024MS103), and the National Natural Science Foundation of China (No. U22A20439). Help on SEM, XPS, TEM and AFM characterizations from the Core Research Facilities and at Southern University of Science and Technology are gratefully appreciated.References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, J. Power Sources, 2013, 226, 272–288 CrossRef CAS.
- J. B. Goodenough, Energy Storage Mater., 2015, 1, 158–161 CrossRef.
- X. Shen, H. Liu, X. B. Cheng, C. Yan and J. Q. Huang, Energy Storage Mater., 2018, 12, 161–175 CrossRef.
- W. B. Lu, Z. P. Li, H. F. Huang and L. F. Jiao, Nano Res., 2023, 16, 8219–8252 CrossRef CAS.
- P. G. Li, Z. F. Liu, Y. T. Peng, S. S. Yang, T. Meng, Y. H. Hu, Y. J. Jiang, H. Sun, Q. Li and X. L. Hu, Nano Res., 2024, 17, 2746–2754 CrossRef CAS.
- H. J. Kim, N. Umirov, J. S. Park, J. H. Lim, J. F. Zhu, S. S. Kim and S. T. Myung, Energy Storage Mater., 2022, 46, 76–89 CrossRef.
- J. Y. Kim, O. B. Chae, G. Kim, W. B. Jung, S. Choi, D. Y. Kim, S. Moon, J. Suk, Y. Kang, M. Wu and H. T. Jung, Energy Environ. Mater., 2022, 6, 12392 CrossRef.
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- D. R. MacFarlane, J. H. Huang and M. Forsyth, Nature, 1999, 402, 792–794 CrossRef CAS.
- R. Rojaee and R. Shahbazian-Yassar, ACS Nano, 2020, 14, 2628–2658 CrossRef CAS PubMed.
- A. Santiago, X. Judez, J. Castillo, I. Garbayo, A. S. de Buruaga, L. Qiao, G. Baraldi, J. A. Coca-Clemente, M. Armand, C. Li and H. Zhang, J. Phys. Chem. Lett., 2020, 11, 6133–6138 CrossRef CAS PubMed.
- C. Y. Cui, C. Y. Yang, N. Eidson, J. Chen, F. D. Han, L. Chen, C. Luo, P. F. Wang, X. L. Fan and C. S. Wang, Adv. Mater., 2020, 32, 906427 Search PubMed.
- H. Xie, C. Yang, Y. Ren, S. Xu, T. R. Hamann, D. W. McOwen, E. D. Wachsman and L. Hu, Nano Lett., 2021, 21, 6163–6170 CrossRef CAS PubMed.
- W. Cao, Q. Li, X. Yu and H. Li, eScience, 2022, 2, 47–78 CrossRef.
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- Y. Liu, D. Lin, P. Y. Yuen, K. Liu, J. Xie, R. H. Dauskardt and Y. Cui, Adv. Mater., 2017, 29, 605531 Search PubMed.
- Y. Z. Chen, A. Elangovan, D. L. Zeng, Y. F. Zhang, H. Z. Ke, J. Li, Y. B. Sun and H. S. Cheng, Adv. Funct. Mater., 2020, 30, 906444 Search PubMed.
- J. R. Wang, M. M. Wang, X. D. He, S. Wang, J. M. Dong, F. Chen, A. Yasmin and C. H. Chen, ACS Appl. Energy Mater., 2020, 3, 7265–7271 CrossRef CAS.
- N. Qin, S. C. Yu, Z. W. Ji, Y. F. Wang, Y. Z. Li, S. Gu, Q. M. Gan, Z. Y. Wang, Z. Q. Li, G. F. Luo, K. L. Zhang and Z. G. Lu, CCS Chem., 2022, 4, 3587–3598 CrossRef CAS.
- T. Zhou, J. D. Shen, Z. S. Wang, J. Liu, R. Z. Hu, L. Z. Ouyang, Y. Z. Feng, H. Liu, Y. Yu and M. Zhu, Adv. Funct. Mater., 2020, 30, 909159 Search PubMed.
- J.-F. Ding, Y.-T. Zhang, R. Xu, R. Zhang, Y. Xiao, S. Zhang, C.-X. Bi, C. Tang, R. Xiang, H. S. Park, Q. Zhang and J.-Q. Huang, Green Energy Environ., 2023, 8, 1509–1530 CrossRef CAS.
- H. C. Hao, T. Hutter, B. L. Boyce, J. Watt, P. C. Liu and D. Mitlin, Chem. Rev., 2022, 122, 8053–8125 CrossRef CAS PubMed.
- F. Liu, Z. Zhang, S. Ye, Y. Yao and Y. Yu, Acta Phys.-Chim. Sin., 2021, 37, 2006021 Search PubMed.
- J.-P. Wang, F. Lang and Q. Li, EcoMat, 2023, 5, 12354 CrossRef.
- S. Ni, M. Zhang, C. Li, R. Gao, J. Sheng, X. Wu and G. Zhou, Adv. Mater., 2023, 35, 209028 Search PubMed.
- A. Sarkar, V. R. Dharmaraj, C.-H. Yi, K. Iputera, S.-Y. Huang, R.-J. Chung, S.-F. Hu and R.-S. Liu, Chem. Rev., 2023, 123, 9497–9564 CrossRef CAS PubMed.
- L. Y. Liu, M. Orbay, S. Luo, S. Duluard, H. Shao, J. Harmel, P. Rozier, P. L. Taberna and P. Simon, ACS Nano, 2022, 16, 111–118 CrossRef CAS PubMed.
- K. M. Yang, K. W. Yang, M. S. Cho, S. Kim and Y. K. Lee, Chem. Eng. J., 2023, 454, 140191 CrossRef.
- M. F. Hamza, H. Mira, Y. Z. Wei, S. M. Aboelenin, E. Guibal and W. M. Salem, Chem. Eng. J., 2022, 441, 135941 CrossRef CAS.
- Z. T. Wondimkun, W. A. Tegegne, J. Shi-Kai, C. J. Huang, N. A. Sahalie, M. A. Weret, J. Y. Hsu, P. L. Hsieh, Y. S. Huang, S. H. Wu, W. N. Su and B. J. Hwang, Energy Storage Mater., 2021, 35, 334–344 CrossRef.
- S. Pyo, S. Ryu, Y. J. Gong, J. Cho, H. Yun, H. Kim, J. Lee, B. Min, Y. Choi, J. Yoo and Y. S. Kim, Adv. Energy Mater., 2022, 13, 203573 Search PubMed.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, 1850854 CrossRef.
- J. Zhu, P. Li, X. Chen, D. Legut, Y. Fan, R. Zhang, Y. Lu, X. Cheng and Q. Zhang, Energy Storage Mater., 2019, 16, 426–433 CrossRef.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03128h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |