
Customized CO2 electroreduction to methane or ethylene by manipulating *H and *CO adsorption on Cu/CeOx catalysts†
Tinghui
Yang
,
Yingbing
Zhang
,
Zichao
Huang
,
Jianping
Yang
 * and
Min
Kuang
*
* and
Min
Kuang
*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: jianpingyang@dhu.edu.cn; mkuang@dhu.edu.cn
First published on 5th July 2024
Abstract
The coverage of *CO and *H intermediates on the surface of a catalyst plays a pivotal role in determining the selectivity towards C1 or C2 products in the electrochemical CO2 reduction reaction (CO2RR). In this study, we engineered two types of interfaces involving copper and rare earth metal oxides, specifically Cu/CeOx and Cu/CuCeOx solid solution, which exhibit enhanced binding affinities for *H and *CO adsorbates in the CO2RR, respectively. As a result, the Cu/CuCeOx catalyst delivered an ethylene faradaic efficiency of 40.2% at a partial current density of −245.7 mA cm−2, whereas the Cu/CeOx catalyst presented a methane faradaic efficiency of 38.6% at a partial current density of −198.3 mA cm−2. Results of theoretical and experimental analyses have demonstrated that the Cu–Ce–Ox solid solution markedly enhances *CO adsorption by stabilizing Cu+ species, thereby favoring its dimerization to ethylene rather than converting to methane through hydrogenation. This investigation elucidates a strategy for directing the selective electroproduction of C1 or C2 compounds from the CO2RR by effectively manipulating *H and *CO adsorption on Cu/CeOx catalysts.
 Min Kuang | Min Kuang is currently a professor in the College of Materials Science and Engineering at Donghua University, China. She received her PhD from the Laboratory of Advanced Materials at Fudan University. Following this, she joined the School of Materials Science and Engineering at Nanyang Technological University as a postdoctoral research associate. Her research focuses on developing advanced electrochemical C1-to-fuel conversion systems and exploring efficient electrocatalysts. |
Introduction
The relentless rise in atmospheric CO2 levels, predominantly driven by anthropogenic activities, has been identified as a principal factor exacerbating global warming, eliciting widespread concern across the globe.1,2 Within this critical context, the electrochemical carbon dioxide reduction (CO2RR), especially when powered by renewable energy sources like solar and wind, has emerged as a vital strategy for carbon balance restoration.3–5 Distinct from the generation of simpler C1 products like formic acid (HCOOH) and carbon monoxide (CO),6 there is an intensified demand for the production of high energy density hydrocarbons, notably methane (CH4) and ethylene (C2H4), which are envisioned as potential sustainable fuels.7–9 To date, copper-based materials, endowed with optimal adsorption energy for the *CO intermediate, have been heralded as the most effective catalysts for facilitating the conversion of CO2 into valuable hydrocarbons.10,11 In pursuit of this, a spectrum of Cu-based electrocatalysts, encompassing metallic Cu, Cu oxides,12–14 Cu-integrated metal–organic frameworks (MOFs),15,16 and Cu-centric molecules,17,18 have been meticulously developed, marking significant strides in the electrocatalytic conversion of CO2 to CH4 or C2H4. Nevertheless, the design of cost-effective and durable catalysts tailored for the targeted transformation of CO2 into high-energy products continues to pose a formidable challenge.The mechanism underlying the CO2RR involves the initial reduction of CO2 to adsorbed *CO, succeeded by *CO hydrogenation and/or C–C coupling processes.8,19 Recent investigations have illuminated that *CO hydrogenation to *CHO represents the rate-limiting step in CH4 generation, while *CHO also acts as a pivotal intermediate for multi-carbon (C2) products.4,20 Moreover, the journey from *CO to hydrocarbons encapsulates a multi-step hydrogenation sequence, with *H emerging as a crucial intermediary for hydrocarbon synthesis. Consequently, fine-tuning the adsorption dynamics of *CO and *H presents a promising avenue for steering the CO2RR towards desired products, particularly for CH4 and C2H4. For example, Zheng et al. showcased that the strategic incorporation of two metal dopants, Zn and Mn, into Cu matrices could simultaneously modulate the coverage of *H and *CO, enabling the fine-tuning of selectivity between CH4 and C2H4.21 Similarly, Sargent et al. effectively manipulated *CO coverage through varying CO2 concentrations, achieving tunable product specificity from C2H4 to CH4.22 In a parallel vein, Wei and colleagues enhanced the adsorption of the CO intermediate by augmenting polyaniline on Cu substrates, thereby activating CO2 molecules and yielding a significant faradaic efficiency (FE) for C2 products.23 Despite these advances, attaining high selectivity shifts between desired products using analogous materials remains elusive, thereby underscoring the impetus for novel catalyst development.
In this report, we introduce an innovative strategy employing functional rare-earth metal oxides to facilitate selectivity switching between CH4 and C2H4 products by modulating *H and *CO adsorption (Fig. 1). Two distinct electrocatalysts, Cu/CeOx and a Cu/CuCeOx solid solution (0 < X < 2), were synthesized, with the CeOx component enhancing *H coverage through expedited water dissociation, while Cu–Ce–Ox solid solutions stabilize Cu+ species, augmenting *CO adsorption and catalyzing the C–C coupling process. Electrochemical CO2RR evaluations revealed that the Cu/CuCeOx catalyst achieved an ethylene FE of 40.2% at a partial current density of −245.7 mA cm−2, in contrast to the Cu/CeOx variant, which exhibited a methane FE of 38.6% at a partial current density of −198.3 mA cm−2. Density functional theory (DFT) calculations further illustrated that Cu–Ce–Ox solid solutions significantly reduce the formation energy of the *COCHO intermediate, thereby enhancing C2H4 production. This paradigm of adjusting CO2RR selectivity via intermediary coverage heralds new avenues for designing innovative Cu-based nanocomposite catalysts.
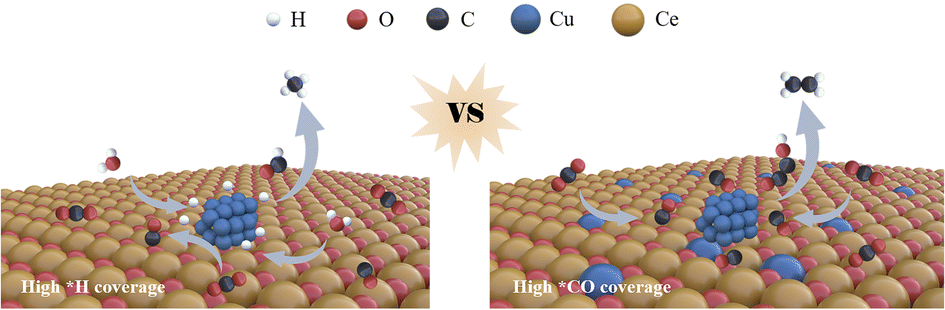 | ||
| Fig. 1 Diagrams of methane and ethylene production on Cu/CeOx and Cu/CuCeOx during the CO2RR, respectively. | ||
Results and discussion
The Cu/CuCeOx catalyst was synthesized utilizing the co-precipitation methodology delineated in previous studies,24 and subsequently subjected to annealing in a mixed atmosphere of 5 vol% H2/Ar (Fig. 2a and S1†). In contrast, the Cu/CeOx catalyst was fabricated through a bifurcated procedure: initially, CeOx was prepared devoid of any Cu precursor via coprecipitation; thereafter, Cu/CeOx was produced by an impregnation technique. The X-ray diffraction (XRD) analysis of the synthesized samples (Fig. 2b) reveals diffraction peaks at 28.55°, 32.08°, 47.48°, 56.34°, 59.09°, 69.42°, 76.70°, 79.08°, and 88.43°, congruent with the established cubic fluorite-like structure of CeO2 (PDF# 43-1002). Additionally, characteristic diffraction peaks of Cu are discernible (indicated by a pentagram at 43.30°, 50.43°, and 74.13°) corresponding to PDF# 04-0836. Notably, the (111) plane of Cu/CuCeOx exhibits a discernible shift towards higher Bragg angles compared to CeOx and Cu/CeOx (Fig. 2c), suggesting the formation of a Cu–Ce–Ox solid solution due to the substitution of a larger cation by a smaller one.25–27 As the Cu content continues to increase, some of the copper precipitates out, forming clustered copper species on the Cu/CuCeOx nanorods.28 And no diffraction peaks corresponding to Cu can be found when the copper loading is below 30 wt%, which may be due to the high dispersion of Cu nanoparticles with too small particle sizes on the surface of the ceria support to be identified by the XRD (Fig. S2†).29,30 Raman spectroscopy of the catalysts reveals an intense band around 459 cm−1 and another at approximately 595 cm−1, corresponding to the F2g vibration pattern and defect-induced mode (Ov) in a cubic fluorite structure, respectively.31 Notably, the F2g bands of Cu/CuCeOx exhibit a redshift to lower frequencies with increasing Cu loading, compared to pure CeOx (Fig. S3†), indicative of Cu atom doping into the CeOx lattice and Cu/CuCeOx interface formation.32,33 This results in extended Ce–O bond lengths at the interface and lattice distortions due to the atomic radius disparity between Cu+ and Ce4+/Ce3+,34,35 corroborating the XRD findings. | ||
| Fig. 2 (a) Synthetic route of the Cu/CuCeOx catalyst by a coprecipitation method. (b) XRD spectra, (c) local amplification of (111) and (d) Raman spectra for CeOx, Cu/CeOx, and Cu/CuCeOx catalysts (CeO2, yellow, PDF# 43-1002; Cu, blue, PDF# 04-0836). (e and f) HRTEM images and (g) EDX elemental distribution mapping images of Cu/CuCeOx. | ||
Transmission electron microscopy (TEM, Fig. S4†) images depict the Cu/CuCeOx as possessing rod-like nanostructures. The dynamic light scattering in Fig. S5† further illustrates that the particle size distribution between 50 and 200 nm is slightly wider than that presented by TEM, which may be caused by partial rupture or aggregation of cerium dioxide. High-resolution TEM (HRTEM) images (Fig. 2e and f) identify the main interplanar spacings of 0.309 nm corresponding to the Cu–Ce–Ox (111), slightly smaller than the Cu/CeOx's CeO2 (111) plane (0.312 nm) observed in Fig. S6.† Other characteristic d-spacings at 0.190 and 0.208 nm correspond to the CeO2 (220) and Cu (111) planes, respectively. Elemental mapping via energy-dispersive X-ray spectroscopy (EDS, Fig. 2g) confirms the uniform distribution of Cu, Ce, and O within Cu/CuCeOx without significant agglomeration. Moreover, inductively coupled plasma atomic emission spectroscopy (ICP-OES) quantified the Ce and Cu composition, aligning closely with theoretical expectations (Table S1†).
The valence states and surface chemistry of Cu, Ce, and O were investigated by X-ray photoelectron spectroscopy (XPS). The Ce 3d XPS spectra for both Cu/CeOx and Cu/CuCeOx nanorods, depicted in Fig. 3a, were deconvoluted into eight peaks corresponding to Ce 3d5/2 (v, 882.4 eV; v′,884.9 eV; v′′, 888.7 eV; v′′′, 898.2 eV) and Ce 3d3/2 states (μ, 900.8 eV; μ′, 903.5 eV; μ′′, 907.2 eV; μ′′′, 916.6 eV).36,37 Peaks designated v, v′′, v′′′, μ, μ′′, and μ′′′ are indicative of Ce4+ species, while the v′ and μ′ peaks are attributed to Ce3+ species, denoting the coexistence of Ce4+ and Ce3+ within the composite matrix.38 The relative abundance of Ce3+ to total cerium species was quantitatively assessed and was summarized in Table S1.† The ratio of Ce3+ in Cu/CeOx and Cu/CuCeOx is 0.114 and 0.128, respectively. A noteworthy observation was the positive shift in the Ce4+ peaks for Cu/CuCeOx (Fig. S7†), suggestive of alterations in the central charge distribution between Cu and Ce.39 The Cu/CuCeOx exhibited a predominance of Ce3+ species, which are postulated to facilitate rapid electron transport and mitigate electron localization at the Cu2+/Cu+ sites within the Cu–Ce–Ox solid solution matrix.40 The O 1s XPS spectra are analyzed in Fig. 3b, which can be resolved into two distinct peaks by deconvolution: one at a lower binding energy of 529.3 eV associated with lattice oxygen (Olatt) and another at 531.3 eV, ascribed to adsorbed oxygen species (Oads).41 The number of oxygen vacancies calculated from the deconvoluted core level O 1s spectra is shown in Table S1.† The Cu 2p XPS spectra (Fig. 3c) exhibit binding energies of 952.7 eV and 932.9 eV, characteristic of Cu+ or Cu0 species.42,43 A minor presence of CuO, attributed to inevitable surface oxidation, was also noted.39,44 Given the challenges in distinguishing between Cu0 and Cu+ based on Cu 2p3/2 spectra alone,29 additional analysis was conducted using Cu LMM Auger kinetic energy spectra (Fig. 3d), where peaks at 570.0 and 565.3 eV substantiate the presence of Cu+ and Cu0 species, respectively.45,46 Herein, most of the Cu on the surface is oxidized to monovalent Cu species. The Cu/CuCeOx sample possesses the most surface monovalent Cu species (0.78), much higher than that of Cu/CeOx (0.56). The presence of monovalent copper is conducive to the adsorption of *CO.47
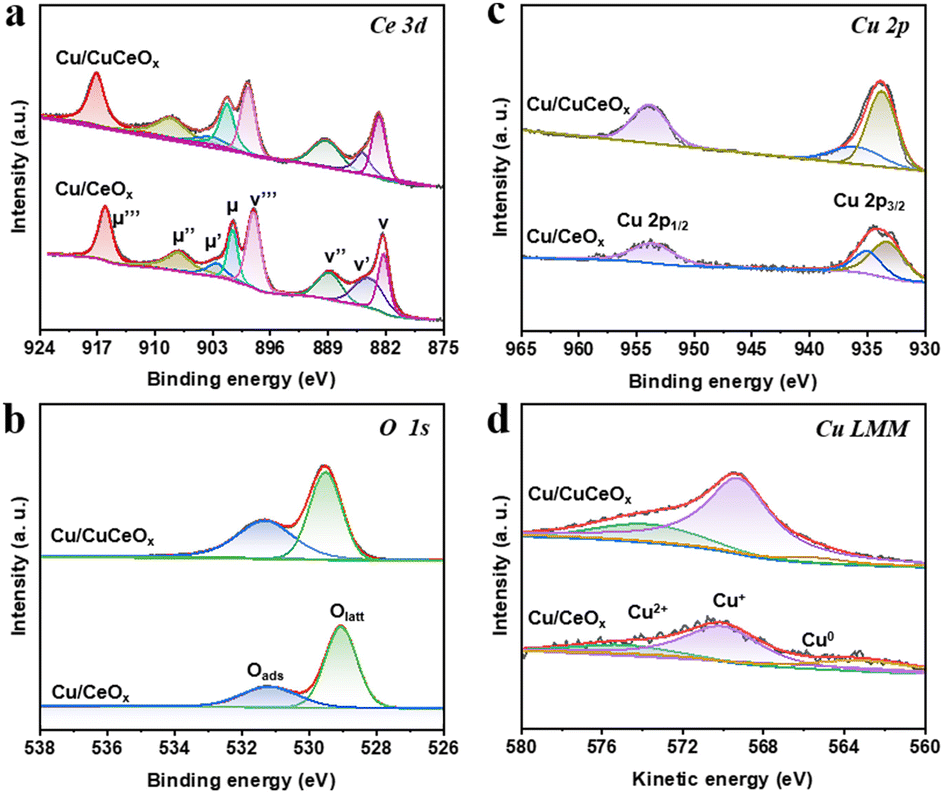 | ||
| Fig. 3 XPS analysis of (a) Ce 3d, (b) O 1s, (c) Cu 2p, and (d) Cu LMM Auger spectra for Cu/CeOx and Cu/CuCeOx. | ||
The CO2RR catalytic performances of Cu/CuCeOx catalysts were tested in a flow cell, in which 1.0 M KOH was used as the electrolyte. In this work, all the applied potentials were converted into a reversible hydrogen electrode (RHE) scale without iR correction. The linear sweep voltammetry curves (LSV) shown in Fig. 4a were recorded by feeding the CO2 gas across the 1.0 M KOH interface. Note that the Cu/CuCeOx catalysts exhibited higher current density responses than those of CeOx and Cu/CeOx catalysts, and we assume that the formation of Cu–Ce–Ox solid solutions may enrich active sites that kinetically favored the CO2RR. To verify the hypothesis, we conducted the cyclic voltammetry (CV) method to estimate the electrochemically active surface area (ECSA) of the catalysts (Fig. S8†). The results show that Cu/CuCeOx had larger ECSA values than the Cu/CeOx catalyst, indicating that the generation of Cu–Ce–Ox solid solutions favors the kinetics of the CO2RR via generating more interfacial active sites, which is consistent with the results of impedance analysis (Fig. S9†).
 | ||
| Fig. 4 (a) LSV spectra of CeOx, Cu/CeOx, and Cu/CuCeOx catalysts. (b–d) FEs of products at different applied potentials in 1.0 M KOH electrolyte for CeOx, Cu/CuCeOx, and Cu/CeOx. (e) C2H4 partial current densities of Cu, Cu/CeOx and Cu/CuCeOx under different potentials. (f) FEC2H4/FECH4 of CeOx, Cu/CeOx, and Cu/CuCeOx at −1.2 VRHE. (g) Stability test of Cu/CuCeOx during 18 h of long-time operation at −600 mA cm−2. | ||
The gaseous and liquid products were quantitatively analyzed by on-line gas chromatography (GC, Fig. S10†) and 1H nuclear magnetic resonance (1H NMR, Fig. S11†), respectively.48 For the pure CeO2 catalyst, H2 was the main product (Fig. 4b). As shown in Fig. 4c, the Cu/CuCeOx catalyst exhibited a high FEC2H4 of 40.2% at −1.2 VRHE. In comparison, the Cu and Cu/CeOx catalysts showed lower FEC2H4 at −1.2 VRHE (Fig. 4d and S12†). For the pure Cu catalyst, the major products were H2, CH4, and C2H4 species, while FECH4 and FEC2H4 were 18.9% and 20.5%, respectively (Fig. S12†). In contrast, for the Cu/CeOx catalyst, CH4 emerged as the predominant product, achieving a FE of 38.6% at a potential of −1.2 VRHE. The comparison of FE for H2 and the CH4 products over those three catalysts further demonstrated that the formation of H2 and the CH4 species was suppressed on the Cu/CuCeOx catalyst. Furthermore, LSV measurements in an Ar-saturated 1.0 M KOH electrolyte without CO2 gas revealed that CeOx exhibited the lowest overpotential at equivalent current densities, further affirming its proficiency in hydrogen evolution (Fig. S13†). Furthermore, the Cu/CuCeOx catalyst demonstrated the highest partial current density for C2H4 production (Fig. 4e), signifying that Cu/CuCeOx has superior catalytic activity for C2H4 synthesis compared to both pristine Cu and Cu/CeOx. Fig. 4f illustrates that the FEC2H4/FECH4 ratio achieved a maximum of 3.77 on Cu/CuCeOx, compared to 1.08 and 0.25 for Cu and Cu/CeOx, respectively, indicating that the formation of Cu–Ce–Ox solid solutions is particularly conducive to C2H4 production. Comparative electrochemical evaluations of Cu0.1/CuCeOx and Cu0.5/CuCeOx, synthesized via the same method, were also performed (Fig. S14†). A 50% increase in Cu loading led to a decline in C2H4 selectivity, potentially due to the formation of larger Cu particles, which diminish the number of Cu active sites directly interacting with Ce, thereby inhibiting C–C coupling.36 The long-term stability of Cu/CuCeOx was assessed through chronopotentiometry, and no significant decrease in Faraday efficiency of C2H4 was observed during 18 h of electrocatalysis at −600 mA cm−2 (Fig. 4g). Post-CO2RR XPS analysis of the electrode confirmed the persistence of Cu+ as the predominant valence state in Cu/CuCeOx, indirectly evidencing the stabilizing influence of the Cu–Ce–Ox matrix on Cu+ species (Fig. S15 and Table S1†). The efficient presence of Cu+ was also demonstrated by in situ Raman spectra of a wide potential window (Fig. S16†), with characteristic peaks around 150, 230 and 500 cm−1.49,50 Moreover, the characteristic peaks of the *CO (2050 cm−1) intermediates were also observed, further indicating the high *CO concentration on the Cu/CuCeOx catalyst surface.
Subsequent density functional theory (DFT) calculations were conducted for Cu/CeOx and Cu/CuCeOx catalysts to rationalize the effects of Cu–Ce–Ox solid solutions on the CO2RR selectivity toward C2H4 (Experimental section in the ESI, Fig. S17 and S18†). Initially, Bader charge analysis was applied to Cu/CuCeOx (Fig. 5a), revealing that copper integration into the cerium dioxide matrix diminished the charge density around the Cu sites, indicating that the charges of Cu and Ce were rearranged.51 The transfer of electrons from Cu to Ce3+ results in higher Cu+ in Cu/CuCeOx, which is conducive to promoting the adsorption of *CO on the surface and enhancing C–C to achieve high selectivity of C2H4. This alteration favorably impacts the protonation of *COCO intermediates, thereby facilitating C2H4 formation. In addition, the hydrogen evolution reaction (HER) performance of both Cu/CeOx and Cu/CeCuOx was assessed (Fig. 5b). The Gibbs free energy (|ΔGH*|) for the HER on Cu/CeOx is 0.13 eV lower than that of Cu/CeCuOx (0.67 eV), thus allowing the promotion of the surface *H coverage and the hydrogenation.
 | ||
| Fig. 5 (a) The Bader charge of Cu/CuCeOx. (b) Desorption energy diagram of hydrogen on Cu/CeOx and Cu/CuCeOx. (c) The free energy change diagram of *CHO or *COCHO generated by *CO over the Cu/CeOx and Cu/CuCeOx. | ||
The free energy diagrams depicting the formation of *CHO or *COCHO from *CO on the Cu/CeOx or Cu/CuCeOx catalysts are presented in Fig. 5c. Moreover, Cu/CuCeOx also exhibits significantly stronger *CO adsorption than Cu/CuOx, enhancing the local *CO concentration and favoring the C–C coupling process on the Cu/CeCuOx catalyst. According to previous reports,52 the formation of CH4 and C2H4 is primarily governed by distinct rate-determining steps: the hydrogenation of *CO to *CHO for CH4, and the coupling of *CO with *CHO for C2H4, respectively.20,53 For the Cu/CeOx catalyst, the ΔG value of the CO hydrogenation is 0.44 eV lower than that of the Cu/CeCuOx catalyst, suggesting that Cu/CeOx has a higher selectivity for producing CH4. In contrast, the formation of the *COCHO intermediate of the Cu/CuCeOx catalyst (0.12 eV) requires less free energy change than that of the Cu/CuOx catalyst (0.45 eV), indicating a more favorable C2H4 pathway. Our computational findings corroborated that surfaces characterized by relatively high *H and low *CO coverage are conducive to the promotion of CH4 production. Conversely, surfaces with a rich presence of *CO and moderate levels of *H are advantageous for the synthesis of C2H4.
Lastly, in situ infrared spectroscopy was employed to analyze potential surface intermediates on Cu/CeOx and Cu/CuCeOx under various potential conditions, further elucidating the catalytic mechanism (Fig. S19†). Not only the peak of CO2 can be observed in a pronounced absorption band of 2400–2300 cm−1,54,55 but also the characteristic peak of CO (2050 cm−1) can also be identified,56 which serves as evidence for the activation and reduction of CO2.57 In addition, a distinct *CHO peak, key intermediate for CH4, was further found at 1226 cm−1.58 In contrast, in the Cu/CuCeOx spectrum, the characteristic peak of *CHO is significantly weakened and replaced by *COCHO at 1550 and 1185 cm−1,59–62 indicating that Cu/CuCeOx catalysts are more inclined to produce C2, which is consistent with the results calculated by DFT.
Conclusions
In summary, this study successfully demonstrates the selective electroreduction of CO2 to CH4 or C2H4 by finely tuning *H and *CO adsorption on Cu/CeOx catalysts. Our innovative approach, involving the manipulation of Cu/CeOx and Cu/CuCeOx interfaces, significantly influences the catalytic selectivity towards desired C1 or C2 products. Notably, the Cu/CuCeOx electrocatalyst demonstrated a peak FEC2H4 of 40.2% and jC2H4 of −245.7 mA cm−2, while the Cu/CeOx electrocatalyst showed good CO2RR performances with a maximum FECH4 of 38.6% and jCH4 of −198.3 mA cm−2. This advancement is underpinned by the enhanced *CO adsorption facilitated by the stabilization of Cu+ species within the Cu–Ce–Ox solid solution. Our findings pave the way for the development of tailored catalysts capable of directing CO2 electroreduction towards specific hydrocarbons, addressing critical needs in CO2 mitigation and sustainable fuel production.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
M. K. proposed, designed, and supervised the project. T. H., Y. B., Z. C., and J. P. wrote the manuscript. T. H., Y. B., and Z. C. performed the experiments and analyzed the data. All authors discussed the results and revised the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Shanghai Pujiang Program (22PJ1400200), the National Natural Science Foundation of China (no. 52122312 and 22209024), Tongcheng R&D Foundation (no. CPCIF-RA-0102), and the State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University.References
- M. Li, F. Zhang, M. Kuang, Y. Ma, T. Liao, Z. Sun, W. Luo, W. Jiang and J. Yang, Nano-Micro Lett., 2023, 15, 238 CrossRef CAS
.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648 CrossRef CAS
- Y. Ma, J. Wang, J. Yu, J. Zhou, X. Zhou, H. Li, Z. He, H. Long, Y. Wang, P. Lu, J. Yin, H. Sun, Z. Zhang and Z. Fan, Matter, 2021, 4, 888 CrossRef CAS
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478 CrossRef CAS
- H. Chen, Z. Wang, X. Wei, S. Liu, P. Guo, P. Han, H. Wang, J. Zhang, X. Lu and B. Wei, Appl. Surf. Sci., 2021, 544, 148965 CrossRef CAS
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef
- W. Zhu, L. Fan, Q. Geng, C. Wang, X. Fan, Y. Zhang and C. Li, Chem. Eng. J., 2024, 489, 151316 CrossRef CAS
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed
- Q.-J. Wu, D.-H. Si, P.-P. Sun, Y.-L. Dong, S. Zheng, Q. Chen, S.-H. Ye, D. Sun, R. Cao and Y.-B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306822 CrossRef CAS
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z.-L. Wang, Small, 2022, 18, 2107450 CrossRef CAS
- H. Shi, L. Luo, C. Li, Y. Li, T. Zhang, Z. Liu, J. Cui, L. Gu, L. Zhang, Y. Hu, H. Li and C. Li, Adv. Funct. Mater., 2024, 34, 2310913 CrossRef CAS
- Y. Sun, J. Xie, Z. Fu, H. Zhang, Y. Yao, Y. Zhou, X. Wang, S. Wang, X. Gao, Z. Tang, S. Li, X. Wang, K. Nie, Z. Yang and Y.-M. Yan, ACS Nano, 2023, 17, 13974 CrossRef CAS PubMed
- L. Ma, Q. Geng, L. Fan, J.-X. Li, D. Du, J. Bai and C. Li, Nano Res., 2023, 16, 9065–9072 CrossRef CAS
- X. Xie, X. Zhang, M. Xie, L. Xiong, H. Sun, Y. Lu, Q. Mu, M. H. Rummeli, J. Xu, S. Li, J. Zhong, Z. Deng, B. Ma, T. Cheng, W. A. Goddard and Y. Peng, Nat. Commun., 2022, 13, 63 CrossRef CAS
- T. Yan, P. Wang and W.-Y. Sun, Small, 2023, 19, 2206070 CrossRef CAS
- Q. Bie, H. Yin, Y. Wang, H. Su, Y. Peng and J. Li, Chin. J. Catal., 2024, 57, 96 CrossRef CAS
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386 CrossRef CAS
- C. Chen, S. Yu, Y. Yang, S. Louisia, I. Roh, J. Jin, S. Chen, P.-C. Chen, Y. Shan and P. Yang, Nat. Catal., 2022, 5, 878 CrossRef CAS
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed
- Y. Chen, N. Lyu, J. Zhang, S. Yan, C. Peng, C. Yang, X. Lv, C. Hu, M. Kuang and G. Zheng, Small, 2024, 2308004 CrossRef CAS
- X. Wang, A. Xu, F. Li, S.-F. Hung, D.-H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525 CrossRef CAS
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152 CrossRef CAS
- X. Guo, W. Ye, Z. a. Chen, A. Zhou, D. Jin and T. Ma, Appl. Catal., B, 2022, 310, 121334 CrossRef CAS
- W. Shan, W. Shen and C. Li, Chem. Mater., 2003, 15, 4761 CrossRef CAS
- W. Liu, X. Liu, L. Feng, J. Guo, A. Xie, S. Wang, J. Zhang and Y. Yang, Nanoscale, 2014, 6, 10693 RSC
- C.-A. Thieu, S. Yang, H.-I. Ji, H. Kim, K. J. Yoon, J.-H. Lee and J.-W. Son, J. Mater. Chem. A, 2022, 10, 2460 RSC
- J. Jiang, H. Yang, H. Jiang, Y. Hu and C. Li, Chem. Eng. J., 2023, 471, 144439 CrossRef CAS
- C. He, Y. Yu, L. Yue, N. Qiao, J. Li, Q. Shen, W. Yu, J. Chen and Z. Hao, Appl. Catal., B, 2014, 147, 156 CrossRef CAS
- P. Bera, K. R. Priolkar, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 3591 CrossRef CAS
- B. Levasseur, A. M. Ebrahim and T. J. Bandosz, Langmuir, 2011, 27, 9379 CAS
- T. Masui, K. Koyabu, K. Minami, T. Egawa and N. Imanaka, J. Phys. Chem. C, 2007, 111, 13892 CrossRef CAS
- S. Zhang, B. Jiang, M. Tong, Y. Yang, Z. Liao, Z. Huang, J. Sun, J. Wang and Y. Yang, Ind. Eng. Chem. Res., 2023, 62, 20667 CrossRef CAS
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493 CrossRef CAS
- H. Sun, H. Wang and Z. Qu, ACS Catal., 2023, 13, 1077 CrossRef CAS
- S. Jin, D. Li, Z. Wang, Y. Wang, L. Sun and M. Zhu, Catal. Sci. Technol., 2022, 12, 7003 RSC
- M. Yusufoğlu, S. Tafazoli, H. Jahangiri, M. B. Yağcı, T. Balkan and S. Kaya, ACS Appl. Mater. Interfaces, 2024, 16, 7288 CrossRef
- C. W. Lee, S.-J. Shin, H. Jung, D. L. T. Nguyen, S. Y. Lee, W. H. Lee, D. H. Won, M. G. Kim, H.-S. Oh, T. Jang, H. Kim, B. K. Min and Y. J. Hwang, ACS Energy Lett., 2019, 4, 2241 CrossRef CAS
- G. Tang, Y. Wu, J. Zhao, H. Zhang, M. Zhou and Y. Wang, Chem.–Eur. J., 2022, 28, e202103459 CrossRef CAS PubMed
- Z. Yang, D. Ji, Z. Li, Z. He, Y. Hu, J. Yin, Y. Hou, P. Xi and C.-H. Yan, Small, 2023, 19, 2303099 CrossRef CAS
- W. Wang, Q. Zhu, F. Qin, Q. Dai and X. Wang, Chem. Eng. J., 2018, 333, 226 CrossRef CAS
- R. Feng, Q. Zhu, M. Chu, S. Jia, J. Zhai, H. Wu, P. Wu and B. Han, Green Chem., 2020, 22, 7560 RSC
- J. Shan, Y. Shi, H. Li, Z. Chen, c. Sun, Y. Shuai and Z. Wang, Chem. Eng. J., 2022, 433, 133769 CrossRef CAS
- K. Cao, H. Liu, W. Li, Q. Han, Z. Zhang, K. Huang, Q. Jing and L. Jiao, Small, 2019, 15, 1901775 CrossRef PubMed
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681 CrossRef CAS PubMed
- N. Martić, C. Reller, C. Macauley, M. Löffler, B. Schmid, D. Reinisch, E. Volkova, A. Maltenberger, A. Rucki, K. J. J. Mayrhofer and G. Schmid, Adv. Energy Mater., 2019, 9, 1901228 CrossRef
- B. Yang, W. Deng, L. Guo and T. Ishihara, Chin. J. Catal., 2020, 41, 1348–1359 CrossRef CAS
- S. Liu, Y. Cao, H. Liu, H. Wang, B. Zhang, Y. Zhang, L. Zhang, S. Zhang and J. Sun, Nanoscale, 2021, 13, 16986 RSC
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS
- M. Wang, Z. Wang, Z. Huang, M. Fang, Y. Zhu and L. Jiang, ACS Nano, 2024, 18, 15303–15311 CrossRef CAS PubMed
- J. Zhang, P. Yu, C. Peng, X. Lv, Z. Liu, T. Cheng and G. Zheng, ACS Catal., 2023, 13, 7170 CrossRef CAS
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809 CAS
- G. N. Vayssilov, M. Mihaylov, P. S. Petkov, K. I. Hadjiivanov and K. M. Neyman, J. Phys. Chem. C, 2011, 115, 23435 CrossRef CAS
- Y. Yan, R. J. Wong, Z. Ma, F. Donat, S. Xi, S. Saqline, Q. Fan, Y. Du, A. Borgna, Q. He, C. R. Müller, W. Chen, A. A. Lapkin and W. Liu, Appl. Catal., B, 2022, 306, 121098 CrossRef CAS
- J. Yin, Z. Gao, F. Wei, C. Liu, J. Gong, J. Li, W. Li, L. Xiao, G. Wang, J. Lu and L. Zhuang, ACS Catal., 2022, 12, 1004 CrossRef CAS
- Y. Katayama, F. Nattino, L. Giordano, J. Hwang, R. R. Rao, O. Andreussi, N. Marzari and Y. Shao-Horn, J. Phys. Chem. C, 2019, 123, 5951 CrossRef CAS
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS
- L. Zhang, J. Feng, L. Wu, X. Ma, X. Song, S. Jia, X. Tan, X. Jin, Q. Zhu, X. Kang, J. Ma, Q. Qian, L. Zheng, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 21945 CrossRef CAS
- Q. Geng, L. Fan, H. Chen, C. Zhang, Z. Xu, Y. Tian, C. Yu, L. Kang, Y. Yamauchi, C. Li and L. Jiang, J. Am. Chem. Soc., 2024, 146, 10599–10607 CrossRef CAS
- L. Fan, Q. Geng, L. Ma, C. Wang, J.-X. Li, W. Zhu, R. Shao, W. Li, X. Feng, Y. Yamauchi, C. Li and L. Jiang, Chem. Sci., 2023, 14, 13851–13859 RSC
- S. Hu, Y. Chen, Z. Zhang, S. Li, H. Liu, X. Kang, J. Liu, S. Ge, J. Wang, W. Lv, Z. Zeng, X. Zou, Q. Yu and B. Liu, Small, 2024, 20, 2308226 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01808g |
| This journal is © The Royal Society of Chemistry 2024 |