
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultinuclear systems for photo-induced production of green fuels: an overview of homogeneous catalysts based on transition metals
Alessandro
Amadeo†
ab,
Emanuele
La Mazza†
a,
Antonino
Arrigo
 a,
Giuseppina
La Ganga
a and
Ambra M.
Cancelliere
*a
a,
Giuseppina
La Ganga
a and
Ambra M.
Cancelliere
*a
aDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche e Ambientali, Interuniversitary Research Center on Artificial Photosynthesis (SOLAR-CHEM, Messina node), University of Messina, Via F. Stagno d'Alcontres 31, 98166 Messina, Italy. E-mail: acancelliere@unime.it
b2DCBB, University of Perugia, Via dell'Elce di sotto, 8, 06123, Perugia, Italy
First published on 14th March 2024
Abstract
In the last century, humankind has based its energy demand on the extensive exploitation of fossil resources with the massive emission of CO2, resulting in problems such as pollution, the greenhouse effect, and, consequently, climate change. However, these resources are limited, and each year, their depletion is getting closer. Therefore, in the last few decades, scientific efforts have been focused on finding alternative ways to use and store green energy sources. The purpose of the present review is to present an overview of the most efficient multinuclear catalytic systems in the field of artificial photosynthesis based on reductive and oxidative mechanisms. In particular, it emphasizes the most recent improvements in homogeneous photocatalysis based on multimetallic complexes, illustrating the advantages of systems consisting of a linked catalyst(s) and photosensitizer(s) that can be employed to achieve high-energy demanding processes, such as the reduction of CO2 and water oxidation.
1. Introduction
As the earth's population grows and time progresses, the energy demand cannot be ignored. Since the Industrial Revolution, the welfare of society has been based on carbon, and later fossil fuels, with a gradually increasing awareness that this regime of non-renewable sources cannot sustain both human beings and the planet in the long term. There are two problems related to our current lifestyle: (i) the prolonged and excessive exploitation of finite fossil fuel reservoirs will inevitably lead to their exhaustion, with a significant overall economic impact due to the continuous increase in the cost of producing and purchasing them1–3 and (ii) the combustion process to obtain energy from fossil fuels involves increasing the concentration of greenhouse gases, such as CO2, in the atmosphere,4 as well as the production of smog, which is harmful to human health (and other living organisms).Accordingly, in the last few decades, efforts have been devoted to making the global energy demand independent of exhaustible and environmentally toxic fuels. Among the potential solutions, the use of clean and renewable energy sources5 such as solar energy has been found to be one of the most promising options. In this case, nowadays, research is focused on the energy-efficient production of H2,6–8 CO, formic acid9–12 and other solar fuels/energy carriers through reactions promoted by visible light. Ideally, the simplest ways to achieve this involve photoinduced water splitting to obtain H2 (reduction process) and O2 (oxidation process)13 and the photoinduced reduction of CO2 into CO and/or formic acid. However, all these reactions are characterized by two common factors: they are associated with reactants that do not absorb visible light and involve multi-electron processes. Thus, it is not possible to realize these processes by simple irradiation. Well-designed photosensitizers require species that may absorb visible light to promote electron-transfer processes and catalysts, i.e. species that may accumulate electrons or holes to achieve the overall redox reaction.14 In this regard, mainly semiconductors (for heterogeneous catalysis) and transition metal complexes (for homogeneous catalysis) have been developed as catalytic systems through several methods such as photocatalysis, electrochemistry and photo-electrochemistry.
This review focuses on the photocatalytic processes to obtain water oxidation and CO2 reduction using multinuclear catalysts in a homogeneous phase. Fundamentally, transition metals are in the spotlight of this research field because they are characterized by various oxidation states, which combine perfectly with the requirement to achieve multi-electron processes;15 in fact, the photoexcitation of a molecule typically leads to a single oxidized or reduced species because the excitation with a single photon can only trigger a one-electron transfer process. Therefore, to achieve a multi-electron reaction, cyclic processes are required, where for every photon absorbed, a charge (positive or negative) is accumulated on the catalyst, eventually leading to a species capable of participating in reactions with high-energy requirements (Fig. 1a and b).
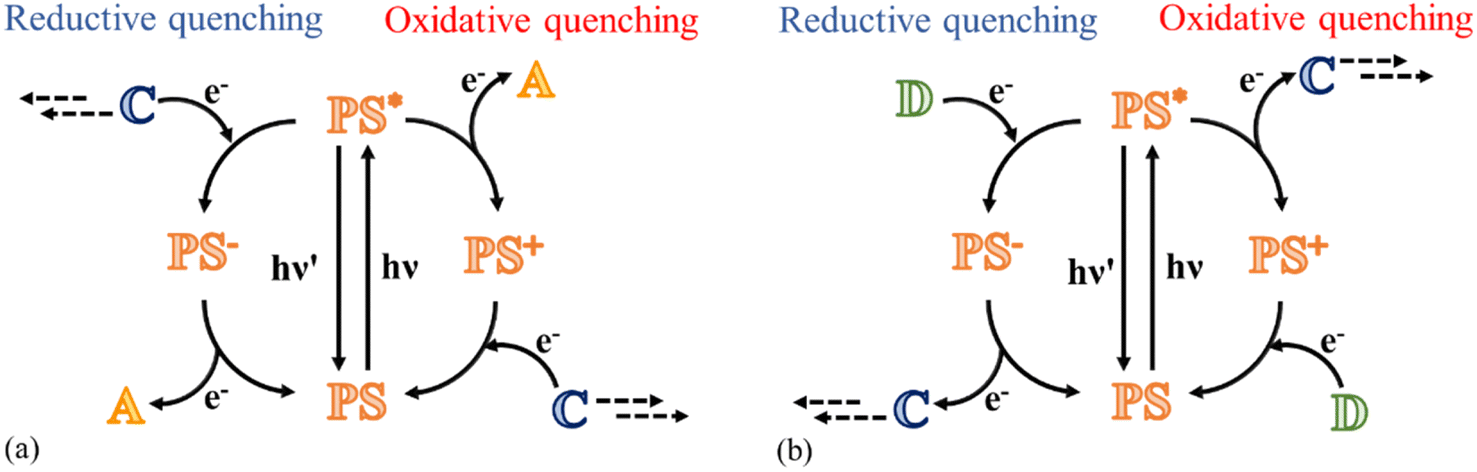 | ||
| Fig. 1 Schematic of photoinduced catalysis for (a) water oxidation and (b) CO2 reduction processes. Reductive and oxidative quenching mechanisms are illustrated (on the left and right side, respectively). A is the electron-acceptor species, D is the electron-donor species, PS is the photosensitizer and C represents the catalyst. | ||
In addition to the photosensitizer and catalyst, to complete the catalytic cycle, a species that acts as an electron donor or acceptor is necessary, depending on the cycle considered, enabling the excited photosensitizer to be reduced or oxidized. Ideally, an optimal sacrificial agent would be the solvent itself, but the Gibbs energy for this type of reaction is generally too high,18 and thus other compounds dissolved in solution are usually used, for example, sodium persulfate or pentaamminechlorocobalt(III)chloride for the oxidative side and BIH (1,3-dimethyl-2-phenylbenzimidazoline) and BNAH (1-benzyl-1,4-dihydronicotinamide) for the reduction processes.16
Multinuclear complexes are species in which two or more metals are connected to each other through bridging ligands.17 If a metal complex catalyst is used, this species may be considered a “supramolecular catalyst”. Supramolecular photocatalysis offers the benefit of fast electron transfer processes between the components, given that diffusion and collision components between subunits are not required, thereby enhancing the overall performance of the photocatalytic system.18–20 Consequently, this results in increased durability of the photosensitizer subunits. In fact, the more rapidly consumed excited and/or reduced state leads to the acceleration of the photocatalytic reaction.21
Based on all these reasons, the aim of this review is to focus attention on the advantage of connecting two or more catalytic subunits to increase the photocatalytic performances of an artificial photosynthetic supramolecular system. Thus, we explore the development in the last decade of the water oxidation part of the system (the actual bottleneck of the entire process), and regarding the reducing part, we will investigate only the processes for CO2 reduction, given that the water reduction to H2 is widely discussed.22
2. Multinuclear species in water oxidation reaction
The bottleneck of all photosynthetic processes is the half-reaction of water oxidation to O2 molecules. The overall water splitting reaction is represented by eqn (1):| H2O (l) → ½ O2 (g) + H2 (g) | (1) |
| 2H+ + 2e− → H2 E° = 0 V vs. SHE | (2) |
| 2H2O → O2 + 4H+ + 4e− E° = 1.23 V vs. SHE | (3) |
In the last few decades, numerous reports have been published on the light-driven production of hydrogen from water,23 and nowadays efforts in this regard are focused on optimizing the process rather than implementing it.
Alternatively, more efforts are required to achieve oxygen evolution from water, given that it requires the formation of an O–O bond, and simultaneously the transfer of 4 electrons and 4H+; consequently, this leads to substantial energy barriers and slow kinetics, making the overall process more challenging to overcome them.24,25 The result is the potential needed to achieve oxygen evolution being greatly affected by the high overpotential, thus requiring a more positive potential than 1.23 V vs. SHE. This potential should be quite high to access using energy provided by light. Therefore, significant effort has been devoted to the realization of catalysts that can facilitate this reaction.
The catalytic oxidation mechanism of water involves several steps (Fig. 2), which can be grouped in three stages, as follows: (i) the formation of a highly-reactive metal-oxo species; (ii) the formation of O–O bonds; and (iii) O2 evolution and catalyst restoration. In the first stage, a single water molecule is bound to the metal center of the catalytic subunit and successively activated in the form of an M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. The second stage is crucial, and generally may follow one of two possible paths, i.e., water nucleophilic attack (WNA) and interaction of two MO units (I2M). In the WNA mechanism, a molecule of water acts as the nucleophilic agent, targeting the electrophilic metal-oxo sites, generating a hydroperoxide species. Instead, in the I2M mechanism, the radical coupling of two metal-oxo sites occurs, leading to their dimerization and generation of an M–O–O–M species. In the case of both mechanisms, the latter step is the oxidation of the intermediate and the evolution of O2.26–29
O species. The second stage is crucial, and generally may follow one of two possible paths, i.e., water nucleophilic attack (WNA) and interaction of two MO units (I2M). In the WNA mechanism, a molecule of water acts as the nucleophilic agent, targeting the electrophilic metal-oxo sites, generating a hydroperoxide species. Instead, in the I2M mechanism, the radical coupling of two metal-oxo sites occurs, leading to their dimerization and generation of an M–O–O–M species. In the case of both mechanisms, the latter step is the oxidation of the intermediate and the evolution of O2.26–29
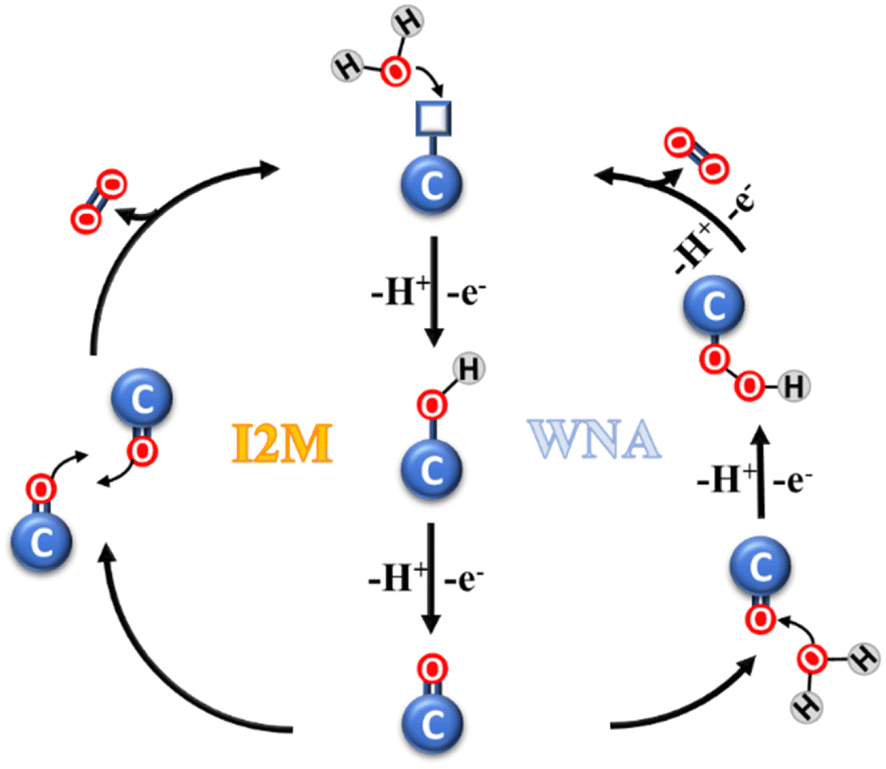 | ||
| Fig. 2 Schematic representation of the two mechanisms of catalytic water oxidation. | ||
In the last few decades, research was centered on developing water oxidation catalysts (WOCs) based on metal oxide materials or molecular transition-metal (Ru, Fe, Mn, Co, Ir, etc.) complexes to improve the selectivity, efficiency and stability of the system.30
Although several examples of first-row transition-metal WOCs have been reported recently, ruthenium complexes are still the most studied systems due to their long-lived 3MLCT state, redox stability and high absorption in the UV-vis spectrum.31 In 2009, Sun and group developed an interesting complex, [Ru(bda)(pic)2] (bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = picoline)32 (see Fig. 3), which showed outstanding efficiency such as TON = 2000 and TOF = 41 s−1, using cerium(IV) ammonium nitrate (CAN) as the oxidizing agent at pH 1.
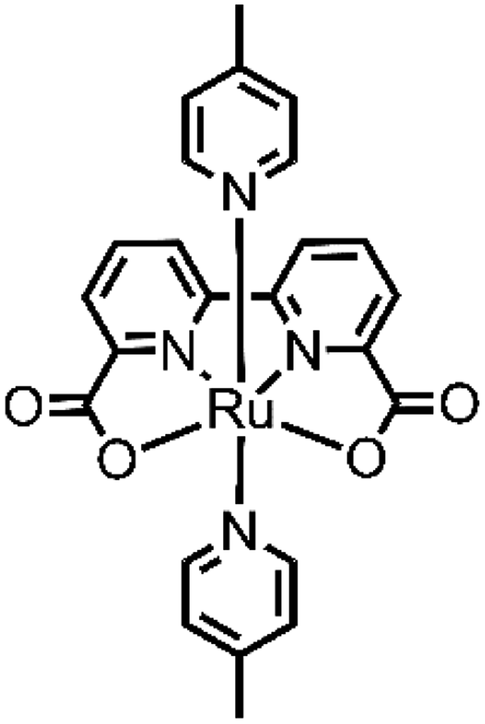 | ||
| Fig. 3 Chemical structure of the [Ru(bda)(pic)2] catalyst. | ||
Since then, numerous new systems based on [Ru(bda)(pic)2] have been studied, trying to achieve better efficiency. Looking to nature (the multimetallic Mn4CaO5 oxygen evolving center) and to the first reported catalysts, such as the so-called “blu dimer”33 and others,34 an interesting approach involving the assembly of multinuclear structures, which include multiple catalytic centers. The positive aspects of type kind of design are the increased stability of the catalytic units, which are now bound together (thus the decomposition of the ligand, one of the most common deactivation pathways of catalysts, is more difficult). Others positive aspects are the increased efficiency due to the electronic communication between the components of the system and the possibility to modulate the shape of this structure and the way they interact each other and with other subunits in the system. Moreover, a multinuclear catalyst sometimes can use a different mechanism from the mononuclear one. This is the case of [Ru(bda)(pic)2], which when free in solution, oxidizes water through an I2M mechanism, and in multinuclear complexes, the mechanism reaction can shift to WNA. The catalytic and photocatalytic performances (with the respective experimental conditions) of all the catalyst metal complexes for the oxygen evolution reported in this review are summarized in Table 1. The comparison with the homologue monomeric species is reported in the main text.
| Catalyst | Chemical catalysis | Photocatalysis | |||||
|---|---|---|---|---|---|---|---|
| TON | TOF | Experimental conditions | TON | TOF | Φ | Experimental conditions | |
| a PS refers to photosensitizer species and SA is the sacrificial agent used in the photocatalytic cycle. b The reference, [Ru(bda)(pic)2], under the same experimental conditions showed a TON of 970 and TOF of 8.4 s−1. | |||||||
| 1 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 206 206 |
34 s−1 | CAN, pH 1 for triflic acid | 369 | PS: [Ru(bpy)2(4,4′-EtCOO2-bpy)](PF)6 | ||
| SA: Na2S2O8a | |||||||
| 2 | 28832 |
42 s−1 | CAN, pH 1 for triflic acid | 535 | |||
| 3 | — | — | >1000b | 14 s−1 | PS: [Ru(deeb)2(bpy)]Cl2 | ||
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7.2) and CH3CN as co-solvent | |||||||
| 4 | 7400b | 155 s−1b | 1255 | 13.1 s−1 | PS: [Ru(bpy)3]Cl2 | ||
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7.2) and CH3CN (1:1, v/v) |
|||||||
| 5 | 5200 | 147 s−1 | CAN, pH 1 (CH3CN 30%) | ||||
| 6 | 2200 | 72 s−1 | CAN, pH 1 for triflic acid | ||||
| 7 | — | 12 s−1 | CAN, pH 1 for triflic acid (CH3CN 50%) | 36 | 1.1 s−1 | PS: [Ru(bpy)3]Cl2 | |
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) and CH3CN (1:1, v/v) |
|||||||
| 8 | — | 26 s−1 | CAN, pH 1 for triflic acid (CH3CN 50%) | 400 | 10 s−1 | PS: [Ru(bpy)3]Cl2 | |
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) and CH3CN (1:1, v/v) |
|||||||
| 9 | — | 42 s−1 | CAN, pH 1 for triflic acid (CH3CN 50%) | 500 | 23 s−1 | PS: [Ru(bpy)3]Cl2 | |
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) and CH3CN (1:1, v/v) |
|||||||
| 10 | 460 | 15.5 s−1 | PS: [Ru(bpy)3]Cl2 | ||||
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) and CH3CN (40%) | |||||||
| 11 | 56 | 0.8 s−1 | [Ru(bpy)3](ClO4)3, pH 8 | 150 | PS: [Ru(bpy)3](ClO4)2 | ||
| SA: Na2S2O8 | |||||||
| Ru4POM | 0.3 | PS: [Ru{(μ-dpp)Ru(bpy)2}3](PF6)8 | |||||
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) | |||||||
| [Co4(H2O)2(PW9O34)2]10− | 0.34 | PS: [Ru(bpy)3]Cl2 | |||||
| SA: Na2S2O8 | |||||||
| [Mn3(H2O)3(SbW9O33)12]12− | 103 | 0.13 | PS: [Ru(bpy)3]Cl2 | ||||
| SA: Na2S2O8 | |||||||
| Sodium borate buffer (pH 9) | |||||||
| [(VIV5VV)O7(OCH3)12]− | 0.2 | PS: [Ru(bpy)3]Cl2 | |||||
| SA: Na2S2O8 | |||||||
| Phosphate buffer (pH 7) | |||||||
| 12 | 178 | 3.6 s−1 | 0.38 | PS: [Ru(bpy)3](ClO4)2 | |||
| SA: Na2S2O8 | |||||||
| Sodium borate buffer (pH 9) | |||||||
| 13 | 0.4 min−1 | SA: Na2S2O8 | |||||
| NaHCO3 buffer (pH 7) | |||||||
| 14 | 1.4 min−1 | SA: Na2S2O8 | |||||
| NaHCO3 buffer (pH 7) | |||||||
2.1 Multinuclear catalysts based on Ru(bda) moieties
An interesting example of a system that connects two or more catalytic subunits with a suitable bridging ligand can be found in the report published by Sun in 2016. Briefly, Sun compared the efficiencies of di- and trinuclear catalysts based on Ru(bda) units linked by bis- or tris(pyridylethyl)benzene35 (1 and 2 in Fig. 4, respectively).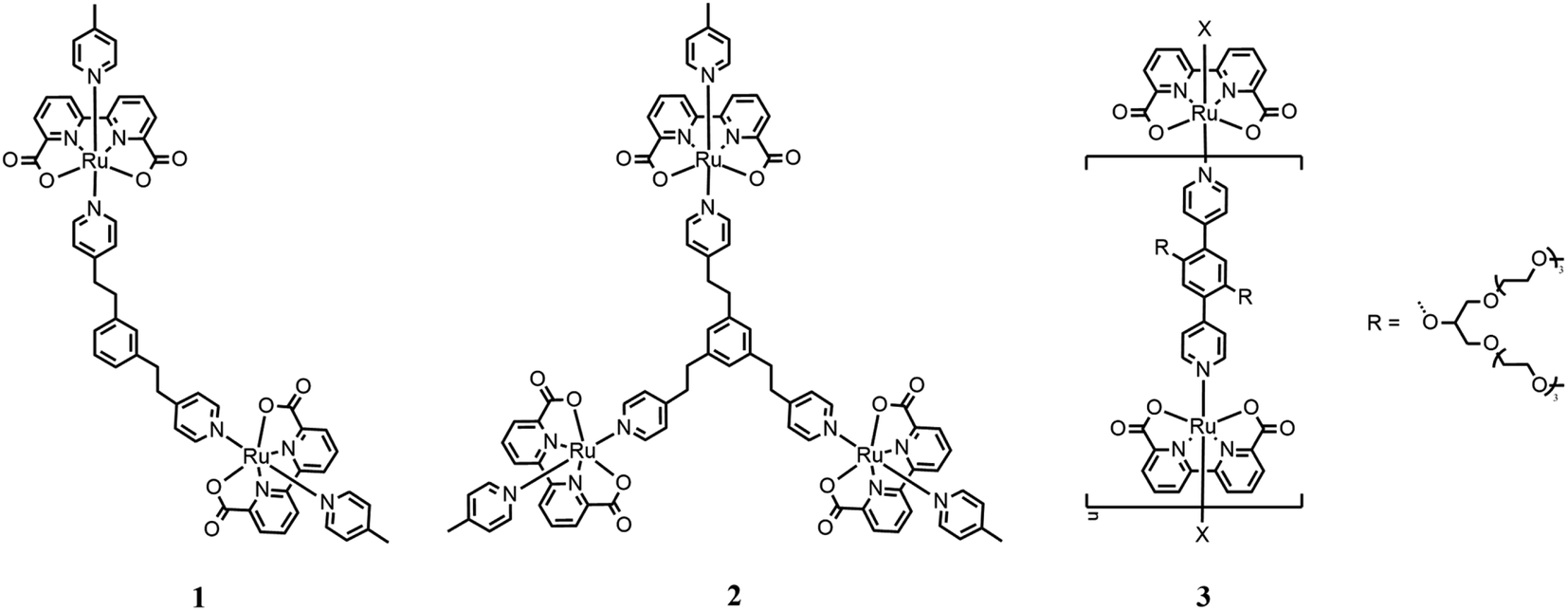 | ||
| Fig. 4 Chemical structures of dinuclear (1) and trinuclear (2) catalysts. Catalyst 3 represents oligomeric species. | ||
These systems have been tested for both the chemical and photochemical water oxidation. In the first case, CAN was used as the chemical oxidant agent in aqueous solution at pH 1 for triflic acid. Good performances were recorded for both systems, where dinuclear catalyst (1) exhibited a turnover number (TON) of 22206 and turnover frequency (TOF) of 34 s−1 per Ru center, while the trinuclear catalyst (2) exhibited a TON of 28832 and TOF of 42 s−1 per Ru unit. Each catalytic unit of these multinuclear complex catalysts shows better performances than the mononuclear catalyst used as the reference (TON of 1460 and TOF of 3.8 s−1). This can be explained by hypothesizing the advantageous cooperation between two metal centers. To collect more information about the catalytic mechanism, kinetic studies have been carried out to monitor the CAN consumption with UV-vis spectrometry. The results obtained by these experiments confirmed a first-order reaction for each the binuclear and trinuclear systems, even if it is not clear whether the pathway is I2M between two metal centers linked together or WNA type. These multinuclear systems have also been tested for light-driven water oxidation using [Ru(bpy)2(4,4′-EtOOC2-bpy)](PF6)2 as the photosensitizer and Na2S2O8 as the sacrificial oxidant. The binuclear catalyst showed a TON of 369, while that of the trinuclear catalyst was 535.
A new way to build multinuclear catalysts consists of assembling many catalytic units in a polymeric arrangement, which are more stable and often easier to produce than mononuclear catalysts. In this frame, Würthner designed, synthesized and studied new oligomeric chains based on Ru(bda), in which the metal centers are linked by 1,4-di(pyridin-4-yl)benzene substituted with oligoethylene glycol groups to enhance the solubility of the oligomeric species (3 in Fig. 4).36
By tuning the synthetic conditions, researchers have been able to build three different categories of species, which differ by the length of their chain. It is difficult to precisely determine the number of units in the chains because different characterization techniques presented contrasting results due to the different methods used to collect the data. These species have been tested for photocatalytic water oxidation using [Ru(deeb)2(bpy)]Cl2 (deeb = diethyl 2,2′-bipyridine-4,4′dicarboxylate, bpy = 2,2′-bipyridine) as the photosensitizer and Na2S2O8 as the sacrificial oxidant in an aqueous solution of phosphate buffer (pH 7.2) and CH3CN as the co-solvent in various ratio. It has been observed that the performances of all three categories of compounds improved with a decrease in the concentration of CH3CN. Moreover, the efficiency of the oligomers increased with chain length, resulting in a TOF of ∼14 s−1 and TON of >1000 for the larger categories of compounds in pure water. Under the same conditions, the smallest compounds showed a lower efficiency with the TOF of 9.5 s−1 and TON of 640. Kinetic investigations were conducted and the results indicated an I2M pathway due to the intra-assembly aggregation of oligomeric fibers, especially with a low amount of CH3CN or in pure water. These materials are very promising due to their high stability and simplicity of their synthesis due to the self-assembly mechanism.
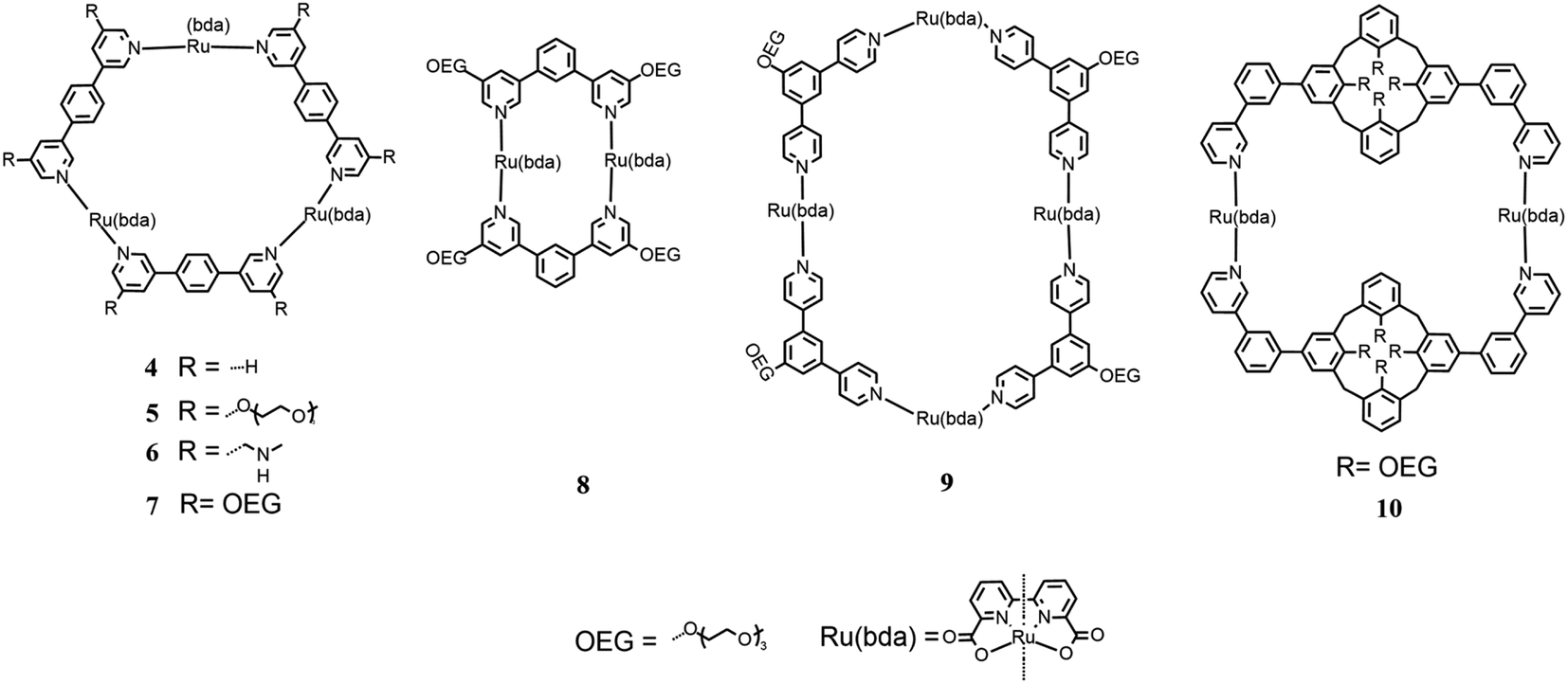 | ||
| Fig. 5 Chemical structures of different macrocyclic catalysts. | ||
This macrocycle was tested as catalyst for water oxidation using CAN as the chemical oxidant in aqueous solution at pH 1. The best performance was achieved using 59% of CH3CN, which is necessary to allow good solubilization. Under these conditions, the efficiency gave a TON of 7400 (∼2500 per Ru unit) and TOF of 155 s−1. These values were compared to that one resulting from the same experiment using the mononuclear [Ru(bda)(pic)2] as a reference, which showed a TON of 970 and TOF of 8.4 s−1, indicating that in the macrocyclic structure, the Ru(bda) units have enhanced activity and increased stability due to the scaffold of the ring. Computational studies displayed the presence of up to ten water molecules preorganized inside the cavity, and this supramolecular effect probably contributes to the improved efficiency of each catalytic unit. These computational studies together with the kinetic studies showed that the mechanistic pathway followed by this macrocycle is WNA instead of I2M. This macrocycle was also tested for photocatalytic water oxidation jointly with [Ru(bpy)3]2+ as the photosensitizer and Na2S2O8 as the sacrificial oxidant in a 1:1 acetonitrile/phosphate buffer (pH 7.2). This system appeared to also be efficient when diluted, e.g. with a concentration of the catalyst of 90 nM, showing a TON of ∼1.255 and TOF of ∼13.1 s−1.
In 2017, Würthner modified this macrocycle substituting a –CH2NMe2 group and triethylene glycol in the meta position of all pyridines (5 and 6 in Fig. 5)38 to increase the solubility of the macrocycle in water and avoid the use of large amounts of CH3CN as the cosolvent, which is one of the principal ways for the deactivation of the catalyst.
Chemical water oxidation with different CH3CN/H2O ratios was tested using CAN as the oxidant. In the case of the macrocycle substituted with triethylene glycol, the CH3CN concentration could be reduced to 30%, obtaining a TOF of 147 ± 9 s−1 and TON of 5.2 × 103. The macrocycle with a –CH2NMe2 group was investigated in water without cosolvent (pH 1 for triflic acid) and a TON of 2.2 × 103 and TOF of 72 ± 6 s−1 were recorded.
In 2021, the photocatalytic performances for the water oxidation of the latter macrocycle were investigated and compared with [Ru(bda)bpb]3 (4 in Fig. 5) using different substituted [Ru(bpy)3]2+ as the photosensitizers,39 as shown in Fig. 6.
 | ||
| Fig. 6 Different substituted photosensitizers. | ||
In the same year, Würthner published a study on new macrocycles similar to [Ru(bda)bpb]3 but containing two and four catalytic units each, [Ru(bda)bpb]2 and [Ru(bda)bpb]4 (8 and 9 in Fig. 5), respectively.40 Each linker between the catalytic units of these supramolecular systems was differently substituted in the ortho position with oligoethylene glycol groups to improve the solubility of the system in water. In this paper, the authors compared the catalytic activity of these systems for water oxidation, focusing on the dependence of the efficiency on the size of the ring. For the first time, the efficiency of these systems toward chemical water oxidation was tested using CAN as the sacrificial oxidant in a solution containing 50% of CH3CN at pH 1 for triflic acid. The kinetic studies proved first-order kinetics according to the WNA mechanism. Macrocycles 7, 8 and 9 in Fig. 5 (with two, three or four catalytic subunits) showed a TOF of 12 s−1, 26 s−1, and 42 s−1 (6 s−1, 8.7 s−1, and 10.5 s−1 per Ru unit), respectively. These results show that the performances increased with the size of the ring but not only because there are more catalytic units but also because each unit enhances the efficiency. The three systems were tested for photocatalytic water oxidation using [Ru(bpy)3]2+ as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor in a solution phosphate buffer/CH3CN 1:1 at pH 7. The macrocycles with 2, 3 and 4 Ru(bda) units showed a TOF of 1.1 s−1, 10 s−1, and 23 s−1 (0.6 s−1, 3.3 s−1, and 5.8 s−1 per Ru) and TON of 36, 400, and 500 respectively. All these systems showed a higher TON per Ru unit than that of [Ru(bda)(pic)2] (under the same experimental conditions, the turnover number was 13), demonstrating the increased stability of the catalyst when it is organized in a supramolecular structure.
In the same year, Würthner tried a different approach for the realization of a new type of supramolecular catalyst using two calix4 arenes, functionalized with oligoethylene glycol groups to enhance the total solubility of the system, and two Ru(bda) units using 2-phenilpyridine as linkers to realize a macrocycle (10 in Fig. 5).41
Photocatalytic experiments were performed using [Ru(bpy)3]Cl2 as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor in phosphate buffer with 40% of CH3CN at pH 7. These tests were performed at different concentrations of the catalyst in the range of 12–200 nM, and the TOF of 15.5 (7.5 per Ru unit) and TON of 460 were reported.
2.2 Catalyst with helicate structure
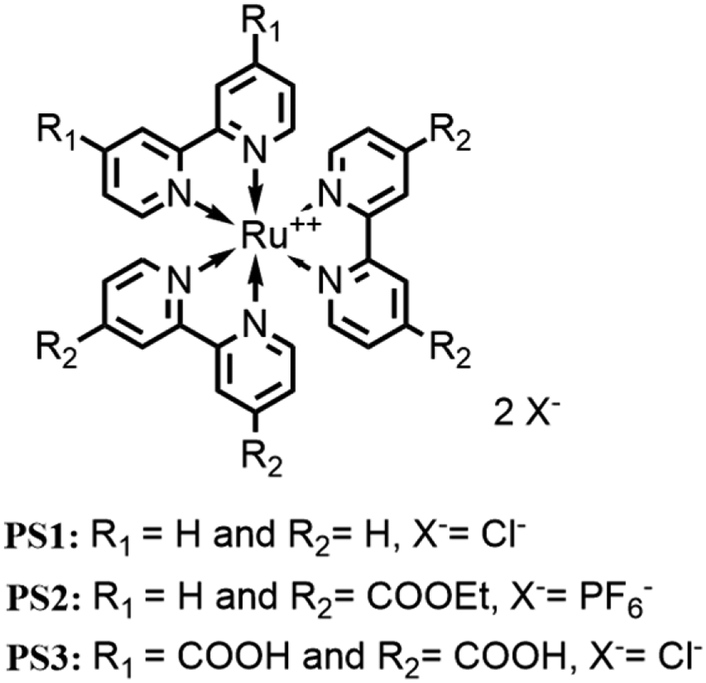
A global process for the conversion and storage of energy should involve abundant, easily available and economic materials to be sustainable from all points of view. In this perspective, recently, new catalysts have appeared in literature based on earth abundant transition metals, which represent a valid alternative for applications in future technologies, although they usually show lower performances than catalysts made of ruthenium.One of the most studied elements for water oxidation is cobalt. It is now well established that CoOx constitutes a good example of a water oxidation heterogeneous catalyst. Many Co complexes have been developed to act as homogeneous catalysts, but they usually act as just pre-catalysts because they are not active themselves but decompose into CoOx. A very intriguing exception is represented by [Co2(spy)2](ClO4)4 (spy = 2,2′:6′,2′′:6′′,2′′′:6′′′,2′′′′:6′′′′,2′′′′′-sexipyridine), a double-helical dicobalt(II), as shown in Fig. 7.42
 | ||
| Fig. 7 Chemical structure of cobalt helicate [Co2(spy)2](ClO4)4. | ||
[Co2(spy)2] is constituted by two Co(II) units linked together by two spy ligands (each metal center is bound to a 2,2′:6′,2′′-terpyridine fragment). This structure leads to a distorted octahedral geometry with an N–C–N angle of about 150°, unlike the ideal 180° that allows the coordination of a water molecule. This was tested as catalyst for water oxidation using [Ru(bpy)3](ClO4)3 as the oxidant at pH 8, obtaining a TON of 56 and TOF of 0.8 s−1. It has also been tested for light-driven water oxidation using [Ru(bpy)3](ClO4)2 as the photosensitizer and Na2S2O8 as the sacrificial agent, obtaining a TON of 150. These results are promising if we consider that analogous Co complexes such as [Co(tpy)2]2+ showed no activity under similar conditions. Further investigations were conducted to exclude that this molecule acts as a precatalyst and the results showed that more than 97% of oxygen was produced by the homogeneous catalyst and not by CoOx.
2.3 Polyoxometalate (POM) catalysts
A new category of species that can be used as water oxidation catalysts is represented by polyoxometalates (POMs), which are completely inorganic species based on earth abundant elements. These structures are composed of a transition metal core linked to inorganic ligands. They show great solubility in water due to their high negative charge and high thermal and chemical stability due to the absence of organic moieties that usually oxidize or degrade.The first paper dealing with POMs in oxygen evolving catalysis was reported in 200443 when Shannon et al.43 observed electrochemical oxygen generation at low potentials (E0 = 0.756 V vs. NHE), from a 2 μM aqueous solution of Na14[Ru2Zn2(H2O)2–(ZnW9O34)2] in phosphate buffer at pH = 8.0 using pulsed voltammetry. In 2008, simultaneously and independently, two groups reported the preparation of a tetraruthenium POM {Ru4(μ-OH)2(μ-O)4(H2O)4[γ-SiW10O36]}10− (Ru4POM) as a high-efficient water oxidation catalyst.44 The Ru4POM species has also been extensively studied in light-driven water oxidation, which is an interesting approach to enhance the photocatalytic performances of POMs by coupling this type of catalyst with efficient photosensitizers. One of the most fascinating examples in this field is the assembled system containing the Ru(II) tetranuclear dendrimeric photosensitizer [Ru{(μ-dpp)Ru(bpy)2}3](PF6)8 (dpp = 2,3-bis(2′-pyridyl)pyrazine)45 and a POM.46 This system was tested for light-driven water oxidation in phosphate buffer with Na2S2O8 and Na2SO4 as sacrificial oxidants. Very good performances were registered with a photoreaction quantum yield of 0.3 (excitation wavelength 550 nm), which means that 60% of the photons absorbed was used for oxygen production. This interesting value was achieved due to the fast hole scavenging, which prevented the decomposition of the antenna system. In 2019, an ‘artificial quantasome’ was reported, which was specifically designed by the template association of light-harvesting perylene bisimides with the polyoxometalate WOC, Ru4POM.47 The resulting [PBI]5Ru4POM complex, taking advantage of the electrostatic interactions and hydrophobic properties of the molecular building blocks, showed interesting oxygen evolution in the presence of the persulfate anion as a sacrificial agent.
An important example of a POM for water oxidation is [Co4(H2O)2(PW9O34)2]10−, which was reported in 2014 by Hill and group.48 Hill tested its catalytic activity for the water oxidation reaction using [Ru(bpy)3]3+ as the oxidant. This system achieved a yield (4 [O2]/[Ru(bpy)3]3+) of about 70% ± 5%49 and an outstanding TOF in the range of 1600–2200 s−1. A system for photocatalytic water oxidation was assembled using [Co4(H2O)2(PW9O34)2]10− to act as the catalyst, [Ru(bpy)3]2+ as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor. Many investigations were conducted to study the stability of this complex. This POM was also unchanged after photocatalytic water oxidation, confirming its excellent features.
Another interesting paper reported a study about the catalytic efficiency of POMs with a huger number of atoms,50 where [Co9(H2O)6(OH)3(PW9O34)3]16−,51 [Co6(H2O)30{Co9Cl2(OH)3(H2O)9(SiW8O31)3}]5− (ref. 52) and [{Co4(OH)3PO4}4(PW9O34)4]28− (ref. 53) were tested for the water oxidation reaction using Na2S2O8 as the sacrificial electron acceptor and [Ru(bpy)3]2+ as the photosensitizer in phosphate buffer at pH 8. These complexes, due to their polyanionic nature, could interact with [Ru(bpy)3]2+, associating in ion pairs that enhance the efficiency of the system.
The mechanistic studies showed that the three systems investigated do not act in the same way. The cluster with 9 Co units can be considered to be composed of three {Co3(m-OH)3(H2O)6} triads linked together by two HPO42− bridges and six external water molecules. The POM with 16 Co units is made up of four {Co4(m-OH)3} distorted cubanes connected through the phosphate linkers. In these two catalysts, a mono-electron transfer occurs from the cobalt core. The POM with 15 Co units had a different structure, in which three {Co3(μ-OH)(H2O)3} triads are bound with six Co(II)(H2O)5 groups positioned on the surface of the POM. In this catalyst, overall oxidation from Co(II) to Co(IV) was shown. Moreover, this configuration facilitated the proton-coupled electron transfer of the water molecules in the core and the external units. These considerations can explain the better performance of the latter complex than other ones analyzed and the great values of TON and TOF reported. Recently, molybdenum-based polyoxometalates containing mono and dicobalt(III) catalyst cores, [CoMo6O24H6]3− and [Co2Mo10O38H4]6−, were also investigated as photocatalysts in the presence of Na2S2O8 as the sacrificial electron acceptor and [Ru(bpy)3]2+ as the photosensitizer in borate buffer solution at pH 8. In this case, the presence of two cobalt centers did not increase the photocatalytic performances.54 Indeed, in the case of [CoMo6O24H6]3−, there is a O–O coupling among the oxygen atoms in the structure, leading to an increase in its photocatalytic performances.55
Other earth abundant-based POMs have been studied, and among them, those containing manganese are of particular interest. In 2017, Zheng reported a study on photocatalytic water oxidation using [Mn3(H2O)3(SbW9O33)12]12− as the catalyst at different concentrations.56 This species, formed by two identical Na9[SbW9O33] Keggin portions, was firstly reported by Krebs.57 Tests were conducted using [Ru(bpy)3]2+, Na2S2O8 as the sacrificial oxidant and sodium borate buffet (pH 9). The best performance was recorded with a catalyst concentration of 10 mM (O2 yield of 13.2% corresponding to a TON of 103). Exceeding this amount of catalyst led to a decrease in the efficiency of the system due to the formation and subsequent precipitation of the couple Dye-POM. After the photocatalytic experiments, the catalyst was reused to run other experiments and it was observed that the O2 yield decreased from 13.2% to 8.0%. The robustness of this catalyst towards hydrolysis and oxidation was tested in different ways and the results of these measurements confirmed the high stability of this POM.
Another earth abundant metal that can form polyoxometallates able to catalyze light-driven water oxidation is vanadium. The first attempt reported in the literature58 is related to the [(VIV5VV1)O7(OCH3)12]− species.59
The peculiarity of this cluster is that the polyoxometalate framework is the active catalytic center itself, while the polyoxometalate is usually a scaffold used to stabilize the metal, acting like the actual catalyst. This species was tested as a light-driven water oxidation catalyst in a system with [Ru(bpy)3]2+ as the photosensitizer and Na2S2O8 as the sacrificial oxidant in phosphate buffer at pH 7. Under these conditions, the system reached a photochemical quantum yield for O2 production of 0.2 (this is a very high value considering that the theoretical yield is 0.5 because the absorption of two photons is needed to obtain one O2 molecule). The rate constant of the hole-scavenging reaction (the electron transfer between the catalyst and the oxidized photosensitizer) was also determined and the calculated value is 2.5 × 108 M−1 s−1. Furthermore, electrochemical water oxidation experiments clarified that [(VIV5VV)O7(OCH3)12]− is not convert into vanadium oxides during the reaction, and thus it is an homogeneous catalyst itself.
2.4 Cubane-like catalysts
An interesting research line focuses on the development of water oxidation catalysts mimicking the oxygen evolving center (Mn4CaO5) of Photosystem II, which contains an Mn3CaO4 cubane structure.60 Many attempts have been made to reproduce the design and the performances of this species using earth abundant metal.One of the most notable, which appeared recently in the literature, is [Cu8(dpk·OH)8(OAc)4](ClO4)4 (dpk·OH = the monoanion of the hydrated, gem-diol form of di-2-pyridyl ketone).61
This structure, 12 in Fig. 8, is formed by two [Cu(II)4O4] units linked by two acetate groups. It was used as a catalyst for water oxidation using [Ru(bpy)3](ClO4) as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor in a borate buffer solution at pH 9. The reported results showed an oxygen yield of 38%, TON of 178 and TOF of 3.6 s−1. Cyclic voltammetry, UV-vis absorption spectrometry, capillary electrophoretic analysis and other techniques were used to exclude the possibility that the cubane catalyst was not the active species but it decomposed into CuOx. The results of these investigations confirmed that [Cu8(dpk·OH)8(OAc)4](ClO4)4 acts as a homogeneous molecular catalyst itself.
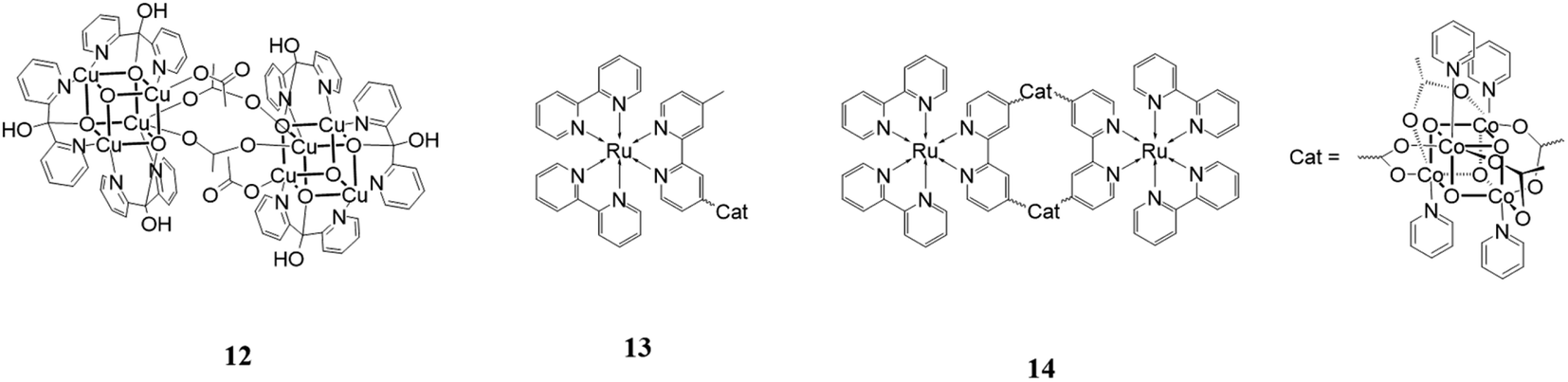 | ||
| Fig. 8 Chemical structures of cubane catalysts. | ||
A cubane catalyst for water oxidation can also be coupled with photosensitizers to build a more complex and efficient supramolecular species. One important example in this field was designed and studied by Sun and coworkers in 2014.62 They assembled Co4O4(OAc)4(py)4 with [Ru(bpy)2 (4-methyl-4′-carboxy-bpy)] (see 13 in Fig. 8) to enhance the electron transfer between the two components of the system. This species was tested as a catalyst for water oxidation in a system composed of NaHCO3 buffer at pH 7.0 containing and persulfate as the sacrificial oxidant. The reported TOF was 0.4 min−1, which is much greater that showed by an analogous system in which the cubane unit is not linked to the photosensitizer. The better result of the former is due to the greater stability of the photosensitizer, easily transferring electrons intramolecularly to the cubane, thus avoiding one of the principal degradation pathways. Another system studied was a macrocycle constituted by two cubane units alternating with two photosensitizers (14 in Fig. 8). This structure displayed a better performance than the linear structure with a TOF of 1.4 min−1 and five times greater O2 production. This data can be explained by the additional stability given by the macrocyclic configuration.
A hypothesis sustained by some researchers involves considering that cubane catalysts are not molecular catalysts themselves but are just a precatalyst that turns into oxides, which act as heterogenous catalysts. Scientists have been debating for a long time about the real catalytic species and a study conducted in 2015 finally answered this question, clarifying that the catalytic activity is conducted by a molecular species.63 In this paper, researchers conducted many experiments on [Co4O4(OAc)4(py)4] (OAC = acetate, py = pyridine),64,65 which they used as an example for all the family of Co-cubanes. In this structure, the alle metal centers are Co(III), cat in Fig. 8.
The test reported the enhanced efficiency of this catalyst with an increase in aging time, which may due to the formation of a more active species. A hypothetic pathway is the hydrolysis of the compound and the consequent release of the Co aqua ions, but 31P-NMR denied this explanation. Kinetic investigations reported a more efficient electron transfer from this new species to [Ru(bpy)3]3+ (hole scavenging), which is about 40 times faster than the same process from [Co4O4(OAc)4(py)4] to [Ru(bpy)3]3+. The great velocity of regeneration of the photosensitizer led to the greater stability of the latter and increased catalytic performance of the system. The photocatalytic system under illumination was also studied using continuous-wave EPR. This analysis showed the formation of Co(IV) in the metallic core due to the photogenerated [Ru(bpy)3]3+. After 20 min of irradiation, the formation of a Co(II), concomitant with a decrease in the Co(IV) signal, was registered. This new species could be the resting state of the active species originating from the cubane.
3. CO2 reduction
The mass consumption of “fossil fuels” has led to an increasing amount of atmospheric CO2, causing serious environmental problems, e.g. global warming66 and the greenhouse effect.67,68 These problems have affected our planet for over a century. In this case, reducing CO2 to obtain energy-rich chemicals, such as CO and HCOOH, can be a solution to shortage of carbon resources environmental problems.69 Although CO2 may be reduced through a one-electron process (eqn (4)), this reaction requires a very low potential (−1.9 V vs. SHE) to be realized, becoming effectively of no interest for the intended purpose.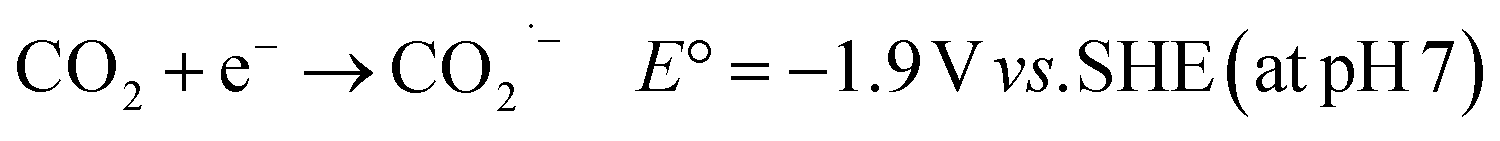 | (4) |
| CO2 + 2H+ + 2e− → CO + H2O E° = −0.52 V vs. SHE (at pH 7) | (5) |
| 2CO2 + 2e− → CO + (CO3)2− E° = −0.64 V vs. SHE (at pH 7) | (6) |
| CO2 + 2H+ + 2e− → HCOOH E° = −0.61 V vs. SHE (at pH 7) | (7) |
Conversely, the CO2 reduction reactions that involve two-electron transfer processes (eqn (5)–(7))70 can be effectively employed at significantly more positive potentials with respect to eqn (4), and also the two-electron product of CO2 reduction are mainly carbon monoxide (CO) and formic acid (HCOOH). CO can be easily converted into liquid hydrocarbon by the Fischer–Tropsch synthesis;71 however, formic acid has been investigated as a potential H2 storage material in recent years, given that it is liquid at room temperature and can be easily converted to CO2 and H2 with suitable catalysts under moderate conditions72 or used directly as fuels in direct formic acid fuel cells (DFAFCs).73
The catalytic cycle to reduce CO2 may follow two different paths, depending on whether the excited photosensitizer undergoes a reductive quenching process or an oxidative quenching process. The starting step of each pathway is the absorption of a photon by the PS subunit with the formation of the excited state of the photosensitizer. In the reductive mechanism, a sacrificial electron donor reduces the excited photosensitizer, and subsequently the reduced photosensitizer transfers the electron to the catalyst to collect negative charges (Fig. 1b). In the oxidative mechanism, excitation of the photosensitizer is followed by a charge separation process, leading to the formation of the oxidized form of photosensitizer and the reduced form of the catalyst; eventually, the photosensitizer is restored by an electron transfer process that involves a sacrificial electron donor (Fig. 1b).
The first studies on photocatalytic CO2 reduction were conducted by Lehn and co-workers in the early 1980s. They employed fac-Re(bpy)(CO)3Cl complexes as photocatalysts74,75. This type of compound has been proven to be selective and efficient for CO2 reduction but showed some drawbacks such as limited absorption in the UV region, low abundance of rhenium, low turnover number and necessary presence of an electron donor.76 Thus, to maximize the efficiency of photocatalysis, it is crucial to introduce a sensitizer to the previously known catalysts. To use visible light as best as possible, in recent years the aim of research has been the introduction of a link (a bridging ligand) between the photosensitizer and the catalyst. Regarding this goal, the photocatalytic reduction of CO2 is performed using multinuclear metal complexes combined with redox photosensitizers (PS), which can mediate photoinduced electron transfer from a reductant (D) to a catalyst, and the catalyst (C) itself, the so-called supramolecular photocatalyst, has been extensively investigated.77,78
In supramolecular photocatalysts, faster electron transfer occurs between the PS and C subunits, which leads to an improvement in the performances of these systems with respect to the separated species in solution due to the increase in the photocatalysis speed and higher durability of the photosensitizer subunit, given that the unstable intermediate state is consumed faster than in separated mixed systems. The photocatalytic performances with their experimental conditions of all the catalyst metal complexes suitable for CO2 reduction reported in this review are summarized in Table 2.
| Catalyst | TON | TOF | Experimental conditions |
|---|---|---|---|
| 17 | 671 | 11.6 min−1 | HCOOH formation |
| BNAH as sacrificial agent | |||
| DMF/TEA (4:1, v/v) |
|||
| 21 | 315 | — | CO formation |
| BNAH as sacrificial agent | |||
| DMF solution | |||
| 22 | 286 | — | CO formation |
| BNAH as sacrificial agent | |||
| DMA/TEOA(5:1, v/v) |
|||
| 31–34 | — | 12–29 h−1 | CO formation |
| TEOA as sacrificial agent | |||
| CH3CN as solvent | |||
| 35 | 60 | 21 h−1 | CO formation |
| TEOA as sacrificial agent | |||
| CH3CN as solvent | |||
| Co(II) cryptate | 16896 |
0.47 s−1 | CO formation |
| TEOA as sacrificial agent | |||
| CH3CN/H2O (4:1, v/v) |
|||
| PS: [Ru(phen)3](PF6)2 | |||
| Co(II)–Zn(II) cryptate | 65000 |
1.8 s−1 | CO formation |
| TEOA as sacrificial agent | |||
| CH3CN/H2O (4:1, v/v) |
|||
| PS: [Ru(phen)3](PF6)2 | |||
| Cu(I)–Co(II) cryptate | 2305 | — | CO formation |
| TEOA as sacrificial agent | |||
| CH3CN/H2O (4:1, v/v) |
|||
| PS: [Ru(phen)3](PF6)2 | |||
| 36 | 829 | — | CO formation |
| Phenol as sacrificial agent | |||
| CH3CN as solvent | |||
| 37 | 766 | — | HCOOH and CO formation (60 and 28% selectivity) |
| CH3CN as solvent | |||
| PS: [Ru(phen)3](PF6)2 | |||
| 38 | 255 | See ref. 96 | |
| Ni(II) S2N2-type complex | 120 | 0.87 min−1 | See ref. 98 |
| [Co5(btz)6(NO3)4(H2O)4] | 2748 | CH3CN as solvent | |
| TEOA as sacrificial agent | |||
| PS: [Ru(bpy)3]Cl2 | |||
| [CoZn(bpbp)(CH3COO)2](CH3COO) | 6680 | 0.19 s−1 | CO formation |
| CH3CN/H2O (4:1, v/v) |
|||
| TEOA as sacrificial agent | |||
| PS: [Ru(phen)3](PF6)2 | |||
| [Fe2Na3(1,2-oxo-4-hydroxyanthracene-9,10-dione)6](TBA)3 | 2625 | — | CO formation (91% selectivity) |
| BIH as sacrificial agent | |||
| DMF as solvent | |||
| 39 | 14956 |
276 min−1 | CO formation |
| TEA as sacrificial agent | |||
| PS: CzIPN |
3.1 Integrated systems: multinuclear species with different functionalities
One of the advantages of multinuclear complexes is the possibility to link various units with different functions within the system with topological control, usually resulting in better overall stability and predetermined photosensitizer/catalyst ratios (due solely to the synthetically-determined structure of the system).Ishitani and co-workers79 synthesized and studied several Ru(II) multinuclear complexes (Fig. 9), where based on the ligands, the Ru(II) centers may act as catalytic subunits (through fragments of the type of [Ru(dmb)2(CO)2]2+) or as photosensitizer subunits ([Ru(dmb)3]2+). They synthetized and investigated four different integrated systems by varying the number of photosensitizers or catalyst moieties, including species 15 (one catalyst and one sensitizer moiety), species 16 (one sensitizer and two catalyst subunits), species 17 (one catalyst and two photosensitizer moieties) and species 18 (one photosensitizer and three catalyst subunits). Studying the CO2 photocatalytic reduction, it was observed that the outcome product of the catalytic cycle is strongly dependent on the ratio between the catalyst and the photosensitizer units, resulting in the favorable formation of HCOOH with a high Ru(photosensitizer)/Ru(catalyst) ratio, and vice versa, obtaining CO as the major product with a low Ru(photosensitizer)/Ru(catalyst) ratio. The experiments were performed in a mixture of DMF/TEOA (v/v = 4:1), with BNAH as the sacrificial reductant, under irradiation at 480 nm. The authors highlighted the important role of TEOA in the catalytic cycle, which was also supported by previous works.80 Specifically, TEOA does not serve as an electron donor, but it acts as proton-acceptor for the one-electron oxidation process involving BNAH, simultaneously suppressing the back-electron transfer process between the reduced photosensitizer and the oxidized BNAH. In addition, it results in the more favorable reduction of CO2 to HCOOH. It was reported that the most efficient photocatalytic system was the complex formed by a single Ru(catalyst) subunit bound to two Ru(photosensitizer) subunits, promoting the formation of HCOOH with a TON of 671 and TOF of 11.6 min−1.
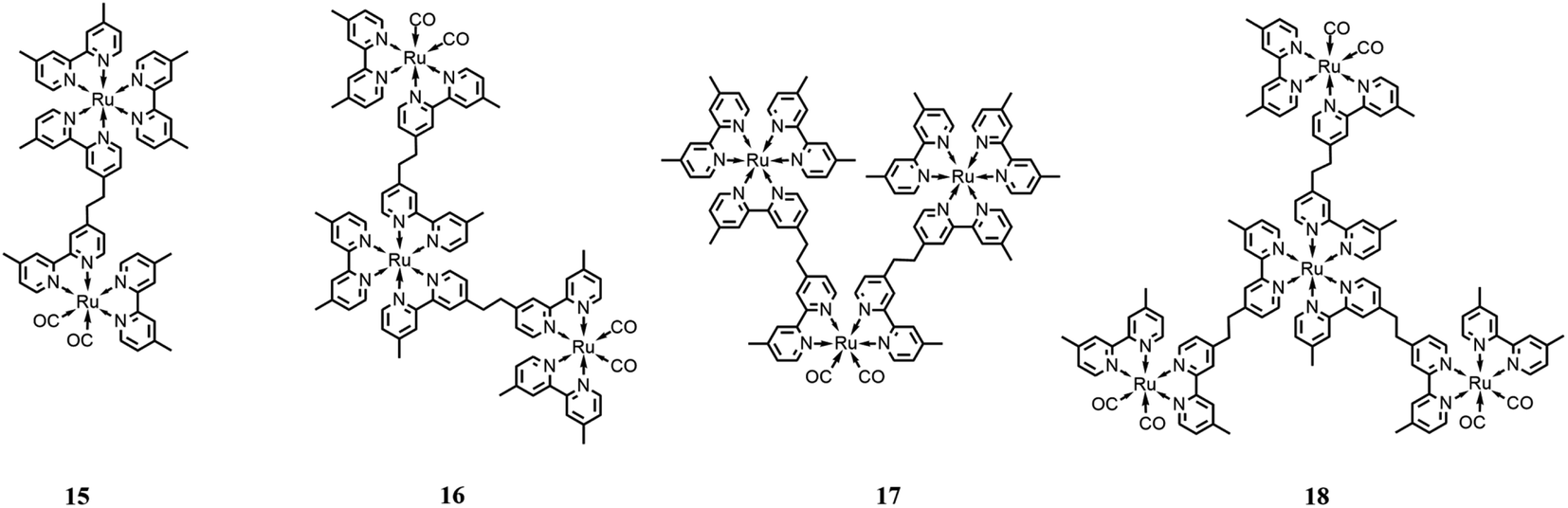 | ||
| Fig. 9 Different integrated systems with the Ru(II)-based photosensitizer and Ru(II)-based catalysts. Charge is omitted for clarity. | ||
Rieger and co-workers81 reported a trinuclear complex consisting of a photosensitizer subunit, [Ru(dmb)3]2+, bound to two catalyst subunits, [Re(dmb)(CO)3Cl] (21 in Fig. 10). It was studied in comparison with model systems incorporating one Ru(II) center and two Re(I) centers with different bridging modes (19 and 20 in Fig. 10). The performance of these trinuclear mixtures was assessed in the photocatalytic reduction of CO2 to examine the influence of covalent bounds connecting each of the individual complexes. The catalytic performances were tested in a DMF solution, with BNAH as the sacrificial electron donor, assisted by TEOA, under 520 nm irradiation, obtaining a TON of 315 for CO. The authors found that the photocatalytic performance of the trinuclear complex is enhanced in comparison with the model systems, facilitating a faster CO2 reduction due to a dinuclear reduction mechanism. Moreover, the covalent bounds between the subunits enable intramolecular electron transfer processes, which lead to a shortened lifetime of the one-electron reduced photosensitizer, making the complex less susceptible to deactivation caused by photobleaching resulting from the excitation of the reduced photosensitizer. Besides, the covalent linkage of the system enabled the Re(I) subunits to act as light antennae, improving the system in its entirety.
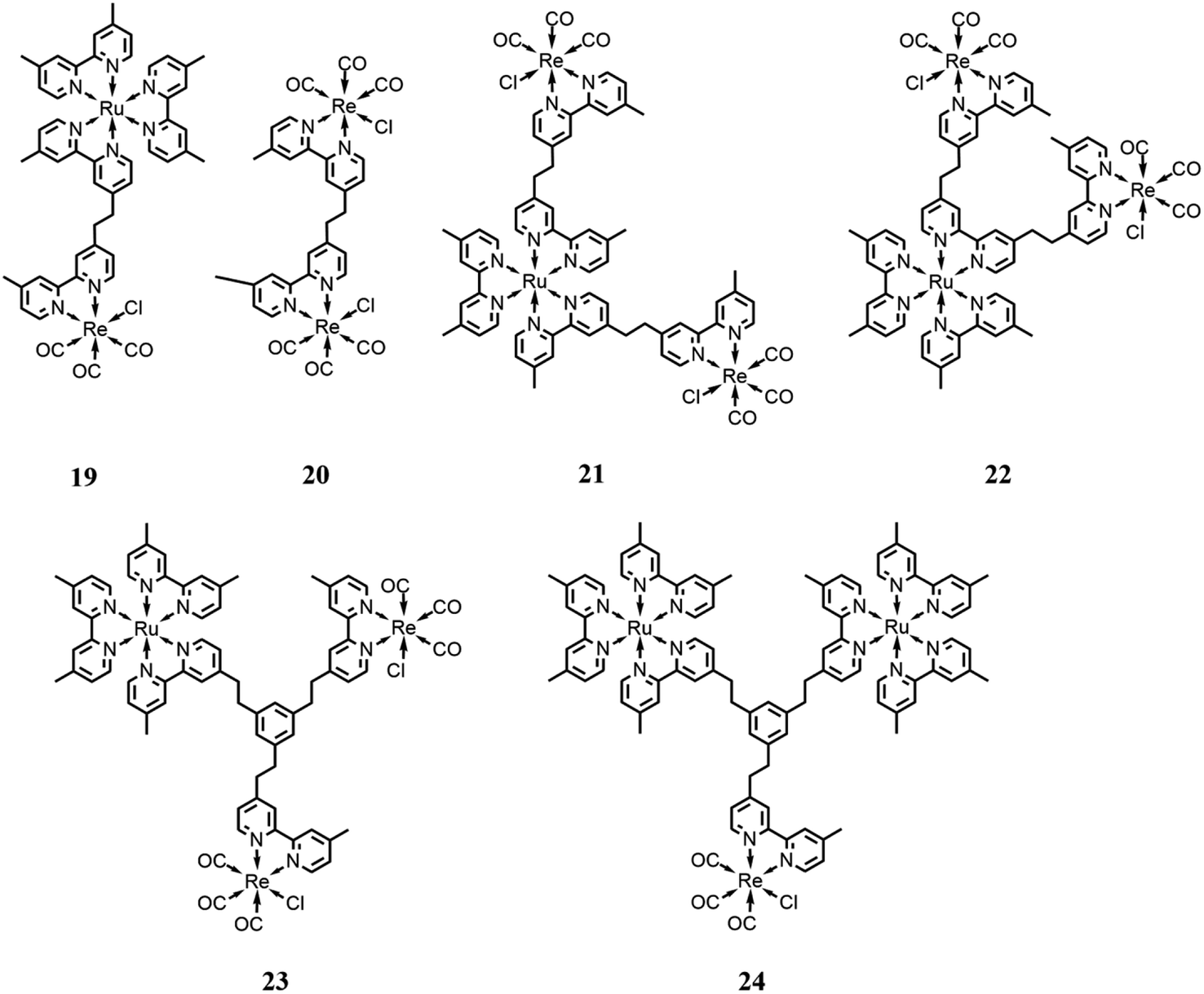 | ||
| Fig. 10 Integrated systems with different ratios and bridging ligands with the Ru(II)-based photosensitizer and Re(I)-based catalysts. Charges are omitted for clarity. | ||
Ishitani and co-workers82 designed and synthesized a trinuclear complex consisting of an Ru(II)-based photosensitizer subunit linked to two Re(I) catalyst subunits through ethylene chains (22 in Fig. 10) and compared its photocatalytic behavior with related mono- and dinuclear Ru- and Re-based complexes, in solution with defined molar ratios. CO2 photoreduction tests were conducted in a DMA/TEOA solution (v/v = 5:1) with BNAH as the sacrificial reductant and an irradiation wavelength of >500 nm. In comparison with the model systems, the trinuclear complex showed enhanced behavior and the decomposition rate of the Ru unit in the 21 system during the photocatalytic reaction occurred at a slower pace, with a TON of 286 and CO formation selectivity of 90%. The better stability, and thus the higher selectivity for CO evolution, was correlated to the suppression of the photochemical ligand substitution, which appears to occur only to the detriment of mononuclear Ru-units, while leaving the ruthenium subunits of the trinuclear and dinuclear complexes intact. This was also associated with the lower localization of the photochemically added electron in the ruthenium subunit caused by the electron transfer process to the two rhenium subunits.
Other developments by the same research group consisted of increasing the distance between the photosensitizer and the catalyst subunits by using different bridging ligands compared to the simplest alkyl chain reported to date in the literature.83 In particular, they used a tris-chelating polypyridine ligand composed of a phenylene ring connected at the 1, 3, 5 positions to the 2,2′-bypiridine moieties by an ethylene chain. This bridging ligand is suitable for the synthesis of trinuclear complexes with different ratios between the photosensitizer (based on Ru(dmb)3-type chromophore) and catalyst (based on Re(dmb)(CO)3-type) subunits (23 and 24 in Fig. 10), respectively. All these systems showed a negligible electronic interaction between the subunits in the ground state, and thus the metal subunits also presented their own photophysical and redox properties in the supramolecular assemblies. These systems were tested in DMA/TEOA solution (v/v = 5:1) with BNAH and BIH as the sacrificial electron donor and an LED (530 nm, 4 mW) as the light source. The results showed that the assemblies presented quite efficient visible light-induced catalytic CO formation with high a turnover number, selectivity and durability when BIH was employed as the sacrificial agent. The better performance obtained by the use of BIH can be rationalized because BIH donates both electrons to the photocatalytic systems required for CO2 reduction.16
The results also showed the better performances in terms of selectivity and turnover number of the system with 2 catalytic and 1 photosensitizer subunits compared with the system with only 1 catalyst and 2 photosensitizers.
Further investigations84 were performed on these systems, where on the catalytic subunits contained the CO2TEOA adduct (known to be an effective catalytic subunit) instead of chloride, which showed an increase in the turnover number and selectivity. This result could be rationalized considering the different distributions of the one-electron reduced form of the supramolecular photocatalysts on the Ru-subunit(s) (leading to decreased CO formation due to a poisoning ligand loss process) and on the Re-subunit(s) and to the presence of chloride ions in solution (inevitable for the species with a chloride ligand), which could interfere with the formation of the CO2TEOA adduct, a requisite for CO-forming catalysis. These results also confirmed the better performance of the 2:1 catalyst-photosensitizer assembly analogously to that observed for the precursor species with the chloride ligand.
Inagaki and co-workers85 achieved the photoinduced hydrogenation of CO2 using a trinuclear iridium hexahydride complex (26 in Fig. 11) as a photo-switchable catalyst. Each Ir center is also complexed by a diphosphine ligand, which acts as a photosensitizer. For comparison, two other analogue complexes were also studied (27 and 29 in Fig. 11). Their photocatalysis performances were evaluated in MeOH, under LED irradiation at 395 nm, in an atmosphere of CO2/H2 (1:1 ratio) and a total pressure of 10 atm. Tests were also performed under dark and light conditions to evaluate the switchability of the complexes. Among the several bases used to achieve HCOOH formation, DBU, thanks to its high basicity, exhibited the highest activity. Overall, the trinuclear complex showed the best catalytic performance, which was enhanced by light irradiation. The authors reported that irradiation facilitated the H2 activation process, which is considered one of the rate-determining steps in the involved reactions and confirmed under a D2 atmosphere by evaluating the H–D exchange reaction. It was explained that the possible effect of light could be the dissociation of the coordinated hydrides, thus leaving unsaturated coordination sites for complexation by CO2. In fact, the CO2 insertion process was also found to be enhanced by light irradiation.
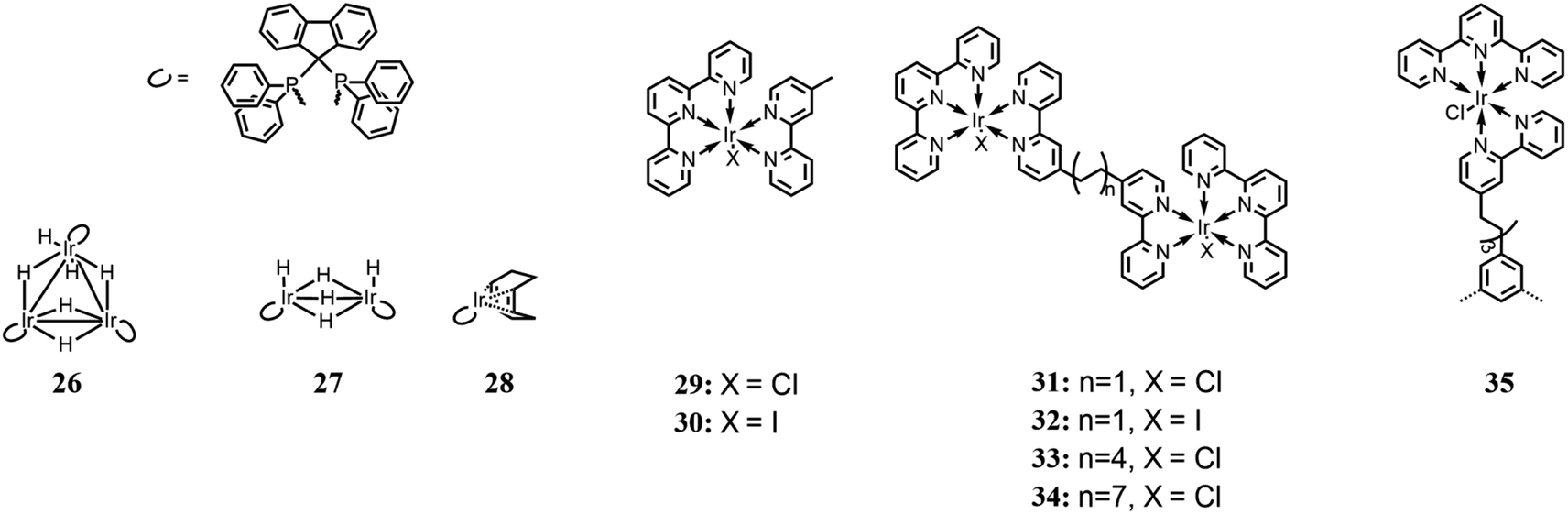 | ||
| Fig. 11 Iridium-based photocatalytic systems. Charge and counter anions are omitted. | ||
A different type of integrated systems is shown below, which was structured not as connected subunits, with some acting as a catalyst and others as a photosensitizer, but as multinuclear complexes, where each unit can act not only as a catalyst but also as a photosensitizer.
Herdtweck and co-workers,86 based on the work of Ishitani et al.,87 designed several dinuclear Ir(III) complexes, whose metallic centers are connected through bis(2-phenylpyridin-4-yl) bridging ligands (from 31 to 34 in Fig. 11). The four dinuclear complexes differ from each other based on the coordinated halides and/or the length of the bridging ligand. Catalytic activity for CO formation was investigated under a 450 nm light source, in CH3CN, with TEOA as sacrificial reductant. No photosensitizer was used because the iridium complexes themselves can act as one. In comparison with analogous Ir(III) mononuclear complexes reported in the same work and shown in Fig. 11 (29 and 30 respectively), the stability of the dinuclear systems appeared to be highly improved, and among the four complexes, TON values in the range of 80–135 were achieved, calculated per molecule of catalyst, and TOF in the range of 12–29 h−1. The difference of catalytic capabilities was attributed to the length of the bridging ligand, which only enabled a weak interaction between the Ir(III) centers for the shortest alkyl chains and no interaction for the longest ones, and to steric hindrance. Besides, differently from the previous work by Ishitani, where the catalyst deactivation was attributed to dimerization processes,87 the authors considered these processes or their minor relevance and believed that the deactivation may be ascribed to the oxidation products of TEOA.
Rieger and co-workers,88 continuing the work of Herdtweck et al.,86 synthesized a trinuclear Ir(III) complex, whose properties and photocatalytic behavior were compared with mono- and dinuclear analogue complexes (35 and 31 in Fig. 11), respectively. The authors reported the selective reduction of CO2 to CO with no other reduction products formed. Photocatalysis tests were performed in CH3CN, with TEOA as the sacrificial electron donor, by irradiating at 450 nm, and similar to the previous work,85 no photosensitizer was added. Among the three complexes, the highest TON value (60, calculated per Ir(III) center) was obtained for the trinuclear complex, with a TOF of 21 h−1. Because both the TOF and quenching constant are situated between the respective values of mono- and dinuclear complexes, it is assumed that there is a strong correlation between the photocatalytic performance and the effect of intramolecular quenching.
3.2 Cryptands
Metal cryptate complexes have received some attention in the field of photocatalysis given that their space orientation allows synergistic catalysis between the coordinated metal centers, leading to a more efficient CO2 reduction with respect to mononuclear systems in terms of efficiency, yield and selectivity.Lu and co-workers89 realized a dinuclear Co(II) cryptate to achieve the photocatalytic reduction of CO2 to CO, investigating the kinetic properties to evaluate the synergistic effect between the two cobalt metal centers. It was found that only one of the two Co(II) ions acts as the actual active site for the catalytic process; the second Co(II) ion reduces the energy barrier for CO2 reduction, with respect to an analogous mononuclear compound, by facilitating the removal of the hydroxyl group in the C–OH fragment that is formed within the catalytic process. These results were supported by the experimental analysis and DFT calculations. In a mixture of CH3CN/H2O (v/v = 4:1) and under 450 nm LED light irradiation, the authors reported a TON of 16896 and a TOF of 0.47 s−1, with a CO selectivity of 98%. [Ru(phen)3](PF6)2 was used as the photosensitizer and TEOA as the sacrificial reductant.
Lu and co-workers90 continued their work studying a dinuclear heterometallic cryptate based on a Co(II) ion and a Zn(II) ion. This complex is equivalent to the homonuclear one studied previously.88 In reference to their precious work, the authors reported an increase in the photocatalytic performances, operating under the same conditions.88 This catalyst achieved CO2 reduction with a TON of 65000, TOF of 1.8 s−1 and selectivity of 98%. The increased efficiency of the process was attributed to the stronger binding affinity of the Zn(II) ion to the hydroxyl group compared to the Co(II) ion, thus enhancing the whole process. Referring to the mechanism, the Co(II) center was reported to act as the actual catalyst site, whereas the Zn(II) center is the assistant catalyst site.
Martinho and co-workers91 realized three dinuclear Co(II) octaazacryptate catalysts, which have well-known photophysical behavior, and studied the influence of the electron-withdrawing and -donor effect of the functionalized in the aromatic rings. They reported that the photocatalytic reduction of CO2, under blue visible light (492–455 nm), was only successful using [Ru(phen)3](PF6)2 as the photosensitizer and TEOA as the sacrificial electron-donor, using a solution of CH3CN/H2O (v/v = 4:1) as the solvent. This behavior was attributed to the stronger oxidizing power of the photosensitizer/sacrificial agent pair compared to the other ones tested. Beyond that, it was reported that at a low catalyst concentration, solely CO was produced but increasing the catalyst concentration or the light-exposure time (30 h), a mixture of CO and CH4 was evolved. A TON of 27311, TOF of 0.25 s−1 and CO selectivity of 92% were achieved.
Su and co-workers92 studied a cryptate trinuclear Re(I) catalyst. A higher catalytic performance compared to the analogous mononuclear Re-bpy complex was recorded, which was attributed to (i) the easier adsorption of CO2 and, consequently, the easier contact between catalytic sites and substrate due to the structure of the cryptands themselves and (ii) to the bridging ligands, which are capable of achieving a faster electron transfer process (point supported by cyclic voltammetry studies). Photocatalysis tests were conducted in a DMSO/H2O solution by using Ir(ppy)3 as the photosensitizer and BIH as the sacrificial electron donor, with an LED light source at a wavelength of >420 nm.
Apfel and co-workers93 synthesized an asymmetric ([(CH2)2SCH2(m-C6H4)CH2NH(CH2)2]3N) dinuclear cryptand based on Cu(I) and Co(II) centers based on the work of Lu et al.89 and taking inspiration from CO2-converting enzymes to opt for a sulphur-rich coordination site. Photocatalytic experiments were performed in CH3CN/H2O (v/v = 4:1) with [Ru(phen)3](PF6)2 as the photosensitizer and TEOA as the sacrificial electron donor, under an LED light (450 nm). Photoinduced reduction tests resulted in a TON of 2305 and CO selectivity of 98%. The authors emphasized the importance of the synergistic effect arising from the presence of the two metal centers, which was proven by comparison with analogue mononuclear complexes and computational studies. The importance of the metal–metal distance was also specified: in fact, increasing this distance led to a lower synergistic effect, resulting in a process catalyzed only by the Co(II) center.
3.3 Multinuclear catalysts using a xanthene bridge
Similarly to metal cryptates, dinuclear complexes based on a molecular structure have been reported, enabling synergistic catalysis, namely, two chelating moieties connected by a xanthene bridge.Robert and co-workers94 reported a dinuclear Co(II) bisquaterpyridine complex (36 in Fig. 12) capable of the visible light-induced selective formation of formate ion or CO. The solvent used for the photocatalysis tests was CH3CN. Although the authors reported that DMF is a better solvent to be used for formate production, they also cited studies where it was affirmed that traces of water or amine may cause hydrolysis products that can poison the catalytic cycle,95 and thus they preferred CH3CN. Besides, several combinations of photosensitizers and sacrificial reductants were involved under 460 nm LED irradiation. It was found that adding phenol, a weak acid, to the solution it was possible to obtain CO formation selectively; according to the authors, this dual activity is caused by the synergy of the two Co centers, which are capable of controlling the two-electron/two-proton CO2 reduction. In the absence of weak acid, formate with a selectivity of 97% and a TON of 821 were obtained; conversely, in the presence of weak acid, CO was highly produced with a selectivity of 99% and TON of 829. Mechanistic studies were also conducted by DFT calculations to propose the catalytic cycle scheme.
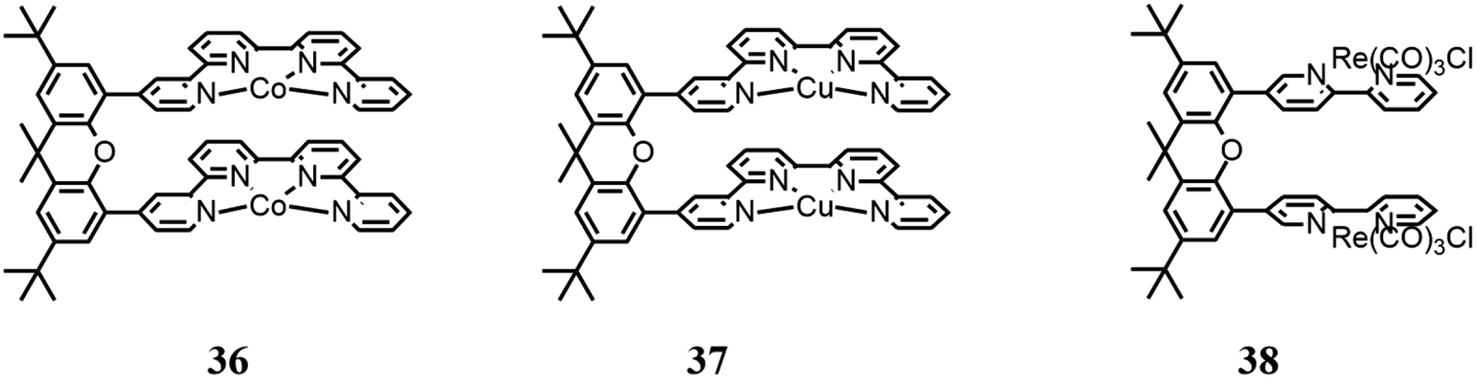 | ||
| Fig. 12 Structures of different systems using the xanthene bridge. | ||
Robert and co-workers96 continued their previously reported study93 using an analogue dinuclear Cu(II) bisquaterpyridine complex (37 in Fig. 12) in CH3CN under irradiation at 420 nm. In this study, a fundamental synergy was also found between the two metallic centers, which in the presence of water as a proton donor, generates a bridging metal-hydride intermediate (M–H–M), thus achieving photoinduced CO2 reduction to formate ion. CO is also produced, derived from CO2 forming an adduct with the reduced catalyst, which was restored following the reduction of CO2 to CO. However, the amount of water did not seem to affect the selectivity of the catalyst, according to the authors, implying that the initial charge transfer steps to the catalysts limit the overall rate. A TON value of 766 was reported, with a selectivity of 60% for formate and 28% for CO, using Ru(phen)32+ as a photosensitizer. The authors also reported a comparative study with an analogous mononuclear Cu complex (Fig. 10b), indicating that by using thus complex as a CO2 catalyst, only CO is formed, and thus the authors hypothesized the presence of the above-mentioned bridging hydride.
Tschierlei and co-workers97 studied a dinuclear Re(I) complex based on Re(bpy)(CO)3Cl units bound to each other through a xanthene bridge (38 in Fig. 12). The authors previously reported a study focused on the related mononuclear complex (Re(bpy)(CO)3Cl),98 and found that a dimer of the mononuclear Re(I) complex is a crucial intermediate for CO2 reduction to CO, and the studies were continued, investigating the aforementioned dinuclear Re(I) complex. Environmentally friendly photosensitizers were used, specifically [Cu(xant)(bcp)](PF6) and [Ir(dFppy)3]. Besides, photocatalysis tests were performed using several sacrificial electron donors (TEA, TEOA and BIH) in various combinations. It was reported that the dinuclear complex exhibited better CO2 activation in comparison with the mononuclear complex, with a higher TON of up to 45 times (255).
3.4 Catalysts based on earth abundant metals
Great efforts have also been devoted in the last decade to synthesizing complexes based on low-cost earth-abundant metals, e.g. Ni, Mg, Fe, Na, Zn and Co, to go beyond the use of noble metals, which are more expensive due to their low abundance and difficult extraction.Kojima and co-workers99 synthesized an Ni(II) complex bearing an S2N2-type tetradentate ligand and two pyridine pendants, which may act as coordination sites for Lewis acids. In this instance, the two pyridine fragments coordinate with an Mg2+ ion, which enhanced the production of CO by photoinduced CO2 reduction in comparison to the Ni(II) complex with no coordinated Lewis acid. The authors supported the idea that the Mg2+ ion greatly helps to trap CO2, leading to easier CO2–metal intermediate formation and efficient catalytic behavior even at a low CO2 concentration. In addition to Mg2+, they reported the dependence of CO evolution related to other Lewis acids (Li+, Na+, Ca2+, Zn2+, Sc3+ and Y3+), but only Ca2+ and Zn2+ ions were capable of exhibiting photocatalytic behavior similar to that recorded for the Ni(II)–Mg(II) complex. All the photocatalysis experiments were performed in a mixture of DMA/H2O (v/v = 9:1) under a 450 nm LED lamp, using [Ru(bpy)3]Cl2 as a photosensitizer and BIH as the sacrificial electron donor. In the case of the Ni(II)–Mg(II) complex, it was reported to show a TON of 120, TOF of 0.87 min−1 and a selectivity value for CO of 99.7%.
Su and co-workers100 explored a pentanuclear Co(II) complex, [Co5(btz)6(NO3)4(H2O)4], for CO2 conversion to syngas (CO and H2). Photocatalysis tests were carried out under a pure flux of CO2 in CH3CN, with [Ru(bpy)3]Cl2 as the photosensitizer and TEOA as the sacrificial reductant, under irradiation from a xenon lamp at 420 nm. Compared with a mononuclear cobalt complex with a similar structure, the syngas produced was 212 times higher, with a TON value of 2748 within 70 h. The catalytic cycle also showed the advantage of granting a wide adjustability of H2/CO ratio by varying the solvent component and the amount of TEOA, ranging from 16:1 to 2:1. The good performance of the complex for syngas evolution was also confirmed for a diluted CO2 flux. DFT calculation was also conducted, showing a low energy barrier for the formation of the photocatalytic intermediate, which supported the experimental evidence of the efficiency for CO2 reduction.
Chen and co-workers101 synthesized and studied the photocatalytic behavior a Co(II)–Zn(II) heterometallic dinuclear complex [CoZn(bpbp)(CH3COO)2](CH3COO) and a Co(II) homometallic dinuclear complex [Co2(bpbp)(CH3COO)2](CH3COO) using [Ru(phen)3](PF6)2 as the photosensitizer and TEOA as the sacrificial reductant, in several solvents and solvent mixtures, obtaining the best performance with CH3CN/H2O (v/v = 4:1) as the solvent. The irradiation was conducted for 10 h under a 450 nm LED light. The authors reported that both complexes acted efficiently as a catalyst for the photoinduced reduction of CO2 to CO, highlighting that the heterometallic complex exhibited the best performances. Based on the DFT studies, they ascribed this result to the lower energy barrier related to the coordination for CO2 from the heterometallic complex with respect to the energy barrier related to the homometallic coordination. In fact, they found that the Co center of the heterometallic complex showed a higher electron density than the Co centers of the homometallic complex, leading to the easier coordination of CO2, which was the rate-limiting step. The TON, TOF and CO selectivity values were reported to be 6680, 0.19 s−1 and 98%, respectively, for the Co–Zn complex.
Han and co-workers102 reported a heterometallic purpurin complex based on earth-abundant elements ([Fe2Na3(1,2-oxo-4-hydroxyanthracene-9,10-dione)6](TBA)3), which was capable of reducing CO2 to CO under irradiation from a 450 nm light with no additional photosensitizer. In fact, photoinduced electron transfer was reported from the excited ligands to the iron centers, which acted as catalytic sites for CO2 reduction. This system achieved a TON of 2625 and 91% selectivity for CO production in DMF, with BIH as the sacrificial electron donor.
Chao and co-workers103 reported a cyclic complex based on six Fe(II) centers with terpyridine-based bridging ligands (Fig. 13).
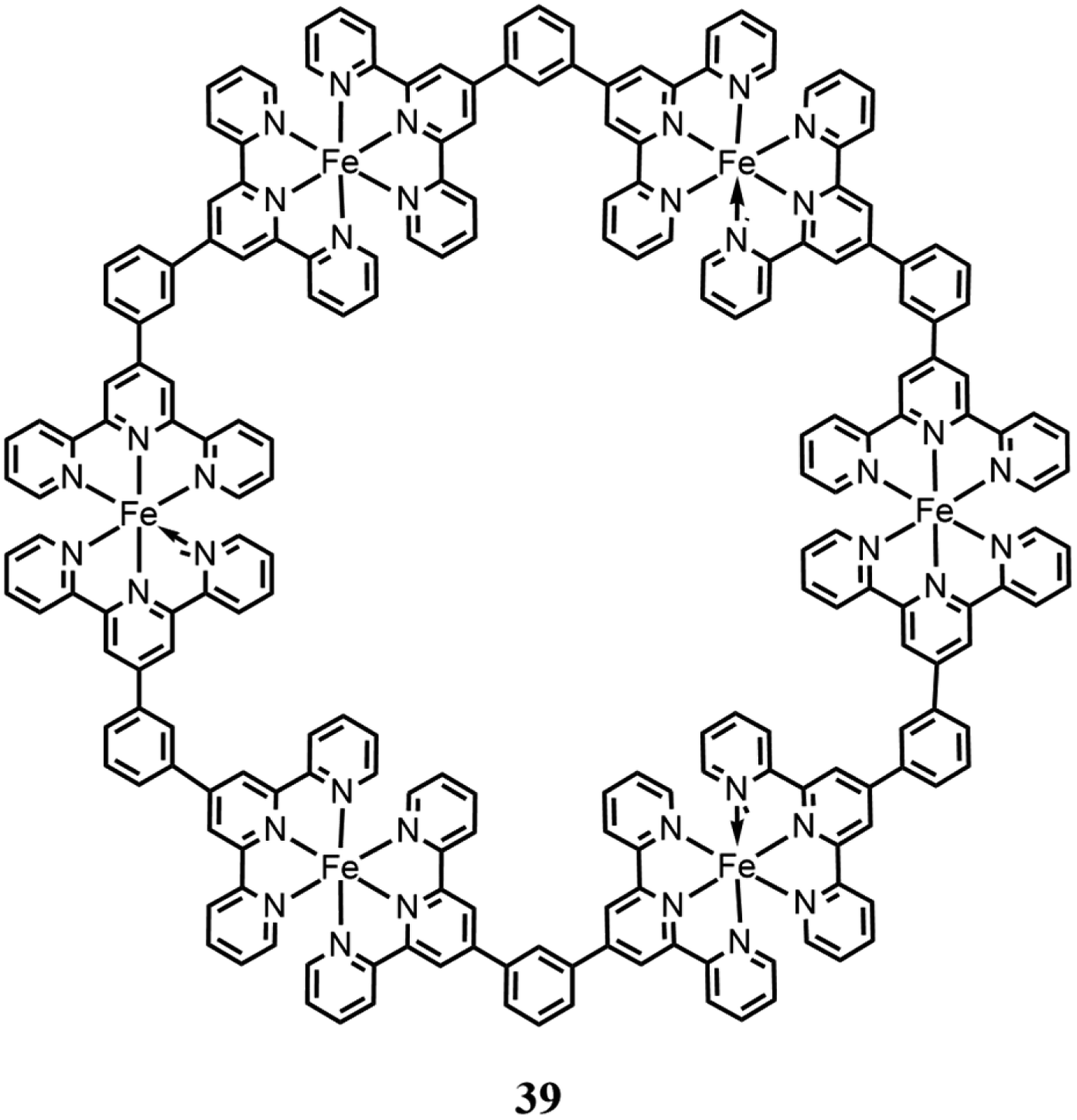 | ||
| Fig. 13 Fe(II) macrocyclic catalyst for CO2 reduction. | ||
They studied the catalytic performances by coupling the complex to 4CzIPN (an organic photosensitizer) and TEA as the sacrificial reagent, under irradiation from a white LED light (420–650 nm). This complex showed catalytic activity for CO evolution with a TON of 14956, TOF of 276 min−1 and CO selectivity of 99.6%. The authors emphasized the importance of the ligands, given that they are redox-active, and consequently contribute significantly to CO2 photoreduction. In fact, they demonstrated that the photosensitizer could reduce the terpyridine fragments, leading to an electron reservoir, and therefore achieving an easier and highly efficient CO2 reduction process.
4. MOFs and COFs as multinuclear photocatalysts
Metal–organic frameworks (MOF) and covalent organic frameworks (COF), in particular show numerous interesting applications such as gas separation and absorption,104 environmental treatment, catalysis105 and energy conversion. In the last decade, a particular class of multinuclear catalysts for artificial photosynthesis is represented by MOFs and COFs.MOFs exhibit a regular crystalline lattice, which is composed of metal ions or metal ion clusters and bridging organic linkers conveniently assembled, leading to a well-defined porous structure, which is the principal responsible for their interesting properties.106 Also, COFs show a crystalline polymeric organic structure with high porosity, in which their building blocks are covalently linked in a giant covalent structure.107 Due to the covalent nature of their intramolecular bonds, COFs present also high stability and high tunability of their properties by changing the building blocks and due to the diversity of their covalent linkage topology.108 Indeed, MOFs and COFs are characterized by the tunability of their properties by changing their base components, offering the opportunity to increase their properties by anchoring active sites inside their pores.
To date, there are many reviews in the literature on the use of MOFs and COFs in photocatalytic water oxidation, CO2 reduction109 and the overall water splitting process110 and this topic is too vast to be exhaustively debate here.
5. Conclusion
In the last few decades, many metal complexes with multiple catalytic subunits have been reported in the literature. The aim of this review was exploring catalysts for oxygen evolution and CO2 reduction, which are composed of multinuclear metal centers. Herein, were discussed multinuclear systems with only catalytic centers and integrated systems with more than one catalytic subunit and photosensitizer moieties. In almost all the reported compounds, both in oxidation and reduction processes, according to the comparison with the homologous mononuclear species, it is possible to observe that there is an increase in stability, durability and overall photocatalytic performances in the presence of more than one active unit. Moreover, in integrated systems, the connection between the catalysts and the photosensitizers leads to a further increase in the photocatalytic performances.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded in part by a grant from the Italian Ministry of Foreign Affairs and International Cooperation (PGR project on Artificial Photosynthesis, collaboration Italy-Japan, Grant Number JP21GR09) and in part by European Union (NextGeneration EU), through the MUR-PNRR project SAMOTHRACE (ECS00000022).Notes and references
- J. Curtin, C. McInerney, B. Ó Gallachóir, C. Hickey, P. Deane and P. Deeney, Renewable Sustainable Energy Rev., 2019, 116, 109402 CrossRef.
- B. Hawkins Kreps, Am. J. Econ. Sociol., 2020, 79, 695–717 CrossRef.
- V. Scott, R. S. Haszeldine, S. F. B. Tett and A. Oschlies, Nat. Clim. Change, 2015, 5, 419–423 CrossRef CAS.
- T. R. Karl and K. E. Treberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renewable Sustainable Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- C.-H. Liao, C.-W. Huang and J. C. S. Wu, Catalysts, 2012, 2, 490–516 CrossRef CAS.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- K. H. Ng, S. Y. Lai, C. K. Cheng, Y. W. Cheng and C. C. Chong, Chem. Eng. J., 2021, 417, 128847 CrossRef CAS.
- F. Puntoriero, G. La Ganga, A. M. Cancelliere and S. Campagna, Curr. Opin. Green Sustainable Chem., 2022, 36, 100636 CrossRef CAS.
- Y. Kuramochi and A. Satake, Catalysts, 2023, 13, 282 CrossRef CAS.
- Y. Pellegrin and F. Odobel, Comptes Rendus Chimie, 2017, 20, 283–295 CrossRef CAS.
- (a) A. Arrigo, F. Puntoriero, G. La Ganga, S. Campagna, M. Burian, S. Bernstorff and H. Amenitsch, Chem, 2017, 3, 494–508 CrossRef CAS; (b) F. Puntoriero, F. Nastasi, G. La Ganga, A. M. Cancelliere, G. Lazzaro and S. Campagna, Multicomponent Supramolecular Photochemistry in Comprehensive Inorganic Chemistry III, Elsevier, Third Edition, 2023, 628–653 Search PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- A. Arrigo, F. Nastasi, G. La Ganga, F. Puntoriero, G. Zappalà, A. Licciardello, M. Cavazzini, S. Quici and S. Campagna, Chem. Phys. Lett., 2017, 683, 96–104 CrossRef CAS.
- A. Arrigo, G. La Ganga, F. Nastasi, S. Serroni, A. Santoro, M.-P. Santoni, M. Galletta, S. Campagna and F. Puntoriero, C. R. Chim., 2017, 20, 209–220 CrossRef CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- (a) H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC; (b) A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS; (c) K. Kitamoto and K. Sakai, Chem. Commun., 2016, 52, 1385–1388 RSC.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331–341 CrossRef CAS.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- H. Zhang, W. Tian, X. Duan, H. Sun, S. Liu and S. Wang, Adv. Mater., 2020, 32, 1904037 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- N. Govindarajan and E. J. Meijer, Inorganics, 2019, 7, 62 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- M. Yagi and M. Kaneto, Chem. Rev., 2001, 101, 21–36 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- (a) C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798–7799 CrossRef CAS PubMed; (b) R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed; (c) Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS PubMed.
- L. L. Zhang, Y. Gao, Z. Liu, X. Ding, Z. Yu and L. C. Sun, Dalton Trans., 2016, 45, 3814–3819 RSC.
- T. Schlossarek, V. Stepanenko, F. Beuerle and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e202211445 CrossRef CAS PubMed.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Würthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS PubMed.
- V. Kunz, M. Schulze, D. Schmidt and F. Würthner, ACS Energy Lett., 2017, 2, 288–293 CrossRef CAS.
- A.-L. Meza-Chincha, D. Schindler, M. Natali and F. Würthner, ChemPhotoChem, 2021, 5, 173–183 CrossRef CAS.
- D. Schindler, A.-L. Meza-Chincha, M. Roth and F. Würthner, Chem.–Eur. J., 2021, 27, 16938–16946 CrossRef CAS PubMed.
- N. Noll and F. Würthner, Chem.–Eur. J., 2021, 27, 444–450 CrossRef CAS PubMed.
- M. Chen, S.-M. Ng, S.-M. Yiu, K.-C. Lau, R. J. Zeng and T.-C. Lau, Chem. Commun., 2014, 50, 14956–14959 RSC.
- A. R. Howells, A. Sankarraj and C. Shannon, J. Am. Chem. Soc., 2004, 126, 12258–12259 CrossRef CAS PubMed.
- (a) A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed; (b) Y. V. Geletii, B. Botar, P. Koegerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896 CrossRef CAS PubMed; (c) N. D. McDaniel, M. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210–217 CrossRef CAS PubMed; (d) J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Cabtree and G. W. Brudvig, Science, 1999, 238, 1524–1527 CrossRef PubMed.
- (a) V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26–34 CrossRef CAS; (b) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- F. Puntoriero, G. La Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725–4727 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- The authors consider the oxygen production in respect of the Ru3+ chemically producted.
- M. Natali, I. Bazzan, S. Goberna-Ferrón, R. Al-Oweini, M. Ibrahim, B. S. Bassil, H. Dau, F. Scandola, J. R. Galán-Mascarós, U. Kortz, A. Sartorel, I. Zaharieva and M. Bonchio, Green Chem., 2017, 19, 2416–2426 RSC.
- (a) T. J. R. Weakley, J. Chem. Soc., Chem. Commun., 1984, 1406 RSC; (b) J. R. Galán-Mascarós, C. J. Gómez-Garcia, J. J. Borrás-Almenar and E. Coronado, Adv. Mater., 1994, 6, 221–223 CrossRef.
- B. S. Bassil, S. Nellutla, U. Kortz, A. C. Stowe, J. van Tol, N. S. Dalal, B. Keita and L. Nadjo, Inorg. Chem., 2005, 44, 2659–2665 CrossRef CAS PubMed.
- M. Ibrahim, Y. Lan, B. S. Bassil, Y. Xiang, A. Suchopar, A. K. Powell and U. Kortz, Angew. Chem., Int. Ed., 2011, 50, 4708 CrossRef CAS PubMed.
- S. Tanakara, M. Annaka and K. Sakai, Chem. Commun., 2012, 48, 1653–1655 RSC.
- N. Taira, K. Yamauchi and K. Sakai, ACS Catal., 2023, 13, 3211–3223 CrossRef CAS.
- L. Yu, Y. Ding and M. Zheng, Appl. Catal., B, 2017, 209, 45–52 CrossRef CAS.
- M. Bösing, A. Nöh, I. Loose and B. Krebs, J. Am. Chem. Soc., 1998, 120, 7252–7259 CrossRef.
- M.-P. Santoni, G. La Ganga, V. Mollica Nardo, M. Natali, F. Puntoriero, F. Scandola and S. Campagna, J. Am. Chem. Soc., 2014, 136, 8189–8192 CrossRef CAS PubMed.
- (a) C. Daniel and H. Hartl, J. Am. Chem. Soc., 2005, 127, 13978–13987 CrossRef CAS PubMed; (b) C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101–5114 CrossRef CAS PubMed.
- J.-R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- J. Lin, X. Liang, X. Cao, N. Weia and Y. Ding, Chem. Commun., 2018, 54, 12515 RSC.
- X. Zhou, F. Li, H. Li, B. Zhang, F. Yu and L. Sun, ChemSusChem, 2014, 7, 2453–2456 CrossRef CAS PubMed.
- A. Genoni, G. La Ganga, A. Volpe, F. Puntoriero, M. Di Valentin, M. Bonchio, M. Natali and A. Sartorel, Faraday Discuss., 2015, 185, 121–141 RSC.
- J. K. Beattie, T. W. Hamblie, J. A. Klepetko, A. F. Masters and P. Turner, Polyhedron, 1998, 17, 1343 CrossRef CAS.
- R. Chakrabarty, S. J. Bora and B. K. Das, Inorg. Chem., 2007, 46, 9450–9462 CrossRef CAS PubMed.
- O. Hodnebrog, M. Etminan, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, K. P. Shine and T. J. Wallington, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- T. R. Karl and K. E. Treberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- H. Akimoto, Science, 2003, 302, 1716–1719 CrossRef CAS PubMed.
- S. C. Roy, O. K. Varghese, M. Paulosea and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- D. Hildebrandt, D. Glasser, B. Hausberger, B. Patel and B. J. Glasser, Science, 2009, 323, 1680–1681 CrossRef PubMed.
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U.S.A., 1982, 79, 701–704 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- S. Das and W. M. A. Wan Daud, RSC Adv., 2014, 40, 20856–20893 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- (a) B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed; (b) Z.-Y. Bian, K. Sumi, M. Furue, S. Sato, K. Koike and O. Ishitani, Dalton Trans., 2009, 983–993 RSC.
- S. Meister, R. O. Reithmeier, A. Ogrodnik and B. Rieger, ChemCatChem, 2015, 7, 3562–3569 CrossRef CAS.
- A. Umemoto, Y. Yamazaki, D. Saito, Y. Tamaki and O. Ishitani, Bull. Chem. Soc. Jpn., 2020, 93, 127–137 CrossRef CAS.
- A. M. Cancelliere, F. Puntoriero, S. Serroni, S. Campagna, Y. Tamaki, D. Saito and O. Ishitani, Chem. Sci., 2020, 11, 1556–1563 RSC.
- A. Santoro, A. M. Cancelliere, K. Kamogawa, S. Serroni, F. Puntoriero, Y. T. S. Campagna and O. Ishitani, Sci. Rep., 2023, 13, 11320 CrossRef CAS PubMed.
- S. Shitaya, K. Nomura and A. Inagaki, Chem. Commun., 2019, 55, 5087–5090 RSC.
- R. O. Reithmeier, S. Meister, B. Rieger, A. Siebel, M. Tschurl, U. Heiz and E. Herdtweck, Dalton Trans., 2014, 43, 13259–13269 RSC.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- R. O. Reithmeier, S. Meister, A. Siebel and B. Rieger, Dalton Trans., 2015, 44, 6466–6472 RSC.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- S. Realista, J. C. Almeida, S. A. Milheiro, N. A. G. Bandeira, L. G. Alves, F. Madeira, M. José Calhorda and P. N. Martinho, Chem.–Eur. J., 2019, 25, 11670–11679 CrossRef CAS PubMed.
- J.-Q. Song, Y.-L. Lu, S.-Z. Yi, J.-H. Zhang, M. Pan and C.-Y. Su, Inorg. Chem., 2023, 62, 12565–12572 CrossRef CAS PubMed.
- J. Jökel, E. B. Boydas, J. Wellauer, O. S. Wenger, M. Robert, M. Römelt and U.-P. Apfel, Chem. Sci., 2023, 14, 12774–12783 RSC.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- (a) A. Paul, D. Connolly, M. Schulz, M. T. Pryce and J. G. Vos, Inorg. Chem., 2012, 51, 1977–1979 CrossRef CAS PubMed; (b) Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed.
- J. Bharti, L. Chen, Z. Guo, L. Cheng, J. Wellauer, O. S. Wenger, N. von Wolff, K.-C. Lau, T.-C. Lau, G. Chen and M. Robert, J. Am. Chem. Soc., 2023, 145(46), 25195–25202 CrossRef CAS PubMed.
- R. Giereth, M. Obermeier, L. Forschner, M. Karnahl, M. Schwalbe and S. Tschierlei, ChemPhotoChem, 2021, 5, 644–653 CrossRef CAS.
- P. Lang, R. Giereth, S. Tschierlei and M. Schwalbe, Chem. Commun., 2019, 55, 600–603 RSC.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- M. Sun, C. Wang, C.-Y. Sun, M. Zhang, X.-L. Wang and Z.-M. Su, J. Catal., 2020, 385, 70–75 CrossRef CAS.
- D. Liu, M. Zhang, H.-H. Huang, Q. Feng, C. Su, A. Mo, J.-W. Wang, Z. Qi, X. Zhang, L. Jiang and Z. Chen, ACS Sustain. Chem. Eng., 2021, 9, 9273–9281 CrossRef CAS.
- H. Yuan, J. Du, M. Ming, Y. Chen, L. Jiang and Z. Han, J. Am. Chem. Soc., 2022, 144, 4305–4309 CrossRef CAS PubMed.
- Y. Wang, L. Chen, T. Liu and D. Chao, Dalton Trans., 2021, 50, 6273–6280 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- (a) A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC; (b) A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed. Engl., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- S. Qui and G. Zhu, Coord. Chem. Rev., 2009, 253, 2891–2911 CrossRef.
- (a) S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed; (b) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- (a) K. Guo, I. Hussain, G. Jie, Y. Fu, F. Zhang and W. Zhu, J. Environ. Sci., 2023, 125, 290–308 CrossRef CAS PubMed; (b) Y. Zhang, H. Liu, F. Gao, X. Tan, Y. Cai, B. Hu, Q. Huang, M. Fang and X. Wang, EnergyChem, 2022, 4, 100078 CrossRef CAS; (c) Y. Yang, Y. Lu, H.-Y. Zhang, Y. Wang, H.-L. Tang, X.-J. Sun, G. Zhang and F.-M. Zhang, ACS Sustain. Chem. Eng., 2021, 9, 13376–13384 CrossRef CAS; (d) Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS; (e) J. Ozdemir, I. Mosleh, M. Abolhassani, L. F. Greenlee, R. R. Beitle Jr and M. H. Beyzavi, Front. Energy Res., 2019, 7, 77 CrossRef.
- S. Navalòn, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. Garcìa, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |