DOI:
10.1039/D4SC04034A
(Edge Article)
Chem. Sci., 2024,
15, 14161-14170
Phospha-bicyclohexene-germylenes exhibiting unexpected reactivity†
Received
19th June 2024
, Accepted 26th July 2024
First published on 30th July 2024
Abstract
Introducing phospha-bicyclohexene (BCH)-germylenes (BCHGe's) as a novel, multifunctional compound class: the title compounds 15–18 are obtained from simple salt metathesis reactions of dipotassium germacyclopentadienediides K2[1] with phosphorusdichlorides. The BCHGe's 15–18 are stabilized by homoconjugation of the germanium(II) centre with the remote C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Despite substantial thermodynamic stabilization, phospha-BCHGe's are reactive and undergo a reductive elimination of elemental germanium to give the corresponding phospholes. The elimination is a nucleophilic, bimolecular process and is prevented by large substituents. The reaction of phospha-BCHGe's with small electrophiles gives the corresponding phosphonium salts. Oxidation with chalcogens takes place at both the germanium and the phosphorus atom, and after elimination of germanium chalcogenides the corresponding phosphole chalcogenides were isolated. The introduced germylenes exhibit strong nucleophilic but also non-neglectable electrophilic properties.
C double bond. Despite substantial thermodynamic stabilization, phospha-BCHGe's are reactive and undergo a reductive elimination of elemental germanium to give the corresponding phospholes. The elimination is a nucleophilic, bimolecular process and is prevented by large substituents. The reaction of phospha-BCHGe's with small electrophiles gives the corresponding phosphonium salts. Oxidation with chalcogens takes place at both the germanium and the phosphorus atom, and after elimination of germanium chalcogenides the corresponding phosphole chalcogenides were isolated. The introduced germylenes exhibit strong nucleophilic but also non-neglectable electrophilic properties.
Introduction
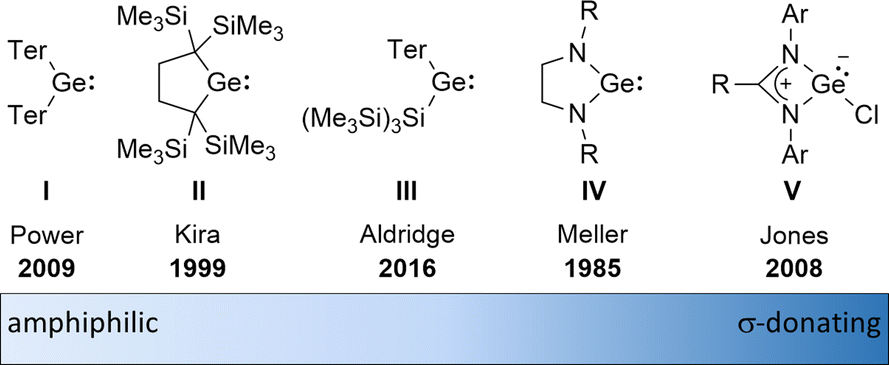
The amphiphilic reactivity of heavy carbene analogues as well as the development of methods for their synthesis and the strategies to control their reactivity are in the focus of modern molecular main group chemistry.1–3 Germylenes are the outriders in this field due to the moderate strength of bonds between germanium and other elements and due to the relative stability of the formal oxidation state +II of germanium.4 For these reasons, germylenes are also attractive goals for catalyst design based on main group elements. The σ-donating and π-accepting properties of germylenes can be tuned applying different stabilisation strategies combined with sophisticated substituent design. This resulted in a variety of germylenes known today, ranging from amphiphilic to solely σ-donating (Fig. 1). The fine tuning of their reactivity enables their application for different purposes such as small molecule activation, bond activation in larger molecules and ligand design.
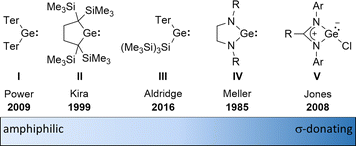 |
| | Fig. 1 Literature-known germylenes exhibiting different properties (Ter = 2,6-dimesitylphenyl).5–9 | |
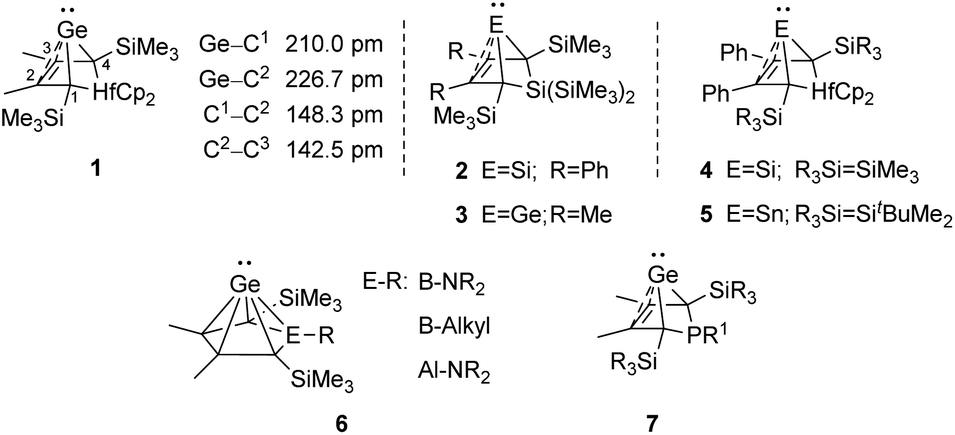
An alternative type of intramolecular stabilisation, previously only known for bicyclic boranes10 and group 14 element cations11–16 and applied for matrix isolation of silylenes,17 was introduced to the chemistry of stable germylenes by our group in 2016.18,19 Germylene 1, with the germanium centre integrated into a bicyclo-[2.1.1]-hexene (BCH) framework, is stabilised by through-space interaction of the germylene centre with the CC double bond in the homoallylic position.20 This homoconjugation leads to a destabilization of the LUMO and to preferentially nucleophilic reactivity of BCHGe 1. Two closely related BCH silylenes (BCHSi's) 2 and 4 were reported shortly after.21,22 Their denotation as bicyclic tetrylenes was justified based on structural and NMR spectroscopic parameters and it was supported by the results of quantum chemical calculations.21,23 It contrasts with the interpretation of the related tin compound 5 as a Sn(0) butadiene complex by Saito and co-workers.24 The very different life times of hafnocena-BCHGe 118 and the sila-BCHGe 222 suggest already a remarkable influence of the second bridging group, the spectator group, of the bicyclic cage on the stability of the germylene. Recently, our group reported on boron- and aluminum-based germa[5]pyramidanes 6.25–27 Despite the close similarity of their topology to the BCHGe's 1 and 3, the inclusion of the electron deficient group 13 elements results in a quite different electronic structure of these nido-clusters. Bearing this significant effect of the spectator group in mind, we introduced here the electron-rich aminophosphanyl group into the BCH framework. This resulted in the formation of germanium(II) compounds with two Lewis basic sides in close neighbourhood: phospha-BCHGe's 7 (Fig. 2).
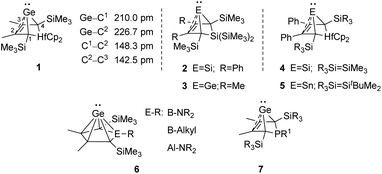 |
| | Fig. 2 Bicyclohexene-type germylenes (BCHGe's) with different bridging groups and related silicon and tin compounds. | |
Results and discussion
The synthesis of aminophospha-bicyclohexene (BCH)-germylenes 15–18 was achieved by double salt metathesis reaction, using dipotassium germolediides K2[8] and amino-dichlorophosphanes 9–13 as starting materials.28,29 The tBuMe2Si-substituted BCHGe's 15b–18b were isolated in yields between 71 and 79% (Scheme 1). In the case of the trimethylsilyl-substituted potassium germolediide K2[8a], the reactions were also selective, but the formation of elemental germanium was observed as a follow-up reaction, giving the corresponding aminophospholes 19a–23a as the final products (Scheme 1). These were isolated after filtration as pure materials. This kind of reductive elimination had previously been reported by our group for a related sila-BCH-germylene 3 (Fig. 2).22 The complete reaction sequence, shown in Scheme 1, describes a germole to phosphole transformation. In the case of phospha-BCHGe's 15a, 16a and 18a, the elimination process is slow, enabling their characterisation by NMR spectroscopy, although they cannot be isolated as pure substances. The elimination proceeds only in solution and is independent of the solvent.
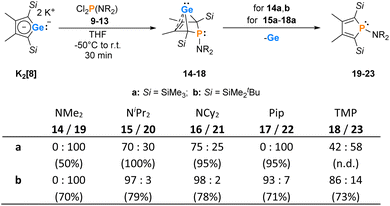 |
| | Scheme 1 Synthesis of BCHGe's 15–18 and phospholes 19–23 (Pip = piperidyl; TMP = 2,2,6,6-tetramethylpiperidyl). The obtained molar ratio, applying standard reaction conditions, was determined by 31P{1H} NMR spectroscopy. The overall yield is given in parentheses. | |
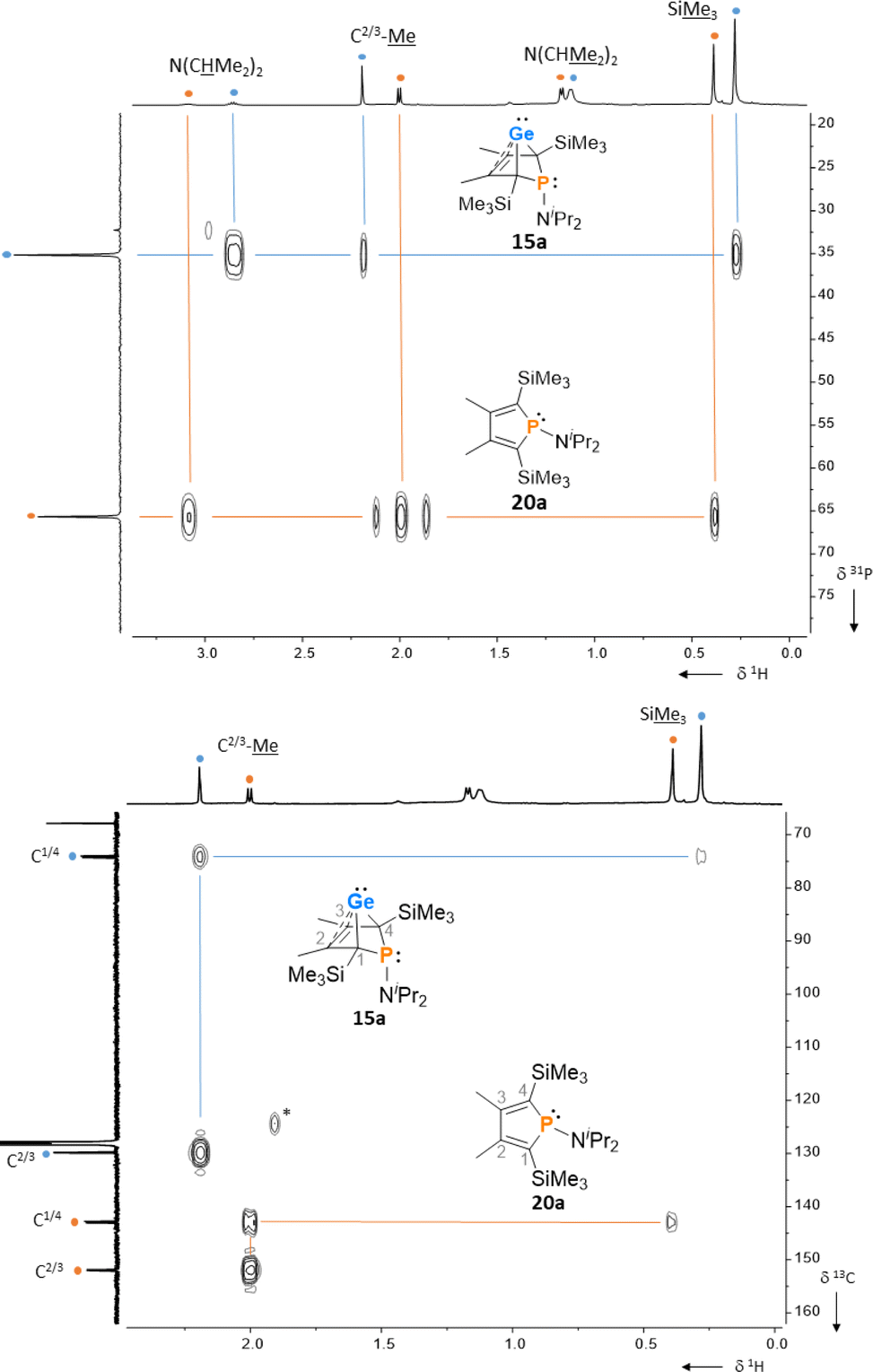
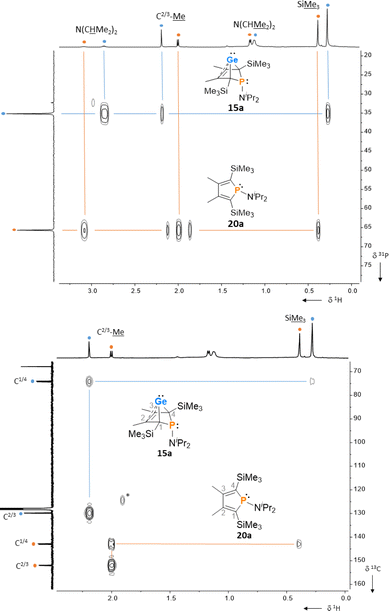
After a standard reaction time of 30 min in THF, the germylene/phosphole mixtures were usually obtained in high yields with the germylenes being the predominant component (Scheme 1). We were not able to detect the BCHGe's 14a and 17a as the elimination of germanium was too fast. We assume the elimination of germanium from the BCHGe's 14–18 to be a bimolecular process, in the case of germylenes 14a–18a induced by the germylenes themselves. This is supported by the following observations: (i) dissolution of the pure crystalline material of germylene 15a immediately led to the formation of small amounts (3%) of phosphole 20a, increasing over time (see ESI: Fig. S13†) and (ii) exchange of the SiMe3 groups for SiMe2tBu substituents enhanced the stability of the BCHGe's 15b–18b. In the latter cases, the elimination of germanium was not observed after their formation and the germylenes were isolated with only small contamination of the corresponding phospholes 20b–23b (see Scheme 1). Solely the NMe2 substituted germylene 14b eluded detection. In a test reaction, the germylene/phosphole mixture 15b/20b obtained under standard conditions was heated in toluene for several hours without any signs of decomposition of the germylene 15b. We attribute the increased stability of the SiMe2tBu substituted germylenes to sterical factors, arising mainly from the bulkier silyl groups and, to a minor extent, also from the amino substituent. We presume that the small amounts of phospholes 20b–23b obtained during the synthesis result from the reaction of already formed BCHGe's 15b–18b with the nucleophilic germole dianion [8b]2− (see ESI: Fig. S119†). Separation of the small amounts of phospholes 20b–23b (Scheme 1) from the corresponding germylenes 15b–18b by crystallization on large scales was not possible due to their similar high solubility in all tested solvents. The sensitivity of germylenes 15b–18bversus air and moisture excluded other separation techniques. Overall, seven different aminophospha-BCHGe's and ten different aminophospholes were identified by NMR spectroscopy (Scheme 1, Table 1 and see ESI†). The reaction sequence shown in Scheme 1 is very selective with germylenes and phospholes being the only products. For that reason, also the mixtures (Scheme 1) obtained with the trimethylsilyl-substituted germolediide [8a]2− were analysed using NMR spectroscopy and both products, phosphole30,31 and germylene, were characterised. An exemplary analysis is shown for the 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of germylene 15a and phosphole 20a. This example demonstrates that BCHGe's, phospholes and their derivatives can clearly be distinguished by heteronuclear NMR spectroscopy. This feature is of importance for the mechanistic studies on the reactivity of the BCHGe's. The 1H NMR spectrum displayed two signals for each functionality: trimethylsilyl protons (SiMe3), isopropyl-methyl protons (N(CHMe2)2), backbone-methyl protons (C2/3–Me) and isopropyl-methine protons (N(CHMe2)2) (Fig. 3, δ1H axis), already indicating the presence of two compounds. Two signals were also displayed in the 31P{1H} NMR spectrum (Fig. 3, top, δ31P axis). The agreement between quantum mechanical predicted 31P NMR chemical shifts for DFT-optimized molecular structures of germylenes 14–18 and phospholes 19–23 and experimental 31P NMR chemical shifts supports our assignment (Table 1).32,331H and 31P NMR spectra indicated the same molar ratio 15a:20a = 7:3. Detailed characterisation of the products was enabled using 2D NMR spectroscopy. After assignment of the 1H NMR signals to the different phosphorus species, using 1H31P HMBC NMR spectra (Fig. 3, top), 1H13C NMR spectra were recorded to determine the structure of the backbone (Fig. 3, bottom).
:30 mixture of germylene 15a and phosphole 20a. This example demonstrates that BCHGe's, phospholes and their derivatives can clearly be distinguished by heteronuclear NMR spectroscopy. This feature is of importance for the mechanistic studies on the reactivity of the BCHGe's. The 1H NMR spectrum displayed two signals for each functionality: trimethylsilyl protons (SiMe3), isopropyl-methyl protons (N(CHMe2)2), backbone-methyl protons (C2/3–Me) and isopropyl-methine protons (N(CHMe2)2) (Fig. 3, δ1H axis), already indicating the presence of two compounds. Two signals were also displayed in the 31P{1H} NMR spectrum (Fig. 3, top, δ31P axis). The agreement between quantum mechanical predicted 31P NMR chemical shifts for DFT-optimized molecular structures of germylenes 14–18 and phospholes 19–23 and experimental 31P NMR chemical shifts supports our assignment (Table 1).32,331H and 31P NMR spectra indicated the same molar ratio 15a:20a = 7:3. Detailed characterisation of the products was enabled using 2D NMR spectroscopy. After assignment of the 1H NMR signals to the different phosphorus species, using 1H31P HMBC NMR spectra (Fig. 3, top), 1H13C NMR spectra were recorded to determine the structure of the backbone (Fig. 3, bottom).
Table 1 Selected NMR spectroscopic data of the synthesised germylenes 15–18 and phospholes 19–23, recorded in benzene-d6, and calculated 31P NMR chemical shifts (italic) (GIAO/M06-L/6-311+G(2d,p)//M06-2X/6-311+G(d,p))
|
|
δ
13C:C1/4 (1JC,P [Hz]) |
δ
13C:C2/3 (2JC,P [Hz]) |
δ
31P |
δ
31P calc. |
|
Data extracted from the 1H13C HMBC NMR spectrum.
|
|
Germylenes
|
|
15a
|
74.4 (36) |
129.9 (8) |
35.4 |
31
|
|
16a
|
74.7 (36) |
129.8 (8) |
39.4 |
37
|
|
18a
|
81.9 (48) |
134.1 (8) |
30.8 |
28
|
|
15b
|
72.8 (41) |
131.1 (7) |
53.8 |
48
|
|
16b
|
73.0 (41) |
131.1 (7) |
57.7 |
50
|
|
17b
|
73.4 (37) |
130.4 (8) |
59.8 |
50
|
|
18b
|
81.6 (53) |
135.1 (8) |
39.7 |
— |
|
|
Phospholes
|
|
19a
|
143.6 (31) |
153.4 (18) |
94.3 |
88
|
|
20a
|
143.0 (33) |
151.9 (19) |
65.6 |
56
|
|
21a
|
143.0 (33) |
151.6 (20) |
63.8 |
61
|
|
22a
|
142.9 (32) |
154.3 (16) |
92.2 |
85
|
|
23a
|
142.7 (34) |
147.0 (26) |
54.2 |
46
|
|
19b
|
141.2 (31) |
154.2 (17) |
98.5 |
— |
|
20b
|
140.7 (36) |
154.0 (16) |
71.7 |
56
|
|
21b
|
141a |
154a |
69.7 |
60
|
|
22b
|
140.2 (34) |
155.1 (16) |
95.9 |
87
|
|
23b
|
139.7 (38) |
148.9 (23) |
61.2 |
— |
 |
| | Fig. 3
1H31P HMBC NMR spectrum (top) and 1H13C HMBC NMR spectrum (bottom) of a mixture of germylene 15a (•) and phosphole 20a (•), (500 MHz, benzene-d6, reaction mixture after exchange of solvent) *impurity. | |
The 13C NMR data of the bicyclohexene and the phosphole carbon backbone are characteristic. Due to the correlation of the trimethylsilyl protons with the C1/4 carbon atoms as well as correlations of the methyl protons with the C1/4 and the C2/3 carbon atoms, triangular patterns are displayed in the 1H13C HMBC NMR spectra. The NMR chemical shifts of the C1/4 carbon atoms allow facile differentiation between the bicyclohexene and the butadiene (phosphole) backbone. The signals of the formally sp3-hybridised bridgehead carbon atoms (δ13C(C1/4) = 72.8–81.9; 15a: δ13C = 74.4) are shifted to lower frequency than those of the sp2-hybridised carbon atoms (δ13C(C2/3) = 129.8–135.1; 15a: δ13C = 129.9). The 13C NMR signals of the butadiene part of the phospholes appear at even higher frequencies (δ13C(C1/4) = 139.7–143.6; 20a: δ13C = 143.0; δ13C(C2/3) = 147.0–155.1; 20a: δ13C = 151.9, see Table 1). Comparison of the data, summarized in Table 1, suggests slight influence of the different silyl and amino groups on the electronic structure of the compounds 15–23. Interestingly, the exchange of the SiMe3 groups for SiMe2tBu groups leads to a significant high frequency shift of the 31P NMR resonances of all phospholes and phospha-BCHGe's. The differences comprise about Δ(δ31P) = 9–18 for the germylenes with the same amino-substituent and about Δ(δ31P) = 4–7 for the phospholes with the same amino substituent.
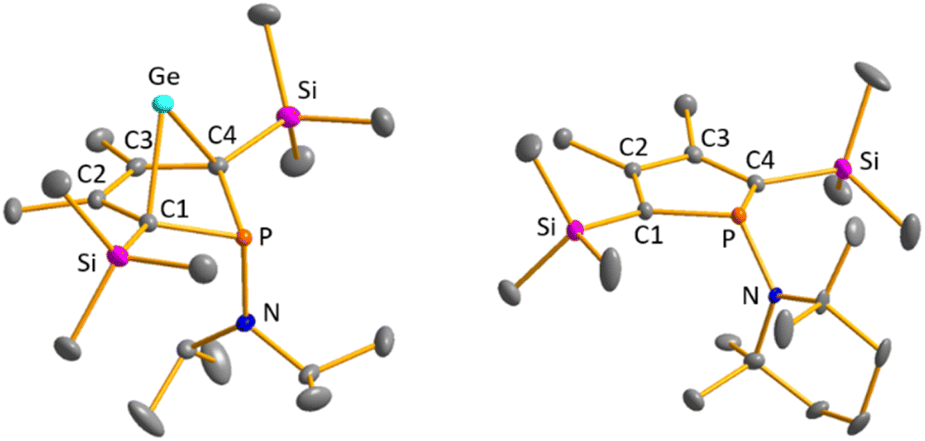
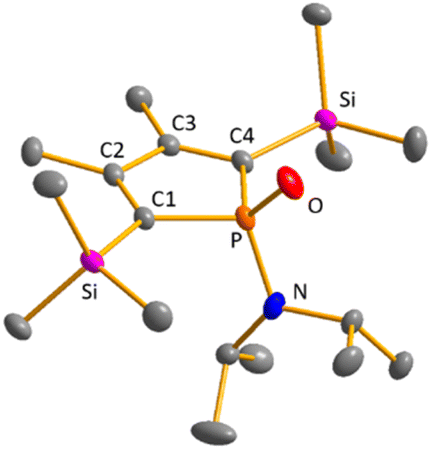
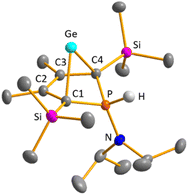
Colourless crystals of germylene 15a, suitable for single crystal X-ray diffraction (sc-XRD), were obtained upon recrystallization from pentane at −30 °C. In agreement with the NMR spectroscopic results, the structure solution revealed a bicyclohexene structure for germylene 15a. The molecule is symmetric, featuring a mirror plane spanned by the Ge, P and N atoms (Fig. 4, left). The C2–C3 bond (142.3 pm) is shorter than the C1–C2 bond (146.5 pm), but still longer than a typical CC double bond (134 pm).34 The Ge–C1 bond of germylene 15a (216.3 pm) is long, elongated by 20 pm compared to the sum of the single bond radii (196 pm).34 The Ge–C2 separation (219.6 pm) is only slightly larger than the Ge–C1 distance, suggesting interaction of the C2–C3 double bond and the germanium atom. The C1–Ge–C4 angle in germylene 15a of α(Ge) = 70.3° is even more acute than that of hafnocena-BCHGe 1 (α(Ge) = 85.0°). Overall, the structural parameters of the GeC4 skeleton are very close to that of the related hafnocena-BCHGe 1 (Fig. 2).18,19 We therefore conclude that in both BCHGe's, 15a and 1, the same stabilising mechanism is operating. Germylene 15a is stabilised by homoconjugation which is delocalisation of π-electrons of the C2C3 bond into the vacant germanium 4p-orbital. The P–C1/4 bonds (184.1 pm) are within the expected range of phosphorus–carbon single bonds (186 pm).34 The coordination sphere of the phosphorus atom is, as typical for tricoordinated phosphorus atoms, pyramidalised (Σ(P) = 302.3°). This underlines the lack of delocalisation of its lone-pair electrons into the backbone. The observed planarisation around the nitrogen atom in germylene 15a (Σ(N) = 360.0°) as well as the short P–N bond (170.1 pm, compared to 182 pm expected for a typical P–N single bond)34 can be assigned to negative hyperconjugation as studied and described by Haaland et al.35,36
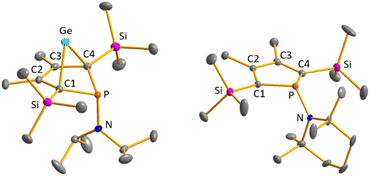 |
| | Fig. 4 Molecular structure of phospha-BCH-germylene 15a (left) in the crystal. Thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. Selected atomic distances [pm] and angles [°]: C1–C2 146.53 (6), C2–C3 142.33 (8), Ge–C1 216.32 (4), Ge–C2 219.64 (5), P–N 170.18 (7), ΣP 302.3, ΣN 360.0. Molecular structure of phosphole 23a (right) in the crystal. Thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. Selected atomic distances [pm] and angles [°]: C1–C2 138.31 (22), C2–C3 145.28 (22), P–N 168.97 (21), ΣP 325.3, ΣN 359.1. | |
Crystals suitable for sc-XRD analysis were obtained from phospholes 21a and 23a.30,31,37 Both molecular structures are very similar, and therefore, only that of the TMP-substituted phosphole 23a will be shortly discussed here (Fig. 4, right). Structural data for aminophosphole 21a are given in the ESI.† Phosphole 23a possesses a slightly folded five-membered ring with a flap angle of α(P) = 15.3°. The phosphorus atom is less pyramidalised (Σ(P) = 325.3°) as in the phospha-BCH-germylene 15a. The backbone consists of two slightly elongated CC double bonds (C1C2 = 138.3 pm) and a slightly shortened C–C single bond (C2–C3 = 145.2 pm), typical for localised butadiene groups. The localisation of the π-electrons and the pyramidalisation of the phosphorus atom correlate with the high s-character of the phosphorus lone pair. As shown for germylene 15a, the coordination sphere of the nitrogen atom of phosphole 23a is trigonal planar and the P–N single bond is shortened (P–N = 168.9 pm). In addition, the dihedral angle of the amino group to the phosphole ring is close to perpendicular (α(N) = 118.6°).
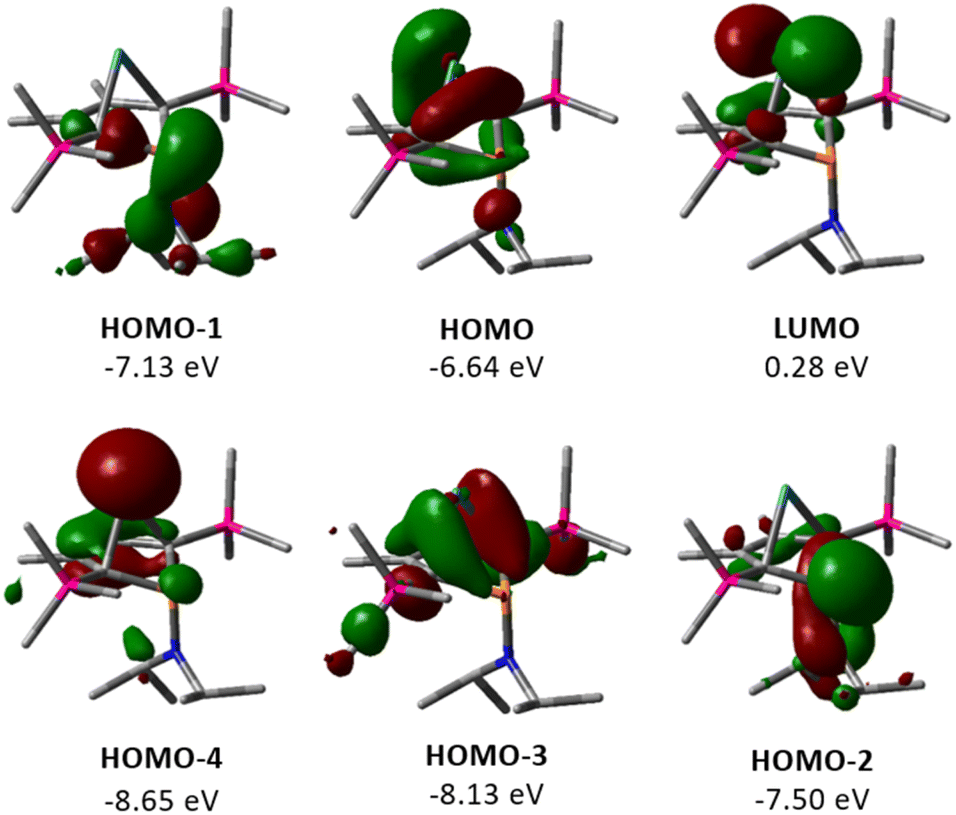
The electronic structure of germylene 15a was further investigated using DFT calculations at the M06-2X/6-311+G(d,p) level of theory.32 The optimized molecular structure of germylene 15a differs only slightly from the experimental structures derived from the solution sc-XRD analysis. Fig. 5 displays selected molecular orbitals. The two frontier orbitals depict the interaction of the empty 4p(Ge) orbital with a filled π-orbital of the butadiene part of the molecule (homoconjugation). The HOMO shows the delocalisation of π-electrons from the butadiene system into the vacant germanium 4p-orbital. The LUMO is mainly the antibonding combination of these orbitals with large contribution from the 4p(Ge) orbital. HOMO−2 and HOMO−1 show contributions from the phosphorus and the nitrogen lone pairs, and from the antibonding P–C1/4 σ-orbitals, which indicates negative hyperconjugation.35 HOMO−3 displays the interaction of the two occupied σ-Ge–C1/4 bonds with the π*-orbital of the C2C3 bond (σ–π*-hyperconjugation). HOMO−4 represents the lone pair at germanium as it shows large contributions from atomic orbitals of the germanium atom. This analysis of the molecular orbitals of germylene 15a identifies it as a carbene analogue. HOMO−4 and the LUMO are the orthogonal occupied and empty orbitals, typical for this class of compounds (Fig. 5).
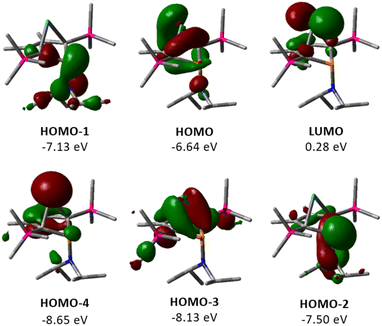 |
| | Fig. 5 Selected frontier molecular orbitals of germylene 15a (M06-2X/6-311+G(d,p); isodensity value 0.04 a.u.). | |
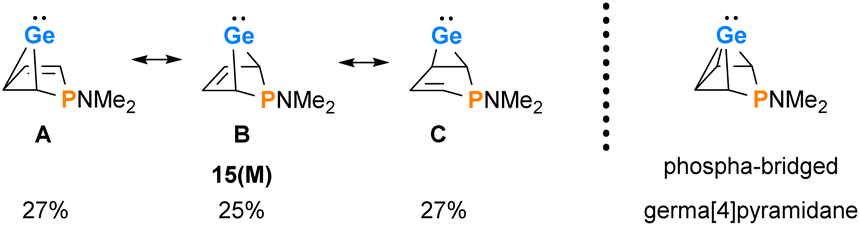
The delocalized electronic structure of germylene 15a is also supported by the results of a natural bond orbital (NBO) analysis of the smaller model compound 15(M).38–40 The analysis reveals significant electron delocalisation from the π(C2C3) bond into an empty 4p Ge orbital (2nd order perturbation energy, ΔE2nd = 4.80 eV) and from the σ-GeC1 and σ-GeC4 bonds into the π*(C2C3) bond (ΔE2nd = 2.54 eV) (see ESI, Fig. S118†). These delocalisations lead to significant covalent bonding between the germanium atom and all four carbon atoms of the butadiene moiety. This is expressed by significant Wiberg bond indices (WBIs)41 between these atoms (WBI(GeC1) = 0.58), WBI(GeC2) = 0.34 vs. WBI(GeC(GeMe4) = 0.83) and by the three dominant resonance structures 15(M)A–C predicted by natural resonance theory (NRT) calculations (see Fig. 6).42
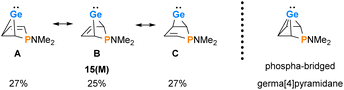 |
| | Fig. 6 Dominant resonance structures A–C of 15(M) according to NRT computations (M06-2X/6-311+G(d,p)) and alternative representation of 15(M) as the arachno-type cluster. | |
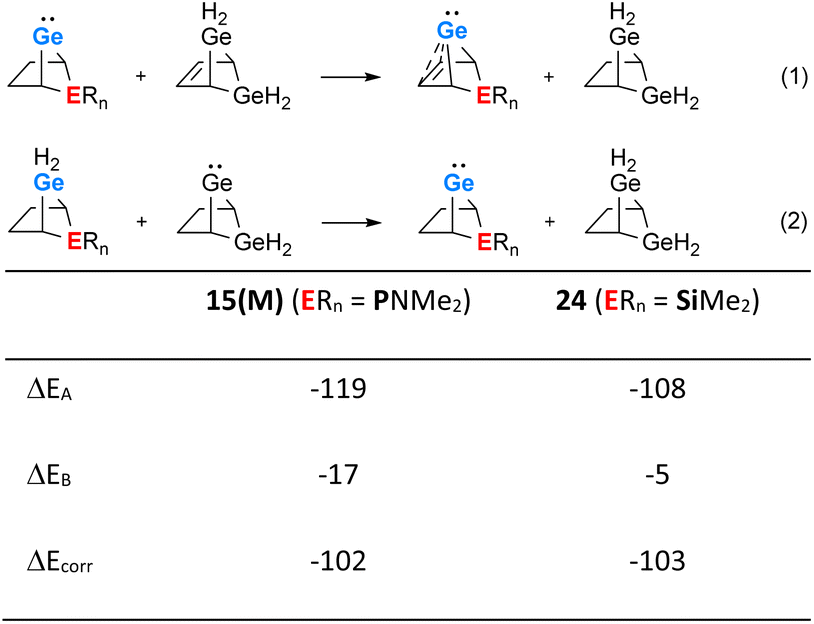
The use of isodesmic reactions allows us to quantify the stabilisation of phospha-BCH-germylene 15(M) by homoconjugation between the germanium atom and the remote π-bond of the bicyclic cyclohexene skeleton (Fig. 7, eqn (1) and (2)). The results for eqn (1) indicate that germylene 15(M) is stabilized through the homoconjugation between the remote CC double bond and the germanium centre by −119 kJ mol−1. The sila-BCH-germylene 24 that was calculated for comparison is slightly less stabilized (−108 kJ mol−1). Eqn (2) takes into account possible interactions of the heteroatom with the germanium(II) centre. These interactions are for both compounds, 15(M) and 24, much smaller. The correction of the stabilisation energy by homoconjugation (eqn (1)) by the effect of the heteroatoms (eqn (2)) leads to both compounds having very similar and large stabilization energies (−102 and −103 kJ mol−1, Fig. 7) due to the cyclohexene cage.
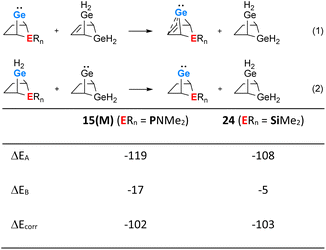 |
| | Fig. 7 Isodesmic eqn (1) and (2) and values calculated for phospha-BCH-germylene 15(M) (ERn = PNMe2) and sila-BCH-germylene 24 (ERn = SiMe2) (M06-2X/6-311+G(d,p)). | |
The delocalised electronic structure suggests for 15(M), and similarly for phospha-BCHGe's 15, also the alternative description as as a phospha-bridged arachno-cluster (4 CH groups and 1 germanium atom with 16 electrons)43–45 with a bridged germa[4]pyramidane structure. Lee and Gapurenko recently suggested this type of structure for the hafnocene derivative 1 (see Fig. 6).46
Reactivity studies
For the reactivity studies of phospha-BCHGe's, we used preferentially the NiPr2- and NCy2-substituted germylenes 15b and 16b as they were formed with only small contamination of the phospholes 20b and 21b (below 5%, see Scheme 1). For comparison purposes, also the trimethylsilyl-substituted derivatives 15a and 16a were tested. In these cases, the reaction time for the preparation was shortened to 5–10 min to minimise the amount of phosphole byproducts 20a and 21a.
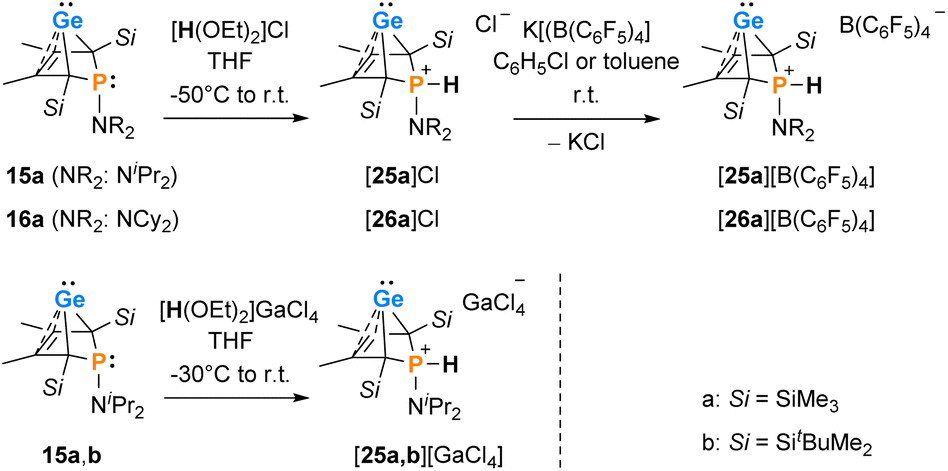
Interestingly, protonation of germylenes 15a/15b and 16a with protonated diethylether exclusively gave the bicyclic phosphonium germylenes [25a/b]+ and [26a]+ in isolated yields up to 80% (Scheme 2). The expected N-protonation did not occur to a sizeable amount.47 The 13C NMR parameters of the products of protonation are very close to those of the starting germylenes, which suggests a bicyclic structure also for the product (Table 2). The NMR spectra displayed characteristic large 1JP,H coupling constants of 566–580 Hz in both the 1H and the hydrogen coupled 31P NMR spectrum. 1H NMR and 31P NMR chemical shifts of the P–H unit depend on the solvent (i.e. for [25a][B(C6F5)4]: δ1H = 6.86 (C6D5Cl) and 7.88 (THF-d8) and δ31P = 14.5 (C6D5Cl) and 28.4 (THF-d8)) as well as on the anion (i.e. for [25a][GaCl4] in C6D6: δ1H = 7.97 and δ31P = 31.0) (Table 2). Interestingly, the trimethylsilyl-substituted phosphonium germylene [25a]+, which was synthesised from in situ prepared germylene 15a, did not undergo the elimination reaction of germanium. It was isolated in 80% yield. In addition, there was no indication for the formation of the corresponding phospholium ions or their follow-up products.48
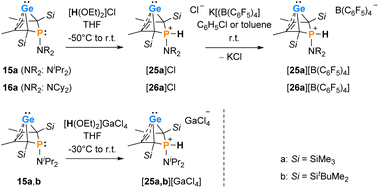 |
| | Scheme 2 Protonation of phospha-BCHGe's 15, 16. | |
Table 2 Selected NMR spectroscopic data of phospha-BCHGe's 15a and 15b and phosphonium-BCHGe's [25a]+, [25b]+ and [26a]+
|
|
Counterion |
Solvent |
δ
13C (C1/4) |
δ
13C (C2/3) |
δ
31P |
δ
1H (P–H) |
|
15a
|
— |
C6D6 |
74.5 |
129.9 |
35.4 |
— |
| — |
THF-d8 |
74.6 |
130.7 |
33.3 |
— |
|
15b
|
— |
C6D6 |
72.8 |
131.1 |
53.8 |
— |
|
[25a]+
|
GaCl4− |
C6D6 |
59.7 |
124.5 |
31.0 |
7.97 |
| [B(C6F5)4]− |
C6D5Cl |
70.2 |
127.7 |
14.5 |
6.86 |
| [B(C6F5)4]− |
THF-d8 |
60.6 |
125.6 |
28.4 |
7.89 |
|
[25b]+
|
GaCl4− |
THF-d8 |
67.8 |
129.3 |
25.7 |
7.78 |
|
[26a]+
|
[B(C6F5)4]− |
C6D5Cl |
69.9 |
127.6 |
13.4 |
6.96 |
Single crystals of phosphonium gallate [25a][GaCl4], suitable for sc-XRD analysis, were obtained upon layering of a benzene solution with pentane (Fig. 8). Comparison of the molecular structure to that of the precursor germylene 15a displays that upon protonation and quarternisation, the s-character of the orbitals involved in the bonding of the phosphorus atom towards the other atoms is enlarged. This results in shortening of the P–C1 and P–N bonds in the phosphonium ion [25a]+ compared to germylene 15a: The C1–P bonds in the phosphonium salt are almost 7 pm (177.4 pm) shorter than in germylene 15a. The P–N bond is shortened by 5 pm (163.9 pm) and almost equals a formal PN double bond (162 pm). The metrics of the but-2-ene backbone of the molecule do not significantly change upon protonation. Both the C1–C2 and the C2–C3 bonds are shortened by less than 1 pm compared to the precursor 15a. The germanium–carbon distances, however, are slightly larger by about 4 pm (C1–Ge) and about 2 pm (C2–Ge), respectively.
 |
| | Fig. 8 Molecular structure of phosphonium-BCH-germylene [25a]+ in the crystal of [25a][GaCl4]. Thermal ellipsoids at 50% probability. Hydrogen atoms (except P–H) and the counterion [GaCl4]− are omitted for clarity. Selected atomic distances [pm] and angles [°]: C1–C2 147.69 (18), C2–C3 141.40 (17), Ge–C1 220.51 (13), Ge–C2 222.41 (12), P–C1 177.91 (14), P–N 163.98 (13), C1–Ge–C4 81.919 (11). | |
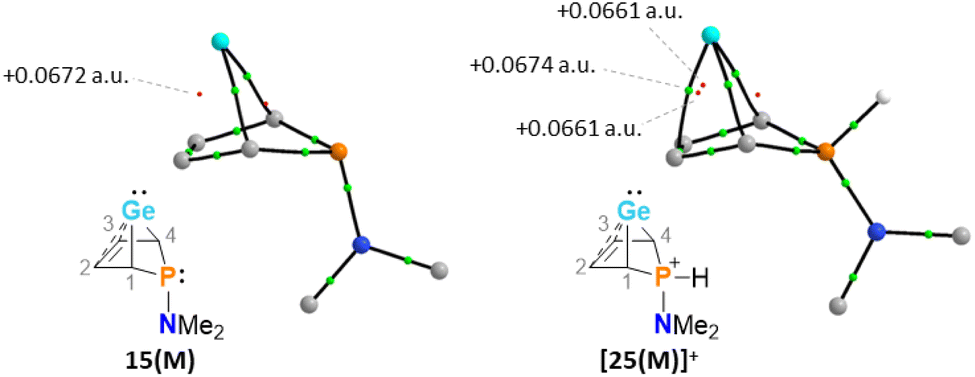
The selective protonation of BCHGe's 15 and 16 at the phosphorus atom is surprising as aminophosphanes are usually protonated at the nitrogen atom.47 The results of quantum mechanical calculations for the different protonation sites of BCHGe 15a reveal that the bicyclic phosphonium ion [25a]+ is by 31 kJ mol−1 less stable than the isomeric ammonium ion [27a]+ (Fig. 9). This suggests that the observed selective protonation at phosphorus is of kinetic origin. The close similarity of the molecular structures of BCHGe 15a and of phosphonium-BCHGe [25a]+ indicates very similar stabilization mechanisms for both types of germylenes. Indeed, the calculated stabilization by homoconjugation (Fig. 7, eqn (1)) of the model compound [25(M)]+ is almost exactly as high as predicted for 15(M) (ΔEA = 119 kJ mol−1).49 Notable are the results of the analysis of the computed electron density of both model compounds, 15(M) and [25(M)]+, based on the quantum theory of atoms in molecules (QTAIM) (Fig. 10).50 For phospha-BCHGe 15(M), the calculated molecular graph displays a dicoordinated germylene with bonds between the germanium and the C1 and C4 carbon atoms. The ring critical point (rcp) of the five-membered C4–Ge ring is located close to a plane spanned by the C2–C3 bond and the germanium atom, indicating the C2C3 → Ge interaction (Fig. 10, left). The calculated molecular graph of phosphonium-BCHGe [25a(M)]+ is different as it displays an additional bonding path between the midpoint of the C2C3 bond and the germanium atom (Fig. 10, right). This T-shaped electron distribution is typical for π-complexes of alkenes with electron deficient centres51–53 and clearly indicates the electron delocalization from the CC double bond to the germanium atom (homoconjugation).22 This obvious difference between the topology of 15(M) and [25(M)]+, however, vanishes during quantitative analysis of the data. The bond critical point (bcp) of the additional bond path of [25(M)]+ is located approximately at the same position where the rcp of the GeC4 ring in phospha-BCH-germylene 15(M) is found (Fig. 10). The rcps of the two thereby emerging rings are located close to this bcp (Fig. 10, right). Additionally, the absolute values of the electron densities at the rcps and the bcp in [25(M)]+ are very similar and furthermore very close to the electron density at the rcp in 15(M) (see Fig. 10). This indicates great similarity of the electron density distribution in both compounds, 15(M) and [25(M)]+, and provides conclusive evidence from the QTAIM analysis for the homoconjugative interaction between the C2C3 double bond and the germanium atom in both compounds.
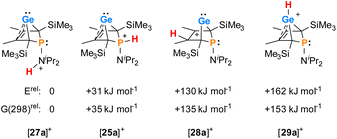 |
| | Fig. 9 Relative energies of isomers of phosphonium-BCHGe [25a]+ at (ICPCM(solvent = THF)/M062X/6-311+G(d,p)//M062X/6-311+G(d,p)). | |
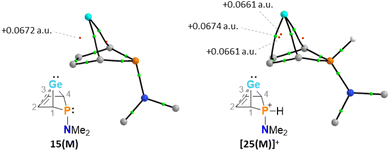 |
| | Fig. 10 Topological graphs of phospha-BCH-germylene 15(M) and phosphonium-BCH-germylene [25(M)]+ according to QTAIM analyses (black lines are bond paths, small green spheres designate the corresponding bond critical points (bcp), which are minima of the electron density along the bond path. Small red spheres indicate ring critical points which represent local minima in rings defined by bond paths; calculated electron densities at critical points are given in black (M06-2X/def2-tzvp//M06-2X/6-311+G(d,p))). | |
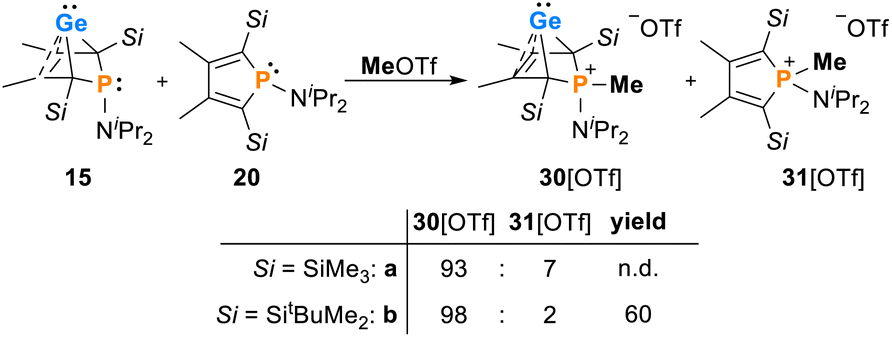
The addition of methyl triflate to solutions of germylenes 15 in pentane resulted in the formation of the methylphosphonium salts [30]OTf which were isolated in up to 60% yield. The product was contaminated with small amounts of the methylphospholium triflates 31, resulting from the methylation of the phosphole byproducts 20 (2–7%, Scheme 3, see the ESI† for identification of [20][OTf]). The methylphosphonium salts [30]OTf were characterised by NMR spectroscopy. In the 1H NMR spectrum, the new P–Me groups feature a doublet signal at δ1H = 2.35 (2JP,H = 12.6 Hz [30a]+) and 2.30 (2JP,H = 11.6 Hz [30b]+). The 13C NMR chemical shifts of the C1/4 bridgehead carbon atoms (δ13C(C1/4) = 67.2 [30a]+, 68.4 [30b]+) are in the typical range for BCHGe's (Tables 1 and 2). The 31P NMR signals were shifted to higher frequency compared to their precursors (δ31P = 54.5 [30a]+, 57.5 [30b]+). Treating the methylphosphonium triflate [30b][OTf] with a second equivalent of methyl triflate did not result in the methylation of the germylene unit. Only the precursor was isolated.
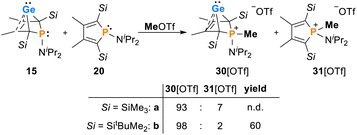 |
| | Scheme 3 Reactivity of phospha-BCHGe's 15 towards MeOTf. | |
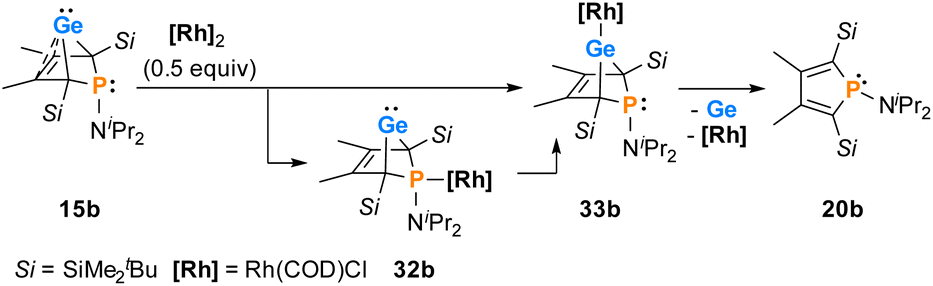
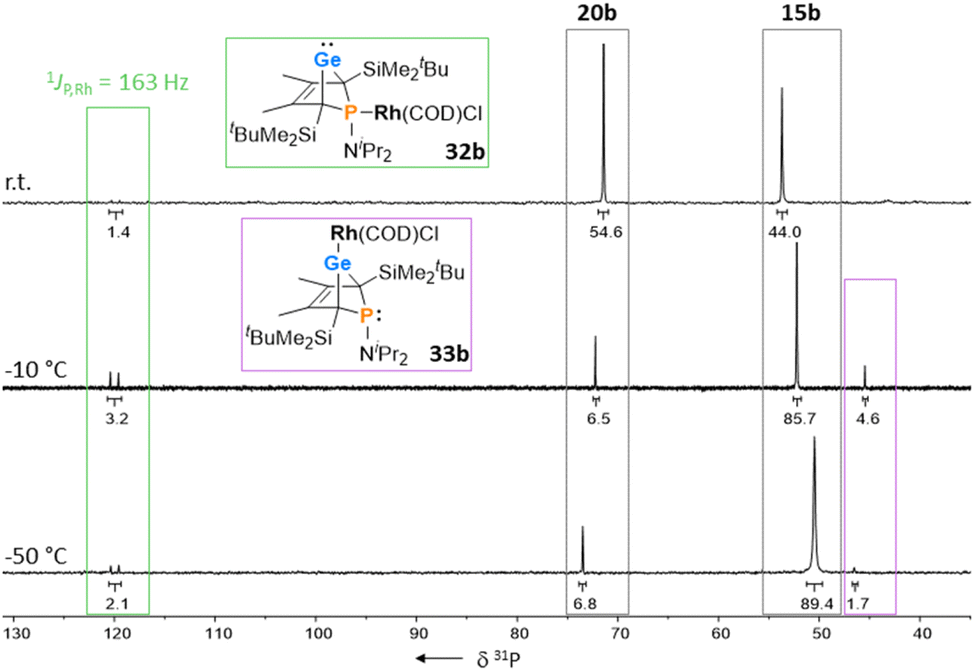
The reactions of phospha-BCHGe's 15 towards various late transition metal complexes were studied to examine their suitability as ligands as well as their preferred coordination mode. In most cases, reductive elimination of germanium occurred and phospholes 20 or their corresponding metal complexes were obtained as final products. The NMR monitoring of the reaction of BCH-germylene 15b with the dimeric cyclooctadiene-rhodium(I)chloride complex [(COD)RhCl]2, [Rh]2, allows a more detailed understanding of the observed reaction sequence. At room temperature, the reaction mixture of germylene 15b and [Rh]2 turned green within five minutes. After an additional five minutes of stirring, the solution turned dark, and a black precipitate was formed. The elimination of germanium and rhodium metal was confirmed by analysis of NMR spectra, recorded from the crude reaction mixture. Almost pure phosphole 20b (δ31P = 71.7) was obtained (Scheme 4). Furthermore, signals of non-coordinated cyclooctadiene (δ1H = 5.53 (CH–), 2.23 (CH2)) were displayed in the 1H NMR spectrum, showing that the rhodium complex decomposed as well during the reaction. To monitor the reaction and detect possible intermediate complexes, the reaction was carried out at room temperature for 5 min and then cooled to T = −60 °C. The reaction was followed by NMR spectroscopy at stepwise increasing temperatures (Fig. 11). Already the first 31P{1H} NMR spectrum, recorded at T = −50 °C, indicated the formation of the phosphole 20b from the germylene 15b (Fig. 11). Additional signals were detected at δ31P = 46.5 and 120.0. The doublet signal at δ31P = 120.0 shows a coupling constant of 1JP,Rh = |163| Hz which is in the reported range of direct 1JP,Rh coupling constants in quadratic planar Rh(I) complexes (1JP,Rh = |110–210| Hz).54,55 Using 2D NMR spectroscopy (see ESI, Fig. S95 and S96†), the signals were assigned to complexes 32b and 33b, both exhibiting a bicyclic backbone structure with the characteristic 13C NMR chemical shift pattern (δ13C(C1/4) = 60.1, δ13C(C2/3) = 129.0 (32b) and δ13C(C1/4) = 57.3, δ13C(C2/3) = 125.3 (33b)). At T = −10 °C, the relative intensity of these two 31P NMR signals slightly increased. At room temperature, they almost vanished, suggesting a short lifetime for these two complexes (Fig. 11).
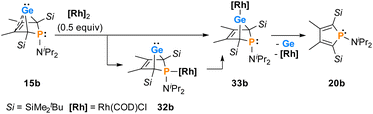 |
| | Scheme 4 Reactivity of germylene 15b towards a Rh(I) complex. | |
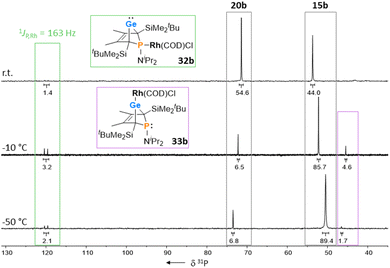 |
| | Fig. 11
31P{1H} VT NMR spectra of the reaction of BCHGe 15b with [(COD)RhCl]2, recorded in toluene-d8. | |
The reactivity studies suggest that germylene 15b coordinates via both the phosphorus (complex 32b) and the germanium atom (complex 33b) to the rhodium centre. In the latter case, irreversible elimination to form phosphole 20b occurs. The phosphane complex 32b isomerises to the germylene complex 33b which then decomposes to give phosphole 20b (Scheme 4). Additional experiments show that phosphole 20b does not form complexes with [(COD)RhCl]2. The results of DFT calculations indicate that the formation of the trimethylsilyl-substituted phosphanyl rhodium complex 32a is more stable than the germylene rhodium complex 33a by ΔE = 9 kJ mol−1 (M062X/6-311+G(d,p)). This small energy difference suggests the possibility for the proposed 32b → 33b isomerisation.
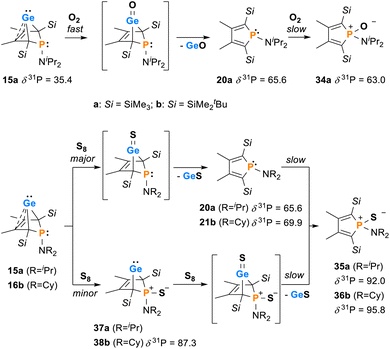
Next, the reactivity of phospha-BCHGe's 15 and 16 towards elemental oxygen and sulphur was examined (Scheme 4). The related hafnocena-BCHGe 1 formed 1,3-digermetanes upon treatment with elemental chalcogens, featuring four-membered Ge–Ch–Ge–Ch rings (Ch = S, Se and Te).56 Exposure to dioxygen led to decomposition, giving the corresponding hafnocenacyclopentadiene and germanium monoxide. Phospha-BCHGe's 15 and 16 showed similar reactivity, preferably reacting with the germylene site towards oxygen and sulphur. Upon exposure of germylene 15a to oxygen, a two-step reaction was observed (Scheme 5, top). In the first, fast step (5 min), pale yellow germanium monoxide was eliminated, giving the phosphole 20a (δ31P = 65.6). The 31P NMR spectrum of the reaction mixture already showed an additional small signal, corresponding to the phosphole oxide 34a (δ31P = 63.0). In the second, slow step (16 h), the phosphorus atom of phosphole 20a was oxidised, quantitatively giving phosphole oxide 34a (Scheme 5, top; identification see ESI †). Phosphole oxide 34a is stable versus Diels–Alder dimerization, a follow-up reaction that is frequently observed for phosphole oxides.30,31 Obviously, the large substituents prevent dimerization in this case. Phosphole oxide 34a was fully analysed by NMR spectroscopy (see ESI †). Additionally, colourless crystals, obtained upon recrystallization from pentane at −30 °C, were analysed by sc-XRD (Fig. 12). The molecular structure of 34a shows the expected localized butadiene group (C1–C2 = 135.42 pm; C2–C3 = 151.37 pm) and a tetracoordinated phosphorus atom with a very short P–O bond (P–O = 148.78 pm). The reaction of BCHGe 15a with elemental sulphur proceeds similarly. Elimination of GeS and formation of the phosphole 20a was observed. Subsequent oxidation by a second equivalent of sulphur gave, after 16 h reaction time, quantitatively phosphole sulphide 35a. The product is characterized by a 31P NMR resonance at δ31P = 92.0 (full characterisation of sulphide 35a, see ESI†). The reaction of the NCy2-substituted phospha-BCHGe 16b with elemental sulphur proceeded slower and revealed a second reaction pathway (Scheme 5, bottom). The predominant pathway was found to be the initial oxidation of the germanium atom and the subsequent elimination of GeS, giving phosphole 21b and finally phosphole sulphide 36b. Additionally, phospha-BCHGe sulphide 38b was detected in the reaction mixture after 20 min at r.t. (ratio 38b:21b = 1:2.5). This suggests that in this case, the oxidation at the phosphorus atom competes with the reaction at germanium (see ESI Fig. S109–S112†). The reaction was completed after 5 days at r.t., giving pure phosphole sulfide 36b.
 |
| | Scheme 5 Reactivity of BCHGe's 15, 16 towards elemental oxygen and sulphur. | |
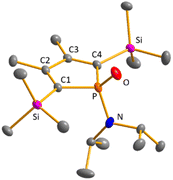 |
| | Fig. 12 Molecular structure of phosphole oxide 34a in the crystal. Thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. Selected atomic distances [pm] and angles [°]: C1–C2 135.42 (10), C2–C3 151.31 (11), P–C1 180.46 (8), P–O 148.78 (6). | |
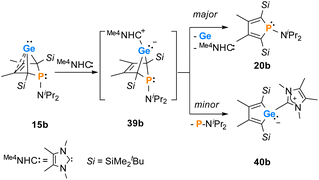
Finally, the reactivity towards nucleophiles was studied. Addition of tetramethylimidazol-2-ylidene (Me4NHC) to a solution of germylene 15b at room temperature induced a colour change from yellow-orange to brownish red as well as the precipitation of a red solid.57,58 Control NMR spectra after 24 h displayed a mixture of germylene 15b, non-coordinated Me4NHC and, as the main product, an increased amount of phosphole 20b. Additionally, we identified the germolylidene-MeNHC complex 40b as the minor byproduct by NMR spectroscopy (see ESI† for NMR data). A small batch of yellow crystals of the stabilized germylene 40b, suitable for sc-XRD analysis, was isolated from the red precipitate and secured the identification of the NHC-stabilized germylene 40b (Fig. 13, Scheme 6). The ratio of phosphole 20b and germolylidene 40b was determined to be 9:1 from 1H NMR spectroscopy. This suggests the following mechanistic scenario: nucleophilic attack of the Me4NHC at the germanium atom forms the bicyclic intermediate 39b. This intermediate eliminates one of the two isolobal fragments which is either the germolylidene NHC-Ge or the phosphinidene iPr2N–P.57,58 Completion of the reaction took seven days with an equimolar amount of Me4NHC and three days with two equivalents of Me4NHC. Weaker nucleophiles (4-dimethylaminopyridine (DMAP) or THF) as well as strong, but sterically more demanding ones (DippNHC, Dipp: 2,6-diisopropylphenyl) did not react with germylene 15b. These results clearly indicate the importance of steric factors for the germanium elimination from the germylenes 15, induced by nucleophiles. According to calculations of the buried volume,59–61Me4NHC is significantly smaller than DippNHC (% Vbur = 25.8 (Me4NHC) vs. 44.0 (DippNHC), see ESI† for details). Interestingly, the size of germylene 15a (% Vbur = 33.5) is comparable to that of Me4NHC, while 15b (% Vbur = 41.9) is almost as large as DippNHC. This comparison supports the above postulated self-induced germanium elimination of SiMe3-substituted phospha-BCHGe's 15a/16a/18a and furthermore gives hint on why this process is not observed for the sterically more encumbered SiMe2tBu substituted phospha-BCHGe's 15b/16b/17b/18b.
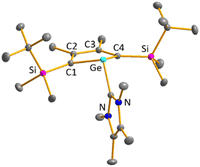 |
| | Fig. 13 Molecular structure of NHC-stabilised germylene 40b in the crystal. Thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. Selected atomic distances [pm] and angles [°]: C1–C2 137.11 (7), C2–C3 148.60 (9), Ge–C(NHC) 203.93 (5), ΣGe 292.8. | |
 |
| | Scheme 6 Reaction of Me4NHC with germylene 15b. | |
Conclusions
Phospha-BCHGe's 15–18 are synthesized by salt metathesis reaction between potassium salts of germole dianions K2[8] and dichloroaminophosphanes 10–13. The size of the flanking silyl groups is of importance for the long-term stability of germylenes. At room temperature in solution, the small SiMe3-substituted germylenes 15a–18a undergo a self-induced elimination of elemental germanium and form the corresponding phospholes 19–23. In contrast, germylenes 15b–18b with larger SiMe2tBu-groups are stable at room temperature. The germylenes 15–18 are stabilized by homoconjugation between the empty 4p orbital at the dicoordinated germanium atom and the remote C2C3 double bond. Therefore, reactivity studies show reduced electrophilicity of the germylenes. Small and strong nucleophiles add to the germanium atom and after elimination of germanium, the corresponding phospholes are formed. Small electrophiles add to the phosphorus atom, forming cationic phosphonium-BCHGe's [25]+, [26]+ and [30]+. The reaction of phospha-BCHGe's with electrophilic transition metal complexes leads to the elimination of germanium and the formation of the corresponding phospholes. Low temperature NMR studies of the reaction of 15b with (COD)RhCl dimer revealed the subsequent formation of a Rh–phosphane 32b and Rh–germylene complex 33b prior to elimination of germanium and generation of the phosphole 20b. Oxidation of phospha-BCHGe's 15 with elemental oxygen and sulphur leads to elimination of GeCh (Ch = O, S) and intermediate formation of the phospholes 20. The final products of these oxidation reactions are the corresponding phosphole chalcogenides 34, 35 and 36.
Overall, the herein introduced phospha-BCHGe's 15–18 exhibit strong nucleophilic but also non-neglectable electrophilic properties. This allows ranking of the reactivity of these germylenes, stabilized by homoconjugation with the remote CC double bond, in between the silyl-substituted germylene III and NHGe IV (Fig. 1).
Data availability
All experimental procedures along with the analytical data are available in the ESI.† The XRD data are deposited in the CCSD database. Original analytical data (source data) are available on request from the corresponding author. Computated molecular structures are given in the ESI† in XYZ coordinates, readable with the CCSD software “Mercury”.
Author contributions
Investigation, data curation, formal analysis, validation: MSW, SK (supporting), TB (supporting); writing: MSW, TM; conceptualization, funding acquisition, project administration, supervision, methodology, resources: TM; XRD: MS.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the DFG (MU-1440/13 and INST 184/227-1). Computations were done at the HPC Clusters, CARL and ROSA, located at the University of Oldenburg, funded by the DFG (INST 184/108-1 FUGG) and the Ministry of Science and Culture (MWK) of the Lower Saxony State.
Notes and references
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
-
N. Nakata, in Organogermanium Compounds, ed. V. Y. Lee, 2023, pp. 387–434 Search PubMed.
- Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS.
- M. Kira, S. Ishida, T. Iwamoto, M. Ichinohe, C. Kabuto, L. Ignatovich and H. Sakurai, Chem. Lett., 1999, 28, 263–264 CrossRef.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem.–Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed.
- A. Meller and C.-P. Gräbe, Chem. Ber., 1985, 118, 2020–2029 CrossRef CAS.
- C. Jones, R. P. Rose and A. Stasch, Dalton Trans., 2008, 2871–2878 RSC.
- P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS.
- T. Laube, J. Am. Chem. Soc., 1989, 111, 9224–9232 CrossRef CAS.
- T. Laube and C. Lohse, J. Am. Chem. Soc., 1994, 116, 9001–9008 CrossRef CAS.
- A. Sekiguchi, T. Matsuno and M. Ichinohe, J. Am. Chem. Soc., 2000, 122, 11250–11251 CrossRef CAS.
- Y. Ishida, A. Sekiguchi and Y. Kabe, J. Am. Chem. Soc., 2003, 125, 11468–11469 CrossRef CAS PubMed.
- C. Gerdes, W. Saak, D. Haase and T. Müller, J. Am. Chem. Soc., 2013, 135, 10353–10361 CrossRef CAS PubMed.
- L. A. Paquette, Angew Chem. Int. Ed. Engl., 1978, 17, 106–117 CrossRef.
- G. Maier and H. P. Reisenauer, Eur. J. Org Chem., 2003, 2003, 479–487 CrossRef.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Angew. Chem., Int. Ed., 2016, 55, 15899–15904 CrossRef CAS.
- Z. Dong, K. Bedbur, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2018, 140, 3052–3060 CrossRef CAS PubMed.
- R. Hoffmann, Acc. Chem. Res., 1971, 4, 1–9 CrossRef CAS.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2017, 139, 7117–7123 CrossRef CAS PubMed.
- C. R. W. Reinhold, Z. Dong, J. M. Winkler, H. Steinert, M. Schmidtmann and T. Müller, Chem.–Eur. J., 2018, 24, 848–854 CrossRef CAS.
- Z. Dong, L. Albers and T. Müller, Acc. Chem. Res., 2020, 53, 532–543 CrossRef CAS PubMed.
- T. Kuwabara, M. Nakada, J. Hamada, J. D. Guo, S. Nagase and M. Saito, J. Am. Chem. Soc., 2016, 138, 11378–11382 CrossRef CAS PubMed.
- P. Tholen, Z. Dong, M. Schmidtmann, L. Albers and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 13319–13324 CrossRef CAS.
- L. Albers, P. Tholen, M. Schmidtmann and T. Müller, Chem. Sci., 2020, 11, 2982–2986 RSC.
- L. Bührmann, L. Albers, M. Beuße, M. Schmidtmann and T. Müller, Angew. Chem., Int. Ed., 2024, 63, e202401467 CrossRef.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- R. B. King and N. D. Sadanani, Synth. React. Inorg. Met.-Org. Chem., 1985, 15, 149–153 CrossRef CAS.
-
L. D. Quin, in Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, 1996, pp. 757–856 Search PubMed.
- F. Mathey, Chem. Rev., 1988, 88, 429–453 CrossRef CAS.
- All computations were done with Gaussian-16. See ESI† for further details..
- The NMR Chemical shifts were calculated using the GIAO method, the M06L functional and the 6-311G(2d,p) basis set for molecular structures optimized at M06-2X/6-311+G(d, p), see ESI† for details..
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef.
- A. V. Belyakov, A. Haaland, D. J. Shorokhov, V. I. Sokolov and O. Swang, J. Mol. Struct., 1998, 445, 303–309 CrossRef CAS.
- P. E. Baskakova, A. V. Belyakov, A. Haaland and H. V. Volden, J. Mol. Struct., 2001, 567–568, 197–202 CrossRef CAS.
- J. Hydrio, M. Gouygou, F. Dallemer, G. G. A. Balavoine and J.-C. Daran, Eur. J. Org Chem., 2002, 2002, 675–685 CrossRef.
- All NBO computations were done with the NBO 7.0 program, see ESI† for details..
- F. Weinhold, Isr. J. Chem., 2022, 62, e202100026 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop and F. Weinhold, J. Comput. Chem., 1998, 19, 628–646 CrossRef CAS.
- K. Wade, J. Chem. Soc. D, 1971, 792–793 RSC.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- R. W. Rudolph, Acc. Chem. Res., 1976, 9, 446–452 CrossRef CAS.
- V. Y. Lee and O. A. Gapurenko, Chem. Commun., 2023, 59, 10067–10086 RSC.
-
M. Alajarín, C. López-Leonardo and P. Llamas-Lorente, in New Aspects in Phosphorus Chemistry, ed. J.-P. Majoral, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 77–106 Search PubMed.
- L. D. Quin, S. E. Belmont, F. Mathey and C. Charrier, J. Chem. Soc., Perkin Trans. 2, 1986, 629–633 RSC.
- The QTAIM computations were done using the AIMAll program, see ESI† for details..
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- I. Krossing and A. Reisinger, Angew. Chem., Int. Ed., 2003, 42, 5725–5728 CrossRef CAS.
- A. Reisinger, N. Trapp, I. Krossing, S. Altmannshofer, V. Herz, M. Presnitz and W. Scherer, Angew. Chem., Int. Ed., 2007, 46, 8295–8298 CrossRef CAS.
- A. Reisinger, N. Trapp, C. Knapp, D. Himmel, F. Breher, H. Rüegger and I. Krossing, Chem.–Eur. J., 2009, 15, 9505–9520 CrossRef CAS.
-
S. Berger, S. Braun and H. O. Kalinowski, NMR-Spektroskopie von Nichtmetallen, Thieme, 1993 Search PubMed.
- J. M. García, E. Ocando-Mavárez, T. Kato, D. S. Coll, A. Briceño, N. Saffon-Merceron and A. Baceiredo, Inorg. Chem., 2012, 51, 8187–8193 CrossRef.
- Z. Dong, M. Schmidtmann and T. Müller, Z. Anorg. Allg. Chem., 2018, 644, 1041–1046 CrossRef CAS.
- Also for the dimer of the aryl-substituted DippNHC-Ge: a red colour is reported. Unfortunately, we cannot provide any resilient evidence for the formation of such a dimer in our case..
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701–9704 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Steffen
Kühn
,
Tobias
Bötel
,
Steffen
Kühn
,
Tobias
Bötel