
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcylhydrazone-based reversibly photoswitchable ion pair transporter with OFF–ON cotransport activity†
Sandip
Chattopadhayay‡
 ,
Paras
Wanjari‡§
and
Pinaki
Talukdar
*
,
Paras
Wanjari‡§
and
Pinaki
Talukdar
*
Department of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pashan, Pune 411008, Maharashtra, India. E-mail: ptalukdar@iiserpune.ac.in
First published on 30th August 2024
Abstract
The cellular membrane transport of physiologically important cations and anions is omnipresent and regulates different physiological functions. Whereas a notable number of cation–anion transporters are being developed to transport salts across the membrane, developing an artificial cation–anion symporter with stimulus-responsive activities is an immense obstacle. Herein, for the first time, we report reversibly photoswitchable acylhydrazone-based transporter 2 that has distinctive OFF–ON cation–anion co-transport abilities. The substituent was modified in 1a–1c and 2, to change the to-and-fro movement of the transporter to enhance the ion transport efficiency. Ion transport experiments across the lipid bilayer membrane demonstrate that 1a has the highest transport activity among the series with irreversible photoisomerization properties, whereas 2 has a unique reversible photoisomerization property. A detailed transport study indicated that the E-conformer of compound 2 facilitates Na+/Cl− transport via the symport process by following the carrier mode of ion transport. 23Na NMR and chloride selective electrode assays confirmed the OFF and ON state of ion transport of compound 2 with photoirradiation. An assembly of [(2E)2 + NaCl] was subjected to geometry optimization to understand the responsible ion binding motif. Geometry optimization followed by the natural bond orbital analysis of 1aZ and 2Z demonstrated that 1aZ forms comparatively stronger intramolecular H-bonding than 2Z, making it accessible for reversible photoisomerization.
1 Introduction
Appropriate concentrations of physiologically important ions (Na+, K+, Cl−, etc.) are crucial for the functioning of different biological processes, including regulating cardiac and neuronal activity, systemic pH, salt and water homeostasis, and cell proliferation, differentiation, and apoptosis.1–3 Structurally and functionally complex transmembrane cation–anion cotransporters (CACs)4–6 are one of the classes of transmembrane proteins that mediate electroneutral transport of cations and anions across the cellular membrane according to the requirement of the cells. Dysfunction of these transmembrane proteins is linked to several pathologies that are generally termed channelopathies, which comprise a plethora of diverse diseases, including epilepsy, migraine, cardiac arrhythmia, asthma, cystic fibrosis, cancer, etc.7–9 Hence, chemical modification of natural transmembrane proteins10–12 or developing small molecule-based synthetic transporters13–15 is a vintage strategy that is still intriguing for giving structural and functional information. Moreover, recent findings revealed that synthetic transporters have the potential for antibacterial16–18 and anticancer19–21 activities. Such an interesting outcome with the synthetic transporter motivated us to design new synthetic ion transporter systems that can translocate ions across the bilayer membrane. Over time, scientists have developed cation22,23 and anion24–26 transporters to mimic the structures and functions of natural ion transporters and their application in various biological contexts. In recent years, cation–anion transporters have also been developed to realize the more complex dual ion selectivity. Synthetic K+–Cl− and Na+–Cl− symporting carriers, based on the salt-binding macrobicycle, were first invented by Smith and coworkers.27 Following this, Kyu-Sung Jeong,28 Philip A. Gale,29 Stefan Matile,30 Yun-Bao Jiang,31 and Nandita Madhavan32 and coworkers have also developed synthetic symporter carriers that can transport cation–anion across the bilayer. Alongside, Madhavan and our group have also made an efficient effort to develop cation–anion symporter channels.19,32,33 Despite the advances in cation–anion transport, the non-stimulus response of these systems is a major limitation for real-life applications.A variety of synthetic gated ion transporters that involve pH, enzymes, voltage, light, redox, ligands, etc., as external stimuli are reported.34–39 Among these, light is a suitable choice as an external stimulus due to its wide control at the molecular level with high spatiotemporal precision.40 To this extent, various photoresponsive transmembrane transporters based on stilbene,41,42 azobenzene,43,44o-nitrobenzyl,45,46 and acyl hydrazone47,48 have been reported in the literature. Although acyl hydrazone-based systems have both cationic and anionic sites, their usage as a photoresponsive cation–anion symporter is unprecedented. In 2017, Zhou et al. showed that an acyl hydrazone-based molecule could be utilized to photoregulate the ion channel formation with different wavelengths of UV light, thus regulating the cation transport.49 Our group also made an appreciable attempt to make an acylhydrazone-based transmembrane carrier that can photocontrol the transport of anions across the lipid bilayer membrane.47,48 But all of these systems either transport cations or anions. Moreover, the use of UV-B region (280 nm–320 nm) of light for the deactivation process or redox stimulus in the reactivation process makes it difficult to use in real-life biological applications. The sparsity of these acyl-hydrazone-based cation–anion transporters with reversible ON and OFF transport activity stimulates us to develop stimulus-responsive small molecule-based cation–anion synthetic transporters.
Herein, we have reported a series of acylhydrazone-based cation–anion transporters 1a–1c and 2 whose transport activity can be controlled spatiotemporally with the help of a specific wavelength of electromagnetic radiations to achieve both ON and OFF state of ion transport (Fig. 1). A series of derivatives were synthesized to modulate the ion transport activity by altering its insertion efficiency into the bilayer. Compound 1a is supposed to show its higher transport activity among the congeners 1a–1c due to its optimum logP value. In investigating the influence of heteroatom X within the chromophoric moiety on the reversible photoswitching process, the design of molecule 2 was implemented.
 | ||
Fig. 1 Chemical structure of compounds 1a–1c and 2 with photoisomerization property and its effect on the ion transport process (A); structural description and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of designed molecules 1a–1c and 2 (B). P values of designed molecules 1a–1c and 2 (B). | ||
2 Results and discussion
2.1 Design and synthesis of 1a–1c and 2
Acylhydrazones are known to bind to cations and anions.50 Here, we designed an acylhydrazone-based molecule that has both cation and anion binding sites in itself. An electron withdrawing nitro group was introduced on the chromophoric moiety to reduce the double bond character of the hydrazone bond by enhancing the extended conjugation, which leads to a rapid photoswitching property. Compounds 1a–1c, and 2 have been synthesized by following Scheme 1. Initially, compounds 3a–3c were reacted with hydrazine monohydrate (NH2NH2·H2O) in methanol solvent under reflux conditions to get compounds 4a–4c. Without further purification of 4a–4c, it was coupled separately either with 5-nitrofuran-2-carbaldehyde or 5-nitrothiophene-2-carbaldehyde in 2:5 water:ethanol mixture at 100 °C to get compounds 1a–1c and 2. All compounds are characterized with the help of 1H, 13C, HRMS, and IR data.
 | ||
| Scheme 1 Synthetic scheme of compounds 1a–1c and 2. | ||
2.2 Photoswitching studies in UV
Initially, the photoswitching efficiency of both compounds 1a and 2 were analyzed through UV-Vis spectroscopy in methanol solvent. Photoirradiation on compound 1a with 365 nm of UV light for 5.5 min showed a significant bathochromic shift of the absorbance peak at 450 nm (Fig. S1A†), indicating the E → Z photoisomerization process. To validate the reversibility of the photoswitching process, the isomerized Z-conformer was further irradiated with 450 nm of LED light. Data revealed that even after 20 min of photoirradiation, reverse isomerization was unfeasible (Fig. S1B†).Notably, the photoswitching study for compound 2 in methanol revealed a very rapid E → Z photoisomerization within 5 s of 365 nm irradiation (Fig. 2A and S3A†). Further, to substantiate the photo reversibility of compound 2, an isomerized Z-conformer was irradiated with 450 nm light. Remarkably, after 9 min of photoirradiation, the Z-conformer reverts to its initial E-conformer (Fig. 2A and S3B†), which validated the photo reversibility of compound 2. The photoisomerization between E and Z-conformer was repeated for four cycles, and no major degradation was observed (Fig. 2B), which revealed that our acylhydrazone-based molecular switch remains stable even after higher photoirradiation cycles. After subjecting compound 2 to photoirradiation at both 365 nm and 450 nm, an insignificant change in the high-resolution mass spectra (HRMS) spectra was observed (Fig. S6†). Notably, the accurate molecular ion peak mass of compound 2 remained constant, indicating an efficient and reversible photoswitching process without any apparent photodegradation of the compound. Further, the presence of the UV-absorbance peak of 2Z conformer in 500 nm wavelength influenced us to investigate the Z → E photoisomerization with 500 nm of LED light. Efficient photoisomerization of the 2Z → 2E conformer was noticed upon photoirradiation with 500 nm of light for 14 min (Fig. S7†), indicating the Z → E photoisomerization can be achieved even with 500 nm of LED light. The stability of compound 2 was investigated by subjecting it to sequential photoirradiation cycles with 365 nm and 500 nm light in MeOH solvent, aiming to assess its photo reversibility (Fig. S8†). No photodegradation was observed even after four cycles, validating compound 2 can be used as a reversible efficient photoswitching system even with longer wavelength of light. The calculated photostationary state (PSS) composition during E → Z isomerization with 365 nm is 2E:2Z = 20:80. Whereas the calculated PSS composition during Z → E isomerization with 450 nm and 500 nm lights is 2E:2Z = 82:18 and 91:9, respectively.
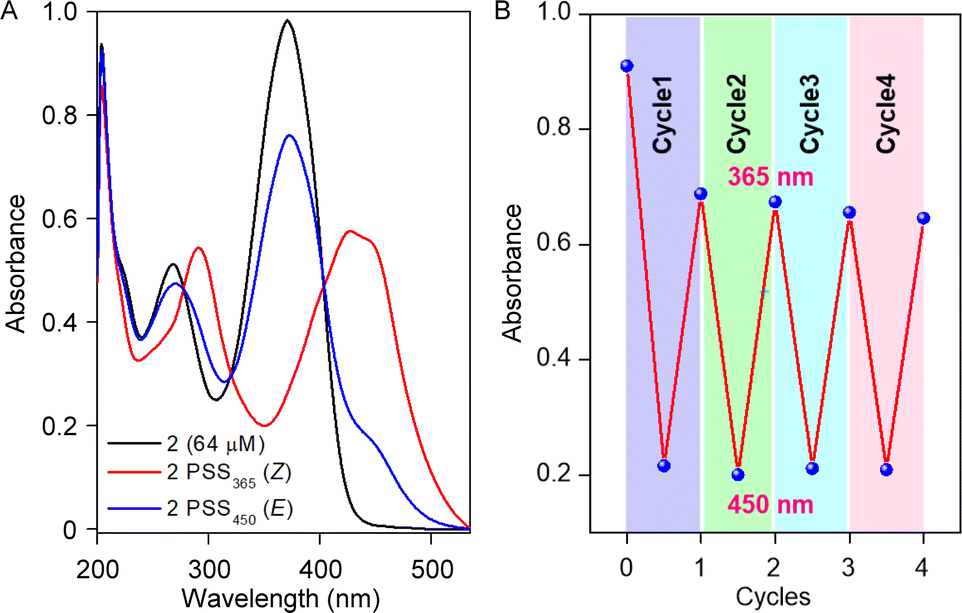 | ||
| Fig. 2 UV-Vis spectrum of compound 2 showing photostationary states for 365 nm and 450 nm (A); change in absorbance value at 365 nm after each cycle of irradiation (B). | ||
To scrutinize the stability of the Z-conformer, time-dependent 1H NMR was recorded in MeOH-d4 (Fig. S9†) and MeOH-d4:D2O (2:1 v/v) solvent (Fig. S10†) systems. Interestingly, a significant enhancement in the stability of 2Z conformer was noticed in the MeOH-d4:D2O solvent (≈48 h) compared to the MeOH-d4 solvent (≈24 h), validating the solvent-dependent thermal stability of the Z-conformer (Fig. S11†). The calculated photostationary state (PSS) of 2Z → 2E thermal relaxation is approximately 80% and 85% in MeOH-d4 and MeOH-d4:D2O solvent stems.
2.3 Ion binding studies
Ion binding efficiency of 1a–1c and 2 were investigated by 1H NMR titration in acetonitrile–d3 with a host concentration of 2 mM.47 Increasing the equivalent of the tetrabutylammonium chloride (TBACl) salt demonstrated a significant downfield shift of the Ha, Hb, and Hc protons, validating the involvement of these three protons in the overall ion binding process by N–H⋯X− and two C–H⋯X− hydrogen bonding interactions. BindFit v0.5 program51 was further used to investigate the binding constant of compounds 1a–1c and 2 with TBACl salt. A 1:1 model of host:guest confirmed that ion binding constants of 1a, 1b, 1c, and 2 with TBACl salt are 995 ± 5% M−1, 662 ± 6% M−1, 429 ± 0.7% M−1, and 935 ± 3% M−1 respectively (Fig. S13–S20†). The photo reversibility nature of compound 2 influenced us to investigate the ion binding process with the tetrabutylammonium halide (TBAX, X− = Br− and I−) salts. A 1:1 model of host:guest confirmed compound 2 has higher binding constants for TBACl (935 ± 3% M−1), followed by TBABr (91 ± 3% M−1). In contrast, the binding constant for TBAI could not be determined due to the insignificant change in the chemical shift with the sequential addition of the TBAI salt (Fig. S23†). To scrutinize the involvement of the cation in the binding process, 1H NMR titration was carried out with compound 2, titrating with NaPF6 (Fig. S24†). Being a larger anion, PF6− is unlikely to fit in the anion binding sites, and hence, changes shown during the 1H NMR titration will be due to the binding of Na+ cation. During the NMR titration Ha, Hb, and Hc peaks are getting upfield shifted with an increase in the equivalent of NaPF6 salt, indicating the Na+ is binding to the respective cation binding sites of compound 2. Further, 1H NMR titration of compound 2 with Na+Cl− ion pair was investigated by mixing an equimolar amount of NaPF6 and TBACl salt (Fig. S25†). Interestingly, during ion pair titration, all of the Ha, Hb, and Hc protons shift towards the downfield region with an increase in the equivalence of the guest. This data indicated compound 2 has the potential to bind both Na+ and Cl− ions in their respective ion binding sites. A 1:1 host:guest binding model by using the BindFit v0.5 program, revealed the binding constant 307 ± 14% M−1 (Fig. S26†).
2.4 Transmembrane ion transport activities of 1a–1c and 2
The rapid, reversible photoswitching process and significant difference in the binding constant with different salts influenced us to scrutinize the ion transport comparisons of the series of compounds 1a–1c and 2. An egg yolk phosphatidylcholine large unilamellar vesicles (EYPC–LUVs), entrapped with a pH-sensitive 8-hydroxypyrene-1,3,6-trisulfonate (HPTS, pKa = 7.2) dye was used to investigate the ion transport activity of compounds 1a–1c and 2. Initially, a 0.8 unit of pH gradient (pHin = 7 and pHout = 7.8) was generated upon the addition of 20 μL of 0.5 M NaOH solution at t = 20 s. A change in the fluorescence intensity was monitored (λem = 510 nm, λex = 450 nm) after the addition of the compounds 1a–1c, and 2 separately. Finally, Titron X–100 was added (t = 300 s) to disintegrate the pH gradient by lysing the vesicles to achieve the complete leakage of HPTS dye.19,52 Ion transport efficiency of compounds 1a–1c, and 2 were investigated at 300 nM concentration with extravesicular and intravesicular NaCl salt. Comparison data divulge the activity sequence of 1a > 2 > 1b > 1c (Fig. 3A). As expected, compounds 1a and 2 showed higher transport efficiency among the congeners due to their optimum logP value (Fig. 1B), which enhanced the to-and-fro movement efficiency of the transporter molecule across the bilayer. HPTS assay was further utilized to investigate the dose-dependent studies of compounds 1a–1c, and 2. Half-maximal effective concentration (EC50) and Hill coefficient (n) values for each analog of compounds were analyzed by the Hill equation. Hill analysis provided the n = 2 for all of the compounds, indicating that two monomeric units of the transporter molecule are actively involved in the overall transport process. Calculation of the EC50 values demonstrated that compound 1a has the lowest EC50 value (71.9 nM ± 2.28, 0.106 mol%) followed by compounds 2 (365 nM ± 17.5, 0.539 mol%), 1b (0.54 μM ± 0.05, 0.798 mol%) and 1c (6.36 μM ± 0.39, 9.40 mol%), which also validate the transport comparison data of the compounds.
 | ||
| Fig. 3 Comparison of ion transport activity of 1a–1c and 2 (A); dose–response study of 2 across EYPC–LUVs ⊃ HPTS (B). | ||
2.5 Ion selectivity studies
Irrespective of the highest ion transport activity of compound 1a, further ion transport studies were performed with compound 2 because of its unique reversible photoswitching nature. To elucidate the involvement of different ions in the transport process, an ion selectivity assay was performed across the EYPC–LUVs ⊃ HPTS. Firstly, the anion selectivity of compound 2 by changing the extravesicular NaX (X− = F−, Cl−, Br−, I−, NO3−, and SCN−) salts was investigated.52,53 A considerable change in transport activity confirmed the involvement of the anions in the transport process with a selectivity order Cl− > NO3− > F− ≈ Br− ≈ I− > SCN− (Fig. 4A). The observed anion selectivity does not follow any of the Hofmeister series,54,55 indicating anion selectivity is controlled by both the hydration energy and the size of the anions (Fig. S33A and B†). Likewise, the involvement of the cations in the overall ion transport process was also checked by varying the extravesicular MCl salts (M+ = Li+, Na+, K+, Rb+, and Cs+).52,53 A conspicuous change in the fluorescence activity upon variation of the extravesicular MCl salt indicated the cation selectivity with an activity sequence Na+ > K+ > Rb+ ≈ Cs+ > Li+ (Fig. 4B). The observed cation selectivity suggested that both the size and hydration energy of the cation regulate the overall cation selectivity (Fig. S33C and D†).54,56 Further, 23Na NMR was utilized for the confirmation of the cation transport property of compound 2. A shift reagent was added externally to distinguish the external and internal Na+ signals. The change in the extravesicular Na+ signal was monitored by increasing the concentration of compound 2. A prominent change in the Na+ ion transport rate was observed with increasing the concentration of compound 2 (Fig. S37†), indicating that compound 2 is capable of transporting Na+ ions across the bilayer membrane. Hence, the aforementioned selectivity studies divulge the involvement of both cation and anion in the overall transport process.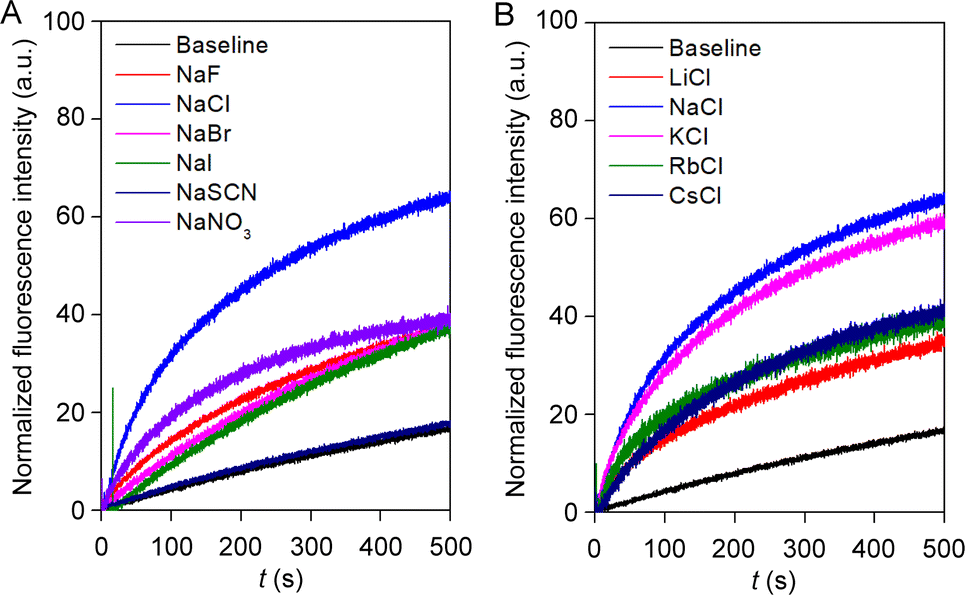 | ||
| Fig. 4 Ion selectivity of compound 2 across EYPC–LUVs ⊃ HPTS towards anion (A); and cation (B) at 300 nM concentration. | ||
2.6 Chloride influx and cation selectivity studies
To ensure the chloride transport activity of compound 2, a halide-sensitive lucigenin dye,57 200 mM NaNO3 salt with pH = 7.0, was encapsulated in EYPC–LUVs. A Cl−/NO3− gradient was created by the addition of 33.3 μL from 2 M NaCl salt in the extravesicular medium, and the change in the fluorescence activity was monitored at λem = 535 nm (λex = 455 nm) with time. At t = 300 s, Titron-X 100 was added to lyse the vesicles.57,58 The increasing concentration of the compound gradually increased the fluorescence quenching of the lucigenin dye, demonstrating the chloride influxing capability of compound 2 (Fig. S35†). Hill analysis provided the EC50 value 2.47 μM ± 0.18 and the Hill coefficient n = 2, reiterating the aforementioned Hill coefficient values of our transporters during the HPTS assay.The cation selectivity of compound 2 was further reconfirmed across the EYPC–LUVs ⊃ lucigenin. A chloride gradient was created by the addition of 33.3 μL of 2 M MCl salts (M+ = Li+, Na+, K+, Rb+, and Cs+) in the extravesicular medium at t = 20 s. A change in the fluorescence quenching activity of lucigenin was monitored after the addition of compound 2 at t = 100 s. Variation in the extravesicular cations conspicuously changed the lucigenin fluorescence quenching activity (Fig. S36†), demonstrating compound 2 is selective towards cations with selectivity order Na+ > K+ > Rb+ > Cs+ ≈ Li+. The obtained cation selectivity sequence coordinates with the selectivity pattern observed during the HPTS assay.
2.7 Mechanistic studies
Increment of the HPTS fluorescence activity in the presence of the transporter molecule can be due to either antiport (OH−/A− or H+/M+ where A− and M+ are anions and cations) or symport (M+/OH−, M+/A− or H+/A−) of ions. To evaluate the operated mechanism by our transporter molecule, different experiments were conducted with compound 2. Initially, the change in the chloride influx across the EYPC–LUVs ⊃ lucigenin was investigated in the presence of valinomycin.59 The analogous chloride influx by compound 2 in the absence and presence of valinomycin confirmed the non-cooperative effect between compound 2 and valinomycin (Fig. S38†). The non-cooperative effect preliminary demonstrated that the compound 2 transports both cations and anions via the symport process. For the confirmation of the mechanistic insight into the transport process, we chose NO3−/SO42− assay.33 Here, 200 mM NaCl salt was encapsulated in the EYPC–LUVs with 1 mM lucigenin, and the pH was adjusted to 7.0. An extravesicular solution of isoosmolar NaNO3 or Na2SO4 was used to create the ionic gradient, and fluorescence intensity was monitored after the addition of compound 2. SO42− is a doubly negatively charged anion, and hence, it is difficult to transport across the bilayer as compared to the NO3− ion. It is expected that if compound 2 follows the antiport process as the transport mechanism, then changing the extravesicular ion from NO3− to SO42− will significantly reduce the fluorescence activity gaining of the lucigenin dye. Irrespective of the extravesicular salt medium, an insignificant difference in ion transport activity for both of the extravesicular salts (Fig. S40†) was observed. This data demonstrated compound 2 can transport ions via a symport mechanism. Further reconfirmation of the transport mechanism was investigated by entrapping the 1 mM lucigenin dye with either isoosmolar NaNO3 salt or Na2SO4 salt. The Cl− ion influx was monitored separately by creating an ionic gradient by adding NaCl salt extravesicularly. It is expected that if compound 2 follows the symport mechanism during ion transport, then changing the intravesicular salt will not affect the Cl− ion influx. However, if it follows the antiport as a transport mechanism, then Cl− ion influx will be negligible in the presence of intravesicular Na2SO4 salt compared to NaNO3 salt. As expected, irrespective of the entrapped salt, a comparable amount of Cl− ion influx was observed by compound 2 (Fig. 5A). Whereas our earlier reported compound60 (RAS02052, Fig. S41C†), which is known to follow the antiport mechanism during the transport process, did not show any Cl− ion influx (compound concentration 2.5 μM) in the presence of intravesicular Na2SO4 salt. Therefore, it is evident that compound 2 transports cation and anion by following the symport mechanism during the transport process. | ||
| Fig. 5 Chloride influx by compound 2 (2.5 μM) across EYPC–LUVs ⊃ lucigenin with entrapped iso-osmolar NaNO3 and Na2SO4 salt in the presence of externally added NaCl ionic pulse (A). U-tube experiment of effective NaCl symport by compound 2 by carrier mechanism (B). | ||
The U-tube experiment was carried out to establish the mode of the ion transport (carrier or channel) by compound 2.61 During the experiment, the source arm (500 mM NaCl, 5 mM phosphate buffer, pH = 7.0) and the receiver arm (5 mM phosphate buffer, pH = 7.0) were separated by a 15 mL chloroform layer dissolved with 1 mM compound 2. The practicability of a supramolecular channel to span the chloroform layer is very unlikely, and hence, it will not increase the Na+ and Cl− concentration in the receiver arm, but a carrier can easily shuttle across the chloroform layer and transport the Na+ and Cl− ion from the source arm to the receiver arm. The experimental result showed a gradual increase in the Na+ and Cl− concentration in the receiver arm (Fig. 5B), indicating that compound 2 follows the carrier mode of ion transport. Subsequently, ANTS–DPX assays were performed to elucidate the occurrence of large pore formation and assess membrane stability in the presence of compound 2.60 The experimental data revealed an indiscernible leakage of ANTS dye across the bilayer membrane in the presence of compound 2 (Fig. S42†). These findings suggest that compound 2 does not induce the formation of large pores within the bilayer and does not compromise membrane integrity.
2.8 Effect of photoswitching in ion transport
The effect of photoirradiation in the transport process was initially investigated across EYPC–LUVs ⊃ HPTS by irradiating the membrane-embedded compound 2 with 365 nm of light at different concentrations. It was evident from the experiment that 2E shows significant transport activity, whereas irradiation with 365 nm of light diminished the ion transport activity due to the 2E → 2Z isomerization (Fig. S44A†). A dose–response study of photoisomerized 2Z indicated that even at high concentrations, it remains inefficient in transporting ions (Fig. S44B†). The presence of higher absorbance intensity at 450 nm and shorter photoswitching time influenced us to scrutinize the impact of photo reversibility on the ion transport process using 450 nm light instead of 500 nm. To investigate the effect of the cation transport during the photoswitching process, a 23Na NMR experiment was carried out with compound 2. 200 mM NaCl salt encapsulated EYPC–LUVs was used during the experiment. A shift regent was added externally during the experiment to differentiate the internal and external Na+ ion peaks. Initially, sodium ion concentrations in intravesicular and extravesicular medium are equal in equilibrium. The addition of 0.2 M sodium tri(poly)phosphate and 0.1 M dysprosium trichloride solution in 2:1 ratio will form Na7Dy(P3O10)2·3NaCl. Noticeably, the addition of membrane-impermeable Dy(P3O10)7− in the extravesicular medium makes ionic concentration unequal across the vesicular membrane. Since the large complex cannot be transported inside the vesicles through the transporter, diffusion of sodium ions through transporter 2 can be visualized by the line broadening observed for the Na+ resonances due to the exchange of the internal and external Na+ ions. Ion transport can be visualized and quantified by the line broadening observed for the Na+ resonances. The exchange rate of the internal and external Na+ ions is directly proportional to the line broadening observed for the Na+ signal. A change in the line width of the external Na+ ion peak was monitored during the photoirradiation of compound 2 with 365 nm and 450 nm of light prior to the addition into vesicles. As expected, a conspicuous decrement in the Na+ transport rate was observed upon photoirradiation with 365 nm light due to the E → Z isomerization. Conversely, compound 2 achieved an initially comparable Na+ transport rate (Fig. 6A) upon photoisomerization of Z → E conformer during exposure to 450 nm of light, demonstrating its stimulus-responsive OFF–ON transport behavior. Further, the effect of photoswitching on the chloride ion transport process was investigated through an ion-selective electrode (ISE) assay. EYPC–LUVs were entrapped with 500 mM NaCl and 10 mM sodium phosphate buffer with pH = 7.0. During the experiment, vesicles were immersed in 500 mM NaNO3 and 10 mM sodium phosphate buffer with pH = 7.0 to create an ionic gradient. A stock solution compound 2 was sequentially irradiated with 365 nm and 450 nm of light and added at 50 s, and the efflux of the chloride ion was monitored by the chloride selective electrode.47 Experimental data revealed that the E-conformer showed significant ion transport activity, but irradiation with 365 nm of light decreased the transport activity due to the closing of the ion binding site in the Z-conformer. To further understand the effect of the reversibility of Z → E conformer in the ion transport, the isomerized Z-conformer was irradiated with 450 nm of light to reconvert to its initial transport active E-conformer. As expected, it regains its initially comparable transport activity, indicating the efficient OFF–ON state of the ion transport property. Three cycles of the photoswitching process were carried out to validate the effect of the reversibility in the ion transport process. Experimental data revealed that OFF–ON transport activity can be retained even after three cycles of photoirradiation (Fig. 6B).
 | ||
| Fig. 6 23Na NMR experiment to investigate the effect of light in Na+ transport rate by compound 2 (3.42 mol% relatives to phospholipids) across EYPC–LUVs (A); effect in Cl− efflux by 2 (4.11 mol% relatives to phospholipids) upon repeating photoirradiation with different wavelength of light (B). | ||
2.9 Theoretical studies
The X-ray crystal structure of compound 2 showed that the molecule is planar and associated with another molecule by forming a hydrogen bond.62 To understand the motifs that are responsible for binding both cation and anion, all possible conformers were initially analyzed by the CONFLEX 8 software program.63,64 1306 number of possible conformations of [(2E)2 + NaCl] assembly complex was obtained in the CONFLEX 8 software program. The highest populated ten conformers (Fig. S48†), along with the dimeric E-conformer of compound 2 were further used to optimize by Gaussian 09 program package65,66 using B3LYP functional and 6-311++G(d,p) basis set. The [(2E)2 + NaCl] complex suggested that the transporter molecule 2 participates in the anion recognition by hydrogen bonding interactions with Ha (Ha⋯Cl− = 2.167 Å), Hb (Hb⋯Cl− = 2.984 Å), and Hc (Hc⋯Cl− = 2.691 Å) protons. Whereas sodium ion binds through the electrostatic interaction between sodium cation and CONH group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Na+ (2.283 Å), hydrazone nitrogen atom N⋯Na+ (2.643 Å) and sulfur atom S⋯Na+ (2.870 Å) (Fig. 7A). The binding energy of the geometrically optimized [(2E)2 + NaCl] complex was calculated to be −28.22 kcal mol−1 (Table S3†).
O⋯Na+ (2.283 Å), hydrazone nitrogen atom N⋯Na+ (2.643 Å) and sulfur atom S⋯Na+ (2.870 Å) (Fig. 7A). The binding energy of the geometrically optimized [(2E)2 + NaCl] complex was calculated to be −28.22 kcal mol−1 (Table S3†).
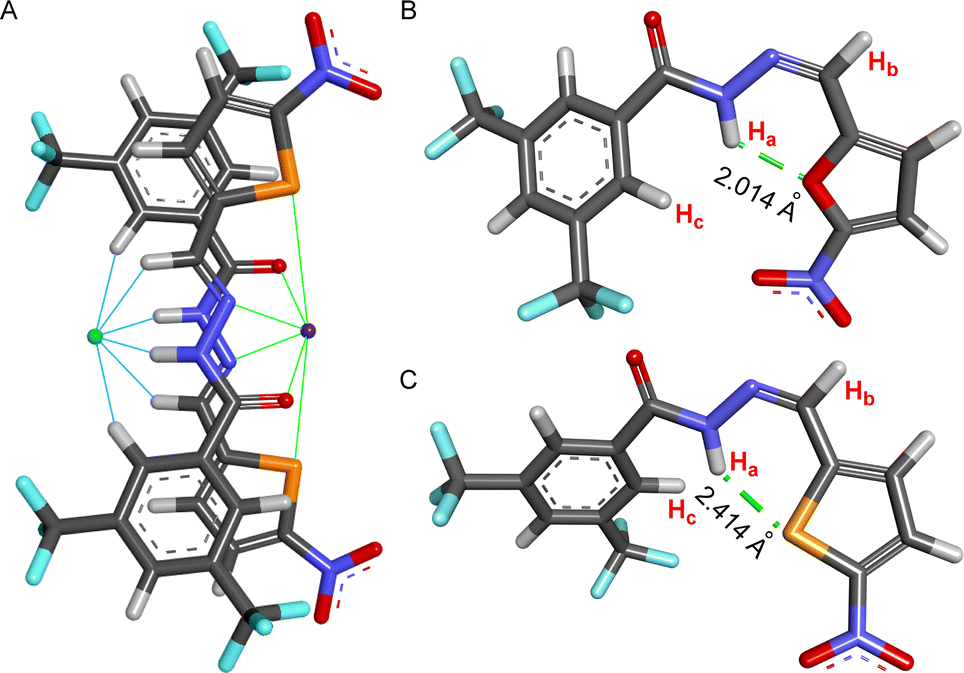 | ||
| Fig. 7 Geometry optimized structure of [(2E)2 + NaCl] complex with different hydrogen bonds and electrostatic interactions to bind anion and cation (A); geometry optimized structure of 1aZ (B), and 2Z with intramolecular hydrogen bond (C). | ||
Further, to unveil the reason for reversibility in the photoswitching of compound 2, the highest populated two conformers of compounds 1aZ and 2Z were geometrically optimized by the Gaussian 09 program with B3LYP functional and 6-311++G(d,p) basis set after analyzing all possible conformers with CONFLEX 8 software program. Geometry optimization of compounds 1aZ and 2Z suggested that the conf-IIZ is the most stable for both of the compounds. Further, natural bond orbital (NBO) analysis67 and the corresponding hydrogen bond interaction energies (E2) were carried out with conf-IIZ of 1aZ and 2Z. Investigation divulged that in 1aZ the oxygen atom (LP 1) in the chromophoric moiety formed an intramolecular H-bond with Ha proton with an interaction energy of 3.92 kcal mol−1 (Fig. 7B), whereas in 2Z the sulfur atom (LP 1) in the chromophoric moiety formed an intramolecular H-bond with Ha proton with comparatively lesser interaction energy of 2.58 kcal mol−1 (Fig. 7C). Further, evidence of the formation of a stronger H-bond in the case of 1aZ also reflects from its shorter H-bonding distance. Lesser H-bonding strength and larger H-bonding length in 2Z, make it a suitable candidate for reversible photoisomerization.
3 Conclusions
We introduced the first example of a reversibly photoswitched acylhydrazone-based ion transporter system that transports cations and anions across the bilayer membrane. A series of molecules were synthesized by varying the lipophilicity. Among the series, although compound 1a showed higher transport activity, compound 2 has unique reversible photoswitching properties. Compound 2 can be reversibly photoswitched by sequential photoirradiation with 365 nm and 450 nm of light as well as 365 nm and 500 nm of light. Ion selectivity studies confirmed its higher selectivity towards Na+ and Cl− ions. The mechanistic study revealed that compound 2 transports ions via the symport mechanism by following the carrier mode of ion transport. Further, the dose–response studies in lucigenin assay demonstrated the chloride influx ability of compound 2. The effect of the photoswitching process in ion transport revealed the OFF–ON transport of compound 2 on sequential repetitive photoirradiation with 365 nm and 450 nm of light. The mode of binding of compound 2 with NaCl as well as the reason for reversible photoisomerization of transporter 2 were finally studied using geometry optimization and natural bond orbital analysis, which strongly validates the experimental evidence. Hence, we believe these acylhydrazone-based small transporter molecules have the potential to be utilized as a photoresponsive ion transport system with reversible OFF–ON transport activity upon photoirradiation. We are certain that in the future, this can open up a new avenue for utilizing these acylhydrazone-based reversibly photoswitchable cation–anion transport systems for real-life biological applications.Data availability
Crystallographic data for compound 2 has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2340801.62 The datasets supporting this article have been uploaded as part of the ESI.† Data for this paper, including synthesis, compound characterization, experimental procedures, and theoretical calculations, are available in the ESI.†Author contributions
P. T. conceived the project and directed the experimental studies. P. W. synthesized and characterized the compounds and investigated the ion binding studies. S. C. performed the ion transport studies and the computational studies. S. C. and P. T. wrote the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PT acknowledges the support of the Science and Engineering Research Board, Govt. of India (Project No. CRG/2022/001640) and IISER Pune. SC thanks the Prime Minister Research Fellowship (PMRF), Government of India, for research fellowships. We acknowledge the computational support and the resources provided by ‘PARAM Brahma Facility’ under the National Supercomputing Mission, Government of India at IISER, Pune. We also thank Mr Sourav Mandal for his help during the theoretical calculation.Notes and references
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouauxt, Science, 1996, 274, 1859–1866 CrossRef CAS PubMed.
- T. E. DeCoursey, K. G. Chandy, S. Gupta and M. D. Cahalan, Nature, 1984, 307, 465–468 CrossRef CAS PubMed.
- S. Y. Chiu and G. F. Wilson, J. Physiol., 1989, 408, 199–222 CrossRef CAS PubMed.
- J. M. Russell, Physiol. Rev., 2000, 80, 211–276 CrossRef CAS PubMed.
- J. A. Payne, C. Rivera, J. Voipio and K. Kaila, Trends Neurosci., 2003, 26, 199–206 CrossRef CAS PubMed.
- P. Blaesse, M. S. Airaksinen, C. Rivera and K. Kaila, Neuron, 2009, 61, 820–838 CrossRef CAS PubMed.
- G. Bernard and M. I. Shevell, Pediatr. Neurol., 2008, 38, 73–85 CrossRef PubMed.
- J. Y. Choi, D. Muallem, K. Kiselyov, M. G. Lee, P. J. Thomas and S. Muallem, Nature, 2001, 410, 94–97 CrossRef CAS PubMed.
- T. J. Jentsch, T. Maritzen and A. A. Zdebik, J. Clin. Invest., 2005, 115, 2039–2046 CrossRef CAS PubMed.
- J.-L. Galzi, A. Devillers-Thiery, N. Hussy, S. Bertrand, J.-P. Changeux and D. Bertrand, Nature, 1992, 359, 500–505 CrossRef CAS PubMed.
- W. Grosse, L.-O. Essen and U. Koert, ChemBioChem, 2011, 12, 830–839 CrossRef CAS PubMed.
- S. Reitz, M. Cebi, P. Reiß, G. Studnik, U. Linne, U. Koert and L.-O. Essen, Angew. Chem., Int. Ed., 2009, 48, 4853–4857 CrossRef CAS PubMed.
- P. Xin, H. Kong, Y. Sun, L. Zhao, H. Fang, H. Zhu, TaoJiang, J. Guo, Q. Zhang, W. Dong and C.-P. Chen, Angew. Chem., Int. Ed., 2019, 58, 2779–2784 CrossRef CAS PubMed.
- W.-L. Huang, X.-D. Wang, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, J. Am. Chem. Soc., 2020, 142, 13273–13277 CrossRef CAS PubMed.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- A. I. Share, K. Patel, C. Nativi, E. J. Cho, O. Francesconi, N. Busschaert, P. A. Gale, S. Roelens and J. L. Sessler, Chem. Commun., 2016, 52, 7560–7563 RSC.
- S. Dey, A. Patel, N. Haloi, S. Srimayee, S. Paul, G. K. Barik, N. Akhtar, D. Shaw, G. Hazarika, B. M. Prusty, M. Kumar, M. K. Santra, E. Tajkhorshid, S. Bhattacharjee and D. Manna, J. Med. Chem., 2023, 66, 11078–11093 CrossRef CAS PubMed.
- M. R. Ghadlri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef PubMed.
- J. A. Malla, R. M. Umesh, A. Vijay, A. Mukherjee, M. Lahiri and P. Talukdar, Chem. Sci., 2020, 11, 2420–2428 RSC.
- S.-H. Park, S.-H. Park, E. N. W. Howe, J. Y. Hyun, L.-J. Chen, I. Hwang, G. Vargas-Zuñiga, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Chem, 2019, 5, 2079–2098 CAS.
- F.-F. Shen, S.-Y. Dai, N.-K. Wong, S. Deng, A. S.-T. Wong and D. Yang, J. Am. Chem. Soc., 2020, 142, 10769–10779 CrossRef CAS PubMed.
- R. Sasaki, K. Sato, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2021, 143, 1348–1355 CrossRef CAS PubMed.
- R. Ye, C. Ren, J. Shen, N. Li, F. Chen, A. Roy and H. Zeng, J. Am. Chem. Soc., 2019, 141, 9788–9792 CrossRef CAS PubMed.
- F. Zeng, F. Liu, L. Yuan, S. Zhou, J. Shen, N. Li, H. Ren and H. Zeng, Org. Lett., 2019, 21, 4826–4830 CrossRef CAS PubMed.
- P. H. Schlesinger, R. Ferdani, J. Liu, J. Pajewska, R. Pajewski, M. Saito, H. Shabany and G. W. Gokel, J. Am. Chem. Soc., 2002, 124, 1848–1849 CrossRef CAS PubMed.
- M. Merritt, M. Lanier, G. Deng and S. L. Regen, J. Am. Chem. Soc., 1998, 120, 8494–8501 CrossRef CAS.
- A. V. Koulov, J. M. Mahoney and B. D. Smith, Org. Biomol. Chem., 2003, 1, 27–29 RSC.
- J. H. Lee, J. H. Lee, Y. R. Choi, P. Kang, M.-G. Choi and K.-S. Jeong, J. Org. Chem., 2014, 79, 6403–6409 CrossRef CAS PubMed.
- C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC.
- A. V. Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef PubMed.
- Z. Zhao, B. Tang, X. Yan, X. Wu, Z. Li, P. A. Gale and Y.-B. Jiang, Front. Chem. Sci. Eng., 2022, 16, 81–91 CrossRef CAS.
- D. Basak, S. Sridhar, A. K. Bera and N. Madhavan, Org. Biomol. Chem., 2016, 14, 4712–4717 RSC.
- S. Chattopadhayay, A. Ghosh, T. K. Mukhopadhyay, R. Sharma, A. Datta and P. Talukdar, Angew. Chem., Int. Ed., 2023, 62, e202313712 CrossRef CAS PubMed.
- L. Lien, D. C. J. Jaikaran, Z. Zhang and G. A. Woolley, J. Am. Chem. Soc., 1996, 118, 12222–12223 CrossRef CAS.
- E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS PubMed.
- Y. R. Choi, B. Lee, J. Park, W. Namkung and K.-S. Jeong, J. Am. Chem. Soc., 2016, 138, 15319–15322 CrossRef CAS PubMed.
- A. Docker, T. G. Johnson, H. Kuhn, Z. Zhang and M. J. Langton, J. Am. Chem. Soc., 2023, 145, 2661–2668 CrossRef CAS PubMed.
- J. A. Malla, R. M. Umesh, S. Yousf, S. Mane, S. Sharma, M. Lahiri and P. Talukdar, Angew. Chem., Int. Ed., 2020, 59, 7944–7952 CrossRef CAS PubMed.
- P. Talukdar, B. Pinaki, M. Guillaume, S. Jiri and M. Naomi, Chem.–Eur. J., 2005, 11, 6525–6532 CrossRef CAS PubMed.
- M. R. Banghart, M. Volgraf and D. Trauner, Biochemistry, 2006, 45, 15129–15141 CrossRef CAS PubMed.
- S. J. Wezenberg, L.-J. Chen, J. E. Bos, B. L. Feringa, E. N. W. Howe, X. Wu, M. A. Siegler and P. A. Gale, J. Am. Chem. Soc., 2022, 144, 331–338 CrossRef CAS PubMed.
- W.-Z. Wang, L.-B. Huang, S.-P. Zheng, E. Moulin, O. Gavat, M. Barboiu and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 15653–15660 CrossRef CAS PubMed.
- T. G. Johnson, A. Sadeghi-Kelishadi and M. J. Langton, J. Am. Chem. Soc., 2022, 144, 10455–10461 CrossRef CAS PubMed.
- T. Liu, C. Bao, H. Wang, Y. Lin, H. Jia and L. Zhu, Chem. Commun., 2013, 49, 10311–10313 RSC.
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed.
- L. E. Bickerton and M. J. Langton, Chem. Sci., 2022, 13, 9531–9536 RSC.
- M. Ahmad, S. Chattopadhayay, D. Mondal, T. Vijayakanth and P. Talukdar, Org. Lett., 2021, 23, 7319–7324 CrossRef CAS PubMed.
- M. Ahmad, N. J. Roy, A. Singh, D. Mondal, A. Mondal, T. Vijayakanth, M. Lahiri and P. Talukdar, Chem. Sci., 2023, 14, 8897–8904 RSC.
- Y. Zhou, Y. Chen, P.-P. Zhu, W. Si, J.-L. Hou and Y. Liu, Chem. Commun., 2017, 53, 3681–3684 RSC.
- Z. Kokan and M. J. Chmielewski, J. Am. Chem. Soc., 2018, 140, 16010–16014 CrossRef CAS PubMed.
- BindFit v0.5, https://www.app.supramolecular.org/bindfit/ Search PubMed.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS PubMed.
- T. Saha, M. S. Hossain, D. Saha, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 7558–7567 CrossRef CAS PubMed.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- E. M. Wright and J. M. Diamond, Physiol. Rev., 1977, 57, 109–156 CrossRef CAS PubMed.
- G. Eisenman and R. Horn, J. Membr. Biol., 1983, 76, 197–225 CrossRef CAS PubMed.
- J. Biwersi, B. Tulk and A. S. Verkman, Anal. Biochem., 1994, 219, 139–143 CrossRef CAS PubMed.
- B. P. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I.-C. Hwang, E. H. Lee, J. H. Roh and K. Kim, J. Am. Chem. Soc., 2017, 139, 7432–7435 CrossRef CAS PubMed.
- S. V. Shinde and P. Talukdar, Angew. Chem., Int. Ed., 2017, 56, 4238–4242 CrossRef CAS PubMed.
- R. Sharma, S. Sarkar, S. Chattopadhayay, J. Mondal and P. Talukdar, Angew. Chem., Int. Ed., 2024, 63, e202319919 CrossRef CAS PubMed.
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed.
- S. Chattopadhayay, P. Wanjari and P. Talukdar, CCDC 2340801: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2jksqm.
- H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950–8951 CrossRef CAS.
- H. Goto, S. Obata, N. Nakayama and K. Ohta, CONFLEX 8, CONFLEX Corporation, Tokyo, Japan, 2017 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02474e |
| ‡ Authors contributed equally. |
| § Present address: Institut Europeen des Membranes, University of Montpellier, ENSCM-CNRS, UMR 5635 Place E. Bataillon CC047, 34095 Montpellier, France. |
| This journal is © The Royal Society of Chemistry 2024 |