
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContracted porphyrins and calixpyrroles: synthetic challenges and ring-contraction effects
Keita
Watanabe
a,
Narendra Nath
Pati
 b and
Yasuhide
Inokuma
*ab
b and
Yasuhide
Inokuma
*ab
aDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8 Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: inokuma@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
First published on 24th April 2024
Abstract
Ring-contracted porphyrin analogues, such as subporphyrins and calix[3]pyrroles, have recently attracted considerable attention not only as challenging synthetic targets but also as functional macrocyclic compounds. Although canonical porphyrins and calix[4]pyrrole are selectively generated via acid-catalyzed condensation reactions of pyrrole monomers, their tripyrrolic analogues are always missing under similar conditions. Recent progress in synthesis has shown that strain-controlled approaches using boron(III)-templating, core-modification, or ring tightening provide access to various contracted porphyrins. The tripyrrolic macrocycles are a new class of functional macrocycles exhibiting unique ring-contraction effects, including strong boron chelation and strain-induced ring expansion. This Perspective reviews recent advances in synthetic strategies and the novel ring-contraction effects of subporphyrins, triphyrins(2.1.1), calix[3]pyrroles, and their analogous.
 From left to right: Keita Watanabe, Narendra Nath Pati, and Yasuhide Inokuma | Keita Watanabe (left) received his BS from Hokkaido University, Japan, in 2022. He is now an MC student under the supervision of Prof. Inokuma at Hokkaido University studying the chemistry of calix[3]pyrroles and related macrocycles. Narendra Nath Pati (center) received his M.Sc. from Banaras Hindu University, India, and PhD from the School of Chemistry, University of Hyderabad, India, under the supervision of Prof. Pradeepta K. Panda. Subsequently, he pursued postdoctoral research under the supervision of Prof. Pradeepta K. Panda at the same university, initially at the School of Chemistry and later at the Advanced Centre of Research in High Energy Materials. After that, he spent a year as a senior scientist in the pharmaceutical sector. He is currently a postdoctoral fellow working under Prof. Y. Inokuma at the Institute for Chemical Reaction Design and Discovery (ICReDD), Hokkaido University. His research focuses on the synthesis and application of contracted and unsymmetrical substituted porphyrionoids. Yasuhide Inokuma (right) is a Professor in the Faculty of Engineering at Hokkaido University and Principal Investigator at ICReDD. He received his PhD from Kyoto University in 2009 under the supervision of Prof. Atsuhiro Osuka. Subsequently, he worked with Prof. Makoto Fujita at the University of Tokyo as an assistant professor from 2009 and as a lecturer from 2014. In 2016, he moved to Hokkaido University, where he started his independent research and was promoted to full professor in 2023. His research interests are focused on the synthesis and applications of discrete polyketone-related molecules, including contracted porphyrinoids such as calix[3]pyrroles. |
1. Introduction
Intrigued by elaborate biomolecular systems such as photosynthesis1 and oxygen delivery,2 chemists have explored the chemistry of the porphyrins that play key roles. In recent decades, research on the synthesis of porphyrin-related compounds has been directed toward the synthesis of artificial chromophores essential to the development of organic functional materials, including photovoltaic materials,3–7 catalysts,8–10 and molecular machines,11 rather than the total synthesis of naturally occurring molecules such as vitamin B12.12 Although enzyme-based biosynthesis has always been used by bacteria, plants, and animals, the chemical synthesis of porphyrins originates in the discovery of an acid-catalyzed condensation between pyrrole and aldehyde by Rothemund in 1935.13 Canonical porphyrins are formed through the oxidation of porphyrinogen intermediates, which are produced through the cyclotetramerization of pyrrole monomers with four meso-sp3-carbon bridges. When macrocyclization occurs with fewer or more than four pyrrole units, contracted or expanded porphyrins are formed. The discovery of sapphyrin by Woodward14 has triggered the development of expanded porphyrins, which exhibit unique structural and optical properties derived from their enlarged inner cavity and π-conjugation.15–17 To date, giant macrocycles with more than 20 pyrrole units have also been developed using Rothemund-type condensation of oligopyrrole precursors.18,19 Despite the considerable development of expanded porphyrin chemistry over the last half century, research on contracted porphyrins is still in its infancy owing to the difficulty of synthesis.Macrocyclic compounds, including calixarenes,20 cyclodextrins,21 and cucurbiturils,22 as well as porphyrins, have optimal and marginal sizes that can be accessed through monomer oligomerization and subsequent macrocyclization. These sizes are typically determined by the balance between enthalpy and entropy factors. The formation of a macrocycle smaller than the optimal size is entropically favorable, although it often requires countering the enthalpy factors arising from steric repulsion and ring strain. Therefore, the synthesis of ever-smaller links in each macrocyclic family requires unconventional and well-designed methods.23–25
Under Rothemund- and modified Rothemund–Lindsey-type conditions, porphyrins are considered the optimal and marginal size of macrocycles composed of repeats of alternating pyrrole and methine carbon units owing to the absence of subporphyrin in the reaction mixture (Fig. 1). Subporphyrin is a contracted porphyrin composed of three pyrrole units bridged by three sp2-hybridized carbon atoms, also known as triphyrin(1.1.1). Until the first synthesis of tribenzosubporphine by Osuka and coworkers in 2006,26 it was unclear whether this type of macrocycle could be synthesized or even existed. This situation is in striking contrast to the fact that subphthalocyanines,27–29 contracted phthalocyanine analogues, have been extensively developed since their first discovery by Meller and Ossko in 1972.30 The emergence of the contracted analogues in the porphyrinoid family has, however, led to the development of synthetic methodologies and discovery of their properties.
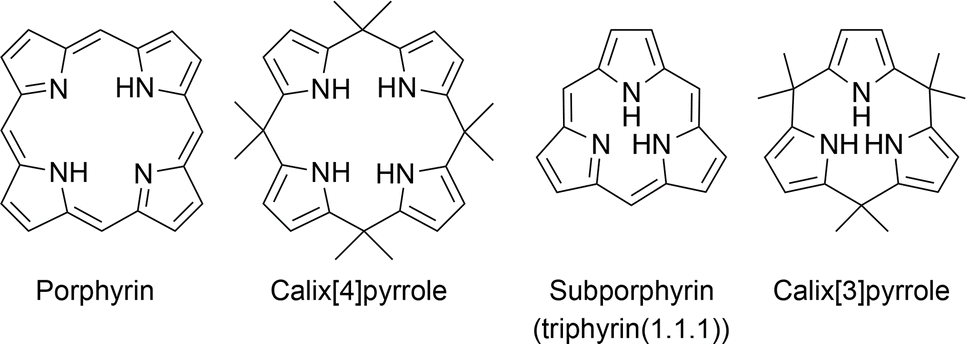 | ||
| Fig. 1 Canonical porphyrin and calix[4]pyrrole accessible by acid-catalyzed pyrrole monomer condensation and their contracted analogues, subporphyrin (also known as triphyrin(1.1.1)) and calix[3]pyrrole. | ||
Recent advances in the synthesis of boron-free subporphyrins31 and calix[3]pyrrole,32 a porphyrinogen-like analogue of subporphyrins, have expanded the potential of contracted porphyrins. Calix[4]pyrroles, tetrapyrrolic counterparts of calix[3]pyrrole, have demonstrated their distinctive supramolecular chemistry owing to the conformational diversity and molecular recognition abilities.33,34 These properties have led to the development of a number of applications, including ion sensors,35 selective ion bindings,36 exchanges,37 transporters38 and anti-cancer drugs.39 Since calix[4]pyrrole is the optimal and marginal size in acid-catalyzed condensation of pyrrole and acetone, similar to the porphyrin synthesis, calix[3]pyrrole has long been inaccessible by conventional methods. The synthetic breakthrough of calix[3]pyrrole has also triggered the discovery of its unique properties derived from the ring-contraction effects. This perspective introduces the recent progress in the synthesis of various ring-contracted porphyrinoids, such as triphyrin(n.1.1), core-modified subporphyrins, and calix[3]pyrrole analogues, according to the three typical classifications of methodology. Their structural, optical, and chemical properties are also discussed in terms of ring-contraction effects derived from the tripyrrolic ring systems. In the case of calix[3]pyrrole ring systems, their ring strain has recently been recognized as a key factor of unusual reactivity and synthetic feasibility. We also discuss our future perspective on the chemistry of contracted porphyrins and related macrocycles.
2. Synthetic approaches
Rothemund–Lindsey-type direct macrocyclization of pyrrole monomers has been unsuccessful in the synthesis of subporphyrins and calix[3]pyrroles. Nevertheless, these macrocycles and related compounds have so far been synthesized using three main approaches: (1) boron(III)-templated synthesis, (2) direct intramolecular macrocyclization enabled by core-modification or organometallic catalysis, and (3) ring tightening to complement strain energy with aromatic stabilization. This section summarizes the pros and cons of these approaches for the synthesis of contracted porphyrins.2.1 Boron-templated synthesis
Boron(III) atoms are suitable templates for the synthesis of subporphyrins and subphthalocyanines, and most of these macrocycles have been reported as boron(III) complexes. Given the absence of other template atoms in their reported synthesis, the atomic radius of boron is likely to fit well into the cavity of the tripyrrolic ring system compared with other metal ions such as Zn2+, Cu2+, and Ni2+, which are frequently used as templates for porphyrin and phthalocyanine synthesis.40–42 In the first synthesis of boron(III)-subphthalocyanine (1), Meller and Ossko used haloboranes, such as BCl3, both as a template and as an acid to activate phthalonitrile (Scheme 1).30 Although the synthetic approach appeared to be applicable to subporphyrins, they have remained undiscoverable for more than 30 years. A possible reason for the failures in the subporphyrin synthesis is that haloboranes, which are strong Lewis's acids, promote hydrolytic B–N(pyrrole) bond cleavage in the presence of water and accelerate the polymerization of pyrrole monomers.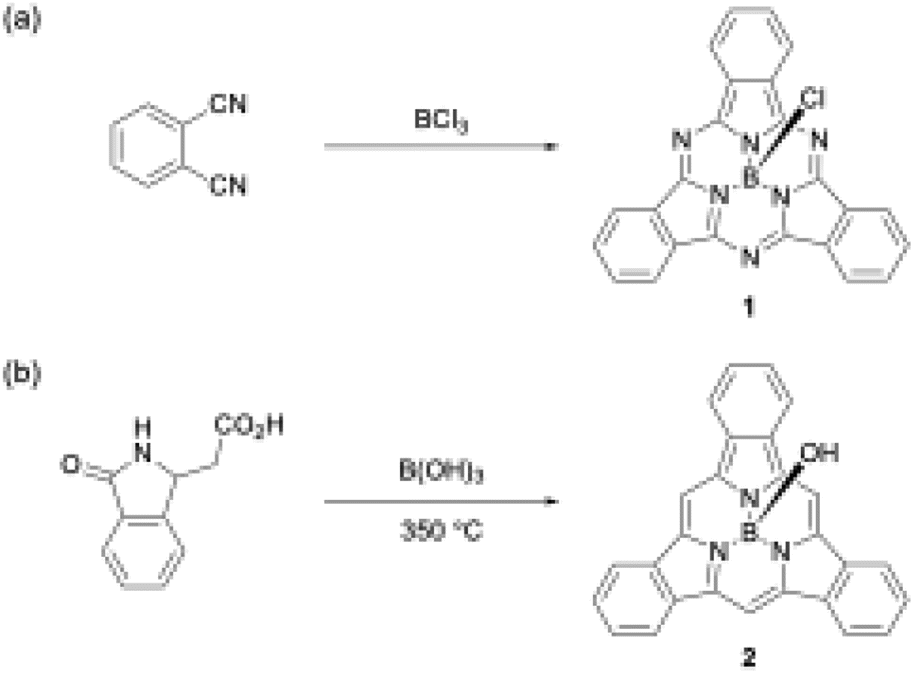 | ||
| Scheme 1 First boron(III)-templated syntheses of (a) subphthalocyanine 1 and (b) tribenzosubporphyne 2. | ||
The first example of a subporphyrin, boron(III)–tribenzosubporphine (2), was synthesised in 2006 by combining Gouterman's tetrabenzoporphyrin synthesis43 with a boron(III)-template. In this synthesis, boric acid was used as a template to promote the cyclotrimerization of an isoindole moiety generated from 2-(3-oxo-2,3-dihydro-1H-isoindol-1-yl)acetic acid at 350 °C (Scheme 1).26 The boron center in 2 is initially coordinated by a hydroxo ligand, which is easily converted into the less-polar methoxo group by dissolving 2 in methanol for separation by silica gel column chromatography. Owing to the ineffective template effect of boric acid, 2 was isolated in only 1.4% yield.
Shortly after the first report of 1, Kobayashi disclosed the synthesis of meso-aryl-substituted subporphyrins 4–6 using tri-N-pyrrolylborane (3)44 as a precursor (Scheme 2). Although the B–N bonds in precursor 3 readily dissociate to release pyrrole in the presence of a strong Brønsted acid, Kobayashi's group used propionic acid as a mild acid for condensation with aldehyde under Adler's conditions to obtain 4 in approximately 5% yields after tedious chromatographic separation.45
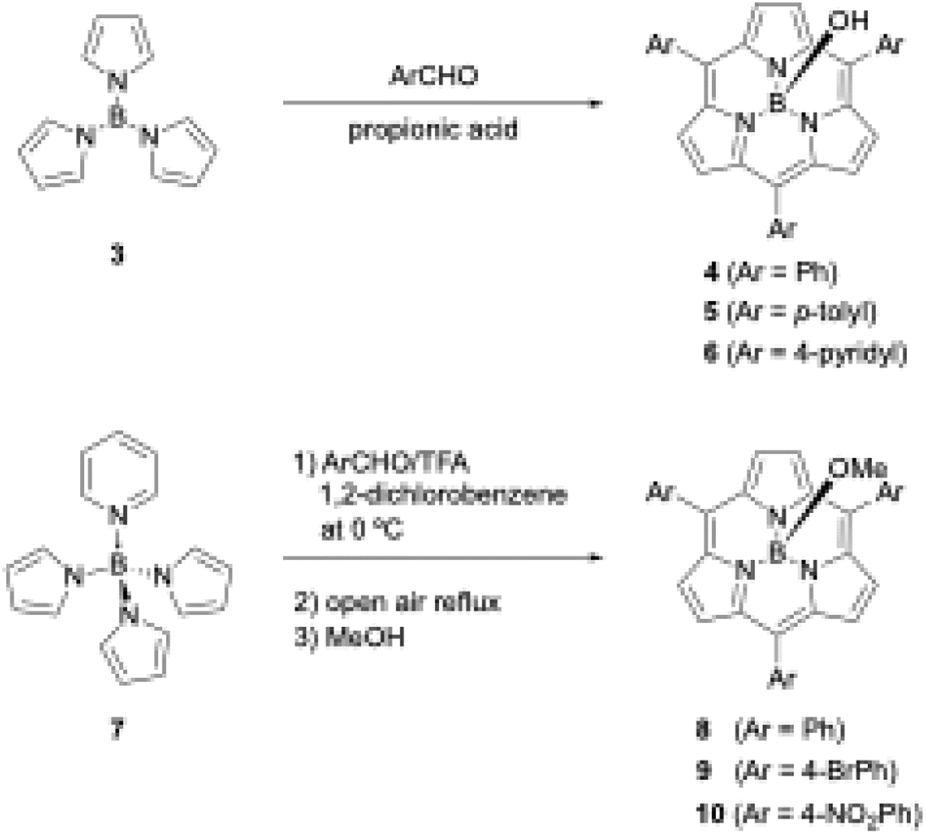 | ||
| Scheme 2 Synthesis of meso-aryl-substituted subporphyrins using tri-N-pyrrolylborane derivatives as precursors. | ||
When pyridine is coordinated to the boron center of 3, the resulting pyridine-tri-N-pyrrolylborane (7)46 can be used as a more soluble and slightly moisture- and acid-stable precursor for subporphyrin synthesis. Osuka and coworkers also successfully synthesized meso-aryl-substituted subporphyrins with various aryl groups, namely 8–10, in 2–6% yields using 7 and trifluoroacetic acid (TFA) as a catalyst (Scheme 2).47 Osuka's group isolated 8–10 in the B–OMe forms, which are quantitatively obtained from the B–OH forms upon heating in methanol and become less polar on silica gel. Aerobic oxidation in the synthesis of meso-aryl-subporphyrins often produces subchlorins, β,β-reduced subporphyrin analogues, as byproducts.48 Oxidation of subchlorins with MnO2 improves the isolate yield of 8 from 3.8% to 6.3%. Although tri-N-pyrrolylborane derivatives allow the introduction of various meso-aryl substituents to the boron(III)-subporphyrin skeletons, synthetic yields remain below 10%, and considerable amounts of tetraarylporphyrin byproducts are always generated as a result of B–N bond cleavage.
Boron(III)-templated synthesis is more versatile and efficient with tripyrrane precursors than with tri-N-pyrrolylboranes. Triethylamine–boron(III)-tripyrromethene 12,49 which is generated in situ from tripyrromethene 11 and BH3·NEt3, gives subporphyrin 8 in 18% yield upon treatment with benzoyl chloride (Scheme 3).50 Precursor 12 is also useful for the synthesis of A2B-type and meso-free subporphyrins 13 and 14, including subporphine 15.51Meso-free subporphyrins provide good scaffolds for the construction of subporphyrin oligomers via bromination with N-bromosuccinimide and subsequent cross-coupling reaction using organometallic reagents.
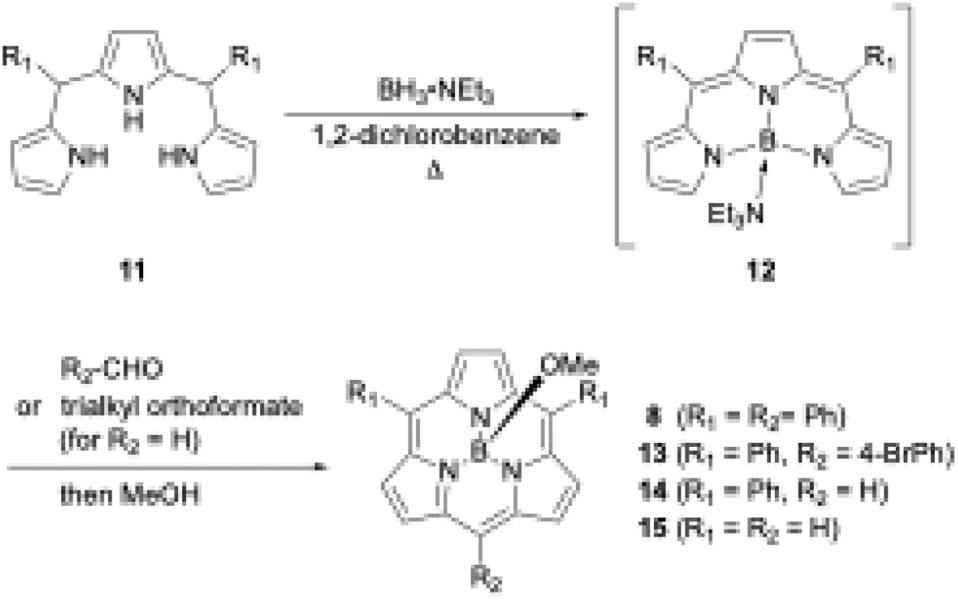 | ||
| Scheme 3 Synthesis of A2B-type subporphyrins including meso-free analogues. | ||
Boron(III)-templated subporphyrin synthesis using N-pyrrolylborane precursors still has an unsolved question. Given the well-known reaction mechanism of porphyrin synthesis,52 boron(III)-subporphyrinogen is considered the most likely intermediate of boron(III)-subporphyrins. However, no such intermediate has been observed during subporphyrin synthesis. Furthermore, the presence of intermediate 12 in the tripyrrane-based synthesis suggests the existence of an alternative reaction pathway leading to subporphyrins that does not involve subporphyrinogen-like intermediates. Understanding the detailed reaction mechanism of boron(III)-templated subporphyrin synthesis may hold the key to further improving the synthetic yields of subporphyrins.
Another unique boron(III)-templated subporphyrin synthesis is the metathesis-like ring splitting reaction of [32]heptaphyrin(1.1.1.1.1.1.1) Cu(II) complex 16 (Scheme 4a).53,54 Upon boron(III) complexation, the heptapyrrolic macrocycle splits into boron(III)-subporphyrin and copper(II)-porphyrin. Although the scope of meso-substituents is limited, meso-pentafluorophenyl- and trifluoromethyl-substituted subporphyrins 17 and 18 are obtained from corresponding heptaphyrins in 36% and 6% yields, respectively.
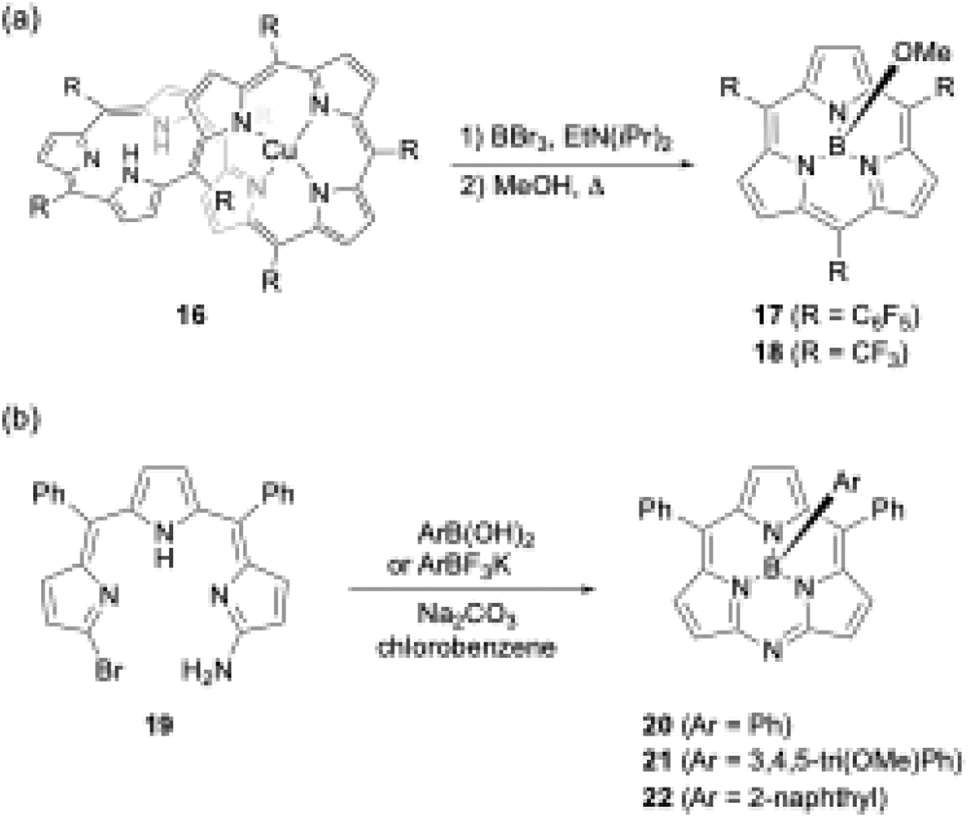 | ||
| Scheme 4 (a) Metathesis-like ring splitting of heptaphyrin 16, and (b) boron-templated synthesis of submonoazaporphyrins. | ||
Recently, a useful method for the high-yield synthesis of boron(III)-submonoazaporphyrins 20–22, a hybrid-type macrocycle of subporphyrin and subporphyrazine,55 was reported by Jiao and co-workers (Scheme 4b).56 Jiao's group used α-amino-α′-bromotripyrromethene precursor 19 for the intramolecular nucleophilic substitution induced by boron(III)-complexation. Because arylboronic acid derivatives can be used as a template, various axial ligands are directly introduced onto the boron in high yields (≥70%), enabling facile perpendicular functionalization of the macrocycles.
2.2 Direct macrocyclization
Organometallic reagents and/or core-modifications have recently enabled the direct macrocyclization of tripyrrolic ring systems without boron templates. Interestingly, incorporation of an additional carbon atom at the meso-position of the triphyrin(1.1.1) skeleton enables acid-catalyzed condensation, which leads directly to triphyrin(2.1.1) macrocycles. Direct macrocyclization of triphyrins(2.1.1) and meso-expanded calix[3]pyrroles also enables the synthesis of various heteroarene-embedded analogues.A recent striking finding in this research field is the Pd-catalyzed synthesis of subporphyrin free-bases by Osuka, Song, and coworkers.31 The group coupled α,α′-diborylated tripyrrane precursor 23 to 9,10-bis(dibromomethylene)-substituted anthracene 24 in the presence of Sphos Pd G2 catalyst. Although the isolated yields were low (6%, 4%, and 2% for 25, 26, and 27 respectively), subporphyrin free-bases 25–27 were fully characterized using NMR and X-ray crystallographic analyses. The 1H NMR spectrum of boron-free macrocycle 25 in CDCl3 showed an up-field shift of the inner NH protons at 3.99 ppm, indicating that its 14π-aromatic nature is comparable with that of boron(III)-coordinated analogues. However, in the crystal structure of 14, the tripyrrolic macrocycle adopts a non-planar, partial cone conformation (see Section 3.1) (Scheme 5).
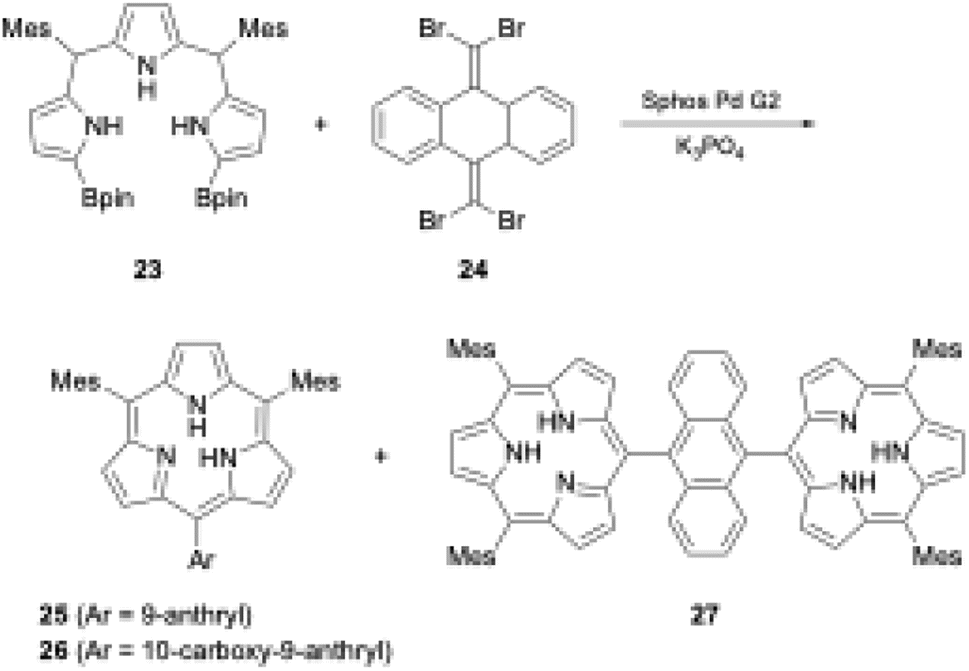 | ||
| Scheme 5 Synthesis of subporphyrin free-bases through Pd-catalyzed cross-coupling reaction. (Bpin: pinacolatoboryl; Mes: mesityl). | ||
Meso-expansion renders contracted porphyrin macrocycles more accessible through both acid-catalyzed condensation and organometallic means. Triphyrin(2.1.1) is a ring-contracted porphyrin bearing three pyrrole units and four meso-carbon atoms. The first examples of [14]triphyrins(2.1.1) 29–31 were serendipitously synthesized in approximately 35% yields by Yamada and coworkers through BF3·OEt2-catalyzed Rothemund-type condensation of bicyclo[2.2.2]octadiene-fused pyrrole 28 and arylaldehyde (Scheme 6).57 Following this first report, Yamada's group found that the amount of BF3·OEt2 is critical for the selectivity of triphyrin(2.1.1) over expected porphyrin.58 Although the detailed reaction mechanism is still unclear, the meso-vinylene bridge is formed by two formyl groups of the arylaldehyde. Bicyclo[2.2.2]octadiene-fused triphyrins(2.1.1) 29–31 were quantitatively converted into tribenzotriphyrins(2.1.1) 32–34 upon heating at 220 °C under reduced pressure, in a similar fashion as tetrabenzoporphyrin synthesis.59
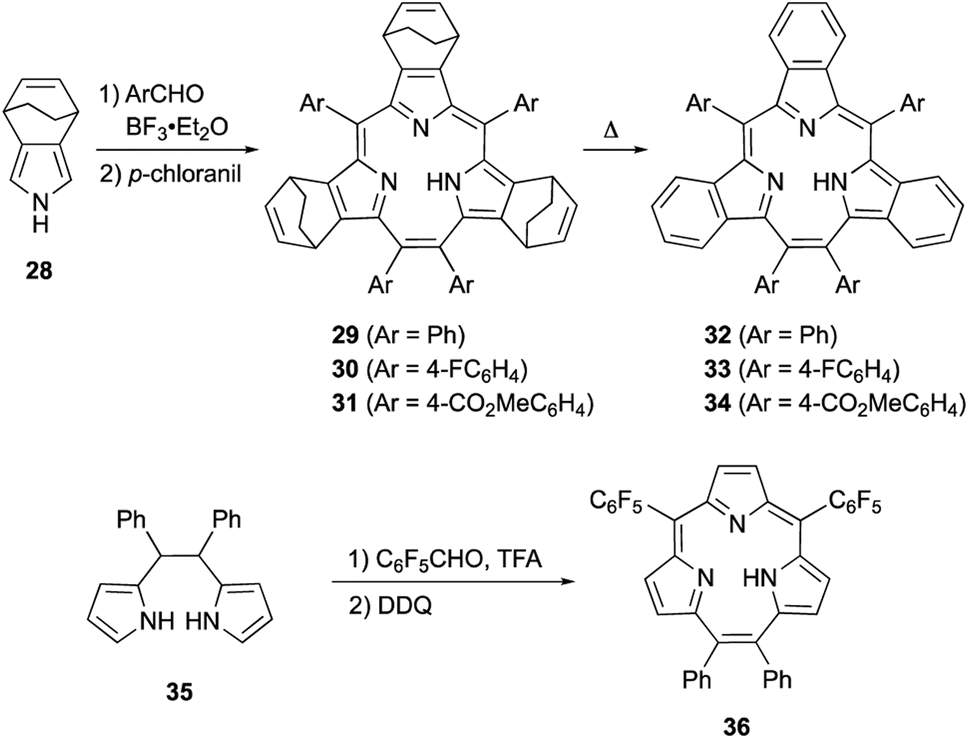 | ||
| Scheme 6 Acid-catalyzed direct macrocyclization of triphyrin(2.1.1) analogues. | ||
Rothemund-type protocols are also applicable to dipyrrolylethane precursor 35. Condensation of 35 with pentafluorobenzaldehyde in the presence of TFA followed by DDQ oxidation furnished triphyrin 36 in 5% yield.60 Without the addition of a pyrrole monomer to the reaction, the third pyrrole unit is likely generated through an acid-catalyzed scrambling process.61
Triphyrins(2.1.1) can also be synthesized through the McMurry coupling reaction. Intramolecular cyclization of α,α′-diformyltripyrrane precursor 37 using TiCl4, Zn, and CuCl and subsequent oxidation of the resulting macrocycle gave triphyrin(2.1.1) 38 in 16% yield (Scheme 7).62 Unlike acid-catalyzed synthesis, the McMurry coupling reaction enables the synthesis of meso-free triphyrins(2.1.1) in moderate yields.62 When the McMurry coupling strategy is applied to dimethylmethylene-linked tripyrrane 39, a meso-expanded calix[3]pyrrole analogue 40 is obtained in 33% yield through Pd/C catalyzed hydrogenation of the vinylene bridge.63
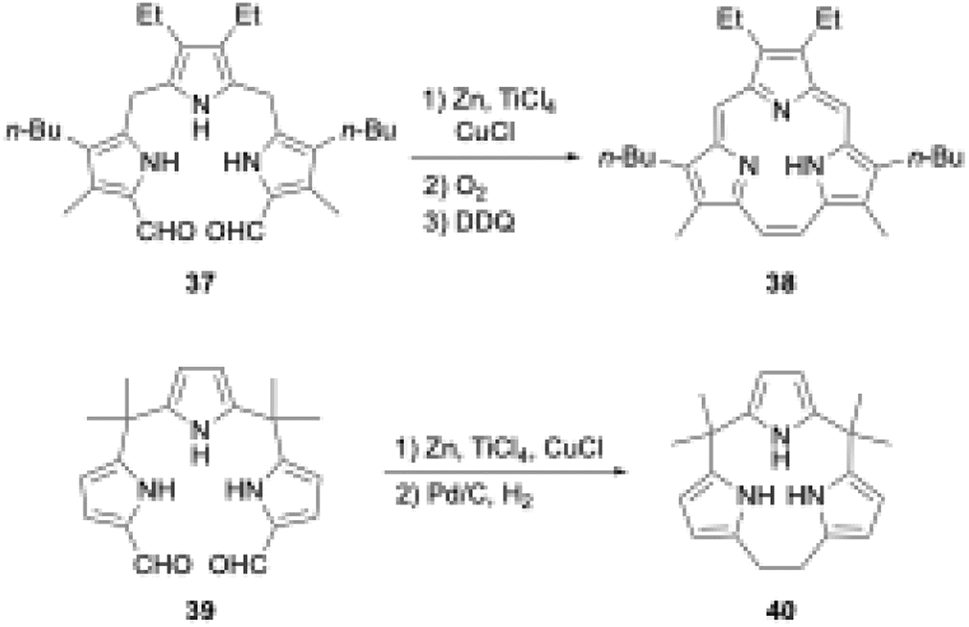 | ||
| Scheme 7 McMurry coupling-based synthesis of triphyrin(2.1.1) and meso-expanded calix[3]pyrrole. | ||
Core-modification enables the direct macrocyclization of triphyrin and calix[3]pyrrole analogs containing various heteroarenes, such as furan and pyridine, instead of pyrrole units. Pyridine-embedded tripyrrane analogue 41 undergoes acid-catalyzed intramolecular cyclization in the carbinol form, which is generated in situ by reduction with NaBH4. Following oxidation with DDQ, subpyriporphyrin 42 is obtained in 6% yield (Scheme 8).64 Unlike subporphyrin free-bases, 42 exhibits non-aromatic characteristics, as indicated by its 1H NMR spectrum, although 42 adopts a slightly ruffled yet planar conformation in the crystalline state. The key to this direct macrocyclization is the reduced number of NH protons and wide connection angles of the pyridine moiety.
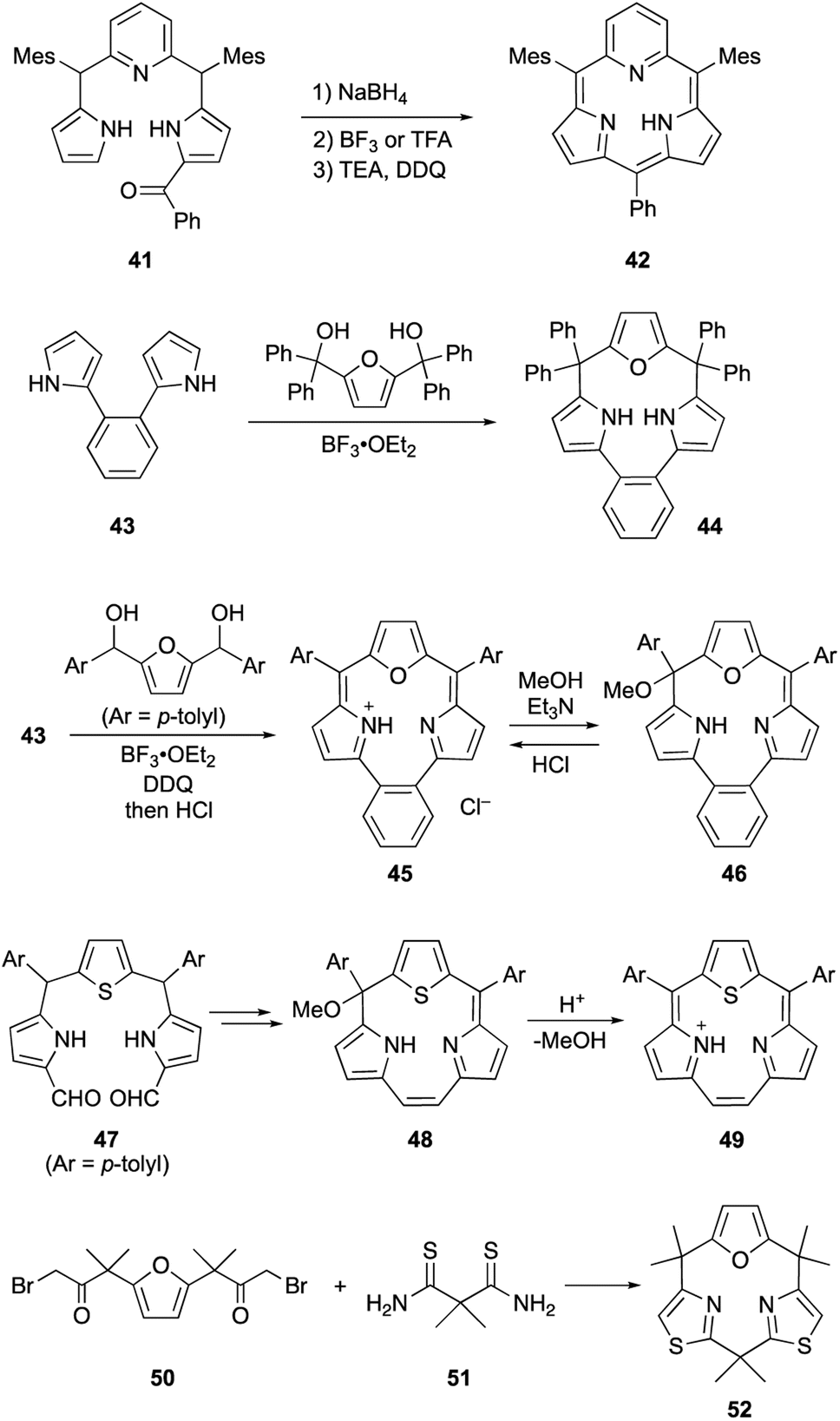 | ||
| Scheme 8 Synthesis of various heteroarene-embedded triphyrin and calix[3]pyrrole analogues. | ||
Latos-Grażyński's group used 1,2-dipyrrolylbenzene 43 to synthesize furan-embedded calix-triphyrin 44 and oxatriphyrin 45.65 BF3·OEt2-catalyzed condensation of 43 with bis(hydroxydiphenylmethyl)furan affords 44 in 65% yield, while that with 2,5-bis(hydroxymethyltolyl)furan followed by DDQ oxidation and counterion exchange using HCl gives protonated 45 in 20% yield. Upon treatment with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Et3N/MeOH, oxatriphyrin 45 was quantitatively converts into phlorin-type macrocycle 46. The transformation between 45 and 46 is reversible because acidic removal of the methoxy group of 46 recovers 45. A similar conversion also occurs with thiophene-embedded triphyrin(2.1.1) analogue 48, which is obtained from tripyrrane 47 using the McMurry coupling reaction.66 Stepwise addition of TFA to a dichloromethane solution of 48 increases the intensity of the Soret-like band at 414 nm in the UV-vis absorption spectrum of 49, indicating the 14π-aromatic nature of 49.
:1 mixture of Et3N/MeOH, oxatriphyrin 45 was quantitatively converts into phlorin-type macrocycle 46. The transformation between 45 and 46 is reversible because acidic removal of the methoxy group of 46 recovers 45. A similar conversion also occurs with thiophene-embedded triphyrin(2.1.1) analogue 48, which is obtained from tripyrrane 47 using the McMurry coupling reaction.66 Stepwise addition of TFA to a dichloromethane solution of 48 increases the intensity of the Soret-like band at 414 nm in the UV-vis absorption spectrum of 49, indicating the 14π-aromatic nature of 49.
Recently, our group achieved direct macrocyclization of a calix[3]pyrrole analogue through strain-based molecular design for the feasible synthesis.67 Although macrocyclic ring strain is considered as a key factor for the macrocyclization of contracted porphyrinoids, it is invisible and often unpredictable in complex ring systems. Our group applied the lowest energy conformation search using the artificial force induced reaction (AFIR)68 to various heteroarene-replaced analogues of calix[3]pyrrole and, furthermore, visualized/evaluated their ring strain using the StrainViz69 analysis developed by Jasti and coworkers. As a result, we found that calix[1]furan[2]thiazole (52) is less strained than the parent calix[3]pyrrole. In fact, a Hantzsch-type thiazole formation reaction between 50 and 51 proceeds under neutral conditions and desired 52 is obtained in 60% yield. This demonstrates the importance of pre-synthetic strain evaluation using theoretical calculations for feasible macrocyclization of contracted porphyrinoids.
2.3 Ring tightening approach
Aromatic rings are typically planar, and thus cyclooligomerization of aromatic subunits tends to produce linear oligomers and less-strained (larger) macrocycles rather than strained macrocycles. However, once precursor macrocycles are preorganized using conformationally flexible, non-aromatic synthons for aromatic subunits, the strain energy of the target macrocycles can be compensated for by the aromatic stabilization energy. Organic chemists have developed useful synthons to obtain elusive macrocycles with this ring tightening approach. As a representative example, [n]cycloparaphenylenes have been synthesized from precursor macrocycles composed of 1,4-diaryl-1,4-dialkoxycyclohexane or its cyclohexadiene analogues as para-phenylene synthons.70–72In porphyrin-related chemistry, Kohnke and coworkers introduced a non-conventional method to synthesize calix[n]pyrroles and hybrid-type calix[n]furan[m]pyrroles using polyketone macrocycles.73 Because 1,4-diketone unit can generate a pyrrole ring via the Paal–Knorr reaction, calix[N]pyrroles are successfully synthesized from cyclic N-mers of 3,3-dimethylpentane-2,4-dione (53).74,75 In this calixpyrrole synthesis, the 5N-membered polyketone rings are contracted to 4N-membered rings. Although this approach was considered applicable to the synthesis of calix[3]pyrroles, promising hexaketone precursors, such as 59 in Scheme 9, were unavailable, because these polyketone precursors have been derived from calix[n]furans (n ≥ 4), which are obtained through acid-catalyzed cyclooligomerization of furans.
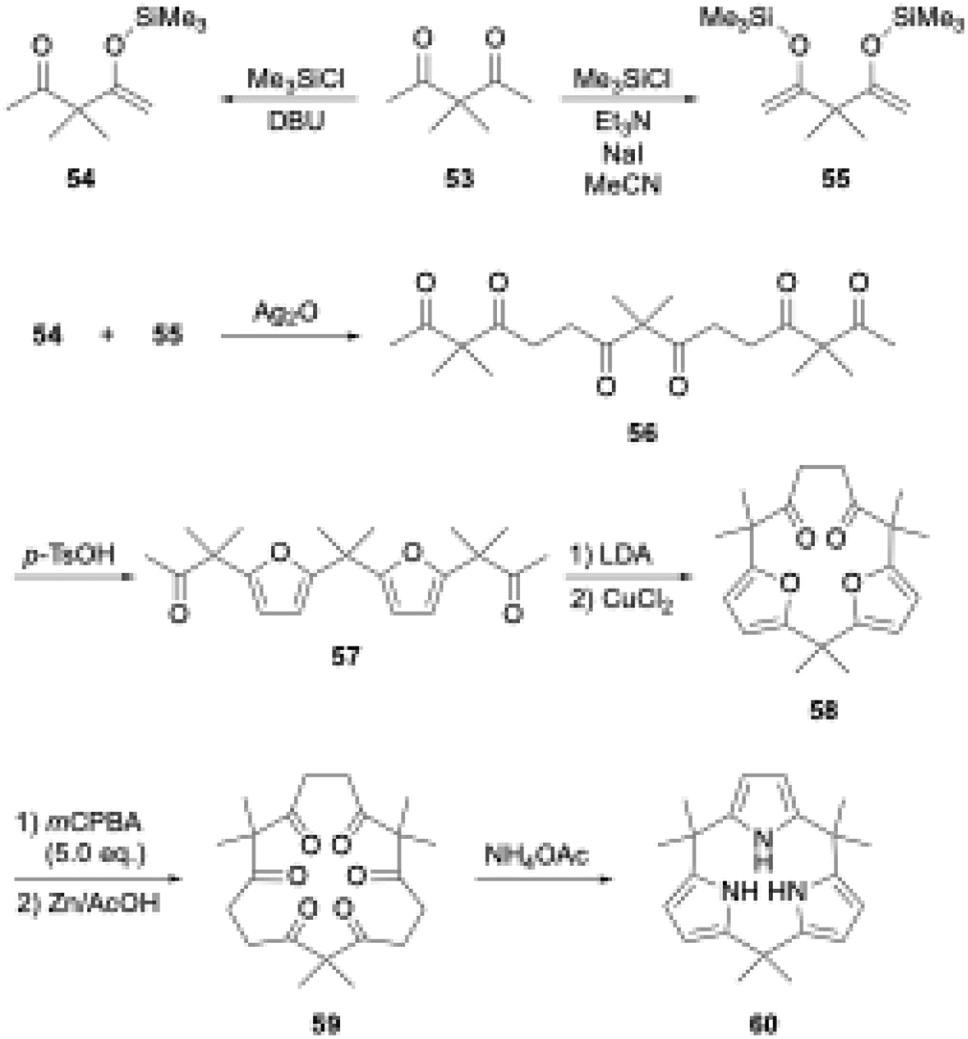 | ||
| Scheme 9 First synthesis of calix[3]pyrrole. | ||
To realize the ring tightening synthesis of calix[3]pyrrole, our group has developed stepwise oligomerization76,77 and macrocyclization78 of acetylacetone derivative 53. Compound 53 undergoes mono- and bis-selective silylation in the presence of 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) and triethylamine to give 54 and 55,79 respectively. Linear trimer 56 is obtained in 27% yield by Ag2O-mediated oxidative coupling between 54 and 55. Although intramolecular cyclization of the terminally bis-silylated analogue of 56 with Ag2O is unsuccessful, protection of the internal ketones by furan formation allows the use of lithium diisopropylamide (LDA)/CuCl2. Intramolecular cyclization of 57 indeed proceeds to give difuran macrocycle 58 in 42% yield. The desired hexaketone precursor 59 is obtained in 28% yield through oxidative furan ring opening reaction of 58 using excess m-chloroperbenzoic acid (mCPBA) followed by Zn reduction.
Calix[3]pyrrole 60 was first synthesized in 41% yield through a triple Paal–Knorr reaction using cyclic hexaketone 59 and ammonium acetate (Scheme 9). An important factor in the successful synthesis of 60 is the use of non-acidic reaction conditions because acids commonly used under Rothemund–Lindsey conditions, such as TFA, trigger rapid, quantitative strain-induced ring expansion of 60, which will be discussed in Section 3.
The ring tightening approach using cyclic oligoketone precursors also enables the synthesis of furan- and thiophene-embedded calix[3]pyrrole analogues (Scheme 10). Dehydrative furan ring formation reaction of 58 using chlorotrimethylsilane and P2O5 in methanol furnishes calix[3]furan (61) in 53% yield,80 which is inaccessible via acid-catalyzed condensation. Pyrrole ring formation reaction of 58 gives calix[2]furan[1]pyrrole (62) in 79% yield. The addition of 3 equivalents of mCPBA to 58 affords monofuran macrocycle 63 in 71% yield after reductive workup using Zn. Macrocycle 63 is further converted into calix[1]furan[2]pyrrole (64) and calix[1]furan[1]pyrrole[1]thiophene (65) using ammonium acetate and Lawesson's reagent.81 The synthesis of calix[3]pyrrole analogues based on cyclic polyketones has provided insight into their ring strain-induced chemical properties.
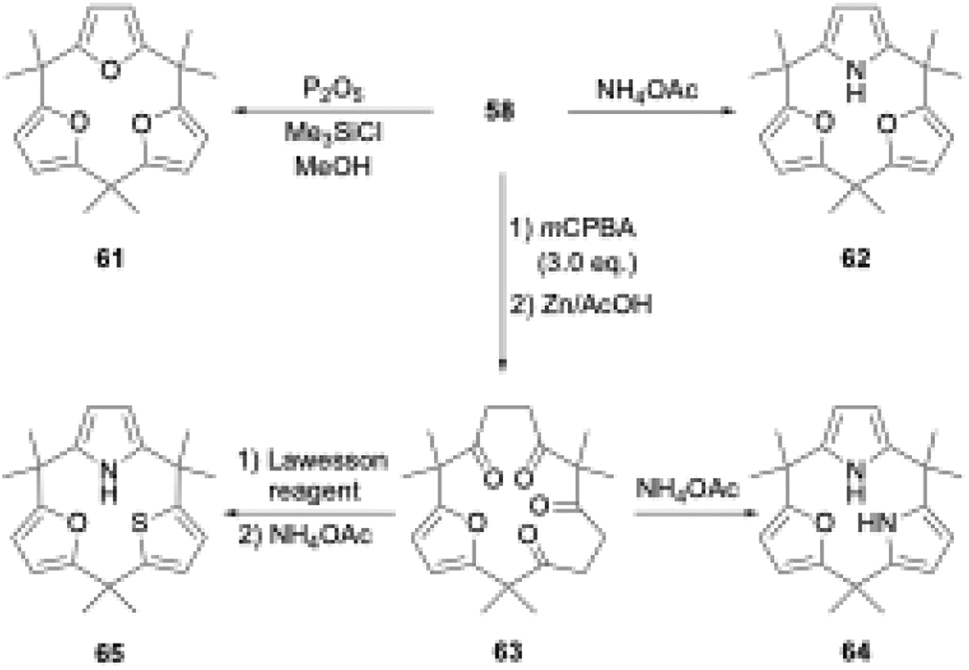 | ||
| Scheme 10 Ring tightening synthesis of calix[3]pyrrole analogues. | ||
3. Ring-contraction effects
Until ring-contracted porphyrins and calixpyrroles were first synthesized, they were mainly recognized as synthetic targets rather than functional molecules. Recent studies on their structural, optical, chemical, and supramolecular properties, however, have gradually revealed the effects of ring-contraction on their porphyrin-like yet tripyrrole-unique macrocycles. This section summarizes the unique properties of contracted porphyrinoids, which are not commonly observed in other porphyrins or expanded porphyrins.3.1 Macrocyclic structures
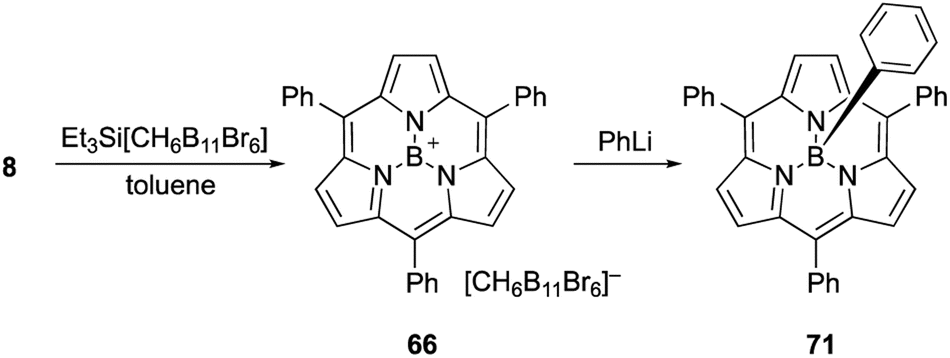
Boron(III)-subporphyrins typically have a bowl-shaped structure owing to the tetrahedral coordination geometry of the boron center. The subporphyrin macrocycle behaves as a dianionic tridentate ligand, and the boron center has an anionic axial ligand that is coordinated perpendicular to the macrocycle, forming a neutral complex. Boron(III)-subporphyrins can adopt a planar conformation with a tricoordinated boron center upon the generation of borenium cation species.82 Borenium cation with an inert carborane counterion has been crystallographically analyzed by Reed's group. Although a similar subphthalocyanine borenium cation83 adopts a bowl-shaped conformation (bowl-depth of 1.073 Å), subporphyrin 66 is almost planar with a bowl-depth of 0.114 Å (Fig. 2, see also Scheme 11 in Section 3.3). NMR and X-ray crystallographic analyses indicated that boron(III)-subporphyrins interconvert between planar borenium and bowl-shaped neutral forms in acidic solutions.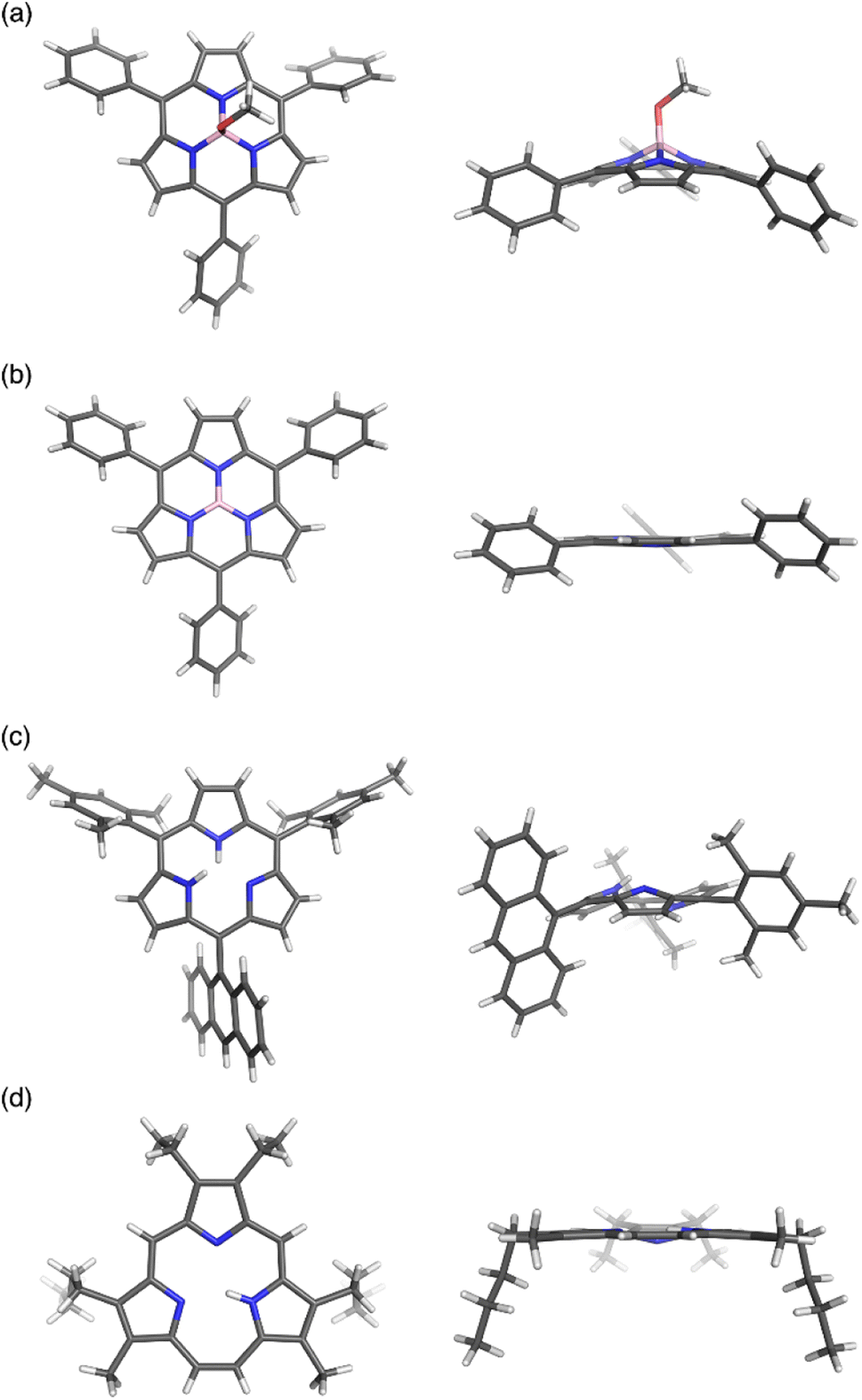 | ||
| Fig. 2 X-ray crystal structures of (a) meso-phenyl subporphyrin 8, (b) subporphyrin borenium cation 66, (c) subporphyrin free-base 25, and (d) triphyrin(2.1.1) 38. Left: top view, right: side view. | ||
 | ||
| Scheme 11 Formation of subporphyrin–borenium cation species for subsequent introduction of axial phenyl ligand. | ||
The remarkable ring-contraction effects also occur in meso-aryl-substituted boron(III)-subporphyrins. Although meso-tetraphenylporphyrin has four phenyl groups arranged almost perpendicular to the porphyrin ring, subporphyrin 8 has phenyl groups tilted at 38 to 48° in the crystalline state.47 This suggests that the meso-aryl groups of subporphyrins experience a decreased rotational barrier and thus increased substituent effects through π-conjugation (discussed in the Section 3.2).
Boron-free subporphyrin 25 adopts a non-planar, partial cone-like conformation, despite its fully conjugated 14π-aromatic nature.31 This can be attributed to the repulsive interactions between the two NH groups inside the narrow cavity. In contrast, planar conformations are adopted by triphyin(2.1.1) free-bases such as 38. Triphyrins(2.1.1) have an NH and two iminic nitrogen atoms in cavities that are slightly larger than those of subporphyrins. Therefore, the NH protons preferentially form hydrogen bonds with iminic nitrogen atoms on the same plane.
Calix[3]pyrrole 60 exhibits an unusually strained macrocyclic structure in the crystalline state. In its monohydrated form, 60 adopts a partial cone conformation, with one pyrrole unit oriented toward the opposite side of the other units (Fig. 3). Notably, the meso-carbon atoms are out of the mean plane of the neighboring pyrrole rings, suggesting a highly strained structure of 60. Their deformation angles α and β,84 which are used to evaluate the displacement around the aromatic rings embedded in a macrocycle, are 2.52° and 9.20°, respectively.32 These values are remarkably larger than those of calix[4]pyrrole (α = 0.38°, β = 1.60°). Theoretical calculations revealed that calix[3]pyrrole 60 is 22.1 kJ mol−1 more unstable per repeat unit than calix[4]pyrrole 67.
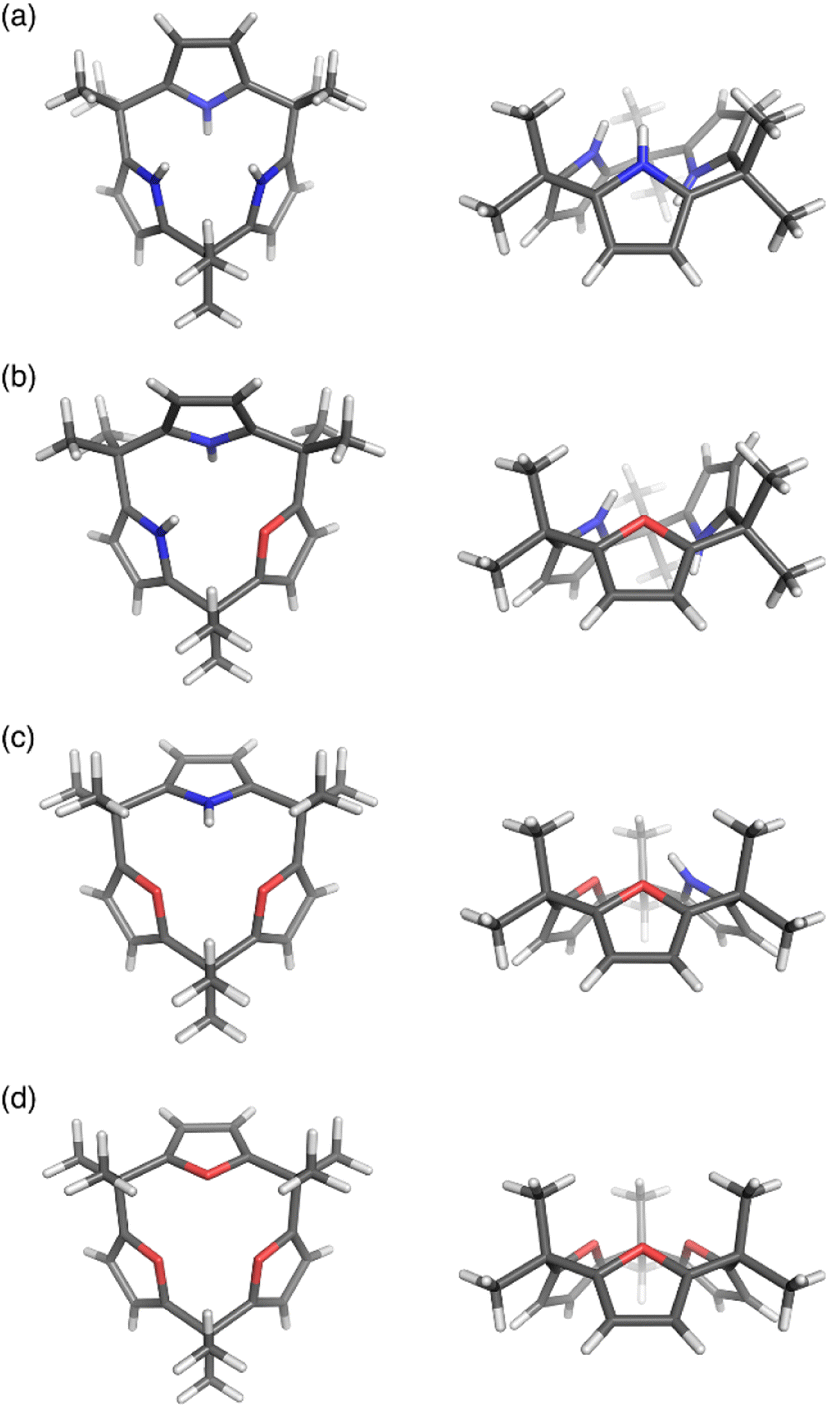 | ||
| Fig. 3 X-ray crystal structures of (a) calix[3]pyrrole 60, (b) calix[1]furan[2]pyrrole 64, (c) calix[2]furan[1]pyrrole 62, and (d) calix[3]furan 61. Left: top view, right: side view. | ||
Pyrrole/furan hybrid type calix[3]pyrrole analogues, calix[2]furan[1]pyrrole 62 and calix[1]furan[2]pyrrole 64, adopt cone and partial cone conformations, respectively. The deformation angles of the pyrrole units, α and β, are respectively 2.48° and 6.17° in 62 and 2.49° and 8.52° in 64. These observations indicate that the displacement of the pyrrole units diminishes as the number of pyrrole units decreases. Calix[3]furan 61 adopts a cone conformation in the crystalline state, and its deformation angles around the furan rings are smaller than those in calix[2]furan[1]pyrrole 62.80
3.2 Optical and electrochemical properties
Subporphyrins are characterized by 14π-conjugated aromatic macrocycles that exhibit larger HOMO–LUMO gaps and blue-shifted absorption/emission bands compared with the corresponding porphyrins comprising 18π-aromatic macrocycles. Meso-triphenylsubporphyrin 8 displays a Soret-like band at 373 nm and Q-like bands at 450–500 nm in CH2Cl2 (Fig. 4). In contrast to the Zn(II) complex of meso-tetraphenylporphyrin (Zn-TPP),85 which is red in solution, 8 is yellowish orange and emits green fluorescence with a fluorescence quantum yield of 13%. The first oxidation and reduction potentials of 8 are observed at 0.78 and −1.82 eV versus the ferrocene/ferrocenium (Fc/Fc+) couple,47 while those of Zn-TPP lie at 0.76 and −1.41 eV, respectively.86 The larger electrochemical HOMO–LUMO gap of 8 (2.60 eV) in comparison to Zn-TPP (2.17 eV) is consistent with the bule-shifted Q-like bands observed in the absorption spectrum of 8.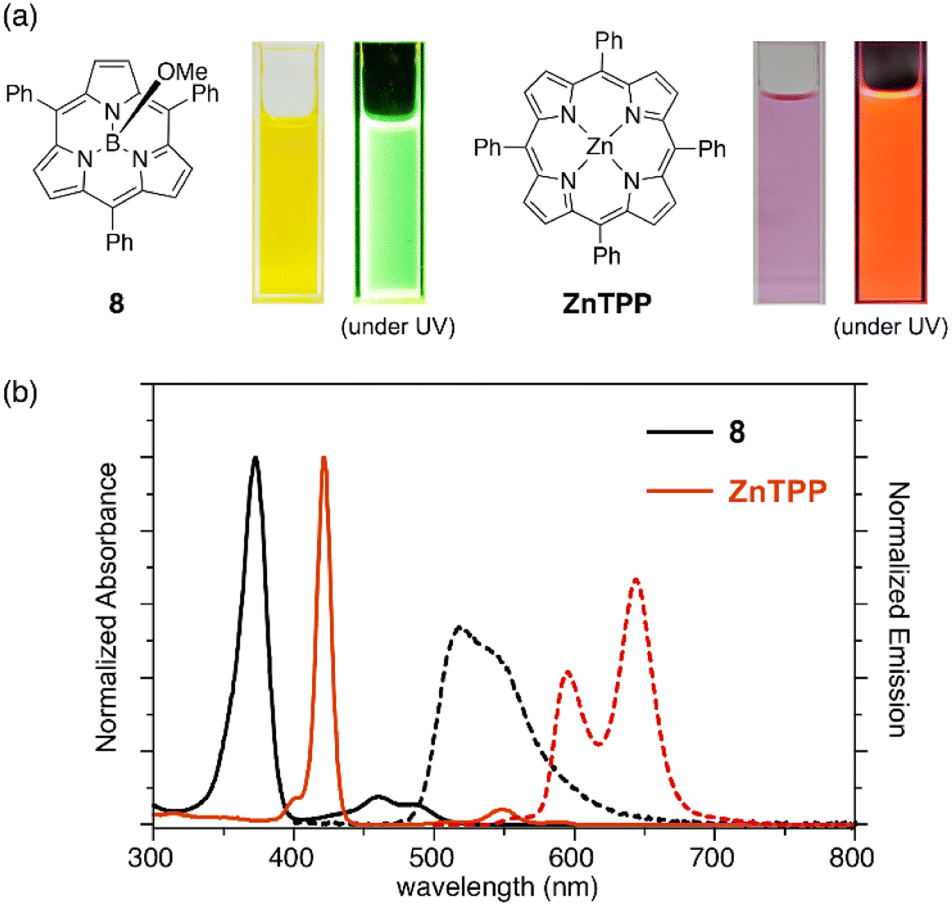 | ||
| Fig. 4 (a) Photographs of CH2Cl2 solutions of 8 and ZnTPP under room light and UV light. (b) UV-vis absorption (solid line) and emission (dashed line) spectra of subporphyrin 8 and tetraphenylporphyrin (ZnTPP) in CH2Cl2. | ||
The optical properties of subporphyrins can be widely modulated by the substituent effects of meso-aryl groups. Substitution of the N,N-dialkylamino group at the p-position of the meso-phenyl group leads to a remarkable red-shift of the absorption and emission bands of 68 with an increased fluorescence quantum yields up to 60%.87 This is attributed to intramolecular charge transfer interactions between the electron-donating amino group and the low-lying LUMO of the subporphyrin core. The substituent effects at the p-position of the meso-phenyl group are typically trivial in tetraphenylporphyrins because of the high rotation barrier and almost perpendicular orientation of the meso-phenyl groups.88 In contrast, the optical properties of meso-aryl subporphyrins are significantly perturbed by the substituents. For example, 4-nitrophenyl-substituted subporphyrin 10 (ref. 47) shows solvent-dependent absorption and fluorescence. Moreover, π-conjugation of the meso-substituents on oligo(1,4-phenyleneethynylene)89 substituted analogue 69 significantly increases the absorption coefficient of the Soret-like band (Fig. 5).
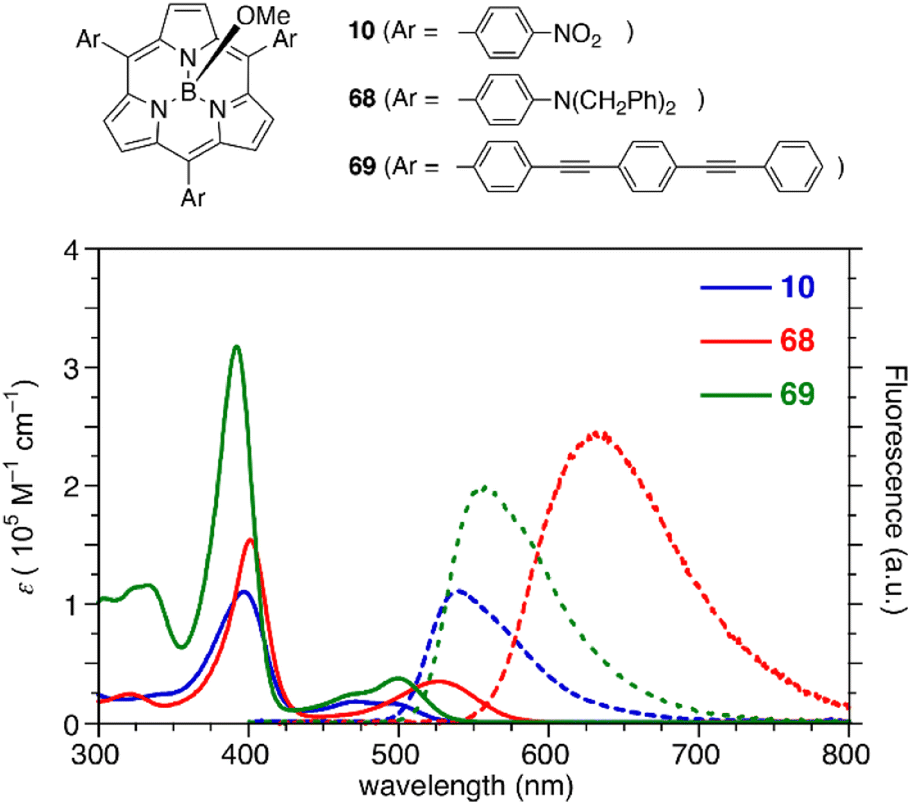 | ||
| Fig. 5 UV-vis absorption (solid line) and fluorescence (dashed line) spectra of subporphyrins 10, 68, and 69 bearing 4-substituted phenyl groups at the meso-positions in CH2Cl2. | ||
Ring contraction also affects the optical properties of calix[3]pyrrole analogues whose π-conjugation is interrupted at the meso-sp3-carbon atoms.90 Although calix[4]pyrrole (67) and calix[6]pyrrole (70) give rise to nearly identical absorption spectra derived from the π–π* transition of the pyrrole units, calix[3]pyrrole 60 exhibits a broad red-shifted band at 265 nm. Non-covalent interaction (NCI) plot analysis indicates that the contracted ring system enhances through-space interactions between neighboring pyrrole rings. Similar intramolecular interactions between the furan and pyrrole units in furan/pyrrole hybrid macrocycles 62 and 64 result in solvent-dependent Stokes shifts, indicating polar excited states. Such optical properties can be used as a probe to distinguish calix[3]-type macrocycles from other larger analogues (Fig. 6).
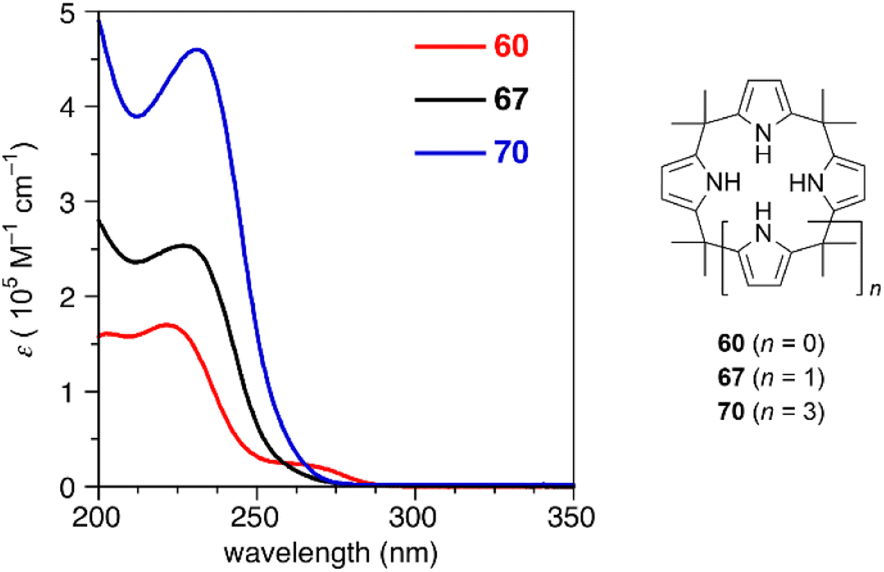 | ||
| Fig. 6 UV-vis absorption spectra of calix[n]pyrroles (n = 3, 4, 6) in acetonitrile. | ||
3.3 Strong boron chelation
Most subporphyrin and triphyrin analogues stably chelate a boron(III) atom in their cavity. Although various harsh conditions, such as heating with strong Lewis/Brønsted acids or treatment with organolithium reagents, have been applied to boron(III)-subporphyrins, removal of the boron atom to give free-base macrocycles has not yet been achieved. However, axial ligands on the boron center readily exchange with hydrogen, oxygen, carbon, and halogen-based monoanionic ligands. Therefore, the boron center in subporphyrins can serve as a rigid scaffold for the orthogonal functionalization of macrocycles.The reaction of boron(III)-subporphyrin 8 with Et3Si[CH6B11Br6], a silylium reagent, generates tri-coordinated borenium cation 66 without deboronation of the subporphyrin ligand (Scheme 11).82 Treatment of 66 with phenyl lithium results in the formation of axially phenyl-substituted 71.
The B–OH and B–OMe forms undergo various axial ligand exchange reactions (Scheme 12). Fluoro- and trifluoroacetoxo-substituted forms 72 and 73 are obtained through the reactions of 4 with BF3·OEt2 and TFA, respectively. These analogues can also be converted back into B–OH form 4 through hydrolysis. Upon heating in the presence of excess alcohol, the hydroxo group in 4 can also be replaced with an alkoxo ligand.91 Methoxo form 8 can react with Grignard reagents, including aryl, alkynyl, and alkyl magnesium bromides, to form an axial B–C bond. For example, treatment of 8 with 20 equivalents of 1-naphthyl magnesium bromide results in the formation of 75 in 91% yield.92 Upon treatment with diisobutylaluminium hydride (DIBAL-H), compound 8 affords subporphyrinato boron(III) hydride 74, which is capable of hydroborating aldehydes and ketimines.93 Borohydride 74 releases the hydride ion upon the addition of a tritylium cation, Ph3C[B(C6F5)4], to give borenium cation species. Air oxidation of 74 slowly proceeds even in the solid state to give a boron peroxide 76, which is also prepared from 8 by treatment with hydrogen peroxide. Interestingly, thermolysis of peroxide 76 in 1,2-dichlorobenzene results in the formation of peroxo-bridged dimer 77. Analogous μ-oxo dimer 78 can be prepared from B–OH form 4 through the reaction with Et3N.94 Reversible and almost quantitative axial ligand exchange reactions have also been used to form molecular assemblies of subporphyrins (see Section 3.6).
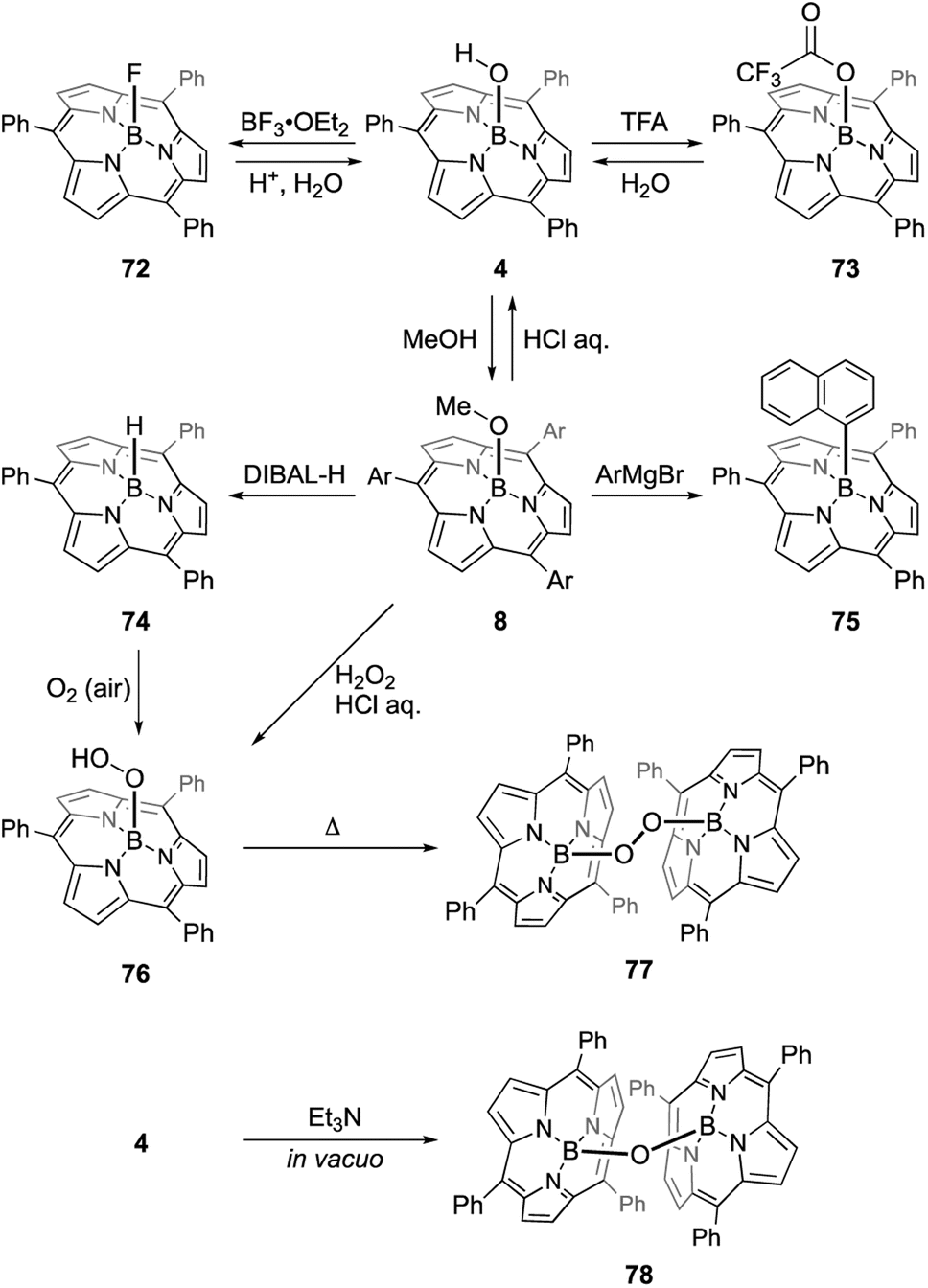 | ||
| Scheme 12 Axial ligand exchange reactions of boron(III)-subporphyrins. | ||
Triphyrin(2.1.1) and calix[3]pyrrole can also form acid-stable boron(III) complexes. Treatment of triphyrin(2.1.1) 38 with BPhCl2 leads to the formation of tetracoordinated boronium cation complex 79 (Scheme 13). Complexation with a boron(III) atom changes the conformation of the macrocycle from planar to bowl-shaped. Boron complexes of triphyrin(2.1.1) are stable to strong acids including concentrated HCl, HPF6, and HBF4 without deboronation.95 Calix[3]pyrrole 60 also forms stable boron(III) complex 80 with a tricoordinated planar boron center. Although tri-N-pyrrolylborane 3 is susceptible to hydrolysis and acidolysis, resulting in the release of pyrrole monomers, boron complex 80 resists deboronation even after treatment with TFA.32
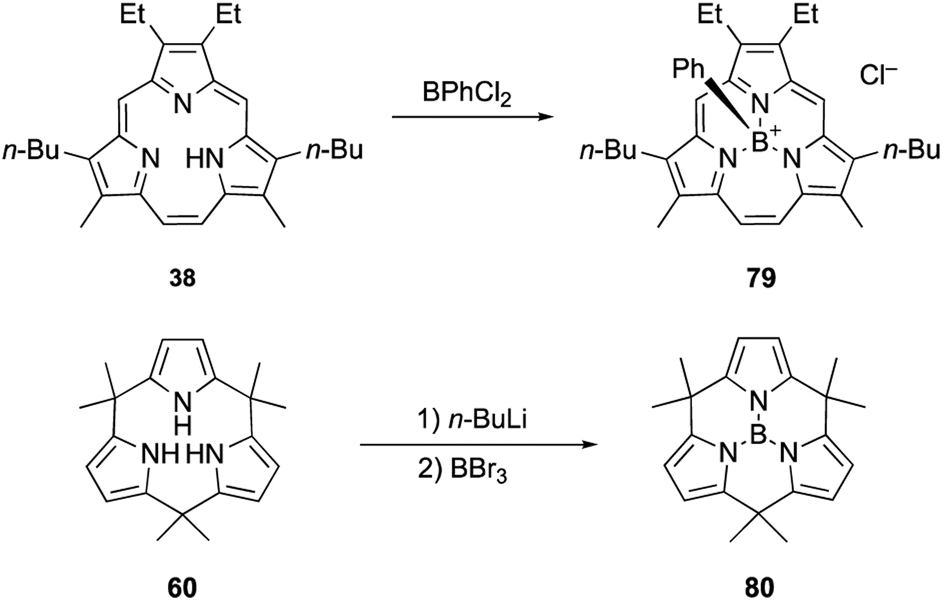 | ||
| Scheme 13 Formation of acid-stable boron(III) complexes. | ||
3.4 Metal complexation
Although the metal coordination of subporphyrin free-bases with boron(III) only has been reported so far, metal complexes of triphyrins(2.1.1) have been extensively studied. Because triphyrins(2.1.1) act as monoanionic tridentate ligands, they can form complexes with low-valent metal ions.Complexation of meso-free triphyrin(2.1.1) 38 with Mn(CO)5Br and Re(CO)5Cl salts, furnish isostructural Mn(I) and Re(I) tricarbonyl complexes 81 and 82, respectively.62 The Mn(I) ion in 81 is coordinated by the three CO ligands and three nitrogen atoms of the triphyrin, resulting in an octahedral coordination geometry (Fig. 7). Triphyrin macrocycles 81 and 82 no longer maintained a planar conformation but adopt a slightly bowl-shaped structure, and the metal ion is not fully incorporated into the cavity. The first oxidation potentials of 81 and 82 are observed at 0.25 and 0.48 eV, respectively, versus Fc/Fc+ couple. The shifted oxidation potentials of these complexes in comparison to that of the free-base analogue 38 (0.63 eV) indicate that the initial oxidation events occur at the metal centers.62 Complexation of tribenzotriphyrin 34 with [{Fe(CO)2(Cp)}2] (Cp: cyclopentadienyl) leads to the formation of sandwich complex 83 bearing a CpFe(II) unit and a bowl-shaped triphyrin ligand. The oxidation potential of the iron complex 83 is lower than that of ferrocene.96
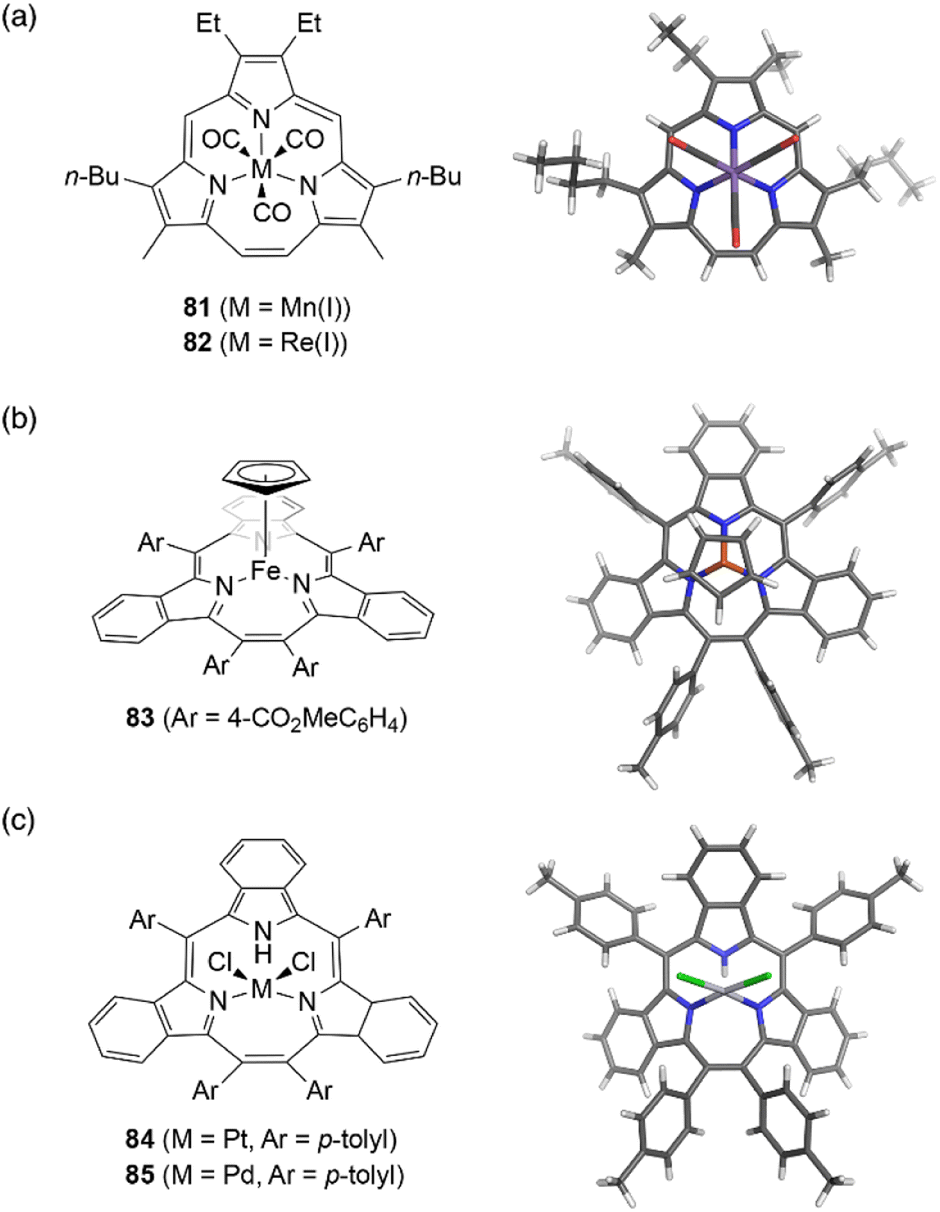 | ||
| Fig. 7 Transition metal complexes of triphyrin(2.1.1). X-ray crystal structures of (a) Mn(I) complex 81, (b) Fe(II) complex 83, and (c) Pt(II) complex 84. | ||
Triphyrin(2.1.1) can also behave as a neutral bidentate ligand. Complexation with PtCl2 and PdCl2 salts results in the formation of complexes 84 and 85, respectively.97 These complexes exhibit a similar coordination, with the triphyrin(2.1.1) macrocycles adopting partial cone-like conformations and an uncoordinated NH site. The square-planar metal center is coordinated to two iminic nitrogen atoms and two chloride anions. Upon oxidation with molecular oxygen, Pt(II) complex 84 converts into a Pt(IV) complex with a bowl-shaped triphyrin conformation.98 This indicates conformational flexibility of the triphyrin(2.1.1) macrocycles despite their 14π-aromatic nature.
Recently, metal complexes of a calix[3]pyrrole analogue, calix[1]pyrrole[2]thiazole 86, have been reported,67 with the ring contraction effect stabilizing the chelation of the ethyl zinc moiety. When 86 is treated with diethylzinc in toluene, ethylzinc complex 87 is obtained in 72% yield. X-ray crystallographic analysis reveals the cone conformation of calix[3]-related ligand 86, which behaves as a monoanionic tridentate ligand. Because the ethylzinc moiety is sterically protected by the macrocyclic ligand, hydrolysis and alcoholysis of 87 are effectively suppressed even in the presence of excess water and alcohol in solution. In contrast, organozinc 87 is used as a catalyst for the polymerization of rac-lactide in refluxing toluene in the presence of benzyl alcohol (Fig. 8).
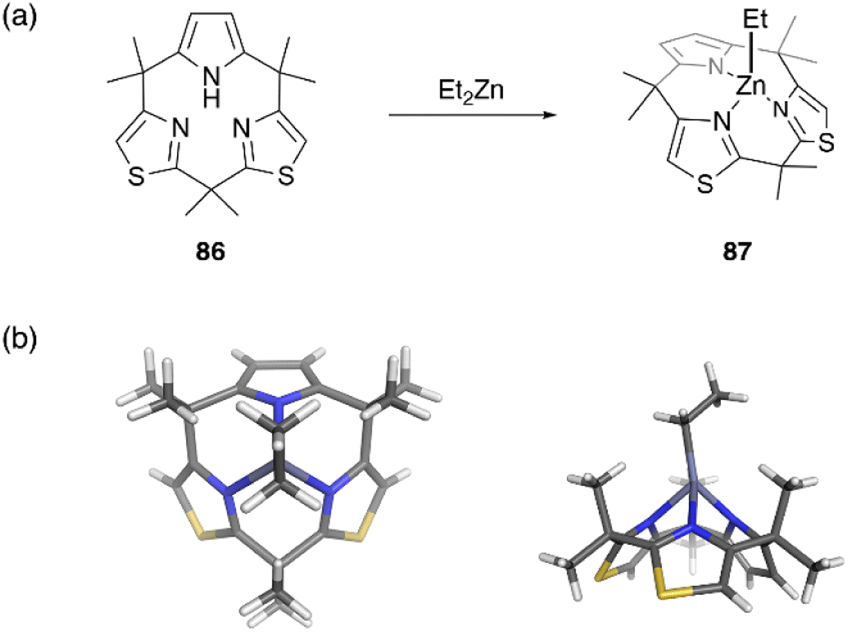 | ||
| Fig. 8 Formation of moisture-stable organozinc complex 87 from calix[3]pyrrole analogue 86. (a) Synthetic scheme and (b) crystal structure of 87. | ||
3.5 Strain-induced ring expansion
The macrocyclic ring strain of calix[3]pyrrole analogues induces unusual ring expansion reactions. Under Rothemund–Lindsey type acidic conditions, calix[3]pyrrole 60 quantitatively converts into calix[6]pyrrole 70 within 30 s (Scheme 14).32 Calix[1]furan[2]pyrrole 64 exhibits similar reactivity to give calix[2]furan[4]pyrrole 88 in 95% yield after 5 min under the Rothemund–Lindsey conditions. In contrast, calix[3]furan 61 and calix[2]furan[1]pyrrole 62 do not undergo such ring expansion owing to their decreased ring strain. Upon N-methylation of the pyrrole moiety, however, methylated form 89 undergoes acid-catalyzed ring expansion, resulting in the formation of calix[6]-type products 91 and 92 in 32 and 18% yields, respectively.80 | ||
| Scheme 14 Strain-induced ring expansion of calix[3]pyrrole analogues. | ||
A detailed study of the reaction mechanism reveals that protonation of the pyrrole-α carbon initiates an irreversible and regioselective ring cleavage of calix[3]pyrrole analogues to give linear tripyrrane-like intermediates, such as 90. The linear intermediates undergo cyclodimerization in a head-to-tail manner, resulting in the formation of calix[6]pyrrole-type products. Rapid ring cleavage explains why calix[3]pyrrole-type macrocycles, including subporphyrinogens, have been missing during porphyrin or calix[4]pyrrole synthesis under Rothemund–Lindsey conditions.
Strain-induced ring expansion can be appled to obtain giant macrocycles, which are larger than the calix[6]pyrrole system. TFA-catalyzed strain-induced ring expansion of calix[1]furan[1]pyrrole[1]thiophene 65 produces calix[9]- and calix[12]-type macrocycles 94 and 95 in 16% and 7% yields, respectively, as well as calix[6]-type product 93 in 30% yield.81 Single crystal X-ray diffraction analysis of 95 reveals its windmill-like conformation and perfect sequence selectivity (Fig. 9).
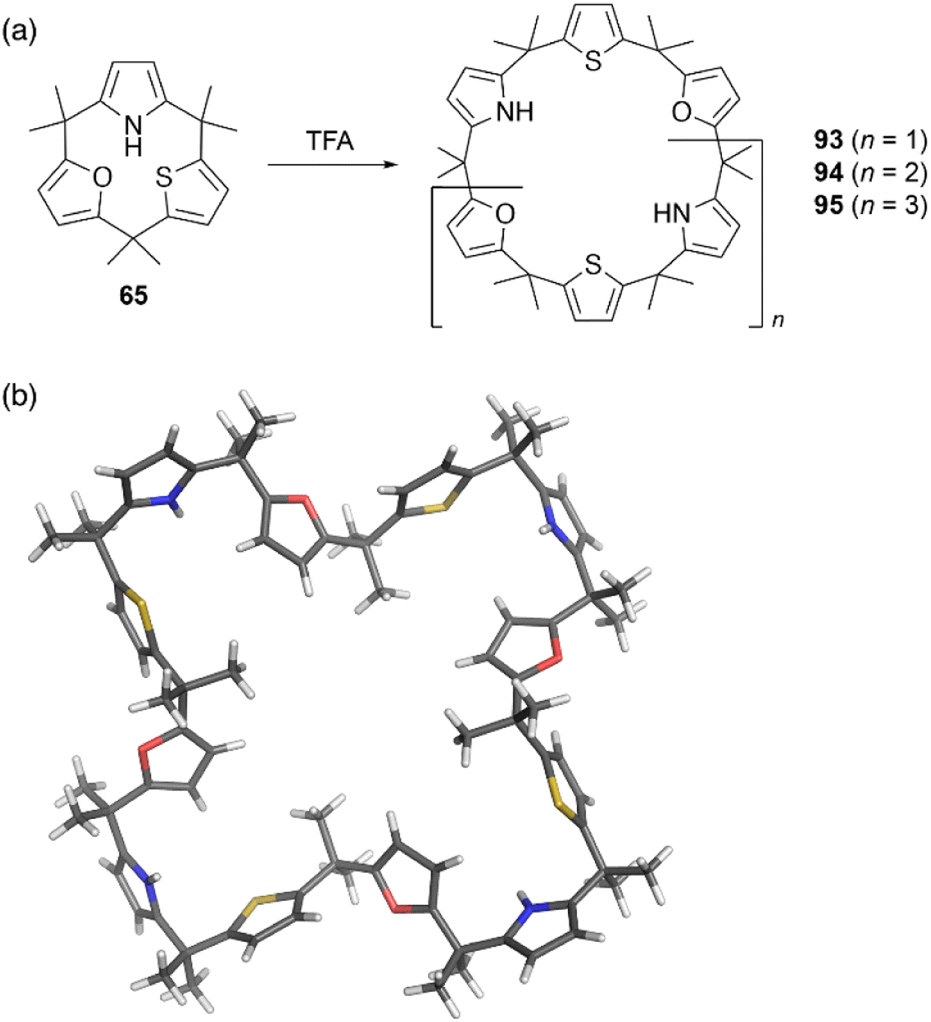 | ||
| Fig. 9 (a) Strain-induced ring expansion of 65 and (b) X-ray crystal structure of calix[12]-type macrocycle 95. | ||
3.6 Supramolecular chemistry
The concave surface of bowl-shaped π-conjugated molecules is advantageous for molecular recognition of fullerenes. Boron(III)-subporphyrins can serve as good scaffolds for capturing fullerenes as well as corannulenes99 and subphthalocyanines100 through π–π interactions. Osuka and coworkers found that tris(1,4-benzodithiino)subporphyrin 96 binds fullerene C60 in solution and in the solid state. Job's and Foster–Fyfe plot analyses of NMR titration results reveal a 1:1 complexation of 96 and C60 in toluene-d8 with an association constant of Ka of 857 ± 58 M−1 at 25 °C. Single crystal X-ray diffraction analysis reveals that all 1,2-benzenedithio groups are located on the concave side of 96, enhancing π–π interactions with C60 (Fig. 10).101 Furthermore, syn-type β,β-(1,4-dithiino)subporphyrin dimer 97 and C60 engage in 1:1 binding in toluene, with a remarkably large association constant Ka of (1.9 ± 0.2) × 106 M−1. In contrast, binding of 97 to C70 occurs with 2:1 stoichiometry characterized by association constants K1 and K2 of (1.6 ± 0.5) × 106 and (1.8 ± 0.9) × 105 M−1, respectively.102
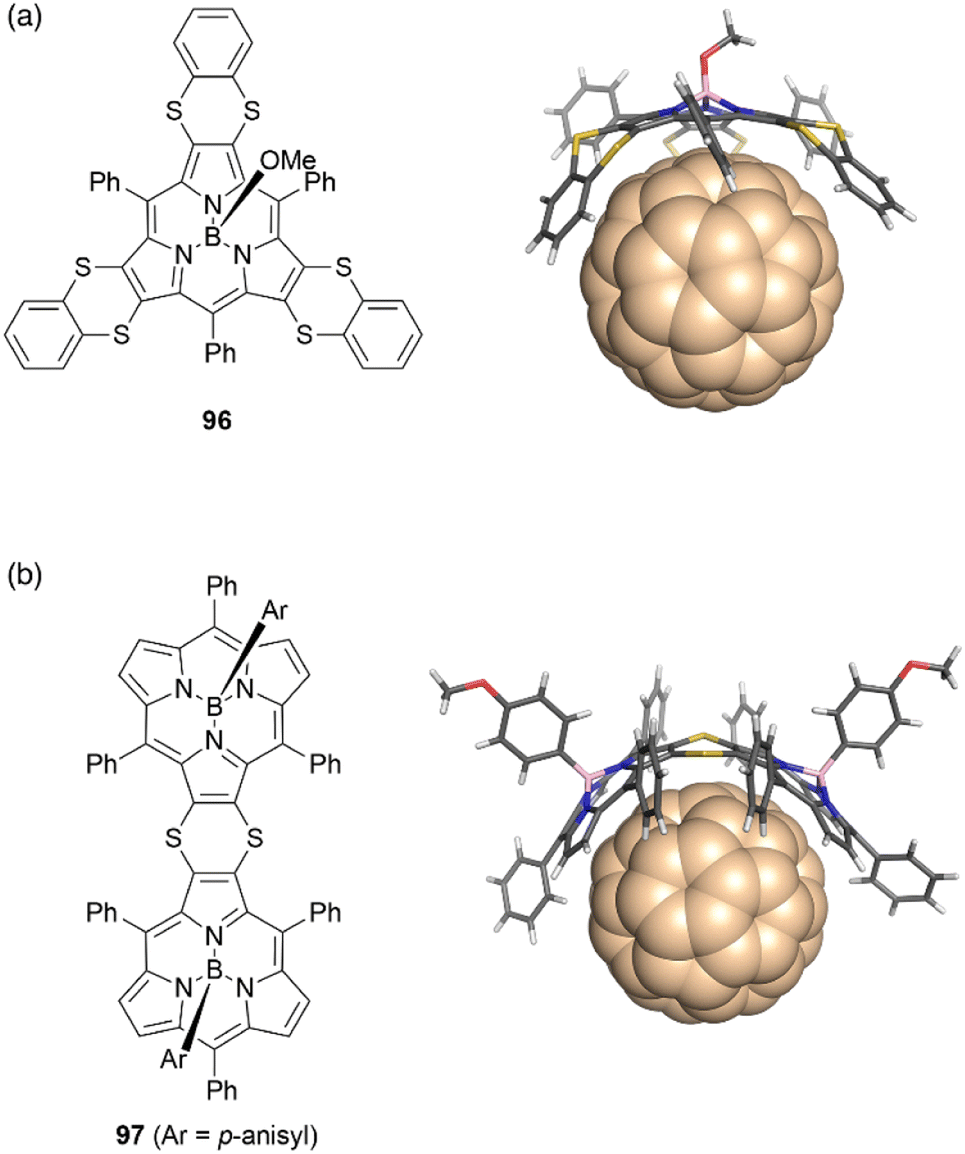 | ||
| Fig. 10 Crystal structures of fullerene complexes (a) 96·C60 and (b) 97·C60. | ||
Axial ligand exchange at the boron(III) center helps to construct supramolecular assemblies of subporphyrins. Because alcohols and carboxylic acids can reversibly replace boron axial ligands, B–O bond formation on boron(III)-subporphyrins can be harnessed for dynamic covalent chemistry. Subporphyrin 98, bearing a 2-carboxyphenyl group, quantitatively forms self-assembled dimer 99 through azeotropic removal of methanol in refluxing toluene (Scheme 15).103 Dimerization of 98 is completed within 3 h at the concentration of 100 μM. Although dimer 99 is resistant to hydrolysis, heating in a mixture of dichloromethane/methanol results in the quantitative recovery of monomer 98.
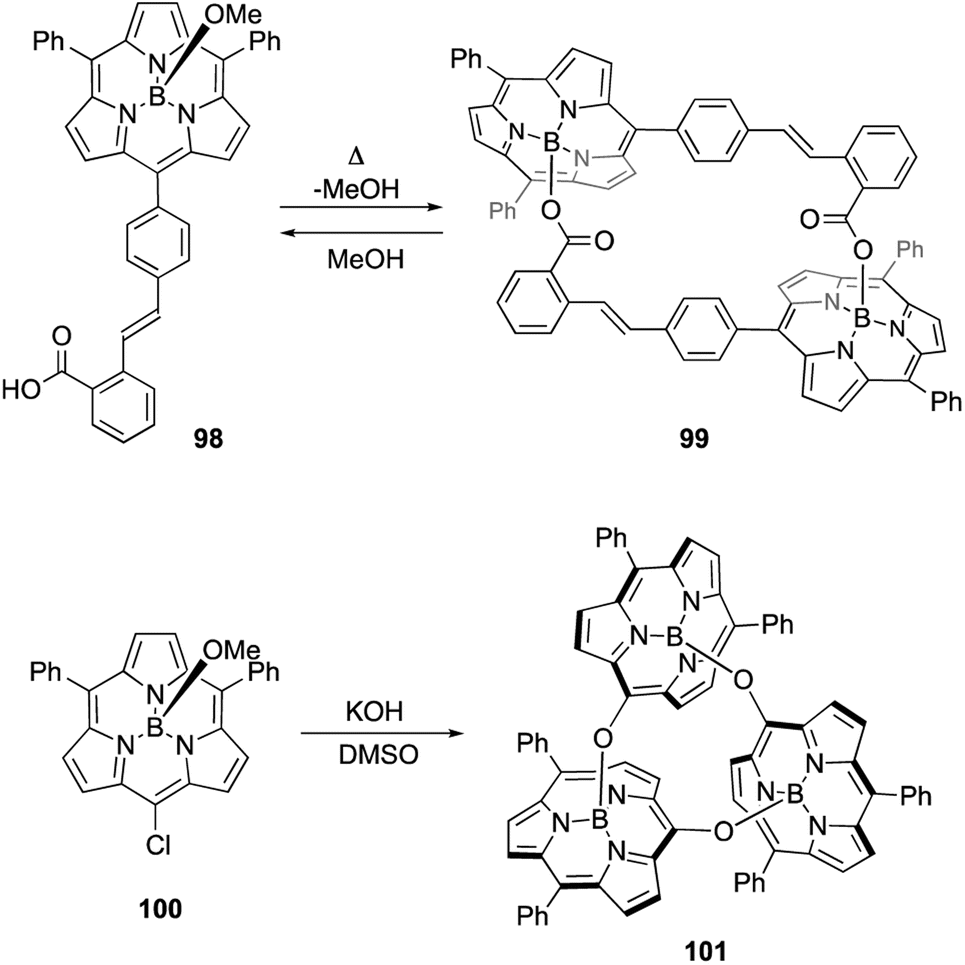 | ||
| Scheme 15 Formation of molecular assemblies through boron axial ligand exchange. | ||
When meso-chloro-substituted subporphyrin 100 was treated with potassium hydroxide in DMSO, cyclic trimer 101 is obtained in 94% yield after aqueous workup (Scheme 15).104 The chlorine atom at the meso-position is replaced with a hydroxy group as a result of the SNAr reaction, and the subsequent axial ligand exchange with the meso-hydroxy group yields 101. Although various cyclic oligomers are derived from meso-hydroxy subporphyrin, trimer 101 is obtained with remarkable selectivity. Trimer 101 dissociates into monomers upon treatment with TFA, potassium t-butoxide, or methanol.
Ring contraction also affects the anion binding properties of calix[n]pyrroles. Calix[4]pyrrole 67 is a well-known host for anion binding, with an association constant of Ka = 17170 ± 900 M−1 in CD2Cl2 for fluoride binding. Upon anion binding, the solid state conformation of 67 changes from 1,3-alternate to cone (Fig. 11).105 In contrast, binding of ring-contracted analogue, calix[3]pyrrole 60, to the fluoride anion is characterized by a markedly small association constant (230 ± 20 M−1). X-ray diffraction analysis of single crystals of 60·F−, obtained as a tetrabutylammonium salt, reveals a cone conformation of 60 in which three NH sites are hydrogen bonded to the fluoride anion.32 In addition to the reduced number of NH sites, the unstable cone conformation likely contributes to the reduced binding constant. Indeed, molecular dynamics (MD) simulations show the rapid and continuous flipping of pyrrole units in 60, which interconverts between partial cone conformations.106 The cone conformation is found to be energetically unfavorable in the absence of an anion.
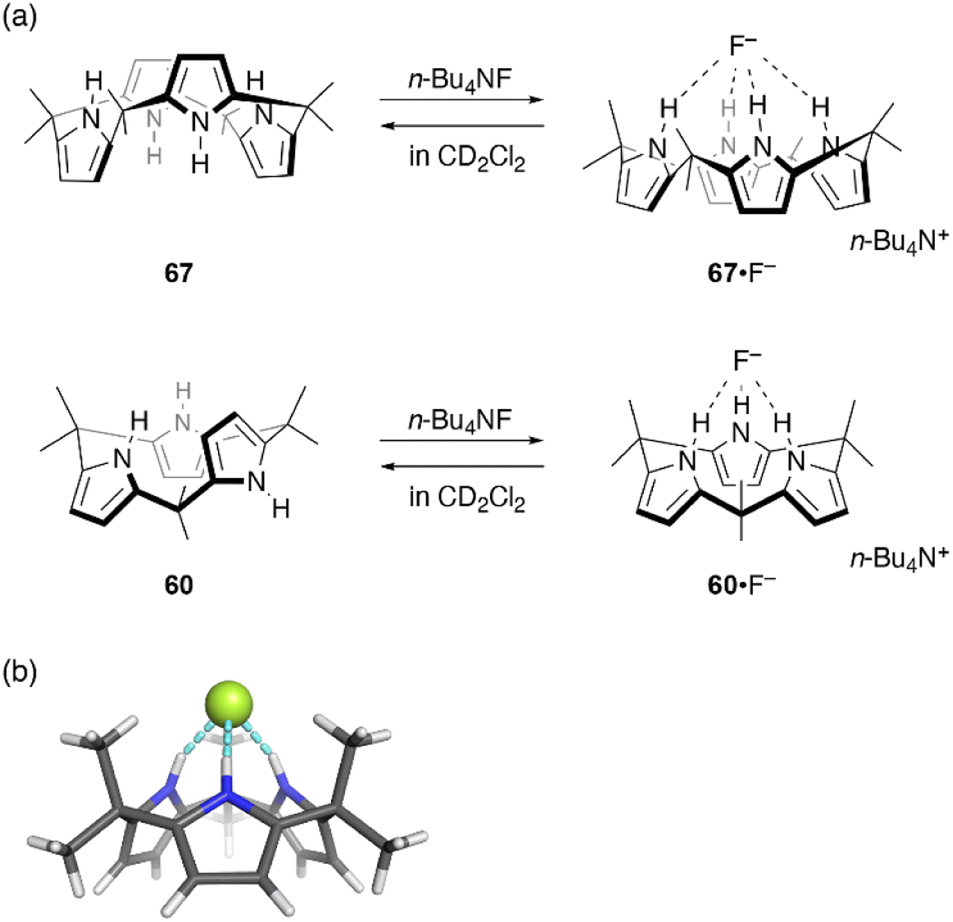 | ||
| Fig. 11 (a) Fluoride binding to calix[4]pyrrole 67 and calix[3]pyrrole 60. (b) Crystal structure of 60·F−. | ||
Restricted flipping of calix[3]pyrroles allows the generation of inherently chiral macrocycles. The inherent chirality is the concept introduced by Böhmer in 1994 for asymmetrically substituted calix[4]arenes.107 Although macrocycles have no stereogenic center, they display chirality because they belong to the C1 point group. In the case of calix[4]pyrroles, rapid ring flipping render asymmetrically substituted analogues virtually achiral. MD calculations indicated that the ring flipping of calix[3]pyrrole analogue 65 is sufficiently slow for enantiomeric separation. Chiral resolution of 65 using an amylose-based chiral HPLC column leads to the separation of enantiomers, although their racemization through ring flipping in hexane/i-PrOH (v/v = 1:1) occurs with a half-life of 2.1 h.81 When 65 is N-methylated using MeI/NaH, resulting 102 does not undergo racemization at room temperature even after standing for a month. The absolute configuration of each enantiomer is unambiguously determined through X-ray diffraction analysis. Enantiomerically pure forms, (Rp,Sp,Sp)-102 and (Sp,Pp,Pp)-102, give rise to mirror-image circular dichroism (CD) spectra (Fig. 12). Furthermore, enantioselective N-methylation of 65 occurs in the presence of a chiral ammonium salt, (R)-4,4-dibutyl-2,6-bis(3,4,5-trifluorophenyl)-4,5-dihydro-3H-dinaphtho[2,1-c:1′,2′-e]azepinium bromide,108 although enantioselectivity is modest (48% yield, 10% ee). This result demonstrates the potential of inherently chiral calix[3]pyrrole analogues for chiral molecular recognition.
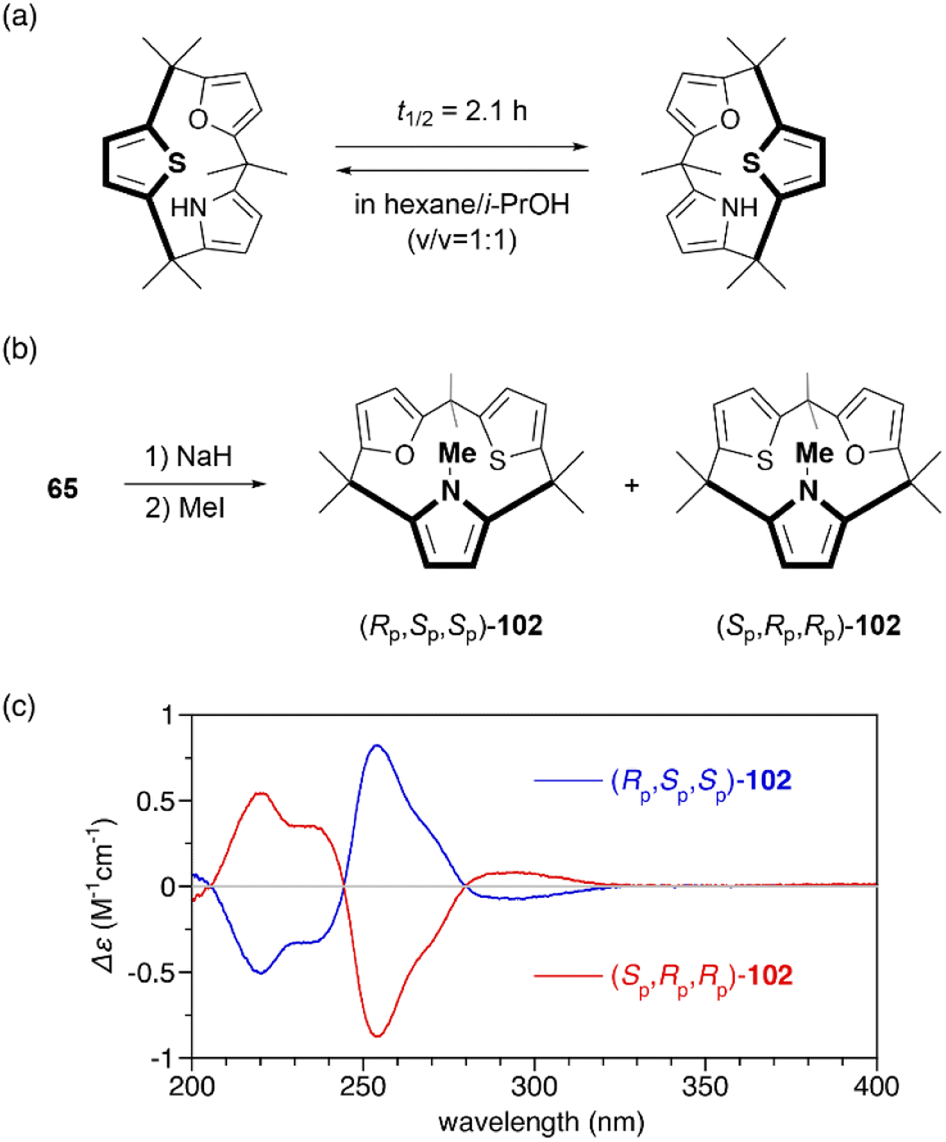 | ||
| Fig. 12 (a) Racemization of chiral calix[3]pyrrole analogue 65. (b) N-Methylation of 65 to suppresses racemization through ring flipping. (c) CD spectra of 102 in n-hexane. | ||
4. Conclusion and future perspectives
Tackling the total synthesis of natural products has led to the development of new synthetic methods.109–112 Similarly, resolving the various challenges in the synthesis of non-natural contracted porphyrins and calixpyrroles has contributed to the discovery of not only new reactions but also functional chromophores, metal complexes, and supramolecules. Controlling the ring strain during macrocyclization is crucial to obtain contracted porphyrinoids with a macrocyclic ring size smaller than the marginal size of compounds obtained via Rothemund–Lindsey protocol. Therefore, unique approaches have been developed, including boron templating, core-modification, and ring tightening. Boron-templated synthesis has led to the discovery of unusual boron-chelation effects in subporphyrins, which allows orthogonal functionalization by axial ligand exchange. Core-modification of the triphyrin(1.1.1) skeleton enables the incorporation of various heteroarenes and additional meso-carbon atoms, which provides scaffolds for the generation of their metal complexes. Furthermore, the ring tightening approach provides a rational route to calix[3]pyrroles, leading to the discovery of strain-induced ring expansion reactions.The ring contraction effects on reactivity, structure, and optical properties have been demonstrated, namely contracted macrocycles have properties that are similar but distinct from those of their parent porphyrinoids. Revealing hidden contraction effects should be a key to advancing this emerging research area. There are still a number of synthetic targets that have not yet been achieved. For example, the synthesis of triphyrin(1.1.0), a corrole-type113 tripyrrole macrocycle, requires unconventional approaches to overcome the significant increase in ring strain during macrocyclization. Highly contracted macrocycles composed of only two pyrrole units might link the chemistry of porphyrins and cyclophanes, although theoretical calculations have shown an unrealistic increase in their strain energy even with the ring tightening approach.114 Furthermore, the development of general methods for the removal of boron templates from boron-subporphyrins is of significant interest, as it will expand the coordination chemistry of subporphyrins. As research on non-natural contracted porphyrins and calixpyrroles transitions from the infant stage (synthetic attempts) to the application stage (development of functional molecules), new aspects of porphyrin-related compounds will be uncovered.
Author contributions
Y. I. outlined all the sections, wrote the first draft of the manuscript, and revised subsequent drafts of the manuscript. K. W. and N. N. P. provided input and feedback on each section and contributed to subsequent drafts of the manuscript. K. W. and Y. I. created the original figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the JSPS Grant-in-Aid for Scientific Research (B) (No. 22H02058) and the JST FOREST Program (No. JPMJFR211H), of which Y. I. is the principal investigator. The Institute for Chemical Reaction Design and Discovery (ICReDD) was established by World Premier International Research Initiative (WPI), MEXT, Japan. Dedicated to Professor Atsuhiro Osuka on his 70th birthday.Notes and references
- L. R. Milgrom, The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, 1997 Search PubMed.
- J. G. Riess, Chem. Rev., 2001, 101, 2797–2920 CrossRef CAS.
- J. M. Cole, P. Giulio, O. K. Al Bahri and C. B. Cooper, Chem. Rev., 2019, 119, 7279–7327 CrossRef CAS.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. D. Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS.
- Y. Zhang, T. Higashino and H. Imahori, J. Mater. Chem. A, 2023, 11, 12659–12680 RSC.
- H. Imahori and M. Akiyama, J. Porphyrins Phthalocyanines, 2024, 28 DOI:10.1142/S1088424624300015.
- K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed.
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS.
- R. B. Woodward, Pure Appl. Chem., 1968, 17, 519–547 CrossRef CAS.
- P. Rothemund, J. Am. Chem. Soc., 1935, 57, 2010–2011 CrossRef CAS.
- R. B. Woodward, Aromaticity: An International Symposium Sheffield, 1966, Special Publication no. 21, The Chemical Society, London, 1966 Search PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef CAS.
- T. Sarma and P. K. Panda, Chem. Rev., 2017, 117, 2785–2838 CrossRef CAS.
- J. Setsune, Y. Katakami and N. Iizuna, J. Am. Chem. Soc., 1999, 121, 8957–8958 CrossRef CAS.
- J. Setsune and S. Maeda, J. Am. Chem. Soc., 2000, 122, 12405–12406 CrossRef CAS.
- A. Dondoni and A. Marra, Chem. Rev., 2010, 110, 4949–4977 CrossRef CAS PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- A. A. Moshfegh, E. Beladi, L. Radnia, A. S. Hosseini, S. Tofigh and G. H. Hakimelahi, Helv. Chim. Acta, 1982, 65, 1264–1270 CrossRef CAS.
- D. Ikuta, Y. Hirata, S. Wakamori, Y. Tomabechi, Y. Kawasaki, K. Ikeuchi, T. Hagimori, S. Matsumoto and H. Yamada, Science, 2019, 364, 674–677 CrossRef CAS PubMed.
- J. Kim, I. Jung, S. Kim, E. Lee, J. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- Y. Inokuma, J. H. Kwon, T. K. Ahn, M. Yoo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 961–964 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–853 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- G. Lavarda, G. Labella, M. V. Martinez-Diaz, M. S. Rodríguez-Morgade, A. Osuka and T. Torres, Chem. Soc. Rev., 2022, 51, 9482–9619 RSC.
- A. Meller and A. Ossko, Monatsh. Chem., 1972, 103, 150–155 CrossRef CAS.
- L. Liu, J. Kim, L. Xu, Y. Rao, M. Zhou, B. Yin, J. Oh, D. Kim, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2022, 61, e202214342 CrossRef CAS PubMed.
- Y. Inaba, Y. Nomata, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda, A. Osuka, J. L. Sessler and Y. Inokuma, J. Am. Chem. Soc., 2021, 143, 12355–12360 CrossRef CAS PubMed.
- P. A. Gale, P. Anzenbecher Jr and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57–102 CrossRef CAS.
- S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525–2536 CrossRef CAS PubMed.
- J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hundnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324–1327 CrossRef CAS.
- I. Saha, J. T. Lee and C.-H. Lee, Eur. J. Org Chem., 2015, 3859–3885 CrossRef CAS.
- S. Peng, Q. He, G. I. Vargas-Zúñiga, L. Qin, I. Hwang, S. K. Kim, N. J. Heo, C.-H. Lee, R. Dutta and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 865 RSC.
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC.
- F. H. Kohnke, Eur. J. Org Chem., 2020, 4261–4272 CrossRef CAS.
- Y. Matano, Org. Biomol. Chem., 2023, 21, 3034–3056 RSC.
- R. K. Sharma, G. Ahuja and I. T. Sidhwani, Green Chem. Lett. Rev., 2009, 2, 101–105 CrossRef CAS.
- D. Langerreiter, M. A. Kostiainen, S. Kaabel and E. Anaya-Plaza, Angew. Chem., Int. Ed., 2022, 61, e202209033 CrossRef CAS.
- L. Edwards, M. Gouterman and C. B. Rose, J. Am. Chem. Soc., 1976, 98, 7638–7641 CrossRef CAS PubMed.
- N. Kobayashi, Y. Takeuchi and A. Matsuda, Angew. Chem., Int. Ed., 2007, 46, 758–760 CrossRef CAS PubMed.
- A. Alder, F. Longo and W. Shergalis, J. Am. Chem. Soc., 1964, 86, 3145–3149 CrossRef.
- P. Szarvas, B. Gyori and J. Emri, Acta Chim. Acad. Sci. Hung., 1971, 70, 1–8 CAS.
- Y. Inokuma, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 4747–4761 CrossRef CAS PubMed.
- E. Tsurumaki, S. Saito, K. S. Kim, J. M. Lim, Y. Inokuma, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 438–439 CrossRef CAS.
- M. Kitano, S. Hayashi, T. Tanaka, H. Yorimitsu, N. Aratani and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 5593–5597 CrossRef CAS.
- T. Tanaka, M. Kitano, S. Hayashi, H. Yorimitsu, N. Aratani and A. Osuka, Org. Lett., 2012, 14, 2694–2697 CrossRef CAS PubMed.
- K. Kise, K. Yoshida, R. Kotani, D. Shimizu and A. Osuka, Chem.–Eur. J., 2018, 24, 19136–19140 CrossRef CAS PubMed.
- O. I. Koifmana and T. A. Ageeva, Russ. J. Org. Chem., 2022, 58, 443–479 CrossRef.
- S. Saito, K. S. Kim, Z. S. Yoon, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2007, 46, 5591–5593 CrossRef CAS PubMed.
- R. Sakamoto, S. Saito, S. Shimizu, Y. Inokuma, N. Aratani and A. Osuka, Chem. Lett., 2010, 39, 439–441 CrossRef CAS.
- M. S. Rodríguez-Morgade, S. Esperanza, T. Torres and J. Barberá, Chem.–Eur. J., 2005, 11, 354–360 CrossRef PubMed.
- Z. Li, L. Zhang, Q. Wu, h. Li, Z. Kang, C. Yu, E. Hao and L. Jiao, J. Am. Chem. Soc., 2022, 144, 6692–6697 CrossRef CAS PubMed.
- Z. Xue, Z. Shen, J. Mack, D. Kuzuhara, H. Yamada, T. Okujima, N. Ono, X. You and N. Kobayashi, J. Am. Chem. Soc., 2008, 130, 16478–16479 CrossRef CAS PubMed.
- Z. Xue, J. Mack, H. Lu, L. Zhang, X. Z. You, D. Kuzuhara, M. Stillman, H. Yamada, S. Yamauchi, T. N. Kobayashi and Z. Shen, Chem.–Eur. J., 2011, 17, 4396–4407 CrossRef CAS PubMed.
- S. Ito, N. Ochi, T. Murashima, H. Uno and N. Ono, Heterocycles, 2000, 52, 399–411 CrossRef CAS.
- K. S. Anju, S. Ramakrishnan and A. Srinivasan, Org. Lett., 2012, 13, 2498–2501 CrossRef PubMed.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS.
- D. Kuzuhara, H. Yamada, Z. L. Xue, T. Okujima, S. Mori, Z. Shen and H. Uno, Chem. Commun., 2011, 47, 722–724 RSC.
- B. S. Kumar, N. N. Pati, K. V. Jovan Jose and P. K. Panda, Chem. Commun., 2020, 56, 5637–5640 RSC.
- R. Myśliborski, L. Latos-Grażyński, L. Szterenberg and T. Lis, Angew. Chem., Int. Ed., 2006, 45, 3670–3674 CrossRef.
- M. Pawlicki, M. Garbicz, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2015, 54, 1906–1909 CrossRef CAS PubMed.
- D. Kuzuhara, Y. Sakakibara, S. Mori, T. Okujima, H. Uno and H. Yamada, Angew. Chem., Int. Ed., 2013, 52, 3360–3363 CrossRef CAS.
- K. Watanabe, K. Shibata, T. Ichino, Y. Ide, T. Yoneda, S. Maeda and Y. Inokuma, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-6nqnq.v1.
- S. Maeda, K. Ohno and K. Mororkuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC.
- C. E. Colwell, T. W. Price, T. Stauch and R. Jasti, Chem. Sci., 2020, 11, 3923–3930 RSC.
- P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404–408 CrossRef CAS PubMed.
- E. Kayahara, V. Kumar Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284–2287 CrossRef CAS.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- G. Cafeo, F. H. Kohnke, G. L. La Torre, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2000, 39, 1496–1498 CrossRef CAS.
- G. Cafeo, F. H. Kohnke, M. F. Parisi, R. P. Nascone, G. L. La Torre and D. J. Williams, Org. Lett., 2002, 4, 2695–2697 CrossRef CAS PubMed.
- G. Cafeo, F. H. Kohnke, G. L. La Torre, M. F. Parisi, R. P. Nascone, A. J. P. White and D. J. Williams, Chem.–Eur. J., 2002, 8, 3148–3156 CrossRef CAS.
- Y. Manabe, M. Uesaka, T. Yoneda and Y. Inokuma, J. Org. Chem., 2019, 84, 9957–9964 CrossRef CAS.
- P. Sarkar, Y. Inaba, H. Shirakura, T. Yoneda and Y. Inokuma, Org. Biomol. Chem., 2020, 18, 3297–3302 RSC.
- N. Ozawa, K. I. Shivakumar, M. Murugavel, Y. Inaba, T. Yoneda, Y. Ide, J. Pirillo, Y. Hijikata and Y. Inokuma, Chem. Commun., 2022, 58, 2971–2974 RSC.
- M. Uesaka, Y. Saito, S. Yoshioka, Y. Domoto, M. Fujita and Y. Inokuma, Commun. Chem., 2018, 1, 23 CrossRef.
- Y. Inaba, Y. Kakibayashi, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda and Y. Inokuma, Chem.–Eur. J., 2022, 28, e202200056 CrossRef CAS.
- Y. Inaba, J. Yang, Y. Kakibayashi, T. Yoneda, Y. Ide, Y. Hijikata, J. Pirillo, R. Saha, J. L. Sessler and Y. Inokuma, Angew. Chem., Int. Ed., 2023, 62, e202301460 CrossRef CAS.
- E. Tsurumaki, S. Hayashi, F. S. Tham, C. A. Reed and A. Osuka, J. Am. Chem. Soc., 2011, 133, 11956–11959 CrossRef CAS PubMed.
- T. Kato, F. S. Tham, P. D. W. Boyd and C. A. Reed, Heteroat. Chem., 2006, 17, 209–216 CrossRef CAS.
- Y. Tobe, K. Ueda, K. Kakiuchi, Y. Odaira, Y. Kai and N. Kasai, Tetrahedron, 1986, 42, 1851–1858 CrossRef CAS.
- P. Rothemund and A. Menotti, J. Am. Chem. Soc., 1948, 70, 1808–1812 CrossRef CAS PubMed.
- K. M. Kadish, E. C. Van and G. Royal, The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 8, pp. 1–219 Search PubMed.
- Y. Inokuma, S. Easwaramoorthi, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 12234–12235 CrossRef CAS PubMed.
- C. Ngaojampa, S. Namuangruk, Y. Surakhot, V. Promarak, S. Jungsuttiwong and N. Kungwan, Comput. Theor. Chem., 2015, 1062, 1–10 CrossRef CAS.
- Y. Inokuma, S. Easwaramoorthi, S. Y. Jang, K. S. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 4840–4843 CrossRef CAS PubMed.
- K. Watanabe, R. Saha, Y. Inaba, Y. Manabe, T. Yoneda, Y. Ide, Y. Hijikata and Y. Inokuma, J. Porphyrins Phthalocyanines, 2023, 27, 157–163 CrossRef CAS.
- S. Shimizu, A. Matsuda and N. Kobayashi, Inorg. Chem., 2009, 48, 7885–7890 CrossRef CAS PubMed.
- S. Saga, S. Hayashi, K. Yoshida, E. Tsurumaki, P. Kim, Y. M. Sung, J. Sung, T. Tanaka, D. Kim and A. Osuka, Chem.–Eur. J., 2013, 19, 11158–11161 CrossRef CAS PubMed.
- E. Tsurumaki, J. Sung, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 1056–1059 CrossRef CAS PubMed.
- E. Tsurumaki, J. Sung, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 2596–2599 CrossRef CAS PubMed.
- D. Kuzuhara, Z. Xue, S. Mori, T. Okujima, H. Uno, N. Aratani and H. Yamada, Chem. Commun., 2013, 49, 8955–8957 RSC.
- Z. Xue, D. Kuzuhara, S. Ikeda, Y. Sakakibara, K. Ohkubo, N. Aratani, T. Okujima, H. Uno, S. Fukuzumi and H. Yamada, Angew. Chem., Int. Ed., 2013, 52, 7306–7309 CrossRef CAS.
- Z. Xue, Y. Wang, J. Mack, Y. Fang, Z. Ou, W. Zhu and K. M. Kadish, Inorg. Chem., 2015, 54, 11852–11858 CrossRef CAS.
- Z. Xue, D. Kuzuhara, S. Ikeda, T. Okujima, S. Mori, H. Uno and H. Yamada, Inorg. Chem., 2013, 52, 1688–1690 CrossRef CAS.
- M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed.
- A. Salazar, M. Moreno-Simoni, S. Kumar, J. Labella, T. Torres and G. de la Torre, Angew. Chem., Int. Ed., 2023, 62, e202311255 CrossRef CAS.
- K. Yoshida and A. Osuka, Chem.–Asian J., 2015, 10, 1526–1534 CrossRef CAS.
- K. Yoshida and A. Osuka, Chem.–Eur. J., 2016, 22, 9396–9403 CrossRef CAS PubMed.
- Y. Inokuma and A. Osuka, Chem. Commun., 2007, 2938–2940 RSC.
- D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6613–6617 CrossRef CAS.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- R. Saha, J. Pirillo, Y. Ide, Y. Inokuma and Y. Hijikata, Theor. Chem. Acc., 2023, 50, 142 Search PubMed.
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- M. Kitamura, S. Shirakawa and M. Maruoka, Angew. Chem., Int. Ed., 2005, 44, 1549–1551 CrossRef CAS.
- J. ApSimon, Total Synthesis of Natural Products, John Wiley & Sons, Inc., 1973 Search PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102–3137 CrossRef.
- T. Sano, Y. Sun, T. Mukai, Y. Inaba, T. Yoneda, Y. Ide, J. Pirillo, Y. Hijikata and Y. Inokuma, J. Porphyrins Phthalocyanines, 2023, 27, 1067–1073 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |