
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer approach by targeted activation of a global inhibitor of sialyltransferases with acrolein†
Takatsugu
Kasahara‡
a,
Tsung-Che
Chang‡
ab,
Hiromasa
Yoshioka
 b,
Sayaka
Urano
b,
Yasuko
Egawa
b,
Michiko
Inoue
c,
Tsuyoshi
Tahara
d,
Koji
Morimoto
b,
Ambara R.
Pradipta
*a and
Katsunori
Tanaka
*ab
b,
Sayaka
Urano
b,
Yasuko
Egawa
b,
Michiko
Inoue
c,
Tsuyoshi
Tahara
d,
Koji
Morimoto
b,
Ambara R.
Pradipta
*a and
Katsunori
Tanaka
*ab
aDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: pradipta.a.aa@m.titech.ac.jp; kotzenori@riken.jp
bBiofunctional Synthetic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan
cLaboratory for Biofunction Dynamics Imaging, RIKEN Center for Biosystems Dynamics Research, 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
dDepartment of In vivo Imaging, Advanced Research Promoting Center, Tokushima University, 3-18-15 Kuramto-cho, Tokushima, Tokushima 770-8503, Japan
First published on 24th May 2024
Abstract
Cells are covered with a thick layer of sugar molecules known as glycans. Abnormal glycosylation is a hallmark of cancer, and hypersialylation increases tumor metastasis by promoting immune evasion and inducing tumor cell invasion and migration. Inhibiting sialylation is thus a potential anticancer treatment strategy. However, targeting sialic acids is difficult because of the lack of selective delivery tools. Here, we present a prodrug strategy for selectively releasing the global inhibitor of sialylation peracetylated 3Fax-Neu5Ac (PFN) in cancer cells using the reaction between phenyl azide and endogenous acrolein, which is overproduced in most cancer cells. The prodrug significantly suppressed tumor growth in mice as effectively as PFN without causing kidney dysfunction, which is associated with PFN. The use of sialylated glycans as immune checkpoints is gaining increasing attention, and the proposed method for precisely targeting aberrant sialylation provides a novel avenue for expanding current cancer treatments.
Introduction
Glycosylation is the most abundant post-translational modification, and cell surface glycans are essential for many biological processes, including infection, cell adhesion, and cell differentiation.1 One of the most remarkable changes in cancer glycosylation is aberrant sialylation due to the marked upregulation of sialyltransferases, which catalyze the addition of sialic acid to growing glycochains on the cell surface to form sialoglycans.2 Sialic acids, which are derivatives of neuraminic acid, are a class of nine-carbon, negatively charged glycans that are typically found at the ends of glycoproteins and glycolipids in almost all vertebrate cells. As shown in Fig. 1A, sialoglycan overexpression promotes cancer development and metastasis via several key pathways, including promoting escape from natural killer (NK) cell-mediated tumor cell death through the recruitment of sialic acid-binding immunoglobulin-like lectin (Siglec) receptors Siglecs-7 and -9,3–5 avoiding immune-mediated apoptosis by inhibiting the internalization of the Fas receptor,6 and promoting tumor invasion by inhibiting integrin-mediated adhesion and enhancing interaction with selectins.7 Desialylation of cancer cells potentiates NK cell-mediated cytotoxicity8,9 and promotes the clearance of cancer cells injected into mice,10,11 suggesting that targeting aberrant sialylation could be developed as an effective anticancer treatment.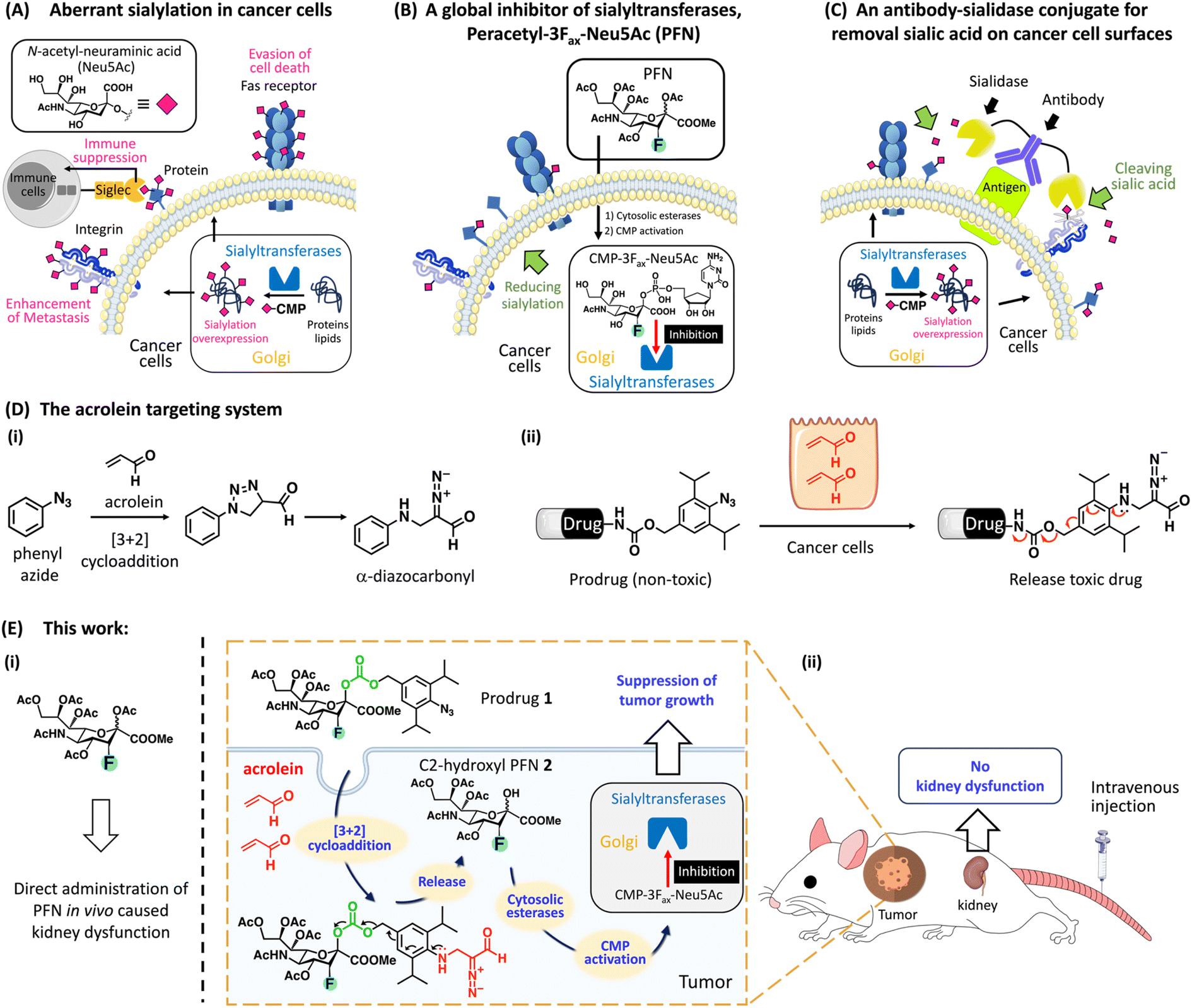 | ||
| Fig. 1 Anticancer approach by targeted activation of a global inhibitor of sialyltransferases with acrolein in cancer. (A) Overview of the roles of sialylation in tumor growth, metastasis, and immune suppression. Therapeutic strategies for modulating sialic acid expression using (B) a metabolic sialyltransferase inhibitor, peracetyl-3Fax-Neu5Ac (PFN), to suppress sialic acid biosynthesis in cancer cells or (C) an antibody–sialidase conjugate, such as trastuzumab (anti-HER2), linked with a sialidase to remove sialic acid from the cell surface. (D) (i) Phenyl azide reacts with acrolein through a [3 + 2] reaction to generate 4-formyl-1,2,3-triazole, which is rearranged to an α-diazocarbonyl derivative. (ii) Drug release strategy using the [3 + 2] cycloaddition between 2,6-diisopropylphenyl azide and acrolein. (E) (i) PFN inhibits the expression of sialic acid in most tissues in mice, but it causes kidney dysfunction. (ii) Schematic of the PFN-based prodrug 1 strategy for the selective release of C2-hydroxyl PFN 2 in tumor sites in vivo through endogenous acrolein to enhance cancer immunotherapy while avoiding kidney dysfunction caused by PFN. | ||
Two important strategies targeting aberrant sialylation in cancer cells are (1) interfering with sialoglycan expression by inhibiting sialyltransferases;12 and (2) enzymatic removal of sialic acids from the cell surface using sialidase,5,13 a sialic acid-cleaving enzyme. More than 20 different Golgi-resident sialyltransferases are involved in the biosynthesis of sialoglycans in human and murine cells; however, they all use the same donor substrate, cytosine monophosphate-N-acetyl-neuraminic acid (CMP-Neu5Ac).14 A cell-permeable peracetylated 3Fax-Neu5Ac (PFN) developed by Paulson et al. effectively inhibited all sialyltransferases via a mechanism involving its intracellular conversion to an active inhibitor, CMP-3Fax-Neu5Ac, thereby reducing overall sialylation in cultured cells (Fig. 1B).15 However, despite the efficacy of PFN in decreasing sialylated glycans in most tissues in the murine model, it causes kidney dysfunction because of the depletion of sialic acids from podocytes, which impairs glomerular filtration.16 To circumvent the deleterious effects in vivo, the group led by Adema pretreated mice with tumor-targeting PFN-encapsulated nanoparticles, which prevented metastasis in a mouse lung cancer model without toxic effects to the kidney.17 These authors also showed that intratumoral injections of PFN significantly suppressed melanoma development in mice by enhancing the effects of tumor immunotherapy; however, kidney dysfunction was still observed at high doses.18
Regarding sialidase treatment, Bertozzi et al. developed antibody–sialidase conjugates to provide target specificity, thereby avoiding unwanted cytotoxicity caused by off-target desialylation (Fig. 1C).19–21 These authors demonstrated that trastuzumab, an antibody against human epidermal growth factor receptor 2 (HER2), linked with a bacterial sialidase desialylated HER2+ breast tumors in mice, thereby increasing NK cell killing effects to inhibit tumor growth.20,21 However, this strategy does not abolish sialic acid formation inside cancer cells, which thus restore sialoglycans. Büll et al. showed that cell surface sialic acid expression is restored within 1 day after sialidase treatment, whereas it requires 2–3 days after PFN treatment.22 These data indicate that the global sialyltransferase inhibitor (PFN) is a more appropriate treatment for aberrant sialylation than sialidase. Nevertheless, a targeted system for the delivery of PFN to cancer sites would be optimal and would prevent kidney toxicity.
Acrolein, a highly reactive α,β-unsaturated aldehyde, is a critical biomarker associated with various oxidative stress-related disorders such as cancer.23 Previous work from our laboratory identified a [3 + 2] cycloaddition reaction between phenyl azide and acrolein that proceeds rapidly and selectively under physiological conditions without the use of a catalyst, yielding an α-diazocarbonyl derivative (Fig. 1D-i).24 Based on this bio–orthogonal reaction, we designed a probe for the intracellular detection of acrolein within cells; the generated α-diazocarbonyl moiety reacts with the nearest organelle and covalently attaches the fluorophore (see ESI Fig. S2A†).25,26 The acrolein-sensing probe revealed that acrolein is generally overproduced in cancer cells and tissues, whereas it is negligible in normal cells,27,28 which is consistent with previous findings.29 We thus hypothesized that acrolein could be used as a new cancer marker. To develop a drug delivery system for targeting acrolein, we created a prodrug system in which a cytotoxic drug was linked to the para-position of 2,6-diisopropylphenyl azide via a cleavable carbamate linker because 2,6-diisopropylphenyl azide reacts more rapidly with acrolein than phenyl azide (Fig. 1D-ii).30 The reaction of the non-toxic prodrug with acrolein regenerated the active form after the self-immolation of the carbamate linker. Based on the described prodrug design, we here present a PFN-derivatized prodrug 1 (Fig. 1E-ii), in which the 2,6-diisopropylphenyl azide moiety of 1 selectively reacted with cancer cell-endogenous acrolein to release C2-hydroxy PFN 2, which was converted to CMP-3Fax-Neu5Ac to inhibit sialyltransferases, thereby enhancing cancer immunotherapy. B16F10 tumor-bearing mice treated with prodrug 1via intravenous administration showed a clear reduction in tumor growth in vivo with no kidney dysfunction caused by PFN (Fig. 1E-i), highlighting the potential of the prodrug for use in clinical trials of cancer immunotherapy.
Results and discussions
Synthesis of prodrug 1 and application of model compound 6 for evaluating drug release kinetics and stability
In this study, we used a carbonate group (Fig. 2A) as a cleavable linker to link C2-hydroxy PFN 2 with the benzyl alcohol moiety of 4 to synthesize the prodrug 1. As depicted in Fig. 2A, after the alcohol of 2 was activated to 4-nitrophenyl carbonate, the reaction with 4 (ref. 30) yielded prodrug 1 at 60% in two steps. To evaluate the acrolein-mediated PFN release kinetics and stability of the carbonate linker in prodrug 1 under physiological conditions, we used umbelliferone, which is easily detectable by UV-visible absorption, to replace 2 for synthesizing model compound 6 (Fig. 2B) using the same method described for the preparation of 1. Moreover, the umbelliferone released by the reaction of 6 with acrolein increased fluorescence intensity, which could thereby be used to monitor the reaction (see ESI Fig. S3†).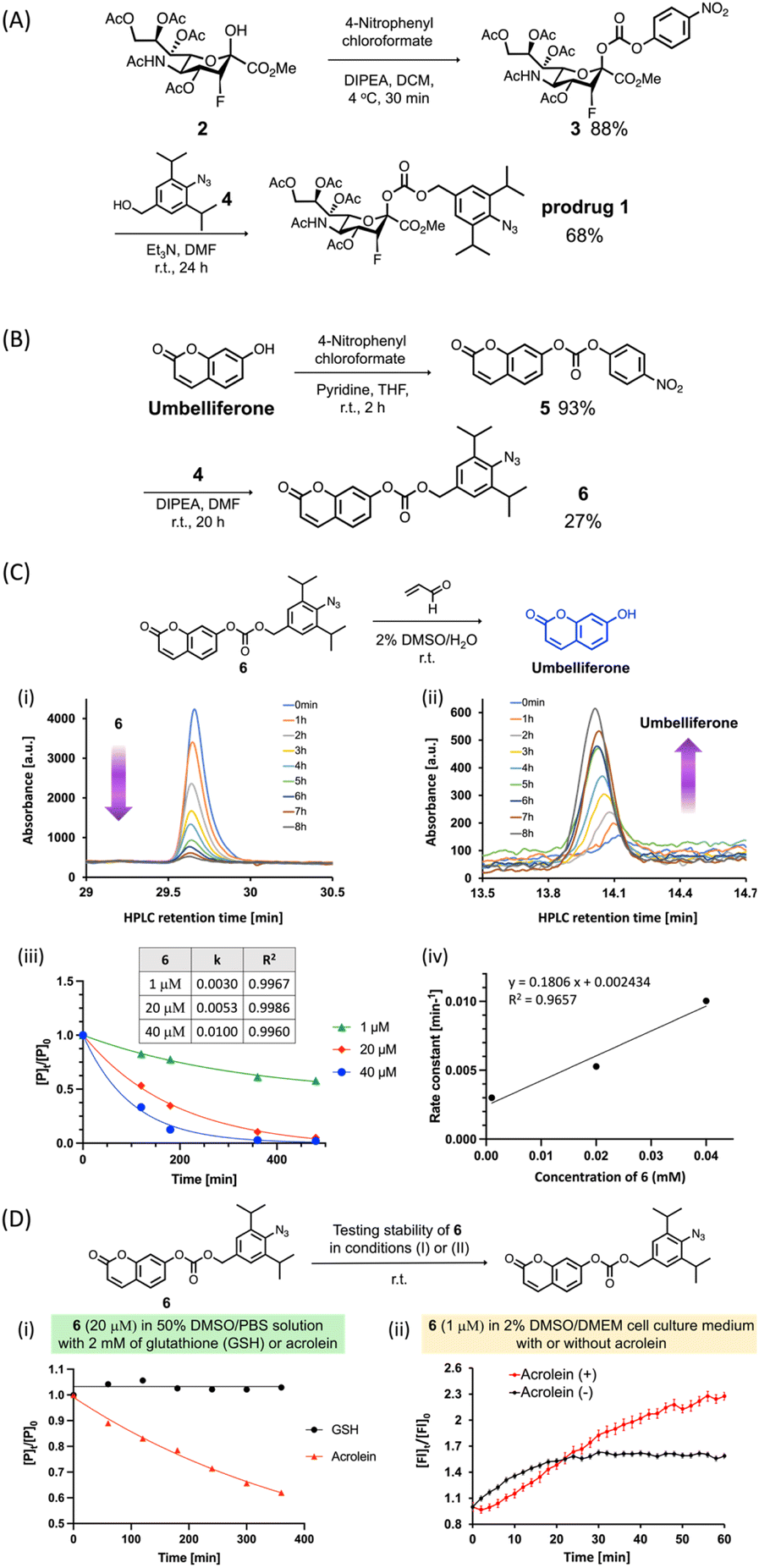 | ||
| Fig. 2 Synthesis of prodrug 1 (A) and model compound 6 (B) using the same synthetic method. (C) The reaction between compound 6 (20 μM) and acrolein (20 mM) in 2% DMSO/H2O at room temperature was monitored at the indicated times by RP-HPLC and UV-visible detection. (i) The peak area of 6 decreased. (ii) The peak area of umbelliferone increased. The umbelliferone release kinetics were measured by monitoring the peak area of compound 6. (iii) The HPLC chromatogram peak area ratios of compound 6 were plotted against reaction time. (iv) The rate constant data were plotted against the concentration of compound 6 to determine the second-order rate constant (k = 18.1 × 10−2 mM−1 min−1). (D) (i) The reactions between compound 6 (20 μM) and glutathione (GSH, 2 mM, black line) or acrolein (2 mM, red line) in 50% DMSO/PBS solution at room temperature were monitored at the indicated times by RP-HPLC. The reactions were observed by monitoring the peak area of compound 6. The HPLC chromatogram peak area ratios of compound 6 were plotted against reaction time. (ii) Compound 6 (1 μM) was incubated in DMEM in the presence (red line) or absence (black line) of acrolein at room temperature. The reaction was observed by monitoring the change in fluorescence intensity of 6. The fluorescence change ratios of compound 6 were plotted against reaction time. Error bars represent the SD of three replicate measurements. [P]t = HPLC chromatogram peak area of 6 at t minute [P]0 = HPLC chromatogram peak area of 6 at 0 minutes. [Fl]t = fluorescence intensity of 6 at t minute [Fl]0 = fluorescence intensity of 6 at 0 minutes. | ||
Incubation of compound 6 (20 μM) with an excess amount of acrolein (1000 equivalents) in a 2% DMSO aqueous solution at room temperature and analysis by RP-HPLC showed that the consumption of 6 (Fig. 2C-i) and the release of umbelliferone (Fig. 2C-ii) increased concomitantly with increasing reaction time, confirming the acrolein-induced drug release in this prodrug system. To investigate the drug release kinetics using the cleavable carbonate group, various concentrations (1, 20, and 40 μM) of compound 6 were incubated with excess amounts of acrolein and results were measured at the indicated reaction times. As shown in Fig. 2C-iii and -iv, the second-order rate constant (k) of the drug release was 18.1 × 10−2 mM−1 min−1, which is consistent with our previous findings using the prodrug system with the carbamate group.30
To examine the stability of the carbonate linker in a biological setting, compound 6 (20 μM) was incubated with 2 mM of glutathione (GSH) or acrolein in a 50% DMSO solution in phosphate-buffered saline (PBS), and the release of umbelliferone was monitored by RP-HPLC. The results showed that compound 6 was not reduced or decomposed by GSH even after 6 h of incubation (Fig. 2D-i, black line), whereas the acrolein-induced consumption of 6 to release umbelliferone increased gradually (Fig. 2D-i, red line). Incubation of compound 6 in Dulbecco's Modified Eagle's Medium (DMEM) solution in the presence or absence of acrolein and measurement of the fluorescence intensity of 6 showed that acrolein rapidly reacted with 6 to release umbelliferone in DMEM solution within 1 hour (Fig. 2D-ii, red line). In the absence of acrolein, the fluorescence signal increased slightly, which was probably due to low levels of carbonate hydrolysis in DMEM solution. Taken together, the results of Fig. 2C and D indicate that the carbonate linker in the prodrug did not affect drug release, and the stability of the linker could minimize off-target effects.
Efficacy of prodrug 1 for inhibiting sialylation examined in cell-based experiments
We next evaluated the efficacy of prodrug 1 for inhibiting sialylation in cancer cells. We previously showed that A549 adenocarcinomic human alveolar basal epithelial cells produce considerable amounts (100 μM–1 mM) of acrolein27 (see ESI Fig. S2B†), and a study reported that this cell line has high expression levels of sialic acids;31 therefore, A549 cells were used as the model cell line. A549 cells were cultured for 3 days in the presence of prodrug 1 or PFN as a control, and the expression of cell surface sialic acids was measured by flow cytometry using specific carbohydrate-binding lectins (Fig. 3A). Dose-effect experiments revealed that treatment with 64 μM PFN significantly reduced α-2,6- and α-2,3-sialylation of A549 cells, as detected by staining with the lectins SNA and MALII, respectively (Fig. 3B and C, blue bars). Prodrug 1 had similar effects on the sialylation of A549 cells, indicating that activation of 1 to release 2 was successfully triggered by endogenous acrolein (Fig. 3B and C, red bars). In B16F10 murine melanoma cells, which show significant expression levels of acrolein (see ESI Fig. S2B†), treatment with prodrug 1 at 64 μM (Fig. 3D and E, red bars) also substantially depleted sialic acid from cells similar to the effect of the PFN control (Fig. 3D and E, blue bars). Decreasing sialic acid expression resulted in exposure to terminal β-galactose residues such as the N-acetyllactosamine (LacNAc) on N-glycans and T antigen (Galβ1,3GalNAcSer/Thr), terminal α-N-acetylgalactosamine, and also extended poly-LacNAc repeats. Due to these facts, the B16F10 cells treated with both PFN and prodrug 1 showed significantly increased binding of the lectins PNA (Fig. 3F), GSL-I (Fig. 3G), and WGA (Fig. 3H). Slight increase in fucosylation was caused by the fact that fucosyltransferases conjugate fucose to N-acetylglucosamine on the extended poly-LacNAc repeats (Fig. 3I). The above findings were in line with a previous report16 by Paulson et al. These outcomes in Fig. 3 clearly demonstrated that prodrug 1 can produce the same glycan profiles as the PFN control when treating cancer cells, indicating that the prodrug design does not influence the bioactivity of the released PFN. As a negative control, prodrug 1 did not alter the sialic acid expression of TIG3 cells (see ESI Fig. S8†) because the cells do not express acrolein.27,28 The viability of both A549 and B16F10 cells was not affected by PFN or prodrug 1 at 64 μM (see ESI Fig. S4†). The results shown in Fig. 3 demonstrate the feasibility of utilizing endogenous acrolein to activate prodrug 1 for the inhibition of sialylation in cancer cells.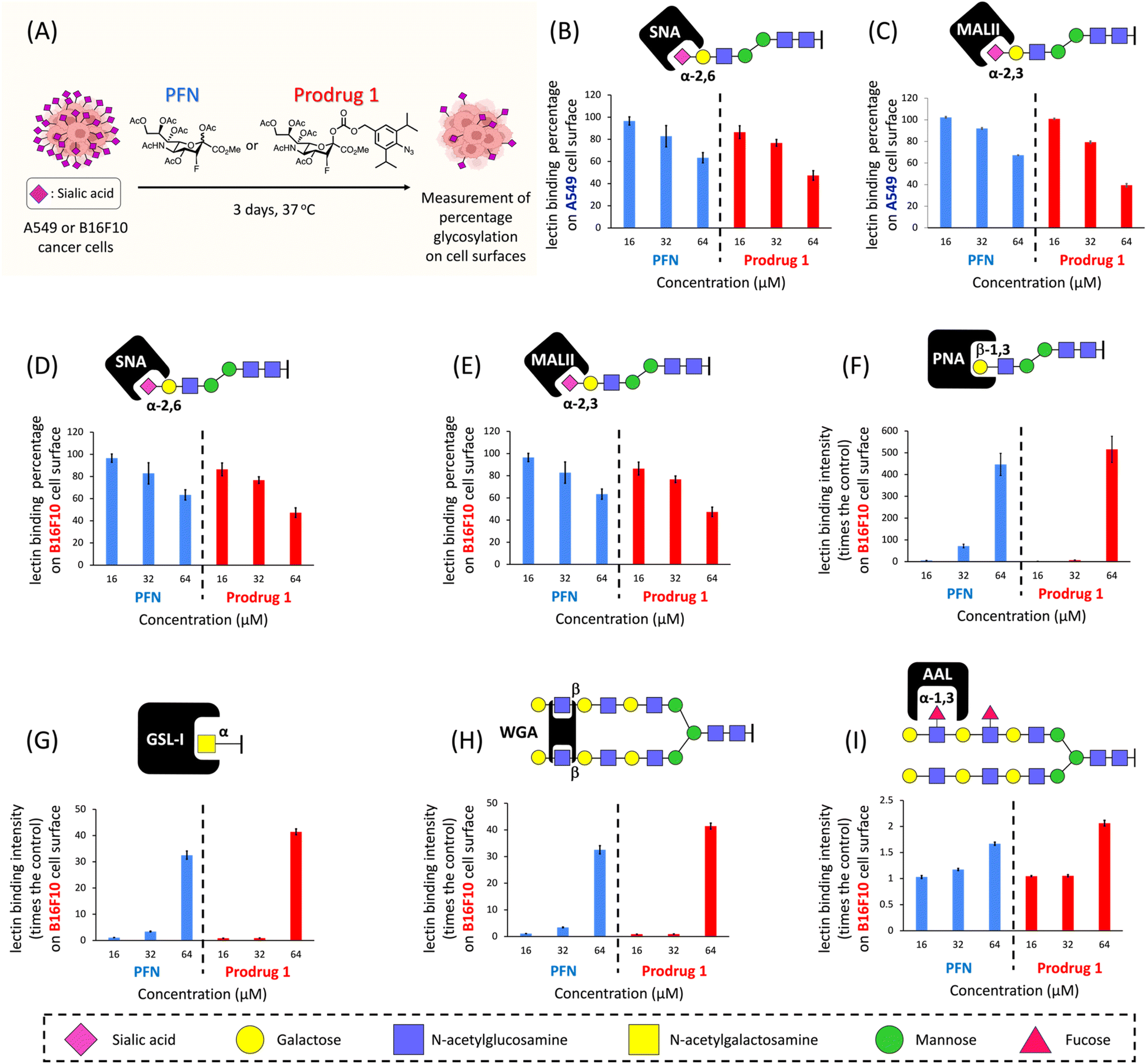 | ||
| Fig. 3 (A) Inhibition of sialic acid expression in A549 and B16F10 cancer cells using PFN and prodrug 1. Schematic of the assay procedure. A549 (B and C) and B16F10 (D–I) cancer cells were treated with different concentrations (16, 32, and 64 μM) of PFN or prodrug 1 for 3 days at 37 °C. Cells were harvested, and various glycan epitopes on the cell surface were detected by lectins via flow cytometry: α-2,6-linked sialic acids by SNA lectin (B and D), α-2,3-linked sialic acids by MALII lectin (C and E), exposure of terminal β-galactose by PNA lectin (F), exposure of terminal α-N-acetylgalactosamine (GalNAc) by GSL-I lectin (G), exposure of β-N-acetylglucosamine (GlcNAc) by WGA lectin (H) and α-1,3 or α-1,6-linked fucose by AAL lectin (I). In B–E, the data were normalized to cells treated with DMSO only, which were considered as 100% sialylated epitope expression, whereas unstained cells were considered as 0%. In F–I, the data were normalized to cells treated with DMSO only as 0%. Error bars represent the SD of three replicate measurements. | ||
NK cells are cytotoxic lymphocytes that possess an innate ability to identify and eradicate malignant cells.32 As mentioned in the introduction, desialylation can increase the susceptibility of cancer cells to NK cell-mediated cytolysis. As shown in Fig. 4A, A549 cells were pretreated for 3 days with PFN or prodrug 1 or DMSO alone as a control, and susceptibility to human NK-92 cell killing was assessed after 4 hours of co-incubation. Compared with the control (Fig. 4B-i), flow cytometry experiments showed a clear increase in NK-92 cell-mediated lysis of desialylated A549 cells, which were pretreated with 64 μM PFN (Fig. 4B-ii). In particular, prodrug 1 had a higher NK cell-mediated cytotoxic effect against A549 cells than PFN treatment (Fig. 4B-iii). This was confirmed by quantification of the cell death percentage in A549 cells (Fig. 4C). The results shown in Fig. 4 indicate that prodrug 1 increases the susceptibility to NK cell-mediated immunosurveillance by suppressing the sialylation of cancer cells as effectively as PFN.
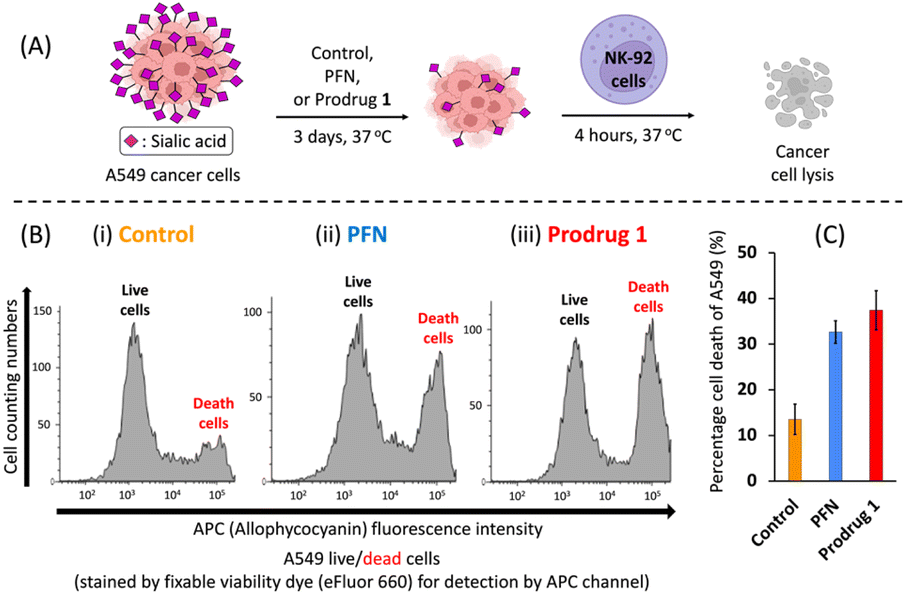 | ||
Fig. 4 Sialylation of cancer cells decreases susceptibility to NK cell-mediated cytotoxicity. (A) Reduction of A549 cell surface sialic acids using PFN and prodrug 1 increases NK cell-mediated cytotoxicity. Schematic diagram of the cytotoxicity assay procedure. A549 cancer cells were treated with 64 μM of either PFN or prodrug 1 or DMSO alone as a control for 3 days at 37 °C, harvested, and co-cultured with NK-92 cells (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, effector/target) for 4 hours at 37 °C. (B) Representative flow cytometry histograms of A549 live/dead cells. (C) The percentage of A549 cell death was quantified. For flow cytometry, a fixable viability dye (eFluor™ 660) was used to stain dead cells and detected at λEX = 633 nm/λEM = 660 nm (allophycocyanin (APC) channel). Error bars represent the SD of three replicate measurements. :1, effector/target) for 4 hours at 37 °C. (B) Representative flow cytometry histograms of A549 live/dead cells. (C) The percentage of A549 cell death was quantified. For flow cytometry, a fixable viability dye (eFluor™ 660) was used to stain dead cells and detected at λEX = 633 nm/λEM = 660 nm (allophycocyanin (APC) channel). Error bars represent the SD of three replicate measurements. | ||
In vivo prodrug 1 activation by acrolein to inhibit tumor growth
As reported by Paulson et al., mice treated with 300 mg kg−1 of PFN develop kidney dysfunction as an adverse effect.16 According to the cell-based results, in the last stage of this study, we investigated the efficacy of prodrug 1 in the treatment of subcutaneous B16F10-xenografted mice and examined whether 1 could be used to avoid the kidney dysfunction caused by PFN. Because of the limited solubility of 1 in saline solution, mice were divided into three groups as follows: vehicle, PFN (60 mg kg−1), and prodrug 1 (60 mg kg−1) via intravenous administration every day for 5 consecutive days for a treatment total of 300 mg kg−1. The control group received a saline solution (vehicle) to replace the compounds in the treatment protocol. Tumor size was monitored for 16 days. The rate of tumor growth significantly decreased in the PFN (Fig. 5B, blue line) and prodrug 1 (Fig. 5B, red line) groups compared with the vehicle group (Fig. 5B, orange line). Mice were sacrificed on day 16 and tumors were extracted (Fig. 5C). The findings shown in Fig. 5B and C demonstrate that PFN and prodrug 1 significantly suppressed tumor growth compared with the control condition (vehicle) by inhibiting sialyation in tumors to enhance the immunotherapy effect.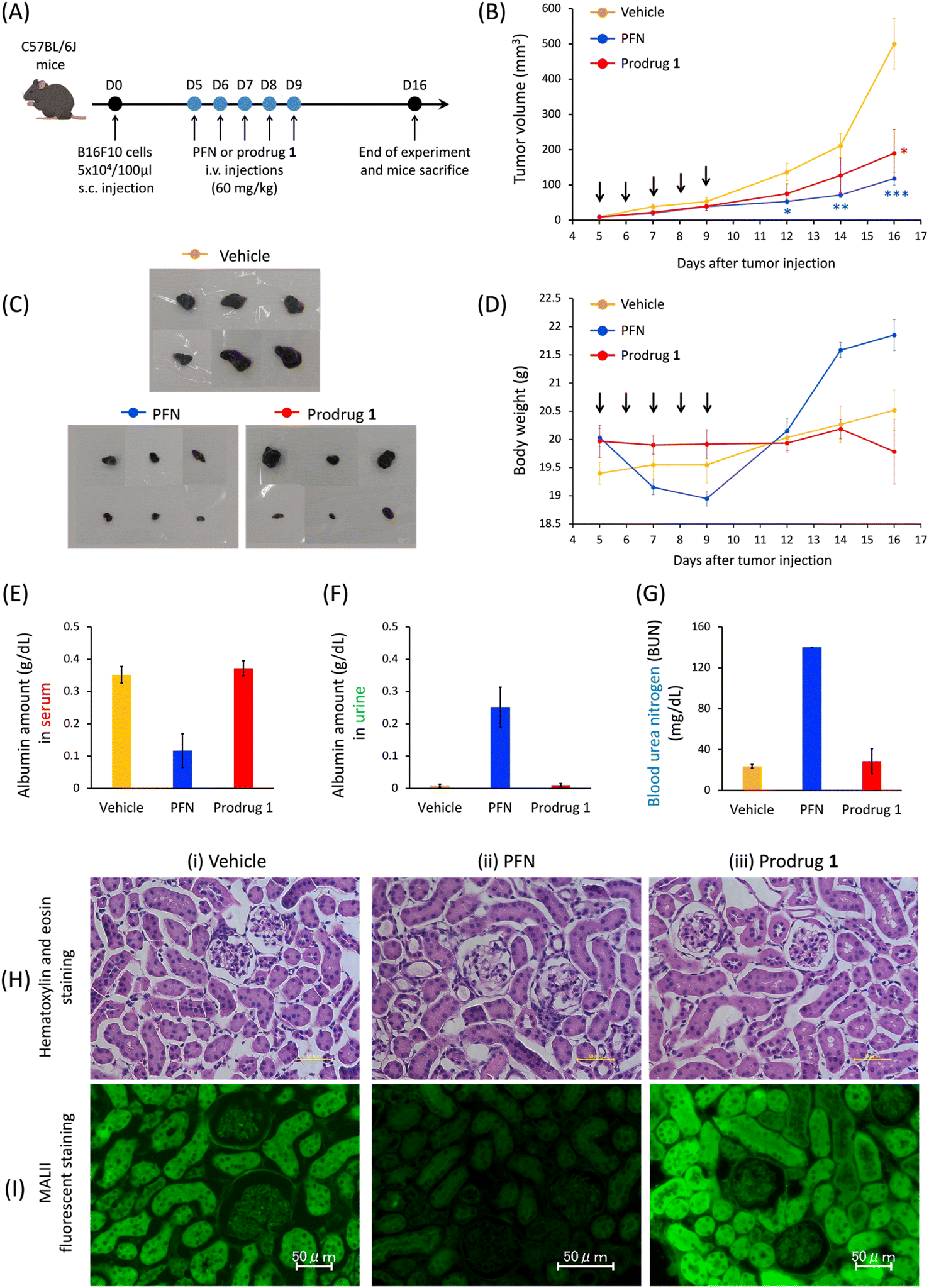 | ||
| Fig. 5 In vivo prodrug 1 activation by acrolein against B16F10 tumor growth in mice. (A) Schematic representation of the in vivo experiment. B16F10 cell xenograft-bearing C57B/6J mice were treated with vehicle, PFN, or prodrug 1. Tumors were initially implanted in mice and developed over 5 days before therapy. When tumor sizes reached 15–20 mm3, the B16F10 tumor-bearing mice were randomly divided into 3 groups: vehicle group (n = 6); PFN treatment (n = 6); prodrug 1 treatment (n = 6). A dose of 60 mg kg−1 was administered in the form of daily injections for 5 days through intravenous injection. The tumor volume and body weight of the mice were recorded until day 16 post-injection. Tumor volume was quantified using an equation of V = W2 × L × 0.4, where W and L represented the minor and major length of the tumor, respectively. On day 16 post-injection, mice were sacrificed, urine and blood were collected, and their tumors were excised, imaged, and weighed. (B) Measurement of tumor size (mm3) in mice over time. (C) Visual comparison of extracted tumors shows the extent of growth inhibition at 11 days after the start of therapy (n = 6). (D) Body weight changes in the different groups of mice. Determination of albumin levels in serum (E) or urine (F) pooled from mice at 11 days after the start of therapy. (G) Determination of blood urea nitrogen (BUN) in blood pooled from mice at 11 days after the start of therapy. (H) Hematoxylin and eosin staining on kidney tissue in various groups of mice at 11 days after the start of therapy. (I) Fluorescence staining by MALII lectin on kidney tissue in various groups of mice at 11 days after the start of therapy. Black arrows under the horizontal axis indicate the day of treatment with the compounds. Data in (B and D-G) are presented as the mean ± SE, n = 6 biological replicates. P values were determined using a two-tailed Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the vehicle group. | ||
PFN causes kidney dysfunction in mice, which manifests as edema, weight gain, excretion of albumin in the urine, and albumin loss from the blood.16,18 In this study, mice receiving PFN showed higher body weight (Fig. 5D, blue line) than those in the vehicle group (Fig. 5D, orange line) at approximately 8 days after the start of treatment, indicating the occurrence of edema. Analysis of the blood and urine in the three groups showed a decrease in blood albumin (Fig. 5E) concomitant with an increase in urine albumin (Fig. 5F) in the PFN group compared with the vehicle group, with a substantial increase in urea nitrogen in the blood (BUN) (Fig. 5G), indicating that PFN caused kidney dysfunction in mice. By contrast, in the prodrug 1 group, these measurements were comparable to those in the vehicle group. In addition, histochemical studies (Fig. 5H and I) were performed on day 16 paraffin-embedded kidney tissue sections from mice treated with vehicle, PFN, and prodrug 1. Although the hematoxylin and eosin staining on the kidney tissues of treated mice in the three groups did not show histological changes (Fig. 5H), the fluorescent lectin staining of these kidney tissues revealed that the PFN group (Fig. 5I-ii) obviously decreased MALII staining relative to the vehicle and prodrug 1 groups (Fig. 5I-i and iii). The data in Fig. 5I demonstrated that prodrug 1 did not alter histochemical characteristics or sialic acid expression in the kidney tissues of mice. These in vivo results of Fig. 5 indicate that activation of prodrug 1 by endogenous acrolein in tumors to release 2-hydroxyl of PFN 2 for inhibiting sialic acid formation may enhance tumor immunotherapy to inhibit tumor growth without causing kidney dysfunction, highlighting the potential of this strategy for future applications in cancer therapy. To apply the prodrug strategy to clinical trial testing in the future, we will proceed with a preclinical study using a patient-derived xenograft (PDX) mouse model to further investigate the effect of prodrug 1 on tumor therapy and evaluate its toxicity.
Conclusions
This study represents a significant advance in research into targeting aberrant sialylation in cancer to enhance cancer immunotherapy. Cell-based experiments showed that endogenous acrolein can be used to activate prodrug 1 to release 2, thereby inhibiting sialylation in cancer cells to increase susceptibility to NK cell-mediated cytolysis. Prodrug 1 significantly suppressed B16F10 tumor growth in mice as effectively as PFN without causing kidney dysfunction. Because the use of sialylated glycans as immune checkpoints is gaining increased attention,33 this system for precisely targeting aberrant sialylation offers another avenue for expanding current cancer immunotherapy.Data availability
All the data have been presented in the manuscript and ESI.†Author contributions
Conceptualization: T.-C. C., A. R. P., and K. T.; methodology: T.-C. C., A. R. P., and K. T.; investigation: T. K., T.-C. C., S. U., Y. E., H. Y., M. I., T. T., and K. M.; visualization: T.-C. C., A. R. P., and K. T.; funding acquisition: A. R. P., and K. T.; project administration: K. T.; supervision: K. T.; writing – original draft: T.-C. C., and K. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (W2019-2-049). This work was financially supported by JSPS KAKENHI (Grant Numbers 23K17971, JP21H02065, JP21K19042, JP21K05269, and JP18H05503), the Naito Foundation, and the Canon Foundation.Notes and references
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346 CrossRef PubMed.
- S. Pietrobono and B. Stecca, Cancers, 2021, 13, 2014 CrossRef CAS PubMed.
- J. V. Rinsum, L. A. Smets, H. V. Rooy and D. H. V. D. Eijnden, Int. J. Cancer, 1986, 38, 915–922 CrossRef.
- J. E. Hudak, S. M. Canham and C. R. Bertozzi, Nat. Chem. Biol., 2014, 10, 69–75 CrossRef CAS PubMed.
- J. Lübbers, E. Rodríguez and Y. V. Kooyk, Front. Immunol., 2018, 9, 2807 CrossRef.
- A. F. Swindall and S. L. Bellis, J. Biol. Chem., 2011, 286, 22982–22990 CrossRef CAS.
- L. A. Coupland and C. R. Parish, Semin. Oncol., 2014, 41, 422–434 CrossRef CAS.
- C. Jandus, K. F. Boligan, O. Chijioke, H. Liu, M. Dahlhaus, T. Démoulins, C. Schneider, M. Wehrli, R. E. Hunger, G. M. Baelocher, H.-U. Simon, P. Romero, C. Münz and S. V. Gunten, J. Clin. Invest., 2014, 124, 1810–1820 CrossRef CAS PubMed.
- P. Rosenstock and T. Kaufman, Cells, 2021, 10, 263 CrossRef CAS PubMed.
- T. Kato, Y. Wang, K. Yamaguchi, C. M. Milner, R. Shineha, S. Satomi and T. Miyagi, Int. J. Cancer, 2001, 92, 797–804 CrossRef CAS PubMed.
- T. Uemura, K. Shiozaki, K. Yamaguchi, S. Miyazaki, S. Satomi, K. Kato, H. Sakuraba and T. Miyagi, Oncogene, 2009, 28, 1218–1229 CrossRef CAS PubMed.
- S. J. L. P. Perez, C.-W. Fu and W.-S. Li, Molecules, 2021, 26, 5673 CrossRef CAS PubMed.
- M. Ghosh, P. Hazarika, S. J. Dhanya, D. Pooja and H. Kulhari, Int. J. Biol. Macromol., 2024, 257, 128415 CrossRef CAS PubMed.
- A. Harduin-Lepers, V. Vallejo-Ruiz, M.-A. Krzewinski-Recchi, B. Samyn-Petit, S. Julien and P. Delannoy, Biochimie, 2001, 83, 727 CrossRef CAS PubMed.
- C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, A. Dell, S. M. Haslam and J. C. Paulson, Nat. Chem. Biol., 2012, 8, 661–668 CrossRef CAS PubMed.
- M. S. Macauley, B. M. Arlian, C. D. Rillahan, P.-C. Pang, N. Bortell, M. C. G. Marcondes, S. M. Haslam, A. Dell and J. C. Paulson, J. Biol. Chem., 2014, 289, 35149–35158 CrossRef CAS PubMed.
- C. Büll, T. J. Boltje, E. A. W. V. Dinther, T. Peters, A. M. A. De Graaf, J. H. W. Leusen, M. Kreutz, C. G. Figdor, M. H. D. Brok and G. J. Adema, ACS Nano, 2015, 9, 733–745 CrossRef PubMed.
- C. Büll, T. J. Boltje, N. Balneger, S. M. Weischer, M. Wassink, J. J. V. Gemst, V. R. Bloemendal, L. Boon, J. V. D. Vlag, T. Heise, M. H. D. Brok and G. J. Adema, Cancer Res., 2018, 78, 3574 CrossRef PubMed.
- H. Xiao, E. C. Woods, P. Vukojicic and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10304–10309 CrossRef CAS PubMed.
- J. Daly, S. Sarkar, A. Natoni, J. C. Stark, N. M. Riley, C. R. Bertozzi, M. Carlsten and M. E. O'Dwyer, Blood Adv., 2022, 6, 3352–3366 CrossRef CAS.
- M. A. Gray, M. A. Stanczak, N. R. Mantuano, H. Xiao, J. F. A. Pijnenborg, S. A. Malaker, C. L. Miller, P. A. Weidenbacher, J. T. Tanzo, G. Ahn, E. C. Woods, H. Läubi and C. R. Bertozzi, Nat. Chem. Biol., 2020, 16, 1376–1384 CrossRef CAS PubMed.
- C. Büll, T. J. Boltje, M. Wassink, A. M. A. De Graaf, F. L. V. Delft, M. H. D. Brok and G. J. Adema, Mol. Cancer Ther., 2013, 12, 1935 CrossRef PubMed.
- J. P. Kehrer and S. S. Biswal, Toxicol. Sci., 2000, 6, 57 Search PubMed.
- A. R. Pradipta, M. Taichi, I. Nakase, E. Saigitbatalova, A. Kurbangalieva, S. Kitazume, N. Taniguchi and K. Tanaka, ACS Sens., 2016, 1, 623 CrossRef CAS.
- A. R. Pradipta, M. Fujii, T. Tanei, K. Morimoto, S. Shimazu, K. Noguchi and K. Tanaka, Bioorg. Med. Chem., 2019, 27, 2228–2234 CrossRef CAS PubMed.
- T. Tanei, A. R. Pradipta, K. Morimoto, M. Fujii, J.-I. Ikeda, E. Morii, S. Noguchi and K. Tanaka, Breast J., 2019, 44, S26–S27 CrossRef.
- T. Tanei, A. R. Pradipta, K. Morimoto, M. Fujii, M. Arata, A. Ito, M. Yoshida, E. Saigitbatalova, A. Kurbangalieva, J.-I. Ikeda, E. Morii, S. Noguchi and K. Tanaka, Adv. Sci., 2019, 6, 1801479 CrossRef.
- A. R. Pradipta, T. Tanei, K. Morimoto, K. Shimazu, S. Noguchi and K. Tanaka, Adv. Sci., 2020, 17, 1901519 CrossRef.
- S. Kato, G. C. Post, V. M. Bierbaum and T. H. Koch, Anal. Biochem., 2002, 305, 251–259 CrossRef CAS.
- A. R. Pradipta, P. Ahmadi, K. Terashima, K. Muguruma, M. Fujii, T. Ichino, S. Maeda and K. Tanaka, Chem. Sci., 2021, 12, 5438 RSC.
- Y. Liang, Q. Hua, P. Pan, J. Yang and Q. Zhang, Biol. Res., 2015, 48, 52 CrossRef.
- T. J. Laskowski, A. Biederstädt and K. Rezvani, Nat. Rev. Cancer, 2022, 22, 557–575 CrossRef CAS PubMed.
- J. Munkley, Cancers, 2022, 14, 4248 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00969j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |