
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering of bespoke photosensitiser–microbe interfaces for enhanced semi-artificial photosynthesis
Imogen L.
Bishara Robertson
 a,
Huijie
Zhang
a,
Erwin
Reisner
b,
Julea N.
Butt
c and
Lars J. C.
Jeuken
*a
a,
Huijie
Zhang
a,
Erwin
Reisner
b,
Julea N.
Butt
c and
Lars J. C.
Jeuken
*a
aLeiden Institute of Chemistry, Leiden University, PO Box 9502, Leiden 2300 RA, the Netherlands
bYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
cSchool of Chemistry and School of Biological Sciences, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
First published on 21st May 2024
Abstract
Biohybrid systems for solar fuel production integrate artificial light-harvesting materials with biological catalysts such as microbes. In this perspective, we discuss the rational design of the abiotic–biotic interface in biohybrid systems by reviewing microbes and synthetic light-harvesting materials, as well as presenting various approaches to coupling these two components together. To maximise performance and scalability of such semi-artificial systems, we emphasise that the interfacial design requires consideration of two important aspects: attachment and electron transfer. It is our perspective that rational design of this photosensitiser–microbe interface is required for scalable solar fuel production. The design and assembly of a biohybrid with a well-defined electron transfer pathway allows mechanistic characterisation and optimisation for maximum efficiency. Introduction of additional catalysts to the system can close the redox cycle, omitting the need for sacrificial electron donors. Studies that electronically couple light-harvesters to well-defined biological entities, such as emerging photosensitiser–enzyme hybrids, provide valuable knowledge for the strategic design of whole-cell biohybrids. Exploring the interactions between light-harvesters and redox proteins can guide coupling strategies when translated into larger, more complex microbial systems.
Introduction
Generating fuels from renewable energy, such as sunlight, is of paramount importance in our urgent transition away from fossil fuels. In its most basic form, three components are required for photoredox catalysis: a catalyst for chemical reduction, a catalyst for chemical oxidation, and a light absorber to harvest solar energy. These components can be synthetic, natural, or a combination of both. Semi-artificial photosynthesis has been previously defined as interfacing biological catalysts, which provide unparalleled catalytic specificity for the production of desirable chemicals, with synthetic light-harvesting materials to create biohybrid systems.1 Here, we focus specifically on the integration of microbial catalysts directly with synthetic nanoparticle light-harvesters. We refer the reader to previous reviews on other systems of semi-artificial photocatalysis, including photoelectrochemical cells (PECs), which are not discussed here.2,3 While the use of microbes as biological catalysts in solution with light-harvesting nanoparticles has been investigated for decades,4 a landmark study in 2016 initiated a renaissance of research into these biotechnologies by demonstrating that nanoparticles could be synthesised and then localised at the surface of the microbe.5 A long-lived physical interface between microbe and photosensitiser eliminates diffusion as a limiting factor. Synthetic materials can have advantages over natural photosystems in terms of resistance to photodamage; some subunits of natural photosystems require repair as often as every 30 minutes.6 Further, microbial approaches exploit the capability of live cells to self-replicate and repair, reducing the input of materials such as purified proteins that require costly and time-consuming production. The ability to genetically program cells also provides opportunities to engineer them as factories for a specific chemical product.7,8While light-harvesters and microbes have been previously reviewed in depth, and whole-cell biohybrids discussed and analysed,2,9–16 the challenge of creating a defined electron transfer interface between photosensitiser and microbe has only been highlighted.17 The nature of this abiotic–biotic interface in existing biohybrids is often unclear and difficult to characterise retrospectively.18 Thus, the question we aim to address here is: how can we establish controllable and characterisable pathways for electron transfer, from photosensitiser to cytoplasmic enzymes, that can be built-in by design? What research should we turn to for creating these biohybrid blueprints, and where are remaining gaps of knowledge that prevent progression? In this perspective, we propose that the rational design of these systems should exploit microbes with natural mechanisms of electron uptake, and involve engineered attachment of the photosensitiser to the biological catalyst. We support this view with a detailed discussion of research that we hope can benefit the ongoing optimisation of whole-cell biohybrids in the future.
Components for semi-artificial photosynthesis
Construction of a controllable, optimisable and scalable biohybrid requires consideration of three important areas: selection of an effective light absorber for photoelectron generation (together with a catalyst for regeneration and preferably the synthesis of an added-value chemical); selection of a suitable microbe for catalysis; and finally, electronic coupling of the two via a defined electron transfer pathway, from (photo)electron generation through to intracellular catalysis (see Fig. 1). Most research has focused on using the microbe as a site of fuel production through reductive catalysis, although some recent studies reversed this concept and instead use the microbe as an electron donor.19 The components discussed in this review are broadly applicable to both designs.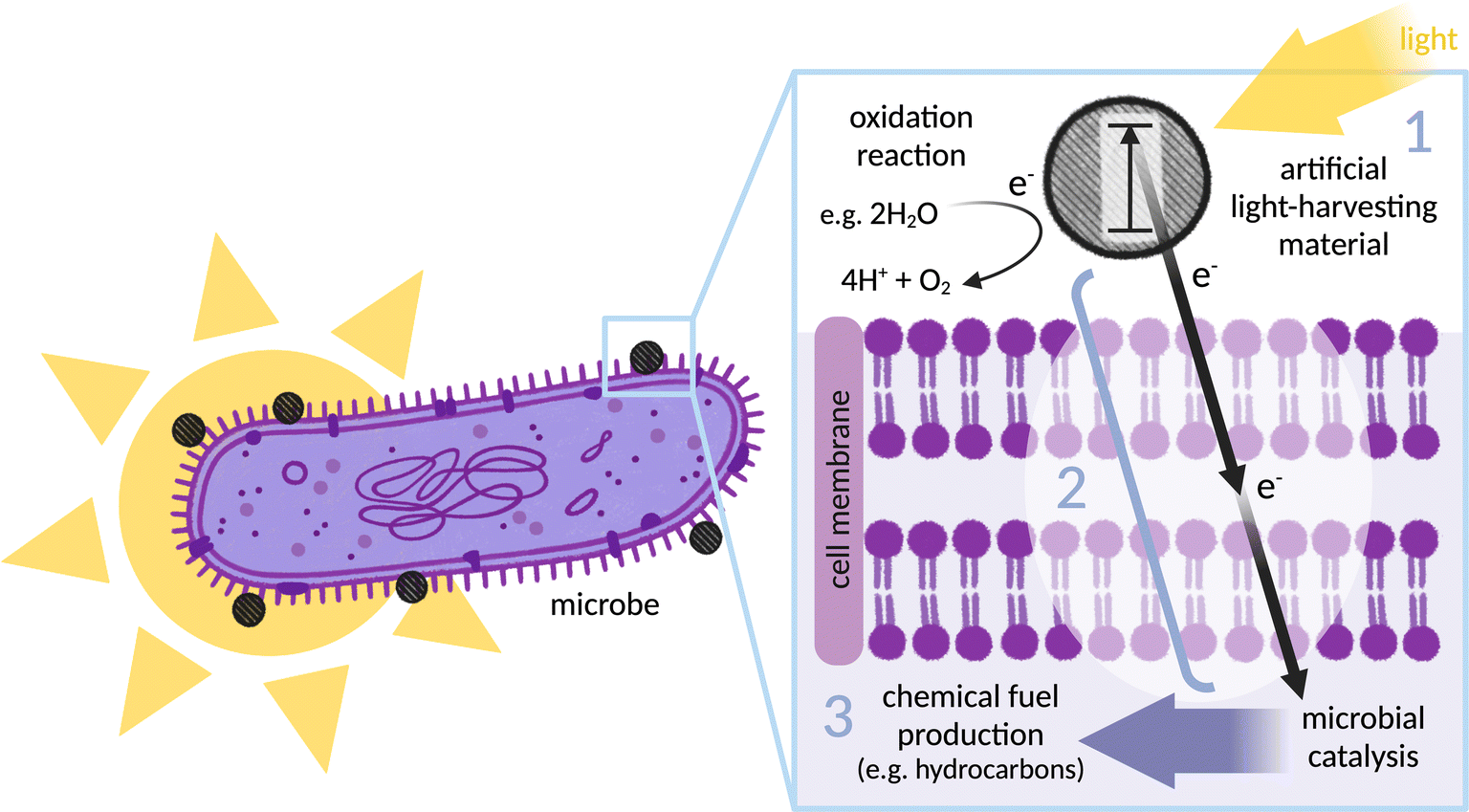 | ||
| Fig. 1 Illustration of the biohybrid concept (in this case using the microbe as a site of reductive catalysis and fuel production): an illuminated (Gram-negative) bacterium, with an inset showing a light-harvesting nanoparticle located on the outer membrane. (1) Artificial light-harvesting material (grey dashed circle) transitions to excited state on illumination: schematically indicated is an electron (e−) that moves into a higher energy orbital (in a molecular photosensitiser) or from the valence band to the conduction band (in a nanoparticle). (2) Electron transfer between the light-harvester and the site of catalysis: photoelectrons must be generated in close proximity to the microbe before passing through the microbial membrane to drive intracellular catalysis. (3) Microbial catalysis: the organism is selected or engineered to express enzymes that catalyse production of a desired chemical product. Finally, the light-harvester must be replenished by an oxidation reaction to complete the redox cycle, here indicated by catalytic water oxidation. | ||
A wide range of artificial light-harvesters have been applied, including organic and inorganic dyes, nanomaterials, and organic semiconductors.12,20,21 It is essential to consider the compatibility of these materials with the microbes involved in each system, along with characteristics such as band gap, redox potential, excited state lifetime, stability, and ease of chemical modification. Designing an interaction between the light-harvester and microbe will rely heavily on the mode by which electrons are able to be internalised. Several successful biohybrid systems for fuel production have not fully characterised their photosensitiser–microbe interface for electron transfer, limiting both controllability and optimisability. Other biohybrid systems make use of redox mediators.22–26 Mediators are membrane soluble electron carrier molecules that can shuttle electrons between the extracellular environment and the cytoplasm. While this concept works in systems that involve a sacrificial electron donor (SED) to regenerate the light-harvesting material and is valuable at the research stage, systems addressing the ultimate vision of catalysing an oxidative half reaction with a reactant other than a SED could be short-circuited in the presence of redox mediator. Separating the oxidative and reductive half-reaction in different compartments with macroscopic (photo)electrodes would present a potential solution to this short-circuiting. However, there are several disadvantages to these assemblies of macroscopic wiring: they are technically more complicated, and rely on electrodes and ion-exchange membranes which can be both toxic and expensive. Maintaining the neutral pH required by biohybrids is difficult in a set-up where mass transport can create large pH gradients at the electrode interface. Moreover, the single compartment aqueous systems discussed in this perspective are intrinsically compatible with tubular photobioreactors,27 making scalable solar light harvesting relatively easy. For a self-generating system, it is also important to consider that growing microorganisms is more controllable in solution than on electrodes.
The case will be presented for exploiting the defined, naturally evolved, characterisable pathways of transmembrane electron transfer in exoelectrogenic bacteria as a simple method for electronically coupling extracellular photoelectrons with intracellular enzymes. Emerging molecular details of these pathways can provide context for informed advances. We argue here that rational design of a specific, engineered interaction at the abiotic–biotic interface is not only superior to interaction by diffusion but also fundamental for systems to be controllable, optimisable and scalable. This perspective presents a range of strategies used to couple photosensitisers with proteins, a discussion that aims to assist in designing the conjugation of photosensitisers to biological components at the surface of microbes.
Current approaches in biohybrid research
Current biohybrid systems can be classified broadly into two themes: (1) synthesised nanomaterials can be directly added to the growth-media of the bacteria and (2) nanoparticles can be bio-precipitated by the bacteria itself upon addition of starting materials. Furthermore and as mentioned above, a distinction can be made on whether or not redox mediators are used to shuttle electrons from the nanomaterial to the intracellular environment of the microbe. Each of these approaches has a distinct effect on the photocatalytic performance (refer to Table 1 for a summary of key features of the systems discussed, including performance parameters).| Photosensitiser | Microbe | Physical attachment | Electron transfer (ET) interface | System performance | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Material | Toxicity | Species | Gram +/− | Reaction | Quantum efficiency (QE)/quantum yield (QY) | Product yield | Light source | |||
| OF/PTP | N/A | R. palustris | − | H2 production | None | MV mediated ET from light-harvester to microbe | N/A | ∼1.25 μmol H2 produced in 2 h (20 μM OF, 20 μM PTP, 0.5 mM MV, OD660 = 1.0) | Visible light, 100 W m−2 | 22 |
| TiO2 NPs | N/A | Recombinant E. coli | − | H2 production | None | MV mediated ET from light-harvester to microbe | Apparent QY: 31.2% (350 nm) | 0.95 mmol H2 produced in 2 h (50 mg semiconductor, 5 mM MV, 100 mg wet cells) | Xenon lamp, 330 W | 23, 24 |
| Eosin Y/Ru(bpy)32+ | N/A | S. oneidensis | − | H2 production/CO2 fixation | None | MV mediated ET from light-harvester to microbe | Apparent QE (H2 production): 0.6 ± 0.1% (Eosin Y, 500 nm)/0.5 ± 0.1% (Ru(bpy)32+, 450 nm) | H2 produced in 40 min: ∼0.17 μmol (Eosin Y)/∼15 nmol (Ru(bpy)32+); (0.11 mM photosensitiser, 0.3 mM MV, OD590 = ∼0.25) | Visible light, 700 W m−2 | 25 |
| Eosin Y/Ru(bpy)32+ | N/A | Recombinant E. coli | − | H2 production | None | MV mediated ET from light-harvester to microbe | Apparent QY: 4.8% (Eosin Y, DQ mediator) | TON (μmol H2 mL−1 OD600−1): 10.2 ± 0.7 (Eosin Y, in 184 h)/6.2 ± 1.0 (Ru(bpy)32+, in 360 h); (100 μM photosensitiser, 1 mM MV, OD600 = 5) | White light, 4000 lx | 26 |
| CdS NPs | N/A | M. thermoacetica | − | CO2 fixation | Self-assembled (associated with microbe) | Undefined ET pathway | QY (acetate formation): 52 ± 17% (435-485 nm, 5 × 1013 photons cm−2 s−1) | ∼1.25 mM acetate produced in 3 days (see reference SI for sample preparation) | Xenon lamp, 75 W | 5 |
| CdS NPs | Activity of D. desulfuricans biohybrid (under illumination) decreased with Cd2+ concentrations above 3 mM | D. desulfuricans, C. freundii, S. oneidensis | − | H2 production | Self-assembled (associated with microbe) | Undefined ET pathway | Apparent QY (D. desulfuricans): 23% (+MV)/4% (no MV) | 55 μmol H2 produced in 100 h (D. desulfuricans); (3 mM Cd2+, 100 μmol Cys, 0.5 mM MV, dry cell weight 5.3 mg) | 445 nm LED, 0.42 W m−2 | 28 |
| CdS NPs | No cytotoxicity found in presence of CdS (tested under dark) | R. palustris | − | N2 fixation | Self-assembled (associated with microbe) | Cross-membrane interaction of nanoparticles proposed as transmembrane ET pathway | N/A | ∼0.6 μmol C2H4 produced (h−1 mg−1 cells) at 18 h (nearly 2-fold compared to natural cells); (see reference for details) | Visible light, 80 W m−2 | 29 |
| CdS NPs | Cell proliferation heavily inhibited by > 100 mM Cd2+ without cysteine, and >300 mM Cd2+ with 1 mM cysteine (light conditions unspecified) | E. coli | − | H2 production | Self-assembled (associated with microbe) | ET proposed directly between membrane-embedded CdS particles and intracellular enzymes | Apparent QE: 7.93% (470 nm)/9.59% (620 nm) | 400 μmol (additional) H2 produced in 3 h (0.3 mM Cd2+, 1 mM Cys) | Visible light, 2000 W m−2 | 30 |
| CdS NPs (+/− N-doping) | N/A | M. barkeri | N/A | CO2 fixation | Self-assembled (associated with microbe) | Transmembrane ET proposed via unidentified membrane proteins | QY (CH4 production): 39.04 ± 1.34% | 0.24 μmol CH4 produced (h−1); (0.6 mM Ni(0.75%): CdS semiconductor incubated with cells of OD600 = ∼0.2) | 395 ± 5 nm, 10 W m−2 | 31,32 |
| CdS NPs | Cytotoxicity was not determined, but significant levels of ROS were detected (under illumination) | S. oneidensis | − | H2 production | Self-assembled (associated with microbe) | Several ET pathways are possible | N/A | 362.44 ± 119.69 μmol H2 produced (mg−1 biohybrid) in 72 h (∼711-fold compared to natural cells); (see reference SI for sample preparation) | Visible light, 1000 W m−2 | 33 |
| CuInS2/ZnS | Cell viability unaffected by nanoparticles until at least 36h (under illumination) | S. oneidensis | − | H2 production | Nanoparticles localised to periplasm (non-specific) | Direct ET from periplasmic nanoparticles to intracellular enzymes | Apparent QE: 15.02% (475 nm) | 491.8 ± 26.6 μmol H2 produced in 9 h (8.6-fold compared to bare QDs); (28.1% cellular uptake efficiency of 100 mg ml−1 Cu QDs, OD600 = 1.0) | Visible light, 2750 W m−2 | 34 |
| CdS NPs | Cell viability in 1 mM CdS is ∼70%, which decreased further to ∼40% upon 24h irradiation (2 W cm−2) | S. ovata | − | CO2 fixation | CdS adhere to and encapsulate the bacteria (non-specific) | Transmembrane ET proposed via membrane-associated flavoprotein and ferredoxin | QY: 16.8 ± 9% | ∼40 mM acetate produced in 4.5 days, rotation of 0.5 day light/dark cycles; (1.0 mM CdS) | 400 ± 5 nm LED, 2.0 W m−2 | 35 |
| CdS NPs | N/A | C. autoethanogenum | + | CO2 fixation | Cell surface associated (non-specific) | Transmembrane ET proposed via metal-ion or flavin-binding proteins | N/A | 12.1 mM acetate produced in 72 h (3.8-fold compared light-deficient conditions); (0.28 mg L−1 CdS) | N/A | 36 |
| CdS NRs | C. necator withstands higher CdS concentration than other microbes (under illumination) | C. necator | − | CO2 fixation for bioplastic production | Direct physical contact observed in aggregates (non-specific) | Unclear ET pathway | N/A | 1.41 ± 0.13 g L−1 PHB produced in 120 h (∼2-fold compared to natural cells); (2 gL−1 CdS NR) | LED, 4200 lx | 37 |
| g-C3N4 | N/A | C. necator | − | CO2 fixation for bioplastic production | Direct physical contact observed in aggregates (non-specific) | Unclear ET pathway | QE (NADPH): 8.74 ± 0.70% | 6.73 ± 0.45 g L−1 PHB produced in 96 h (1.4-fold compared to natural cells); (0.5 g L−1 g-C3N4) | LED, 4200 lx | 38 |
| Cu2O/RGO nanosheet | N/A | S. oneidensis | − | H2 production | Nanoparticles and microbes networked via RGO (non-specific) | RGO provides ET pathway from nanoparticles to microbe; transmembrane ET via membrane cytochromes | N/A | 322.0 μmol H2 produced (g−1 Cu2O) in 4 h | Xenon lamp, 300 W | 39 |
| Photocatalytic sheet (see text) | N/A | S. ovata | − | CO2 fixation | Bacteria grow in close contact with nanoparticle clusters (non-specific) | Photocatalytic sheet generates H2 (utilised by bacteria) and electrons (transferred into bacteria via undefined pathway) | Apparent QY (acetate formation): 21.3% (420 ± 15 nm) | ∼9 mM acetate produced in 15 h (OD600 = 0.6) | Solar illumination, 1000 W m−2 | 40 |
Due to a lack of electronic interaction between photosensitisers and microbes, some systems require a redox mediator such as methyl viologen (MV) for electron transfer into bacteria. For example, a biohybrid system has been constructed in which the phototropic bacterium Rhodopseudomonas palustris is further photosensitised with conjugated polymers, oligofluorene (OF) and polythiophene (PTP).22 The natural H2 generation by these bacteria was increased when MV was added to mediate electron transfer from the conjugated polymers to the intracellular enzymes. MV has also been used to shuttle electrons between a TiO2 nanoparticle photosensitiser suspended in bacteria culture media with a recombinant strain of Escherichia coli. Within the E. coli microbe, electrons were transmitted to an exogenously expressed [FeFe]-hydrogenase derived from Clostridium acetobutylicum NBRC 13948 to produce H2.23,24 Photosensitisers Eosin Y and ruthenium-tris-2-2′-bipyridine (Ru(bpy)32+) have been coupled to Shewanella oneidensis MR-1 as a biocatalyst using MV, driving H2 generation along with the reduction of fumarate, pyruvate and CO2 to, respectively, succinate, lactate and formate.25 An E. coli system, expressing [FeFe]-hydrogenases for H2 production and using Eosin Y and Ru(bpy)32+ as photosensitisers, was enhanced by redox mediators MV and diquat derivative 1,1′-(1,3-propylene)-5,5′-dimethyl-2,2′-bipyridinium (DQ).26 Both mediators significantly improved the performance of the system although, due to its more negative reduction potential, a faster reaction rate was achieved using DQ.
Use of mediators is not only unsustainable due to potential toxicity, but also the lack of a defined interface between components. Moreover, as mentioned, when an oxidative half reaction is coupled to the biohybrid system to close the redox cycle in place of an SED, the free redox mediator can cause back reactions and short-circuit the electron for the reductive reaction. Therefore our view is that redox mediators, although a useful research tool, limit optimisation of electronic coupling and thus the longevity of the system.
Biohybrids based on the self-assembly of CdS nanoparticles precipate Cd2+ and S2− at the surface of bacteria. The first system to pioneer this method used self-photosensitisation of Moorella thermoacetica to drive solar-powered reduction of CO2 to acetic acid.5 Similar systems constructed with Desulfovibrio desulfuricans, Citrobacter freundii and S. oneidensis MR-1 were used for light-driven H2 generation, with D. desulfuricans-CdS displaying the highest activity.28 Quantum yields of 23% and 4% were obtained for this biohybrid, with and without the addition of MV respectively. The photoheterotrophic bacterium R. palustris was coated with CdS nanoparticles for N2 fixation.29 In this example, CdS has been shown to form clusters that are able to cross the cell membrane, leading to the assumption that this provides a feasible electron transfer pathway within the cell. In the E. coli-CdS biohybrid for H2 generation,30 nanoparticles are localised across the periplasmic space of the E. coli membrane. It was proposed that the intracellular surface of the CdS particles could interact with bioactive components and integrate into the hydrogen production pathway. Methanosarcina barkeri-CdS biohybrids were able to reduce CO2 to methane.31 The CH4 yield increased by approximately 250% when CdS were doped with Ni.32 The introduction of Ni dopant was proposed to suppress the recombination of electron–hole pairs in the biohybrid, leading to enhanced electron transfer. Additionally, Ni induces alterations in the metabolic state of M. barkeri, leading to increased expression of various proteins involved in electron transfer, energy conversion, and CO2 fixation. It was proposed in this study that photoelectron transfer for CO2 reduction occurred through membrane-bound proteins. However, the type of membrane protein was not identified. In a recent study involving S. oneidensis MR-1-CdS biohybrids for H2 generation, four possible electron transfer pathways were proposed.33 Hydrogen could be produced directly by the light-harvesting nanoparticles themselves, by hydrogenase enzymes, by nanoparticle-hydrogenase enzyme hybrids, or by electron transfer from the nanoparticles to intracellular hydrogenases through the outer membrane proteins. The complexity of these systems, and lack of clarity due to multiple possible routes, makes it difficult to ascertain precise electron transfer pathways.
Instead of nanoparticles self-assembled by the bacteria upon addition of starting materials, pre-synthesised nanoparticles (or other nano-scale materials) can be added directly to the bacteria media. For example, CuInS2/ZnS quantum dots (QDs) have been added to a S. oneidensis strain expressing periplasmic hydrogenases.34 The QDs can be taken up into the bacterial periplasm where they interact directly with the hydrogenases for H2 production. A Sporomusa ovata-CdS hybrid was used to convert CO2 to acetic acid. An electron transfer pathway has been proposed35 where photoelectrons are simultaneously accepted by the membrane-associated flavoprotein and ferrodoxin before being shuttled to the intracellular space via menaquinone. In a Clostridium autoethanogenum-CdS hybrid for CO2 reduction, it has been proposed that electrons generated from nanoparticles are largely transported to the intracellular matrix via metal ion or flavin-binding proteins.36 CdS nanorods37 and graphitic-carbon nitride (g-C3N4)38 were combined with Cupriavidus necator for the production of the bioplastic polyhydroxybutyrate (PHB) from CO2 or fructose. It was found that synthesised CdS nanorods with optimised shape and photoactivity improved the performance of the biohybrid system. However, the electron transfer pathway from photosensitiser to bacterium remains unclear. For other systems, more information about transmembrane electron transport is known. In a Cu2O/reduced graphene oxide (RGO)-S. oneidensis MR-1 biohybrid for H2 generation, Cu2O acted as the semiconductor for harvesting solar energy.39 RGO nanosheets provided a surface to enable efficient photoelectron collection from Cu2O and efficient electron distribution to the cells. When metal reducing proteins such as MtrA, MtrB, and MtrC/OmcA (described in detail below) were not expressed, H2 generation was significantly inhibited. This suggested that, in this case, these specific outer membrane proteins provided the main electron transfer pathway between the semiconductor and bacterium.
Most existing biohybrid systems are designed for reductive reactions that require SEDs to regenerate the photosensitiser. Systems capable of catalysing an oxidative reaction that do not require a costly SED are rare (see Fig. 1). A pioneering biohybrid system was recently developed which performs H2O oxidation and CO2 reduction simultaneously, without sacrificial reagents or organic mediators.40 In this system, the CO2-fixing acetogenic bacterium S. ovata is combined with a photocatalytic sheet of nanoparticle clusters (Cr2O3/Ru–SrTiO3:La,Rh|ITO|RuO2–BiVO4:Mo). Acetate and oxygen are produced by the biohybrid system using only sunlight, CO2 and H2O. A solar-to-acetate conversion efficiency of 0.7% was achieved at ambient conditions. S. ovata used both H2 and electrons derived directly from the photocatalytic sheet to reduce CO2 to acetate.
The formation of biohybrids using a variety of light-harvesters and many different microbes reflects positively on the robustness of this concept. However, a fundamental disadvantage of some biohybrid systems discussed in this section is the challenge to explain non-specific binding and unclear electron transfer pathways across microbial membranes. Systems for which these pathways are not built in by design require extensive characterisation to understand and improve function, and are at risk of operating in ways other than the desired solar-driven pathway. Our perspective is that this can be mitigated by rationally designing a known electron transfer route to reach the site of catalysis. Microorganisms that naturally contain transmembrane electron transfer machineries provide an elegant design solution for cellular internalisation of electrons, advancing biohybrid systems for which the electron transfer pathway can be more easily defined, understood, and optimised.
Natural electron transport pathways across the microbial membrane
In nature, exoelectrogenic microbes can respire using extracellular material as a terminal electron acceptor, while electrotrophs accept electrons from the environment for reductive metabolism.41,42 This highly specific functionality has allowed exoelectrogenic bacteria, archaea, microalgae and fungi to occupy extreme environmental niches, for example those rich in minerals and subject to anoxic conditions or fluctuating availability of terminal electron acceptors.42 Species across these different kingdoms of life have independently evolved mechanisms and machineries for the exchange of electrons between the intracellular and extracellular environments, commonly involving cross-membrane transport by redox mediators (Fig. 2a) and/or a chain of conductive cytochromes that act as wires through the membrane(s) and peptidoglycan cell wall (Fig. 2b and c). This natural function provides an excellent opportunity to exploit transmembrane electron transfer by coupling exoelectrogens to extracellular electron donors such as electrodes or photosensitisers. Exoelectrogens have been used extensively in systems of microbial electrosynthesis to power reductive metabolism for chemical production, and also within microbial fuel cells (MFCs), which harness electrons released from oxidative catabolism to produce an electrical current.43,44 Electron transfer mechanisms in bioelectrochemical systems have been comprehensively reviewed elsewhere.45,46 Here, we focus on the mechanistic details of electron transmission within examples of representative species, with an outlook towards integration into biohybrid solar fuel production systems.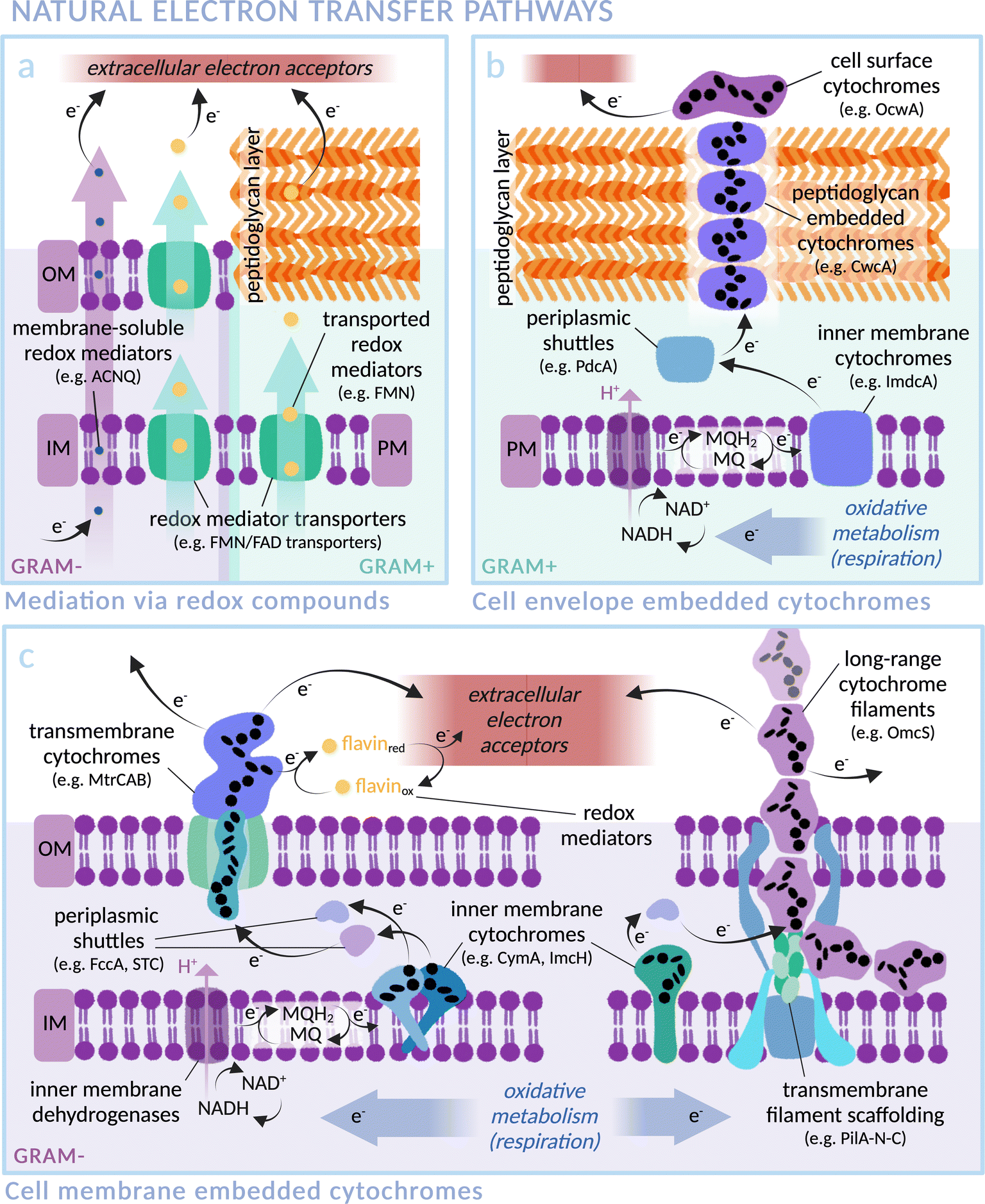 | ||
| Fig. 2 Transmembrane electron transfer in nature: electron transport machineries embedded in (Gram-negative) microbial cell membranes and (Gram-positive) peptidoglycan cell walls. Proteins are arranged according to current literature, with heme groups of select cytochromes indicated as black ellipses. (IM: Inner Membrane, OM: Outer Membrane, PM: Plasma Membrane). (a) Mediated electron transfer via redox compounds: membrane soluble compounds (i.e. phenazines in Pseudomonas aeruginosa,76,77 ACNQ in S. oneidensis78) or via transmembrane transporters (i.e. FAD/FMN transporters; RibU, FmnA, FmnB and PpIA in Gram-positive Listeria monocytogenes,79,80 periplasmic FccA and STC in Gram-negative S. oneidensis81). (b) Representative extracellular electron transfer pathway of the Gram-positive Thermincola ferriacetica (i.e. surface cytochrome OcwA, peptidoglycan-embedded CwcA filament, periplasmic PdcA and inner membrane ImdcA),82 with functional homology to e.g. TherJR proteins in T. potens and OmhA in carboxydothermus ferrireducens.72–75,82,83 (MQ = menaquinone; MQH2 = menaquinol) (c) Representative extracellular electron transfer pathway of the Gram-negative S. oneidensis (i.e. left: outer membrane MtrCAB, periplasmic FccA and STC, inner membrane CymA)47–49,81,84 and G. sulfurreducens (i.e. right: long-range OmcZ/OmcS cytochrome filaments, periplasmic PpcA and inner membrane ImcH).58,63,85 | ||
The exoelectrogen and facultative anaerobe S. oneidensis MR-1 is a versatile species able to respire using a wide range of terminal electron acceptors.47–49 With a fully sequenced and annotated genome,50 it has been used extensively in MFC studies, leading to a detailed characterisation of the protein machinery between the inner membrane quinone pool and the extracellular space (see Fig. 2c).43 The outer membrane spanning Mtr complex, made up of protein components MtrA, MtrB and MtrC, is integral within this pathway. Electrons from the quinone pool are first passed to inner membrane c-type cytochrome CymA. They are then shuttled by periplasmic electron carriers to cytochrome MtrA, a decaheme chain encased within the insulating pore of outer membrane spanning β-barrel MtrB, before emerging on the outside of the cell via lipid-anchored decaheme cytochrome MtrC.51 MtrC interacts with extracellular electron acceptors and passes electrons laterally to both other MtrC proteins and OmcA, a similar cytochrome in terms of structure and function. A specific arrangement of heme groups was revealed by the crystal structure of MtrC: the shape of a ‘staggered cross’.52 Eight heme groups form a roughly linear chain perpendicular to the cell membrane, with the two central hemes also part of an orthogonal tetraheme chain. Rapid electron exchange between these heme groups enables the “nanowire” function.53,54 The junction where the heme chains intersect, comprising co-planar and T-shaped heme stacking motifs, allows electrons derived from inside the cell to travel in three possible directions to surface cytochromes or extracellular acceptors with approximately equal efficiency.55 Controlling expression levels of MtrCAB has been shown to significantly alter extracellular electron transfer efficiency.56
Of key importance for biohybrid development is the possibility to reverse the direction of transmembrane electron flow in S. oneidensis; the membrane machinery can be induced to pass electrons from the extracellular space into the cytoplasm.52 The energetics of this reversed electron transfer through the MtrCAB pathway were analysed by measuring the electron flux between a counter electrode and a bacteria-coated working electrode, using the cathodic current caused by reduction of fumarate to succinate as a read-out.57 Electrons were successfully transferred to periplasmic fumarate reductase FccA. Situated at the surface, MtrC provides a potential entry point for injection of extracellular photoelectrons into the Mtr pathway within a biohybrid system.
In another well studied exoelectrogen Geobacter sulfurreducens,58 the protein complexes and transmembrane electron transport mechanisms are less well characterised, with two tandem four-gene clusters coding for an assortment of periplasmic and outer-membrane cytochromes thought to be similar in function to the Mtr proteins of S. oneidensis.59,60 Outer membrane proteins OmcZ, OmcS and OmcE are found in networks of protein filaments extending from the cell surface that facilitate long-range (>10 μm) interaction with extracellular electron acceptors (see Fig. 2c).61–65 Similarly to MtrC and OmcA of S. oneidensis, these Geobacter proteins have hemes packed in approximately linear chains. OmcE forms thinner filament structures than OmcS and OmcZ, and undergoes a greater number of post-translational modifications, indicating a variation in properties between filament types.65 The T-stacked hemes of OmcZ have been shown to pack at a closer distance than those of other Omc proteins in G. sulfurreducens, a feature hypothesised to be responsible for the exceptionally high electronic conductivity of this protein (OmcZ filaments have 1000-fold higher conductivity than OmcS). Additionally, a branched arrangement of heme groups is likely integral to the cross-linking function of OmcZ filaments in biofilm formation.61,62,66 Despite contrasting amino acid sequences, the physical arrangement of heme groups in an unbranched structure is highly conserved between OmcS and OmcE.65 Similarly, conductive filament structures recently noted in two major orders of archaea also exhibit non-homologous protein sequences yet comparable heme arrangements, indicating the convergent evolution of heme structures both within and across multiple species for optimised electron transfer.67 In the context of biohybrid design, it could be possible to wire photosensitisers directly to these filaments.
In contrast to S. oneidensis, for which only inner membrane cytochrome CymA is linked to extracellular electron transport, G. sulfurreducens expresses various different inner membrane cytochromes depending on the types and redox states of available electron acceptors.68 ImcH, for example, is required for electron transfer to high redox potential acceptors, but not those below −0.1 V versus standard hydrogen electrode (SHE). The opposite response pattern was found for CbcL,69 and others such as CbcBA are required for very specific redox potential ranges.70 Multiple available electron transfer pathways allow rapid switching in a fluctuating environment. Recently, the formation of intracytoplasmic membrane structures has been discovered.71 It is proposed that these extra inner membrane folds provide a larger surface area for electron transfer proteins under low energy conditions, in order to increase the quantity of electrons able to be transmitted from the cytoplasm. This native versatility makes G. sulfurreducens a highly promising candidate for scalable biohybrid systems. Multi-heme membrane cytochromes are also found in several Gram-positive bacteria, such as Thermincola potens, where a stacked chain of electron conduit proteins are hypothesised to transfer electrons through the cell wall (see Fig. 2b).72–75 Many exoelectrogenic archaea are also reliant on cytochromes for transmembrane electron transfer although, intriguingly, there are examples of archaea that are able to pass electrons to other microbes but do not express multi-heme proteins, indicating alternative electron transfer pathways in nature.42 Despite the gaps in knowledge that remain for many exoelectrogenic species, the certainty that there exists a natural transmembrane electron transfer pathway to be harnessed makes this class of microbes a promising choice for biohybrid systems.
Though the mechanistic details of electron transfer in exoelectrogens are continually emerging, injecting electrons into the cell is not so simple as directly reversing the natural pathway; some research has shown that there may be routes which bypass the Mtr pathway.86 Engineering exoelectrogens to enhance transmembrane electron transport could improve future systems.87 As well as further characterisation of these machineries,57,88 the interface between photosensitiser and microbe should become a major complimentary research focus. Direct electron injection is a crucial and under-researched stage of electron transfer in biohybrid design. Rational design of this interface is required, taking into account both physical attachment and electron transfer between components.
Rational design of the photosensitiser-protein interface
Many current biohybrid systems are limited by a lack of molecular design and control at the photosensitiser–microbe interface.17 Here, we highlight a range of research that would be valuable to explore when designing this essential interface: hybrid proteins. These are systems in which a photosensitiser has been specifically bound to a protein, often an isolated redox enzyme, to create a direct and defined path of electron transfer.16,89 Fast electron transfer is necessary for the optimisation of biohybrid systems. This needs precise engineering at the molecular level; according to the Moser–Dutton ruler, a distance of ≤14 Å is required between the photosensitiser and the distal redox co-factor of the enzyme.90[NiFeSe]-hydrogenase enzymes91,92 from Desulfomicrobium baculatum, among others, have been photosensitised using ruthenium-dye-sensitised titanium oxide nanoparticles.93 Attachment of TiO2 nanoparticles to hydrogenase enzymes was inspired by the stable electrochemistry observed when the enzyme was adsorbed onto a TiO2-ITO electrode in comparison to graphite electrodes.94 Coupling TiO2 nanoparticles to a ruthenium photosensitiser expanded the range of photoabsorption into the visible range, with TiO2 extending the lifetime of the charge separated state and acting as an attachment surface for the hydrogenase. There are several systems which use the ability of TiO2 to adsorb enzymes, including a CO2-reducing enzyme (CODH) from Carboxydothermus hydrogenoformans95 and formate dehydrogenase (FDH) from Desulfovibrio vulgaris.96 TiO2-adsorbed CODH has been coupled to light absorbing Ag nanoclusters stabilised with polymethacrylic acid.97 In addition to Ru-sensitised TiO2, biohybrid systems are established for polymeric carbon nitride (CNX), CNX-TiO2 and Eosin Y as photosensitisers.98–100 A [NiFeSe]-hydrogenase system photosensitised with CNX, despite a weaker photosensitiser-enzyme interaction than for Ru-sensitised TiO2, demonstrated much higher stability and remained active for 2 days compared to 8 hours.94,98 The interaction was enhanced by adding TiO2 as a surface for adsorption (CNX-TiO2).99 However, TiO2 systems have the disadvantage of generating toxic reactive oxygen species (ROS) in the presence of O2. To overcome this limitation, Eosin Y was introduced as a photosensitiser. This organic dye actively protects the enzyme by reacting with O2 to create a pseudo-anaerobic environment under air. Even in atmospheric conditions, the light-driven hydrogenase was able to maintain ∼11% of the photoactivity measured under anaerobic conditions.100 In an alternative approach, CNX has been recently demonstrated to exhibit a strong electrostatic interaction with an [FeFe]-hydrogenase from Clostridium pasteurianum101 without the need for TiO2.
Light-harvesting nanoparticles such as carbon dots (CDs) have been functionalised with surface groups to engineer the nature of their interaction with an enzyme. Likely involving hydrogen bonding, FDH was found to bind CDs functionalised with either positive (-NHMe2+) or negative (–COO−) groups in equivalent amounts.102 However, the CD surface chemistry affects their localisation on the protein surface and thereby (presumably) the kinetics of photoelectron transfer: CDs with positive surface groups were able to drive CO2 reduction to formate in the presence of a sacrificial electron donor, while CDs with negative surface groups were not. This demonstrates how integral the positioning of the photosensitiser component is for successful electron transfer in biohybrid systems. Another study investigated the interaction of functionalised CDs with FccA and [NiFeSe]-hydrogenase enzymes and drew a similar conclusion.103 In a different example, the electrostatic interaction between CDs and an [FeFe]-hydrogenase enzyme was disrupted by the addition of ethylenediaminetetraacetic acid (EDTA) as an SED, with hydrogen evolution after 1 hour around 4.4-fold higher when replaced by a different SED: triethanolamine (TEOA).104
The nature of electrostatic self-assembly between QDs and enzymes has been analysed in detail for CdS and CdTe nanoparticles coupled to hydrogenase enzymes.105 Functionalisation of these nanoparticles with a self-assembled monolayer of 3-mercaptopropionic acid (3-MPA) allowed solvation in an aqueous environment and, due to the negative charge of exposed carboxylate groups, facilitated interaction with positively charged areas of the protein surface. A C. acetobutylicum [Fe–Fe]-hydrogenase, with surface-localised [FeS] clusters accessible for direct electron transfer, was adsorbed to 3-MPA-functionalised CdS nanocrystals.106 The rate of catalysis was shown to increase with hydrogenase only up to a certain enzyme:CdS ratio before declining, an effect attributed to back electron transfer due to steric hindrance of the hydrogenase crowding. An optimised ratio of 0.67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for hydrogenase:CdS achieved ∼20% quantum yield for H2 production. This value remained constant over a range of light intensities, indicating that the limiting step was not the rate of electron transfer or catalysis, but rather the rate of photoelectron generation. The quantum yield could thus be improved in this example by increasing the absorbance range of the photosensitiser component, but the electron transfer interface itself is considered highly effective. 3-MPA-functionalised CdS nanorods/nanocrystals and other QDs have also been adsorbed to an Azotobacter vinelandii nitrogenase [Mo–Fe] catalytic centre.107,108 The production yield when using CdS nanorods as a photosensitiser far outcompeted that of Ru-based compounds for this example, indicating the extent to which the photosensitiser component can affect the functionality of a catalyst.109 As described for hydrogenase systems, the nature of physical attachment between functionalised CdS photosensitisers and the protein is attributed to self-assembly via surface electrostatics.106,110 Physical binding of CdS to the enzyme in solution was demonstrated using dynamic light scattering (DLS), with the hybrid exhibiting a significantly higher average hydrodynamic diameter than either individual component. Characterisation of the electron transfer interface was investigated extensively in terms of quantum efficiencies and kinetics, with formation of a single complex concluded to be integral for efficient electron transfer and product formation.111–113 3-MPA-coated CdS nanorods have also been coupled to an oxidoreductase enzyme from Magnetococcus marinus MC-1.114
:1 for hydrogenase:CdS achieved ∼20% quantum yield for H2 production. This value remained constant over a range of light intensities, indicating that the limiting step was not the rate of electron transfer or catalysis, but rather the rate of photoelectron generation. The quantum yield could thus be improved in this example by increasing the absorbance range of the photosensitiser component, but the electron transfer interface itself is considered highly effective. 3-MPA-functionalised CdS nanorods/nanocrystals and other QDs have also been adsorbed to an Azotobacter vinelandii nitrogenase [Mo–Fe] catalytic centre.107,108 The production yield when using CdS nanorods as a photosensitiser far outcompeted that of Ru-based compounds for this example, indicating the extent to which the photosensitiser component can affect the functionality of a catalyst.109 As described for hydrogenase systems, the nature of physical attachment between functionalised CdS photosensitisers and the protein is attributed to self-assembly via surface electrostatics.106,110 Physical binding of CdS to the enzyme in solution was demonstrated using dynamic light scattering (DLS), with the hybrid exhibiting a significantly higher average hydrodynamic diameter than either individual component. Characterisation of the electron transfer interface was investigated extensively in terms of quantum efficiencies and kinetics, with formation of a single complex concluded to be integral for efficient electron transfer and product formation.111–113 3-MPA-coated CdS nanorods have also been coupled to an oxidoreductase enzyme from Magnetococcus marinus MC-1.114
H2 production was achieved in a similar electrostatically conjugated hydrogenase system, with 3-MPA-functionalised CdTe nanocrystals as the light-harvesting component.110 The CdTe-hydrogenase hybrid was confirmed to form a stable single complex using Native Polyacrylamide Gel Electrophoresis (PAGE). With this technique, CdTe nanocrystals were clearly seen to form a fluorescent hybrid species with hydrogenase on the gel. The kinetics of adsorption were analysed: despite being weaker than a true covalent bond, the interaction was concluded to form a stable complex. Electron transfer between components was also investigated. Contrary to expectations, it was found that mixing at a lower hydrogenase:CdTe ratio resulted in much higher electron transfer efficiency and light-driven H2 production rate. These observations indicate that in this case, a lower ratio of enzyme per nanocrystal made for a more optimal system, a result attributed to high enzyme coverage forcing hydrogenases into an orientation less effective for electron transfer.110 A study involving uncoated CdS nanomaterials coupled to CODH additionally highlights the effect nanoparticle shape and size can have on catalytic activity.115 Due to the importance of this interface for electron transfer efficiency, the position, size and quantity of light-harvesters per microbe are important factors to optimise.
Closing the redox cycle is an important challenge to address. Very few examples of this have been achieved across any system of biohybrid photocatalysis, with water oxidation an obvious target when considering fuel production on a global scale.40 However, other substrate candidates aside from water are feasible for oxidation in a scaled-up system, for instance biomass (lignocellulose),116 which is already widely available and circular, or plastic117 (which will likewise become circular).118,119 Recently, the reductive chemistry of a nanoparticle-enzyme biohybrid was successfully coupled to a value-adding oxidative reaction: TiO2-adsorbed FDH enzymes catalysed reduction of CO2 to formate, while the TiO2 photosensitiser was regenerated through oxidation of glucose to arabinose and formate.116 This example neatly demonstrates the ability to design a system which couples two redox reactions and generates multiple desirable products, eliminating the need for a costly and thus uneconomical SED. Compartmentalised photocatalysis becomes highly important in these systems to eliminate back-reactions.
While the examples above are successful electron transfer interfaces, coupling methods that rely on non-specific adsorption are not inherently transferable to whole-cell biohybrids due to the complexity of the microbial surface.120,121 An approach where the photosensitiser is covalently tethered to the protein might solve this issue, and has two major advantages: (a) the method can be more easily applied to any system with chemically adjustable components (e.g. chemical dyes and functionalised nanoparticles), and (b) it is possible to target photosensitisers to a specific site, thus allowing optimisation of the electron transfer interface. CdS/CdSe quantum rods coated with carboxylic acid-containing ligands have been covalently bound to a [NiFe]-hydrogenase from Aquifex aeolicus via amide bond formation with lysine residues.122 Biohybrid complex formation was verified by both colocalisation of nanoparticle and enzyme on native PAGE and peptide mass fingerprint. Expanding this method to bind a targeted site on the enzyme would allow further optimisation of the electron transfer interface (see Fig. 3). A cysteine-binding Ru-based photosensitiser has been covalently bound to an engineered cysteine residue in cytochrome P450 BM3 from Bacillus megaterium, driving integration of atmospheric O2 into hydrocarbons within the heme-based catalytic centre of the enzyme.123–125 Targeting the Ru electron donor site to an optimised location for electron transfer required comparing cysteine mutants at several positions on the enzyme, each of which significantly affected catalytic rate. This highlights the sensitivity of the abiotic–biotic interface on electron transfer, and reiterates the importance of designing and investigating an optimal physical interaction for efficient electron transfer.
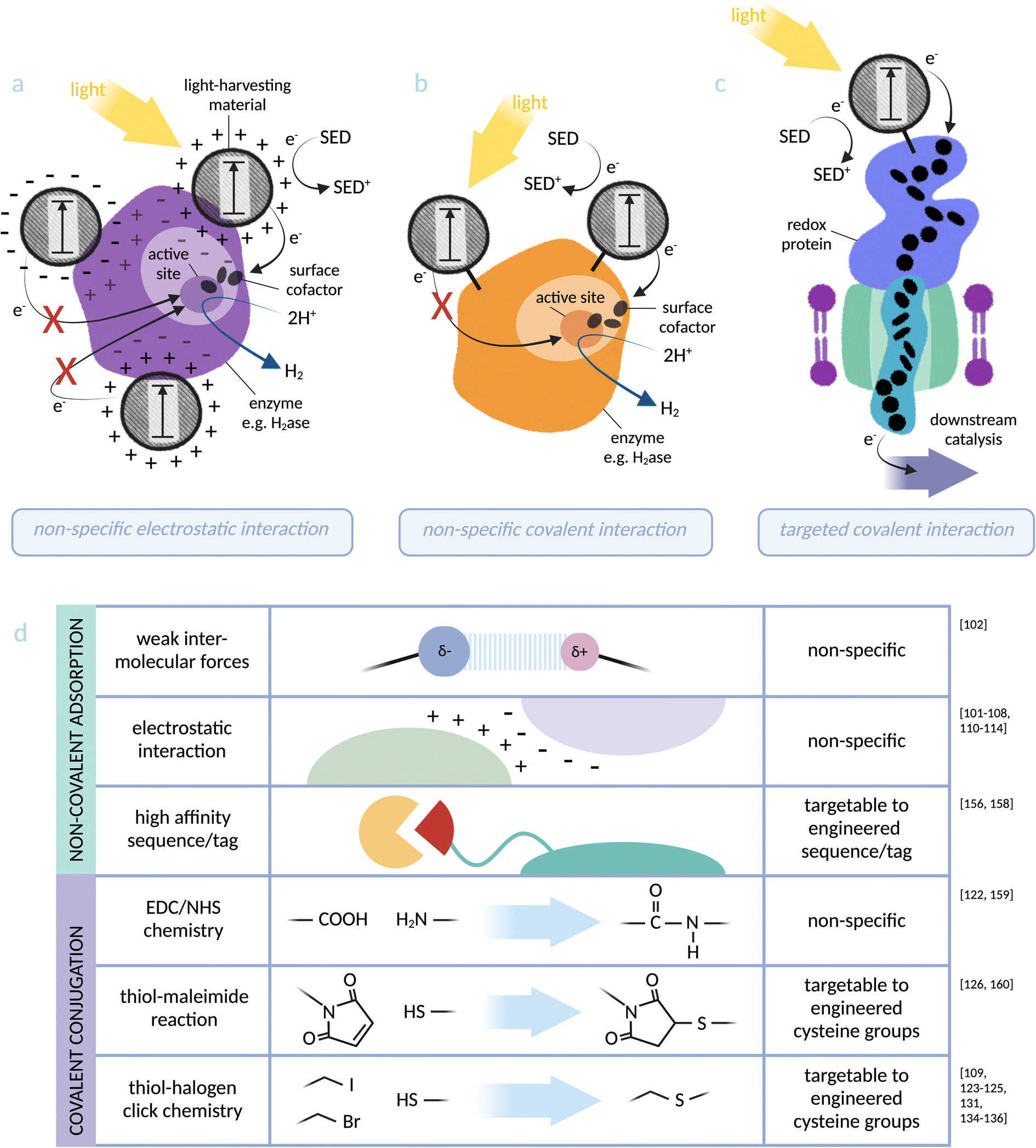 | ||
| Fig. 3 Photosensitiser-protein hybrid interfaces: (a) non-specific electrostatic interaction between a light-harvesting material and an enzyme (based on [NiFeSe]-hydrogenase):103 distance dependency of electron transfer to the catalytic centre makes catalytic activity directly dependent on the location of the nanoparticle, which can be investigated but not controlled using this binding method. (b) Non-specific covalent binding of a light-harvesting material to lysine residues at an enzyme surface (based on [NiFe]-hydrogenase):122 proximity to the active site can again be investigated but not controlled via this method. (c) Specific covalent conjugation between a light-harvesting material and a transmembrane cytochrome (based on MtrCAB from S. oneidensis):126 binding directly via an engineered cysteine residue allows targeting to an optimal site for electron injection into the redox protein. (d) Summary of the physical photosensitiser-protein binding methods discussed in this perspective. Note: some studies91–93,95–99,115,116,127,128,130 do not distinguish between weak intermolecular forces (such as van der Waals and hydrogen bonding) and stronger electrostatic interactions and these have not been included in the figure. | ||
Coupling to membrane cytochromes of exoelectrogenic microbes
Building on the concept of interfacing redox enzymes with photosensitisers, a small number of studies have demonstrated binding light-harvesting materials directly to the membrane cytochromes of exoelectrogenic bacteria. While these membrane proteins are not enzymes themselves, the concept of creating an electron transfer interface from light absorber to redox protein for downstream catalysis is translatable between the two research areas. Coupling transmembrane cytochromes to light-harvesters and achieving electron transfer provides a convincing proof-of-principle for the ultimate application of photosensitising microbes.As discussed, exoelectrogenic bacteria have the ability to transmit electrons between intracellular and extracellular environments.48 The transmembrane protein complex MtrCAB from S. oneidensis MR-1 provides the point of entry for extracellular electrons to access intracellular electron transfer pathways.43,52 A selection of light-harvesting nanoparticles were tested for adsorption with MtrC, the outward-facing protein subunit, assembled as a monolayer on electrode surfaces: RuP–TiO2–COO− (negatively charged), RuP–TiO2–NH3+ (positively charged), CdS–COO– and CdS–N(CH3)2H+.127,128 It was found that binding of MtrC was highest and most rigid for RuP–TiO2–COO−. The photocurrent was linearly dependent on light intensity, indicating that the electron transfer interface was not limiting. However, variation in binding properties between these different nanoparticles demonstrates the difficulty in predicting interactions between components. Non-specific adsorption (see Fig. 3) is inherently difficult to predict or control. These results reinforce the perspective that binding methods with built-in specificity, such as targeted covalent binding, are essential for guaranteeing an efficient electron transfer interface.
A recent study from our group has addressed this challenge by rationally designing the covalent conjugation of MtrC with light-harvesting CD nanoparticles.126,129 Previously, we coupled MtrC to Ru-based photosensitisers (a) adsorbed on TiO2 nanoparticles130 and (b) via site-specific covalent labelling.131 The latter achieved ultrafast electron transfer on the ps timescale. CDs were introduced to this biohybrid design as a comparably sustainable source of light-harvester. The surface groups of CDs are amenable to chemical modification, and they have been previously demonstrated to pass electrons to the MtrCAB complex when mixed free in solution.132 Covalent binding was achieved through functionalisation of carboxylic acid residues at the CD surface with maleimide groups, followed by reaction with an engineered cysteine residue of MtrC. The covalent conjugation of photosensitiser-protein components was verified on native PAGE, where the conjugated protein was visualised to carry CD fluorescence and migrate as a wider, more diffuse band (indicating addition of the heterogeneously sized CD particles). Electron (or energy) transfer was shown to occur between components: the fluorescence lifetime of the CD became shorter when interfaced with MtrC. This chemical modification of MtrC demonstrates the use of sustainably produced, low-cost components to create a system that could in theory be translated into a whole-cell biohybrid system using S. oneidensis MR-1 for solar fuel production.132
PpcA, a periplasmic cytochrome from exoelectrogen G. sulfurreducens,133 has been conjugated to cysteine-binding Ru-based photosensitisers for the same purpose. In this study, site-directed mutagenesis was used to introduce non-native cysteine residues to PpcA as a model for targeted conjugation.134 The aim was to form chiral linkages with photosensitiser components, allowing sterically specific conformations that mimic the physically constrained photosystem structures in nature. Structural aspects of the conjugates were analysed alongside rates of photo-induced electron transfer (PET). With PET rates of 6 ps, 130 ps and 35 ns for 3 different mutants, a clear preference for location of electron injection was demonstrated. It was also shown that certain mutants favoured one enantiomer of the chiral photosensitisers, indicating different environmental constraints at each position. A further study validated the conclusion that the tertiary structure of the conjugate directly controls efficiency of electron transfer, this time focussing on comparing cysteine positions introduced directly into heme binding motifs.135 Similar conclusions were reached in a study of Ru-dye sensitised STC, a periplasmic cytochrome from S. oneidensis MR-1.136 These detailed analyses of geometries, interactions and flexibility at the site of conjugation are invaluable for rational design of biohybrids such as these going forwards. While PpcA and STC are less likely to be candidates for translating into a microbial biohybrid system, being located in the periplasm, these proof-of-principle studies lay the foundations for fundamental design principles: precise structural control of the point of electron injection from light-harvesting materials to the heme chains of cytochrome proteins.
The examples discussed above have been chosen to highlight advantages and disadvantages of various protein conjugation methods for translation into whole-cell biohybrid systems. Interfacing photosensitisers with redox proteins requires nuance and rational design. An optimum conjugation method will depend greatly on the nature of the components and the desired interaction. To achieve competitive electron transfer efficiencies in a biohybrid system, an evidence-based approach to directing the site of electron exchange between components is required. While it could be argued that there currently exists too little characterisation for strategic design in this manner, we propose that research should continue to pursue a deeper understanding of prospective biohybrid proteins and their interactions with photosensitisers to accelerate the innovation of successful system designs in the future.
Toxicity of non-biological components
The toxicity of synthetic light-harvesters on bacteria, and their effect on the environment when applied at a large scale, both play a crucial role in ensuring longevity, sustainability and feasibility of biohybrid systems. This aspect has received limited attention in other reviews on biohybrid systems and is not consistently mentioned in studies. As discussed previously, many different light-harvesters have been employed in biohybrid systems (Table 1). Toxicity is dependent on the type of light-harvester, the concentration, the species of microorganism, and conditions such as light intensity. In this perspective, we aim to provide a more thorough discussion of this critical factor (see Table 1 for a summary of known photosensitiser toxicities under specific conditions, including light/dark, for the examples of solar-driven biohybrids discussed).Biohybrid systems using water soluble dyes as a photosensitiser such as Eosin Y and ruthenium complexes often use a redox mediator to shuttle the electron into the bacteria.22–25,100 MV has been identified as toxic to microorganisms. Alongside other disadvantages previously discussed, this factor makes it unsuitable for long term use. Additionally, organic dye Eosin Y has been found to be harmful for both microorganisms and human health, exhibiting increased production of ROS under visible light illumination.137,138 Ruthenium photosensitisers are specifically used for anticancer therapy and as antimicrobial agents due to their cytotoxicity.139,140 Metal-based photosensitisers in general can often result in the incidental dissolution and accumulation of toxic metal ions. The toxicity of these components in the context of specific microbial biohybrid systems, however, is not well researched.
QDs are semiconducting nanoparticles that have unique photophysical properties. The nature of the primary material, the size and shape of the nanoparticles and their specific surface functionalisation are all factors that play an essential role in their potential toxicity.141 However, the precise toxicity of nanomaterials to microorganisms and the natural environment is a serious concern that remains largely undetermined, despite extensive research in this area.142,143 Cadmium-containing QDs such as CdS and CdTe are most commonly used and have been applied in biohybrid systems involving both redox enzymes and bacteria.5,33,106,107,110 Cd2+ exhibits high toxicity to a range of microorganisms including bacteria, algae and fungi.144 CdS nanoparticles can either be directly added to the bacteria, or formed via bio-precipitation on the bacteria in the presence of cysteine. Cell proliferation for E. coli was shown to be heavily inhibited with a Cd2+ concentration higher than 0.1 mM in the absence of cysteine.30 On addition of cysteine, cell growth significantly increased for Cd2+ concentrations up to 0.4 mM. However, despite this reduction in toxicity, cysteine addition to biohybrid systems has disadvantages: due to relying on excess thiol for stabilisation, there is no obvious way to couple bio-precipitated CdS systems to a meaningful oxidative reaction, and additionally cysteine can itself be metabolised by some microbial systems.145
Various QDs added directly in solution (CdTe/CdS/TGA, CdTe/CdS/cysteamine, CdTe and CuInS2/ZnS/PMAL) have been evaluated for their nanotoxicology with S. oneidensis MR-1, using a colony-forming units method and a fluorescence viability assay.146 CdTe and CuInS2/ZnS/PMAL QDs displayed no toxicity, but CdTe/CdS/TGA, CdTe/CdS/cysteamine QDs significantly inhibited the bacteria growth. In another study, seven different core–shell QDs were tested for toxicity to non-photosynthetic bacteria (A. vinelandii and C. necator) for the purpose of solar fuels and chemicals generation.147 The surfaces of the QDs were each capped with ligands that affected surface charge: negatively charged MPA, positively charged cysteamine (CA), and zwitterionic cysteine. The Cys-capped QDs showed no inhibition on bacteria growth. However, MPA-capped QDs impaired growth moderately, and CA-capped QDs strongly, clearly illustrating how surface chemistry affects the toxicity of nanoparticles. It has also been demonstrated that activity in D. desulfuricans-CdS biohybrids decreased when cadmium concentrations surpassed 3 mM, likely due to toxicity.28 In the S. ovata-CdS hybrid system, a colony-forming unit assay showed that cell viability in the CdS-containing biohybrid was decreased compared to that of cells without CdS, further indicating toxicity of Cd-based photosensitisers.35 Toxicity of CdS was tested on the microorganisms C. necator, Saccharomyces cerevisiae, Gluconacetobacter xylinus, and Anabaena sp. PCC7120 using different concentrations of CdS nanorods, with toxicity observed for all except C. necator.37 Nanoparticles can also have toxic effects on the metabolism of different microbes, including protein expression. An example is Xanthobacter autotrophicus in which 727 proteins were upregulated and 53 downregulated upon addition of CdTe nanoparticles.148 Proteomic and metabolic analyses show that significant alterations occurred for electron transport chain and redox signalling pathways. Expression of N2-fixation related proteins such as nitrogenases was increased, while expression of ATP synthase and overall ATP concentration was lowered. Interaction between S. oneidensis MR-1 and several nanoparticles including CdS has been shown to increase biofilm formation.149 Addition of TiO2 has been shown to increase riboflavin secretion.150 In conclusion, it is difficult to predict the toxic or metabolic effects of different light-harvesting materials on different organisms.
Gold nanoclusters (AuNCs), considered to be a less toxic material than cadmium-containing QDs, were used as photosensitisers (accumulating intracellularly) for the acetogenic bacterium M. thermoacetica.151 Both with and without the presence of AuNC, the M. thermoacetica system could maintain a relatively high proliferation rate and cell viability until the fourth day, demonstrating biocompatibility of these AuNCs. Recently, carbon-based nanomaterials such as CNX and CDs have been applied to biohybrid systems for photobiocatalysis.98,99,103,104,152 CDs are generally considered to be environmentally benign. Biocompatibility with S. oneidensis MR-1 was evaluated by assessing the viability of bacteria incubated with CDs. Cell viability was found to be maintained >96% after 94 h, confirming the high compatibility and low cytotoxicity of the CDs for this bacterium. They have, however, been implicated in increased intracellular signaling.153 Organic semiconductors such as perylene diimide derivative (PDI) and poly(fluorene-co-phenylene) (PFP) are also considered non-toxic as a photosensitiser for biohybrid semi-artificial photosystems.154
In comparison to organic dyes, nanoparticles offer greater potential for adjustment of both their band gap and surface chemistry. Their high stability and durability are advantageous. However, the impact of nanoparticles both on the environment and the bacteria remains uncertain, with toxicity varying depending on the species of microbe involved. To create a more sustainable system, it is imperative to focus on development of benign photosensitisers such as gold nanoclusters, organic semiconductors, and carbon-based nanomaterials which exhibit biocompatibility and lower toxicity.
Outlook
The aim of this perspective is to pitch a forward-looking “road-map” for the future of whole-cell solar biohybrids, highlighting a range of research areas that can be explored for solutions to limitations of current systems. Exploiting microbes with natural pathways of transmembrane electron transfer provides a characterisable route for internalisation of photoelectrons, and strategies for conjugating photosensitisers to enzymes can be explored for transferable electron transfer pathways that facilitate a controllable and optimisable interface. Rather than providing a conclusive set of studies, we propose that the field continues to examine research across these areas for inspiration, harnessing new materials, components and conjugation methods from a wide range of studies for application in the whole-cell biohybrid vision.The main focus has been to highlight methodology used to anchor two redox components together in a such a way that electron transfer is achieved. However, it is also possible to examine protein conjugation methods for purposes such as energy transfer. While electron transfer requires a distance of ≤14 Å between redox centres (according to the Moser–Dutton ruler), energy transfer requires a distance of a 100–500 Å. Physical conjugation methods can nonetheless be explored. A light-harvesting reaction centre (RC) from Rhodobacter sphaeroides has been hybridised with CdTe QDs, enabling energy transfer from QDs to occur in a similar way to antennae pigments in nature.155–157 CdTe QDs were bound with high affinity to an engineered His-tag with a modifiable linker sequence at the RC surface.156,158 Methods to conjugate light-absorbing organic dyes and fluorophores have been established that exploit the presence of available cysteine and lysine residues on the RC. A synthetic aryleneethynylene fluorophore, designed to act as an antenna pigment, was covalently bound to lysine residues via a succinimidyl ester group at a ratio of 4.1 ± 0.3 fluorophores per protein.159 In another study, fluorescent dyes functionalised with maleimide groups were targeted to engineered cysteine residues.160 These types of studies highlight further translatable conjugation methods for targeting light-harvesters to specific locations on a protein.
In a different approach, Texas Red (TR)-modified phospholipid has been used for energy transfer to light-harvesting complex II (LHCII) from spinach.161,162 In this system, the light-havester is not coupled to a protein, but to the membrane. Further studies expand the concept to include dye-conjugated phospholipids ATTO647N-DOPE and Cyanine-7-DOPE, and lipophilic dialkylcarbocyanine dyes DiI and DiR.163 This type of interaction demonstrates the possibility of designing an energy (or electron) transfer interface between membrane-anchored components via proximity rather than direct physical conjugation. However, organic dyes do not absorb a broad spectrum of light, and are less durable than alternative photosensitisers such as semi-conducting nanoparticles. COOH-coated semi-conducting graphitic CDs have been modified with a phospholipid.164 Chemically functionalising membrane components with nanoparticles such as this could be highly applicable to the overall biohybrid concept.
An alternative to labelling natural membrane components is to create synthetic membrane-insertable photosensitisers such as amphipathic fluorescent nanoparticles. Lipophilic CDs have been developed that undergo self-assembly into structures similar to liposomes (CDsomes).165 They have been used for selective labelling of membrane structures in live mammalian cells, demonstrating high biocompatibility and negligible cytotoxicity, along with the key ability to insert into natural cellular membrane environments. Due to their ability to photoluminesce, these CDs have additionally been investigated for use in photocatalysis.166 At 365 nm, CDsomes exhibit a fluorescent on-state, and at 530 nm a non-fluorescent off-state. Using this photo-switchability, it was found that in the on-state catalysis of O2 to H2O2 was driven, and in the off-state ROS were generated. The principle of using a synthetic membrane-embedded CD for photochemistry is fascinating for further research. In another study, a synthetic transmembrane photosystem I mimic has been developed that, on excitation by visible light, is able to pass electrons to an organic dye.167 The oligofluorene chromophore structure spans the width of an average phospholipid bilayer, situating itself in a perpendicular orientation that mimics natural transmembrane structures. Photons are channelled to an energy acceptor, generating photoelectrons that can be transferred to acceptors at the membrane surface. Energy transfer between the synthetic light-harvester and organic dye Eosin Y was demonstrated in liposomes by measuring fluorescence emission quenching. Another system uses a perylene diamine chromophore that imbeds in liposomal membranes.168 Use of liposomes as a platform for energy transfer, electron transfer and chemical conversion is a branch of its own within the field of developing bio-based systems for solar fuel generation.169,170 Within the context of designing whole-cell systems, the relevance of synthetic membrane components such as these can only be judged by their ability to insert into the biological membranes of microbes and interact with native components. For many of these synthetic chromophores, this remains to be seen.
In conclusion, we propose that a broad range of research be used to inform the strategic design of biohybrid systems. The route of a photoelectron, from generation to intracellular catalysis, can be broken down into two main optimisable stages: the interface between photosensitiser and microbe, and internalisation of the electron by the microbe. Delving into the fundamental interactions between photosensitiser and redox protein components will reduce the unknowns when translated into larger, more complex systems. Using known methods for directly tethering key biohybrid components can allow control and optimisation of their interaction. In many cases, there is not yet enough understanding of how the building blocks interact: it is therefore of high importance to continue investigating and engineering these materials and interfaces, and to ultimately use this accumulated knowledge to establish blueprints for the future rational design of solar-driven biohybrids for fuel production.
Author contributions
Imogen L. Bishara Robertson: conceptualization, writing – original draft, writing – review & editing, and visualization; Huijie Zhang: conceptualization, writing – original draft, writing – review & editing, and visualization; Erwin Reisner: conceptualization, writing – review & editing, supervision, project administration and funding acquisition; Julea N. Butt: conceptualization, writing – review & editing, supervision, project administration, and funding acquisition; Lars J. C. Jeuken: conceptualization, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the UK Biotechnology and Biological Sciences Research Council for funding (No. BB/S002499/1, BB/S00159X/1, and BB/S000704/1). ER acknowledges a UKRI/ERC Advanced Grant (No. EP/X030563/1).References
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Interfacing nature's catalytic machinery with synthetic materials for semi-artificial photosynthesis, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- J. Ye, A. Hu, G. Ren, M. Chen, S. Zhou and Z. He, Biophotoelectrochemistry for renewable energy and environmental applications, iScience, 2021, 24, 102828 CrossRef CAS.
- L. T. Wey, J. M. Lawrence, X. Chen, R. Clark, D. J. Lea-Smith, J. Z. Zhang and C. J. Howe, A biophotoelectrochemical approach to unravelling the role of cyanobacterial cell structures in exoelectrogenesis, Electrochim. Acta, 2021, 395, 139214 CrossRef CAS.
- A. A. Krasnovsky and V. V. Nikandrov, The photobiocatalytic system: Inorganic semiconductors coupled to bacterial cells, FEBS Lett., 1987, 219, 93–96 CrossRef.
- K. K. Sakimoto, A. B. Wong and P. Yang, Self-photosensitization of nonphotosynthetic bacteria for solar-to-chemical production, Science, 2016, 351, 74–77 CrossRef CAS.
- V. M. Johnson and H. B. Pakrasi, Advances in the understanding of the lifecycle of photosystem II, Microorganisms, 2022, 10, 836 CrossRef CAS.
- P. Majidian, M. Tabatabaei, M. Zeinolabedini, M. P. Naghshbandi and Y. Chisti, Metabolic engineering of microorganisms for biofuel production, Renewable Sustainable Energy Rev., 2018, 82, 3863–3885 CrossRef CAS.
- P. P. Peralta-Yahya, F. Zhang, S. B. del Cardayre and J. D. Keasling, Microbial engineering for the production of advanced biofuels, Nature, 2012, 488, 320–328 CrossRef CAS PubMed.
- J. A. Gralnick and D. R. Bond, Electron transfer beyond the outer membrane: putting electrons to rest, Annu. Rev. Microbiol., 2023, 77, 517–539 CrossRef CAS PubMed.
- N. Wu, M. Xing, Y. Li, Q. Xu and K. Li, Recent advances in microbe-photocatalyst hybrid systems for production of bulk chemicals: a review, Appl. Biochem. Biotechnol., 2023, 195, 1574–1588 CrossRef CAS.
- P. C. Sahoo, D. Pant, M. Kumar, S. K. Puri and S. S. V. Ramakumar, Material-microbe interfaces for solar-driven CO2 bioelectrosynthesis, Trends Biotechnol., 2020, 38, 1245–1261 CrossRef CAS PubMed.
- G. Okoro, S. Husain, M. Saukani, C. Mutalik, S. Yougbaré, Y.-C. Hsiao and T.-R. Kuo, Emerging trends in nanomaterials for photosynthetic biohybrid systems, ACS Mater. Lett., 2023, 5, 95–115 CrossRef CAS.
- S. Xu, Q. Shen, J. Zheng, Z. Wang, X. Pan, N. Yang and G. Zhao, Advances in biomimetic photoelectrocatalytic reduction of carbon dioxide, Adv. Sci., 2022, 9, 2203941 CrossRef CAS.
- L. Li, Z. Xu and X. Huang, Whole-cell-based photosynthetic biohybrid systems for energy and environmental applications, ChemPlusChem, 2021, 86, 1021–1036 CrossRef CAS PubMed.
- S. Cestellos-Blanco, H. Zhang, J. M. Kim, Y.-x. Shen and P. Yang, Photosynthetic semiconductor biohybrids for solar-driven biocatalysis, Nat. Catal., 2020, 3, 245–255 CrossRef CAS.
- X. Fang, S. Kalathil and E. Reisner, Semi-biological approaches to solar-to-chemical conversion, Chem. Soc. Rev., 2020, 49, 4926–4952 RSC.
- K. K. Sakimoto, N. Kornienko, S. Cestellos-Blanco, J. Lim, C. Liu and P. Yang, Physical biology of the materials-microorganism interface, J. Am. Chem. Soc., 2018, 140, 1978–1985 CrossRef CAS PubMed.
- N. Kornienko, K. K. Sakimoto, D. M. Herlihy, S. C. Nguyen, A. P. Alivisatos, C. B. Harris, A. Schwartzberg and P. Yang, Spectroscopic elucidation of energy transfer in hybrid inorganic-biological organisms for solar-to-chemical production, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11750–11755 CrossRef CAS.
- J. Liu, X. Guo, L. He, L.-P. Jiang, Y. Zhou and J.-J. Zhu, Enhanced photocatalytic CO2 reduction on biomineralized CdS via an electron conduit in bacteria, Nanoscale, 2023, 15, 10755–10762 RSC.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Semiconducting quantum dots for artificial photosynthesis, Nat. Rev. Chem, 2018, 2, 160–173 CrossRef CAS.
- Y. Yang, L.-N. Liu, H. Tian, A. I. Cooper and R. S. Sprick, Making the connections: physical and electric interactions in biohybrid photosynthetic systems, Energy Environ. Sci., 2023, 16, 4305–4319 RSC.
- Z. Wang, D. Gao, H. Geng and C. Xing, Enhancing hydrogen production by photobiocatalysis through Rhodopseudomonas palustris coupled with conjugated polymers, J. Mater. Chem. A, 2021, 9, 19788–19795 RSC.
- Y. Honda, M. Watanabe, H. Hagiwara, S. Ida and T. Ishihara, Inorganic/whole-cell biohybrid photocatalyst for highly efficient hydrogen production from water, Appl. Catal., B, 2017, 210, 400–406 CrossRef CAS.
- Y. Honda, H. Hagiwara, S. Ida and T. Ishihara, Application to photocatalytic H2 production of a whole-cell reaction by recombinant Escherichia coli cells expressing [FeFe]-hydrogenase and maturases genes, Angew. Chem., Int. Ed., 2016, 55, 8045–8048 CrossRef CAS.
- S. F. Rowe, G. Le Gall, E. V. Ainsworth, J. A. Davies, C. W. J. Lockwood, L. Shi, A. Elliston, I. N. Roberts, K. W. Waldron, D. J. Richardson, T. A. Clarke, L. J. C. Jeuken, E. Reisner and J. N. Butt, Light-driven H2 evolution and C=C or C=O bond hydrogenation by Shewanella oneidensis: a versatile strategy for photocatalysis by nonphotosynthetic microorganisms, ACS Catal., 2017, 7, 7558–7566 CrossRef CAS.
- M. T. Gamache, R. Charaf, L. Kurth, D. T. Filmon, M. Senger, N. Plumeré, L. Hammarström and G. Berggren, Elucidating electron transfer kinetics and optimizing system performance for Escherichia coli-based semi-artificial H2 production, ACS Catal., 2023, 13, 9476–9486 CrossRef CAS.
- R. Sirohi, A. Kumar Pandey, P. Ranganathan, S. Singh, A. Udayan, M. Kumar Awasthi, A. T. Hoang, C. R. Chilakamarry, S. H. Kim and S. J. Sim, Design and applications of photobioreactors - a review, Bioresour. Technol., 2022, 349, 126858 CrossRef CAS.
- M. Martins, C. Toste and I. A. C. Pereira, Enhanced light-driven hydrogen production by self-photosensitized biohybrid systems, Angew. Chem., Int. Ed., 2021, 60, 9055–9062 CrossRef CAS PubMed.
- B. Wang, K. Xiao, Z. Jiang, J. Wang, J. C. Yu and P. K. Wong, Biohybrid photoheterotrophic metabolism for significant enhancement of biological nitrogen fixation in pure microbial cultures, Energy Environ. Sci., 2019, 12, 2185–2191 RSC.
- B. Wang, C. Zeng, K. H. Chu, D. Wu, H. Y. Yip, L. Ye and P. K. Wong, Enhanced biological hydrogen production from Escherichia coli with surface precipitated cadmium sulfide nanoparticles, Adv. Energy Mater., 2017, 7, 1700611 CrossRef.
- J. Ye, J. Yu, Y. Zhang, M. Chen, X. Liu, S. Zhou and Z. He, Light-driven carbon dioxide reduction to methane by Methanosarcina barkeri-CdS biohybrid, Appl. Catal., B, 2019, 257, 117916 CrossRef CAS.
- J. Ye, G. Ren, L. Kang, Y. Zhang, X. Liu, S. Zhou and Z. He, Efficient photoelectron capture by Ni decoration in Methanosarcina barkeri-CdS biohybrids for enhanced photocatalytic CO2-to-CH4 conversion, iScience, 2020, 23, 101287 CrossRef CAS PubMed.
- H.-X. Han, L.-J. Tian, D.-F. Liu, H.-Q. Yu, G.-P. Sheng and Y. Xiong, Reversing electron transfer chain for light-driven hydrogen production in biotic–abiotic hybrid systems, J. Am. Chem. Soc., 2022, 144, 6434–6441 CrossRef CAS PubMed.
- B. Luo, Y.-Z. Wang, D. Li, H. Shen, L.-X. Xu, Z. Fang, Z. Xia, J. Ren, W. Shi and Y.-C. Yong, A periplasmic photosensitized biohybrid system for solar hydrogen production, Adv. Energy Mater., 2021, 11, 2100256 CrossRef CAS.
- Y. He, S. Wang, X. Han, J. Shen, Y. Lu, J. Zhao, C. Shen and L. Qiao, Photosynthesis of acetate by Sporomusa ovata–CdS biohybrid system, ACS Appl. Mater. Interfaces, 2022, 14, 23364–23374 CrossRef CAS PubMed.
- S. Jin, Y. Jeon, M. S. Jeon, J. Shin, Y. Song, S. Kang, J. Bae, S. Cho, J.-K. Lee, D. R. Kim and B.-K. Cho, Acetogenic bacteria utilize light-driven electrons as an energy source for autotrophic growth, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2020552118 CrossRef.
- M. Xu, P.-L. Tremblay, R. Ding, J. Xiao, J. Wang, Y. Kang and T. Zhang, Photo-augmented PHB production from CO2 or fructose by Cupriavidus necator and shape-optimized CdS nanorods, Sci. Total Environ., 2021, 753, 142050 CrossRef CAS PubMed.
- M. Xu, P.-L. Tremblay, L. Jiang and T. Zhang, Stimulating bioplastic production with light energy by coupling Ralstonia eutropha with the photocatalyst graphitic carbon nitride, Green Chem., 2019, 21, 2392–2400 RSC.
- H. Shen, Y.-Z. Wang, G. Liu, L. Li, R. Xia, B. Luo, J. Wang, D. Suo, W. Shi and Y.-C. Yong, A whole-cell inorganic-biohybrid system integrated by reduced graphene oxide for boosting solar hydrogen production, ACS Catal., 2020, 10, 13290–13295 CrossRef CAS.
- Q. Wang, S. Kalathil, C. Pornrungroj, C. D. Sahm and E. Reisner, Bacteria–photocatalyst sheet for sustainable carbon dioxide utilization, Nat. Catal., 2022, 5, 633–641 CrossRef CAS.
- L. Shi, H. Dong, G. Reguera, H. Beyenal, A. Lu, J. Liu, H. Q. Yu and J. K. Fredrickson, Extracellular electron transfer mechanisms between microorganisms and minerals, Nat. Rev. Microbiol., 2016, 14, 651–662 Search PubMed.
- D. R. Lovley and D. E. Holmes, Electromicrobiology: the ecophysiology of phylogenetically diverse electroactive microorganisms, Nat. Rev. Microbiol., 2022, 20, 5–19 Search PubMed.
- A. Kouzuma, T. Kasai, A. Hirose and K. Watanabe, Catabolic and regulatory systems in Shewanella oneidensis MR-1 involved in electricity generation in microbial fuel cells, Front. Microbiol., 2015, 6, 609 Search PubMed.
- J. Li, H. Han, Y. Chang and B. Wang, The material-microorganism interface in microbial hybrid electrocatalysis systems, Nanoscale, 2023, 15, 6009–6024 RSC.
- B. S. Thapa, T. Kim, S. Pandit, Y. E. Song, Y. P. Afsharian, M. Rahimnejad, J. R. Kim and S. E. Oh, Overview of electroactive microorganisms and electron transfer mechanisms in microbial electrochemistry, Bioresour. Technol., 2022, 347, 126579 CrossRef CAS.
- M. Edel, L.-A. Philipp, J. Lapp, J. Reiner and J. Gescher, Electron transfer of extremophiles in bioelectrochemical systems, Extremophiles, 2022, 26, 31 CrossRef PubMed.
- A. S. Beliaev, D. M. Klingeman, J. A. Klappenbach, L. Wu, M. F. Romine, J. M. Tiedje, K. H. Nealson, J. K. Fredrickson and J. Zhou, Global transcriptome analysis of Shewanella oneidensis MR-1 exposed to different terminal electron acceptors, J. Bacteriol., 2005, 187, 7138–7145 CrossRef CAS PubMed.
- L. Shi, T. C. Squier, J. M. Zachara and J. K. Fredrickson, Respiration of metal (hydr)oxides by Shewanella and Geobacter: a key role for multihaem c-type cytochromes, Mol. Microbiol., 2007, 65, 12–20 CrossRef CAS PubMed.
- C. Liu, Y. A. Gorby, J. M. Zachara, J. K. Fredrickson and C. F. Brown, Reduction kinetics of Fe(III), Co(III), U(VI), Cr(VI), and Tc(VII) in cultures of dissimilatory metal-reducing bacteria, Biotechnol. Bioeng., 2002, 80, 637–649 CrossRef CAS PubMed.
- J. F. Heidelberg, I. T. Paulsen, K. E. Nelson, E. J. Gaidos, W. C. Nelson, T. D. Read, J. A. Eisen, R. Seshadri, N. Ward, B. Methe, R. A. Clayton, T. Meyer, A. Tsapin, J. Scott, M. Beanan, L. Brinkac, S. Daugherty, R. T. DeBoy, R. J. Dodson, A. S. Durkin, D. H. Haft, J. F. Kolonay, R. Madupu, J. D. Peterson, L. A. Umayam, O. White, A. M. Wolf, J. Vamathevan, J. Weidman, M. Impraim, K. Lee, K. Berry, C. Lee, J. Mueller, H. Khouri, J. Gill, T. R. Utterback, L. A. McDonald, T. V. Feldblyum, H. O. Smith, J. C. Venter, K. H. Nealson and C. M. Fraser, Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis, Nat. Biotechnol., 2002, 20, 1118–1123 CrossRef CAS PubMed.
- R. S. Hartshorne, B. N. Jepson, T. A. Clarke, S. J. Field, J. Fredrickson, J. Zachara, L. Shi, J. N. Butt and D. J. Richardson, Characterization of Shewanella oneidensis MtrC: a cell-surface decaheme cytochrome involved in respiratory electron transport to extracellular electron acceptors, J. Biol. Inorg. Chem., 2007, 12, 1083–1094 CrossRef CAS PubMed.
- X. Jiang, B. Burger, F. Gajdos, C. Bortolotti, Z. Futera, M. Breuer and J. Blumberger, Kinetics of trifurcated electron flow in the decaheme bacterial proteins MtrC and MtrF, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3425–3430 CrossRef CAS PubMed.
- M. Breuer, K. M. Rosso and J. Blumberger, Electron flow in multiheme bacterial cytochromes is a balancing act between heme electronic interaction and redox potentials, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 611–616 CrossRef CAS PubMed.
- G. F. White, Z. Shi, L. Shi, Z. Wang, A. C. Dohnalkova, M. J. Marshall, J. K. Fredrickson, J. M. Zachara, J. N. Butt, D. J. Richardson and T. A. Clarke, Rapid electron exchange between surface-exposed bacterial cytochromes and Fe(III) minerals, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6346–6351 CrossRef CAS PubMed.
- J. H. van Wonderen, K. Adamczyk, X. Wu, X. Jiang, S. E. H. Piper, C. R. Hall, M. J. Edwards, T. A. Clarke, H. Zhang, L. J. C. Jeuken, I. V. Sazanovich, M. Towrie, J. Blumberger, S. R. Meech and J. N. Butt, Nanosecond heme-to-heme electron transfer rates in a multiheme cytochrome nanowire reported by a spectrally unique His/Met-ligated heme, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2107939118 CrossRef PubMed.
- Y. Cao, M. Song, F. Li, C. Li, X. Lin, Y. Chen, Y. Chen, J. Xu, Q. Ding and H. Song, A synthetic plasmid toolkit for Shewanella oneidensis MR-1, Front. Microbiol., 2019, 10, 410 CrossRef PubMed.
- D. E. Ross, J. M. Flynn, D. B. Baron, J. A. Gralnick and D. R. Bond, Towards electrosynthesis in Shewanella: energetics of reversing the Mtr pathway for reductive metabolism, PLoS One, 2011, 6, 16649 Search PubMed.
- D. R. Bond and D. R. Lovley, Electricity production by Geobacter sulfurreducens attached to electrodes, Appl. Environ. Microbiol., 2003, 69, 1548–1555 CrossRef CAS PubMed.
- Y. Liu, Z. Wang, J. Liu, C. Levar, M. J. Edwards, J. T. Babauta, D. W. Kennedy, Z. Shi, H. Beyenal, D. R. Bond, T. A. Clarke, J. N. Butt, D. J. Richardson, K. M. Rosso, J. M. Zachara, J. K. Fredrickson and L. Shi, A trans-outer membrane porin-cytochrome protein complex for extracellular electron transfer by Geobacter sulfurreducens PCA, Environ. Microbiol. Rep., 2014, 6, 776–785 CrossRef CAS PubMed.
- Y. Liu, J. K. Fredrickson, J. M. Zachara and L. Shi, Direct involvement of ombB, omaB, and omcB genes in extracellular reduction of Fe(III) by Geobacter sulfurreducens PCA, Front. Microbiol., 2015, 6, 1075 Search PubMed.
- Y. Gu, M. J. Guberman-Pfeffer, V. Srikanth, C. Shen, F. Giska, K. Gupta, Y. Londer, F. A. Samatey, V. S. Batista and N. S. Malvankar, Structure of Geobacter cytochrome OmcZ identifies mechanism of nanowire assembly and conductivity, Nat. Microbiol., 2023, 8, 284–298 CrossRef CAS PubMed.
- S. E. Yalcin and N. S. Malvankar, The blind men and the filament: understanding structures and functions of microbial nanowires, Curr. Opin. Chem. Biol., 2020, 59, 193–201 CrossRef CAS PubMed.
- Y. Gu, V. Srikanth, A. I. Salazar-Morales, R. Jain, J. P. O'Brien, S. M. Yi, R. K. Soni, F. A. Samatey, S. E. Yalcin and N. S. Malvankar, Structure of Geobacter pili reveals secretory rather than nanowire behaviour, Nature, 2021, 597, 430–434 CrossRef CAS PubMed.
- F. Wang, Y. Gu, J. P. O'Brien, S. M. Yi, S. E. Yalcin, V. Srikanth, C. Shen, D. Vu, N. L. Ing, A. I. Hochbaum, E. H. Egelman and N. S. Malvankar, Structure of microbial nanowires reveals stacked hemes that transport electrons over micrometers, Cell, 2019, 177, 361–369 CrossRef CAS PubMed.
- F. Wang, K. Mustafa, V. Suciu, K. Joshi, C. H. Chan, S. Choi, Z. Su, D. Si, A. I. Hochbaum, E. H. Egelman and D. R. Bond, Cryo-EM structure of an extracellular Geobacter OmcE cytochrome filament reveals tetrahaem packing, Nat. Microbiol., 2022, 7, 1291–1300 CrossRef CAS PubMed.
- F. Wang, C. H. Chan, V. Suciu, K. Mustafa, M. Ammend, D. Si, A. I. Hochbaum, E. H. Egelman and D. R. Bond, Structure of Geobacter OmcZ filaments suggests extracellular cytochrome polymers evolved independently multiple times, eLife, 2022, 11, 81551 CrossRef PubMed.
- D. P. Baquero, V. Cvirkaite-Krupovic, S. S. Hu, J. L. Fields, X. Liu, C. Rensing, E. H. Egelman, M. Krupovic and F. Wang, Extracellular cytochrome nanowires appear to be ubiquitous in prokaryotes, Cell, 2023, 186, 2853–2864 CrossRef CAS PubMed.
- C. E. Levar, C. H. Chan, M. G. Mehta-Kolte and D. R. Bond, An inner membrane cytochrome required only for reduction of high redox potential extracellular electron acceptors, Mbio, 2014, 5, 2034 CrossRef PubMed.
- L. Zacharoff, C. H. Chan and D. R. Bond, Reduction of low potential electron acceptors requires the CbcL inner membrane cytochrome of Geobacter sulfurreducens, Bioelectrochemistry, 2016, 107, 7–13 CrossRef CAS PubMed.
- K. Joshi, C. H. Chan and D. R. Bond, Geobacter sulfurreducens inner membrane cytochrome CbcBA controls electron transfer and growth yield near the energetic limit of respiration, Mol. Microbiol., 2021, 116, 1124–1139 CrossRef CAS PubMed.
- E. Howley, A. Mangus, D. Williams and C. I. Torres, Intracytoplasmic membranes develop in Geobacter sulfurreducens under thermodynamically limiting conditions, npj Biofilms Microbiomes, 2023, 9, 18 CrossRef CAS PubMed.
- H. K. Carlson, A. T. Iavarone, A. Gorur, B. S. Yeo, R. Tran, R. A. Melnyk, R. A. Mathies, M. Auer and J. D. Coates, Surface multiheme c-type cytochromes from Thermincola potens and implications for respiratory metal reduction by Gram-positive bacteria, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1702–1707 CrossRef CAS PubMed.
- N. L. Costa, B. Hermann, V. Fourmond, M. M. Faustino, M. Teixeira, O. Einsle, C. M. Paquete and R. O. Louro, How thermophilic gram-positive organisms perform extracellular electron transfer: characterization of the cell surface terminal reductase OcwA, Mbio, 2019, 10, 1210–1219 CrossRef PubMed.
- T. V. Tikhonova, E. M. Osipov, N. I. Dergousova, K. M. Boyko, I. M. Elizarov, S. N. Gavrilov, M. G. Khrenova, F. T. Robb, A. Y. Solovieva, E. A. Bonch-Osmolovskaya and V. O. Popov, Extracellular Fe(III) reductase structure reveals a modular organization enabling S-layer insertion and electron transfer to insoluble substrates, Structure, 2023, 31, 174–184 CrossRef CAS PubMed.
- M. J. Edwards, D. J. Richardson, C. M. Paquete and T. A. Clarke, Role of multiheme cytochromes involved in extracellular anaerobic respiration in bacteria, Protein Sci., 2020, 29, 830–842 CrossRef CAS PubMed.
- A. Chukwubuikem, C. Berger, A. Mady and M. A. Rosenbaum, Role of phenazine-enzyme physiology for current generation in a bioelectrochemical system, Microb. Biotechnol., 2021, 14, 1613–1626 CrossRef CAS PubMed.
- S. H. Saunders, E. C. M. Tse, M. D. Yates, F. J. Otero, S. A. Trammell, E. D. A. Stemp, J. K. Barton, L. M. Tender and D. K. Newman, Extracellular DNA promotes efficient extracellular electron transfer by pyocyanin in Pseudomonas aeruginosa biofilms, Cell, 2020, 182, 919–932 CrossRef CAS PubMed.
- E. Mevers, L. Su, G. Pishchany, M. Baruch, J. Cornejo, E. Hobert, E. Dimise, C. M. Ajo-Franklin and J. Clardy, An elusive electron shuttle from a facultative anaerobe, eLife, 2019, 8, 48054 CrossRef PubMed.
- S. H. Light, L. Su, R. Rivera-Lugo, J. A. Cornejo, A. Louie, A. T. Iavarone, C. M. Ajo-Franklin and D. A. Portnoy, A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria, Nature, 2018, 562, 140–144 CrossRef CAS PubMed.
- R. Méheust, S. Huang, R. Rivera-Lugo, J. F. Banfield and S. H. Light, Post-translational flavinylation is associated with diverse extracytosolic redox functionalities throughout bacterial life, eLife, 2021, 10, 66878 CrossRef PubMed.
- E. D. Brutinel and J. A. Gralnick, Shuttling happens: soluble flavin mediators of extracellular electron transfer in Shewanella, Appl. Microbiol. Biotechnol., 2012, 93, 41–48 CrossRef PubMed.
- M. M. Faustino, B. M. Fonseca, N. L. Costa, D. Lousa, R. O. Louro and C. M. Paquete, Crossing the wall: characterization of the multiheme cytochromes involved in the extracellular electron transfer pathway of Thermincola ferriacetica, Microorganisms, 2021, 9, 293 CrossRef CAS PubMed.
- S. N. Gavrilov, D. G. Zavarzina, I. M. Elizarov, T. V. Tikhonova, N. I. Dergousova, V. O. Popov, J. R. Lloyd, D. Knight, M. Y. El-Naggar, S. Pirbadian, K. M. Leung, F. T. Robb, M. V. Zakhartsev, O. Bretschger and E. A. Bonch-Osmolovskaya, Novel extracellular electron transfer channels in a gram-positive thermophilic bacterium, Front. Microbiol., 2020, 11, 597818 CrossRef PubMed.
- Y. A. Gorby, S. Yanina, J. S. McLean, K. M. Rosso, D. Moyles, A. Dohnalkova, T. J. Beveridge, I. S. Chang, B. H. Kim, K. S. Kim, D. E. Culley, S. B. Reed, M. F. Romine, D. A. Saffarini, E. A. Hill, L. Shi, D. A. Elias, D. W. Kennedy, G. Pinchuk, K. Watanabe, S. Ishii, B. Logan, K. H. Nealson and J. K. Fredrickson, Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11358–11363 CrossRef CAS PubMed.
- A. I. Pimenta, C. M. Paquete, L. Morgado, M. J. Edwards, T. A. Clarke, C. A. Salgueiro, I. A. C. Pereira and A. G. Duarte, Characterization of the inner membrane cytochrome ImcH from Geobacter reveals its importance for extracellular electron transfer and energy conservation, Protein Sci., 2023, 32, 4796 CrossRef PubMed.
- A. R. Rowe, F. Salimijazi, L. Trutschel, J. Sackett, O. Adesina, I. Anzai, L. H. Kugelmass, M. H. Baym and B. Barstow, Identification of a pathway for electron uptake in Shewanella oneidensis, Commun. Biol., 2021, 4, 957 CrossRef CAS PubMed.
- J. Zhang, F. Li, D. Liu, Q. Liu and H. Song, Engineering extracellular electron transfer pathways of electroactive microorganisms by synthetic biology for energy and chemicals production, Chem. Soc. Rev., 2024, 53, 1375–1446 RSC.
- N. Heidary, N. Kornienko, S. Kalathil, X. Fang, K. H. Ly, H. F. Greer and E. Reisner, Disparity of cytochrome utilization in anodic and cathodic extracellular electron transfer pathways of Geobacter sulfurreducens biofilms, J. Am. Chem. Soc., 2020, 142, 5194–5203 CrossRef CAS PubMed.
- Z. Wang, Y. Hu, S. Zhang and Y. Sun, Artificial photosynthesis systems for solar energy conversion and storage: platforms and their realities, Chem. Soc. Rev., 2022, 51, 6704–6737 RSC.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Natural engineering principles of electron tunnelling in biological oxidation-reduction, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- C. Wombwell, C. A. Caputo and E. Reisner, [NiFeSe]-Hydrogenase Chemistry, Acc. Chem. Res., 2015, 48, 2858–2865 CrossRef CAS PubMed.
- E. Reisner, Solar hydrogen evolution with hydrogenases: from natural to hybrid systems, Eur. J. Inorg. Chem., 2011, 2011, 1005–1016 CrossRef.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, Visible light-driven H2 production by hydrogenases attached to dye-sensitized TiO2 nanoparticles, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Catalytic electrochemistry of a [NiFeSe]-hydrogenase on TiO2 and demonstration of its suitability for visible-light driven H2 production, Chem. Commun., 2009, 550–552 RSC.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Efficient and clean photoreduction of CO2 to CO by enzyme-modified TiO2 nanoparticles using visible light, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. C. Pereira and E. Reisner, Interfacing formate dehydrogenase with metal oxides for the reversible electrocatalysis and solar-driven reduction of carbon dioxide, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS PubMed.
- L. Zhang, M. Can, S. W. Ragsdale and F. A. Armstrong, Fast and selective photoreduction of CO2 to CO catalyzed by a complex of carbon monoxide dehydrogenase, TiO2, and Ag nanoclusters, ACS Catal., 2018, 8, 2789–2795 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Photocatalytic hydrogen production using polymeric carbon nitride with a hydrogenase and a bioinspired synthetic Ni catalyst, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Carbon nitride–TiO2 hybrid modified with hydrogenase for visible light driven hydrogen production, Chem. Sci., 2015, 6, 5690–5694 RSC.
- T. Sakai, D. Mersch and E. Reisner, Photocatalytic hydrogen evolution with a hydrogenase in a mediator-free system under high levels of oxygen, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- Y. Liu, C. Pulignani, S. Webb, S. J. Cobb, S. Rodríguez-Jiménez, D. Kim, R. D. Milton and E. Reisner, Electrostatic [FeFe]-hydrogenase–carbon nitride assemblies for efficient solar hydrogen production, Chem. Sci., 2024, 15, 6088–6094 RSC.
- V. M. Badiani, C. Casadevall, M. Miller, S. J. Cobb, R. R. Manuel, I. A. C. Pereira and E. Reisner, Engineering electro- and photocatalytic carbon materials for CO2 reduction by formate dehydrogenase, J. Am. Chem. Soc., 2022, 144, 14207–14216 CrossRef CAS PubMed.
- G. A. Hutton, B. Reuillard, B. C. Martindale, C. A. Caputo, C. W. Lockwood, J. N. Butt and E. Reisner, Carbon dots as versatile photosensitizers for solar-driven catalysis with redox enzymes, J. Am. Chem. Soc., 2016, 138, 16722–16730 CrossRef CAS PubMed.
- K. Holá, M. V. Pavliuk, B. Németh, P. Huang, L. Zdražil, H. Land, G. Berggren and H. Tian, Carbon dots and [FeFe] hydrogenase biohybrid assemblies for efficient light-driven hydrogen evolution, ACS Catal., 2020, 10, 9943–9952 CrossRef.
- J. K. Utterback, J. L. Ruzicka, H. R. Keller, L. M. Pellows and G. Dukovic, Electron transfer from semiconductor nanocrystals to redox enzymes, Annu. Rev. Phys. Chem., 2020, 71, 335–359 CrossRef CAS PubMed.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, Characterization of photochemical processes for H2 production by CdS nanorod-[FeFe] hydrogenase complexes, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Light-driven dinitrogen reduction catalyzed by a CdS:nitrogenase MoFe protein biohybrid, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- K. A. Brown, J. Ruzicka, H. Kallas, B. Chica, D. W. Mulder, J. W. Peters, L. C. Seefeldt, G. Dukovic and P. W. King, Excitation-rate determines product stoichiometry in photochemical ammonia production by CdS Quantum Dot-Nitrogenase MoFe protein complexes, ACS Catal., 2020, 10, 11147–11152 CrossRef CAS.
- L. E. Roth, J. C. Nguyen and F. A. Tezcan, ATP- and iron-protein-independent activation of nitrogenase catalysis by light, J. Am. Chem. Soc., 2010, 132, 13672–13674 CrossRef CAS PubMed.
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, Controlled assembly of hydrogenase-CdTe nanocrystal hybrids for solar hydrogen production, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed.
- J. L. Ruzicka, L. M. Pellows, H. Kallas, K. E. Shulenberger, O. A. Zadvornyy, B. Chica, K. A. Brown, J. W. Peters, P. W. King, L. C. Seefeldt and G. Dukovic, The kinetics of electron transfer from CdS nanorods to the MoFe protein of nitrogenase, J. Phys. Chem., 2022, 126, 8425–8435 CAS.
- J. K. Utterback, M. B. Wilker, K. A. Brown, P. W. King, J. D. Eaves and G. Dukovic, Competition between electron transfer, trapping, and recombination in CdS nanorod-hydrogenase complexes, Phys. Chem. Chem. Phys., 2015, 17, 5538–5542 RSC.
- J. K. Utterback, M. B. Wilker, D. W. Mulder, P. W. King, J. D. Eaves and G. Dukovic, Quantum efficiency of charge transfer competing against nonexponential processes: the case of electron transfer from CdS nanorods to hydrogenase, J. Phys. Chem. C, 2019, 123, 886–896 CrossRef CAS.
- H. Hamby, B. Li, K. E. Shinopoulos, H. R. Keller, S. J. Elliott and G. Dukovic, Light-driven carbon−carbon bond formation via CO2 reduction catalyzed by complexes of CdS nanorods and a 2-oxoacid oxidoreductase, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 135–140 CrossRef CAS PubMed.
- Y. S. Chaudhary, T. W. Woolerton, C. S. Allen, J. H. Warner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Visible light-driven CO2 reduction by enzyme coupled CdS nanocrystals, Chem. Commun., 2012, 48, 58–60 RSC.
- E. Lam, M. Miller, S. Linley, R. R. Manuel, I. A. C. Pereira and E. Reisner, Comproportionation of CO2 and cellulose to formate using a floating semiconductor-enzyme photoreforming catalyst, Angew. Chem., Int. Ed., 2023, 62, 15894 CrossRef PubMed.
- S. Bhattacharjee, C. Guo, E. Lam, J. M. Holstein, M. Rangel Pereira, C. M. Pichler, C. Pornrungroj, M. Rahaman, T. Uekert, F. Hollfelder and E. Reisner, Chemoenzymatic photoreforming: a sustainable approach for solar fuel generation from plastic feedstocks, J. Am. Chem. Soc., 2023, 145, 20355–20364 CrossRef CAS PubMed.
- E. Reisner, When does organic photoredox catalysis meet artificial photosynthesis?, Angew. Chem., Int. Ed., 2019, 58, 3656–3657 CrossRef CAS PubMed.
- S. Bhattacharjee, S. Linley and E. Reisner, Solar reforming as an emerging technology for circular chemical industries, Nat. Rev. Chem, 2024, 8, 87–105 CrossRef PubMed.
- T. Lithgow, C. J. Stubenrauch and M. P. H. Stumpf, Surveying membrane landscapes: a new look at the bacterial cell surface, Nat. Rev. Microbiol., 2023, 21, 502–518 CrossRef CAS PubMed.
- L. Karygianni, Z. Ren, H. Koo and T. Thurnheer, Biofilm matrixome: extracellular components in structured microbial communities, Trends Microbiol., 2020, 28, 668–681 CrossRef CAS PubMed.
- C. Hamon, A. Ciaccafava, P. Infossi, R. Puppo, P. Even-Hernandez, E. Lojou and V. Marchi, Synthesis and enzymatic photo-activity of an O2 tolerant hydrogenase-CdSe@CdS quantum rod bioconjugate, Chem. Commun., 2014, 50, 4989–4992 RSC.
- N.-H. Tran, N. Huynh, T. Bui, Y. Nguyen, P. Huynh, M. E. Cooper and L. E. Cheruzel, Light-initiated hydroxylation of lauric acid using hybrid P450 BM3 enzymes, Chem. Commun., 2011, 47, 11936–11938 RSC.
- N. H. Tran, N. Huynh, G. Chavez, A. Nguyen, S. Dwaraknath, T. A. Nguyen, M. Nguyen and L. Cheruzel, A series of hybrid P450 BM3 enzymes with different catalytic activity in the light-initiated hydroxylation of lauric acid, J. Inorg. Biochem., 2012, 115, 50–56 CrossRef CAS PubMed.
- N. H. Tran, D. Nguyen, S. Dwaraknath, S. Mahadevan, G. Chavez, A. Nguyen, T. Dao, S. Mullen, T. A. Nguyen and L. E. Cheruzel, An efficient light-driven P450 BM3 biocatalyst, J. Am. Chem. Soc., 2013, 135, 14484–14487 CrossRef CAS PubMed.
- H. Zhang, C. Casadevall, J. H. van Wonderen, L. Su, J. N. Butt, E. Reisner and L. J. C. Jeuken, Rational design of covalent multiheme cytochrome-carbon dot biohybrids for photoinduced electron transfer, Adv. Funct. Mater., 2023, 33, 2302204 CrossRef CAS.
- E. T. Hwang, K. L. Orchard, D. Hojo, J. Beton, C. W. J. Lockwood, T. Adschiri, J. N. Butt, E. Reisner and L. J. C. Jeuken, Exploring step-by-step assembly of nanoparticle:cytochrome biohybrid photoanodes, ChemElectroChem, 2017, 4, 1959–1968 CrossRef CAS PubMed.
- E. T. Hwang, K. Sheikh, K. L. Orchard, D. Hojo, V. Radu, C. Y. Lee, E. Ainsworth, C. Lockwood, M. A. Gross, T. Adschiri, E. Reisner, J. N. Butt and L. J. Jeuken, A decaheme cytochrome as a molecular electron conduit in dye-sensitized photoanodes, Adv. Funct. Mater., 2015, 25, 2308–2315 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Enhancing light absorption and charge transfer efficiency in carbon dots through graphitization and core nitrogen doping, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS PubMed.
- E. V. Ainsworth, C. W. Lockwood, G. F. White, E. T. Hwang, T. Sakai, M. A. Gross, D. J. Richardson, T. A. Clarke, L. J. Jeuken, E. Reisner and J. N. Butt, Photoreduction of Shewanella oneidensis extracellular cytochromes by organic chromophores and dye-sensitized TiO2, ChemBioChem, 2016, 17, 2324–2333 CrossRef CAS PubMed.
- J. H. van Wonderen, D. Li, S. E. H. Piper, C. Y. Lau, L. P. Jenner, C. R. Hall, T. A. Clarke, N. J. Watmough and J. N. Butt, Photosensitised multiheme cytochromes as light-driven molecular wires and resistors, ChemBioChem, 2018, 19, 2206–2215 CrossRef CAS PubMed.
- A. Stikane, E. T. Hwang, E. V. Ainsworth, S. E. H. Piper, K. Critchley, J. N. Butt, E. Reisner and L. J. C. Jeuken, Towards compartmentalized photocatalysis: multihaem proteins as transmembrane molecular electron conduits, Faraday Discuss., 2019, 215, 26–38 RSC.
- P. R. Pokkuluri, Y. Y. Londer, X. Yang, N. E. Duke, J. Erickson, V. Orshonsky, G. Johnson and M. Schiffer, Structural characterization of a family of cytochromes c7 involved in Fe(III) respiration by Geobacter sulfurreducens, Biochim. Biophys. Acta, 2010, 1797, 222–232 CrossRef CAS PubMed.
- N. S. Ponomarenko, O. Kokhan, P. R. Pokkuluri, K. L. Mulfort and D. M. Tiede, Examination of abiotic cofactor assembly in photosynthetic biomimetics: site-specific stereoselectivity in the conjugation of a ruthenium(II) tris(bipyridine) photosensitizer to a multi-heme protein, Photosynth. Res., 2020, 143, 99–113 CrossRef CAS PubMed.
- D. R. Marzolf, A. M. McKenzie, M. C. O'Malley, N. S. Ponomarenko, C. M. Swaim, T. J. Brittain, N. L. Simmons, P. R. Pokkuluri, K. L. Mulfort, D. M. Tiede and O. Kokhan, Mimicking natural photosynthesis: designing ultrafast photosensitized electron transfer into multiheme cytochrome protein nanowires, Nanomaterials, 2020, 10, 2143 CrossRef CAS PubMed.
- J. H. van Wonderen, C. R. Hall, X. Jiang, K. Adamczyk, A. Carof, I. Heisler, S. E. H. Piper, T. A. Clarke, N. J. Watmough, I. V. Sazanovich, M. Towrie, S. R. Meech, J. Blumberger and J. N. Butt, Ultrafast light-driven electron transfer in a Ru(II)tris(bipyridine)-labeled multiheme cytochrome, J. Am. Chem. Soc., 2019, 141, 15190–15200 CrossRef CAS PubMed.
- Y. Liu, Y. Chen, Y. Shi, D. Wan, J. Chen and S. Xiao, Adsorption of toxic dye Eosin Y from aqueous solution by clay/carbon composite derived from spent bleaching earth, Water Environ. Res., 2021, 93, 159–169 CrossRef CAS PubMed.
- B. Fan, W. Peng, Y. Zhang, P. Liu and J. Shen, ROS conversion promotes the bactericidal efficiency of Eosin Y based photodynamic therapy, Biomater. Sci., 2023, 11, 4930–4937 RSC.
- A. C. Munteanu and V. Uivarosi, Ruthenium complexes in the fight against pathogenic microorganisms. An extensive review, Pharmaceutics, 2021, 13, 874 CrossRef CAS PubMed.
- P. Konda, L. M. Lifshits, J. A. Roque Iii, H. D. Cole, C. G. Cameron, S. A. McFarland and S. Gujar, Discovery of immunogenic cell death-inducing ruthenium-based photosensitizers for anticancer photodynamic therapy, Oncoimmunology, 2021, 10, 1863626 CrossRef PubMed.
- A. Sukhanova, S. Bozrova, P. Sokolov, M. Berestovoy, A. Karaulov and I. Nabiev, Dependence of nanoparticle toxicity on their physical and chemical properties, Nanoscale Res. Lett., 2018, 13, 44 CrossRef PubMed.
- A. K. Suresh, D. A. Pelletier and M. J. Doktycz, Relating nanomaterial properties and microbial toxicity, Nanoscale, 2013, 5, 463–474 RSC.
- Z. Wang and M. Tang, The cytotoxicity of core-shell or non-shell structure quantum dots and reflection on environmental friendly: a review, Environ. Res., 2021, 194, 110593 CrossRef CAS PubMed.
- J. T. Trevors, G. W. Stratton and G. M. Gadd, Cadmium transport, resistance, and toxicity in bacteria, algae, and fungi, Can. J. Microbiol., 1986, 32, 447–464 CrossRef CAS PubMed.
- L. Göbbels, A. Poehlein, A. Dumnitch, R. Egelkamp, C. Kröger, J. Haerdter, T. Hackl, A. Feld, H. Weller, R. Daniel, W. R. Streit and M. C. Schoelmerich, Cysteine: an overlooked energy and carbon source, Sci. Rep., 2021, 11, 2139 CrossRef PubMed.
- A. M. Wroblewska-Wolna, A. J. Harvie, S. F. Rowe, K. Critchley, J. N. Butt and L. J. Jeuken, Quantum dot interactions with and toxicity to Shewanella oneidensis MR-1, Nanotechnology, 2020, 31, 134005 CrossRef CAS PubMed.
- Y. Ding, J. R. Bertram, C. Eckert, R. R. Bommareddy, R. Patel, A. Conradie, S. Bryan and P. Nagpal, Nanorg microbial factories: light-driven renewable biochemical synthesis using quantum dot-bacteria nanobiohybrids, J. Am. Chem. Soc., 2019, 141, 10272–10282 CrossRef CAS PubMed.
- X. Guan, S. Ersan, X. Hu, T. L. Atallah, Y. Xie, S. Lu, B. Cao, J. Sun, K. Wu, Y. Huang, X. Duan, J. R. Caram, Y. Yu, J. O. Park and C. Liu, Maximizing light-driven CO2 and N2 fixation efficiency in quantum dot-bacteria hybrids, Nat. Catal., 2022, 5, 1019–1029 CrossRef CAS PubMed.
- E. H. Edwards, J. Jelušić, R. M. Kosko, K. P. McClelland, S. S. Ngarnim, W. Chiang, S. Lampa-Pastirk, T. D. Krauss and K. L. Bren, Shewanella oneidensis MR-1 respires CdSe quantum dots for photocatalytic hydrogen evolution, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, 2206975120 CrossRef PubMed.
- M. A. Maurer-Jones, I. L. Gunsolus, B. M. Meyer, C. J. Christenson and C. L. Haynes, Impact of TiO2 nanoparticles on growth, biofilm formation, and flavin secretion in Shewanella oneidensis, Anal. Chem., 2013, 85, 5810–5818 CrossRef CAS PubMed.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Bacteria photosensitized by intracellular gold nanoclusters for solar fuel production, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Carbon dots as photosensitisers for solar-driven catalysis, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- C. Yang, H. Aslan, P. Zhang, S. Zhu, Y. Xiao, L. Chen, N. Khan, T. Boesen, Y. Wang, Y. Liu, L. Wang, Y. Sun, Y. Feng, F. Besenbacher, F. Zhao and M. Yu, Carbon dots-fed Shewanella oneidensis MR-1 for bioelectricity enhancement, Nat. Commun., 2020, 11, 1379 CrossRef CAS PubMed.
- P. Gai, W. Yu, H. Zhao, R. Qi, F. Li, L. Liu, F. Lv and S. Wang, Solar-powered organic semiconductor–bacteria biohybrids for CO2 reduction into acetic acid, Angew. Chem., Int. Ed., 2020, 59, 7224–7229 CrossRef CAS PubMed.
- Y. J. Kim, H. Hong, J. Yun, S. I. Kim, H. Y. Jung and W. Ryu, Photosynthetic nanomaterial hybrids for bioelectricity and renewable energy systems, Adv. Mater., 2021, 33, 2005919 CrossRef CAS PubMed.
- J. Liu, J. Mantell, N. Di Bartolo and M. R. Jones, Mechanisms of self-assembly and energy harvesting in tuneable conjugates of quantum dots and engineered photovoltaic proteins, Small, 2019, 15, 1804267 CrossRef PubMed.
- W. Hillier and G. T. Babcock, Photosynthetic reaction centers, Plant Physiol., 2001, 125, 33–37 CrossRef CAS PubMed.
- G. Amoruso, J. Liu, D. W. Polak, K. Tiwari, M. R. Jones and T. A. A. Oliver, High-efficiency excitation energy transfer in biohybrid quantum dot-bacterial reaction center nanoconjugates, J. Phys. Chem. Lett., 2021, 12, 5448–5455 CrossRef CAS PubMed.
- F. Milano, R. R. Tangorra, O. Hassan Omar, R. Ragni, A. Operamolla, A. Agostiano, G. M. Farinola and M. Trotta, Enhancing the light harvesting capability of a photosynthetic reaction center by a tailored molecular fluorophore, Angew. Chem., Int. Ed., 2012, 51, 11019–11023 CrossRef CAS PubMed.
- P. K. Dutta, S. Lin, A. Loskutov, S. Levenberg, D. Jun, R. Saer, J. T. Beatty, Y. Liu, H. Yan and N. W. Woodbury, Reengineering the optical absorption cross-section of photosynthetic reaction centers, J. Am. Chem. Soc., 2014, 136, 4599–4604 CrossRef CAS PubMed.
- A. M. Hancock, S. A. Meredith, S. D. Connell, L. J. C. Jeuken and P. G. Adams, Proteoliposomes as energy transferring nanomaterials: enhancing the spectral range of light-harvesting proteins using lipid-linked chromophores, Nanoscale, 2019, 11, 16284–16292 RSC.
- A. M. Hancock, M. Son, M. Nairat, T. Wei, L. J. C. Jeuken, C. D. P. Duffy, G. S. Schlau-Cohen and P. G. Adams, Ultrafast energy transfer between lipid-linked chromophores and plant light-harvesting complex II, Phys. Chem. Chem. Phys., 2021, 23, 19511–19524 RSC.
- A. M. Hancock, D. J. K. Swainsbury, S. A. Meredith, K. Morigaki, C. N. Hunter and P. G. Adams, Enhancing the spectral range of plant and bacterial light-harvesting pigment-protein complexes with various synthetic chromophores incorporated into lipid vesicles, J. Photochem. Photobiol., B, 2022, 237, 112585 CrossRef CAS PubMed.
- S. Nandi, R. Malishev, S. K. Bhunia, S. Kolusheva, J. Jopp and R. Jelinek, Lipid-bilayer dynamics probed by a carbon dot-phospholipid conjugate, Biophys. J., 2016, 110, 2016–2025 CrossRef CAS PubMed.
- R. S. Wu, Y. S. Lin, A. Nain, B. Unnikrishnan, Y. F. Lin, C. R. Yang, T. H. Chen, Y. F. Huang, C. C. Huang and H. T. Chang, Evaluation of chemotherapeutic response in living cells using subcellular organelle–selective amphipathic carbon dots, Biosens. Bioelectron., 2022, 211, 114362 CrossRef CAS PubMed.
- Y. F. Lin, Y. S. Lin, T. Y. Huang, S. C. Wei, R. S. Wu, C. C. Huang, Y. F. Huang and H. T. Chang, Photoswitchable carbon-dot liposomes mediate catalytic cascade reactions for amplified dynamic treatment of tumor cells, J. Colloid Interface Sci., 2022, 628, 717–725 CrossRef CAS PubMed.
- A. Pannwitz, H. Saaring, N. Beztsinna, X. Li, M. A. Siegler and S. Bonnet, Mimicking photosystem I with a transmembrane light harvester and energy transfer-induced photoreduction in phospholipid bilayers, Chem, 2021, 27, 3013–3018 CrossRef CAS PubMed.
- N. Sinambela, R. Jacobi, D. Hernandez-Castillo, E. Hofmeister, N. Hagmeyer, B. Dietzek-Ivansic, L. Gonzalez and A. Pannwitz, Alignment and photooxidation dynamics of a perylene diimide chromophore in lipid bilayers, Mol. Syst. Des. Eng., 2023, 8, 842–852 RSC.
- A. Pannwitz, D. M. Klein, S. Rodríguez-Jiménez, C. Casadevall, H. Song, E. Reisner, L. Hammarström and S. Bonnet, Roadmap towards solar fuel synthesis at the water interface of liposome membranes, Chem. Soc. Rev., 2021, 50, 4833–4855 RSC.
- N. Sinambela, J. Bosking, A. Abbas and A. Pannwitz, Recent advances in light energy conversion with biomimetic vesicle membranes, ChemBioChem, 2021, 22, 3140–3147 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |