
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of N-heterocyclic carbene (NHC)-supported heavier nitrile ylides†
Debotra
Sarkar
a,
Sayan
Dutta
 b,
Franziska
Hanusch
a,
Debasis
Koley
*b and
Shigeyoshi
Inoue
*a
b,
Franziska
Hanusch
a,
Debasis
Koley
*b and
Shigeyoshi
Inoue
*a
aTUM School of Natural Sciences, Department of Chemistry, Institute of Silicon Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstraße 4, 85748, Garching, Germany. E-mail: s.inoue@tum.de
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741 246, India. E-mail: koley@iiserkol.ac.in
First published on 9th January 2024
Abstract
The synthesis and isolation of stable heavier analogues of nitrile ylide as N-heterocyclic carbene (NHC) adducts of phosphasilenyl-tetrylene [(NHC)(TerAr)Si(H)PE14(TerAr)] (E14 = Ge 1, Sn 2; TerAr = 2,6-Mes2C6H3, NHC = IMe4) are reported. The delocalized Si–P–E14 π-conjugation was examined experimentally and computationally. Interestingly, the germanium derivative 1 exhibits a 1,3-dipolar nature, leading to an unprecedented [3 + 2] cycloaddition with benzaldehyde, resulting in unique heterocycles containing four heteroatoms from group 14, 15, and 16. Further exploiting the nucleophilicity of germanium, activation of the P–P bond of P4 was achieved, leading to a [(NHC)(phosphasilenyl germapolyphide)] complex. Moreover, the [3 + 2] cycloaddition and the σ-bond activation by 1 resemble the characteristics of the classic nitrile ylide.
Introduction
1,3-Dipolar cycloadditions with nitrile ylides, a central theme in Huisgen's chemistry, offer a versatile approach for heteroatom incorporation into five-membered rings.1–3 Nitrile ylides, with a C–N–C framework and delocalized π-electrons, exhibit diverse structures from bent to linear.1–4 These highly reactive species, generated in situ, engage in cycloadditions with dienophiles, yielding valuable N-heterocycles for applications in materials science and natural product synthesis.2,5 The electronic configurations of nitrile ylides reveal a rich diversity, categorized into six forms (I–VI, Scheme 1a), each imparting distinct reactivity.6–10 Noteworthy, among these are nitrile ylides possessing allyl-zwitterionic (V) and carbenic character (VI), exhibiting dual ambiphilicity reminiscent of the ‘imino carbene’ concept.11,12 This unique characteristic allows these species to engage in both 1,3-dipolar additions and carbene-type reactivity, including [3 + 2] cycloaddition, σ-bond activation, or dimerization via the carbene carbon (Scheme 1b).10–12 However, the isolation of the elusive free carbenic nitrile ylide (type VI, Scheme 1a) remains a formidable task, with only one structurally characterized nitrile ylide known, possessing a propargylic structure.13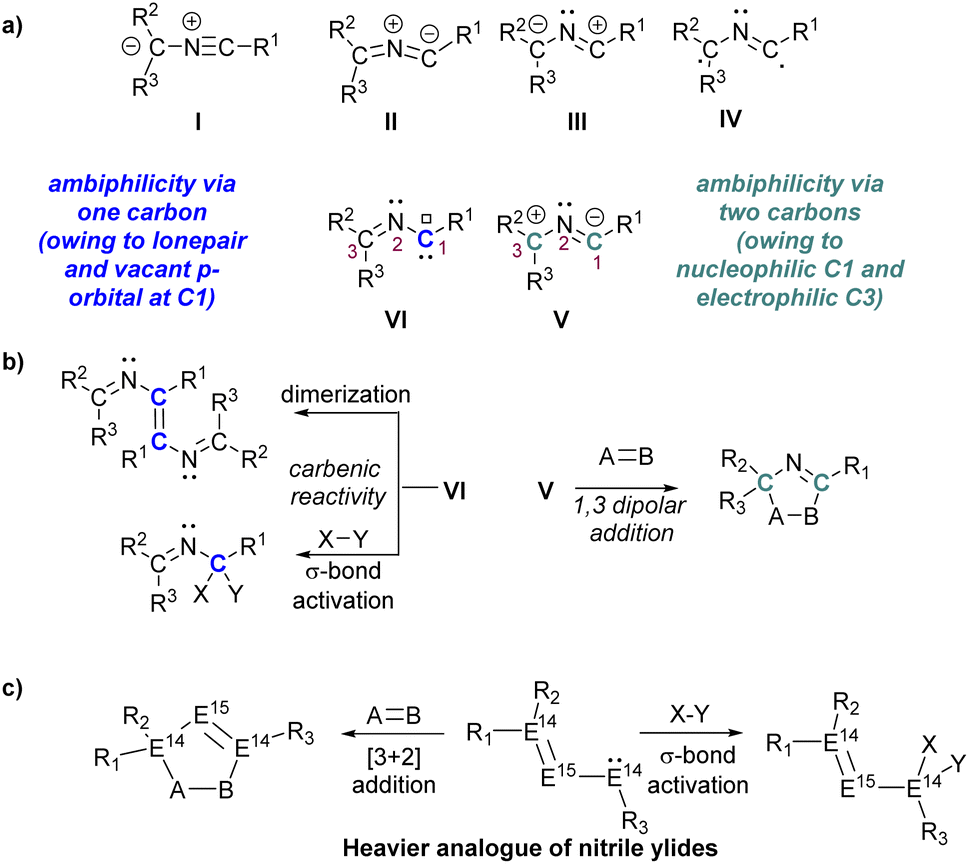 | ||
| Scheme 1 (a) Resonance structures of nitrile ylides, (b) reactivity of nitrile ylides, (c) reactivity of heavier analogues of nitrile ylides. | ||
In recent years, significant progress has been made in exploring heavier main group compounds, particularly those with multiple bonds between group 14–15 elements and their derivatives.14–33 This advancement is attributed to their unique synthetic potential and growing applications, ranging from small molecule activation to catalysis. While the chemistry of phosphasilenes (R2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PR) is well-established within group 14–15 multiple bonded compounds,14 those featuring multiple bonds between heavier group-14 elements (E14, where E14 = Ge–Sn) and phosphorus remain relatively scarce.16–18,22,24,30–33 The inherent polarity of the E14P bond often results in intriguing 1,2-dipolar behaviour, leading to cycloadditions or addition onto the E14P bonds. Notably, among E14–P multiple bonded compounds, the homo or hetero-allenic heavier analogues of nitrile ylides (R2E14E15E14R, E14 = Si–Pb, E15 = P, Scheme 1c) remains elusive, despite the known constitutional isomers of “heavy heteroallenes” (R2E14E14=E15R) that behave as 1,3-dipoles.34–37 The persistent elusiveness of these compounds underscores the intricate challenges associated with their synthesis, highlighting uncharted territories within the expansive field of heavy main group chemistry.38,39
PR) is well-established within group 14–15 multiple bonded compounds,14 those featuring multiple bonds between heavier group-14 elements (E14, where E14 = Ge–Sn) and phosphorus remain relatively scarce.16–18,22,24,30–33 The inherent polarity of the E14P bond often results in intriguing 1,2-dipolar behaviour, leading to cycloadditions or addition onto the E14P bonds. Notably, among E14–P multiple bonded compounds, the homo or hetero-allenic heavier analogues of nitrile ylides (R2E14E15E14R, E14 = Si–Pb, E15 = P, Scheme 1c) remains elusive, despite the known constitutional isomers of “heavy heteroallenes” (R2E14E14=E15R) that behave as 1,3-dipoles.34–37 The persistent elusiveness of these compounds underscores the intricate challenges associated with their synthesis, highlighting uncharted territories within the expansive field of heavy main group chemistry.38,39
In this study, we introduce N-heterocyclic carbene (NHC)-coordinated bent nitrile ylides 1 and 2, offering preliminary insights into the structural, bonding, and reactive characteristics of these intriguing heavier nitrile ylide analogues. Compound 1, demonstrating both 1,3-dipolar addition and classical carbene reactivity, emerges as a compelling analogue reminiscent of type VI and type V nitrile ylides, respectively. The inorganic ‘Huisgen’-type 1,3-dipolar additions of 1 towards the dienophile (PhCHO) resulted in the formation of an elusive cyclic five-membered phosphagermene (3), featuring the four heteroatoms Si, P, Ge, and O, including three heavier main group elements. Meanwhile, the activation of P4 by 1 led to the formation of the NHC-phosphasilenyl germapolyphide (4).
Results and discussion
The hypothesis that P-metallo-phosphasilenes (R2SiP[M], M = metal fragment) could exhibit 1,3-dipolar behaviour was grounded in the contribution of the zwitterionic B form to the electronic ground state (Scheme 2a).40,41 Achieving the B form requires a coordinated effort involving electronic push on silicon and pull through the metal centre (M). This coordination is facilitated by a metal with an energetically accessible p-orbital associated with a ligand providing a ‘pull’ effect, such as aryl.16 Therefore, we envisioned that the introduction of an electron-donating NHC-stabilized bulky (aryl)phosphasilene to the tetrylene centre via trimethylsilyl chloride (TMSCl) elimination could lead to the heavier nitrile ylides [(NHC)(R2SiP–E14–R)], where the coordination of the NHC to silicon not only provides thermodynamic stabilization but also pushes the electron density from the SiP moiety to the vacant p-orbital of the tetrylene centre.14,42 Using this incentive, we treated aryl tetrylene chloride [TerArE14Cl, E14 = Ge, Sn]43 with one equivalent of [(NHC)(TerAr)Si(H)PTMS]44 at room temperature (Scheme 2b). This reaction resulted in the elimination of TMSCl and the selective formation of stable NHC-phosphasilenyl-tetrylene compounds 1 and 2. Both compounds were isolated in good yields as red and orange crystals, exhibiting solubility in polar solvents while displaying limited solubility in nonpolar organic solvents such as benzene or toluene. Analysis of their 31P NMR spectra in C6D6 revealed signals relatively downfield at 150.7 ppm (1; calc. 165.3 ppm) and 94.5 ppm (2; calc. 141.1 ppm; 1J(119Sn,P) = 1554 Hz) compared to the starting material [(NHC)(TerAr)Si(H)PTMS] (δ(31P) = −323 ppm; calc. −341.6 ppm).44 These values fell within the range reported for phosphagermenes (R2GePR, 87–416 ppm)21,24–26,30,32 and phosphastannenes (R2SnPR, −25.1–204 ppm)17,20,30 The 119Sn NMR spectrum of 2 displayed a doublet at 1691 ppm (1J(119Sn,P) = 1554 Hz), falling within the range reported for bis(aryl)stannylenes (980–2323 ppm) and significantly downfield-shifted compared to phosphastannenes signals (499–658 ppm), indicating pronounced tetrylene character.16–18 The 1J(119Sn,P) coupling constant, positioned between typical values for Sn–P single (ca. 1000 Hz) and double bonds (2208–2295 Hz),30 suggested the multiple bond character of the Sn–P bond. The 29Si NMR spectra of 1 and 2 displayed distinct signals at −25.5 (calc. −26.5 ppm) and −22.7 ppm (calc. −23.9 ppm), respectively. The Si–P coupling constants for 1 (137 Hz) and 2 (142 Hz) fell within the range observed for NHC-coordinated phosphasilenes [(NHC)(R2SiPR)], 1J(29Si,P) = 120–157 Hz).14
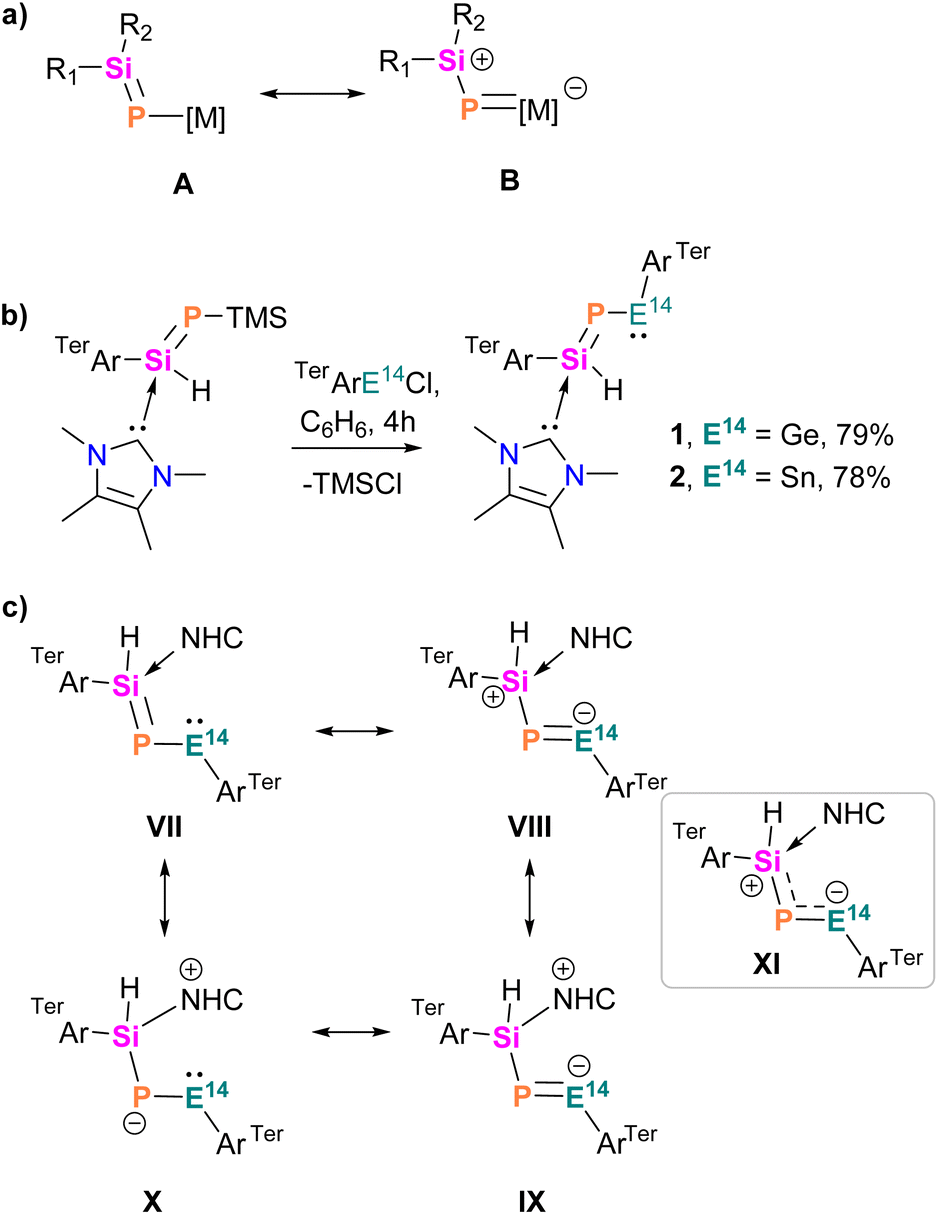 | ||
| Scheme 2 (a) Resonance structures of P-metallo-phosphasilenes, (b) synthesis of NHC-phosphasilenyl-tetrylene complexes 1 and 2, (c) possible resonance structures of 1 and 2. | ||
Single crystal X-ray diffraction (SC-XRD) analysis provided detailed insights into the structural features of compounds 1 and 2 (Fig. 1). In both compounds, the heavier metal centres (Ge and Sn) are di-coordinated and bound by a phosphasilene moiety [(NHC)(TerAr)Si(H)P] and an m-terphenyl group (TerAr). The bond angles around the group 14 metals are (∢P1–E14–C1: 102.6°(5) at Ge, and 95.8°(8) at Sn), larger than monomeric tetrylene-phosphinidenes TerArE14P(IDipp) (E14 = Ge, Sn; 89.5° at Ge, 86.6° at Sn)16 and germylidenylpnictinidenes (93.0° at Ge) but within the range of acyclic bis(aryl)- and bis(phosphido)-tetrylenes.18–20,45 The E14–P bond distances in 1 (Ge1–P1: 2.236(6) Å) and 2 (Sn1–P1: 2.451(2) Å) are shorter than typical Ge–P and Sn–P single bonds and those in (aryl)(phosphido)-tetrylenes (TippAr)[(Me3Si)2P]E14 (Ge–P: 2.291 Å, Sn–P: 2.527 Å; TippAr = 2,4-(2,4,6-iPr3C6H2)2C6H3),46 cyclic phospha(germylene) (Ge–P: 2.247 Å),25 and digerma-2,4-diphosphacyclobutadiene (avg. Ge–P: 2.26 Å).32,47 The Ge1–P1 and Sn1–P1 bond distances in 1 and 2 are comparable to those in bis(phosphido)tetrylenes [Dipp2P]2E14 (Ge–P: 2.234 Å, Sn–P: 2.447 Å),19,20TerArE14P(IDipp) (Ge–P: 2.236 Å, Sn–P: 2.456 Å), germylidenylpnictinidenes (Ge–P: 2.242 Å),18 and cyclic phosphagermene (Ge–P: 2.22 Å),21 but longer than those in acyclic-phosphagermenes (2.138–2.174 Å)22,24,31 and phosphastannenes (2.342 Å).17 The Si1–P1 bond lengths in 1 (2.201(6) Å) and 2 (2.191(2) Å) are shorter than Si–P single bonds (average 2.26 Å) and slightly longer than NHC-stabilized aryl-phosphasilene (2.15–2.16 Å).14,48,49 Overall, this suggests effective π-delocalization across the Si–P–E14 motif due to the donation from the NHC to Si (push effect) and the π-electron acceptance of the vacant p-orbital of the divalent germanium or tin atoms (pull effect).
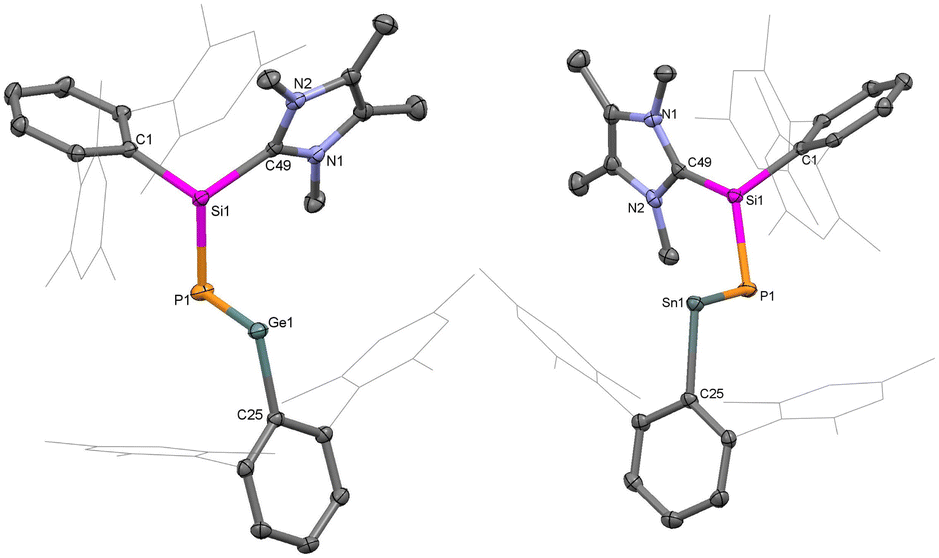 | ||
| Fig. 1 Molecular structures of compound 1 (left) and 2 (right) in the solid state. Ellipsoids are set at the 50% probability level; hydrogen atoms are omitted for clarity. | ||
Theoretical calculations confirmed the electronic ground states of 1 and 2 to be singlet with singlet–triplet energy gaps (ΔES–T) of 32.4 and 35.1 kcal mol−1, respectively. Inspection of the molecular orbitals reveal that the HOMOs in 1 and 2 represent E14–P σ-orbitals, while the π(E14–P) symmetry with a major contribution from the more electronegative phosphorus is found in the HOMO−1 (Fig. 2 and S27†). The LUMOs possess vacant pπ-orbitals on the carbene carbons. The Wiberg bond indices (WBI) indicate partial double bond character for Si–P (1.118/1.170) and E14–P (1.338/1.146) in 1/2. Natural bond orbital (NBO) analysis locates lone pairs of electrons each on P (1.940/1.938 e) and E14 (1.946/1.967 e) centres in 1/2. The bent geometries of the compounds are attributed to the participation of almost pure p-orbitals of phosphorus in the formation of Si–P and E14–P bonds (Table S2†).50 The positive Laplacian [∇2ρ(r)] values at (3, −1) bond critical points of the Si–CNHC bonds (+0.158/+0.155 in 1/2), as suggested by quantum theory of atoms in molecules (QTAIM) calculations, indicate its donor–acceptor nature (Table S3†). In addition, the negative Laplacian values of the Si–P bonds (−0.136/−0.124) indicate true covalent interactions,51 whereas the E14–P bonds (−0.019/+0.042) exhibit highly polarized covalent nature. Therefore, both compounds can be better represented by the resonance form XI. Furthermore, comparison of the formation energies of 1 (−16.7 kcal mol−1) and 2 (−10.9 kcal mol−1) with their carbon (−56.6 kcal mol−1) and silicon (−17.0 kcal mol−1) analogues indicate that the lighter congeners might also be considered as viable targets for syntheses (Fig. S28†).
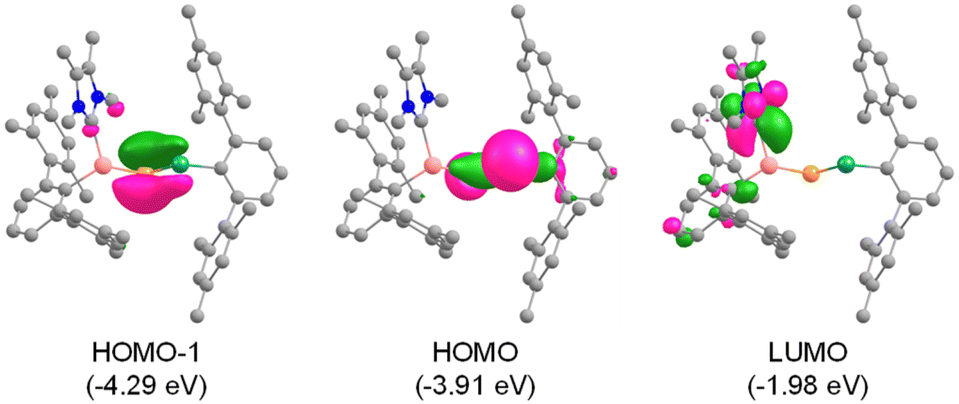 | ||
| Fig. 2 Selected molecular orbitals of 1 (isosurface = 0.05 a.u.). Hydrogen atoms are omitted for clarity. | ||
The electronic features of 1 and 2 differ significantly from the P-plumbyleniophosphasilene [tBu3Si(R)SiP–PbL, L = β-diketiminate, R = 2,4,6-iPr3C6H2],52 where the π-delocalization across Si–P–Pb is perturbed due to the N-donation of N,N-substituted β-diketiminato (NacNac) to the vacant p-orbital of Pb and the poor (Pb)pπ–(P)pπ interaction, leading to a preference for the A type of electronic structure in the ground state (Scheme 2a). In contrast, a facile π-delocalization observed across the Si–P–E14 motif in 1 and 2, led to the electronic form B.
The reactivity of P-metallo-phosphasilenes remains unexplored. To investigate the 1,3-dipolar nature of 1 and 2, the compounds were treated with one equivalent amount of dipolarophile (such as PhCHO, Scheme 3), resulted in the formation of oxaphosphasilagermole 3 in the case of 1. The reaction led to an immediate colour change from orange to yellow, and the subsequent analysis by multinuclear NMR confirmed the quantitative conversion of 1 to 3. The 31P NMR of 3 displayed a distinct signal at −279 ppm, shifted upfield compared to 1. The 29Si NMR of 3 showed a downfield shift compared to 1, and the Si–P coupling constant indicated a single bond between Si and P. SC-XRD analysis of 3 revealed a nonplanar five-membered ring, representing the first molecular snapshot of true heterocycles containing four heteroatoms of group 14, 15, and 16, with a low-valent phosphorus centre (Fig. 3). The Ge1–P1 bond length in 3 (2.248(1) Å) was shorter than the average Ge–P single bonds, indicating its partial multiple bond character. Therefore, compound 3 stands as a unique example of a base-stabilized cyclic five-membered phosphagermene. This finding is noteworthy, particularly in the context of 1,3-dipolar additions involving main group ylides with heavier elements, which are barely observed.34–36,53–55
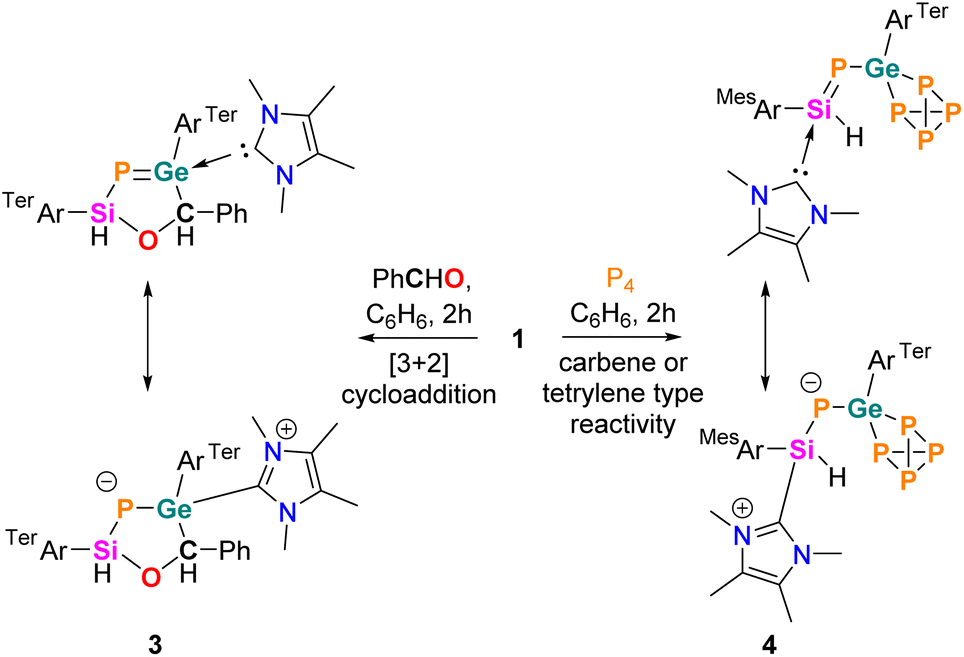 | ||
| Scheme 3 Synthesis of compounds 3 and 4 and their possible zwitterionic structures. | ||
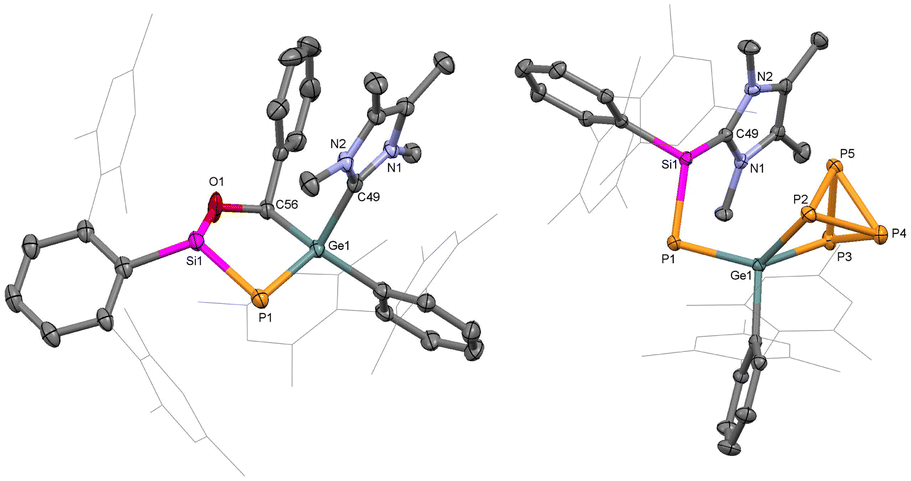 | ||
| Fig. 3 Molecular structures of compounds 3 (left) and 4 (right) in the solid state. Ellipsoids are set at the 50% probability level; hydrogen atoms are omitted for clarity. | ||
The HOMO−1 and HOMO in 3 possess lone pair orbitals located on phosphorus, whereas the LUMO shows the vacant pπ-orbital on the carbene carbon (Fig. S29†). The partial double bond character in the Ge–P bond with calculated WBI of 1.140 in 3 gets significantly reduced compared to 1. Moreover, the formation of 3 demonstrates the nucleophilicity of germanium and the electrophilicity of the silicon centre in 1, like the 1,3-dipolar nature of nitrile ylides. Additionally, in 3, the migration of NHC from silicon to the germanium centre suggests a donor–acceptor type interaction between Si and NHC in 1. Notably, the reactivity of 1 with PhCHO markedly differs from the known reactivity of carbonyl compounds (R2CO) with EP multiple bonded complexes or with terylenes (R2E, E = Si–Pb). For example, reactivity of phosphasilene [R2SiPSiiPr3, R = 2,4,6-iPr3(C6H2)], phosphagermene, and TerArE14P(IDipp) with carbonyl led to the [2 + 2] cycloaddition product,16,56,57 whereas the reactivity of a silylene with carbonyl led to the [1 + 2] cycloaddition product58 or forms the silacarbonyl ylide.59,60 The reactivity of 1 with PhCHO could resemble the intramolecular vicinal frustrated P/B Lewis pair (FLP) [e.g. (C6F5)2P–CHEt–B(C6F5)2]-mediated activation of PhCHO, where, in the case of 1, the germanium centre acts as a Lewis base and silicon behaves as a Lewis acid centre.61,62
Mechanistic investigations using a truncated model with the bulkier m-terphenyl group replaced with 2,6-dimethylphenyl,63,64 suggest that the [2 + 2] cycloaddition of PhCHO into the Ge–P bond in 1M demands an energy barrier of 8.2 kcal mol−1 (Fig. 4). Similarly, the [1 + 2] cycloaddition of PhCHO at the Ge centre requires an energy barrier of 9.3 kcal mol−1. Instead, the [3 + 2] cycloaddition of PhCHO needs to surmount a remarkably lower barrier of 5.3 kcal mol−1, resulting in slightly more stable INT-4 with a noticeably longer Si–CNHC bond (2.236 Å). The subsequent de-coordination of carbene from the Si centre via the transition state TS-5 leads to INT-5. Finally, a barrierless re-coordination of NHC to the Ge centre in INT-5 furnishes 3M. Importantly, substantial stabilization of 3M compared to [2 + 2] and [1 + 2] cycloaddition intermediates indicates that the reaction is thermodynamically governed.
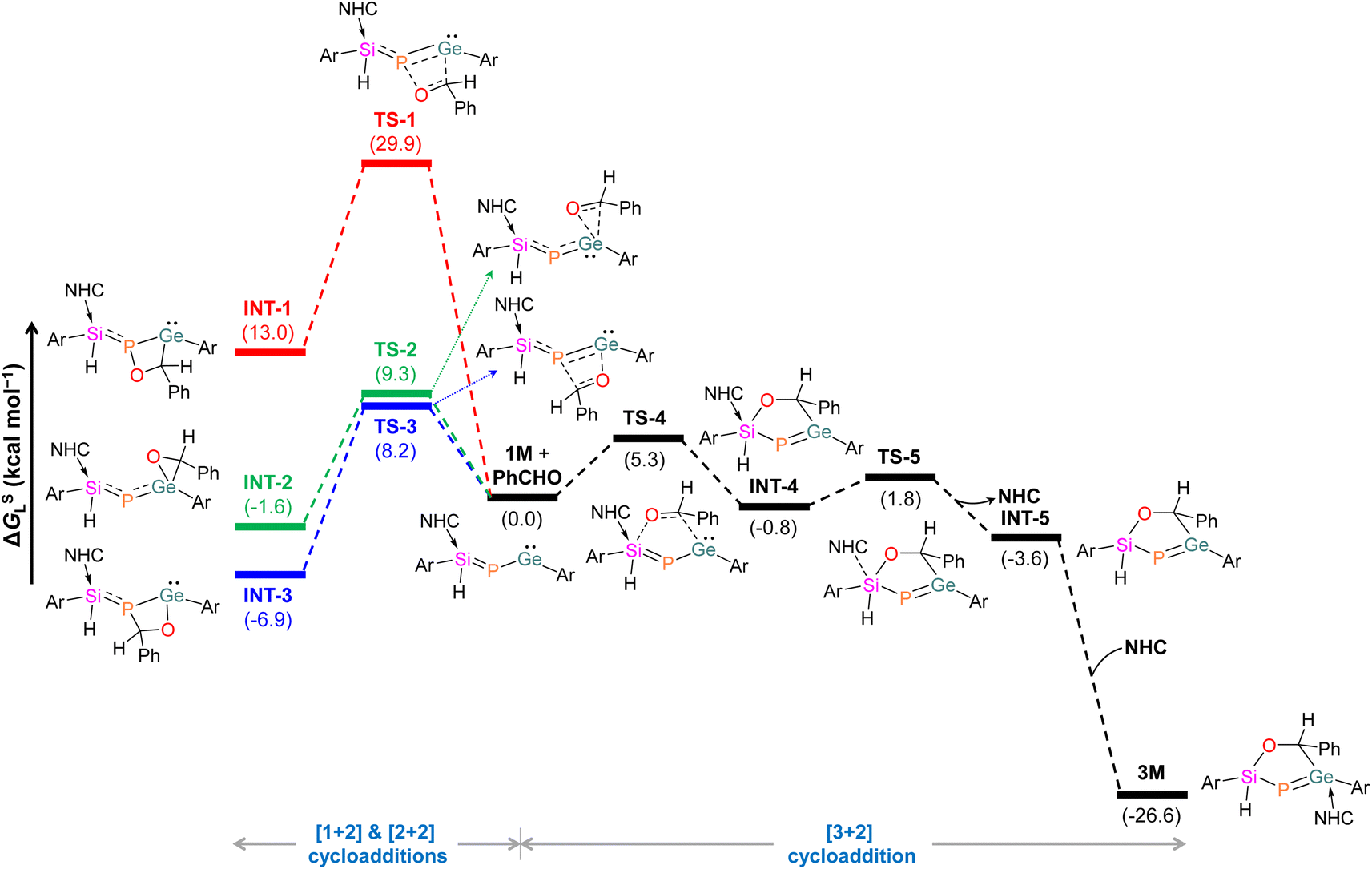 | ||
| Fig. 4 Energy profile for the formation of 3M. Ar = 2,6-dimethylphenyl; NHC = 1,3,4,5-tetramethylimidazol-2-ylidene. | ||
Thermally induced reversible addition of carbonyl across the E14–P bond is known. Therefore, compound 3 was heated at elevated temperature (80 °C) under vacuum to verify reversibility. However, no evidence of reformation of 1 or elimination of PhCHO from 3 was observed, as confirmed by 1H and 31P NMR. In contrast, no reaction was observed between 2 and PhCHO, possibly due to the low nucleophilicity of tin. Reactivity of 1 and 2 with ketones such as acetone, benzophenone, etc., was also tested, but no reaction was observed. This is possibly attributed to steric hindrance of the bulky aryl group attached to the group 14 metal centre. Further experimental studies revealed the inertness of 1 and 2 towards small molecules such as H2, CO, HCCPh, H2CCH2, HBpin, and PhSiH3 despite appreciably lower HOMO–LUMO energy gaps (ΔEH–L: 1.93/2.04 eV in 1/2).
Interestingly, the reaction of P4 with 1 afforded selective insertion of Ge into the P–P bond, leading to germanium polyphosphide 4. The 31P NMR spectrum revealed resonances for five distinct 31P nuclei (X, X′, A, A′ and Y) at δ = 197.7 (m, PX), 186.4 (m, PX′), −125.9 (m, PY), −280.6 (m, PA), −287.7 (m, PA′). A similar type of P4 activation was observed with seven-membered cyclic-benzamido-carbene leading to a carbene-P4 adduct.65 The isolation of 4 confirmed the carbenic nitrile ylide-like reactivity of 1. Importantly, regioselective activation of P4 by tetrylenes is rare.66–68 Compound 4 is isostructural with reported LSiP4 (L = β-diketiminate),69 (MesTer)2GeP4,70 and [MesTerSn(P4)SitBu3] complexes.71 In 4, the Ge1–P1 bond (2.245(9) Å) is slightly longer than the Ge1–P1 bond in 1 but sufficiently shorter than the Ge1–P2 (2.372(9) Å) and Ge1–P3 (2.396(9) Å) single bonds (Fig. 3). Furthermore, the Si1–P1 (2.165(1) Å) bond in 4 is shorter than in 1 (Si–P: 2.20 Å) and lies in the range of NHC-stabilized aryl-phosphasilenes (2.15–2.16 Å). Thus, 4 represents the first example of an [(NHC)phosphasilenyl germapolyphide]. Moreover, P4 activation by the Ge lone pair in 1M results in 4M with an energy barrier of 6.9 kcal mol−1, indicating the strong germylene nature of 1 (Fig. S30†).
The reactivity of 1 with P4 is contrary to silyl-phosphasilene-mediated P4 activation. For example, Trip2SiPR, (R = SiMe2tBu, SiiPr3) was shown to activate P4via SiP bond cleavage.72 Moreover, the reactivity of 2 with P4 led to a complicated mixtures of phosphorus-containing products. Notably, TerArEP(IDipp) (E = Ge, Sn) did not activate P4.16 Attempts to abstract NHC by treating 1 and 2 with Lewis acids (e.g., BPh3 and B(C6F5)3) to generate donor-free phosphasilene-tetrylenes resulted in decomposed mixtures. This emphasizes the crucial role of NHC in stabilizing 1 and 2. Additionally, photolysis of (1, 2, and 4) and proton scavenging from 1 and 2 with the trityl cation [Ph3C][B(C6F5)4] led to inconclusive mixtures.
Conclusions
In conclusion, the study has demonstrated the synthesis, electronic structure, and reactivity of NHC-phosphasilenyl-stabilized germylene 1 and stannylene 2. These compounds contain a heteroleptic delocalized π-bonding motif (Si–P–E14, E14 = Ge, Sn) and provide access to the zwitterionic or carbenic form of the heavier nitrile-ylide analogues, as demonstrated for the germanium derivative. The synthesis of the ‘true heterocycle’733 from 1 represents the first example of the ‘inorganic Huisgen-type [3 + 2] cycloaddition’ reported for the group 14–15 multiple bonded complexes. Further coordination chemistry and catalytic applications of these intriguing compounds are currently under investigation.Author contributions
D. S. carried out the synthetic and reaction studies, S. D. carried out the computational analyses, F. H. conducted the crystallographic studies. D. S., S. D., D. K. and S. I. wrote the manuscript. D. K. and S. I. managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from WACKER Chemie AG and DAAD (Fellowship D. S.). S. D. acknowledges the CSIR, India for the Senior Research Fellowship (SRF) and IISER Kolkata for the computational facility. D. K. acknowledges the funding from MoE-STARS (MoE-STARS/STARS-1/255) scheme. Open access funding enabled and organized by Projekt DEAL.Notes and references
- R. Huisgen, J. Org. Chem., 1976, 41, 403–419 CrossRef CAS.
- M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
- K. Livingstone, G. Little and C. Jamieson, Synthesis, 2021, 53, 2395–2407 CrossRef CAS.
- P. Caramella and K. N. Houk, J. Am. Chem. Soc., 1976, 98, 6397–6399 CrossRef CAS.
- C. Wentrup, S. Fischer, H.-M. Berstermann, M. Kuzaj, H. Lüerssen and K. Burger, Angew Chem. Int. Ed. Engl., 1986, 25, 85–86 CrossRef.
- D. Bégué, C. Addicott, R. Burgard, P. Bednarek, E. Guille, I. Baraille and C. Wentrup, J. Org. Chem., 2014, 79, 2148–2155 Search PubMed.
- H. Inui and S. Murata, J. Am. Chem. Soc., 2005, 127, 2628–2636 CrossRef CAS PubMed.
- O. L. Chapman and J. P. Le Roux, J. Am. Chem. Soc., 1978, 100, 282–285 CrossRef CAS.
- G. Maier, C. Schmidt, H. P. Reisenauer, E. Endlein, D. Becker, J. Eckwert, B. H. Andes Jr and L. J. Schaad, Chem. Ber., 1993, 126, 2337–2352 CrossRef CAS.
- S. Fergus, S. J. Eustace and A. F. Hegarty, J. Org. Chem., 2004, 69, 4663–4669 CrossRef CAS PubMed.
- Y. K. Loh, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2023, 145, 2064–2069 CrossRef CAS PubMed.
- Y. K. Loh, M. Melaimi, M. Gembicky, D. Munz and G. Bertrand, Nature, 2023, 623, 66–70 CrossRef CAS PubMed.
- E. P. Janulis, S. R. Wilson and A. J. Arduengo, Tetrahedron Lett., 1984, 25, 405–408 CrossRef CAS.
- V. Nesterov, N. C. Breit and S. Inoue, Chem.–Eur. J., 2017, 23, 12014–12039 CrossRef CAS PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed.
- V. Nesterov, R. Baierl, F. Hanusch, A. E. Ferao and S. Inoue, J. Am. Chem. Soc., 2019, 141, 14576–14580 Search PubMed.
- M. Fischer, M. M. D. Roy, L. L. Wales, M. A. Ellwanger, A. Heilmann and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 8908–8913 CrossRef CAS PubMed.
- Y. He, C. Dai, D. Wang, J. Zhu and G. Tan, J. Am. Chem. Soc., 2022, 144, 5126–5135 CrossRef CAS PubMed.
- K. Izod, D. G. Rayner, S. M. El-Hamruni, R. W. Harrington and U. Baisch, Angew. Chem., Int. Ed., 2014, 53, 3636–3640 Search PubMed.
- K. Izod, P. Evans, P. G. Waddell and M. R. Probert, Inorg. Chem., 2016, 55, 10510–10522 CrossRef CAS PubMed.
- Y. Wu, Z. Zhao, T. Chen, J. Tan, Z.-W. Qu, S. Grimme, Y. Zhao and D. W. Stephan, Chem.–Eur. J., 2022, 28, e202200666 CrossRef CAS PubMed.
- D. Raiser, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2021, 27, 14073–14080 Search PubMed.
- M. Zweigart, C. Wenzel, K. Eichele, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2023, 62, e202304200 CrossRef CAS PubMed.
- V. Y. Lee, M. Kawai, A. Sekiguchi, H. Ranaivonjatovo and J. Escudié, Organometallics, 2009, 28, 4262–4265 Search PubMed.
- N. Del Rio, M. Lopez-Reyes, A. Baceiredo, N. Saffon-Merceron, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 1365–1370 CrossRef CAS PubMed.
- P. Coburger, F. Masero, J. Bösken, V. Mougel and H. Grützmacher, Angew. Chem., Int. Ed., 2022, 61, e202211749 CrossRef CAS PubMed.
- V. Y. Lee, M. Kawai, O. A. Gapurenko, V. I. Minkin, H. Gornitzka and A. Sekiguchi, Chem. Commun., 2018, 54, 10947–10949 RSC.
- C. Präsang, M. Stoelzel, S. Inoue, A. Meltzer and M. Driess, Angew. Chem., Int. Ed., 2010, 49, 10002–10005 CrossRef PubMed.
- S. Yao, Y. Grossheim, A. Kostenko, E. Ballestero-Martínez, S. Schutte, M. Bispinghoff, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7465–7469 CrossRef CAS PubMed.
- C. Couret, J. Escudié, J. Satge, A. Raharinirina and J. D. Andriamizaka, J. Am. Chem. Soc., 1985, 107, 8280–8281 CrossRef CAS.
- M. Draeger, J. Escudié, C. Couret, H. Ranaivonjatovo and J. Satge, Organometallics, 1988, 7, 1010–1013 CrossRef CAS.
- S. Yao, Y. Xiong, T. Szilvási, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 4781–4785 CrossRef CAS PubMed.
- M. J. Reveley, J. Feld, D. Temerova, E. S. Yang and J. M. Goicoechea, Chem.–Eur. J., 2023, 29, e202301542 CrossRef CAS PubMed.
- D. Ghereg, E. André, J.-M. Sotiropoulos, K. Miqueu, H. Gornitzka and J. Escudié, Angew. Chem., Int. Ed., 2010, 49, 8704–8707 CrossRef CAS PubMed.
- D. Ghereg, E. André, H. Gornitzka, J. Escudié, F. Ouhsaine, N. Saffon, K. Miqueu and J.-M. Sotiropoulos, Chem.–Eur. J., 2011, 17, 12763–12772 CrossRef CAS PubMed.
- D. Ghereg, J.-M. Sotiropoulos, J. Escudié, K. Miqueu, D. Matioszek, S. Ladeira and N. Saffon-Merceron, Organometallics, 2012, 31, 930–940 CrossRef CAS.
- J. Escudié, H. Ranaivonjatovo and L. Rigon, Chem. Rev., 2000, 100, 3639–3696 CrossRef PubMed.
- C. Weetman, Chem.–Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed.
- P. P. Power, Organometallics, 2020, 39, 4127–4138 Search PubMed.
- M. Driess, H. Pritzkow and U. Winkler, J. Organomet. Chem., 1997, 529, 313–321 CrossRef CAS.
- M. Driess, S. Block, M. Brym and M. T. Gamer, Angew. Chem., Int. Ed., 2006, 45, 2293–2296 Search PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- R. S. Simons, L. Pu, M. M. Olmstead and P. P. Power, Organometallics, 1997, 16, 1920–1925 CrossRef CAS.
- D. Sarkar, V. Nesterov, T. Szilvási, P. J. Altmann and S. Inoue, Chem.–Eur. J., 2019, 25, 1198–1202 CrossRef CAS PubMed.
- P. Wilfling, K. Schittelkopf, M. Flock, R. H. Herber, P. P. Power and R. C. Fischer, Organometallics, 2015, 34, 2222–2232 CrossRef CAS.
- B. P. Johnson, S. Almstätter, F. Dielmann, M. Bodensteiner and M. Scheer, Z. Anorg. Allg. Chem., 2010, 636, 1275–1285 CrossRef CAS.
- Y. Wu, L. L. Liu, J. Su, J. Zhu, Z. Ji and Y. Zhao, Organometallics, 2016, 35, 1593–1596 CrossRef CAS.
- M. K. Bisai, T. Das, K. Vanka, R. G. Gonnade and S. S. Sen, Angew. Chem., Int. Ed., 2021, 60, 20706–20710 CrossRef CAS PubMed.
- C.-W. So, H. W. Roesky, P. M. Gurubasavaraj, R. B. Oswald, M. T. Gamer, P. G. Jones and S. Blaurock, J. Am. Chem. Soc., 2007, 129, 12049–12054 Search PubMed.
- S. Dutta, B. Maity, D. Thirumalai and D. Koley, Inorg. Chem., 2018, 57, 3993–4008 Search PubMed.
- S. Kundu, C. Mohapatra, P. P. Samuel, J. Kretsch, M. G. Walawalkar, R. Herbst-Irmer, D. Stalke, S. De, D. Koley and H. W. Roesky, Chem. Commun., 2017, 53, 192–195 RSC.
- S. Yao, S. Block, M. Brym and M. Driess, Chem. Commun., 2007, 3844–3846 RSC.
- L. Zhu and R. Kinjo, Angew. Chem., Int. Ed., 2022, 61, e202207631 CrossRef CAS PubMed.
- R. Guo, J. Jiang, C. Hu, L. L. Liu, P. Cui, M. Zhao, Z. Ke, C.-H. Tung and L. Kong, Chem. Sci., 2020, 11, 7053–7059 RSC.
- L. L. Liu, J. Zhou, L. L. Cao and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 16971–16982 Search PubMed.
- N. C. Breit, T. Szilvási and S. Inoue, Chem.–Eur. J., 2014, 20, 9312–9318 CrossRef CAS PubMed.
- H. Ranaivonjatovo, J. Escudié, C. Couret and J. Satgé, J. Organomet. Chem., 1991, 415, 327–333 CrossRef CAS.
- D. Gau, R. Rodriguez, T. Kato, N. Saffon-Merceron, F. P. Cossío and A. Baceiredo, Chem.–Eur. J., 2010, 16, 8255–8258 CrossRef CAS PubMed.
- W. Ando, K. Hagiwara and A. Sekiguchi, Organometallics, 1987, 6, 2270–2271 CrossRef CAS.
- L. E. Bourque and K. A. Woerpel, Org. Lett., 2008, 10, 5257–5260 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- A. Stute, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2013, 42, 4487–4499 RSC.
- D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS PubMed.
- D. Sarkar, S. Dutta, C. Weetman, E. Schubert, D. Koley and S. Inoue, Chem.–Eur. J., 2021, 27, 13072–13078 CrossRef CAS PubMed.
- C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486–4488 RSC.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 Search PubMed.
- S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169–2179 RSC.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511–4513 CrossRef CAS PubMed.
- J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem.–Eur. J., 2014, 20, 6739–6744 CrossRef CAS PubMed.
- D. Sarkar, C. Weetman, D. Munz and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 3519–3523 Search PubMed.
- M. Drieß, Angew Chem. Int. Ed. Engl., 1991, 30, 1022–1024 CrossRef.
- P. Cheshire, A. M. Z. Slawin and J. D. Woollins, Inorg. Chem. Commun., 2002, 5, 803–804 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2300364–2300367. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06430a |
| This journal is © The Royal Society of Chemistry 2024 |