
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling aggregation-induced emission by supramolecular interactions and colloidal stability in ionic emitters for light-emitting electrochemical cells†
Alba
Sanz-Velasco
 a,
Olivia
Amargós-Reyes
b,
Aya
Kähäri
a,
Sophia
Lipinski
b,
Luca M.
Cavinato
a,
Rubén D.
Costa
*b,
Mauri A.
Kostiainen
a and
Eduardo
Anaya-Plaza
*a
a,
Olivia
Amargós-Reyes
b,
Aya
Kähäri
a,
Sophia
Lipinski
b,
Luca M.
Cavinato
a,
Rubén D.
Costa
*b,
Mauri A.
Kostiainen
a and
Eduardo
Anaya-Plaza
*a
aDepartment of Bioproducts and Biosystems, Aalto University, Kemistintie 1, 02150 Espoo, Finland. E-mail: eduardo.anaya@aalto.fi
bTechnical University of Munich, Campus Straubing for Biotechnology and Sustainability, Schulgasse 22, 94315 Straubing, Germany. E-mail: ruben.costa@tum.de
First published on 25th January 2024
Abstract
Chromophores face applicability limitations due to their natural tendency to aggregate, with a subsequent deactivation of their emission features. Hence, there has been a fast development of aggregation induced emission (AIE) emitters, in which non-radiative motional deactivation is inhibited. However, a fine control of their colloidal properties governing the emitting performance is fundamental for their application in thin film optoelectronics. In addition, ion-based lighting devices, such as light emitting electrochemical cells (LECs), requires the design of ionic AIE emitters, whose structure allows (i) an easy ion polarizability to assist charge injection and (ii) a reversible electrochemical behavior. To date, these fundamental questions have not been addressed. Herein, the hydrophilic/hydrophobic balance of a family of cationic tetraphenyl ethene (TPE) derivatives is finely tuned by chemical design. The hydrophilic yet repulsive effect of pyridinium-based cationic moieties is balanced with hydrophobic variables (long alkyl chains or counterion chemistry), leading to (i) a control between monomeric/aggregate state ruling photoluminescence, (ii) redox behavior, and (iii) enhanced ion conductivity in thin films. This resulted in a LEC enhancement with the first ionic AIE emitters, reaching values of 0.19 lm W−1 at ca. 50 cd m−2. Overall, this design rule will be key to advance ionic active species for optoelectronics.
Introduction
Organic fluorophores have drawn intensive attention during last decades for their practical applications, named medical imaging, light emitting diodes (LEDs), or chemical sensors.1–4 However, their use finds limitations due to the partial or complete emission quenching derived from aggregation, leading to the ‘aggregation caused quenching’ (ACQ). This is consequence of their usually large aromatic structures that lead to π–π stacking, and subsequently deactivates the photoexcited state(s) through non-radiative pathways.5 This phenomenon typically occurs at high concentrations, aqueous media given the high hydrophobicity of these derivatives, or the condensed/solid state.5,6 Thus, aggregation limits their applicability as high payload imaging agents, phototherapeutic agents, or in lighting devices (e.g., OLEDs or solar cells), respectively. While chemical design of non-aggregating dyes,7,8 or directed aggregation9,10 has been pursued with success, a radically different approach has been explored to turn dye aggregation into a desirable feature.Aggregation induced emission (AIE) is a photophysical phenomenon coined in 2001, in which aggregate formation enhances the light emitting process.11,12 AIE emitters (AIEgens) in their monomeric state suffer from emission quenching caused by intramolecular vibrations and rotations in e.g., organic solvents, leading to emission quenching. In contrast, rigidification caused by aggregation blocks intramolecular motion, recovering their emitting properties.13–15 Consequently, AIEgens exhibit emitting properties in aggregate state. Such exceptional characteristics have made them suitable for fields where traditional photosensitizers performance is hindered.16–18
Despite the plethora of AIEgen families, ionic AIE structures are understudied, likely given the a priori counterproductive properties: charged molecules will present enhanced aqueous solubility that, together with the coulombic repulsion, hinder self-assembly and, therefore, the AIE properties.19 For instance, ionic AIE probes have been used as protein labelling agents, with the AIE properties arising upon biomolecular recognition.20 However, a deep understanding of factors influencing aggregation and monomeric states of ionic AIEgens in aqueous media and solid-state is crucial for controlling system forces and then, expanding applications (e.g. ultra-sensitive mechanochromic luminescent materials).21 In this context, ionic AIEgens present a unique potential for ionic-based optoelectronics, such as light-emitting electrochemical cells (LECs).22 They are considered the simplest thin film lighting device, in which the presence of ions in the active layer allows (i) an efficient charge injection from air stable electrodes, and (ii) a controlled electrochemical doping of the emitter forming a stable p-i-n junction, in which exciton radiative recombination occurs. Recently, Edman's group showed for the first time that neutral AIE emitters with a rigid and emissive structure will be compatible with ionic electrolytes, realizing better devices than analogous emitters with typical ACQ.23 However, the use of ionic AIE emitters as well as the design principles to simultaneously merge desired photophysical, electrochemical, and ion transport for LECs remains unanswered.
Herein we present a new versatile platform of positively charged structures derived from tetra-(4-pyridylphenyl)-ethylene 1,1,2,2-tetrakis(4-(pyridin-4-yl)phenyl)ethene (TPPE) that exhibits AIE effect upon aggregation.24–28 They present a propeller-like structure formed by four phenyl groups moving freely in the monomeric state. The hydrophilic/hydrophobic balance governing the AIE effect is finely tuned by the length of alkyl chain and the counterion: on one hand, the hydrophilicity is introduced by the positively charged pyridinium moieties that increase the solubility in aqueous media. On the other hand, the alkyl chain and the counterion nature modulate the hydrophobicity, and thus, overcoming electrostatic repulsions and promoting self-aggregation. We demonstrate the impact of the mentioned forces on the optical properties in organic solvent (monomeric) and aqueous media (monomeric or aggregate) at different concentrations in the series of halide TPPE-alkyl salts and hexafluorophosphate (PF6−) TPPE-alkyl salts. The length of the alkyl chains plays a key role on self-aggregation for TPPE-alkyl salts: C12 chains are needed in order to turn on the AIE effect. PF6TPPE-alkyl salts, however, exhibit an enhanced AIE at lower concentrations and with shorter alkyl chains as well as a more reversible electrochemical behavior. Furthermore, we demonstrate the impact of the molecular structure in the electrochemical behavior and ion conductivity in thin films. This series was further applied to LECs nicely showing that (i) the emission features of the films enhance with the AIE behavior, (ii) the ion-assisted mechanism is fully operative and (iii) a linear trend to increase device figures of merit upon increasing the alkyl chain. This resulted in the first LECs with ionic AIE emitters, exhibiting a yellow emission associated to ca. 50 cd m−2 and 0.19 lm W−1, which is in the range of small molecules LECs.29
Results and discussion
Aggregation induced emission of cationic TPPE derivatives
 | ||
| Scheme 1 Synthesis of the family of TPPE-alkyl salts via Suzuki cross-coupling reaction and alkylation. | ||
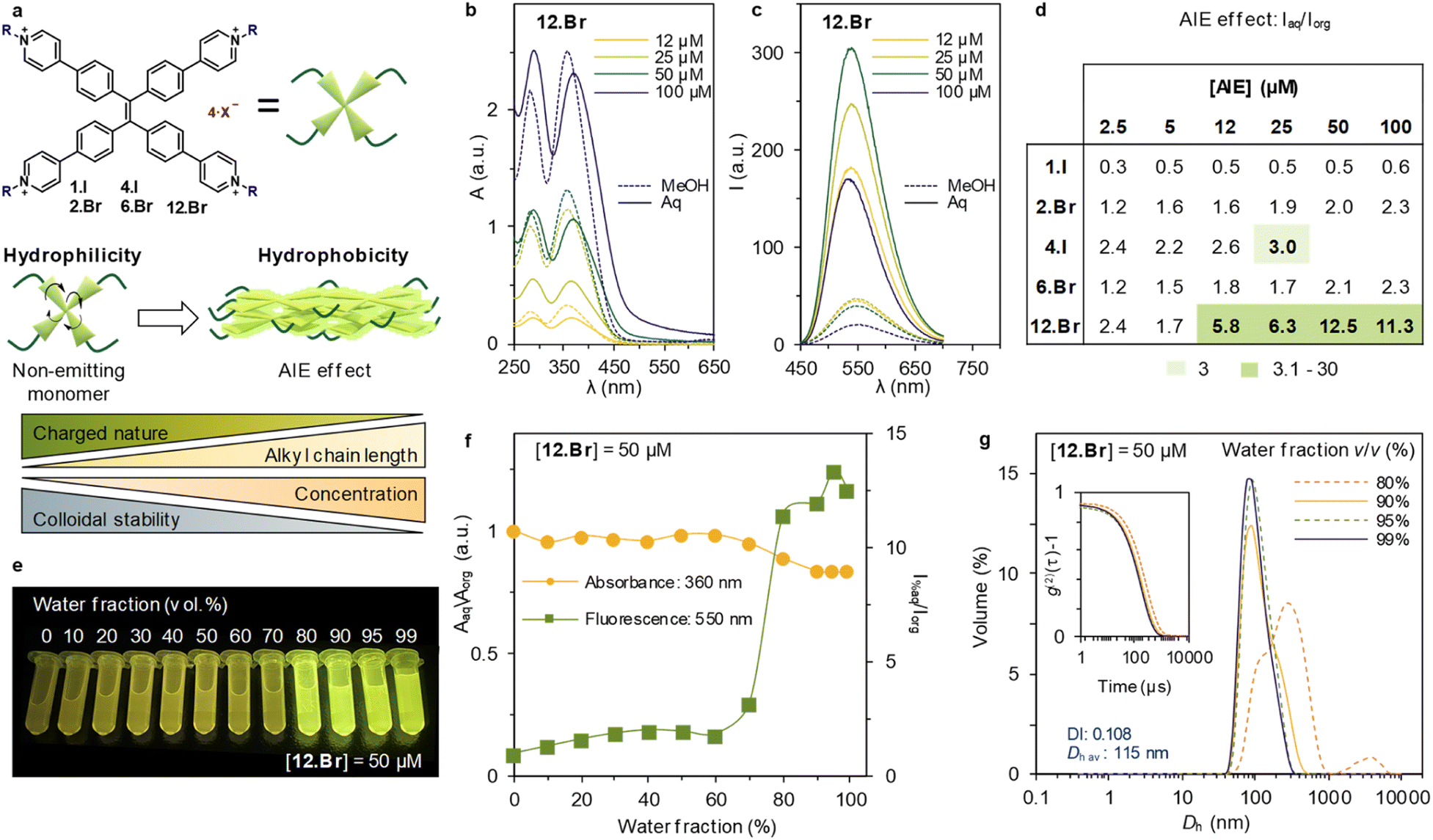 | ||
| Fig. 1 (a) Schematic representation of the TPE derivatives, as well as the key design features and supramolecular driving forces leading to monomeric or aggregating state in aqueous media. (b) UV-vis absorbance spectra of 12.Br at different concentrations (full and dashed lines for sample in aqueous and organic media, respectively). (c) Emission spectra of 12.Br at different concentrations (full and dashed lines for sample in aqueous and organic media, respectively) upon excitation at 360 nm. (d) Ratio Iaq/Iorg at maximum of emission (550 nm) at varying concentrations of the series. In light green and green are marked results with a ratio of 3 and 3.1–30, respectively. (e) Image of 12.Br ([12.Br] = 50 μM) titration at different water fraction content, showing increased emission above 80% of water. (f) Titration of [12.Br] = 50 μM at varying water percentage. Absorbance (orange line) at 360 nm and emission (green line) at 550 nm upon excitation at 360 nm. A sharp increase can be found above 80% water/MeOH mixture. (g) Volume-averaged particle size distribution of [12.Br] = 50 μM measured by DLS, showing high dispersity at 80% and size stabilization at 90% and above. No data was obtained for solutions below 80% water, indicating lack of aggregates. Inset: second order normalized autocorrelation function of the selected samples. Dispersity index (DI) and volume-averaged hydrodynamic diameter (Dh av.) correspond to average obtained after three repetitions of 12.Br at 99% water/MeOH mixture. | ||
First, the absorbance spectra of the halide TPPE-alkyl salts are recorded at different concentrations, both in organic and aqueous media (Aorg and Aaq, respectively), meaning aqueous media 99% of water content in the sample (ESI Fig. S62†). The synthetized family of compounds shows consistent absorption maxima, centered at ca. 280 and 360 nm (Fig. 1b) with minor changes between organic and aqueous solvent. However, 12.Br shows a minor shift in aqueous media, that becomes more evident with the increase in concentration (Fig. 1b). Further analysis of the A360 dependence with the concentration (see S62†) shows linearity, in good agreement with the Lambert–Beer law, for the whole series.
Subsequently, the emission spectra upon excitation at 360 nm is recorded in organic and aqueous media (Iorg and Iaq, respectively). The photoluminescence intensity is shown at different concentrations for 12.Br in Fig. 1c and S62† for the rest of the series. The general trend shows enhanced emission in aqueous media (Iaq > Iorg), which is in agreement with the AIE effect. In order to determine the extent of this increase through the series, Fig. 1d shows the ratio between Iaq and Iorg at different concentrations. 4.I shows limited solubility in organic solvents, and therefore cannot be dissolved in a concentration higher than 2.5 mM, limiting the concentration to 25 μM.
Furthermore, we consider a minimum threshold of three-fold increase (marked as light green on the table) to be a relevant AIE effect. This threshold is a desirable noise-to-signal ratio in e.g., microscopy. The table at Fig. 1d highlights the interplay between the alkyl side chains and the electrostatic repulsions: only 12.Br above 12 μM shows an emission increase of 5.8, whereas 6.Br at the same concentration only displays an increase of 1.8 times. This exemplifies the role of alkyl chains in the formation of emitting aggregates. Therefore, the optimized conditions of the series are obtained for [12.Br] = 50 μM, and thus, a thorough study at different water percentages was carried out at this concentration under these conditions.
First, the emitting properties at decreasing organic fraction solvents are recorded. The AIE effect as described above is turned on when the sample contains over 80% of water content (Fig. 1e). Notwithstanding, once aggregates are formed, the emission obtained for the sample at 99% of water is almost 13 times higher compared to in organic solvent (Fig. 1f). The relationship between aggregation and emitting properties is further confirmed by monitoring the size distribution measured by DLS at different water fractions. It was observed that aggregates start forming at 80% of water content in the sample (see ESI Fig. S66†). After, structures reduce the diameter from 185 nm (80% water, DI: 0.178) to 115 nm (99% water, DI: 0.108).
Counterion chemistry as aggregation inducing emission parameter
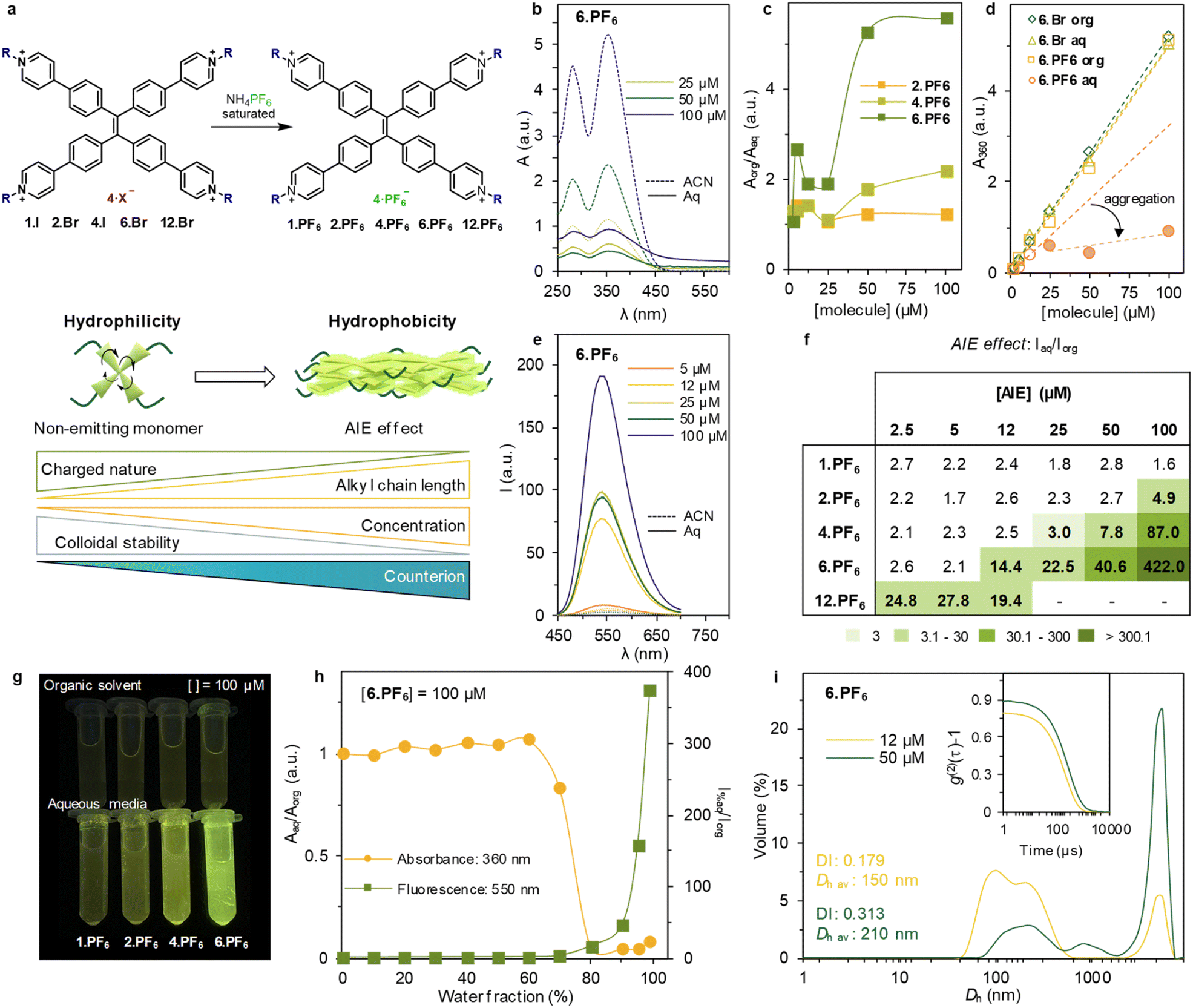 | ||
| Fig. 2 (a) Synthetic path for ion exchange of the series. Scheme of the effect of the counterion modification in supramolecular driving forces that lead to monomeric or aggregated state in aqueous media. (b) UV-vis absorbance spectra of 6.PF6 at different concentrations (full and dashed lines for sample in aqueous and organic media, respectively). (c) Ratio Aorg/Aaq at 360 nm at different concentrations for 2.PF6, 4.PF6 and 6.PF6, showing a sharp increase due to aggregation for 6.PF6. (d) A360 at different concentrations for 6.Br in organic solvent (blue rhombus), 6.Br in aqueous media (green triangles), 6.PF6 in organic solvent (yellow squares) and 6.PF6 in aqueous media (red circles). The trendlines are marked with the same colour. In the case of 6.PF6, there is a trendline based on the three first concentrations and a second trendline (light red) corresponding to the results at 25, 50 and 100 μM highlighting the self-aggregation at these concentrations. (e) Emission spectra of 6.PF6 at different concentrations (full and dashed lines for samples in aqueous and organic media, respectively) upon excitation at 360 nm. (f) Ratio Iaq/Iorg at maximum of emission (550 nm) at varying concentrations of the PF6-family compounds. Different green intensities have been used to emphasize the AIE effect. At [12.PF6] > 12 μM sample not soluble in water. (g) Images of 1.PF6, 2.PF6, 4.PF6 and 6.PF6 (left to right) at 100 μM in organic solvent (upper row) and aqueous media (lower row). (h) Titration of [6.PF6] = 50 μM varying water percentage. Absorbance results at 360 nm and fluorescence results at 550 nm by excitation at 360 nm. (i) Volume-averaged particle size distribution of [6.PF6] = 12 μM (yellow) and 50 μM (green) in aqueous media measured by DLS. Inset: second order normalized autocorrelation function of the selected samples. DI and Dh av. correspond to average obtained after three repetitions at 99% water/MeOH mixture. | ||
| Square wave voltammetry | |||
|---|---|---|---|
| E ox (V)/EHOMO (eV) | E red (V)/ELUMO (eV) | ΔE (HOMO–LUMO) (eV)/(nm) | |
| a Calculated from the onset of the oxidation–reduction potential.33 | |||
| 1.PF6 | 1.3, 1.6/−6.3a | −1.4, −1.6/−3.8a | 2.5/496 |
| 2.PF6 | 1.1, 1.3, 1.5/−6.3a | −1.4, −1.6/−3.8a | 2.5/496 |
| 4.PF6 | 1.1, 1.5, 1.9/−6.2a | −1.4, −1.6/−3.8a | 2.6/477 |
| 6.PF6 | 1.1, 1.3, 1.5/−6.2a | −1.4, −1.6/−3.8a | 2.6/477 |
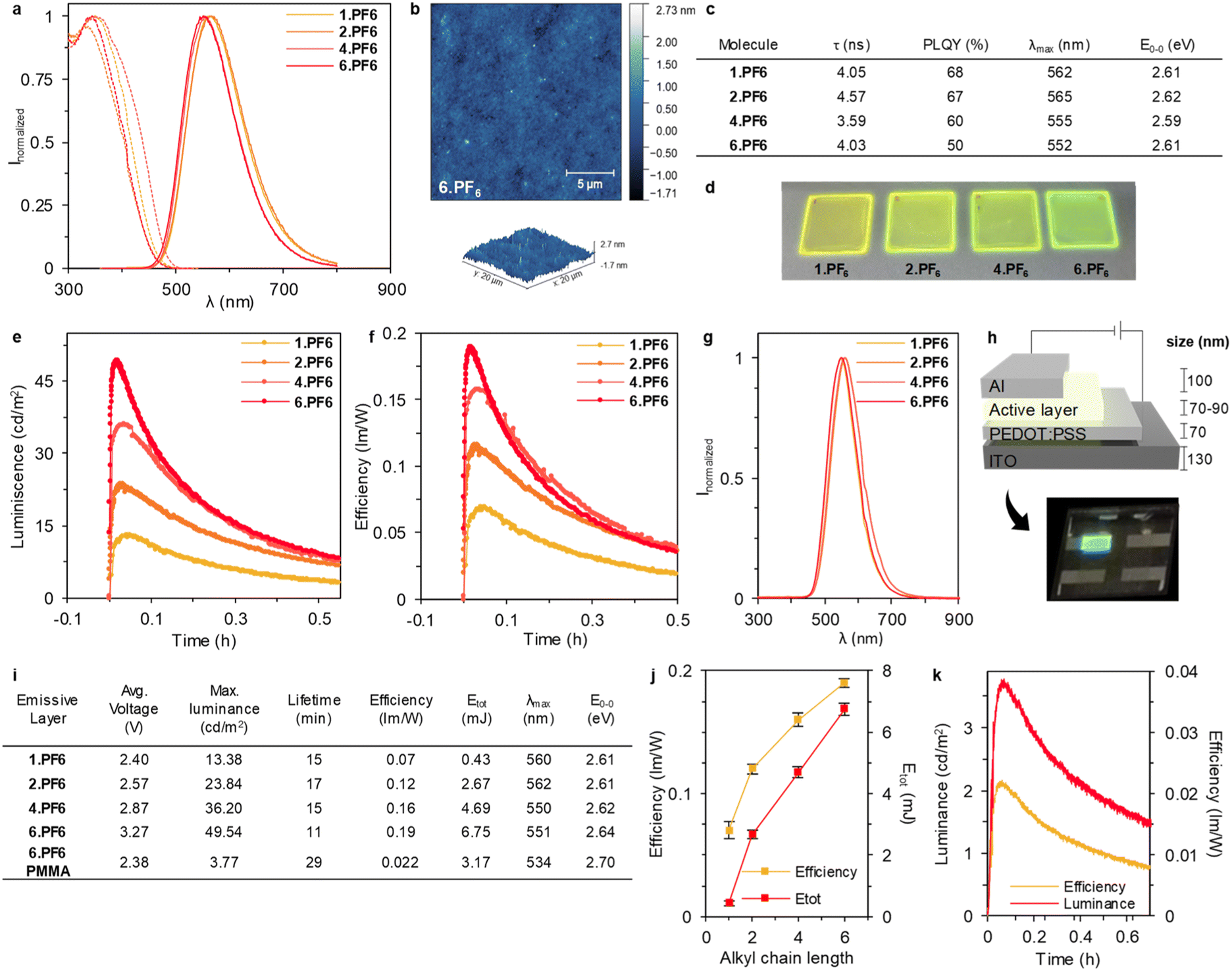 | ||
| Fig. 3 (a) AFM topography of the 6.PF6 film dried under vacuum treatment, and its profile (bottom). (b) Excitation and emission spectra (dashed and full lines, respectively) in thin films. (c) Photoluminescence features of the films. (d) Pictures under UV illumination (305 nm) of the thin films deposited on quartz. (e) Luminance and (f) efficiency of the LECs. (g) Normalized electroluminescence spectra of the devices. (h) Schematic of the device's architecture, in addition to an optical image of a device's pixel in operando. (i) Devices performance at pulsed current density driving of 100 mA cm−2. LECs prepared with 6.PF6 mixed with PMMA (6.PF6-PMMA) to disrupt aggregation. (j) Efficiency (yellow) and total emitted energy (red) in relationship with alkyl chains length. (k) Luminance (red) and efficiency (yellow) of LECs made with 6.PF6-PMMA. | ||
Next, we studied the electroluminescent behaviour of the LECs at pulsed current density of 100 mA cm−2. As expected, all the devices exhibited a reduction of the applied voltage along with an increase of luminance response (Fig. 3e, f and S70†). Indeed, an instantaneous brightness response associated to a broad emission spectrum centred at 550 nm and 560 nm for 1.PF6/2.PF6 and 4.PF6/6.PF6, respectively. Thus, photo-/electroluminescence band shapes and the maximum emission wavelength remains without notable variation for each molecule, indicating that the same excited state is involved regardless of the photo/electrical excitation process over time. (E0–0 and λmax in Fig. 3c (PL) and Fig. 3i (EL). While the emission band is stable for 4.PF6/6.PF6 devices, those of 1.PF6/2.PF6 slightly red-shifts over time due to the increase of the device temperature to ca. 45 °C – vide supra. Overall, the device chromaticity corresponds to a greenish emission as corroborated by the x/y CIE colour coordinates of 0.40–4/0.51–4. More interesting, the luminous efficiency and device brightness gradually increases with the length of the alkyl chain, reaching values of 0.07, 0.12, 0.16, and 0.19 lm W−1 and 13.38, 23.84, 36.20, and 49.54 cd m−2 for 1.PF6, 2.PF6, 4.PF6 and 6.PF6 devices (Fig. 3e and f). This is nicely reflected in the device stability studied using the total emitted energy (Etot) that is calculated by integrating the radiant flux curve until the time needed to reach one-fifth of the maximum luminance as an upper limit definition (Fig. 3i).22 Here, Etot gradually increases from 0.47 mJ to 6.75 mJ upon increasing the alkyl length chain (Fig. 3i). Indeed, Fig. 3j displays a clear enhancement of the device efficiency and stability with the increase of the alkyl chain as expected by the higher and stable thin-film PLQY under working conditions – vide supra.
However, a high PLQY does not directly imply the best performing device, since charge trapping and/or electrochemical doping critically alter the preferred composition of the active material.41 In this context, the enhanced reversibility of the electrochemical processes keeping a good ion polarizability in thin films upon increasing the alkyl chain is also key towards circumventing hole injection/transport limitations.
To further confirm the benefits of the AIE behaviour, we prepared another set of devices using an inert polymer matrix to disrupt aggregation, that is, adding 20 wt%. of polymethylmethacrylate (PMMA) with respect to the best emitting molecule 6.PF6 (see Experimental section for details).42 As showed in Fig. 3k, the device performance with PMMA decreases considerably with a luminance from 49.54 (pristine) to 3.77 (with PMMA) cd m−2, and an efficiency from 0.19 (pristine) to 0.022 (with PMMA) lm W−1, corroborating that the aggregation induced emission is, indeed, crucial for this type of emitters.
Conclusions
In summary, two different families of positively charged structures derived from TPPE were successfully synthetized, and the one with optimized performance applied in ion-based lighting devices, such as LECs, to rationalize the effect of the hydrophobic/hydrophilic forces (represented by the positively charged pyridinium moieties and the increasing chain length and counterion, respectively) that govern the AIE behaviour. For instance, halide AIE series require C12 side chain lengths to overcome the inherent electrostatic repulsions and self-aggregation, while PF6− AIE series increases the hydrophobic nature promoting the aggregate formation and gradually enhancing their emitting properties from C2 up to C6 side chains. This was nicely corroborated by the gradual PLQY increase going from 1.PF6 to 6.PF6 films. In addition, a reasonable electrochemical behaviour that allows their integration in LECs was also noted in the PF6− series. Here, the devices featured a gradually enhanced brightness, efficiency and stability going from 1.PF6 to 6.PF6 emitters, indicating that the AIE feature is beneficial. Indeed, EIS assays corroborated that the lock structure of AIE emitters allows and efficient ion polarization regardless of their neutral or ionic nature;23 a question that has always discouraged the community to apply this type of materials in LECs. Finally, we also state that the AIE feature is key towards meeting high performance by using PMMA additive that disrupt the formation of aggregates LECs and, in turn, dramatically reduced the device performance of 6.PF6 AIE emitter. Overall, this work rationalizes a design guideline for ionic AIE emitters with fine-tuned colloidal stability, encouraging their future application in water-based applications such nanomedicine or sensor or, as in this case, light-management technologies like lighting emitting electrochemical cells.Data availability
A initial version of the manuscript was deposited on ChemRxiv, DOI: https://10.26434/chemrxiv-2023-v464k.Author contributions
The experimental work was carried out mainly by ASV (synthesis and structural characterization of molecules, in solution optical properties and size studies of the aggregates by DLS), OA-R (thin films characterization, device fabrication), AK (spectroscopy), SL (electrochemical characterization), and LMC (electrochemical impedance spectroscopy). Project conceptualization and main funding acquisition: EA-P and RDC. Project discussion, experimental design, data analysis: EA-P, MAK and RDC. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from the Academy of Finland (341057 and 346632). We acknowledge as well, the Academy of Finland Centers of Excellence Program (2022–2029) in Life-Inspired Hybrid Materials (LIBER) 346110. OA-R and RDC thanks the funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 899987. LMC and RDC acknowledged the European Union's MSCA-ITN programme under grant agreement STiBNite No. 956923. We are also thankful to the Aalto Internal Funding Call 2022 – For Cooperation Initiatives with Technical University of Munich (TUM). We acknowledge the provision of facilities and technical support by Aalto University Bioeconomy and Raw Materials Research Infrastructure (RAMI) facilities.References
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed
.
- M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed
- Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517–4546 RSC
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed
-
J. B. Birks, Photophysics of Aromatic Molecules, 1971 Search PubMed
- T. Förster and K. Kasper, Z. Elektrochem. Angew. Phys. Chem., 1955, 59, 976–980 CrossRef
- J. Kollar, M. Machacek, M. Halaskova, J. Lenco, R. Kucera, J. Demuth, M. Rohlickova, K. Hasonova, M. Miletin, V. Novakova and P. Zimcik, J. Med. Chem., 2020, 63, 7616–7632 CrossRef CAS PubMed
- M. Halaskova, A. Rahali, V. Almeida-Marrero, M. Machacek, R. Kucera, B. Jamoussi, T. Torres, V. Novakova, A. De La Escosura and P. Zimcik, ACS Med. Chem. Lett., 2021, 12, 502–507 CrossRef CAS PubMed
- A. Rahali, A. Shaukat, V. Almeida-Marrero, B. Jamoussi, A. De La Escosura, T. Torres, M. A. Kostiainen and E. Anaya-Plaza, Bioconjugate Chem., 2021, 32, 1123–1129 CrossRef CAS PubMed
- E. Anaya-Plaza, A. Aljarilla, G. Beaune, Nonappa, J. V. I. Timonen, A. de la Escosura, T. Torres and M. A. Kostiainen, Adv. Mater., 2019, 31, 1902582 CrossRef PubMed
- J. Luo, Z. Xie, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC
- B. Z. Tang, X. Zhan, G. Yu, P. P. Sze Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974–2978 RSC
- Z. Zhou, S. Xie, X. Chen, Y. Tu, J. Xiang, J. Wang, Z. He, Z. Zeng and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 9803–9807 CrossRef CAS PubMed
- Y. Tu, J. Liu, H. Zhang, Q. Peng, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 14911–14914 CrossRef CAS PubMed
- Q. Li and L. Blancafort, Chem. Commun., 2013, 49, 5966–5968 RSC
- N. Zhao, M. Li, Y. Yan, J. W. Y. Lam, Y. L. Zhang, Y. S. Zhao, K. S. Wongd and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 4640–4646 RSC
- X. Chen, X. Y. Shen, E. Guan, Y. Liu, A. Qin, J. Z. Sun and B. Zhong Tang, Chem. Commun., 2013, 49, 1503–1505 RSC
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC
- X. Liu, C. Zhu and B. Z. Tang, Acc. Chem. Res., 2022, 55, 197–208 CrossRef CAS PubMed
- Y. Tian, X. Yan, M. L. Saha, Z. Niu and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 12033–12036 CrossRef CAS PubMed
- W. Yin, Z. Yang, S. Zhang, Y. Yang, L. Zhao, Z. Li, B. Zhang, S. Zhang, B. Han and H. Ma, Mater. Chem. Front., 2021, 5, 2849–2859 RSC
- L. M. Cavinato, G. Millán, J. Fernández-Cestau, E. Fresta, E. Lalinde, J. R. Berenguer and R. D. Costa, Adv. Funct. Mater., 2022, 32, 2201975 CrossRef CAS
- S. Tang, Z. Wang, Y. Xu, H. Ma, J. Wang, C. Larsen, D. Dang, E. Wang and L. Edman, Angew. Chem., Int. Ed., 2023, 62, e202302874 CrossRef CAS PubMed
- L. Li, M. Zhang, K. Kang and C. Xiao, Inorg. Chem., 2022, 61, 19933–19943 CrossRef CAS PubMed
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed
- Y. Qi, L. Yu, C. Liu, N. Wang, X. Xu and X. Xin, J. Mol. Liq., 2023, 386, 122504 CrossRef CAS
- A. Rananaware, A. N. Abraham, D. D. La, V. Mistry, R. Shukla and S. V. Bhosale, Aust. J. Chem., 2017, 70, 652–659 CrossRef CAS
- S. Kanagaraj, A. Puthanveedu and Y. Choe, Adv. Funct. Mater., 2020, 30, 1907126 CrossRef CAS
- C. M. Gordon, J. D. Holbrey, A. R. Kennedy and K. R. Seddon, J. Mater. Chem., 1998, 8, 2627–2636 RSC
- L. Xu, Y. Mo, N. Su, C. Shi, N. Sun, Y. Zhang, L. Duan, Z. H. Lu and J. Ding, Nat. Commun., 2023, 14, 1678 CrossRef CAS PubMed
- Z. Lian, J. He, L. Liu, Y. Fan, X. Chen and H. Jiang, Nat. Commun., 2023, 14, 2752 CrossRef CAS PubMed
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed
- E. Fresta, J. Dosso, J. Cabanillas-Gonzalez, D. Bonifazi and R. D. Costa, ACS Appl. Mater. Interfaces, 2020, 12, 28426–28434 CrossRef CAS PubMed
- E. Fresta, J. Dosso, J. Cabanillas-González, D. Bonifazi and R. D. Costa, Adv. Funct. Mater., 2020, 30, 1906830 CrossRef CAS
- M. D. Weber, M. Viciano-Chumillas, D. Armentano, J. Cano and R. D. Costa, Dalton Trans., 2017, 46, 6312–6323 RSC
- M. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J. L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef
- E. Fresta, M. D. Weber, J. Fernandez-Cestau and R. D. Costa, Adv. Opt. Mater., 2019, 7, 1900830 CrossRef CAS
- L. D. Bastatas, M. D. Moore and J. D. Slinker, Chempluschem, 2018, 83, 266–273 CrossRef CAS PubMed
- M. H. Bowler, T. Guo, L. D. Bastatas, M. D. Moore, A. V. Malko and J. D. Slinker, Mater. Horiz., 2017, 4, 657–664 RSC
- P. Lundberg, Y. Tsuchiya, E. M. Lindh, S. Tang, C. Adachi and L. Edman, Nat. Commun., 2019, 10, 5307 CrossRef CAS PubMed
- C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2021, 10, 4456–4482 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05941c |
| This journal is © The Royal Society of Chemistry 2024 |