
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiaryl hypervalent bromines and chlorines: synthesis, structures and reactivities
Matteo
Lanzi
*a and
Joanna
Wencel-Delord
 *ab
*ab
aLaboratoire d'Innovation Moléculaire etApplications (UMR CNRS 7042), Université deStrasbourg/Université deHaute Alsace, ECPM, 67087 Strasbourg, France. E-mail: matteo.lanzi1@studenti.unipr.it; joanna.wencel-delord@uni-wuerzburg.de
bInstitute of Organic Chemistry, JMU Würzburg Am Hubland, Würzburg, Germany
First published on 3rd January 2024
Abstract
In the field of modern organic chemistry, hypervalent compounds have become indispensable tools for synthetic chemists, finding widespread applications in both academic research and industrial settings. While iodine-based reagents have historically dominated this research field, recent focus has shifted to the potent yet relatively unexplored chemistry of diaryl λ3-bromanes and -chloranes. Despite their unique reactivities, the progress in their development and application within organic synthesis has been hampered by the absence of straightforward, reliable, and widely applicable preparative methods. However, recent investigations have uncovered innovative approaches and novel reactivity patterns associated with these specialized compounds. These discoveries suggest that we have only begun to tap into their potential, implying that there is much more to be explored in this captivating area of chemistry.
Introduction
Although the octet rule is a fundamental principle in chemistry, certain main group elements (Groups 13–18) are prone to violate the Lewis octet rule by accommodating more than eight electrons in their valence shell, thus reaching the hypervalent state.1–3 Accordingly, hypervalent molecules exhibit a three-center–four-electron (3c–4e) bonding and show a peculiar T-shape structure as a result of a pseudo-Jahn–Teller effect (Fig. 1).4,5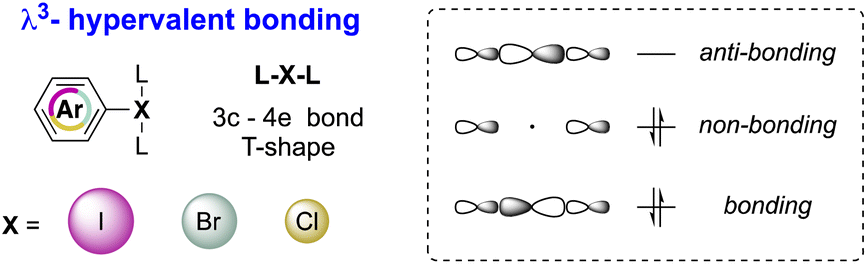 | ||
| Fig. 1 Halogen-based hypervalent molecules feature a 3c–4e bonding. | ||
Such a unique electronic structure of hypervalent compounds translates into unusual reactivity, as breaking of X–L bonds occurs particularly easily. Therefore, hypervalent elements may be considered as extremely powerful leaving groups for example. Besides, the reactivity behaviour of hypervalent compounds shares numerous similarities with the reactivity of transition metals.6 Indeed, reactions involving hypervalent iodine reagents are frequently analysed using concepts such as oxidative addition, ligand exchange, reductive elimination, and ligand coupling, all of which are characteristic features of transition metal chemistry.7,8
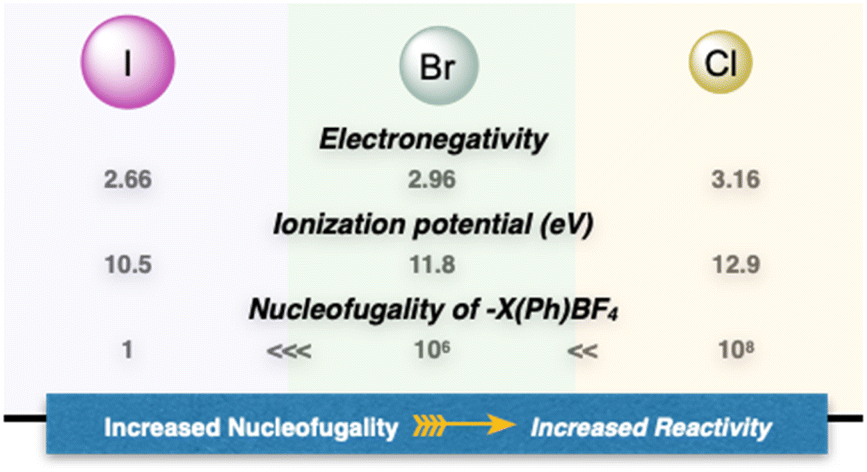
In contemporary organic chemistry, hypervalent compounds have become essential tools for synthetic chemists, finding applications in both academia and industry.9,10 These compounds play pivotal roles as oxidants, electrophiles, and initiators of radical reactions.11,12 Among the hypervalent reagents, iodanes have received extensive attention, due to their simple synthesis, stability, commercial availability and diversity of applications.13–15 In marked contrast and despite their higher nucleofugality, ionization potential and stronger electrophilicity, the chemistry of hypervalent bromines and chlorines has not been thoroughly explored for several decades (Fig. 2).16–18 The primary reason that hampered their diffusion lies in the lack of simple, general, and efficient synthetic methods to access these compounds, as a consequence of the innate greater ionization potential and therefore challenging oxidation.19,20 Consequently, despite their possible increased and complemental reactivity with respect to the iodine congeners, their applications remain largely unexplored.4,21
 | ||
| Fig. 2 Periodic table properties of halogens. | ||
Pivotal contributions by Ochiai, Miyamoto, and Uchiyama have significantly influenced the development of these areas, thus revealing and advancing the synthesis22,23 and the unique reactivity of hypervalent bromines and chlorines as oxidants,24–27 arylating agents28 and as metal-free aminating agents of aliphatic alkanes and alkenes.29,30
Although numerous reviews have been published discussing various aspects of the chemistry of λ3- and λ5-iodanes,6,10,21,31,32 articles summarizing other hypervalent compounds are rare.16–20
We aim to contribute to this field with the following review focusing our attention on the most recent development in the fields of diaryl λ3-hypervalent bromines and chlorines. Particular attention will be focused on the synthesis, structural characterization, and their complemental reactivity, with the intent of providing the readers with a complete overview of this interesting and yet unexplored chemistry.
Synthesis of hypervalent diaryl bromines and chlorines
First reported by Sandin and Hay in 1952, cyclic hypervalent bromines and chlorines have been obtained via in situ formation and thermal decomposition of the corresponding diazonium salts with sodium nitrite in boiling HCl aqueous solution (Scheme 1).33 Despite the interesting stability of the cyclic diaryl bromanes 2 and chloranes 4, harsh reaction conditions and the low yields did not incite further investigations of this field.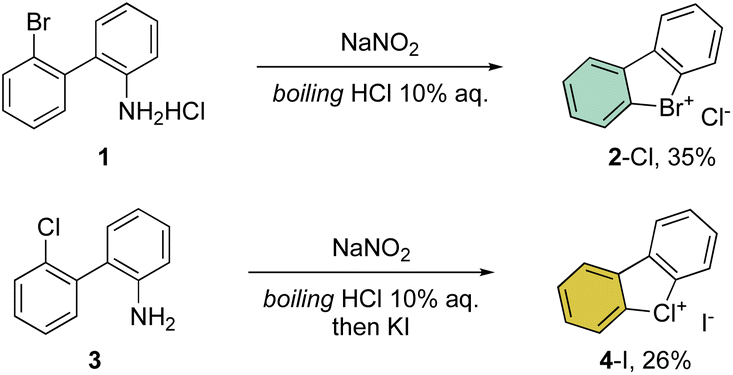 | ||
| Scheme 1 Synthesis of cyclic hypervalent bromines and chlorines. | ||
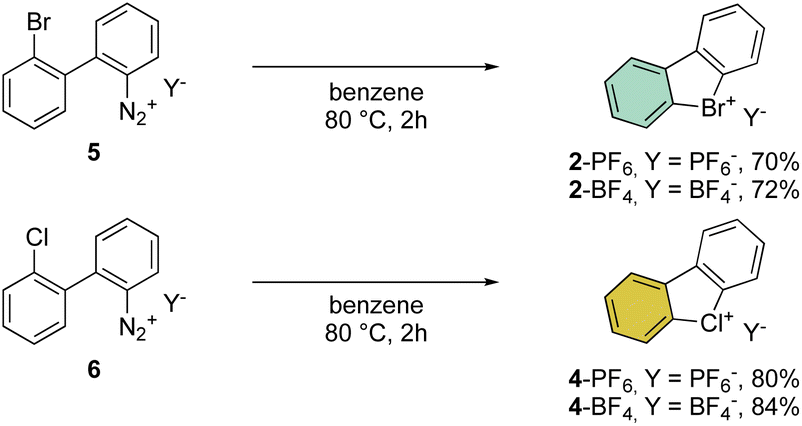
A few years later, Less and Heaney succeeded in preparing tetrafluoroborate (BF4−) and hexafluorophosphate (PF6−) salts of 2 in high yield, by directly engaging corresponding 2,2′-halo diazonium salts as the starting materials. The thermal decomposition yielded the corresponding cyclic λ3-bromanes and chloranes in excellent isolated yields (Scheme 2).34
 | ||
| Scheme 2 Synthesis of tetrafluoroborate (BF4−) and hexafluorophosphate (PF6−) salts of 2. | ||
In clear contrast, the synthesis of acyclic λ3-bromanes 6 and chloranes 7 turned out to be more challenging. The thermal decomposition strategy using diazonium salt 7 in the presence of an excess of aryl bromide, studied in 1957 by Nesmeyanov and Tolstaya, delivered the expected diaryl λ3-bromanes 8 and 9 and chloranes 10, albeit only in low yields and with particular counter anions (Scheme 3 Cond. A).35,36 In order to improve the stability of Br(III) and Cl(III) products, Olah and co-workers have also exploited such a strategy using tetraphenylborate (Ph4B−) as the counter anion (Scheme 3 Cond. B).37
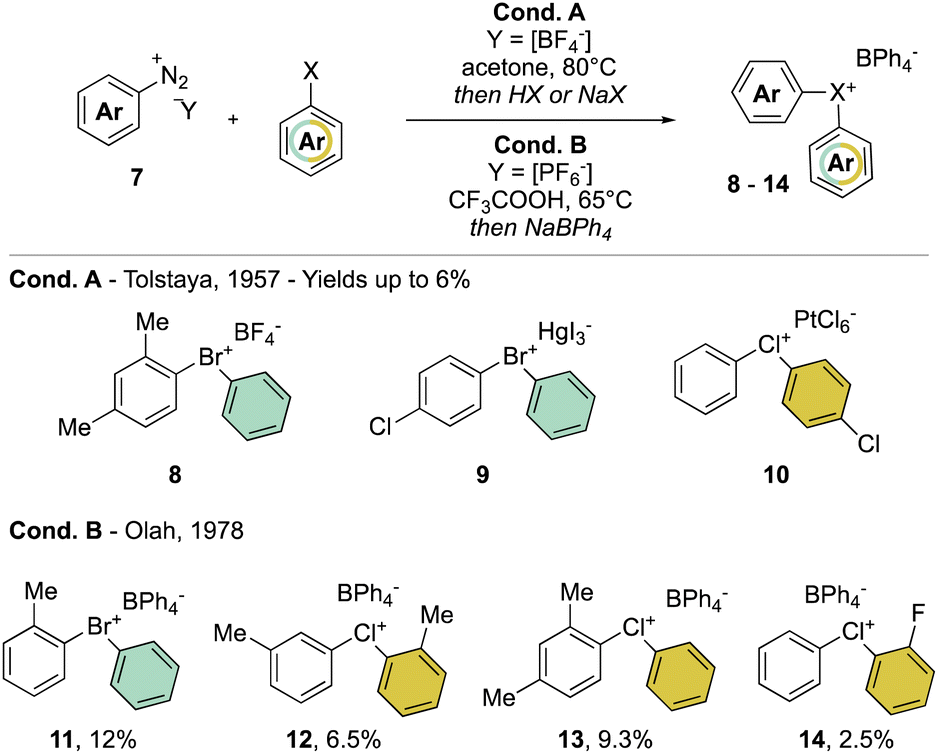 | ||
| Scheme 3 Synthesis of diaryl λ3-bromanes and chloranes. | ||
A straightforward and simple approach for the synthesis of 2 remains the direct oxidation of 15 or 16; although reports have been presented in the literature announcing moderate yields, the need for strong oxidants and acid conditions clearly hampered its application for a synthesis of a diversified library of hypervalent compounds (Scheme 4).38
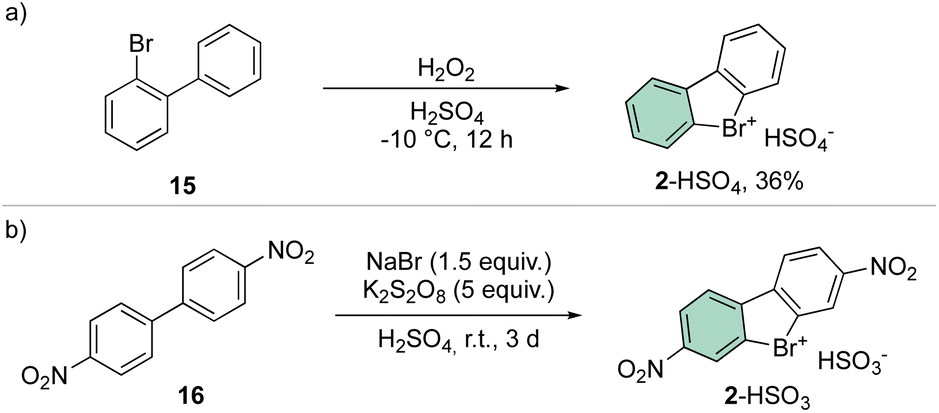 | ||
| Scheme 4 Direct oxidation of 15 and 16. | ||
A remarkable progression of this field of chemistry took place when Nesmeyanov and his team devised a more versatile method to access the diaryl λ3-bromanes (Scheme 5a) involving the use of inorganic hypervalent bromide, bromine trifluoride, (BrF3).39 Although it is a corrosive, toxic and highly reactive liquid, it easily undergoes double ligand exchange on the bromine center when reacted with tetraarylstannane (Ar4Sn) or diarylmercury (Ar2Hg) compounds in the presence of BF3 OEt2.
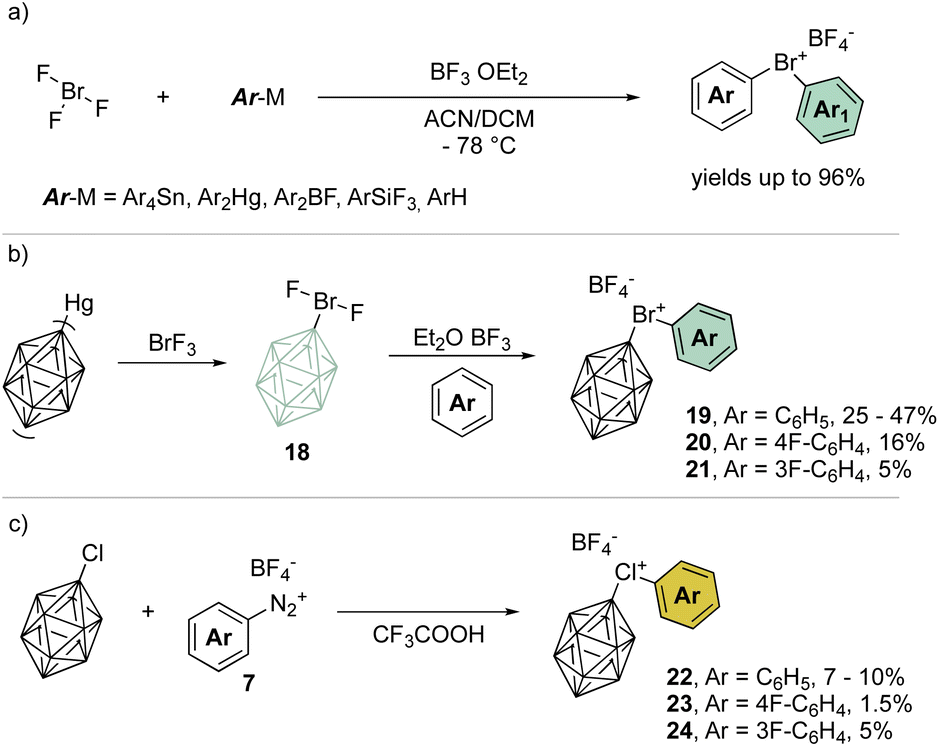 | ||
| Scheme 5 Preparation of λ3-bromanes and chloranes. | ||
Such a strategy was then also extended to simple aryl and aryl silanes respectively by Nesmeyanov and by Frohn groups,40–45 delivering also the difluoro aryl λ3-bromane (ArBrF2), known as Frohn's reagent, a widely used precursor for highly adorned hypervalent bromanes.46,47
The first X-ray structure of a diaryl hypervalent bromine was reported in 1974 by Tolstaya and co-workers for the compound 17 and subsequently corroborated in 1996 by Frohn and co-workers.42,48 The analysis of the crystal structure shows a C–Br–C angle between 98.08 and 97.79° and a Br–F distance of 2.79(6) Å, distinctive for a 3-center–4-electron bond T-shape (Fig. 3).
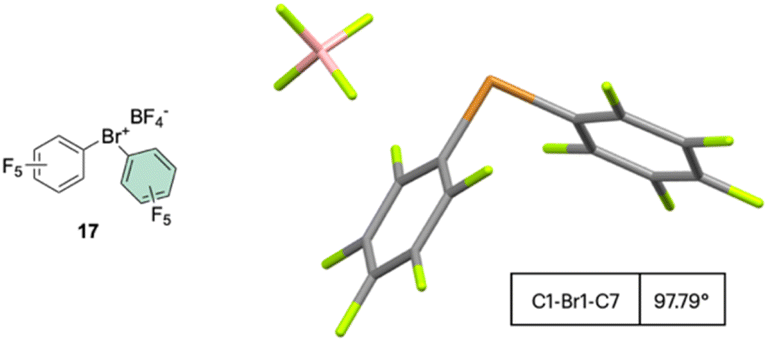 | ||
| Fig. 3 X-ray structure of 17. | ||
Taking advantage of this procedure, Grushin and co-workers have explored the synthesis of peculiar hypervalent bromine and chlorine compounds bearing a unique 3D-aromatic49–51 carborane structure (Scheme 5b).52–54 To access the bromonium derivative, closo-carborane was introduced via double ligand exchange using its mercury precursor and BrF3. The presence of Lewis acid influences the outcome of the reaction leading to the difluoro carborane or to the biscarborane bromonium salts. A subsequent ligand exchange from the difluoro carborane bromane, using the simple arene as solvent, provides the diaryl λ3-bromanes 19–21 in moderate to low yields (Scheme 5b). On the other hand, chloronium compounds were obtained in low yields following the diazonium salt thermal decomposition in the presence of closo-carborane chloride (Scheme 5c).
While the use of highly reactive and corrosive reagents such as BrF3 warrants high yielding reactions, this strategy suffers from three major drawbacks, (1) it is not applicable for the preparation of a large library of products due to the strong acidic conditions; (2) highly limited availability of BrF3 (not commercially available or difficult to purchase); and finally (3) aryl compounds, such as Ar2Hg and Ar4Sn, are toxic.
In 2021, Miyamoto, Uchiyama and Ochiai contributed to the chemistry of diaryl hypervalent bromines by making use of Frohn's reagent (25)55 as a pivotal platform for the synthesis of various bromane structures.56 Following a multistep synthetic approach involving an imino-λ3-bromane30,57 the unique diphenyl λ3-bromane 28 was obtained in excellent yield via ligand exchange with phenyl stannate (Scheme 6).
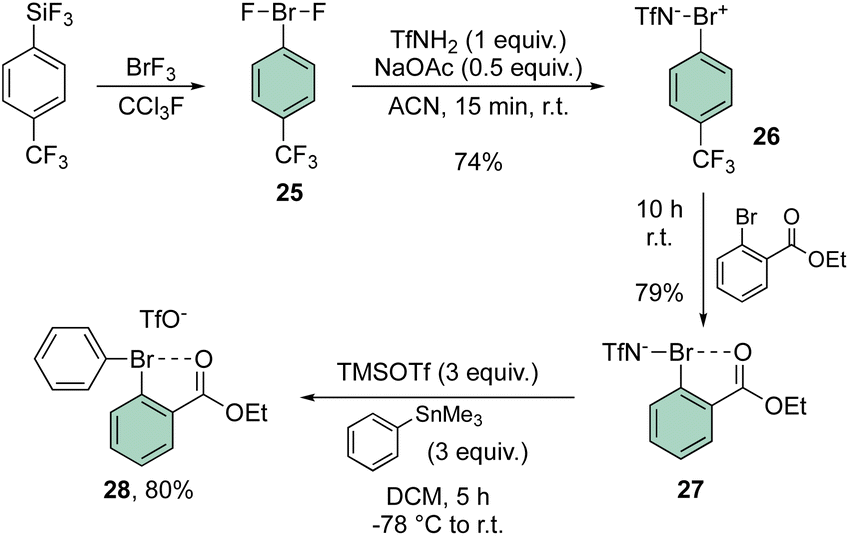 | ||
| Scheme 6 Synthesis of diaryl λ3-bromane 28. | ||
The X-ray structure analysis of the hypervalent bromine 28 disclosed a peculiar and yet interesting interaction between the Br(III) and the carbonyl oxygen, while a comparable C–Br–C angle is observed when compared with the Ph2BrOTf structures (Fig. 4).
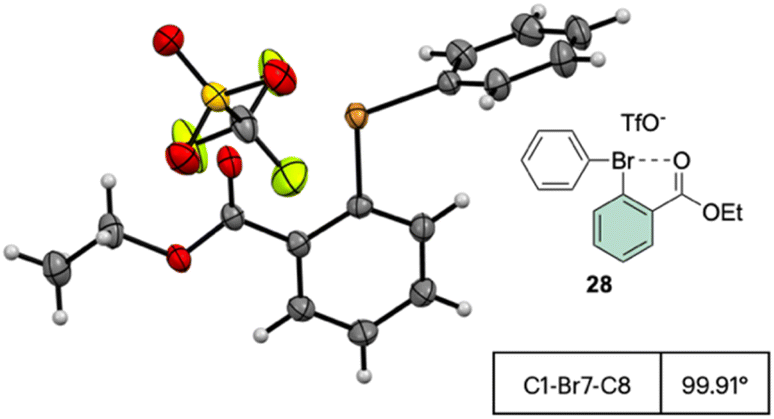 | ||
| Fig. 4 X-ray structure of 28. | ||
Electrochemistry has nowadays ushered fresh opportunities for organic synthesis, emerging as an innovative and eco-friendly platform with promising sustainability prospects for the future.58 In this context, electrochemical synthesis of hypervalent iodines, using electric power as the cheapest, most environmentally benign and sustainable oxidant, has been largely investigated.59–62 Recently, Francke, Suna and co-workers have reported the synthesis of dialkoxyarylbrominane using Martin's reagent63,64via electrochemical oxidation of aryl bromide.65,66
More recently, Suna and co-workers have also disclosed a methodology toward the electrochemical preparation of 29. Remarkably, 29 can be considered as a precursor of a wide range of hypervalent bromines, such as the peculiar diaryl λ3-bromane 30 (Scheme 7).67
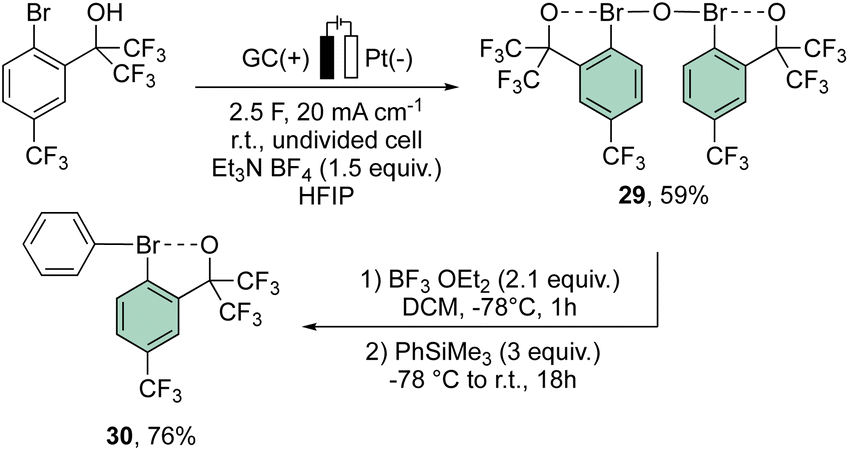 | ||
| Scheme 7 Synthesis of diaryl λ3-bromane 30. | ||
Interestingly, the analysis of the crystal structure of 30 revealed a smaller C–Br–C angle (97.79°) compared to the anionic analog 28, suggesting a diaryl semi-perpendicular structure (Fig. 5).
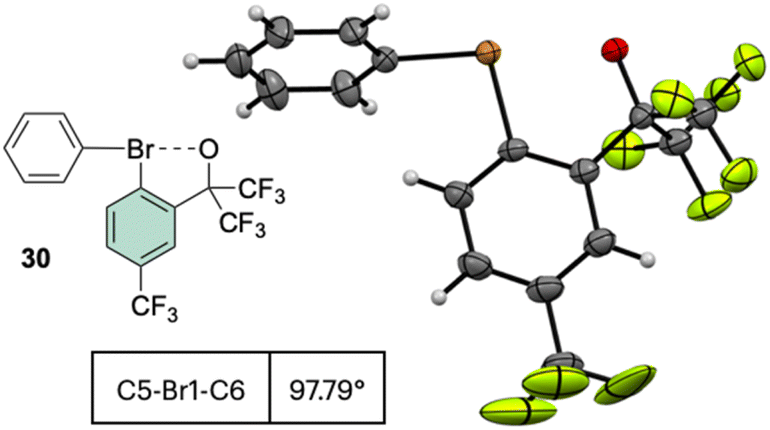 | ||
| Fig. 5 X-ray structure of 30. | ||
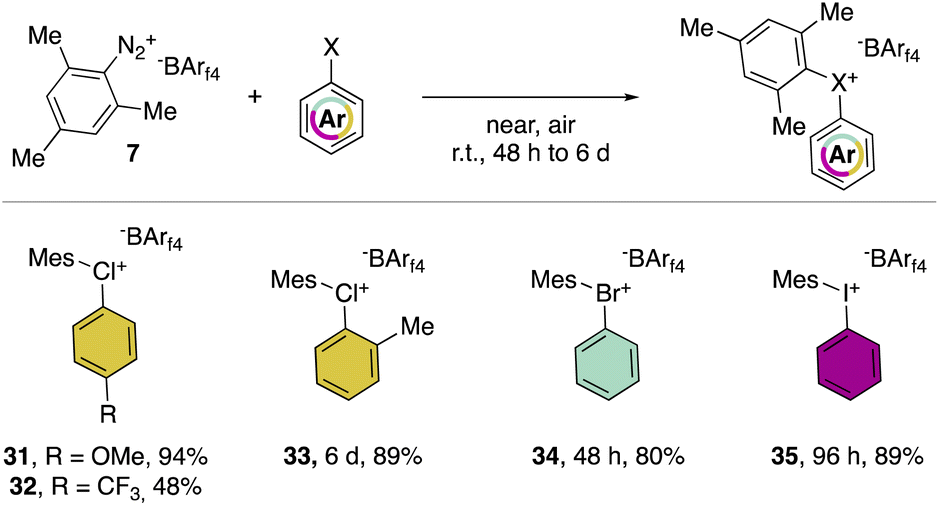
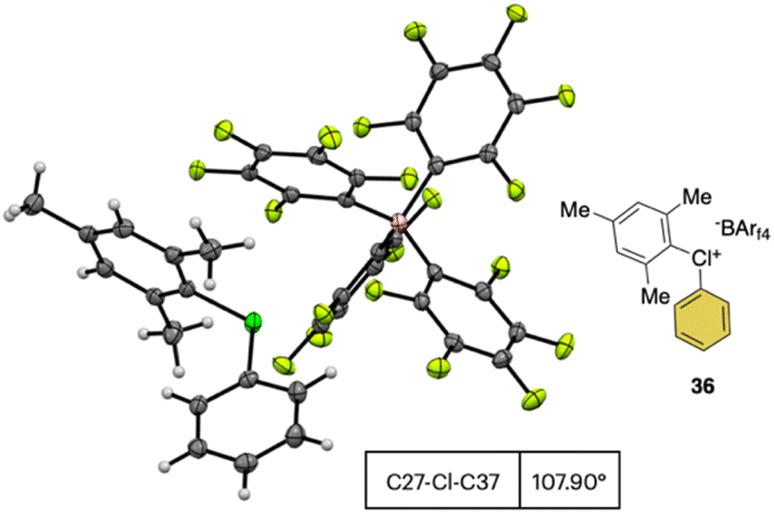
In 2019, Miyamoto, Uchiyama and co-workers uncovered the first simple and high yielding approach for the preparation of diaryl λ3-hypervalent chlorines (Scheme 8).28 The previously described thermal decomposition of the diazonium salts has been used by the authors, while the introduction of the mesityl moiety and the bulky counter ion, [B(C6F5)4], enabled reaching high yields. The unprecedently mild reaction conditions (room temperature), allowed isolation and characterization of a series of bench stable Cl(III) compounds and scaling up their synthesis up to 1 mmol. Interestingly, the authors also extended the method to the synthesis of the corresponding λ3-iodane and bromane compounds. The X-ray analysis of such a rare diaryl λ3-hypervalent chlorine 36 revealed a hybrid nature of the C–Cl–C bonds (Fig. 6).68 Indeed, the angle between C27–Cl1–C37 was 107.30° indicating a partial distortion from the 3-center–4-electron bond, although the median distance between the Cl1 and the fluorine atoms (3.075 Å) of the [B(C6F5)4] was in line with a hypervalent structure compound.
 | ||
| Scheme 8 Preparation of diaryl λ3-halogens. | ||
 | ||
| Fig. 6 X-ray structure of 36. | ||
Recently McCormick, Stuart, Valente and co-workers have analysed the influence of the AOs in the composition of the MOs of the diaryl halonium salts and their related effect on the compound structure.2 The characteristic 3c–4e bond and T-shape structure is dictated by the MOs of the compounds, and iodonium salts are correctly described as p-AO based. However, a contribution of the s- and p-AO has been identified for the other halogens (Scheme 9). A larger contribution of s-AO with respect to the p-induces a larger L–X–L angle as observed moving from the I(III) to the Cl(III). Moreover, ligands and the identity of the counter anion also influence the structure of the diaryl halonium salts; less coordinating counter anions such as BF4 and PF6 induce a higher s-character thus influencing the bond angle.
 | ||
| Scheme 9 Structure and orbital correlation. | ||
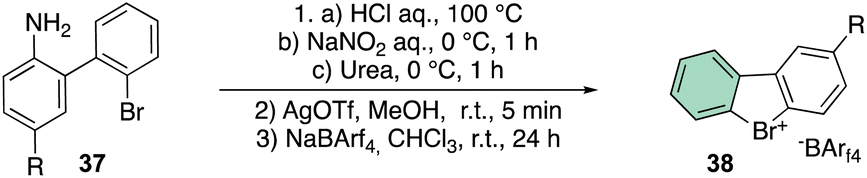
The beginning of this decade was clearly marked by a regain of interest in the chemistry of rare halocompounds. Yoshida and co-workers prepared, although in quite low yields, a few examples of cyclic diaryl λ3-bromane compounds (Scheme 10) by treating the 1-bromo, 1′-amino biaryls with HCl, NaNO2 and urea.69
 | ||
| Scheme 10 Synthesis of cyclic diaryl λ3-bromanes. | ||
To further improve the efficiency of this reaction, a laborious counter ion exchange enables the preparation of the desired bromonium salt 38. It is worth noting that counter ion exchange enhanced the bromonium salt stability towards the decomposition, especially in the case of the bulky BArF.
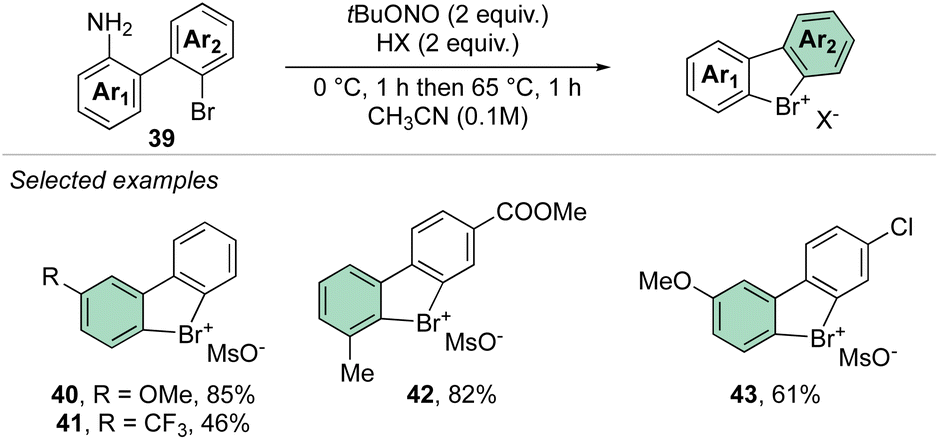
In 2021, our group disclosed a simple and high yielding approach to prepare cyclic diaryl λ3-bromanes (Scheme 11).70 Inspired by the previously described formation of the diazonium precursors, our method introduces the use of an organic oxidant, namely tert-butyl nitrite (TBN) allowing a crucial improvement to the reaction conditions and outcome. Indeed, milder reaction conditions were discovered, and only 2 equivalents of oxidant and Brønsted acid were required to achieve high yields for a large family of bench, air, and moisture stable products. These new compounds can be stored at room temperature for months without erosion or alteration of their chemical properties.
 | ||
| Scheme 11 Synthesis of cyclic diaryl λ3-bromanes. | ||
Subsequently, Yoshida and co-workers, taking into account the use of TBN as an oxidant, reported the preparation of the first chiral λ3-bromane salts (Scheme 12).71 Although, similar reaction conditions were used, only low yields were reported, presumably due to the higher molecular complexity of the substrates and the presence of an oxidable amino group. The method could be further extended to the isoelectronic iodine and chloride compounds. The X-ray structure analysis of 46 shows a C–Br–C bond angle of 87° and a torsion angle between the naphthyl moieties of 73° (Fig. 7).
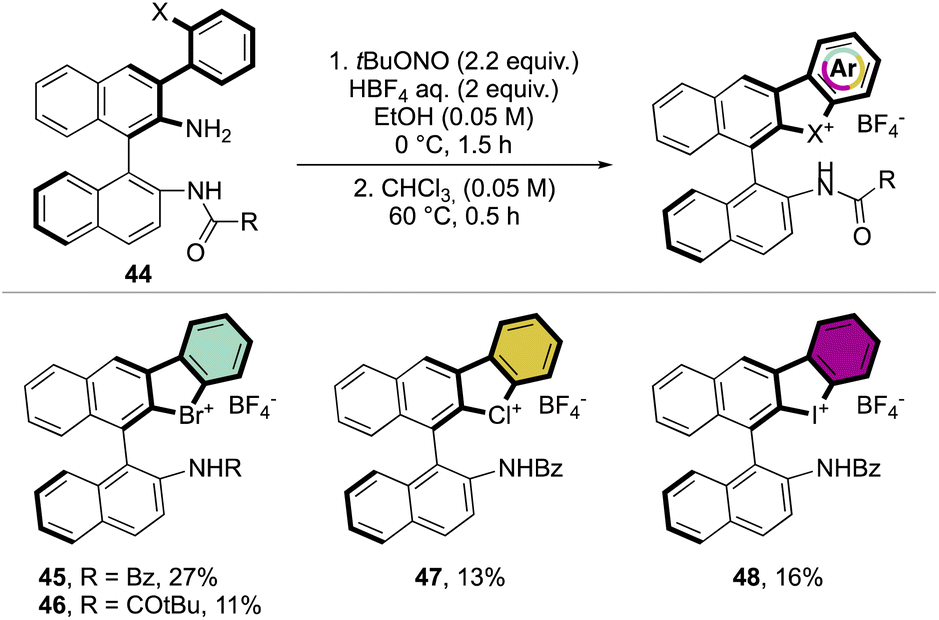 | ||
| Scheme 12 Synthesis of chiral cyclic diaryl λ3-bromanes. | ||
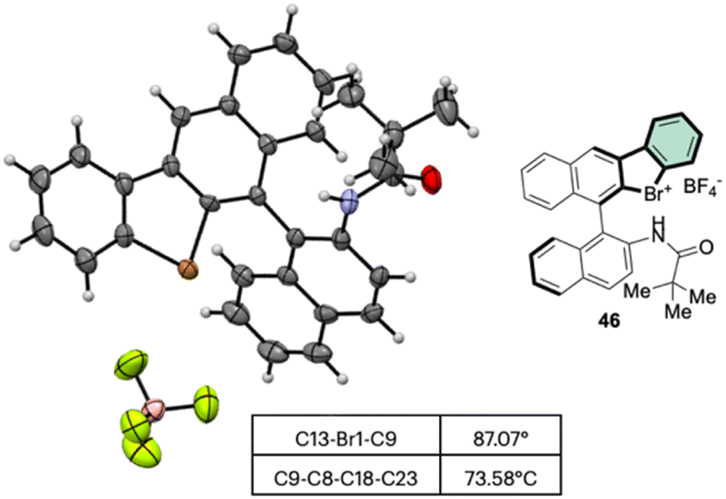 | ||
| Fig. 7 X-ray structure of 46. | ||
Following a similar retrosynthetic pathway, Yoshida and co-workers reported the synthesis of chiral cyclic diary halonium salts bearing an N-nitrosamine moiety (Scheme 13).72 The increment of the organic oxidant equivalents led the authors to indirectly oxidize the aryl bromide and simultaneously introduce the N-nitrosamine function, albeit only in low yields.
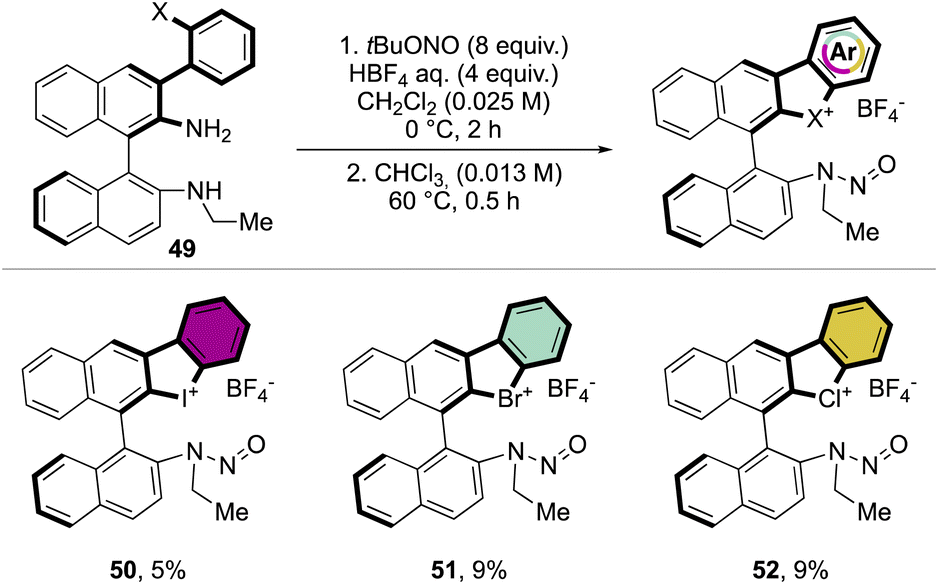 | ||
| Scheme 13 Synthesis of chiral cyclic diaryl λ3-halogens. | ||
Inspired by our first report on the preparation of hypervalent bromanes, we recently uncovered the synthesis of cyclic diaryl λ3-chloranes (Scheme 14).73 Such a strategy relies on the use of 1,1′-chloro(amino)biphenyl 53 and mild reaction conditions for the in situ formation and thermal degradation of the diazonium salt. Such a simple and practical synthetic path enabled the preparation of a plethora of functional new diaryl λ3-chloranes (54–57) in moderate to excellent yields (46–99%).
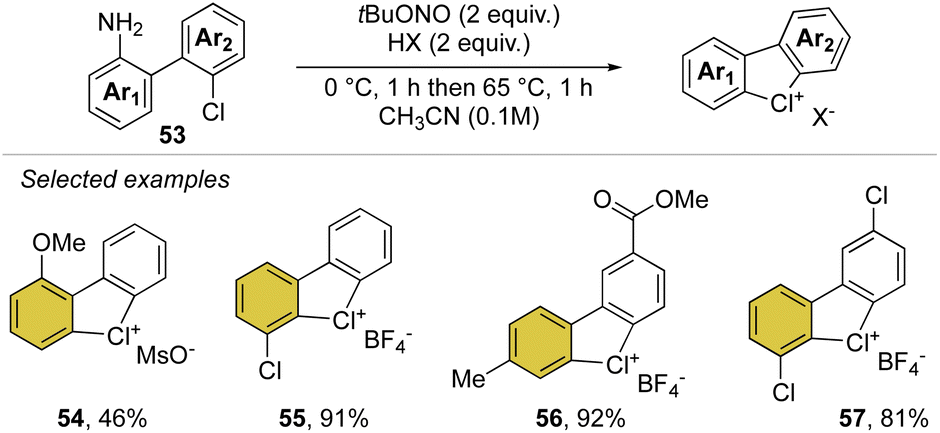 | ||
| Scheme 14 Synthesis of cyclic diaryl λ3-chloranes. | ||
Particularly interesting, varied functional moieties that encompass electron donating and withdrawing groups, like methoxy 54 and methyl esters 56 as well as fluoro-contain groups, were well tolerated under these conditions. Additionally, the procedure turned out to be scalable and robust, allowing a gram-scale synthesis of such elusive structures.
The crystallographic information pertaining to the Cl(III), Br(III), and I(III) unveils an intriguing and distinctive structural pattern (Fig. 8). Not surprisingly, the distance between the carbon and the hypervalent halogen atom demonstrates an ascending trend in the sequence Cl(III) < Br(III) < I(III). This progression leads to a notable expansion of the C–X–C angle, resulting in an almost perfect T-shaped configuration for the chlorane-based compound (I(III), 81.9°; Br(III), 86.9°; Cl(III), 92.0°), thus revealing the characteristic three-centre–four-electron bond structure (3c–4e) of λ3-hypervalent compounds.
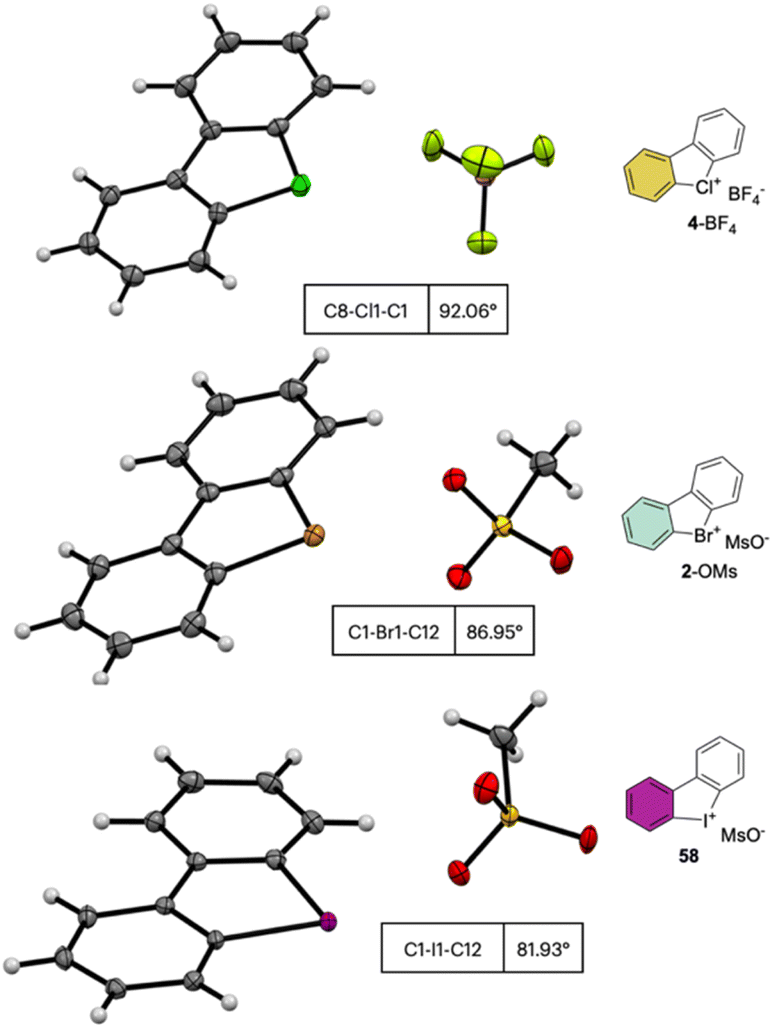 | ||
| Fig. 8 X-ray structures of 4-BF4, 2-OMs and 58. | ||
More recently, Cuenca, Shafir and co-workers have announced the first examples of hetero di-λ3-diarylhalonium structures by reporting the iodine(III)–bromine(III) and iodine(III)–chlorine(III) bis-hypervalent compounds (Scheme 15).74 The synthetic strategy relies on a series of peculiar steps, an initial formation of the diaryl hypervalent iodane 59, followed by one-pot three consecutive steps of deprotection, oxidation and cyclization reactions to generate the second hypervalent species. Despite the moderate efficiency of this methodology, the originality of the new compounds 60 and 61 opens new perspectives in the field.
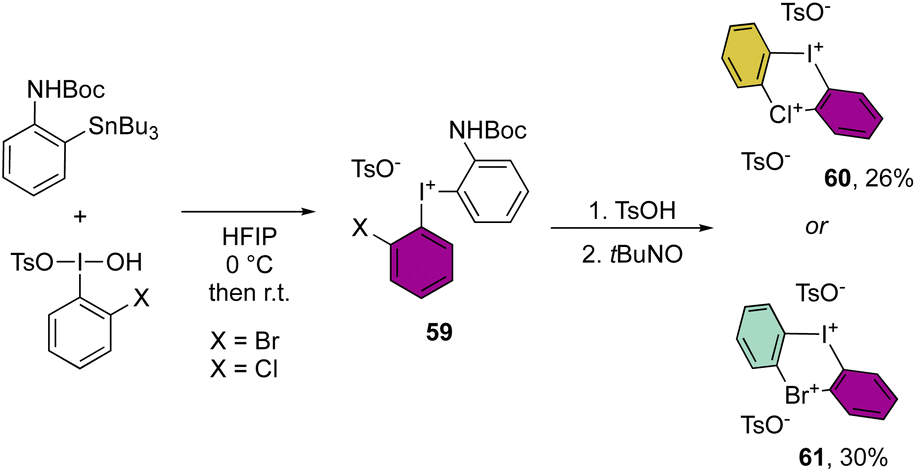 | ||
| Scheme 15 Synthesis of cyclic diaryl bis λ3-halogenum. | ||
The X-ray analysis of these rare compounds 60 and 61 revealed a particularly interesting feature (Fig. 9). Indeed, the measured angles between the aryl carbon and the hypervalent atoms are 89.99°, 93.47° and 98.55° respectively for the C–I–C, C–Br–C and C–Cl–C, thus belonging to the three center–four-electron bond characteristic structures of hypervalent compounds and generating an almost pure perpendicular aryl containing surface.
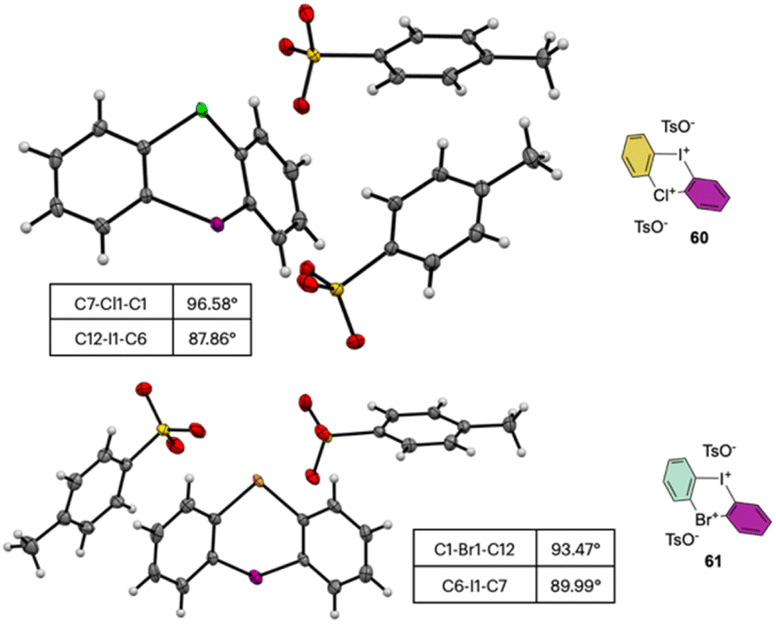 | ||
| Fig. 9 X-ray structures of 60 and 61. | ||
Reactivity of non-cyclic diaryl hypervalent bromine and chlorine compounds
Although considerably less explored, substantial differences have been described regarding the chemical properties of hypervalent λ3-bromane and chlorane compounds with respect to the corresponding hypervalent iodines. Herein, we aim to discuss the reactivity of these uncommon reagents by focusing our attention on their orthogonal chemical behaviour.First, following the fundamental reactivity of hypervalent iodines as arylating agents, the potential of diaryl λ3-bromanes and chloranes compounds in combination with a wide range of heteroatom nucleophiles, including O, N, S, halides, under gentile reaction conditions was surveyed.17,18,20
A preliminary insightful comparison study between the diaryl λ3-bromane and iodane for the arylation of alkolates was conducted in 1972, by Lubinkowski and McEwen (Scheme 16a).75 The reaction with sodium ethoxide led to the phenyl ethyl ether in only 17% with the iodane compound and an exceptional 96% yield with the corresponding bromane, demonstrating the superior reactivity of the latter. Subsequently, in 1978, Olah and co-workers extended such comparative studies focusing on nucleophilic aromatic substitution with NaNO2 (Scheme 16b).37 The reactions proceed smoothly leading to similar yields for the bromane and chlorane compounds while only low conversion was observed in the case of the iodane analog, further corroborating the superior reactivity of Br(III) and Cl(III) derivatives. Moreover, the use of substituted hypervalent chlorines results in the formation of the ipso-functionalized nitro compounds as unique products, thus declaiming the formation of the benzyne intermediate (Scheme 16c). The nitration of unsymmetric chloranes delivers preferentially the functionalization on the more electron poor aryl moiety, while the steric effect of a methyl in 2-position destabilized the C–Cl bond, thus being more prone towards the nucleophilic substitution (Scheme 16d); analogous results were also obtained for the reaction of Br(III).
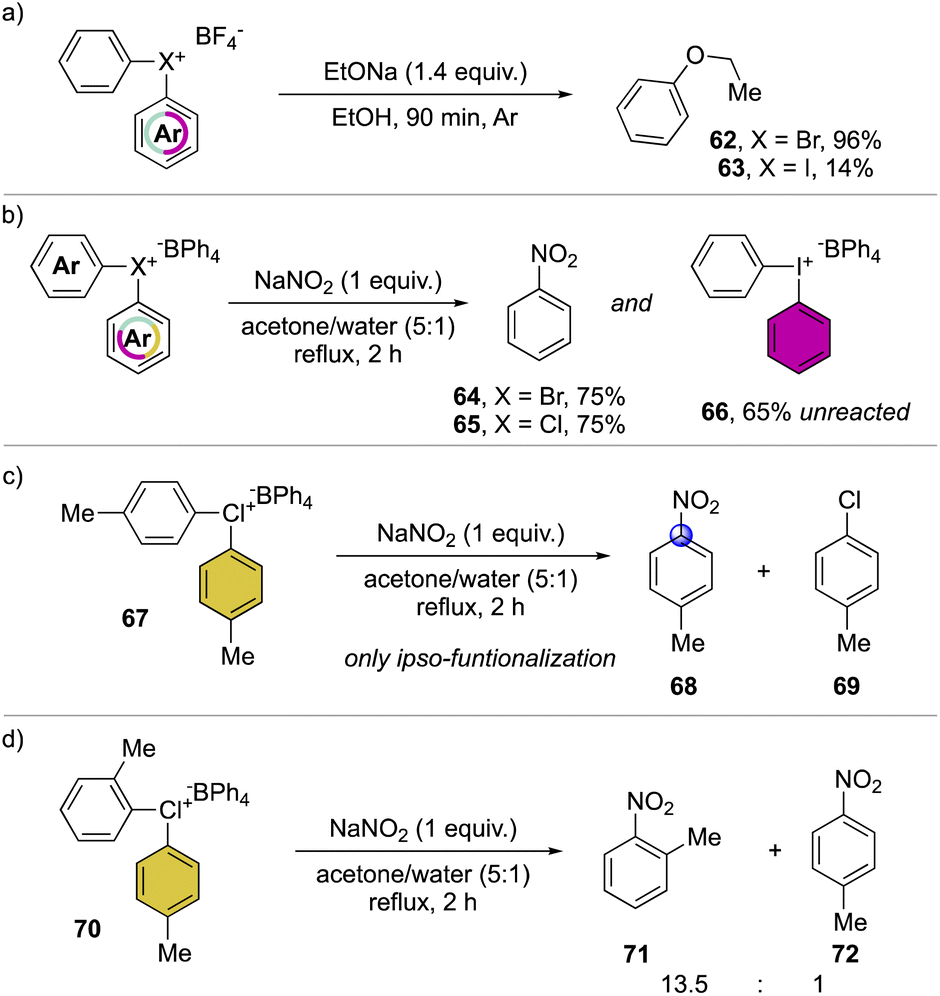 | ||
| Scheme 16 Reactivity of diaryl λ3-bromanes and chloranes. | ||
Further investigation on the complemental activity of hypervalent compounds was carried out by investigating their reactivity versus rhodanide and benzenesulfinate anions in biphasic media (Scheme 17).76 The enhanced reactivity is revealed while considering the reaction time, whilst high yields of 73–78 were obtained in all of cases, the Br(III) and Cl(III) led to the desired product in a considerably reduced reaction time.
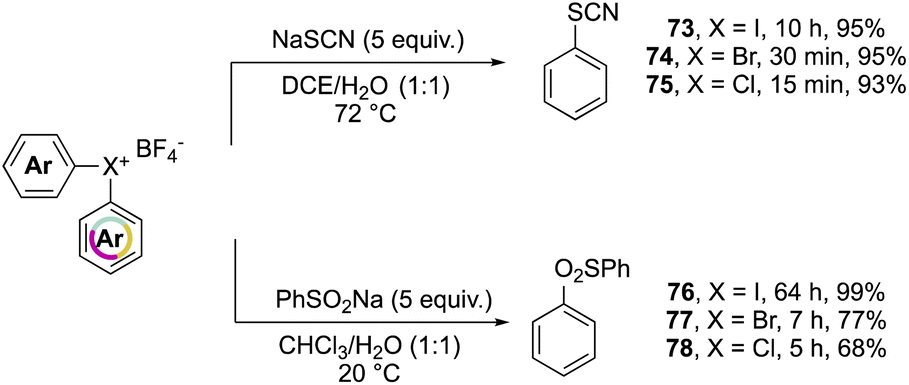 | ||
| Scheme 17 Reactivity of diaryl bis λ3-halogens with rhodanide and benzenesulfinate. | ||
In 1990, Grushin and co-workers investigated the reactivity of the peculiar aryl(m-carboran-9-yl)halonium compounds towards various nucleophiles (Scheme 18a).77,78 Fascinating discoveries have shown that two competitive reaction pathways can be envisioned: a nucleophilic aromatic substitution (SNAr) or a single electron reduction (SET). In the reaction of aryl(m-carboran-9-yl)halonium compounds with sodium azide only the nucleophilic substitution pathway can be awaited, and the formation of a mixture of different products A + A′ and B + B′ clearly indicates how the nature of the onium halogen atom can influence the SNAr reactivity. For instance, nucleophilic substitution occurred in the presence of the azide anion and led to the formation of the product A in 99%, 95% and 65% respectively for I(III), Br(III) and Cl(III) (Scheme 18b). Interestingly, the chemoselectivity decreases moving from the iodane to the chlorane with concomitant formation of the product B.
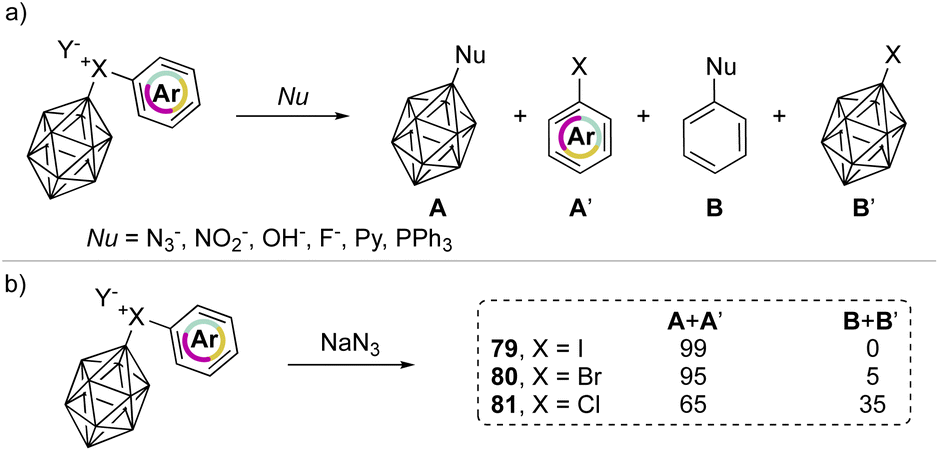 | ||
| Scheme 18 Reactivity of phenyl(m-carboran-9-yl)halogen compounds with various nucleophiles. | ||
On the other hand, aryl(m-carboran-9-yl)halonium compounds react with PPh3via a radical pathway, and thus visible light initiated reaction of I(III) and Br(III) proceeds smoothly delivering PPh4BF4 respectively in 7.5 and 10 hours (Scheme 19). In contrast, chlorane compounds required 38 hours and higher energetic light (UV light). The SET reduction step initiates the reaction, and the relative rate followed the series: I(III) > Br(III) > Cl(III) as a result of the decrement of the positive charge on the halogen atom.
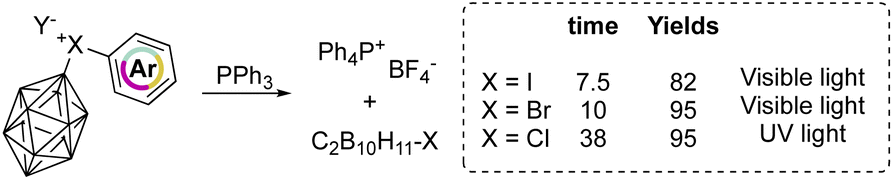 | ||
| Scheme 19 Reactions of phenyl(m-carboran-9-yl)halogen tetrafluoroborates with PPh3. | ||
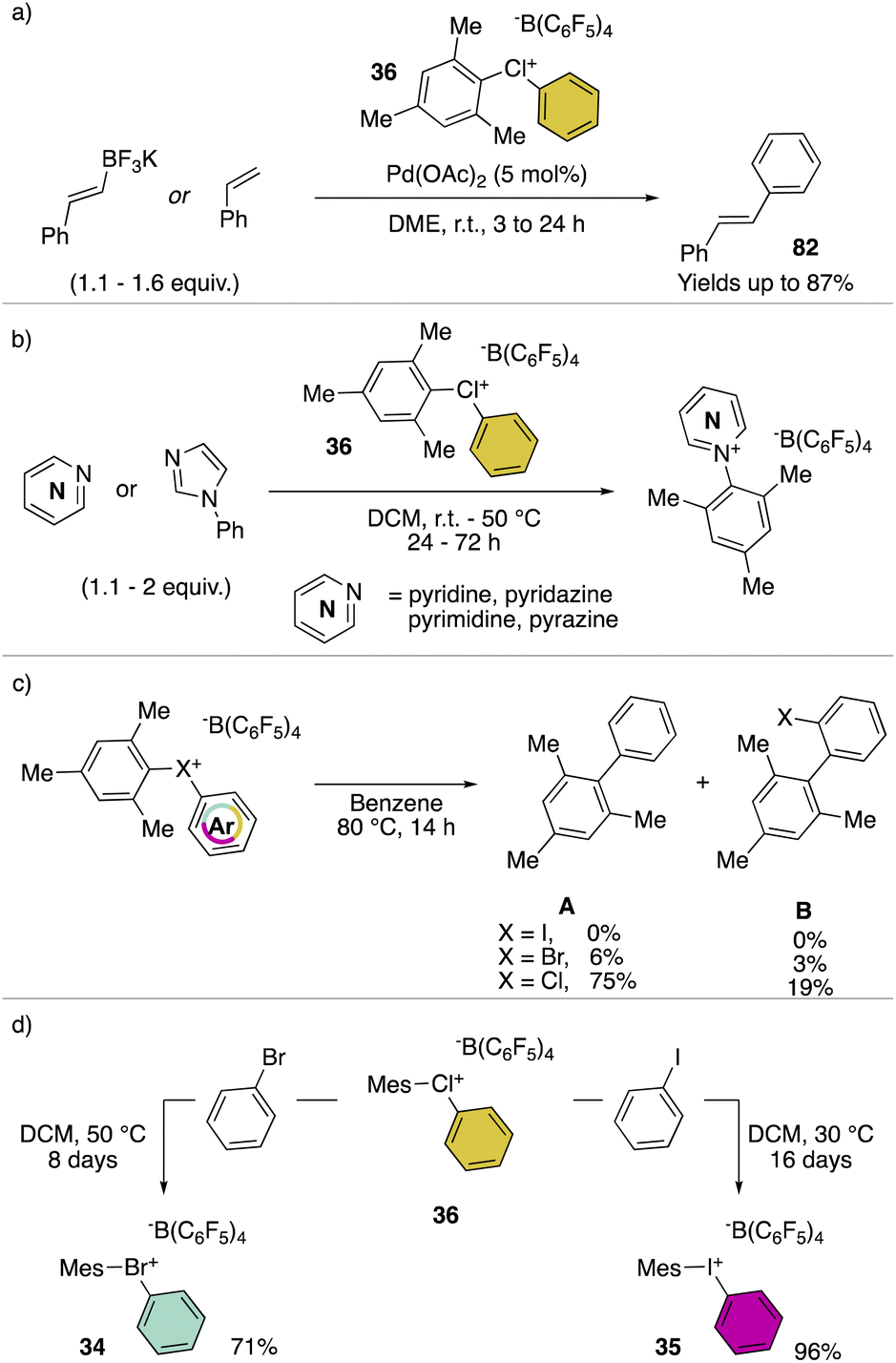
Hypervalent chlorines have also been found active in transition metal-catalysed transformations, as described by Miyamoto, Uchiyama and co-workers in 2019 (Scheme 20a).28 Indeed, palladium catalysed Heck, Suzuki–Miyaura and phosphonium formation using 36-[B(C6F5)4] disclosed the ability of these compounds to act as electrophiles. Phenylation of an activated carbon atom such as enolate was also reported by the authors proceeding in 62% yield under mild reaction conditions. Additionally, weak nucleophiles such as pyridine, pyrimidine and their isomers were arylated using 36-[B(C6F5)4], although in these cases the most electron rich aryl fragment (mesitylene) was transferred (Scheme 20b). Mechanistic investigation for the reaction of hypervalent halogens and pyridine has been recently reported by McCormick, Stuart, Valente and co-workers.2 The arylation involving diaryl hypervalent compounds is commonly described proceeding via ligand coupling or direct ipso-substitution.21 The kinetic studies related to 34, 35 and 36 showed a relatively slower reactivity for the 34 with respect to 36 while 35 was inactive under these metal-free conditions. This pattern aligns with the s-character of the MOs associated with halogens. Therefore, when the X–C bond undergoes heterolytic cleavage, the electron pair relocating to the halogen leaving group is better stabilized due to the presence of the orbitals with an increased s-character. Such reactivity trend was also confirmed for the cyclic diaryl hypervalent compounds.
 | ||
| Scheme 20 Reactivity of diaryl λ3-chloranes with various nucleophiles. | ||
The thermal decomposition of the different hypervalent compounds carried out in benzene enabled the arylation of the solvent by the mesityl moiety, yielding arylated aromatics (A) as the major product, although, following the reductive recombination mechanism also the 2-halo-biphenyl (B) was observed (Scheme 20c). The reaction yields follow the series Cl(III) > Br(III) > I(III), as expected by the nucleofugality of the corresponding halogens.75,78 Furthermore, the ability of the 36-[B(C6F5)4] as the oxidant was tested over the oxidation of bromo-benzene and iodo-benzene leading to the corresponding hypervalent bromine 34 and iodine 35 (Scheme 20d).
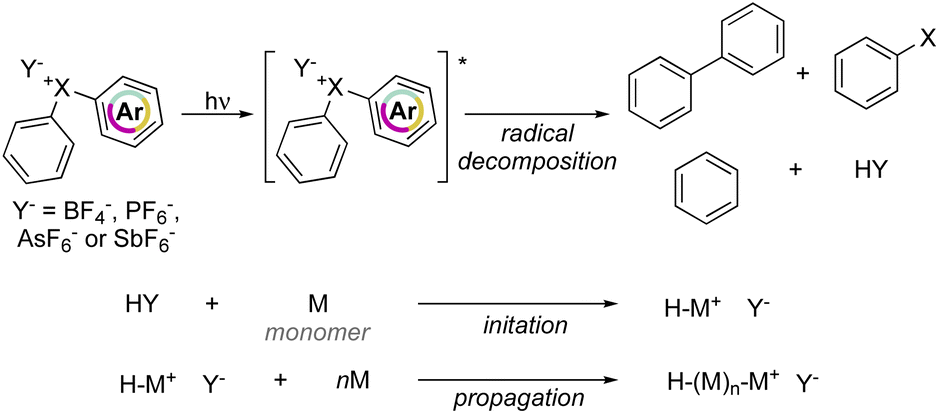
Crivello and Lam have explored the effectiveness of hypervalent compounds in the photo-induced cation polymerization of 2-chloroethyl vinyl ether, THF and cyclohexene oxide (Scheme 21).79–81 Photolysis was conducted under UV-light leading to a mixture of arene, haloarene and recombination products. In concomitance with the photo-degradation of the hypervalent compounds an equivalent of HPF6 (HY) acid is generated, thus initiating the cationic polymerization by protonation of electron-rich monomers.
 | ||
| Scheme 21 Photo-initiated polymerization using diaryl λ3-halogen compounds. | ||
Interestingly, photoinitiated polymerization of cyclohexene oxide and THF respectively with Br(III) and Cl(III) required a short light irradiation time and provided high conversion of the monomers within 5 min and 4 hours respectively.
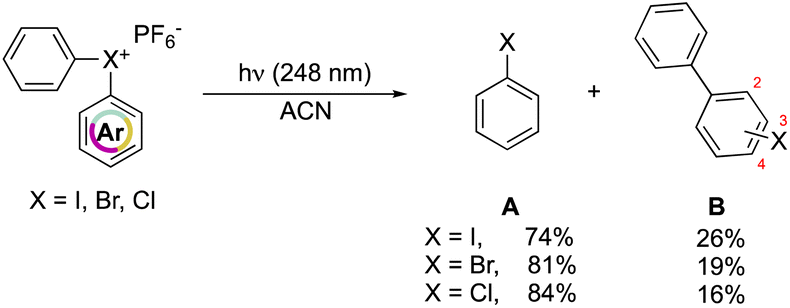
In 1991, Hacker and Dektar compared the behavior of diaryl hypervalent compounds under light irradiation.82 The direct irradiation of I(III), Br(III) and Cl(III) led to the decomposition of the compounds with the concomitant formation of the corresponding halobenzene, as the major product, together with a minor amount the combination of the extruded aryl with the halobenzene (Scheme 22). Interestingly, differences in the recombined distribution of the products were observed. Indeed, a mixture of 2- to 3- to 4-halobiphenyl was formed in 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13:14, 51:30:19 and 48:31:21 ratio respectively for I(III), Br(III) and Cl(III).
:13:14, 51:30:19 and 48:31:21 ratio respectively for I(III), Br(III) and Cl(III).
 | ||
| Scheme 22 Photo-degradation of diaryl λ3-halogen compounds. | ||
At a mechanistic level, upon direct irradiation, the aryl halonium salts generate the corresponding singlet excited state, which can undergo a heterolytic cleavage, resulting in the formation of the aryl cation–haloarene pair. Alternatively, it can be engaged in the intersystem crossing (ISC) pathway leading to the generation of the triplet excited state. Following this, the triplet excited state could proceed to undergo homolysis, forming the triplet aryl radical–haloarene pair. The observed products can be ascribed to the homolytic and heterolytic cleavage reaction pathways and follow the trend of the related stability of the intermediates. Moreover, the quantum yield of the disappearance of hypervalent compounds upon irradiation at 248 nm is in accordance with the series of chloronium, bromonium, and iodonium salts (0.79, 0.82, 0.54 respectively). Nevertheless, the direct photolysis of hypervalent compounds in acetonitrile also provides, in all the cases, the formation of acetanilide, thus supporting the aryl cation intermediate formation.
Reactivity of cyclic diaryl hypervalent λ3-bromane and chlorane compounds
Despite being the first examples of hypervalent compounds prepared, the reactivity of cyclic diaryl λ3-bromanes and chloranes has been scarcely investigated, and only recently a renewed interest in this field has emerged. Seminal investigation on the reactivity of hypervalent bromines and chlorines was reported in 1968 by Lees and Heaney and subsequently by Sato and co-workers in 1974 for their thermal decomposition (Scheme 23).34,83 The pyrolysis of 4-Br and 4-I led to the formation of the 2-chloro, 2′-iodo diphenyl 83 and 2-chloro, 2′-bromo diphenyl 84.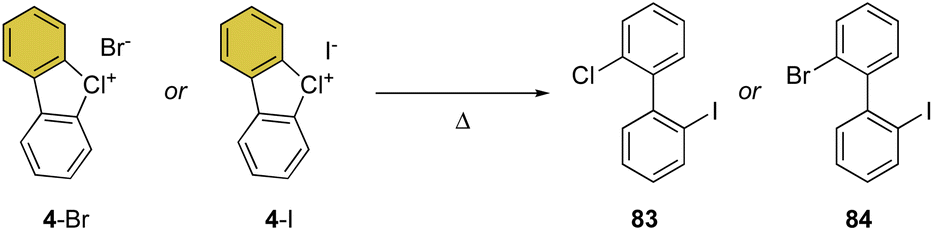 | ||
| Scheme 23 Pyrolysis of diaryl λ3-bromane and chlorane compounds. | ||
Our group has recently discovered a new reactivity for the cyclic diaryl hypervalent bromines and chlorines.70,73 Unexpectedly, these compounds in the presence of a weak base at room temperature serve as aryne precursors, releasing the corresponding 2-halo benzyne under unparallel mild reaction conditions thus obviating the need for fluorinating agents or strong bases (Scheme 24).84,85
 | ||
| Scheme 24 Mechanistic investigation of the reactivity of cyclic diaryl λ3-bromane and chlorane compounds. | ||
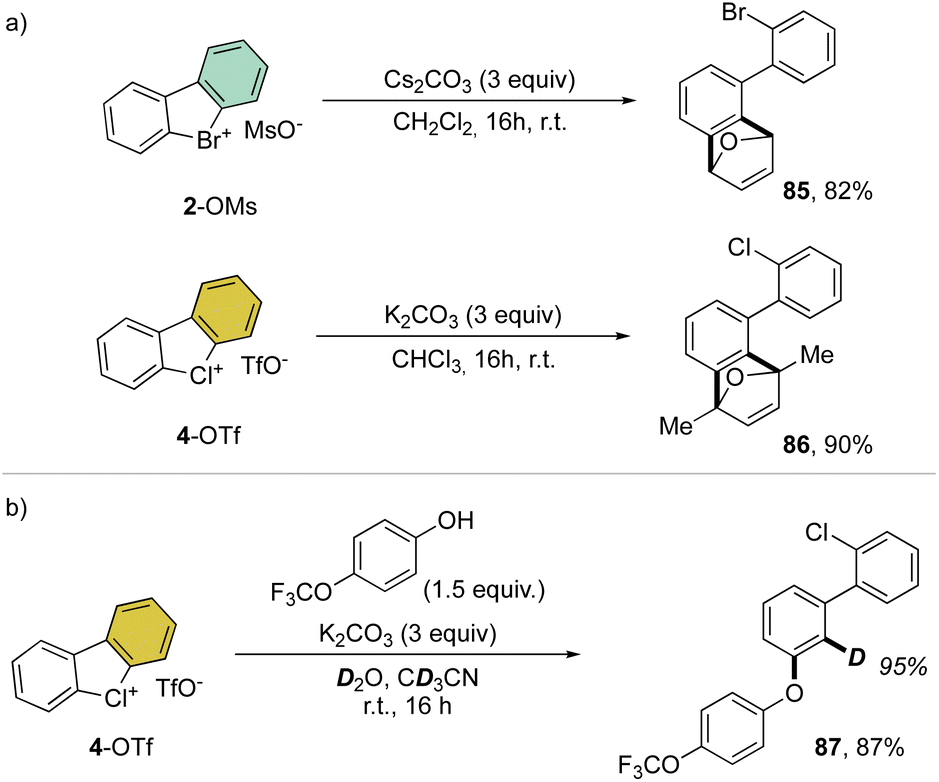
Interestingly, this reactivity turns out to be unique for hypervalent bromines and chlorines while the iodane derivatives remained inert. Experimental and DFT mechanistic investigations were carried out in collaboration with the group of Houk to elucidate the different reactivity observed and their chemoselectivity in the reaction with phenols. Indeed, a pronounced effect of the nature of the base and the solvent has been observed on these reactions, and the hypervalent bromines and chlorines, thus allowing a chemoselective control of the reactions. Initially, reacting 2-OMs and 4-OTf with furans led to the corresponding [4 + 2] products in excellent yields. Then, deuterated hypervalent compounds and/or deuterated coupling partners were used, thus aiming for deuterium labelling. The reaction of 4-OTf in D2O and CD3CN delivered the product 87 in good yield with high deuterium incorporation (up to 95%) at the ortho-position. These experiments proved the formation of an aryne intermediate during the reaction. DFT calculations provide us with a clear understanding of the aryne intermediate formation, which proceeds via an initial anion exchange, followed by a base-mediated deprotonation and concurrent C–X bond cleavage (Fig. 10). The complemental reactivity of these hypervalent compounds can be attributed to the larger stabilization of the newly formed iodonium salt [I(III)CO3]. A larger activation energy is then required to deprotonate the stabilised iodonium species B–I which translates into the lack of reactivity of this species. On the other hand, the lower energy barriers result in mild reaction conditions for the reaction of bromane and chlorane compounds, with the latter showing an enhanced reactivity dictated by the “barrierless” activation energy. Interestingly, under our reaction conditions, nucleophilic attack occurs selectively at the meta-position, and as rationalized by experimental and DFT models, the ortho-position was sterically demanding and less stabilized.86–90
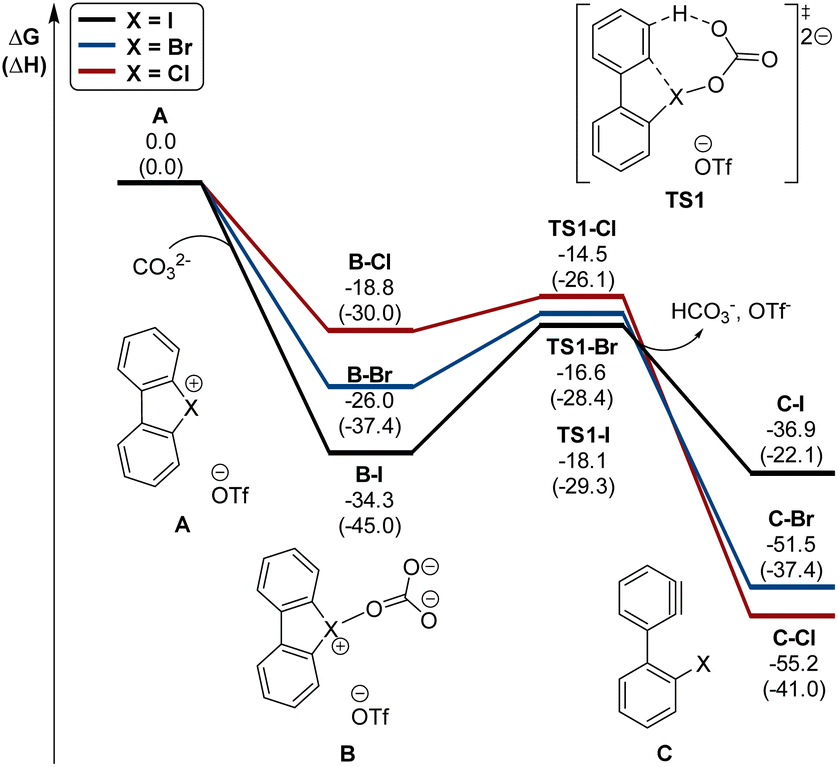 | ||
| Fig. 10 Calculated energy diagram (in kcal mol−1) for the formation of arynes from hypervalent halonium. | ||
Introduction of substituents into the hypervalent chlorine structure affects the regioselectivity of the reaction enabling the generation of the aryne intermediate on the more electron poor aryl fragment and additionally altering the meta-/ortho- selectivity of the nucleophilic addition.
Taking advantage of the mild reaction conditions for the generation of the aryne intermediate a diversity of reactions could be designed with success, including [4 + 2] (88, 89), 1,3-dipolar (90–92) and [2 + 2] cycloadditions (93–95) (Scheme 25). Additionally, rare [4 + 2] reactions involving the β-bromo styrenes were successfully developed yielding interesting tetraphenyl compounds (96–98).91 Carboxylic acids, anilines, aliphatic amines and phenols were also reacted with hypervalent bromines and chlorines, resulting in a vast, diverse, and unprecedented library of compounds (Scheme 26).70,73 Sterically demanding anilines and amines were well tolerated (99, 100) and carboxylic acids were particularly meta-selective. Conditions-driven chemoselective reaction between phenol and hypervalent chlorines has shown a large functional group tolerance with high regioselectivity towards the formation of triphenyl phenol (103, 104) or highly decorated diphenyl ether (105, 106).
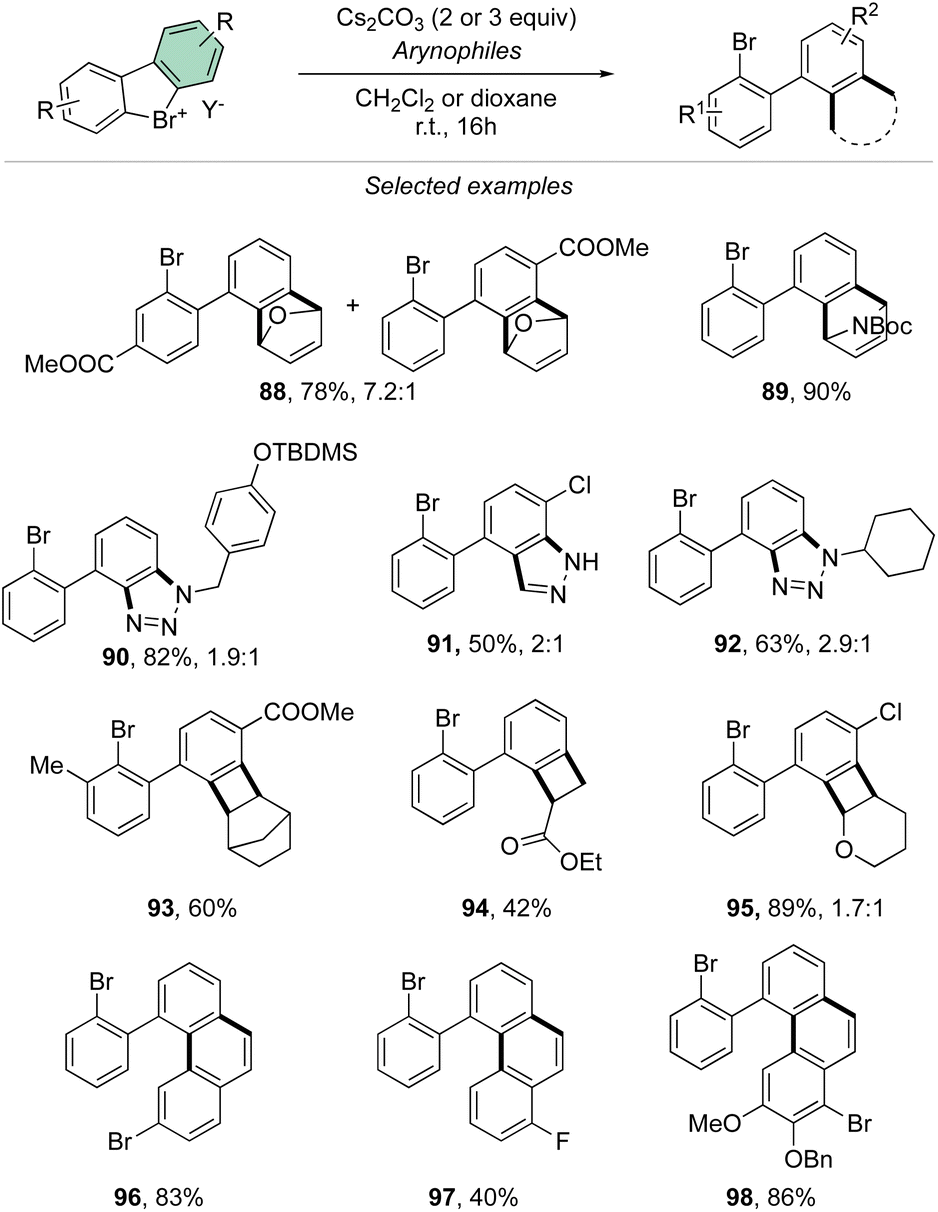 | ||
| Scheme 25 Cycloadditions with cyclic diaryl λ3-bromanes. | ||
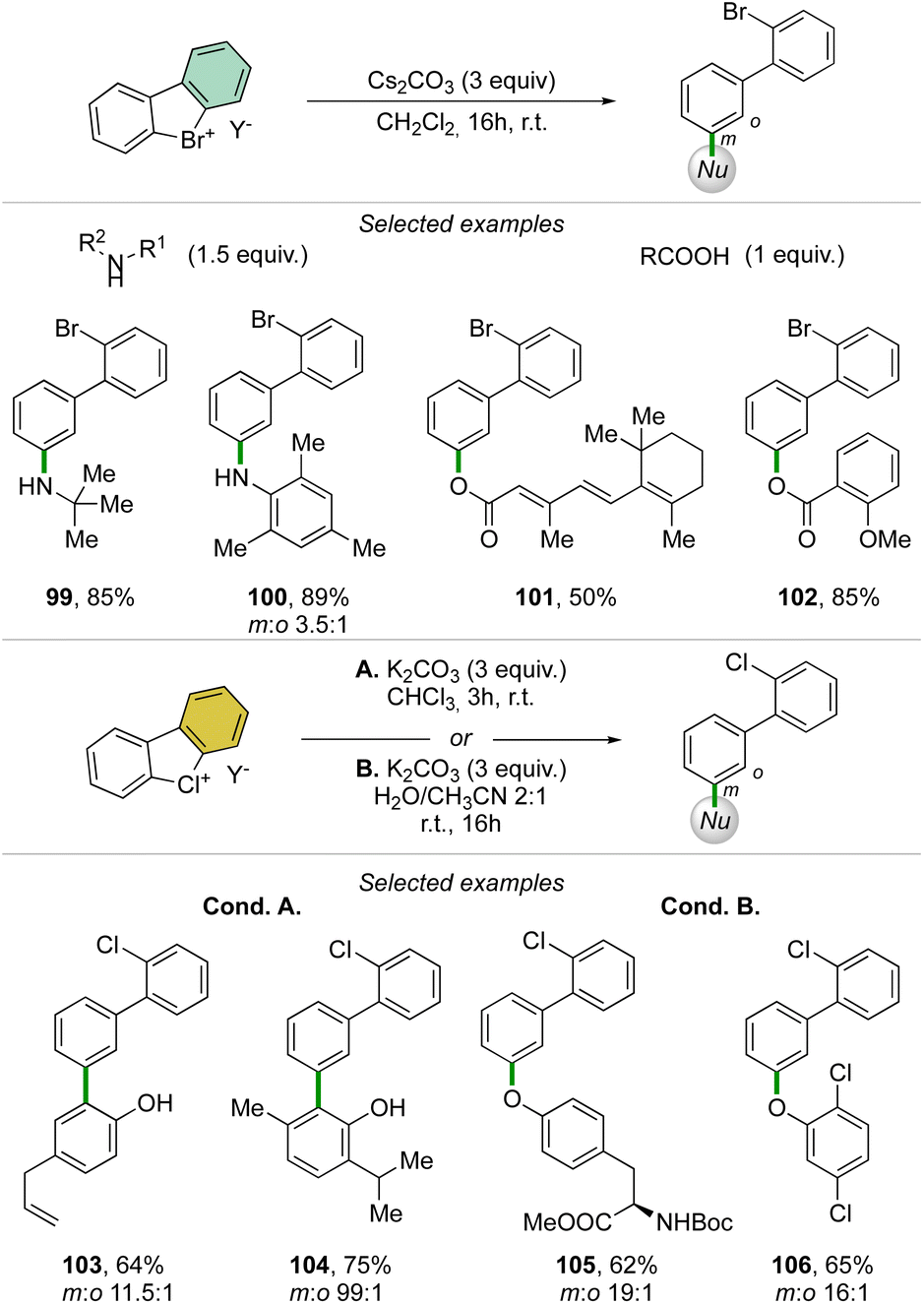 | ||
| Scheme 26 Reactivity of cyclic diaryl λ3-bromanes and chloranes towards carboxylic acids, anilines, amines and phenols. | ||
These metal-free C–O and C–N couplings occur under surprisingly mild reaction conditions offering an appealing alternative to the commonly used palladium and copper catalysed Buchwald-Hartwig and Ullmann couplings.
Taking advantage of the previously described reaction conditions, Li and co-workers have extended the scope of nucleophiles in the reaction of cyclic diaryl hypervalent bromines, thus exploiting the formation of C–O bonds using aliphatic alcohols.92 The reaction proceeds smoothly with a large variety of primary, secondary, and tertiary alcohols delivering the corresponding aryl ethers in good to excellent yields.
Interestingly, the competition experiment using NaOTs, NaOMe and NaOAc (1:1:1 equiv. vs. 1 equiv. of 2) with 2-BF4 delivered a mixture of products (Scheme 27). The distribution can be correlated with the nucleophilicity of the anions, thus following the order: OMe− > OAc− > OTs−.
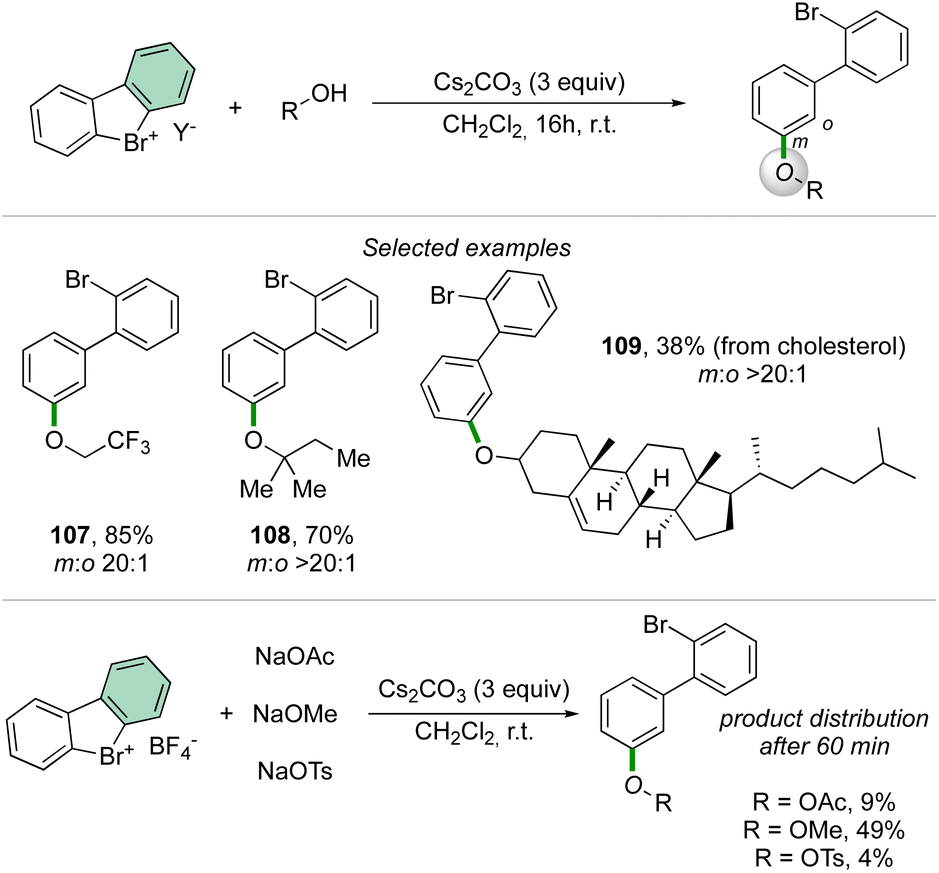 | ||
| Scheme 27 Reactivity of cyclic diaryl λ3-bromanes towards alcohols. | ||
Deuteration experiments further corroborate the formation of an aryne intermediate, thus leading to a high level of deuteration at the ortho-position. Besides acting as a reagent, the catalytic activity of these compounds could also be expected.93–95 Following this hypothesis, Yoshida and co-workers have reported the first halogen-bonding catalysis using cyclic diaryl λ3-bromane 110.69 The larger electron withdrawing ability of the bromane, in comparison with the iodane counterpart, enabled the Michael reaction between indoles and the trans-crotonophenone (Scheme 28).94
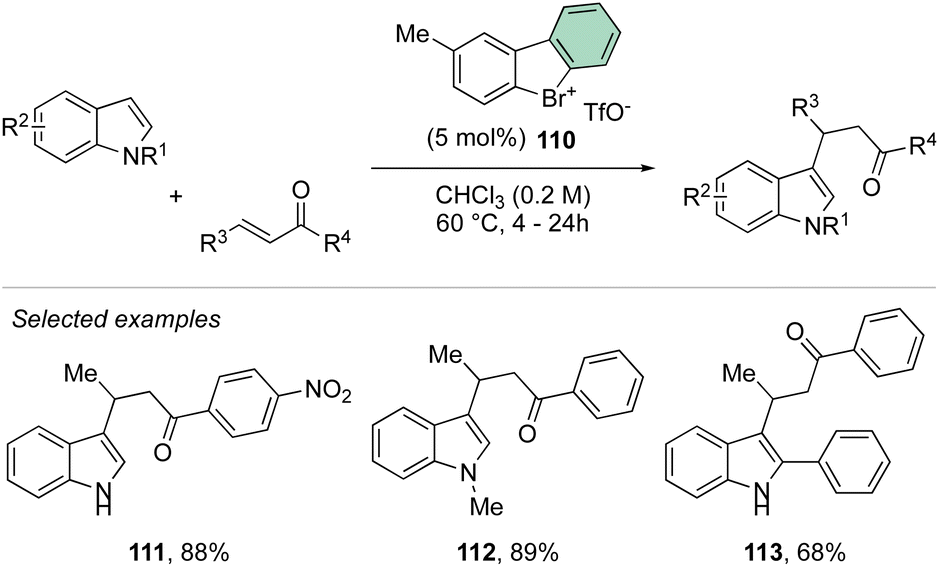 | ||
| Scheme 28 Cyclic diaryl λ3-bromane catalysed Michael reaction. | ||
Indeed, while Br(III) leads to complete conversion within 90 min, the corresponding iodane compound turned out to be completely inactive under these reaction conditions. The counter anion of the bromanes affected the catalytic activity, while indeed the strongly coordinating chlorine anion led to a low-yielding reaction, the triflate and BArF4 accelerated the reaction rate and boosted the product formation to high yields (up to 96%). Subsequently, Yoshida and co-workers described the first enantioselective halogen-bonding catalysis using a naphyl-based chiral cyclic diaryl λ3-bromane to pursue the vinylogous Mannich reaction of cyanomethyl coumarins with ketimines (Scheme 29).71 Interestingly, the screening of halogen catalysts (45–48) revealed the superior catalytic efficiency of bromane derivatives achieving 91% yield and 90% ee. On the other hand, chlorane and iodane led to similar enantio-induction although in moderate and low yields respectively.
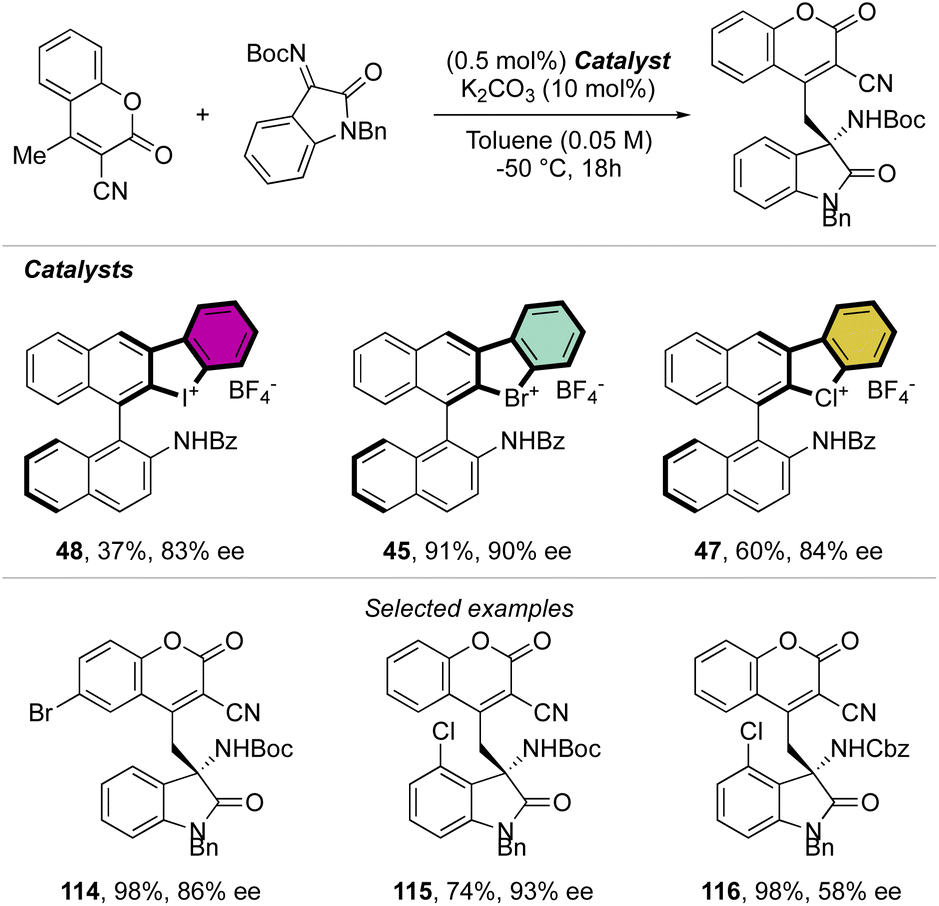 | ||
| Scheme 29 Enantioselective Mannich reaction catalysed by cyclic diaryl λ3-bromanes. | ||
More recently, Yoshida and co-workers have also explored the use of chiral cyclic diaryl λ3-halogens bearing a N-nitrosamine moiety for the Mannich reaction of malonic esters and ketimines (Scheme 30).72 Halogen nitroamine catalysts promoted this transformation, although only the bromane catalyst provided high yields and enantioselectivity. In contrast, low or no enantioselectivity was observed while using iodane and chlorane catalysts.
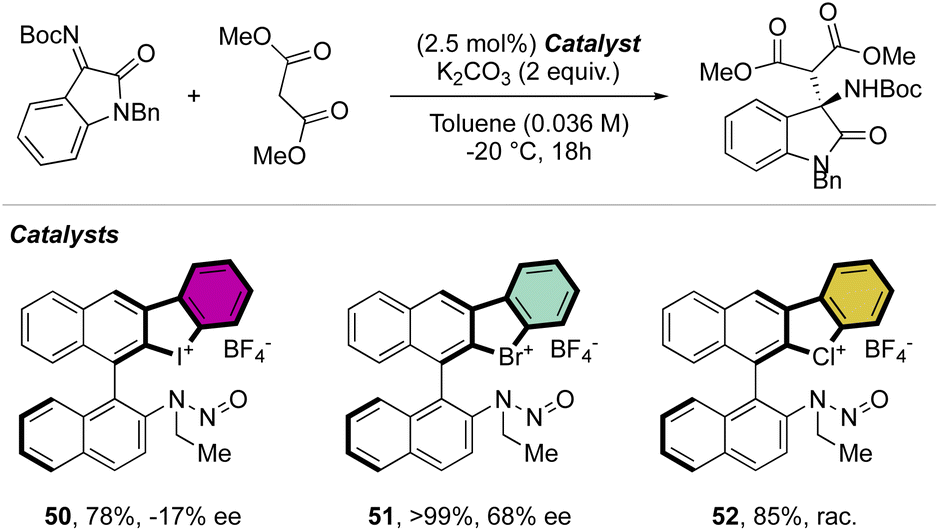 | ||
| Scheme 30 Enantioselective Mannich reaction catalysed by N-nitrosamine cyclic diaryl λ3-bromanes. | ||
Conclusions
As Lubinkowsky and McEwen announced in 1972, “the bromonium salts will not achieve a preeminent status in synthesis until a better method of preparation of the salts than the one presently available is discovered.”, these words remain currently true.75 Indeed, while recent investigations reported innovative and more efficient synthetic approaches and new reactivity patterns for these niche compounds, we may have only uncovered the thin end of the wedge. Within this perspective, we have analysed and compared the reactivity of diaryl hypervalent λ3-iodines, bromines and chlorines with a particular focus on the relative differences. The bromane and chlorane compounds are undoubtedly superior arylating agents with respect to the iodanes, and their enhanced reactivity emerges from the better-leaving group ability of Br(III) and Cl(III), as also expressed by the Hammet value, as a direct consequence of the intrinsic electronegativity of the elements. New opportunities in the field can be envisioned either in the use of these compounds as reagents, for instance as oxidants or aryne precursors for the functionalization and/or construction of more complex structures, as well as catalysts and as fundamental components in crystal engineering. We are strongly convinced that further experimental efforts combined with mechanistic studies will lead to uncovering new reactivity pathways for hypervalent chlorines and bromines, thus expanding the synthetic toolbox. We may have only uncovered the thin end of the wedge. We hope to have shed light on a relatively uncharted hypervalent chemistry of bromane and chlorane compounds, thus providing the readers with a complete overview of the field.Author contributions
ML and JWD have designed and prepare the present review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is funded by EC ERC SG “AlCHIMIE”, Grant agreement ID: 949804.Notes and references
- A. Bauer and N. Maulide, Chem. Sci., 2021, 12, 853–864 RSC.
- S. S. Karandikar, A. Bhattacharjee, B. E. Metze, N. Javaly, E. J. Valente, T. M. McCormick and D. R. Stuart, Chem. Sci., 2022, 13, 6532–6540 RSC.
- N. C. Norman and P. G. Pringle, Chemistry, 2022, 4, 1226–1249 CrossRef CAS.
- H. P. de Magalhães, O. Sala and H. P. Lüthi, in PATAI'S Chemistry of Functional Groups, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–29 Search PubMed.
- G. Sean McGrady and J. W. Steed, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Atwood and R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2006, p. ia094 Search PubMed.
- V. V. Zhdankin, Hypervalent iodine chemistry: preparation, structure, and synthetic applications of polyvalent iodine compounds, John Wiley & Sons, Inc, Chichester, West Sussex, 2014 Search PubMed.
- T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef CAS PubMed.
- Hypervalent iodine chemistry: modern developments in organic synthesis, ed. T. Wirth, Y. Kita and Y. Kita, Springer, Berlin Heidelberg, 2003 Search PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- L. F. Silva, Jr. and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- X. Peng, A. Rahim, W. Peng, F. Jiang, Z. Gu and S. Wen, Chem. Rev., 2023, 123, 1364–1416 CrossRef CAS PubMed.
- X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712–1724 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS.
- F. Sousa e Silva, T. Anthony and S. Wengryniuk, Molecules, 2017, 22, 780 CrossRef PubMed.
- B. Winterson, T. Patra and T. Wirth, Synthesis, 2022, 54, 1261–1271 CrossRef CAS.
- K. Miyamoto and M. Uchiyama, Chem. Lett., 2021, 50, 832–838 CrossRef CAS.
- K. Miyamoto, in Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2018, pp. 1–25 Search PubMed.
- U. Farooq, A.-H. A. Shah and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 1018–1020 CrossRef CAS PubMed.
- M. Ochiai, Synlett, 2009, 2009, 159–173 CrossRef.
- B. Olofsson, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing, Cham, 2015, vol. 373, pp. 135–166 Search PubMed.
- M. Ochiai, N. Tada, K. Miyamoto and M. Shiro, Heteroat. Chem., 2011, 22, 325–330 CrossRef CAS.
- M. M. Hoque, K. Miyamoto, N. Tada, M. Shiro and M. Ochiai, Org. Lett., 2011, 13, 5428–5431 CrossRef CAS PubMed.
- M. Ochiai, A. Yoshimura, Md. M. Hoque, T. Okubo, M. Saito and K. Miyamoto, Org. Lett., 2011, 13, 5568–5571 CrossRef CAS PubMed.
- M. Ochiai, A. Yoshimura, K. Miyamoto, S. Hayashi and W. Nakanishi, J. Am. Chem. Soc., 2010, 132, 9236–9239 CrossRef CAS PubMed.
- M. Ochiai, A. Yoshimura and K. Miyamoto, Tetrahedron Lett., 2009, 50, 4792–4795 CrossRef CAS.
- M. Ochiai, A. Yoshimura, T. Mori, Y. Nishi and M. Hirobe, J. Am. Chem. Soc., 2008, 130, 3742–3743 CrossRef CAS.
- M. Nakajima, K. Miyamoto, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2019, 141, 6499–6503 CrossRef CAS.
- M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448–451 CrossRef CAS PubMed.
- M. Ochiai, K. Miyamoto, S. Hayashi and W. Nakanishi, Chem. Commun., 2010, 46, 511–521 RSC.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- R. B. Sandin and A. S. Hay, J. Am. Chem. Soc., 1952, 74, 274–275 CrossRef CAS.
- H. Heaney and P. Lees, Tetrahedron, 1968, 24, 3717–3723 CrossRef CAS.
- A. N. Nesmeyanov, L. G. Makarova and T. P. Tolstaya, Tetrahedron, 1957, 1, 145–157 CrossRef CAS.
- A. N. Nesmeyanov, I. N. Lisichkina, A. S. Kulikov, A. N. Vanchikov, T. P. Tolstaia and V. A. Chertkov, Dokl. Akad. Nauk SSSR, 1978, 243, 1463–1466 CAS.
- G. A. Olah, T. Sakakibara and G. Asensio, J. Org. Chem., 1978, 43, 463–468 CrossRef CAS.
- Z. Hou, Y. Zhu and Q. Wang, Sci. China, Ser. B: Chem., 1996, 39, 260 CAS.
- A. N. Nesmeyanov, A. N. Vanchikov, I. N. Lisichkina, V. V. Grushin and T. P. Tolstaia, Dokl. Akad. Nauk SSSR, 1980, 255, 1386 CAS.
- H. J. Frohn and M. Giesen, J. Fluorine Chem., 1998, 89, 59–63 CrossRef CAS.
- A. N. Nesmeyanov, I. N. Lisichkina, A. N. Vanchikov and T. P. Tolstaya, Russ. Chem. Bull., 1976, 25, 224 CrossRef.
- H. J. Frohn, M. Giesen, D. Welting and G. Henkel, Eur. J. Solid State Inorg. Chem., 1996, 33, 841 CAS.
- A. N. Nesmeyanov, A. N. Vanchikov, I. N. Lisichkina, V. V. Lazarev and T. P. Tolstaya, Dokl. Akad. Nauk SSSR, 1980, 255, 1136 CAS.
- H. J. Frohn, M. Giesen, D. Welting and G. Henkel, Eur. J. Solid State Inorg. Chem., 1999, 33, 841 Search PubMed.
- A. N. Nesmeyanov, A. N. Vanchikov, I. N. Lisichkina, N. S. Khruscheva and T. P. Tolstaya, Dokl. Akad. Nauk SSSR, 1980, 254, 652 CAS.
- M. Ochiai, Y. Nishi, S. Goto, M. Shiro and H. J. Frohn, J. Am. Chem. Soc., 2003, 125, 15304–15305 CrossRef CAS PubMed.
- M. Ochiai, Y. Nishi, S. Goto and H. J. Frohn, Angew. Chem., Int. Ed., 2005, 44, 406–409 CrossRef CAS PubMed.
- A. N. Nesmeyanov, T. L. Khotsyanova, V. V. Saat-sasov, T. P. Tolstaya and L. S. Isaeva, Dokl. Akad. Nauk SSSR, 1974, 218, 140 CAS.
- T. L. Chan and Z. Xie, Chem. Sci., 2018, 9, 2284–2289 RSC.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1184 CrossRef PubMed.
- R. B. King, Chem. Rev., 2001, 101, 1119–1152 CrossRef CAS PubMed.
- I. I. Demkina, A. N. Vanchikov, V. V. Grushin and T. P. Tolstaya, Russ. Chem. Bull., 1985, 34, 858–861 CrossRef.
- V. V. Grushin, T. P. Tolstaya and I. N. Lisichkina, Izv. Akad. Nauk SSSR, Ser. Khim., 1982, 2412 CAS.
- A. I. Yanovsky, Yu. T. Struchkov, V. V. Grushin, T. P. Tolstaya and I. I. Demkina, Zh. Strukt. Khim., 1988, 29, 89 Search PubMed.
- H. J. Frohn and M. Giesen, J. Fluorine Chem., 1984, 24, 9–15 CrossRef CAS.
- K. Miyamoto, M. Saito, S. Tsuji, T. Takagi, M. Shiro, M. Uchiyama and M. Ochiai, J. Am. Chem. Soc., 2021, 143, 9327–9331 CrossRef CAS PubMed.
- M. Ochiai, T. Kaneaki, N. Tada, K. Miyamoto, H. Chuman, M. Shiro, S. Hayashi and W. Nakanishi, J. Am. Chem. Soc., 2007, 129, 12938–12939 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- M. Elsherbini and W. J. Moran, Org. Biomol. Chem., 2021, 19, 4706–4711 RSC.
- M. Elsherbini and T. Wirth, Chem. - Eur. J., 2018, 24, 13399–13407 CrossRef CAS PubMed.
- R. Francke, Curr. Opin. Electrochem., 2019, 15, 83–88 CrossRef CAS.
- J. D. Haupt, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 242–245 CrossRef CAS PubMed.
- T. T. Nguyen and J. C. Martin, J. Am. Chem. Soc., 1980, 102, 7382–7383 CrossRef CAS.
- T. T. Nguyen, S. R. Wilson and J. C. Martin, J. Am. Chem. Soc., 1986, 108, 3803–3811 CrossRef CAS.
- I. Sokolovs, N. Mohebbati, R. Francke and E. Suna, Angew. Chem., Int. Ed., 2021, 60, 15832–15837 CrossRef CAS.
- N. Mohebbati, I. Sokolovs, P. Woite, M. Lõkov, E. Parman, M. Ugandi, I. Leito, M. Roemelt, E. Suna and R. Francke, Chem. – Eur. J., 2022, 28, e202200974 CrossRef CAS PubMed.
- I. Sokolovs and E. Suna, Org. Lett., 2023, 25, 2047–2052 CrossRef CAS PubMed.
- E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2010, 132, 4062–4063 CrossRef CAS PubMed.
- Y. Yoshida, S. Ishikawa, T. Mino and M. Sakamoto, Chem. Commun., 2021, 57, 2519–2522 RSC.
- M. Lanzi, Q. Dherbassy and J. Wencel-Delord, Angew. Chem., Int. Ed., 2021, 60, 14852–14857 CrossRef CAS PubMed.
- Y. Yoshida, T. Mino and M. Sakamoto, ACS Catal., 2021, 11, 13028–13033 CrossRef CAS.
- Y. Yoshida, T. Ao, T. Mino and M. Sakamoto, Molecules, 2023, 28, 384 CrossRef CAS PubMed.
- M. Lanzi, T. Rogge, T. S. Truong, K. N. Houk and J. Wencel-Delord, J. Am. Chem. Soc., 2023, 145, 345–358 CrossRef CAS PubMed.
- W. W. Chen, M. Artigues, M. Font-Bardia, A. B. Cuenca and A. Shafir, J. Am. Chem. Soc., 2023, 145, 13796–13804 CrossRef CAS PubMed.
- J. J. Lubinkowski and W. E. McEwen, Tetrahedron Lett., 1972, 13, 4817–4820 CrossRef.
- V. V. Grushin, M. M. Kantor, T. P. Tolstaya and T. M. Shcherbina, Russ. Chem. Bull., 1984, 33, 2130–2135 CrossRef.
- V. V. Grushin, I. I. Demkina and T. P. Tolstaya, Inorg. Chem., 1991, 30, 1760–1765 CrossRef CAS.
- V. V. Grushin, Acc. Chem. Res., 1992, 25, 529–536 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Polym. Lett. Ed., 1978, 16, 563–571 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307–1315 CrossRef CAS.
- J. L. Dektar and N. P. Hacker, J. Org. Chem., 1991, 56, 1838–1844 CrossRef CAS.
- T. Sato, K. Shimizu and H. Moria, J. Chem. Soc., Perkin Trans. 1, 1974, 1537 RSC.
- J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892–4044 CrossRef CAS.
- Z. J. P. Hou and L. X. H. He, Chin. Chem. Lett., 2002, 13, 189–192 CAS.
- Z. Liu and R. C. Larock, J. Org. Chem., 2006, 71, 3198–3209 CrossRef CAS PubMed.
- P. M. Tadross, C. D. Gilmore, P. Bugga, S. C. Virgil and B. M. Stoltz, Org. Lett., 2010, 12, 1224–1227 CrossRef CAS PubMed.
- H. Yoshida, S. Sugiura and A. Kunai, Org. Lett., 2002, 4, 2767–2769 CrossRef CAS PubMed.
- S. M. Bronner, J. L. Mackey, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 13966–13969 CrossRef CAS.
- J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798–15805 CrossRef PubMed.
- M. Lanzi, R. A. Ali Abdine, M. De Abreu and J. Wencel-Delord, Org. Lett., 2021, 23, 9047–9052 CrossRef CAS PubMed.
- Y. Wang, Y.-N. Tian, S. Ren, R. Zhu, B. Huang, Y. Wen and S. Li, Org. Chem. Front., 2023, 10, 793–798 RSC.
- F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830–3833 CrossRef CAS PubMed.
- G. Cavallo, J. S. Murray, P. Politzer, T. Pilati, M. Ursini and G. Resnati, IUCrJ, 2017, 4, 411–419 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |