
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-efficiency non-thermal plasma synthesis of imine macrocycles†
Patrycja
Roszkowska‡
 a,
Abbie M.
Scholes‡
a,
James L.
Walsh
*b,
Timothy L.
Easun
*c and
Anna G.
Slater
*a
a,
Abbie M.
Scholes‡
a,
James L.
Walsh
*b,
Timothy L.
Easun
*c and
Anna G.
Slater
*a
aDepartment of Chemistry and Materials Innovation Factory, School of Physical Sciences, University of Liverpool, UK. E-mail: anna.slater@liverpool.ac.uk
bYork Plasma Institute, School of Physics, Engineering & Technology, University of York, Heslington, York, YO10 5DQ, UK. E-mail: james.l.walsh@york.ac.uk
cSchool of Chemistry, Haworth Building, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: t.l.easun@bham.ac.uk
First published on 8th April 2024
Abstract
Macrocycles are candidates for wide-ranging applications, yet their synthesis can be low-yielding, poorly reproducible, and resource-intensive, limiting their use. Here, we explore the use of Non-Thermal Plasma (NTP) as an efficient method for the synthesis of imine macrocycles at the gram scale. NTP-mediated macrocyclisations consistently achieved high yields of up to 97% in reduced reaction times compared to the standard non-plasma method, and were successfully carried out with a range of different aldehyde substrates. Control experiments were performed to explore the origin of the observed improvements. The results indicate that NTP methods could be advantageous for macrocycle synthesis, particularly for substrates that are sensitive to elevated temperature, and other materials formed via imine condensation.
Macrocycles are ubiquitous across many fields of chemistry due to their precise arrangement of functional groups around a well-defined central cavity.1 However, the inherent competition between oligo- or polymeric vs. cyclic products in macrocyclisation limits their large-scale production and use. Several synthetic approaches have emerged to overcome this challenge.2–6 Such approaches include high-dilution methods, where large volumes of solvent are used, or metal templating methods, which require the use of additional reagents and steps to remove the template from the desired macrocycle.7 Neither approach is sustainable or economically desirable, particularly on an industrial scale.
Imine macrocycles are a class of compounds that have been shown to form porous materials capable of industrially relevant separations.8–13 In the case of imine macrocycles, the reversible nature of the condensation is exploited to form the thermodynamic product in an example of dynamic covalent chemistry.14–16 Depending on the substituents, the thermodynamically most stable product could be a [2 + 2], [3 + 3] (Fig. 1), or larger macrocycle, or, if multiple macrocycles with similar stabilization energies are possible, a mixture of multiple macrocyclic products.17
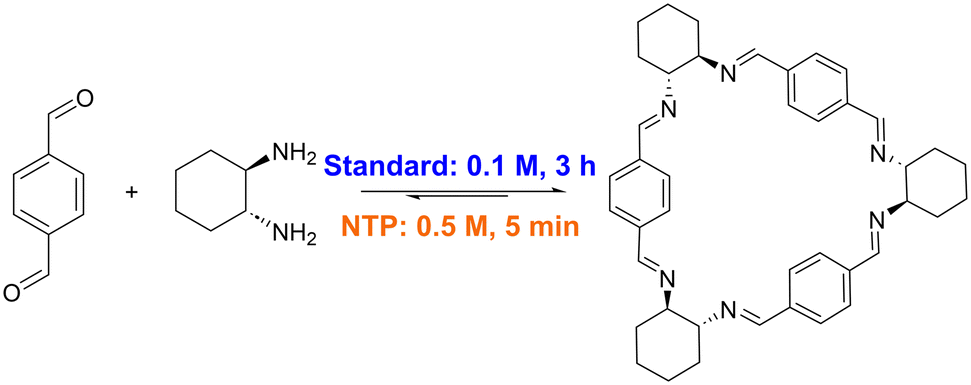 | ||
| Fig. 1 Conditions for synthesis of trianglimine 1, with reported standard conditions (blue) compared to this work, using non-thermal plasma (orange). | ||
Strategies for selective formation of a specific imine macrocycle include templating, or tuning reaction conditions to isolate a specific macrocycle via crystallisation.18–21 For these systems, there have also been limited reports of more sustainable methods of synthesis: López-Periago et al. used supercritical carbon dioxide (scCO2) as a green solvent to synthesise an unsubstituted trianglimine22 and microwave-assisted synthesis has been reported for Schiff base macrocycles.23–25
However, a general method for imine macrocycle synthesis has not been developed, and the success of such strategies can be challenging to predict and control. Imine formation is influenced not only by the substrate, but by concentration gradients, mixing effects, and variable solvent water content, which may make scale-up challenging and contribute to poor reproducibility. As a consequence, reported imine macrocycle formation conditions vary considerably in terms of time, temperature, and resultant yield.17
A simple, transferrable protocol for forming imine macrocycles would thus be desirable. We have recently developed reactors in which non-thermal plasma (NTP) can be generated and mixed with solvents, exemplified via the efficient degradation of methylene blue.26 Here, we use the NTP reactor to accelerate the formation of imine macrocycles, optimising conditions for the NTP synthesis of macrocycle 1 (Fig. 1) and demonstrating that these conditions can be used for the high-yielding and rapid formation of a range of macrocycles with otherwise disparate reported syntheses (Fig. 2).18,22,27–33
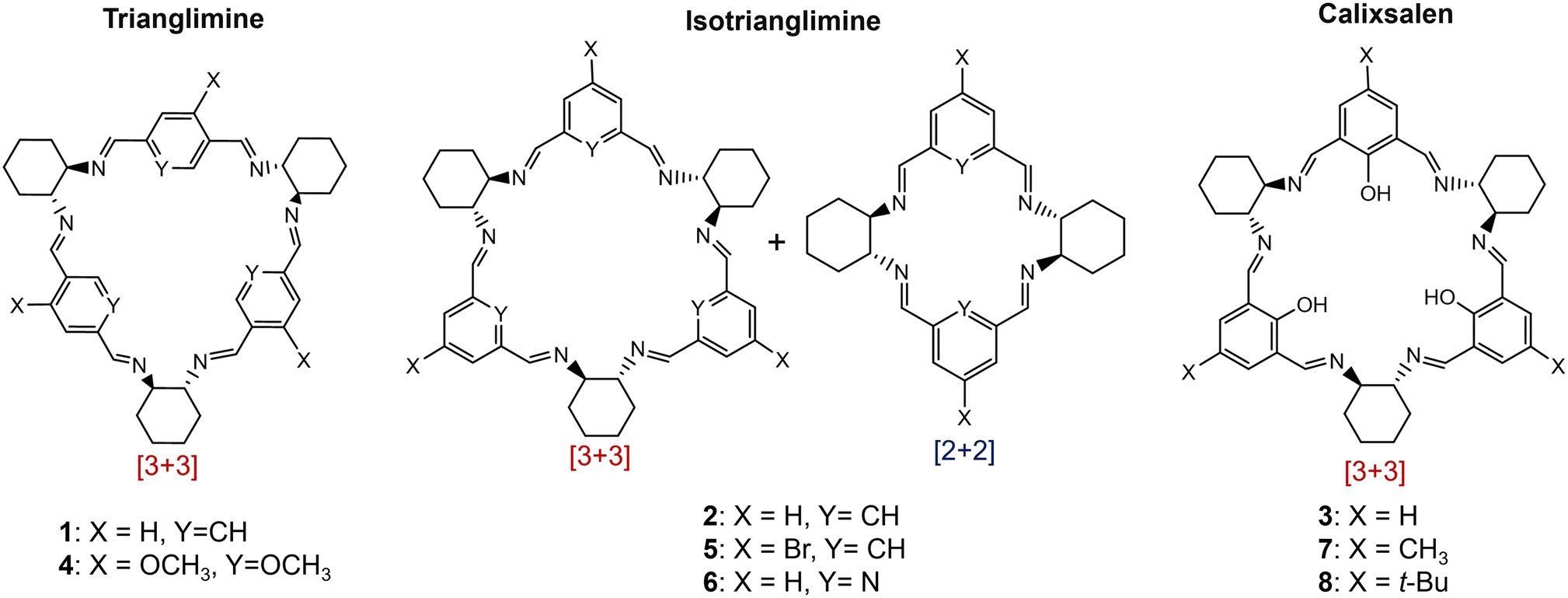 | ||
| Fig. 2 Imine macrocycles synthesised by NTP in this work: trianglimine, isotrianglimine and calixsalens. | ||
Non thermal plasma (NTP) is the fourth state of matter generated by the application of energy to a neutral gas causing its ionisation.34 It offers a highly reactive environment that can be generated at atmospheric pressure and ambient temperature, generating no waste. These unique properties mean NTP has found use in the chemical industry to modify catalytic supports,35 for CO2 conversion and utilisation,36 surface modification37 and beyond.38 Recently, more examples of NTP in contact with liquid are emerging, where the benefits of NTP are transferred to solution-phase chemical reactions. For example, NTP in contact with water was used to initiate radical reactions to dehalogenate iodo-substituted benzoates with high yields and mild conditions.39 A study using NTP in contact with MeOH and water was used to form carbon–carbon bonds via pinacol coupling.40 The NTP-liquid interaction has also been exploited under continuous conditions: a microfluidic NTP setup was used to aminate benzene with ammonia without the need for catalyst,41 and for direct N-acylation of amines by esters.42
Within the last 5 years, NTP assisted synthesis has been extended to the formation of metal-organic43–45 and covalent organic frameworks (COFs).46 The latter example demonstrates the potential of NTP to accelerate acid-catalysed reactions that form stable solid products, e.g., boronate ester, boroxine, and Schiff base COFs, but this has not yet been demonstrated for discrete molecular species such as macrocycles.
Despite these advances, it is still challenging to optimise NTP-reactions, in part due to the extremely complex nature of NTP in contact with liquid.47 Plasma can interact with both solvent and reagents, leading to complex reactive mixtures of short- and long-lived species, some of which can be detected by optical emission spectroscopy (OES).26,48 Detecting these species, or tracking the rates at which they form, is challenging, thus the mechanisms for plasma-derived chemical reactions are often unknown.
We recently reported two non-thermal plasma reactors, one microfluidic, and one batch reactor, which were used to treat chloroform, DCM, and water with argon-fed NTP.26 Here, we use the NTP batch reactor to study the use of NTP for imine macrocyclisation reactions. This work focuses on a range of macrocycles formed from R,R-cyclohexyldiamine (R,R-CHDA) selected from those reported in the literature, prioritising those with commercially available reagents. Each macrocycle differs by the structure of the dialdehyde unit, and 3 classes of imine macrocycle are represented: trianglimines, calixsalens and isotrianglimines (Fig. 2). For trianglimines and calixsalens, the sole product of the reaction is a [3 + 3] macrocycle, however, for isotrianglimines, both [3 + 3] and [2 + 2] macrocycles are formed.27
The range of macrocycles studied covered a) macrocycles known to form only one, thermodynamic, product; b) macrocycles known to form two or more stable products, and c) macrocycles that are difficult to synthesise, i.e., those requiring high temperatures and/or high dilution. The results were benchmarked against the reported syntheses and control experiments were performed to explore the origins of the observed differences.
Experimental
The batch reactor was set up as previously reported (ESI,† Fig. S1).26 Briefly, the plasma was generated via a single metallic electrode connected to a custom-built HV sinusoidal power source operating at 20 kHz. A CT4026 HV probe and Pearson 2877 current probe enabled the voltage/current signals to be monitored on a Keysight EDUX1052A oscilloscope, at a constant plasma discharge power of 3 W (unless stated otherwise). The plasma was operated with an argon feed gas (99.999%) at constant flow of 490 ml min−1 controlled by mass flow controllers (Aalborg GFC 17). While there is no ground electrode in the set-up, the plasma was capacitively coupled to ground via the grounded metallic clamp, as shown in Fig. 3.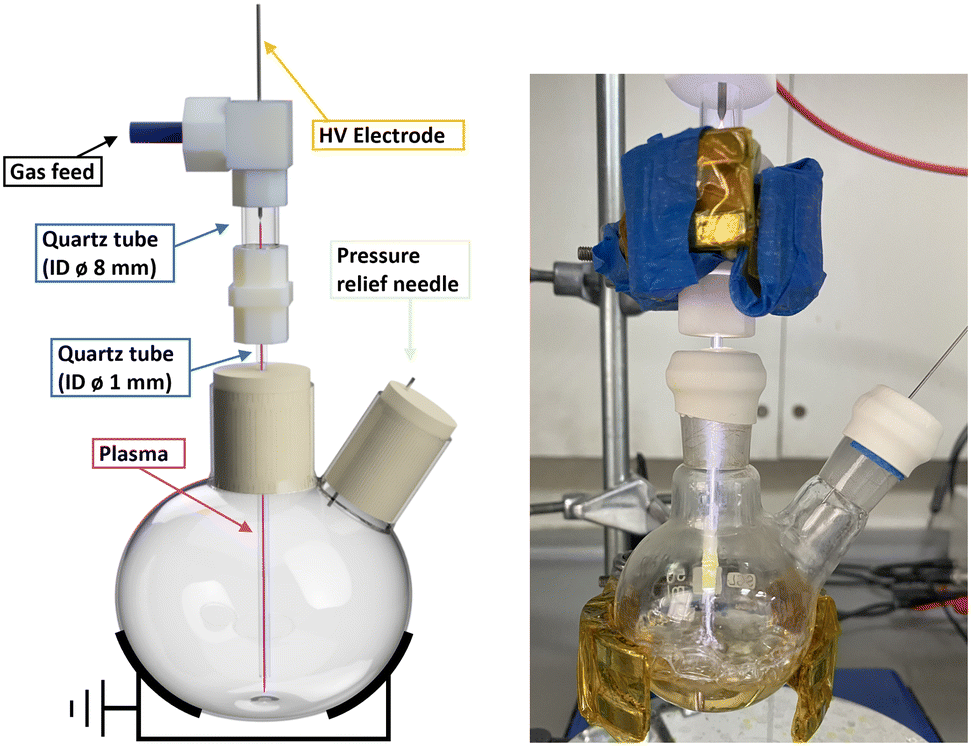 | ||
| Fig. 3 Scheme of NTP setup for chemistry reactions prior to the addition of liquid (left) and the setup in use (right). | ||
In a typical experiment, a two-neck 50 ml round bottom flask with a stirrer bar was used. One neck was sealed with a rubber septum that had been pierced by a needle for pressure relief, while the other neck was sealed by a rubber septum through which a small aperture had been cut to house the inlet for argon as the plasma feeding gas. The argon gas was introduced through a quartz tube measuring 5 cm in length, with an outside diameter (OD) of 10 mm and an inside diameter (ID) of 8 mm, and was connected at the top to the plasma-generating electrode. This tube was connected to narrower and longer tube of 12 cm length and with 1.2 mm internal and 3 mm external diameter via a PTFE connector. The narrow tube was passed through the septum until the tubing end reached close to the bottom of the flask (Fig. 3 and S1†). Prior to the reaction, the flask was exposed to an argon flow at a rate of 490 ml min−1 for a minimum of 2 minutes to ensure complete removal of any remaining air. The plasma discharge remained in contact with the reaction solution throughout the entire plasma exposure other than for one non-contact control experiment.
The relevant aldehyde and R,R-CHDA were separately dissolved in anhydrous chloroform and purged with nitrogen for 15 minutes. Then, the degassed aldehyde solution was transferred to the reaction flask using a syringe under a constant stream of argon. The magnetic stirrer was set to 300 rpm. The argon gas was kept at a consistent flow rate throughout all studies to ensure that the type and density of species within the plasma remained constant.49 When the plasma discharge started (t = 0), the solution of R,R-CHDA was added dropwise via syringe over 4 minutes.
For NMR analysis, aliquots were taken at 5 minutes and then at intervals until the reaction was stopped at 30 minutes. The aliquots taken at all time points were immediately evaporated to dryness to prevent the reaction progressing further, and the solid samples were subsequently dissolved in CDCl3. 1H NMR analysis allowed for the assessment and comparison of the success of each reaction by examining a) the consumption of the initial dialdehyde and b) the presence of impurities or side products (Table 1, ‘NMR Analysis’).
| Entry | Synthetic chemistry parameters | NTP | NMR analysis | ||||
|---|---|---|---|---|---|---|---|
| Concentration [M] | Volumea [ml] | Mass totalb [g] | Power [W] | SM aldehyde [normalised%] | Impurities | ||
| 5 min | 30 min | ||||||
| a Total volume of reactants. b Total mass of reagents used. c Standard conditions. d Final time was 10 minutes. | |||||||
| B | 0.5 | 30 | 1.86 | n/a | 0.1 | 0.1 | Yes |
| 1 | 0.05 | 30 | 0.18 | 3.2 | 3.8 | 3.1 | Yes |
| 2c | 0.5 | 30 | 1.86 | 3.2 | 0.5 | 0 | No |
| 3 | 1.25 | 30 | 4.65 | 3.2 | 0.9 | 1.9 | Yes |
| 4 | 0.5 | 15 | 0.93 | 3.2 | 1.0 | 2.5 | Yes |
| 5 | 0.5 | 45 | 2.79 | 3.2 | 1.8 | 3.4 | Yes |
| 6 | 0.5 | 30 | 1.86 | 1.4 | 7.0 | 5.8d | Yes |
| 7 | 0.5 | 30 | 1.86 | 4.5 | 5.9 | 11.5d | Yes |
The time of reaction (plasma exposure), volume and concentration of reaction solutions, and plasma parameters were varied to explore the effect on yield and selectivity for macrocycle 1 (Table 1), and the optimised conditions were tested on macrocycles 2–8. All macrocycles screened have been previously made via standard methods.27–33 Full experimental details and characterization can be found in the ESI.†
Results and discussion
The synthesis of macrocycle 1, trianglimine, was first used as a case study to investigate the impact of changing reagent concentration, total reaction volume, and plasma power (Table 1).The synthesis of macrocycle 1 is reported to take 2–3 h to reach full conversion at room temperature and a concentration of 0.1 M, giving a 90% yield.27 For comparison with NTP-assisted synthesis, reported batch conditions were carried out for 30 minutes. At 30 minutes, intermediates were observed by NMR (ESI,† Fig. S2), and the crude yield was 90%. By contrast, the NTP-assisted synthesis of macrocycle 1 gave high conversion of starting materials to macrocycle within 5 minutes (Table 1 and Fig. 4) with fewer intermediate species observed by NMR (ESI,† Fig. S3).
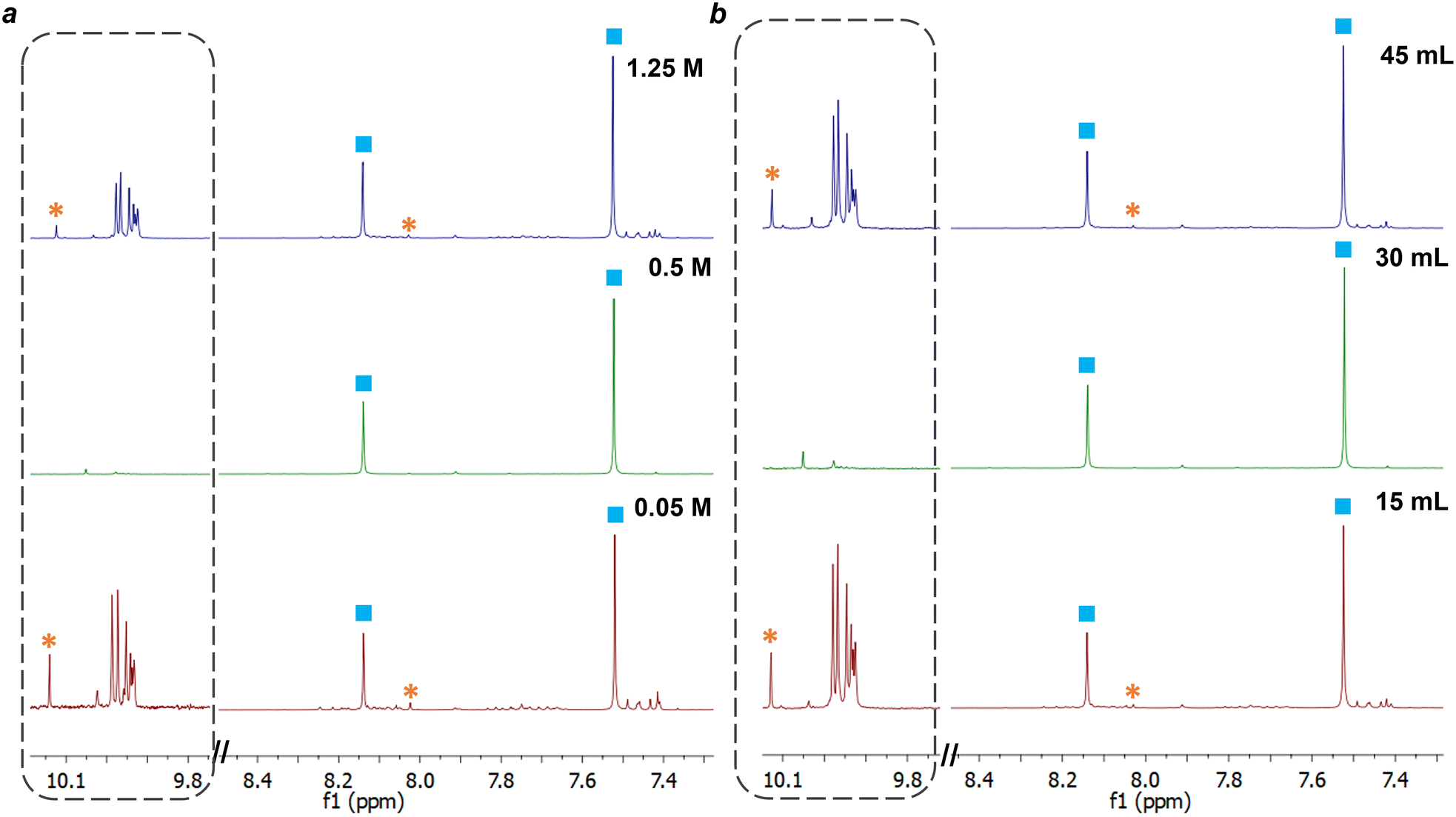 | ||
| Fig. 4 1H NMR spectra (CDCl3, 10.1–7.3 ppm) of NTP synthesis of macrocycle 1 at 5 minutes (a) for differing concentrations of 0.05 M, 0.5 M, and 1.25 M (b) for differing volumes 15 mL, 30 mL, and 45 mL. 9.8–10.1 ppm is magnified approximately 10× for all 3 spectra to highlight aldehyde peaks. Product peaks are labelled with a blue square and peaks from aldehyde starting material with an orange asterix. All other peaks are unassigned impurities, for example 7.4–7.5 ppm and 9.9–10.1 ppm. | ||
A key factor affecting the synthesis outcome of macrocycle 1 proved to be the total reagent mass. This can be seen in experiments where either reaction concentration (Table 1, entry 1–3) or volume (Table 1, entry 4–5) was changed. At a concentration of 0.05 M (entry 1) fast conversion was observed with the NMR spectra at 5 minutes showing the formation of macrocycle 1 alongside a small amount of starting aldehyde (3.8%) and minor impurity peaks (ESI,† Fig. S8). These impurities are likely to be intermediate oligomeric species involved in the formation of the macrocycle.50 As the reaction progressed to 30 minutes, the amount of terephthalaldehyde increased alongside the formation of other aldehyde species, suggesting hydrolysis of macrocycle 1. At a concentration of 1.25 M (entry 3), the NMR spectrum at 5 minutes appeared similar to that of the other concentration reactions, with a slight reduction in the percentage of aldehyde starting material remaining compared to the reaction at 0.05 M. Yet, unlike the lowest concentration of 0.05 M, as the reaction proceeded to 30 minutes, the intermediate peaks disappeared as the reaction progressed (see ESI,† Fig. S9). The most optimal results, however, were observed at a concentration of 0.5 M. In this scenario, <1% of starting aldehyde and no intermediate peaks were present at 5 minutes, and this absence continued throughout the entire reaction till 30 minutes (ESI,† Fig. S3).
As a result, 0.5 M was determined to be the optimal concentration for the synthesis of macrocycle 1.
Altering the volume of the reaction while keeping the concentration fixed at 0.5 M had a similar impact on the reaction. In contrast to a total volume of 30 mL, both smaller (15 mL) and larger (45 mL) total reaction volumes exhibited minor amounts of intermediate and their associated aldehyde peaks in the NMR spectra, albeit less than observed when varying concentrations (Fig. 4b). Interestingly, the quantity of intermediates and aldehydes did not appear to fluctuate as the reaction progressed (ESI,† Fig. S10 & S11). Increasing or decreasing the power used to generate the plasma had a negative effect, with more aldehyde peaks observed at 5 and 10 minutes for both 1.4 and 4.5 W than for 3.2 W.
We therefore established that for synthesis of macrocycle 1 the optimal conditions were 30 mL of total volume, at 0.5 M and 3.2 W, giving 1.48 g of 1 (93%). This reaction was repeated in triplicate to give isolated yields of 94.1%, 93.4% and 94.5% after 20 minutes of plasma exposure.
Based on these results, we hypothesise that the hydrolysis seen at low concentrations is caused by the relatively high ratio of reactive plasma-generated species and solvent to the reagent present in the flask. This hypothesis is supported by the fact that similar hydrolysis was observed under increased power conditions for plasma generation (i.e., a higher proportion of plasma-generated species compared to reagent), with the amount of starting aldehyde present in the NMR increasing from 1 minute to 8 minutes (ESI,† Fig. S13). Under lower power, the presence of aldehyde and intermediate peaks observed by NMR did not change during the progress of the reaction (ESI,† Fig. S12).
Overall, the results above show that the presence of NTP accelerates the synthesis of macrocycle 1, giving high conversion within minutes. It is important, however, to establish that the noted reactivity is only observed for the reactions where NTP is in direct contact with the solution. An alternative set-up of the probe was used to investigate the effect of NTP when the plasma was generated above the treated reaction solution. The results closely resembled those obtained using the batch conditions with no plasma; impurity peaks could be seen, and the crude yield was 85% (ESI,† section 2.1 & 2.3).
Macrocyclic scope
To investigate the effects of NTP on a reversible system where more than one product is formed, the optimised conditions for 1 were employed to synthesise macrocycle 2, an isotrianglimine, for comparison. Comparable results were observed, with both the [2 + 2] and [3 + 3] macrocycles being formed within 1 minute (ESI,† Fig. S14).With the suitability of the standard conditions established, further investigation was conducted to determine whether varying the aldehyde substrate had any impact on (i) optimal reaction time, (ii) product purity, and (iii) yield. Macrocycles 3–8 were synthesised using NTP (reaction details in ESI,† section 4) and monitored by 1H NMR at 5, 10, and 20 minutes. At 20 minutes, the solid was isolated via solvent evaporation to obtain the crude yield (Table 2) and characterised by 1H NMR.
| Macrocycle | Literature examples | Non-thermal plasma | |||
|---|---|---|---|---|---|
| Batch conditions | Literature yield [%] | Best NTP time point [min] | SM aldehyde at 20 minutes [normalised %] | NTP crude yield at 20 minutes [%] | |
| NTP conditions: 20 kV, 50 mM, 20 mL, RT.a Yield after purification.b Purification required. | |||||
| 2 (ref. 22) | 3 h, RT | 90a | 20 | 0 | 90b |
| 3 (ref. 28) | 24 h, RT | 98a | 5 | 0 | 99 |
| 4 (ref. 29) | 3 h, RT | 90a | 5 | 4.5 | >99b |
| 5 (ref. 17 and 30) | 5 h, RT | 69a | 5 | 7.5 | 82b |
| 6 (ref. 20) | 3 h, reflux | 70a | 5 | 0 | 90b |
| 7 (ref. 21) | 3 h, reflux | 90 | 5 | 0 | 94b |
| 8 (ref. 22) | 12 h, 50 °C | 80a | 5 | 0 | 91b |
In all cases, the cleanest NMR spectra were obtained at 5 minutes (ESI,† S18–S29) and the majority product in each case was the desired macrocycle. Macrocycle 3 underwent full conversion and would not require any further purification; no aldehyde peaks were observed at any of the time points. Evidence of partial hydrolysis was observed for macrocycle 4 and 5 at 10 and 20 minutes, albeit in low quantities, suggesting that shorter reaction times would be beneficial for these species, potentially even shorter than the first measurement at 5 minutes.
For macrocycles 6, 7 and 8, minor aldehyde peaks were observed at all time points, in some cases attributed to partially hydrolysed species (ESI,† Fig. S24, S26 & S28 respectively). As discussed for 1, dilute reaction conditions with NTP can lead to hydrolysis as the reaction proceeds; thus, optimising either plasma power or reagent concentration/reaction volume would potentially improve these results.
Despite the presence of minor impurities that are readily removed by recrystallisation, in most cases the unoptimized yields achieved using NTP represent an improvement compared to literature yields. The NTP reactions took minutes rather than hours and, for the case of 6 and 8, did not require the reported elevated temperatures to enable the reaction.
For isotrianglimines, whilst the [3 + 3] product is most commonly reported, both [2 + 2] and [3 + 3] macrocycles are generally produced during batch synthesis, depending on the substituents present.18,27,31 NTP synthesis of the brominated isotrianglimine 5 also produced both the [2 + 2] and [3 + 3] macrocycles. However, it is interesting to note that the ratio of products is shifted heavily towards the [2 + 2] macrocycles, a phenomenon not seen in the standard batch synthesis of this compound (ESI,† Fig. S23).18 This observation opens the possibility that the distribution of macrocycle products could be shifted by tuning the NTP parameters, a hypothesis that is under investigation in our lab. Recrystallisation is required for 5, as the same oligomeric intermediates that can be seen in batch reactions are present, and it is here recrystallisation can be used to shift to [3 + 3]–5 if desired.18,51 Similarly for 6, 1H NMR spectroscopy and mass spectrometry confirmed the presence of the [3 + 3] macrocycle (ESI,† Fig. S25). However, as observed in previous traditional syntheses,31,52 the 1H NMR spectrum showed additional NMR peaks corresponding to what is likely a mixture of oligomeric product and other sized macrocycle peaks; mass spectrometry also shows the presence of the [4 + 4] macrocycle.
Overall, we have shown that NTP-driven synthesis of imine macrocycles is rapid and high-yielding for a range of substrates. The reactions for macrocycles 2–8 could be further optimised to improve yields, for example by tuning plasma power, reagent concentration, or exposure time. In the case of the isotrianglimines, further exploration of reaction parameters is needed to allow selective synthesis of specific macrocyclic products and target sizes that are less accessible with traditional thermal methods.
While the presented results suggest that plasma interactions with reaction solution can accelerate imine condensation, improve conversion, and, in the case of 5, alter the product distribution compared to batch reactions, the precise chemistry occurring between NTP and reaction solution is not yet fully understood. Findings from control experiments (ESI,† section 2) demonstrate that direct contact of NTP and reaction solution produces the greatest difference from the standard, non-plasma synthesis. We have previously used an IR temperature probe to monitor chloroform while exposed to NTP, and did not observe any fluctuations in temperature, thus we hypothesise that increased temperature does not play a major role in accelerating the imine condensation observed for NTP conditions.26 Imines are typically synthesised via acid catalysed condensation (Fig. S30†); although we were not able to definitively confirm at this stage, we hypothesise that plasma interactions with solvent may be generating acidic species that catalyse this reaction via activation of the aldehyde. Grande et al. observed that water washings of NTP-treated chloroform had reduced pH and higher Cl− concentrations compared to water washings of chloroform that had not been exposed to plasma,53 suggesting that acid was produced via NTP interactions with the solvent, alongside ˙CCl3 radicals and resultant chlorinated C2 species.53 A hypothesis of in situ acid generation was also put forward by He et al. to explain the formation of COFs via NTP.46 Further investigations, such as in situ Raman and/or GC-MS, would be helpful to elucidate the mechanism.
Conclusion
NTP in contact with liquid has the potential to carry out reactions under mild conditions. In this work, we show that the use of NTP allows for the rapid formation of imine macrocycles in high yield. This method was applied to nine macrocycles covering three classes of compounds: trianglimines, isotrianglimines and calixsalens. Although the mechanism of the reaction is not yet fully understood, the results obtained highlight the importance of direct NTP contact with the reaction solution vs. indirect NTP discharges. The observation of different selectivity for macrocycles capable of forming multiple sized species opens intriguing possibilities to use NTP techniques to tune production of kinetic species, and this is under investigation in our lab. Understanding NTP-solvent and NTP-reagent interactions using in situ characterisation techniques will be crucial to explore the underpinning mechanism. We anticipate that these methods will in future enable the scalable and efficient NTP-mediated formation of other molecular materials based on imine condensation, such as COFs, organic cages, and interlocked species.Author contributions
Conceptualisation and funding acquisition was carried out by AGS. Supervision carried out by AGS, TLE and JLW. Experiments and methodology were carried out by PR and AMS. All authors contributed to experimental design, analytical measurement, and interpretation of results. PR and AMS produced the first draft and all authors contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TLE thanks the University of Birmingham, and gratefully acknowledges the Royal Society for the award of a University Research Fellowship (6866 and URF\R\201028). JW would like to acknowledge EPSRC grant EP/S025790/1. AGS thanks the Royal Society and the Engineering and Physical Sciences Research Council (EPSRC) for a Royal Society-EPSRC Dorothy Hodgkin Fellowship (DH150156), the Royal Society for a University Research Fellowship (URF\R1\201168) and a Research Grant (RSG\R1\180357), AGS and AS thank the Royal Society for an Enhancement Award (RGF\EA\180194) that supported this work and a PhD studentship, and AGS and PR thank the University of Liverpool for a PhD studentship via the Doctoral Training Partnership allocation. This work made use of equipment from the Analytical Services/Department of Chemistry at the University of Liverpool as well as shared equipment at the Materials Innovation Factory (MIF) created as part of the UK Research Partnership Innovation Fund (Research England) and co-funded by the Sir Henry Royce Institute.References
- Z. Liu, S. K. M. Nalluri and J. Fraser Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC
.
- J. C. Collins and K. James, MedChemComm, 2012, 3, 1489–1495 RSC
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed
- V. Martí-Centelles, Tetrahedron Lett., 2022, 93, 153676 CrossRef
- C. D. Jones, L. J. Kershaw Cook, D. Marquez-Gamez, K. V. Luzyanin, J. W. Steed and A. G. Slater, J. Am. Chem. Soc., 2021, 143, 7553–7565 CrossRef CAS PubMed
- F. Esteve, B. Altava, S. V. Luis and E. García-Verdugo, Catal. Today, 2024, 426, 114407 CrossRef CAS
- D. He, C. Zhao, L. Chen, M. A. Little, S. Y. Chong, R. Clowes, K. McKie, M. G. Roper, G. M. Day, M. Liu and A. I. Cooper, Chem. – Eur. J., 2021, 27, 10589–10594 CrossRef CAS PubMed
- D. He, L. Zhang, T. Liu, R. Clowes, M. A. Little, M. Liu, M. Hirscher and A. I. Cooper, Angew. Chem., Int. Ed., 2022, 61, 8 Search PubMed
- A. Dey, S. Chand, M. Ghosh, M. Altamimy, B. Maity, P. M. Bhatt, I. A. Bhat, L. Cavallo, M. Eddaoudi and N. M. Khashab, Chem. Commun., 2021, 57, 9124–9127 RSC
- Y. Ding, A. Dey, L. O. Alimi, P. M. Bhatt, J. Du, C. Maaliki, M. Eddaoudi, J. Jacquemin and N. M. Khashab, Chem. Mater., 2022, 34, 197–202 CrossRef CAS
- Y. Ding, L. O. Alimi, J. Du, B. Hua, A. Dey, P. Yu and N. M. Khashab, Chem. Sci., 2022, 13, 3244–3248 RSC
- X. Zhu, G. Zhang, L. O. Alimi, B. M. Moosa, A.-H. Emwas, F. Fang and N. M. Khashab, Chem. Mater., 2023, 35(21), 9160–9166 CrossRef CAS
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed
- J. Lisowski, Molecules, 2022, 27, 4097 CrossRef CAS PubMed
- A. M. Scholes, L. J. Kershaw-Cook, F. T. Szczypiński, B. D. Egleston, R. L. Greenaway and A. G. Slater, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-xc820. This content is a pre-print and has not been peer-reviewed.
- C. M. Taylor and N. L. Kilah, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 543–555 CrossRef CAS
- K. Ziach and J. Jurczak, Chem. Commun., 2015, 51, 4306–4309 RSC
- K. Ziach and J. Jurczak, Cryst. Growth Des., 2015, 15, 4372–4376 CrossRef CAS
- A. M. López-Periago, C. A. García-González and C. Domingo, Chem. Commun., 2010, 46, 4315–4317 RSC
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46–79 CrossRef CAS PubMed
- S. Srimurugan, B. Viswanathan, T. K. Varadarajan and B. Varghese, Tetrahedron Lett., 2005, 46, 3151–3155 CrossRef CAS
- S. Srimurugan, P. Suresh and H. N. Pati, J. Inclusion Phenom. Macrocyclic Chem., 2007, 59, 383–388 CrossRef CAS
- P. Roszkowska, A. Dickenson, J. E. Higham, T. L. Easun, J. L. Walsh and A. G. Slater, Lab Chip, 2023, 23, 2720–2728 RSC
- J. Gawroński, H. Kołbon, M. Kwit and A. Katrusiak, J. Org. Chem., 2000, 65, 5768–5773 CrossRef PubMed
- K. Tanaka, R. Shimoura and M. R. Caira, Tetrahedron Lett., 2010, 51, 449–452 CrossRef CAS
- N. Kuhnert, C. Straßnig and A. M. Lopez-Periago, Tetrahedron: Asymmetry, 2002, 13, 123–128 CrossRef CAS
- A. Troć, J. Gajewy, W. Danikiewicz and M. Kwit, Chem. – Eur. J., 2016, 22, 13258–13264 CrossRef PubMed
- N. Kuhnert, G. M. Rossignolo and A. Lopez-Periago, Org. Biomol. Chem., 2003, 1, 1157–1170 RSC
- S. R. Korupoju, N. Mangayarkarasi, S. Ameerunisha, E. J. Valente and P. S. Zacharias, J. Chem. Soc., Dalton Trans., 2000, 2845–2852 RSC
- A. Sarnicka, P. Starynowicz and J. Lisowski, Chem. Commun., 2012, 48, 2237–2239 RSC
- A. Fridman, A. Chirokov and A. Gutsol, J. Phys. D: Appl. Phys., 2005, 38, R1–R24 CrossRef CAS
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella, F. Azzolina-Jury, H.-H. Kim, A. B. Murphy, W. F. Schneider, T. Nozaki, J. C. Hicks, A. Rousseau, F. Thevenet, A. Khacef and M. Carreon, J. Phys. D: Appl. Phys., 2020, 53, 443001 CrossRef CAS
- A. George, B. Shen, M. Craven, Y. Wang, D. Kang, C. Wu and X. Tu, Renewable Sustainable Energy Rev., 2021, 135, 109702 CrossRef CAS
- J. Yang, H. Ding, Z. Zhu, Q. Wang, J. Wang, J. Xu and X. Chang, Appl. Surf. Sci., 2018, 454, 173–180 CrossRef CAS
- F. Rezaei, P. Vanraes, A. Nikiforov, R. Morent and N. De Geyter, Materials, 2019, 12, 2751 CrossRef CAS PubMed
- Y. Gorbanev, D. Leifert, A. Studer, D. O'Connell and V. Chechik, Chem. Commun., 2017, 53, 3685–3688 RSC
- J. Wang, N. B. Üner, S. E. Dubowsky, M. P. Confer, R. Bhargava, Y. Sun, Y. Zhou, R. M. Sankaran and J. S. Moore, J. Am. Chem. Soc., 2023, 145, 10470–10474 CrossRef CAS PubMed
- V. Dupont, S. Ognier, G. Morand, C. Ollivier, L. Fensterbank and M. Tatoulian, Chem. – Eur. J., 2023, 29, 1–8 CrossRef PubMed
- E. Abedelnour, S. Ognier, M. Zhang, L. Schio, O. Venier, J. Cossy and M. Tatoulian, Chem. Commun., 2022, 58, 7281–7284 RSC
- F. Gorky, A. Nambo and M. L. Carreon, J. CO2 Util., 2021, 51, 101642 CrossRef CAS
- X. Jiang, Z. Lin, X. Zeng, J. He, F. Xu, P. Deng, J. Jia, X. Jiang, X. Hou and Z. Long, Chem. Commun., 2019, 55, 12192–12195 RSC
- Q. Wei, S. Xue, W. Wu, S. Liu, S. Li, C. Zhang and S. Jiang, Chem. Rec., 2023, 23, e202200263 CrossRef CAS PubMed
- J. He, X. Jiang, F. Xu, C. Li, Z. Long, H. Chen and X. Hou, Angew. Chem., Int. Ed., 2021, 60, 9984–9989 CrossRef CAS PubMed
- V. V. Kovačević, G. B. Sretenović, B. M. Obradović and M. M. Kuraica, J. Phys. D: Appl. Phys., 2022, 55, 473002 CrossRef
- F. Rezaei, Y. Gorbanev, M. Chys, A. Nikiforov, S. W. H. Van Hulle, P. Cos, A. Bogaerts and N. De Geyter, Plasma Processes Polym., 2018, 15, 1700226 CrossRef
- K. A. Aadim, S. N. Mazhir, N. K. Abdalameer and A. H. Ali, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 987, 012020 CAS
- H. F. Nour, A. M. Lopez-Periago and N. Kuhnert, Rapid Commun. Mass Spectrom., 2012, 26, 1070–1080 CrossRef CAS PubMed
- A. Janiak, J. Gajewy, J. Szymkowiak, B. Gierczyk and M. Kwit, J. Org. Chem., 2022, 87, 2356–2366 CrossRef CAS PubMed
- A. González-Álvarez, I. Alfonso, F. López-Ortiz, Á. Aguirre, S. García-Granda and V. Gotor, Eur. J. Org. Chem., 2004, 5, 1117–1127 CrossRef
- S. Grande, F. Tampieri, A. Nikiforov, A. Giardina, A. Barbon, P. Cools, R. Morent, C. Paradisi, E. Marotta and N. De Geyter, Front. Chem., 2019, 7, 1–11 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00061g |
| ‡ Roszkowska and Scholes contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |