
A multiscale investigation of polypropylene glycol polymer upcycling to propionaldehyde via catalytic cracking on acid sites of mesoporous Y zeolites†
Abraham
Martinez‡
a,
Kanan
Shikhaliyev‡
 b,
Xuemin
Li
a,
Jinyi
Han
c,
Kaustav
Chaudhuri
c,
Son-Jong
Hwang
d,
Jagoda M.
Urban-Klaehn
e,
Alexander
Kuperman
c,
Anne
Gaffney
e,
Jochen
Lauterbach
*b and
Alexander
Katz
*a
b,
Xuemin
Li
a,
Jinyi
Han
c,
Kaustav
Chaudhuri
c,
Son-Jong
Hwang
d,
Jagoda M.
Urban-Klaehn
e,
Alexander
Kuperman
c,
Anne
Gaffney
e,
Jochen
Lauterbach
*b and
Alexander
Katz
*a
aDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, Berkeley, CA, USA. E-mail: askatz@berkeley.edu
bDepartment of Chemical Engineering, University of South Carolina, Columbia, SC, USA. E-mail: lauteraj@cec.sc.edu
cChevron Technology Center, Richmond, CA, USA
dDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, USA
eEnergy Environment S&T Directorate, Idaho National Lab, Idaho Falls, USA
First published on 1st July 2024
Abstract
We investigate acid-catalyzed upcycling of PPG polymer, emphasizing crucial features on multiple length scales that span reaction engineering on macroscopic length scales down to zeolite catalyst design on the nanoscale. We modified a previously described semi-batch reactor configuration to minimize coking and enhance recovered selectivities by incorporating rapid quenching of reaction products (instead of slower quenching with a condenser, which facilitates sequential coupling reactions), and decreased the initial carrier-gas residence time in the bed consisting of mixed catalyst and PPG polymer, further reducing the deposition of solid residues in the used catalyst. Our results highlight the importance of tight interfacial contact between the catalyst surface and the initial PPG polymer reactant, which is achieved via a pretreatment that removes adsorbed water, for drastically increasing the propionaldehyde selectivity, particularly for the large surface-area mesoporous catalysts. Our best catalyst consisted of mesoporous Y zeolite synthesized at an alkalinity of 0.16 M and exhibited nearly the same high propionaldehyde selectivity of approximately 95% (86% propionaldehyde yield) for a PPG polymer with molecular weights of 425 and 2000 Daltons (Da), suggesting the absence of mass transport restrictions. We also deconvolute the catalyst attribute between extra-framework aluminum (AlEF) content and mesopore external surface area that most sensitively controlled propionaldehyde selectivity. This was performed by synthetically incorporating AlEF content into our optimum catalyst, at a high and low alumina dispersion. The high dispersion alumina catalyst consisted of a uniform 10 nm-thick alumina layer covering the interior pores of the mesoporous Y catalyst, whereas the low dispersion alumina catalyst had a completely phase-separated alumina phase, commensurate in size to the zeolite particles. Our results demonstrate that AlEF content in the catalyst decreases propionaldehyde yield by increasing the amount of solid residues in the catalyst post reaction, and had a minor effect on the propionaldehyde selectivity. These results point to a Brønsted rather than Lewis acid-catalyzed mechanism of catalysis for PPG polymer upcycling to propionaldehyde. In summary, our study demonstrates the most sensitive controlling attribute of the zeolite catalyst for selective propionaldehyde synthesis is its mesoporosity (as reflected in the mesopore volume and surface area) and that the multiscale details of the catalyst and reactor design also have profound consequences in achieving high propionaldehyde selectivity and yield.
Introduction
As plastics production increases globally (e.g., doubling between 2000 and 2019), new approaches for repurposing plastic waste are required.1 Currently, only about 9% of the total plastic waste generated is recycled.1 Mechanical recycling methods are the most common form of plastic recycling;2,3 however, these approaches are limited by the inevitable degradation of polymer performance properties due to oxidation.2–4 These difficulties contribute to the unfortunate reality that most plastic waste ends up either landfilled (46%) or incinerated (17%), with deleterious environmental consequences.1 This situation underscores the high priority for developing alternatives for plastic-waste upcycling consisting of value-added methodologies, which can pave the way for significantly prolonged product lifecycles.Chemical approaches for upcycling polymers aim to synthesize value-added chemical products and thereby improve the circulatory lifecycle of polymers. Historically, these approaches have involved pyrolysis,2 but a key challenge has been the synthesis of a diverse set of products (including those that lead to fouling), which cannot be separated in an economically feasible fashion.5–8 More selective chemical upcycling of polymers has been achieved with catalytic cracking and hydrocracking, and this has enabled the synthesis of more functional, value-added classes of products – not as broad as through pyrolysis.9–13 Yet while significant progress has been made in the chemical upcycling of polymers via hydrocracking leading to lubricants, surfactants, refrigerants, and other high-quality liquid products, one of the remaining challenges is the difficulty of increasing selectivity further, towards one desired chemical compound.10,12,14–16 Notable exceptions include the highly selective (>80% yield) synthesis of propane-2,2-diyldicyclohexane, a jet fuel range C-15 dicycloalkane, from polycarbonate – an oxygenated polymer – via hydrocracking using bifunctional supported metal–acid sites.17–19
An additional challenge in chemical upcycling of polymers involving bifunctional sites has been its heavy reliance on precious metals.12,16,20–25 One approach obviating the use of precious metals has been to increase the selectivity of acid-catalyzed cracking processes with the shape-selective microporous environments of zeolites – crystalline aluminosilicates.26–30 A particularly notable success in this regard involves the chemical upcycling of polypropylene glycol (PPG) – a high-volume (9.4 M metric tons per year scale that represents the second largest use of propylene, after polypropylene) oxygenated polymer.31,32 Urgent approaches are necessary for upcycling this particular polymer and related polyurethane foams derived from it due to their low density, causing them to occupy a large volume. This chemical upcycling process involves the acid-catalyzed cracking of PPG polymer to propionaldehyde in 81% yield within a fluidized bed reactor with (H)ZSM-5 as catalyst.32 The remarkable feature of this process is its high selectivity to a single product, which shares in common the example above involving the hydrocracking of polycarbonate (another oxygenated polymer) to a single chemical compound.17–19 The propionaldehyde produced as product by the depolymerization of PPG polymer can be subsequently used for the synthesis of methyl methacrylate, a key building block for another commodity oxygen-containing polymer.31
Guided by an interest in using thermally and oxidatively stable zeolite catalysts (which are widely used in the petrochemical industry for fluid catalytic cracking, hydroisomerization, and alkylation) for the cracking of oxygenated polymers, we recently designed and demonstrated a semi-batch reactor (see Fig. 1 of ref. 33 and S1a, ESI†) for the acid-catalyzed chemical upcycling of PPG polymer.33 Our previous results demonstrate the benefit of acid sites in (H)ZSM-5 zeolite catalysts, which selectively form propionaldehyde with a selectivity of nearly 80%, reproducing similar results in the fluidized bed system.32 These results emphasize the benefit of the catalyst enhancing selectivity to propionaldehyde versus a multitude of different compounds as products when no zeolite catalyst was added in the control. The results also demonstrated that both the external (mesopore) surface area and aluminum content (i.e., the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratio) of the (H)ZSM-5 catalysts affected propionaldehyde selectivity.33 The positive benefit of the mesopore surface area has been previously noted in other chemical upcycling of polymers, including in the catalytic cracking of polyethylene.34–39 However, in our previous study, it remained unclear whether mesopore surface area or the nature of the aluminum sites within the zeolite (framework Brønsted-acid sites versus extra-framework Lewis-acid sites) were responsible for propionaldehyde synthesis. In other cracking-type catalysis, the effect of extra-framework aluminum (AlEF) content has been shown to be profound.40,41
:Al ratio) of the (H)ZSM-5 catalysts affected propionaldehyde selectivity.33 The positive benefit of the mesopore surface area has been previously noted in other chemical upcycling of polymers, including in the catalytic cracking of polyethylene.34–39 However, in our previous study, it remained unclear whether mesopore surface area or the nature of the aluminum sites within the zeolite (framework Brønsted-acid sites versus extra-framework Lewis-acid sites) were responsible for propionaldehyde synthesis. In other cracking-type catalysis, the effect of extra-framework aluminum (AlEF) content has been shown to be profound.40,41
Here, in this study, we initially leverage these previous efforts with the same semi-batch reactor configuration as before, to understand the zeolite properties that control propionaldehyde selectivity. To investigate the effect of mesopore surface area, we chose to study the mesoporous Y (MY) family of zeolites, which are synthesized by a previously reported alkali treatment of parent USY zeolite (H)CBV720 in the presence of long-chain organic quaternary ammonium ion surfactants.42–44 We varied the alkalinity of the MY synthesis in order to control the mesopore surface area, without changing the pore size as shown in Scheme 1a and b.44,45
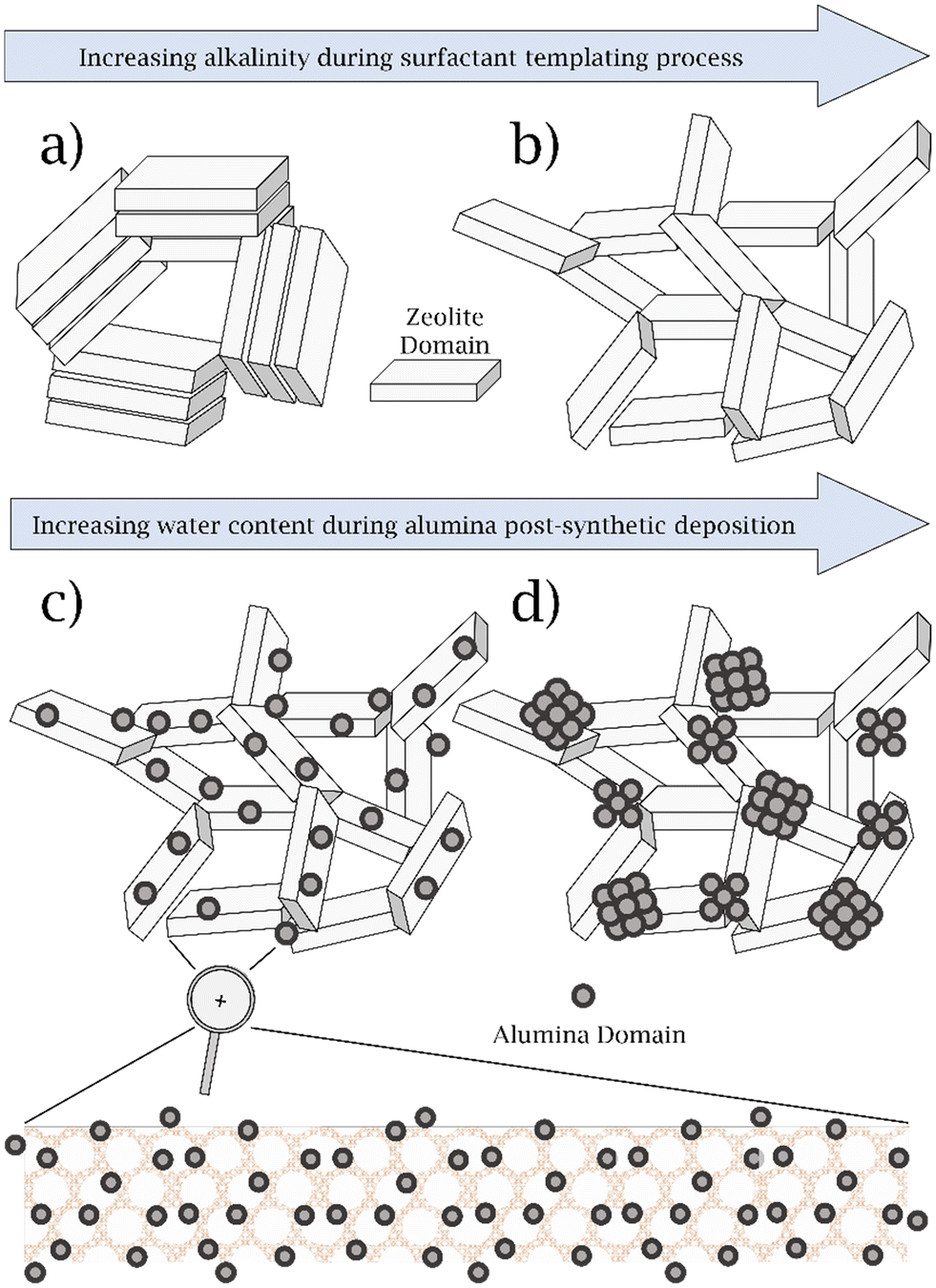 | ||
| Scheme 1 Schematic representation of the MY catalysts synthesized. a) MY-0.09 M, b) MY-0.16 M, c) MY-0.16 M-DA, d) MY-0.16 M-WA. | ||
In addition, we investigated the effect of post-synthetic modification of our optimum MY catalyst with alumina, to deconvolute the effect of AlEF from mesopore surface area, on the propionaldehyde selectivity of the PPG polymer cracking reaction. This was performed by varying the dispersion of the incorporated alumina by leveraging previously described synthetic approaches.46 Relying on a rigorously dry alumina-incorporation approach, we synthesize a highly dispersed alumina phase, which has a high degree of intimacy with internal acid sites within the pores of the zeolite (i.e. not just on the external surface), as shown in Scheme 1c. In contrast, wet alumina incorporation conditions led to a catalyst with a phase-separated alumina phase, as shown in Scheme 1d. We also explore modifications of the reaction conditions, catalyst pretreatment procedures (which play an unexpectedly crucial role), and reactor configurations to understand the controlling variables that affect propionaldehyde selectivity across these multiple length and time scales.
Experimental methods
Materials
PPG polymer having a number-average molecular weight (MW) of 425 and 2000 Daltons (Da) was purchased from Sigma-Aldrich and was used as a reactant. Commercial USY (H)CBV720 zeolite (Si/Al = 15) was supplied by Zeolyst and used as received unless otherwise specified.Synthesis of mesoporous Y (MY) zeolite
The synthesis of MY zeolites was performed according to literature precedent.42,44 Thus, a 0.XX M NaOH solution was prepared in a plastic Nalgene HDPE container. 120 mL of this NaOH solution and 3 grams of cetyltrimethylammonium ammonium bromide (CTAB) surfactant were added to a 250 mL plastic (HDPE) round-bottom flask. The solution in the flask was preheated in an oil bath at 90 °C for 25 minutes, after which 6 grams of parent material (H)CBV720 were added to the preheated solution. The resulting dispersion was allowed to stir for 6 hours at 90 °C. Afterwards, the suspended solids were washed and filtered with deionized water (using about 2.5 L of deionized water) until the filtrate was pH neutral and was subsequently dried in an oven at 60 °C for a period of 12 h. This as-synthesized MY (MY-0.XX M-As) zeolite was calcined at 580 °C for 5 hours in flowing air (MY-0.XX M). MY zeolites synthesized using alkali concentrations of 0.09 M and 0.16 M were compared in this study and were ammonium ion exchanged and calcined to synthesize the H-form zeolite (see below).Synthesis of a low dispersion alumina phase on mesoporous Y zeolite
Synthesis of phase-separated alumina clusters/aggregates on MY catalysts using aluminum isopropoxide (Scheme 1d) was performed according to literature precedent.46 Thus, the required amount of alumina source was dispersed in 40 mL of anhydrous hexane using air-free techniques, and was stirred until the majority of the aluminum source was dissolved. For this study, a loading of 4.6 wt% alumina content (by weight relative to MY-0.16 M-As) was used. Thus, 5 grams of MY-0.16 M-As powder, intentionally with no pretreatment, was added to the mixture, and the resulting slurry was stirred under reflux for 1 hour. Subsequently, the solvent was removed by evacuation. Upon complete solvent removal, the solid products were dried at 120 °C for 2 h under vacuum and were calcined at 580 °C for 5 hours in air. The final calcined material was denoted as MY-0.16 M-WA. In contrast to the literature, emphasis here is taken to assure that the synthesis of this low-dispersed, aggregated alumina phase is due to the water content of the untreated MY-0.16 M-As. We emphasize that the solvents and methodologies described here were performed using air-free techniques, in contrast to the procedure in the literature.46Synthesis of a high dispersion alumina nanoscale layer in the pores of mesoporous Y zeolite
Synthesis of a highly dispersed alumina nanoscale layer in the pores of MY catalyst using aluminum isopropoxide (Scheme 1c) was performed according to literature precedent.46 Thus, 5 grams of MY-0.16 M-As was pretreated in a vacuum furnace at 250 °C for 12 hours to remove water (and some CTA+ species). The resulting material was collected in a glovebox in a separate flask. The required amount of aluminum source (4.6 wt%) was dispersed in 40 mL of anhydrous THF using air-free techniques and was allowed to stir at 60 °C until the majority of the aluminum source was dissolved while the zeolite material was suspended in 15 mL of THF. Using air-free hot filtration, the mixture was filtered to remove a minor amount of undissolved aluminum-containing impurities and subsequently introduced into the slurry solution with the zeolite material. The slurry mixture was stirred under reflux conditions for 1 hour. Subsequently, the solvent was removed by evacuation. Upon complete solvent removal, the solid products were dried at 120 °C for 2 h under vacuum and calcined at 580 °C for 5 h in air. The final calcined material was denoted as MY-0.16 M-DA.Ammonium ion exchange
All calcined catalysts were converted to an ammonium form via ion exchange prior to catalysis runs. Aqueous ion exchange was performed with a 1.0 M NH4NO3 aqueous solution in a 50:1 mass ratio of solution to zeolite in a round bottom flask. This slurry mixture was heated in an oil bath at 60 °C for 24 hours under stirring. The slurry was washed and filtered with deionized water until the filtrate was pH neutral, and it was dried in an oven at 60 °C for 12 hours. This ion-exchange was repeated two additional times (i.e., three total ion exchanges were performed). The material, after ion exchange, was calcined at 550 °C for 5 hours in air. The resulting catalysts upon ion exchange were affixed with an ‘(H)’ at the beginning of the material name (i.e. (H)MY-0.16 M-DA) to emphasize that they were in the proton form.
PPG polymer upcycling to propionaldehyde via catalytic cracking
Our initial experimental reactor configuration is described in detail in a previous publication and was used as a departure point for this study (see Fig. S1a, ESI†).33 Select catalysts were activated by a drying at 200 °C in a convection oven for 1 h, which served to remove excess adsorbed water. The catalysts were premixed with PPG polymer at a mass ratio of unity, which corresponds to the maximum amount of PPG polymer that results in a solid mixed catalyst–polymer bed (attempts to increase the relative amount of PPG polymer to catalyst resulted in an undesirable flowable slurry in the bed of our semi-batch reactor; see Fig. 1b in ref. 33 and S1b, ESI†). In principle, this limitation on the maximum amount of PPG polymer could be circumvented with a continuous flow reactor. Such a continuous flow reactor (e.g., fluidized bed) could also overcome the drastic changing nature of the ratio of catalyst to PPG polymer during the course of the cracking reaction (i.e., currently this ratio increases to infinity during the course of reaction, from its initial value of unity). To monitor the temperature of the reaction, a thermocouple was placed in contact with the catalyst bed into the reactor, adjusted from the top of the reactor and positioned about 200 mm above the bottom of the bed of the reactor with temperature fluctuations not exceeding ±5 °C. The reaction was carried out under different nitrogen-carrier flowrates corresponding to residence times (referenced to the initial bed volume) of 1.5–4.5 s.Two different reactor configurations were evaluated in this study, as explained below. One of these was our original reactor, in which liquid products were collected from a condenser (externally cooled with a water–ethylene glycol mixture at 0 °C).33 These were subsequently analyzed by gas-chromatography mass-spectrometry (2010 Shimadzu GC-MS equipped with an RTX-1701 column), as explained in detail below. Liquid samples were dissolved in methanol, and 500 nL of each sample was injected into the GC-MS column with a split ratio of 30:1. The column temperature was maintained at 40 °C for 3 min. Then, it was increased to 80 °C (2.5 min hold) and to 200 °C (5 min hold), all at a constant ramp rate of 20 °C min−1. A solvent cut time from 0 to 1.35 min was implemented to avoid MS detector saturation by methanol and water, and MS analysis was conducted from 14 to 380 m/z for all analyses in this manuscript. The GC-MS detected more than twenty different species in a typical reaction mixture analysis, and the main products, including propionaldehyde, were calibrated using external calibration standards. The selectivity of a product was calculated based on the total mass of recovered products in the liquid mixture (excluding the minor amount of gas-phase products and remaining residues on the catalyst). The yield of a product was calculated based on the total mass of recovered products in the liquid mixture plus the remaining residues on the catalyst. The total mass balance closure was greater than 97% within an uncertainty of 1% and no more than 3%. For all catalysis experiments, the total liquid product yields ranged between values of 80–93%, in which the remaining yields were determined to be solid residuals on the catalysts surface, as detailed in Table S2, ESI.† In a modified reactor configuration implemented in this manuscript, instead of the condenser, the products were directly quenched in a −30 °C methanol bath. The solution was analyzed as described above, by GC-MS, and gas products were also analyzed by the same method as above.
N2 physisorption at 77 K
N2 adsorption isotherms at 77 K were measured on a Micromeritics ASAP2020 adsorption instrument with the mesopore (external) surface area and micropore volumes determined by the t-plot method and the mesopore volume determined by the Barrett–Joyner–Halenda (BJH) method. Prior to sample analysis, samples were degassed at 350 °C for 4 hours under vacuum to remove residual water species prior to measurement.Powder X-ray diffraction (PXRD)
PXRD patterns were collected on a Rigaku MiniFlex diffractometer using a Cu Kα radiation (40 kV, 15 mA) ranging from 3 to 60° with a step size of 0.01° with a scan rate of 5° min−1.SEM imaging
Images were recorded on a Zeiss Crossbeam 550 using a secondary electrons secondary ions (SESI) detector. Probe voltage was set at 1 kV, with the current was set to 100 pA, and a working distance of 5 mm was employed. SESI and InLens signal mixing option was used at 82% mixed signal.27Al MAS NMR spectroscopy
Solid-state 27Al MAS NMR spectra of zeolite catalysts were recorded on a Bruker DSX500 spectrometer and a Bruker 4 mm MAS probe. Free induction decay signal was collected after a short RF pulse (0.5 μs-π/18 flip angle) and strong 1H decoupling pulses in order to characterize isolated tetrahedral framework aluminum and AlEF species within the zeolites. The sample spinning rate was 13 kHz at ambient conditions. Chemical shifts were calibrated externally using 1 M Al(NO3)3 aqueous solution at 0 ppm for the 27Al nucleus.ICP-OES
ICP measurements were performed at Galbraith Laboratories to determine Si, Al and Na contents on proton form catalysts.SEM (Scanning Electron Microscopy)–EDX (Energy-Dispersive X-ray Spectroscopy) characterization
Data were acquired with a JEOL JSM 6700F fitted with an Oxford Instruments UltimMax 100 mm detector. Approximately 0.25 mg of the sample was transferred on to conductive carbon tape on an aluminum stub. To further enhance the conductivity, the sample was then coated with a ∼30 nm layer of tungsten. More than 50 spot EDX analyses were collected from different particles of each type of morphology and each analysis had an acquisition time of 60 seconds. A total net counts of ∼500000 was acquired for each spectrum.
TEM (Transmission Electron Microscopy)/STEM (Scanning Transmission Electron Microscopy)/Energy Dispersive X-ray Spectroscopy (EDX) characterization
High-resolution transmission electron microscopy (HRTEM) images were captured using a JEOL JEM 2010 microscope in low-dose mode operating at 200 kV accelerating voltage and equipped with a LaB6 electron gun. Samples were prepared by embedding and curing in an epoxy resin followed by cutting thin sections (∼30–50 nm) with a Leica EM UC7 ultramicrotome. The sections were floated onto 300 mesh Cu grids with a thin (20–30 nm) lacey carbon support film. Images were captured using a Gatan OneView 4k × 4k camera, ∼100 pA probe current, using a 2 μs per pixel dwell time. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were collected on a Talos F200X instrument operating at 200 kV accelerating voltage with a field emission gun. EDX spectroscopy and elemental mapping was performed on the Talos instrument equipped with a SuperX Energy Dispersive Spectrometry (SuperX EDS) system with four Silicon Drift Detectors (SDD) with a solid angle of 0.9 srad. Images were captured using a 4k × 4k Ceta 16M CMOS camera.Positron Annihilation Lifetime Spectroscopy (PALS)
The experimental approach was identical to that previously described.47 Briefly, PALS measurements of the specimens were carried out with an APV8702RU spectrometer, TechnoAP spectrometer with a timing resolution of about 170 ps. The PALS instrument uses two fast scintillator detectors with BAF2 cylindrical with built-in photomultiplier tubes H3378-51 manufactured by Hamamatsu Photonics. The Na-22 radioactive source, with a half-life of 2.6 years, serves as a positron generator with examined samples in the sandwich arrangement. The Na-22 source emits positron in coincidence with high energy γ-rays with the energy of 1.27 MeV. The high energy gamma signal is employed as a “start” for opening the timing gate, while the positron annihilation event is marked by detecting one of 511 keV gamma peaks from the annihilation of positrons, and the electron records the “stop”. The sealed source was produced by Eckert & Ziegler Isotope Products, with an activity of 50 mCi (1.85 MBq) on Dec. 4, 2019. It is a disk with an active diameter of 9.83 mm (9.275′′) enclosed in thin titanium layers of 0.0005′′ diameter.Thermogravimetric Analysis (TGA)
Quantities of adsorbed water and reaction residue (coke) on used catalysts were measured with a Shimadzu TGA-50H equipped with a TA-60WS thermal analyzer, a GC-60A flow control unit, and a BLW-50 cooling blower. About 10–15 mg of zeolite sample was used. TGA was conducted from room temperature to 600 °C at a fixed heating rate of 10 °C min−1 with a hold of 10 min at 600 °C in air, with a fixed flow rate of 200 mL min−1 to investigate the residue amounts. To investigate water adsorption, the same process was repeated up to 220 °C, with the same ramping and hold times.Results and discussion
Zeolite catalyst characterization
Data from N2 physisorption at 77 K for all catalysts are shown in Table 1 (see also Fig. S2, ESI† for N2 physisorption isotherms and pore-size distribution). These data demonstrate that commercial USY zeolite (H)CBV720 has the least amount of mesopore volume. This industrial zeolite is synthesized from microporous Y zeolite by a steaming process known to cause significant dealumination.48 This results in a material with only a small, minor population of large mesopores (>10 nm in diameter) and establishes our baseline material consisting of the expected least accessibility to catalytic acid sites for large polyol reactants, of the catalysts used here in this work.| Zeolite | Meso SA (m2 g−1) | Micro PV (cm3 g−1) | Meso PV (cm3 g−1) | AlEF/Altot | Si/Al | Na/Al |
|---|---|---|---|---|---|---|
| (H)CBV720 | 233 | 0.26 | 0.24 | 0.34 | 14.5 | 0.01 |
| MY-0.09 M | 381 | 0.21 | 0.35 | 0.47 | 14.5 | 0.01 |
| MY-0.16 M | 626 | 0.15 | 0.63 | 0.50 | 12.9 | 0.01 |
| MY-0.16 M-WA | 533 | 0.14 | 0.53 | 0.61 | 5.2 | 0.01 |
| MY-0.16 M-DA | 491 | 0.15 | 0.49 | 0.60 | 6.1 | 0.01 |
Data in Table 1 demonstrate that MY catalysts corresponding to MY-0.09 M and MY-0.16 M (where the molarity denotes the NaOH concentration used during the surfactant templating process) exhibit increases in mesoporosity relative to the parent material (H)CBV720. MY-0.09 M has a 1.6-fold increase in mesoporous surface area (and 1.5-fold increase in mesopore volume), and a 0.81-fold decrease in micropore volume, relative to (H)CBV720. In accord with previous studies,44,45 we observe the same 3.5 nm mesopore size at a higher alkalinity of the surfactant templating process in MY-0.16 M as observed for MY-0.09 M. These 3.5 nm mesopores are completely absent in the pore-size distribution for parent material (H)CBV720, and the minor amount of large mesopores in (H)CBV720 (>10 nm described above) disappears in the MY variants (see Fig. S2 (right), ESI†). Data in Table 1 show that MY-0.16 M has a 2.7-fold higher mesopore surface area and 2.6-fold higher mesopore volume (as well as a further 0.58-fold reduced micropore volume) relative to MY-0.09 M, as a result of the increased alkalinity during the surfactant templating process.
We also performed Positron Annihilation Lifetime Spectroscopy (PALS) to corroborate the characterization of the catalysts above, because PALS (as explained below) is particularly well suited to probe micropore length scales. The PALS data were used to investigate (H)CBV720, (H)MY-0.09 M, and (H)MY-0.16 M in the fresh state, before catalysis, and have been discussed in detail elsewhere.47 Briefly, PALS data reflect the positron lifetime in the material before it becomes annihilated. Positron lifetimes depend on positrons being trapped in either a low electron density vacancies or voids or as formed positronium (a hydrogen-like positron–electron state). The lifetime of the positronium depends on the size of the cavity it is trapped in, but the relationship is linear only for radii up to 1 nm; therefore, it is more useful for smaller cavities up to 1 nm in diameter compared with larger ones. Therefore, PALS spectroscopy is especially sensitive to atomic and nanometer scale traps in the range of micropore length scales.
For the three fresh catalysts mentioned above, PALS spectra were deconvoluted into 5 lifetimes, and these are shown in Table 2. The two longest lifetimes were interpreted as a positronium annihilation lifetime within micropores (represented by T4 and I4), whereas mesopores are represented by T5 and I5. The corresponding products (T4 × I4 and T5 × I5) are also shown in Table 2, as they amplify the effects of the total micropore and mesopore fractional volumes. Specifically, the product of the lifetime and intensity reflects the size of the pore multiplied by its frequency and can be interpreted as a fractional volume. The lifetime T4 in the range of 14–17 ns corresponds to a pore radius of 0.70–0.78 nm (assuming a spherical pore), while the lifetime in the range of 58–65 ns corresponds to pore sizes that are well above 1 nm and include mesopores. PALS data in Table 2 demonstrate that mesoporosity increases and microporosity decreases with increasing alkalinity during the surfactant templating process on parent material (H)CBV720. These PALS results corroborate the N2 physisorption at 77 K described above, and are consistent with previous literature reports characterizing similar MY catalysts.42,44,45
| Zeolite | T 4 (ns) | I 4 (%) | T 4 × I4 | T 5 (ns) | I 5 (%) | T 5 × I5 |
|---|---|---|---|---|---|---|
| (H)CBV720 | 15.2 | 4.7 | 72.0 | 58.6 | 9.6 | 560.9 |
| (H)MY-0.09 M | 17.2 | 2.8 | 48.1 | 64.3 | 11.5 | 737.4 |
| (H)MY-0.16 M | 14.4 | 1.7 | 23.7 | 64.7 | 12.4 | 803.5 |
Data in Table 1 demonstrate that our MY catalysts retain the majority of micropore volume present in parent (H)CBV720 zeolite, and we infer that a large portion of the crystallinity in the latter is still present in the former catalysts. PXRD data demonstrate retention of all peaks corresponding to Y zeolite and support our hypothesis of retention of crystallinity in all catalysts, although, as observed in the literature, we do observe a decrease in the peak intensity with increasing alkalinity of the MY surfactant templating process (see Fig. S3, ESI†).43,44 These results are further corroborated by SEM images of the parent (H)CBV720 and MY-0.16 M catalysts, which demonstrate no obvious surface morphological changes within the material (see Fig. S4, ESI†). Data in Table 1 summarize elemental analyses measured by ICP-OES. Results show that while (H)MY-0.09 M shows virtually no change in composition relative to parent (H)CBV720, (H)MY-0.16 M synthesized at higher alkalinity shows a slightly reduced Si/Al of 12.9 (versus 14.5 in (H)CBV720 and (H)MY-0.09 M).
We infer that syntheses at higher alkalinity result in slightly desilicated mesoporous catalysts, resulting in more aluminum-rich catalysts. We quantitatively characterized the amount AlEF in all catalysts via27Al MAS NMR spectroscopy. These data are shown in Fig. 1 and demonstrate the presence of tetrahedral (AlO4−) framework aluminum (AlF) resonance at 60 ppm, as well as distorted (defective) penta-coordinated AlO5 species located at ∼28 ppm and octahedral extra-framework AlF AlO6 located at 0 ppm for all catalysts. In general, extra-framework resonances are attributed to an alumina phase (i.e., Al–O–Al bonds that are not present in zeolites by the Lowenstein rule), which can vary in size from small clusters that are less than a nanometer in diameter up to macroscopic alumina particles, whereas framework aluminum represents aluminum atoms within a tetrahedral oxygen coordination environment surrounded by silicon next nearest neighbors of the zeolite.49,50 Data in Table 1 demonstrate that (H)CBV720 has the lowest fraction of AlEF species (AlEF/Altot) of 0.34, and that this fraction further increases as the alkalinity of the MY synthesis increases, to values of 0.47 and 0.50 for (H)MY-0.09 and (H)MY-0.16, respectively.
 | ||
| Fig. 1 Solid state 27Al NMR spectrum of the parent zeolite and synthesized materials; a) (H)MY-0.09 M; b) (H)MY-0.16 M; c) (H)MY-0.16 M-WA; d) (H)MY-0.16 M-DA. | ||
We also characterized catalysts with an intentionally synthesized AlEF phase. These catalysts consist of MY-0.16M into which alumina has been incorporated (in the as-made form, i.e., without calcination to remove organic surfactant protecting internal microporosity) under either wet (denoted as MY-0.16M-WA) or dry (denoted as MY-0.16M-DA) synthesis conditions.46 Data from PXRD demonstrate no significant change in zeolite crystallinity compared to the parent MY-0.16 M material as a result of alumina incorporation (see Fig. S3, ESI†), and this is consistent with previous work.46
The aluminum distribution for alumina-incorporated catalysts as measured via27Al MAS NMR spectroscopy is shown in Fig. 1 and demonstrates that these catalysts have increased levels of AlEF species, with both catalysts having a 1.2-fold increase relative to the parent (H)MY-0.16 M, as shown in Table 1. These data show that our syntheses successfully added AlEF into the MY catalysts. Furthermore, the distribution of the aluminum species within alumina-incorporated catalysts is different from the parent MY-0.16 M material. Alumina-incorporated catalysts exhibit more: (i) distorted tetrahedral AlO4− species (shoulder growth at 48 ppm), (ii) penta-coordinated AlO5 (28 ppm), and (iii) octahedral AlO6 species (3.0-0 ppm). Both alumina-incorporated catalysts share similar aluminum distributions as shown in the 27Al MAS NMR spectrum of Fig. 1, and have similar integrated areas of resonances comprising these distributions (see Table S1, ESI†). Data from elemental analyses by ICP-OES in Table 1 demonstrate that both alumina-incorporated catalysts exhibit decreased Si/Al ratios relative to the MY-0.16 M catalyst that they are derived from. In particular, (H)MY-0.16 M-WA exhibits a Si/Al ratio of 5.2 whereas MY-0.16 M-DA has a Si/Al ratio of 6.1 (relative to a Si/Al ratio of 12.9 for MY-0.16 M). The slight differences in aluminum content for MY-0.16 M-DA and MY-0.16 M-WA are likely the result of removal of a minority of insoluble aluminum-containing impurities when conducting the synthesis of MY-0.16 M-DA, which involves air-free hot filtration of the aluminum isopropoxide solution prior to reaction with MY-0.16 M.
SEM images of MY-0.16 M-WA in Fig. 2a–c show macroscopic alumina particles that are commensurate in size to the zeolite particles (see Fig. S4, ESI;† these zeolite particles are the same size via SEM as in (H)CBV720 and MY-0.16 M). This macroscopic alumina phase was synthesized on the zeolite external surface during the reaction of the molecular precursor (aluminum isopropoxide) with residual water in the wet MY-0.16 M-As zeolite. Their size is similar to previous reports of alumina phases synthesized in MY catalysts under wet conditions.46
 | ||
| Fig. 2 SEM images of alumina deposited catalysts. a–c) MY-0.16 M-WA and d–f) MY-016 M-DA. | ||
The SEM images corresponding to MY-0.16 M-DA are shown in Fig. 2d–f, and, in contrast to those for MY-0.16 M-WA described above, they show no evidence of an additional phase beyond the zeolite. We infer that alumina incorporation was achieved uniformly on the SEM length scale in MY-0.16 M-DA. This is consistent with SEM data on similar MY catalysts post-synthetically modified with alumina under the same rigorously dry conditions used for MY-0.16 M-DA.46 The contrast between the SEM data for MY-0.16 M-DA and MY-0.16 M-WA is all the more compelling given that both catalysts share similar aluminum contents (vide supra).
To further investigate the uniformity of alumina incorporation in MY-0.16 M-DA, we performed SEM/EDX characterization of MY-0.16 M-DA and control MY-0.16 M in order to investigate aluminum concentration semi-quantitatively locally on the SEM length scale. Based on SEM/EDX data from different regions in both catalysts (see Fig. S5, ESI†), we estimate a 6.2 ± 0.8 fold increase of aluminum in MY-0.16 M-DA relative to MY-0.16 M. This increase is consistent with the amount of aluminum isopropoxide added to MY-0.16 M during synthesis of MY-0.16 M-DA, as well as results from ICP-OES above. We surmise that the alumina incorporated in MY-0.16 M-DA is both compositionally and morphologically uniform on the SEM length scale (i.e. no Si:Al gradients and no alumina-rich phases detected by SEM/EDX), consistent with previously reported characterization by SEM and high-resolution TEM for this material.46
We characterized microtomed cross sections of samples of MY-0.16 M-DA using STEM–EDX to further understand the degree of aluminum penetration in this material at higher resolution. Elemental mapping data are in Fig. 3 clearly demonstrate an alumina nanoscale layer within the pores of the zeolite. This is evident by the localized sections of nearly uniform coverage of this alumina layer on the microtomed cross sections of zeolite particles in MY-0.16 M-DA in the EDX data of Fig. 3. Minor differences in alumina layer thickness are observed, and likely reflect the complex heterogeneity in MY catalysts.44
 | ||
| Fig. 3 TEM–EDX images of microtomed MY-0.16 M-DA material under different magnifications. The appearance of uniform alumina species emphasizes its penetration throughout the zeolite particle interior and exterior surface. | ||
We conclude that the alumina nanoscale layer penetrates deep inside of the zeolite particle, as described previously for related catalysts, where it was demonstrated by high-resolution TEM imaging to have a thickness of approximately 10 nm (see Fig. S6, ESI†).46 We previously demonstrated that this nanoscale layer on the interior and exterior surfaces of MY-0.16 M-DA controls the zeta potential of the alumina-modified zeolite particles. This sharply contrasts the SEM images characterizing MY-0.16 M-WA in Fig. 2, where the alumina aggregates to a size ranging from 200 nm to 1 μm. In summary, when comparing MY-0.16 M-DA and MY-0.16 M-WA, both catalysts consist of added alumina relative to MY-0.16 M. In MY-0.16 M-DA, we synthesized a catalyst consisting of a highly dispersed nanoscale alumina layer on the internal and external surfaces of MY, consistent with our previously described characterization of such an alumina layer.46 In contrast, in MY-0.16 M-WA, the alumina is present at low dispersion, as its own phase having similar dimensions as the zeolite particles.46 We note that our SEM data shows no change in the morphology of the zeolite particles as a result of alumina incorporation, which further reaffirms our conclusions relating to the retention of zeolite crystallinity as a result of this incorporation from PXRD (vide supra).
We performed N2 physisorption on the alumina-incorporated catalysts in order to gain additional insight into the nature of the alumina phase added to MY. Data in Table 1 demonstrate slightly decreased mesopore surface areas of 85% and 78% for MY-0.16 M-WA and MY-0.16 M-DA, respectively, relative to MY-0.16 M. Both alumina-incorporated catalysts retain nearly the same micropore volume as the parent MY-0.16 M material (within >90%), consistent with previously reported syntheses of similar catalysts.46 We infer from the lack of observed change in the microporosity of these catalysts (i.e., with a lack of zeolite micropore blockage) that the alumina modification process did not plug micropores in MY-0.16 M-WA and MY-0.16 M-DA. The similar magnitude of the surface areas for both latter two catalysts within 8% of each other makes them excellent comparators for the effect of alumina-phase dispersion on catalysis, while keeping the textural properties of the catalysts the same. This will be exploited in the catalysis section below in order to understand the catalytic role of AlEF working in conjunction with acid sites in MY.
In summary, we synthesized several MY zeolite catalysts at differing alkalinities and with/without alumina incorporation, which possess 3.5 nm mesopores. We observe increases in the mesopore surface area (measured via N2 physisorption at 77 K) and fraction of aluminum that is extra-framework (measured via27Al MAS NMR spectroscopy) upon increasing the alkalinity of the MY surfactant templating process, while still retaining zeolite crystallinity and morphology as measured via PXRD and SEM. We also incorporated alumina into MY-0.16 M (highest mesopore surface area MY chosen for this post-synthetic modification), under both wet and dry conditions. In MY-0.16 M-WA, synthesized under wet conditions, we observed phase-separated alumina aggregates as measured by SEM (consistent with Scheme 1d); in contrast, MY-0.16 M-DA, synthesized under rigorously dry conditions, produced a material with a uniform alumina nanoscale layer on the interior and external surfaces of the zeolite based on and STEM–EDX data, with no apparent phase separation as measured by SEM images (consistent with Scheme 1c). Below, we investigate these catalysts for the catalytic upgrading of PPG polymer with changes to engineering parameters, with an emphasis on understanding the effect of pretreatment, the effect of AlEF species and mesopores have on this reaction.
Selective catalytic upcycling of polypropylene glycol to propionaldehyde in original reactor configuration: activation study & effect of zeolite catalyst
We posited that when dealing with zeolites with high surface areas, as summarized above in Table 1, after exposure to ambient atmosphere, there may be significant amounts of adsorbed water, which can in principle interfere with the interaction between reactant and catalyst due to competitive adsorption. In order to investigate the catalytic consequences of this adsorbed water layer, we compared catalysts that underwent activation via pre-drying (200 °C for 1 h) versus those that did not. We note here that all catalysts are in their proton form prior to any catalytic runs. TGA data revealed that (H)CBV720, (H)MY-0.09 M, and (H)MY-0.16 M exhibited approximately 3.5 wt%, 3.9 wt%, and 6 wt% adsorbed water, respectively, at the same ambient relative humidity (see Fig. S7, ESI†). We conclude that the amount of adsorbed water reasonably increases as the mesoporous surface area of the material increases as shown in Table 1.We subsequently reacted these six catalysts (i.e., the three catalysts above both with and without pre-drying) with 425 Da MW PPG polymer in a previously described reactor configuration (see Fig. S1a, ESI†).33 The total mass balance closure was greater than 97% within an uncertainty between 1% and 3%, with total liquid product yields detailed in Table S2, ESI.† Results shown in Fig. 4 for catalysts without pretreatment demonstrate (H)CBV720 to be the most selective catalyst for propionaldehyde synthesis, despite it having the lowest mesopore surface area among the three studied in Table 1. However, after catalyst activation via pre-drying (i.e. water removal), as shown by data in Fig. 4, (H)CBV720 became the least selective catalyst for propionaldehyde synthesis. For all catalysts, the propionaldehyde selectivity increased as a result of pre-drying, but the magnitude of this increase in Fig. 4 was disproportionately larger for the MY zeolites, leading (H)MY-0.16 M to become the most selective catalyst after pre-drying. The observed propionaldehyde selectivity in the pretreated catalyst series in Table 1 follows the mesopore surface areas of the catalysts. We previously have studied the role of steam in this reaction, and our results demonstrated an inverse relationship between the steam concentration and propionaldehyde selectivity with the use of (H)ZSM-5 catalysts.33 We surmise that, in our current work, activation of the zeolite catalyst via pre-drying reduced the amount of competitive adsorption from water, and this must have facilitated direct interactions between the large organic-molecule reactants and active sites on the catalyst. We also investigated the amount of residue in the used catalyst for all three catalysts (see Fig. S7 and Table S2, ESI†). TGA data demonstrate that the amount of residue on spent catalysts did not vary on any individual catalyst with and without pre-drying (the most sensitive controlling variable for the amount of residue was the nitrogen carrier flow rate as we discuss further below).
 | ||
| Fig. 4 (Bar graph) Propionaldehyde selectivity changes over (line plot) mesoporous surface area and AlEF content of measured catalysts. (Grey) Selectivity of propionaldehyde measured with catalyst that underwent pretreatment, (dashed pattern) selectivity of propionaldehyde measured with catalyst that had no pretreatment materials, (numbers inside the bar graph) AlEF content of measured catalysts. Original reaction setup conducted at 450 °C, 1:1:PPG:catalyst ratio, 425 Da MW PPG polymer with 10-minute reaction time, under N2 atmosphere. | ||
In summary, our catalyst pretreatment data above emphasize the importance of activating the catalyst by removing water initially before mixing the polypropylene glycol reactant and catalyst. They motivate our use of drying pretreatment for all subsequent reactions investigated in this manuscript. This effect is unintuitive given the high temperatures of catalysis, which would be expected to easily desorb water during the beginning of the reaction. We rationalize our observations by the fact that the lack of water initially must be crucial to steering the cracking reaction to desired products and, by inference, that intimate contact between the polymer reactant and catalyst surface is required for this. We also cannot rule out the possibility that the adsorbed water altered the intrinsic activity of the catalytic sites, as previously invoked on the basis of electronic structure calculations and Fourier-transform infrared spectroscopy,51 and such an interpretation would be consistent with the previously observed inverse relation of increased steam concentration and reduced propionaldehyde selectivity.33
Fig. 4 shows changes to the propionaldehyde selectivity with zeolite mesoporous surface area for the activated (i.e., predried to remove moisture) catalysts. We observe a significant increase in the selectivity for propionaldehyde upon increasing the mesopore surface area of the Y zeolite. This trend is consistent with previous observations for this reaction with ZSM-5 zeolite catalysts.33
However, other differences exist between the catalysts being compared in Fig. 4, such as the AlEF content. More specifically, commensurate with the increase in mesopore surface for the three catalysts described above, these catalysts also exhibit increases in their content of AlEF (see Table 1), as summarized in Fig. 4. We sought to deconvolute these two possible effects (mesoporosity versus extra-framework aluminum content) by intentionally adding AlEF to MY through catalyst synthesis, and examining the kinetic consequences of such addition on the propionaldehyde selectivity. Such a perturbation allows us to investigate the effect of AlEF content that works in conjunction with Brønsted acid catalysis in MY catalysts on the propionaldehyde selectivity, while keeping the textural properties approximately unchanged (vide infra).
Reactor design for selective catalytic upcycling of polypropylene glycol to propionaldehyde
Our subsequent efforts were focused on the design of the reactor and process, in addition to understanding catalyst attributes that led to high propionaldehyde selectivity in acid-catalyzed PPG polymer upcycling via cracking. From the perspective of reaction engineering, we improved the reactor described above (see Fig. S1a, ESI†), leading to a new reactor configuration illustrated in Fig. 5, which features rapid cryogenic capture of products at −30 °C in a methanol quench bath. This bath aimed to reduce undesired reactions, which we previously observed as sequential reactions to coupling products consisting of six carbon atoms (versus three carbon atoms for our target propionaldehyde product).33 We posited that these side reactions occurred primarily in the condenser of our original reactor setup (see Fig. S1a, ESI†), which prompted us to replace the condenser with the methanol quench bath described above. Furthermore, our reactor in Fig. 5 also features an increased nitrogen flow rate, which decreases the residence time of gas through the polyol-catalyst bed by three-fold (i.e., from a residence time of 4.5 s in Fig. S1a, ESI† to a new one of 1.5 s in Fig. 5). We hypothesize that these improvements in the reactor configuration (decreased residence time of gas flow stream in the reactor as well as fast quench of products) would serve to reduce undesirable sequential reactions, thereby increasing propionaldehyde selectivity. | ||
| Fig. 5 Process diagram of modified reactor setup. | ||
The data presented in Fig. 6 compare the selectivity of propionaldehyde in the methanol quench bath using the new, modified reactor configuration versus the original reactor. Both configurations were operated at a constant reaction temperature of 450 °C, and catalysts were pretreated as described above in order to maximize interaction between the catalyst surface and the PPG polymer reactants. Some reaction is expected to occur during the temperature ramping process and not just at the fixed soak temperature of 450 °C. The total mass balance closure was greater than 97% within an uncertainty of 1% (and the uncertainty was never higher than 1% in the modified reactor configuration). When closing the mass balance with the sum of remaining compounds detected gravimetrically on the catalyst and with the captured liquid products, approximately 1–3 wt% of products leave the system as a gas (see total recovered yield of solid and liquid products (%) row in Table S2, ESI†). Mass spectrometry indicates that these gaseous products primarily consist of hydrogen and a small amount of carbon monoxide. Propionaldehyde selectivity data for the catalytic cracking of PPG polymer with a MW of 425 Da in both the modified reactor configuration as well as the previously reported original reactor are shown in Fig. 6 for (H)MY-0.16 M catalyst. These data demonstrate an increase in the propionaldehyde selectivity from approximately 83% in the original reactor to 95% in the modified configuration.
 | ||
| Fig. 6 (Bar graph) Propionaldehyde selectivity changes over (line plot) residue deposits on spent (H)MY-0.16 M material with changes in the reactor setup. Reactions conducted at 450 °C, 1:1:PPG:catalyst ratio, 425 Da MW PPG polymer with 10-minute reaction time, under N2 atmosphere. | ||
An investigation into the remaining residues (including coke and long-chain hydrocarbons strongly adsorbed in the spent porous catalyst) on the catalysts after the reaction using TGA was conducted, with corresponding data shown in Fig. 6. These data indicate that the reduction in residence time, as well as the incorporation of the quenching process, led to a decrease in deposited residues on the spent (H)MY-0.16 M catalyst, dropping from 11.8 wt% (for the original reactor, Fig. S1a) to 6.5 wt% (for the modified reactor, Fig. 5) for (H)MY-0.16 M catalyst. This decrease impacted an increase in the propionaldehyde yield in the modified reactor configuration (see Table S2, ESI†). Selectivity to other products detected in the methanol bath (i.e. in the liquid phase) was also calculated (see Table S2, ESI†). Total liquid product yield for (H)MY-0.16 M catalyst in the modified reactor configuration was 92% (see Table S2, ESI†).
In summary, we modified our reactor configuration to include: (i) a three-fold decrease in residence time, which was accomplished by increasing the nitrogen carrier flowrate, and (ii) replacement of the original reactor's tubular condenser (operating with an external coolant at around 0 °C) with a −30 °C methanol-quench bath, in which products underwent much more rapid quenching instead of undergoing undesired consecutive reactions. In the studies described below, we use this new, modified reactor together with the catalyst drying described above as a pretreatment, to understand the desired catalyst attributes for PPG polymer upcycling via acid-catalyzed cracking.
Selective catalytic upcycling of polypropylene glycol to propionaldehyde in modified reactor configuration: effect of polymer molecular weight
We investigated the effect of PPG polymer molecular weight on selectivity. Our approach relied on polymer reactant molecular weights (MWs) of 425 and 2000 Da, and data in Fig. 7 (for MW of 2000 Da) show the corresponding propionaldehyde selectivities for all catalysts. The selectivity trends between (H)MY-0.16 M versus (H)CBV720 mirror those shown in Fig. 6 for the low MW polymer reactant in the original reactor. Crucially, in the new, modified reactor, data in Fig. 6 and 7 demonstrate that the propionaldehyde selectivity remains nearly unchanged for the (H)MY-0.16 M catalyst as the MW of the polymer reactant changes from 425 to 2000 Da. This catalyst exhibits 95% selectivity to propionaldehyde for polymer reactant having a MW of 425 Da, and approximately 93% selectivity for polymer reactant having a MW of 2000 Da. The nearly identical selectivity's for both the 425 and 2000 Da MWs PPG polymer reactants suggests that reaction of neither one of these is limited by mass transport. The reasoning for this conclusion is as follows. If mass transport limitations did exist, we would expect a drop in selectivity, because a significant amount of time would be spent for mass transport, without interaction with an acid catalyst site (i.e. on route to an acid site). Under such conditions, we have previously demonstrated much less selective PPG polymer cracking to products outside of propionaldehyde.33 The synthesis of such products would drop the propionaldehyde selectivity. We conclude that for the PPG reactants used in this work under our conditions in the modified reactor setup, there is no mass transport limitation. At the same time, we have already observed the importance of catalyst activation via drying pretreatment in order to obtain good interfacial contact between PPG polymer reactants and acid sites on the zeolite external surface (vide supra). We presume that this external surface is the location of initial thermal cracking events of the initial, large PPG polymeric reactant, and that smaller PPG oligomers rapidly diffuse into the internal acid sites based on the logic described above. The possible controlling variables for achieving high selectivity could be either the larger mesoporosity or the presence of AlEF sites in (H)MY-0.16 M. Below we perform further studies to deconvolute these two possible effects. | ||
| Fig. 7 Propionaldehyde selectivity with measured catalyst using modified reactor setup. Reaction conducted at 450 °C, 1:1:PPG:catalyst ratio, 2000 Da MW PPG polymer with 10-minute reaction time, under N2 atmosphere. | ||
Selective catalytic upcycling of polypropylene glycol to propionaldehyde in modified reactor configuration: effect of catalyst AlEF content
Our goal in this section was to elucidate the catalytic consequences of AlEF for the upcycling of PPG polymer via acid-catalyzed cracking. Our approach involved introducing two perturbations in the catalyst synthesis, to assess these catalytic consequences. One of these involves the intentional incorporation of alumina into the MY-0.16 M catalyst under wet conditions, which, as shown above, led to a separate alumina phase on the sub-micron length scale (via supra) in resulting catalyst (H)MY-0.16 M-WA, as shown in Scheme 1d and Fig. 2a–c. The second involves synthesis of (H)MY-0.16 M-DA consisting of a uniform nanoscale alumina layer under rigorously dry conditions, in which the dispersion of the alumina phase is significantly higher than in (H)MY-0.16 M-WA and covers the interior as well as external surfaces of the zeolite, as shown in Scheme 1c and Fig. 2d–f and 3. As summarized in Table 1, both catalysts had similar mesopore and micropore volumes, as well as mesopore surface area. Data in Fig. 7 demonstrate that these two catalysts had essentially the same propionaldehyde selectivity, with only a slight decrease in propionaldehyde selectivity for the highly dispersed alumina catalyst (H)MY-0.16 M-DA. Bulk alumina (Catalox SBA 200) was also measured as a control catalyst for PPG polymer reaction, and it was intrinsically unselective for propionaldehyde synthesis (see Table S2, ESI†). We infer that the slight performance difference between (H)MY-0.16 M-WA and (H)MY-0.16 M-DA samples above may be understood on the basis of the greater alumina dispersion in the dry catalyst. We emphasize that this effect of AlEF was minor and was partially offset by a slightly lower mesopore volume in the dry sample. Consequently, we conclude that the major driver for the observed differences in catalysts characterized here is mesopore volume rather than AlEF content.We also investigated the effect of AlEF content on residues in the zeolite after reaction. These data from gravimetric analysis are summarized in Fig. 8. A comparison of (H)CBV720 with the alumina-incorporated MY zeolites shows that addition of AlEF increased the amount of solid residues. This result cannot be due to mesopore surface area effects because we observe lower amounts of solid residues in (H)MY-0.16 M compared with (H)CBV720. We conclude that higher mesopore volume in the catalyst tends to disfavor solid residues after reaction. By that reasoning, we would have expected fewer residues in MY-0.16 M-WA and MY-0.16 M-DA compared to (H)CBV720, as reflected by the higher mesopore surface areas of the MY catalysts, as summarized in Table 1. The slight increase in residues for dry versus wet alumina-incorporated MY catalysts is consistent with AlEF controlling residue formation in MY catalysts since the alumina dispersion for the former is higher than for the latter (vide supra). We conclude that minimizing AlEF content (and the converse maximizing AlF content, for synthesizing Brønsted acid sites) is desirable both from the standpoint of maximizing propionaldehyde yield as well as minimizing residuals on spent catalysts (these trends are related to one another in the sense that less residual products allows greater yields of propionaldehyde product, see Table S2, ESI†).
 | ||
| Fig. 8 Residue deposits on spent catalysts using modified reactor setup. Reaction conducted at 450 °C, 1:1:PPG:catalyst ratio, 2000 Da MW PPG polymer with 10-minute reaction time, under N2 atmosphere. | ||
We emphasize that the selective formation of propionaldehyde via PPG polymer depolymerization is a Brønsted rather than Lewis acid catalyzed reaction. We make this conclusion on the basis on the lack of effect of AlEF on the high observed propionaldehyde selectivity for our MY-0.16 M-based catalysts, which is a logic that has been previously used to identify purely Brønsted-acid catalyzed reactions, with no role for Lewis Acidity.52 Leveraging the role of these Brønsted acidic sites in activating ether oxygen of the PPG polymer reactant, we propose the propagation sequence shown in Scheme 2. This proposed propagation sequence involves the protonation of the oxygen-anion on the polymer chain by an acid site, followed by carbonyl formation and hydride migration to release propionaldehyde. The key role of the Brønsted acid site is to act as a hydrogen bond donor to the oxygen anion and thereby decrease the energy of this intermediate. The initiation of such an oxygen anion from a OH end group on the PPG polymer chain is at this time unclear. It may be facilitated by the greater acidity of hydroxyl compounds as the temperature increases (i.e. consider the greater acidity of water at and above supercritical conditions).53 We propose the propagation sequence in Scheme 2 because of its simplicity in terms of the number of steps involved, which facilitates the high propionaldehyde selectivity observed here (95%). At the same time, given the high temperatures involved, we also cannot discount the possibility of free-radical contributions to the propagation sequence, where acid catalysis is involved only in directing the transformation of broken up fragments of the PPG polymer via radical reactions, as contemplated previously.33
 | ||
| Scheme 2 Proposed propagation sequence for PPG polymer cracking via Brønsted acidic sites on zeolite materials. | ||
Conclusion
This multi-scale study underscores the pivotal role of MY zeolite mesoporosity in controlling selectivity to propionaldehyde, with the influence of AlEF playing a minor role. This understanding motivates the synthesis of high-surface area zeolites for selective polyol upcycling to propionaldehyde. On the reactor length scale, this study demonstrates the benefit of lower sweep-gas residence times through the semi-batch catalyst bed comprising polymer and catalyst, as well as rapid quenching of the reaction products as soon as they are formed, to prevent undesired sequential side reactions. Bridging the catalyst and reaction engineering length scales of the PPG upcycling process, our data also demonstrate the importance of activating the catalyst by pre-drying, which promotes interfacial contact between the catalyst surface and PPG polymer reactant by reducing the role of excess water as a competitive adsorbate. The highest propionaldehyde selectivity for PPG polymer cracking was observed for MY catalyst (95% selectivity, 86% yield), and was independent of polymer molecular weight (in the range of 425 and 2000 Da). The latter led us to infer the absence of mass transport limitations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Special thanks to Dr. Misun Kang and the staff at the University of California Berkeley Electron Microscope Laboratory for advice and assistance in SEM sample preparation and data collection. The authors gratefully acknowledge funding from the CeRCaS NSF IUCRC. JL and KS also acknowledge funding from the South Carolina Smartstate Center for Strategic Approaches to the Generation of Electricity. STEM/EDX work was performed in part at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-2025064). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI).References
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, Organisation for Economic Co-operation and Development, Paris, 2022 Search PubMed.
- K. P. Pandey, U. R. Jha, J. Kushwaha, M. Priyadarsini, S. U. Meshram and A. S. Dhoble, J. Mater. Cycles Waste Manage., 2023, 25, 1249–1266 CrossRef CAS.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 Search PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- C. E. Brewer, K. Schmidt-Rohr, J. A. Satrio and R. C. Brown, Environ. Prog. Sustainable Energy, 2009, 28, 386–396 Search PubMed.
- M. S. Qureshi, A. Oasmaa, H. Pihkola, I. Deviatkin, A. Tenhunen, J. Mannila, H. Minkkinen, M. Pohjakallio and J. Laine-Ylijoki, J. Anal. Appl. Pyrolysis, 2020, 152, 104804 Search PubMed.
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. M. Van Steenberge, D. R. D'hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef.
- P. F. Britt, G. W. Coates, K. I. Winey, J. Byers, E. Chen, B. Coughlin, C. Ellison, J. Garcia, A. Goldman, J. Guzman, J. Hartwig, B. Helms, G. Huber, C. Jenks, J. Martin, M. McCann, S. Miller, H. O'Neill, A. Sadow, S. Scott, L. Sita, D. Vlachos and R. Waymouth, Report of the Basic Energy Sciences Roundtable on Chemical Upcycling of Polymers, USDOE Office of Science (SC), United States, 2019 Search PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS PubMed.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- H. Chang, I. Bajaj, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2020, 22, 5285–5295 RSC.
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed.
- D. P. Serrano, J. Aguado and J. M. Escola, ACS Catal., 2012, 2, 1924–1941 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 Search PubMed.
- H. Tang, Y. Hu, G. Li, A. Wang, G. Xu, C. Yu, X. Wang, T. Zhang and N. Li, Green Chem., 2019, 21, 3789–3795 RSC.
- L. Wang, G. Li, Y. Cong, A. Wang, X. Wang, T. Zhang and N. Li, Green Chem., 2021, 23, 3693–3699 Search PubMed.
- A. K. Manal, G. V. Shanbhag and R. Srivastava, Appl. Catal., B, 2023, 338, 123021 CrossRef CAS.
- A. Tennakoon, X. Wu, M. Meirow, D. Howell, J. Willmon, J. Yu, J. V. Lamb, M. Delferro, E. Luijten, W. Huang and A. D. Sadow, J. Am. Chem. Soc., 2023, 145, 17936–17944 CrossRef CAS PubMed.
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Chem Catal., 2021, 1, 437–455 CrossRef CAS.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- P. Yan, G. Bryant, M. M.-J. Li, J. Mensah, E. Kennedy and M. Stockenhuber, Microporous Mesoporous Mater., 2020, 309, 110561 CrossRef CAS.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- M. Chia, Y. J. Pagán-Torres, D. Hibbitts, Q. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS PubMed.
- A. J. Foster, J. Jae, Y.-T. Cheng, G. W. Huber and R. F. Lobo, Appl. Catal., A, 2012, 423–424, 154–161 CrossRef CAS.
- L. Dai, N. Zhou, K. Cobb, P. Chen, Y. Wang, Y. Liu, R. Zou, H. Lei, B. A. Mohamed, Y. Cheng and R. Ruan, Appl. Catal., B, 2022, 318, 121835 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and Á. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS.
- R. Bagri and P. T. Williams, J. Anal. Appl. Pyrolysis, 2002, 63, 29–41 CrossRef CAS.
- Y.-H. Lin and H.-Y. Yen, Polym. Degrad. Stab., 2005, 89, 101–108 CrossRef CAS.
- K. Nagai, Appl. Catal., A, 2001, 221, 367–377 CrossRef CAS.
- United States, US5462971A, 1995.
- K. Shikhaliyev, T. Onsree, A. H. Jaeschke, S. M. Ghoreishian, K. Shariati, A. Martinez, A. Katz, S. Hwang, A. Gaffney, J. M. Urban-Klaehn and J. Lauterbach, Appl. Catal., B, 2023, 337, 122991 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola and J. M. Rodríguez, J. Anal. Appl. Pyrolysis, 2005, 74, 353–360 CrossRef CAS.
- J. Aguado, D. P. Serrano, J. M. Escola and A. Peral, J. Anal. Appl. Pyrolysis, 2009, 85, 352–358 Search PubMed.
- V. P. S. Caldeira, A. Peral, M. Linares, A. S. Araujo, R. A. Garcia-Muñoz and D. P. Serrano, Appl. Catal., A, 2017, 531, 187–196 CrossRef CAS.
- K. A. Tarach, K. Pyra, S. Siles, I. Melián-Cabrera and K. Góra-Marek, ChemSusChem, 2019, 12, 633–638 CrossRef CAS PubMed.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- H. Zhang, Y. Ma, K. Song, Y. Zhang and Y. Tang, J. Catal., 2013, 302, 115–125 CrossRef CAS.
- N. Xue, A. Vjunov, S. Schallmoser, J. L. Fulton, M. Sanchez-Sanchez, J. Z. Hu, D. Mei and J. A. Lercher, J. Catal., 2018, 365, 359–366 CrossRef CAS.
- Y. Zhang, R. Zhao, M. Sanchez-Sanchez, G. L. Haller, J. Hu, R. Bermejo-Deval, Y. Liu and J. A. Lercher, J. Catal., 2019, 370, 424–433 CrossRef CAS.
- N. Linares, E. O. Jardim, A. Sachse, E. Serrano and J. García-Martínez, Angew. Chem., Int. Ed., 2018, 57, 8724–8728 CrossRef CAS PubMed.
- A. Sachse, A. Grau-Atienza, E. O. Jardim, N. Linares, M. Thommes and J. García-Martínez, Cryst. Growth Des., 2017, 17, 4289–4305 CrossRef CAS.
- D. Mehlhorn, J. Rodriguez, T. Cacciaguerra, R.-D. Andrei, C. Cammarano, F. Guenneau, A. Gedeon, B. Coasne, M. Thommes, D. Minoux, C. Aquino, J.-P. Dath, F. Fajula and A. Galarneau, Langmuir, 2018, 34, 11414–11423 CrossRef CAS PubMed.
- A. Chawla, N. Linares, J. D. Rimer and J. García-Martínez, Chem. Mater., 2019, 31, 5005–5013 CrossRef CAS.
- L. Xu, A. Okrut, T. Rea, K. Chaudhuri, J. Han, A. Kuperman and A. Katz, Cryst. Growth Des., 2023, 23, 5601–5613 CrossRef CAS.
- J. Urban-Klaehn, K. Shikhaliyev, A. Gaffney, A. Martinez, R. Zaleski, J. Lauterbach, A. Katz, A. Jaeshke and X. Li, ChemCatChem, 2024, 16, e202301282 CrossRef CAS.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 32, 2003264 CrossRef CAS PubMed.
- W. Loewenstein, Am. Mineral., 1954, 39, 92–96 CAS.
- S. González-Gallardo, V. Jancik, A. A. Delgado-Robles and M. Moya-Cabrera, Inorg. Chem., 2011, 50, 4226–4228 CrossRef PubMed.
- A. Jentys, G. Warecka, M. Derewinski and J. A. Lercher, J. Phys. Chem., 1989, 93, 4837–4843 CrossRef CAS.
- R. Gounder, A. J. Jones, R. T. Carr and E. Iglesia, J. Catal., 2012, 286, 214–223 CrossRef CAS.
- P. E. Savage, Chem. Rev., 1999, 99, 603–621 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00001c |
| ‡ Equal first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |