
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrothermal liquefaction of plastics: a survey of the effect of reaction conditions on the reaction efficiency†
Matthijs Justin
Boel
a,
Hongqi
Wang
 a,
Ahmad
AL Farra
b,
Laura
Megido
c,
José Manuel
González-LaFuente
c and
N. Raveendran
Shiju
*a
a,
Ahmad
AL Farra
b,
Laura
Megido
c,
José Manuel
González-LaFuente
c and
N. Raveendran
Shiju
*a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, 1090 GD Amsterdam, The Netherlands. E-mail: n.r.shiju@uva.nl
bTRTG-TotalEnergies Research & Technology Gonfreville, BP 27, 76700 Harfleur, France
cCOGERSA S.A.U., Carretera de Cogersa, 1125, La Zoreda, Serín, 33697 Gijón, Spain
First published on 15th March 2024
Abstract
Plastic waste is a major global issue. Recycling has not made a big impact as hoped; most of the waste is still landfilled or incinerated. Hydrothermal liquefaction (HTL) is a promising technique for plastic recycling. In broad terms, it converts wet carbon-containing feedstocks (e.g., wet biomass or plastics) back into simpler molecules using elevated temperature and pressure. Sub- and supercritical water is used as solvent, reagent, and catalyst, although additional catalysts may be added. HTL efficiency depends on several factors, making optimization potentially complex. We evaluated prior literature on HTL of plastics to summarize the reaction conditions for the optimal results for several types of plastics, such as PE, PP, PET, PS, PC, PVC, and plastic mixtures. Furthermore, the proposed mechanisms were examined and summarized. The polymers with heteroatoms in the main chain, PET and PC, had maximum liquefaction efficiency at subcritical temperatures, separating into their substituent monomers, while those without had maximum liquefaction efficiency at supercritical temperatures. The polyolefins with branches, PS, PP, PVC, and LDPE, liquefied at lower temperatures than that of the branchless HDPE. Plastic and plastic–biomass mixtures showed synergy at subcritical temperatures with maximal yields of around 30%.
Introduction
The continual production of plastics as well as the increase of plastic waste is one of the significant problems plaguing our world's ecosystem, alongside greenhouse gas emissions causing temperature increases and ocean acidification. Microplastics especially have had a significant impact among all ecosystems.One additional prominent issue regarding plastics is the requirement of fossil fuels to produce the majority of them, given that fossil fuels are a limited resource, and our society is highly dependent on plastics. Some plastics can be produced through biological means, but this only accounts for a small amount. These problems make plastic recycling necessary for the future.
Currently, several recycling options for plastics already exist, classified as primary, secondary, tertiary, and quaternary recycling, which is based on how far removed the eventual products are from the original waste. Directive 2008/98/EC on waste talks about reuse, recycling and recovery (“Article 3. Definitions”: CL2008L0098EN0030020.0001_cp 1.1 (https://www.europa.eu)).
The primary recycling involves putting a plastic product back to use (‘reuse’ according to Directive 2008/98/EC), which is not possible in a number of cases. Secondary recycling involves the melting and remolding of plastic waste (defined as ‘recycling’, Directive 2008/98/EC). This method works only on thermoplastic polymers, and the mixing of several types of plastic causes a decrease of quality compared to the virgin plastics. As sorting plastic waste streams can be laborious or simply too difficult to accomplish, it is preferred to use methods that require the least amount of sorting. Tertiary recycling involves the depolymerization of plastic waste, then to be used as either a fuel source (‘recovery’ according to Directive 2008/98/EC) or monomer resource of new plastic production (‘recycling’ or ‘material recovery’ according to the Directive). Lastly, quaternary recycling involves the combustion of the plastics to regain the stored energy (typically known as waste-to-energy or ‘incineration coupled to energy recovery’). Note that we prefer to use combustion rather than incineration as the latter does not always imply “energy recovery or recycling”. There is a great controversy regarding this. Considering EU framework on waste, “inefficient incineration” is a “disposal” option (like landfilling) and “efficient incineration” is a “recovery” option, but less desirable than “material recovery”. We suggest to unlink “incineration” (with or without efficient energy recovery) from the “recycling” concept. At the moment, non-recycling options like dumping plastic in landfills or water sources, and incineration, which produces toxic gases and greenhouse gases, are the most used options of plastic disposal.1
Given these four types of recycling, tertiary recycling could potentially be applied to the broadest selection of plastics, allowing new opportunities in terms of circular economy while avoiding further negative impacts on nature. One such method of tertiary recycling that is currently being researched is hydrothermal liquefaction (HTL). HTL is a type of hydrothermal treatment where feedstock, typically wet biomass, is exposed to elevated temperatures and pressures, causing the organic molecules to react and decompose into smaller products, mainly oil.2 A benefit of HTL that is lacking in other tertiary recycling options is the use of water, which functions as a solvent, a catalyst, and a reagent simultaneously. Under typical reaction conditions water is found in its sub- or supercritical state, which significantly increases its degree of solvation towards organic compounds due to the states' lower dielectric constants. Subcritical water (scw) reacts more through ionic mechanisms, while supercritical water (SCW) participates in radical mechanisms. Because of this, the critical point of water, 374 °C and 22.1 MPa, is an important value to keep in mind when deciding on reaction conditions. The use of water also avoids the need to dry any feedstock before the process, unlike pyrolysis, where feedstock is heated in an oxygen-deprived environment, saving a significant amount of energy. This method also allows for much milder reaction conditions compared to pyrolysis, although high pressure is the major cost for this process.
HTL also overlaps with two other hydrothermal treatments, namely hydrothermal carbonization (HTC) and hydrothermal gasification (HTG).3 In general, these three types use the same general procedure but are separated by the reaction temperatures used, with HTC going from 180 to 250 °C, HTG above 374 °C, and HTL in between. In the literature, however, several procedures that have been termed HTL fall in the HTC or HTG conditions. For the sake of clarity, in most papers concerning hydrothermal treatment, the focus for those working with HTC is towards the production, maximization, and composition of solid hydrochar from feedstock, while HTL focuses on oil products, and HTG focuses on the production of gas, typically referred to as syngas. All three procedures often yield some amount of the undesired phases as by-product, or even have one of the other phases as the major product, despite the reaction conditions falling outside the relevant treatment, making this distinction more useful than the reaction condition definition.
As stated above, the HTL of feedstock yields a variety of hydrocarbons, distributed in the separate phases after completion. The solid phase typically contains unreacted feedstock, larger hydrocarbons, and insoluble products, including inorganic impurities. The liquid phase consists of an oil product, which is considered to be the most important in HTL, and an aqueous phase, both originating from washings of the reactor with organic solvent and the remaining water, respectively. And lastly, the shortest hydrocarbons as well as carbon dioxide gas and hydrogen gas are present in the gas phase. The entire HTL procedure is shown in Scheme 1.
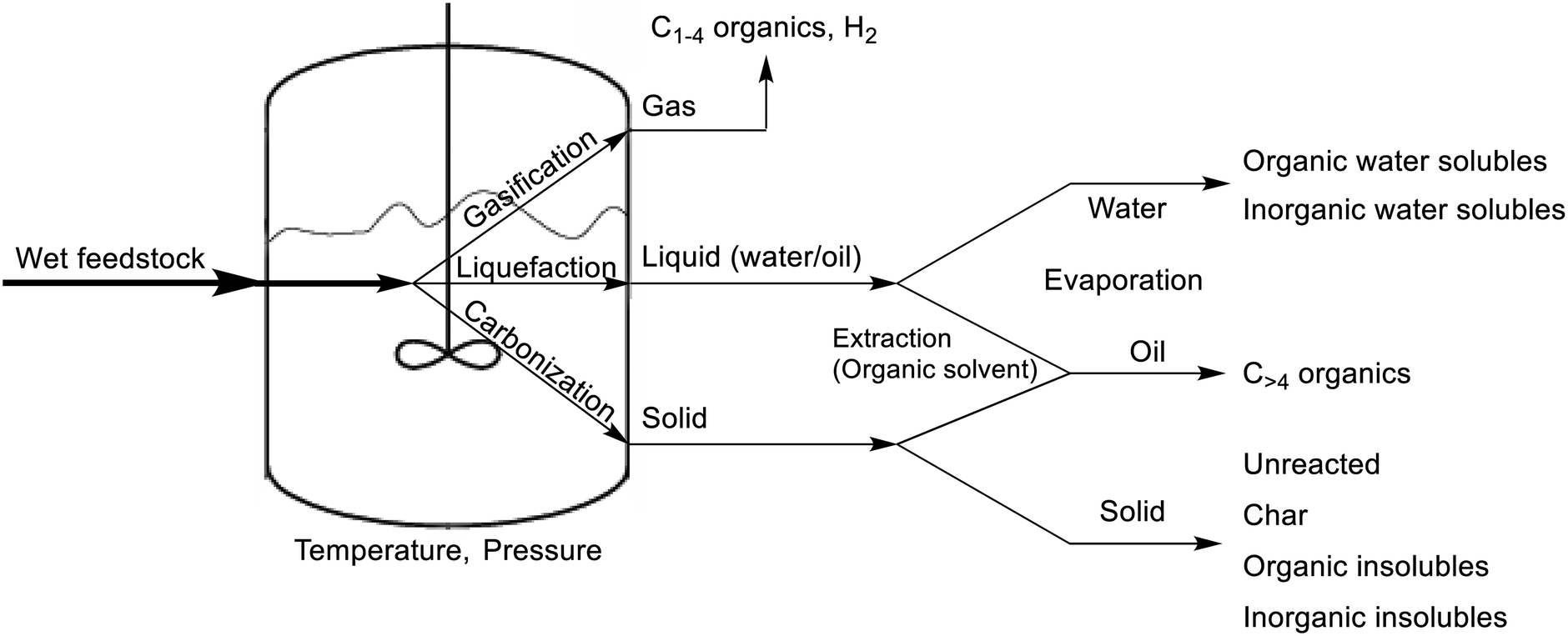 | ||
| Scheme 1 HTL procedure with reactor and product distribution. | ||
HTL has already been widely used for the liquefaction of a wide array of biomass into oil, but the application of HTL towards the chemical recycling of plastics, either through the recovery of monomers or the production of higher energy value fuels, has gained attention only in recent years. In line with this research, the liquefaction of the most common types of plastics has been explored by numerous papers (Table S1†).
Recently, Gopinath et al. compared several disposal methods for plastic waste, taking into account economic, energy, and environmental factors, and concluded that HTL of plastics could have equal efficiency to pyrolysis and was overall more useful than primary recycling and landfills.4 Similarly, Okoro et al. simulated the use of a continuous HTL processing plant for polypropylene and showed that an operating cost of $0.38 per kg of plastic would be needed, with all investment being returned after two years of operation, being significantly more viable economically than other processes of recovering (Directive 2008/98/EC) plastic waste.5
In 2015, the most common plastics, both in production rates and current percentage of total amount in circulation, were, in descending order, polypropylene (PP) at 68 Mt per year, low- and high-density polyethylene (LDPE/HDPE) at 64 and 52 Mt per year, polyvinyl chloride (PVC) at 38 Mt per year, polyethylene terephthalate (PET) at 33 Mt per year, polystyrene (PS) and its butadiene copolymer high-impact variant (HIPS) at 25 Mt per year, and polycarbonate (PC).1 One last type of common polymer is polyurethane (PUR), being produced at 27 Mt per year. There are also other types of plastics, classified as polyesters, polyamides, and acrylic polymer (PP&A), those last three together making up 14.5% of global plastic production at 59 Mt per year. However, unlike the previously mentioned plastics, these are mere umbrella terms for families of plastics, making it difficult to draw any conclusions for processing as they can span a wide range of chemical diversity. All percentages for the global production and the polymer structures are shown in Scheme 2.
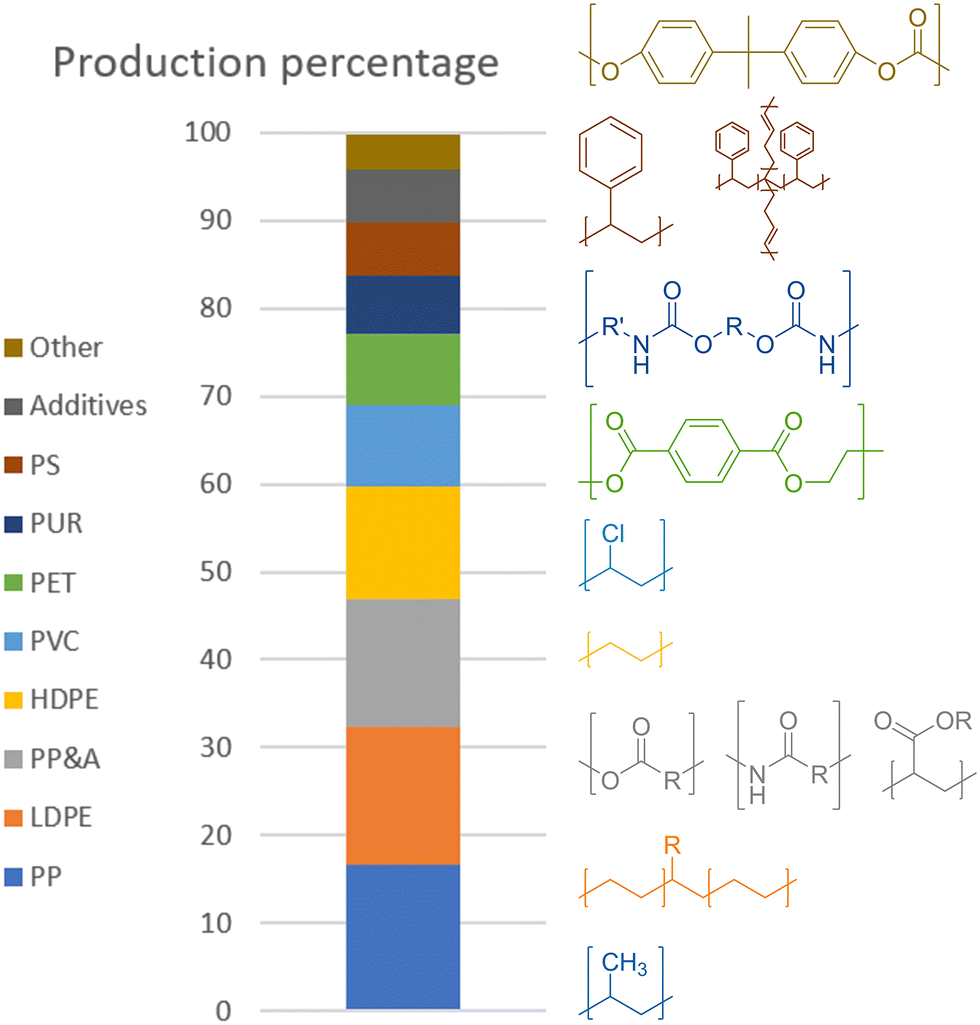 | ||
| Scheme 2 Annual plastic production percentage (2015) with structures.1 | ||
The difficulty in processing plastic waste through HTL lies in the sheer variety of synthetic polymers mixed in. Reaction rate, polymer conversion, product distribution, and oil quality are dependent on a range of relevant factors, mainly temperature, pressure, reaction time, heating rate, feedstock-to-water ratio, feedstock composition, and the type of reactor, mainly its size, reactor lining, and whether it operates in batch, semi-batch, or continuous form.2 Each type of polymer will have its own optimal conditions, and testing for all variables is a laborious task. Furthermore, different polymers reacting simultaneously may create an additional layer of interactions that need to be taken account of, in addition to type specific, additional concerns.
These conditions and interactions require sufficient exploration to determine the most cost- and energy-efficient parameters for the recycling of plastic through HTL as well as potential additional measures, such as which plastic types should be separated for additional efficiency. This literature study summarizes the currently known conditions and interactions of the HTL of the more common types of plastic as well as their mixtures and the underlying decomposition mechanisms.
Polyolefins
Polyethylene (H/LDPE)
Polyethylene, both the high- and low-density variants, makes up the majority of all plastics, adding up to 28.5% of all plastic in circulation back in 2015.1 As such, several studies involving the liquefaction of polyethylene have been conducted. Only a few of these papers studied the liquefaction of pure PE under differing conditions but showed a few important trends.In general, HDPE requires supercritical temperatures to decompose in HTL. Temperatures of 425, 450, and 460 °C yielded a maximum of 90, 87, and 91 wt% conversions of polymer into oil, respectively.6–9 While dos Passos et al. and Sugano et al. showed that typical medium-size pellets (mm scale) did not decompose into oil at all at lower temperatures of 350 or 400 °C, on a small scale (powder) they gave high yields similar to that under supercritical conditions, reaching 68, 86, and 99 wt%, respectively.10,11 Zhang et al. instead employed a continuous-type reactor, which showed that much higher temperatures are necessary to successfully perform HTL.12 At 500 °C and 25 MPa, only 15 wt% oil was produced in two minutes, while a batch process at 460 °C produced over 90 wt% in one minute. The threshold temperature for this continuous process was between 520 and 525 °C, where the oil yield increased from 20 to 65 wt%. At 530 °C, the highest yield of 79 wt% was obtained with the continuous process. At 550 °C, 95 wt% of the solid had decomposed, although the yield of oil was 75 wt%, the remaining 20 wt% having decomposed further into gas. Zhang et al. speculates that flowing supercritical water quenches primary radicals formed from initial hydrocarbon cracking more than stationary SCW, decreasing any further reactions with other hydrocarbons in the continuous process. Please note that as most HTL studies were performed in batch-type reactors, unless otherwise specified, the reactor type is batch in the following discussion (see also Table S1†).
Interestingly, Jin et al. performed a modified HTL procedure in scw at low pressure (1.55 MPa) with supercritical temperatures (450 °C) by significantly reducing the water-to-plastic ratio, going from a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (1.75) ratio, yielding 87 wt% oil at 425 °C, to a 2:7 ratio (0.29), producing a similar yield of 89 wt%, indicating that criticality is not necessarily the driving factor behind HDPE degradation, but instead merely the temperature.8 This was also supported by Sugano et al. who did not observe any degradation with HTL in SCW at 400 °C.11 Jin et al. also performed an experiment with the same procedure but without any water, which gave an 84.5 wt% oil yield, slightly lower than the 89 wt% yield in low water runs, with the gas yield having increased by 3.5 wt% from further decomposition of the oil, showing that the radical mechanism present in pyrolysis is the major factor in HTL of HDPE compared to ionic interactions with water.8 This was to be expected, given that HDPE has no heteroatoms in its main chain to meaningfully interact with water in ionic mechanisms.
:4 (1.75) ratio, yielding 87 wt% oil at 425 °C, to a 2:7 ratio (0.29), producing a similar yield of 89 wt%, indicating that criticality is not necessarily the driving factor behind HDPE degradation, but instead merely the temperature.8 This was also supported by Sugano et al. who did not observe any degradation with HTL in SCW at 400 °C.11 Jin et al. also performed an experiment with the same procedure but without any water, which gave an 84.5 wt% oil yield, slightly lower than the 89 wt% yield in low water runs, with the gas yield having increased by 3.5 wt% from further decomposition of the oil, showing that the radical mechanism present in pyrolysis is the major factor in HTL of HDPE compared to ionic interactions with water.8 This was to be expected, given that HDPE has no heteroatoms in its main chain to meaningfully interact with water in ionic mechanisms.
Sugano et al. also performed a modified HTL procedure by turning the HDPE feedstock into powder (μm) through a steam explosion process before feeding it into the reactor.11 This pretreatment enabled the degradation of HDPE at 400 °C and below, producing a maximum of 99 wt% oil yield at 400 °C. This process also promoted degradation at subcritical temperatures (300 and 350 °C), though with lesser conversion of 68 and 86 wt%, respectively. Whether the increased rate of degradation arises from the more finely divided feedstock, from partial degradation during the pretreatment through mechanochemical processes, or both is undiscernible at the moment. The optimal supercritical region for temperature is seen in Fig. 1, which summarizes the results on HTL of HDPE.
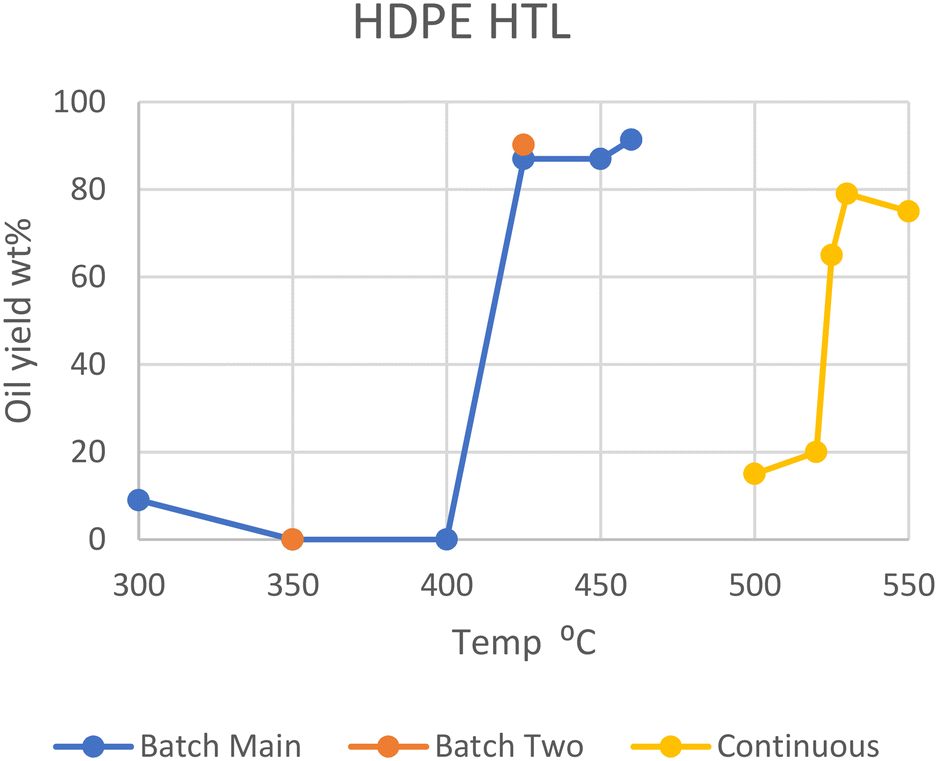 | ||
| Fig. 1 Summary of literature yields of HDPE.11 | ||
Zhang et al. further researched oil yield dependency on other parameters.12 Pressures ranging between 20 and 30 MPa did not have any significant effect on the phase yields, remaining around 80 wt%. However, the alkene-to-alkane ratio in both the oil and gas phases increased with pressure, going from 2.83 to 5.13, and from 1.62 to 2.04, respectively. The heavier hydrocarbons also increased in the oil, the C19–24 fraction increasing from 25.5 to 34.1 wt% of the oil, and the C>24 fraction increasing from 13.3 to 15.4. Zhang et al. speculate that higher pressures increase the density of SCW, which in turn can isolate and quench radicals more easily, coined the ‘cage effect’, which prevents conversion of alkene to alkane.
LDPE shows a similar trend. At 350 °C, Passos et al. showed that only 1 wt% of oil was produced after 20 minutes, with only ∼7 wt% being converted in total.10 However, at 400, 425, and 450 °C in SCW, LDPE gave 87, 88, and 96 wt% oil, respectively.13,14 Wong et al. showed that liquefaction at subcritical temperatures was possible with longer reaction times, as it took two hours at 300 °C and 8 MPa to gain a 28 wt% oil yield.15 They showed it was also possible to obtain ∼14 wt% yield at 152 °C and 1 MPa pressure after one minute, as long as the water-to-plastic ratio is rather large (33.0). The optimal temperature range sits in supercritical conditions, similarly to HDPE, and can be seen in Fig. 2, which also shows the outlier from the study by Passos et al. at 350 °C. The most likely reason for this divergence is the Teflon coating used, while the other studies used metal alloys or quartz. Either the Teflon, which is a highly fluorinated polyolefin, negatively influences the reaction rates, or the metal alloys in the other reactors are positively influencing reaction rates. This may be from differing heat conductivity or chemical interactions.
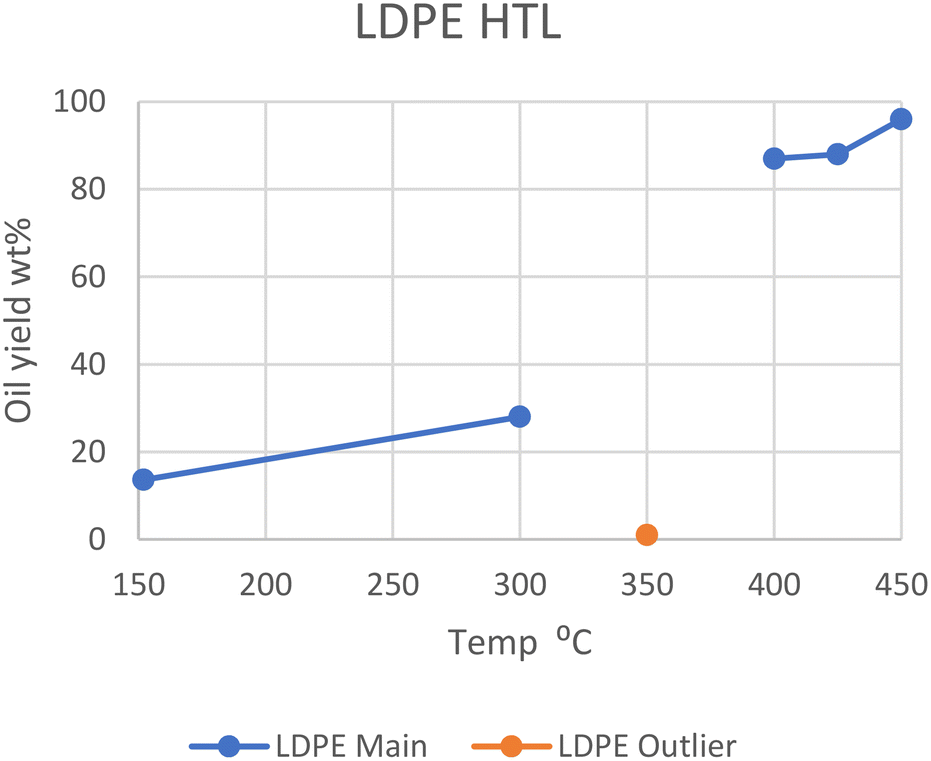 | ||
| Fig. 2 Summary of yields reported for the liquefaction of LDPE.10,13–15 | ||
In general, the degradation mechanism of polyethylene, depicted in Scheme 3, starts with the cracking of the main chain, producing several longer hydrocarbons which are further decomposed into saturated and unsaturated compounds by hydrogen abstraction and β-scission, respectively.8 Moriya et al. reported hydrocarbon products on the C20–C70 range in oil product, with paraffins being present in slightly larger quantities than olefins, though longer reaction times of three hours instead of two showed a shift to the shorter hydrocarbons, with olefins being converted into paraffins, and paraffins further decomposing into smaller chains.6 Zhang et al. also reported the presence of dienes in the oil in their continuous reactor process.12 They also showed that at supercritical conditions higher temperatures increased the unsaturated hydrocarbon content while decreasing the saturated hydrocarbon content, contrary to results in batch processes. From 500 to 550 °C the alkene-to-alkane ratio from HDPE products increased from 2.7 to 4.2, though most of the alkene increase came from alkadienes, their percentage increasing from 17 to 23 wt%. Reactions between decomposing olefins and paraffins are suppressed by supercritical water, even more so with flowing supercritical water removing the olefins before they can further react, which suppresses further conversion of olefins into paraffins.
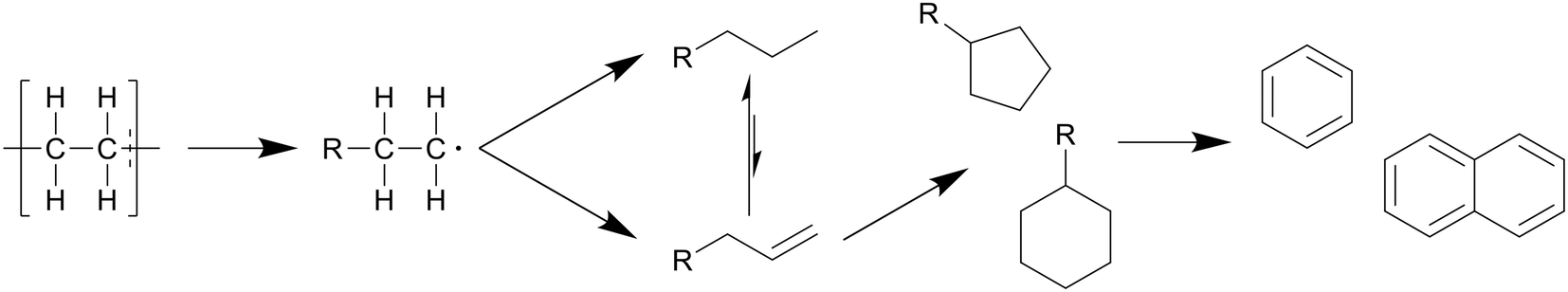 | ||
| Scheme 3 Proposed decomposition pathway of PE.8 The main chain is cracked first, producing several longer hydrocarbons which are decomposed further into saturated and unsaturated compounds by hydrogen abstraction and β-scission, respectively. | ||
In general, olefins also further react to form cyclics and aromatics, thanks to cyclisation through double bonds and further dehydrogenation as cyclics and aromatics are more stable. This is shown by the amount of aromatic protons increasing over time, the ratio of aromatic to aliphatic hydrogen in 1H NMR going from 0.01 to 0.9 between 30 and 120 minutes.6 The lightest molecules became saturated paraffins in the gas phase (C1–C4) at 425 °C, with only a small amount of olefins at an alkene-to-alkane ratio of 0.11 and 0.06 after two and three hours, respectively. Carbon monoxide and dioxide made up only 3.8 mol% of the gas phase, with 9.6 mol% being hydrogen gas. Olefins can also readily react with water to form alcohol. In general, smaller hydrocarbons dissolved in and subsequently reacted with water to form oxygenated compounds like acetaldehyde, ketones, alcohols, and acids, although oxygen gas being present could also allow for these reactions to occur. Whether the water or oxygen plays the key role in conversion to oxygenated compounds has not been researched yet.
Even in short reaction times at 520 °C with the continuous process, significant degradation occurs.12 Within the first minute, almost half of all heavy hydrocarbons (C>17) were converted. Then, over the period of 85 seconds to three minutes, the percentage further decreased from 53.5 to 23.2 wt%, with only 15.3 wt% in the oil remaining after the fourth minute.
Jin et al. also provided product compositions for their low pressure, supercritical temperature approach.8 Low weight paraffins were most predominant, making up 31 wt% of the oil, followed by olefins and cyclics, 40 wt% combined, with aromatics and iso-paraffins having the smallest presence at 21 and 8 wt%, respectively. This composition did not change with their varied pressures.
For subcritical temperatures, Wong et al. showed a lower alkene-to-alkane ratio in the oil fraction of 0.5 with LDPE HTL, compared to 2.7 for HDPE at 500 °C, postulating that β-scission was reduced under subcritical conditions, thus lessening the conversion of larger chains into alkenes while not influencing the production of alkanes through hydrogen abstraction.15 Wong et al. also reported an 8 wt% fraction of oxygenated compounds like acids and ketones in their oil product, and a 2.1 wt% fraction of carbon dioxide and carbon monoxide in their gaseous product. Their reactor was purged of oxygen gas by argon, though they pointed out that the presence of some remaining oxygen gas after the purge cannot be excluded as an explanation. These results also cannot be compared to those in the non-purged experiment by Moriya et al., as they did not provide concentrations of the oxygenated compounds, while Wong et al. did not provide the chemical composition of their aqueous phase.
In supercritical temperatures of 400 °C, however, Zhao et al. reported a similarly low alkene-to-alkane ratio of 0.52 with LDPE HTL.13 This low amount of alkenes also meant a lower amount of cyclic and aromatic products. Additionally, LDPE follows similar degradation mechanisms to HDPE; Čolnik et al. showed similar product compositions in the gas phase between HDPE and LDPE under the same conditions, with similar alkene-to-alkane ratio evolution in the gas products at different temperatures and times.14 The ratio decreased from 0.73 to 0.21 from 15 to 240 minutes at 425 °C, and from 0.46 to 0.02 in the same time span at 450 °C.
Polypropylene (PP)
Polypropylene, the most common type of plastic when taking the two polyethylene types into account separately, shows similar behavior to degradation as the polyolefins. Passos et al. reported 7 wt% oil yield in scw at 350 °C, though the peak yield may not have passed within the limited set time of 20 minutes.10 Additionally, the Teflon coating may be producing lower results in general compared to quartz and steel reactor experiments. Indeed, Zhao et al. showed a 36 wt% conversion of the solid under the same conditions by extending the reaction time to an hour.16In SCW, oil yield already reached 87 wt% after an hour at 400 °C.13 At 425 °C, Seshasayee et al. saw a full conversion into a maximum 34 wt% oil yield after half an hour, with the remainder having converted into gas, and higher temperatures converting more oil into gas with further prolongation.17 A similar study by Chen et al. however showed a maximum 92 wt% yield after two hours under the same conditions.18 The yields and reaction times of these maxima do not match up with each other, and the gas production reported by Seshasayee et al. is much higher. The main distinguishable difference between these two experiments is the heating rate, which was significantly lower in Chen et al.'s study (39 °C min−1), which showed the higher oil yield compared to the other study (135 °C min−1). The higher heating rate may have caused the oil to have passed its optimal point before the heating had finished and the reaction time had started, allowing further conversion into gas. However, at lower temperatures most of the PP had not yet converted, showing 90 wt% solid. Instead, it should be noted that Chen et al. performed their analysis on the oil without solvent evaporation, while Seshasayee et al. evaporated DCM first, reporting that the more volatile, lighter products had evaporated, meaning the oil yields reported from analyses afterwards include only the heavier oil products. Some other differences that may explain the conflicting results less satisfactorily include the purging of the reactor with nitrogen gas prior to HTL in Chen et al.'s study, which was lacking in the other, as well as the addition of a stirrer. However, in Zhao et al.'s study at 400 °C there was no purging performed either, and they reported the already mentioned 86 wt% oil yield, though a stirrer was employed.
One final difference would be the water-to-plastic ratio, which is at 8:1 in Seshasayee et al.'s study, but is only defined as ‘proper amounts of deionized water’ by Chen et al. Without an actual value it is impossible to compare, however. The amount of water can determine how much a plastic can decompose through radical or ionic mechanisms, with less water encouraging radical conversions. Another is pellet size, which also was not specified by Chen et al.
The same study by Jin et al. with their modified HTL procedure showed that temperature, and thus the radical mechanism, is of more influence on the degradation of PP, gaining an 89 wt% oil yield at 450 °C and 1.55 MPa by reducing the water-to-plastic ratio to 2:7, compared to a 90 wt% yield under standard supercritical conditions of 450 °C and 23 MPa.8 Another run lacking any water further indicated that radical cracking is the major mechanism of depolymerization, giving a full conversion of solid with 80.5 wt% oil yield. Jin et al. also showed that prolonging the reaction time under standard supercritical conditions decreases oil yields, going down to 82 wt% compared to 90 wt%.
In general, however, elevated temperatures of 425 °C provide the greatest yield of oil in batch reactors, as can be seen in Fig. 3. The outliers from Passos et al., Zhao et al., and Seshasayee et al. are also seen here.
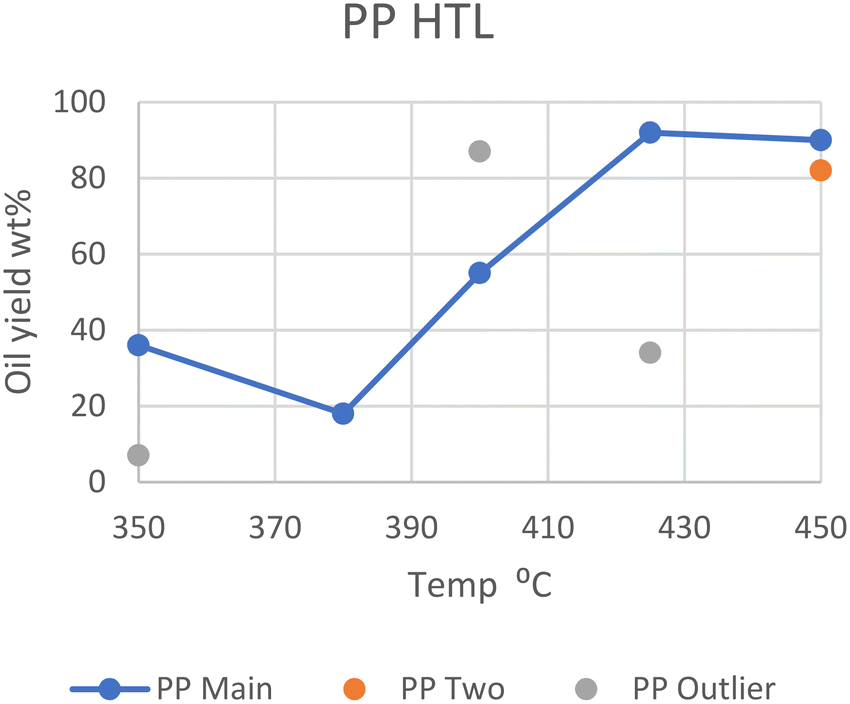 | ||
| Fig. 3 Summary of literature yields of PP (data provided in Table S2†). | ||
Polypropylene undergoes a similar degradation mechanism to that of polyethylene, shown in Scheme 4, though certain reactions in said pathway occur more often compared to polyethylene degradation.8 For example, due to the presence of low bond enthalpy C–CH3 (335 kJ mol−1) bonds, which is lower than that of the main chain C–C bonds (347–377 kJ mol−1), these methyl side groups will readily split from the main chain during degradation, causing higher rates of β-scission and providing a larger alkene-to-alkane ratio throughout the degradation, giving a maximum at 400 °C after an hour. This splitting of methyl side groups also enhances the conversion into alcohols through the interaction with water, which can be dehydrated back to alkenes at higher temperatures, preventing any reactions between the alkenes, now alcohols, and alkanes.13 This then allows for a much higher formation of cyclics and aromatics. Seshasayee et al. showed that from 400 °C and up, cyclics and aromatics were the most prominent components in the oil, with a 40% aromatic proton fraction in 1H NMR, and at 450 °C almost 70% of the lighter oil was composed of multicyclic aromatics like naphthalene and fluorene.17 Zhao et al. corroborated these results, reporting a chemical composition at 400 °C, where 75% of the oil was olefin, with a 5% paraffin and 14% cyclic makeup, compared to LDPE, which had a 63% paraffin, 34% olefin, and 2% cyclic content.16
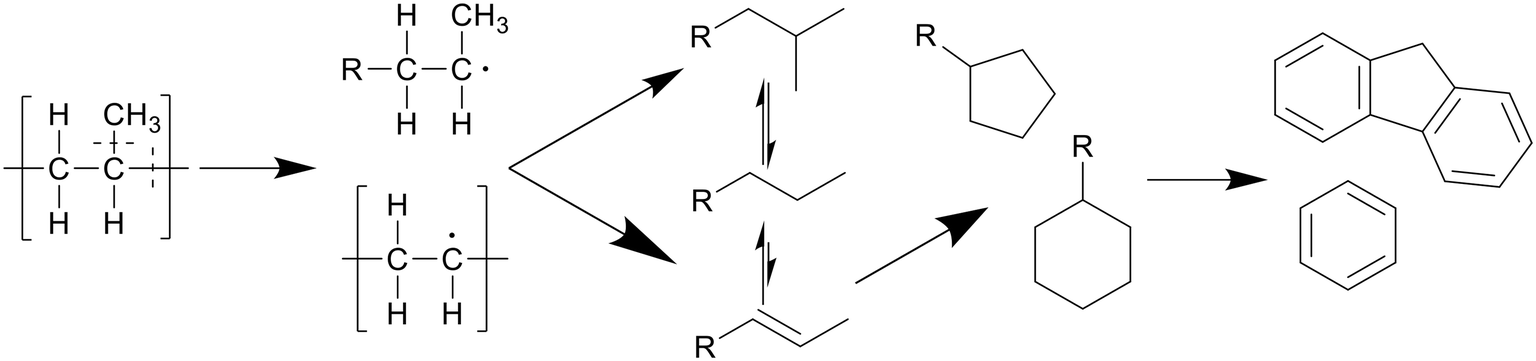 | ||
| Scheme 4 Proposed decomposition pathway of PP.8 | ||
Chen et al. showed that longer reaction times at 425 °C caused further conversion of alkenes into cyclics and aromatics, olefins decreasing from 93% to 49% with reaction times increasing from 30 minutes to 2 hours, while cyclics increased by 35%.18 A similar progression was seen with increasing temperatures at two hour reaction times, olefins decreasing from 95% to 30% from 380 to 450 °C, while cyclics increased by 40% and aromatics increased by 15%.
Polystyrene (PS/HIPS)
Polystyrene is at the lower end of the most commonly produced plastics and shows a similar trend to the previous polyolefins. Sugano et al. employed the same steam-explosion pretreatment to show that PS, like HDPE, does not decompose under subcritical conditions unless it is powdered, with pellets of medium size giving no conversion of solids, while powdered feedstock yielded ∼90 wt% oil yields under the same conditions.11 Passos et al. corroborated this, as 2 mm PS pellets at 350 °C in scw for 20 minutes only gave 3 wt% oil, with 20 wt% of the feedstock converting to aqueous products.10 All plastics in the study by Passos et al. yielded lower amounts of oil compared to near identical conditions in other studies. Given that the only notable difference between Passos et al.'s study and all others are the presence of a Teflon coating in the reactor, this could be due to the unfavorable interactions between the tested plastics and Teflon, which is a fluorinated polyolefin. The exception here is Seshasayee et al., who reported an 86 wt% yield at the same temperatures with 2 mm size pellets.17 Between the study of Seshasayee et al. and those of Sugano et al. and Passos et al., there are no common differences that can explain this anomalous result. Taking the Teflon coating of the latter's study into account, the notable difference between Passos et al. and Seshasayee et al.'s experiments was the presence of mechanical stirring, which the former lacked.At higher temperatures, Kwak et al. showed that PS did not fully convert over time in scw, entering in equilibrium after 5 minutes at 80 wt% at 370 °C.19 PS in SCW eventually fully converted to oil, with full conversion happening in 15 minutes at 380 °C and 3 minutes at 390 °C. Kwak et al. employed a semi batch type reactor for this study. However, Sugano et al. showed the same results with a batch type reactor, with 400 °C reaching full conversion within an hour, though samples at earlier times were not taken.11 Higher temperatures were shown to be disadvantageous by Seshasayee et al., with half of the feedstock being converted into gas at 425 and 450 °C after 30 minutes.17
For high-impact polystyrene (HIPS), where butadiene polymer branches intersperse with the styrene monomers, a different optimal temperature is shown. At 250 °C, HIPS yields 7 wt% oil in an hour, which steadily increases to 40 wt% at 300 °C and peaks around 90 wt% at 350 °C.20 However, decomposition intriguingly decreased under supercritical conditions, yielding only 38 wt% oil at 400 °C and 77 wt% at 490 °C.21 The cause for this dip at 400 °C could be an unexpected difference in interaction of HIPS with sub- and supercritical water, or some other variables. However, Jin et al. did not provide a water-to-feedstock ratio or the solid or gas yields; therefore nothing can be concluded without further research, although the butadiene copolymer may be the cause here.
In Fig. 4, the unusual behavior of HIPS when transitioning between sub- and supercritical conditions is visible. Furthermore, the outliers from Passos et al. and Sugano et al. are also shown. It has already been discussed how Teflon may negatively influence oil yields for Passos et al., but the discrepancies between Sugano et al.'s results and the similar conditions of Seshasayee et al. are less trivially explained. Seshasayee et al. reports a lower yield than that of Sugano et al.; thus evaporation of more volatile products may again be a viable explanation.11,17 Sugano et al. also evaporated the solvent before providing yields, although they used n-hexane as extraction solvent instead of DCM, and their high yield of 98 wt% oil discounts the notion of any volatile oil products having evaporated prior to analysis.
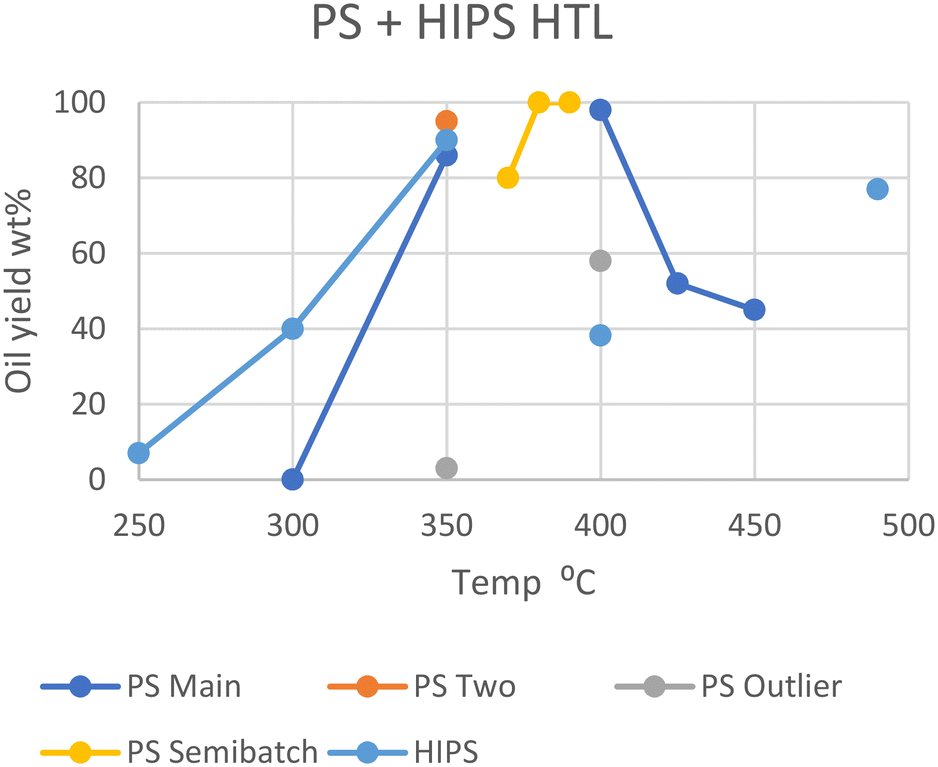 | ||
| Fig. 4 Summary of literature yields of HTL of PS & HIPS (data in Table S3†). | ||
Similar to polypropylene, polystyrene has a weaker C–C bond enthalpy thanks to its side group, the benzylic C–C bond enthalpy being only 333 kJ mol−1, which is even lower than that of the C–CH3 bonds in polypropylene (337 kJ mol−1).20 This allows PS to decompose at lower temperatures than polypropylene, which can in turn degrade at lower temperatures than polyethylene. The benzene groups themselves are too stable to be affected; therefore PS mostly cracks between the main chain and a benzene group, or cracks at the main chain, as shown in Scheme 5. This mostly produced styrene monomers and dimers, 1,3-diphenyl propane, which then formed alkyl-substituted benzenes, which can be further methylated through a radical mechanism.21 Kwak et al. showed that at 400 °C and 28 MPa, styrene monomers, dimers, and trimers made up 45%, 23%, and 10% of the oil, respectively, at the end of the heating period (taken as at zero time), and then decreased to 10%, 0%, and 0%, respectively, over an hour.19 These intermediates were mostly converted into toluene and ethyl benzene, which reached 21% and 18%, respectively, although in regular PS there would mostly be styrene instead of ethyl benzene due to the lack of butadiene monomers during decomposition, which provide more free radicals for random chain scission. They also showed that pressure had a slight influence on how much of each styrene monomer, dimer, and trimer was produced within the first 10 minutes at 400 °C, with the lower degree styrenes being more prominent at higher pressures. At higher temperatures, benzene rings start fusing together into multicyclic aromatics like naphthalene. At temperatures of 500 °C and above the radical mechanism takes over and produces char from the aromatics.
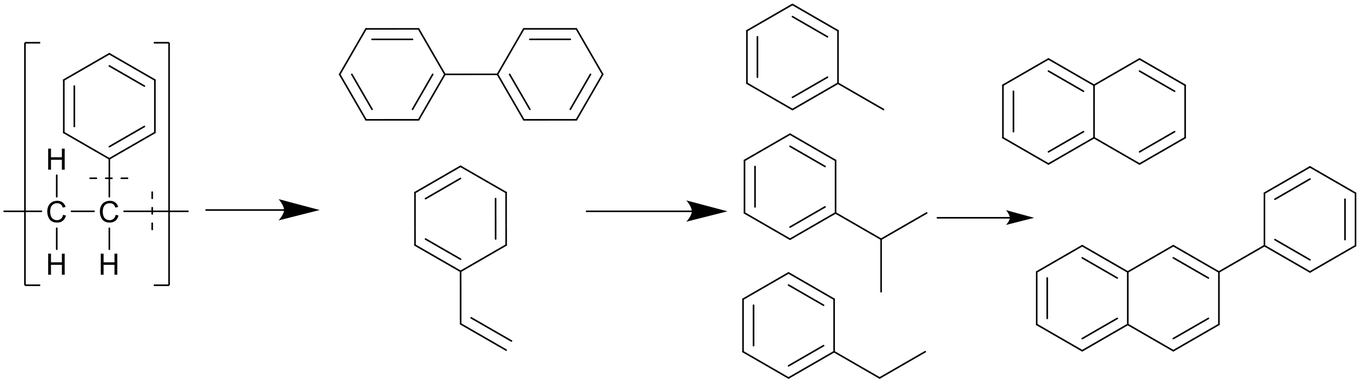 | ||
| Scheme 5 Proposed decomposition pathway of PS.21 | ||
Polyvinyl chloride (PVC)
Compared to the prior polyolefins, there is a significantly lower number of studies on the HTL of polyvinyl chloride (PVC) due to concerns of organochlorides being formed in the process and hydrochloric acid promoting the formation of char as well as it and chlorine gas damaging the reactor over time.Two studies on scw HTL of PVC at 300 and 350 °C showed similar oil yields of 12 and 10 wt%, respectively, under subcritical conditions.10,22 Both processes showed significant dechlorination into the aqueous phase as hydrochloric acid, close to 100% at 300 °C and above, but in both cases significant charring occurred due to the acidic environment. Passos et al. experimented with adding alkali catalyst to the feedstock to counteract the acidification, but this did not reduce the charring.10 Instead, it converted the hydrochloric acid in the aqueous phase into chlorine gas, while also reducing the amount of dechlorination of the solid, making it an unviable solution. In Takeshita et al.'s study, all solid residue was converted in SCW at 400 °C.22
Gandon-Ros et al. reported a significant dechlorination as well, reaching 99.1% after 4 hours at merely 250 °C in scw with the addition of 0.025 M K2CO3 as catalyst. The aqueous phase and gas products were not analyzed to determine where the chlorine transferred to. Most of the product was solid char.23
In an earlier study, Takeshita et al. reported that phenol, benzene, and acetic acid were the most common compounds in the oil at 450 °C, while simple paraffins are predominant in the gas phase, though specific amounts were not given.24 At higher temperatures of 600 °C, the most common compounds in the oil phase were all aromatics like phenol, benzene, and naphthalene. They presented a mechanism where the chlorine atoms are removed through dehydrochlorination, producing hydrochloric acid, and leaving behind a highly unsaturated, conjugated polyene. This then cracks and turns into smaller olefin chains that close to form aromatic compounds through intra- and intermolecular Diels–Alder reactions. The solid mostly consisted of long polyenes, as elemental analysis showed that 88.8 wt% of the residual solid was carbon, with only 7.5 wt% hydrogen, giving a 12:1 mass ratio, which gives a 1:1 molar ratio. Compared to the degradation of PVC by pyrolysis, no organochlorine compounds were detected in the products. However, they also found the presence of several oxygenated compounds, like acetic acid, phenol, benzoic acid, acetone, benzaldehyde, and hydroquinone, in the oil phase, despite having purged the reactor with argon, though small amounts of oxygen were detected, dissolved in the aqueous phase.
In a later study, Nagai et al. proposed that water played a bigger role in the reactions, performing dechlorination through hydroxide nucleophilic substitution, forming polyols, which are then cracked and hydrolyzed to form the polyenes, as depicted in Scheme 6.25 The nucleophilic substitution increases the dissociation of PVC in SCW. At lower amounts of water the radical pathway plays a bigger part. No specific values were reported.
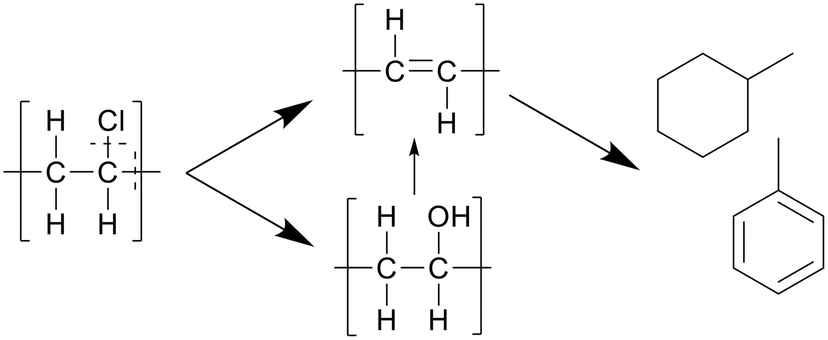 | ||
| Scheme 6 Decomposition pathway of PVC proposed by Nagai et al.25 The hydroxide nucleophilic substitution leads to dechlorination, forming polyols, which are then cracked and hydrolyzed to form the polyenes. | ||
While the mechanism suggested by Nagai et al. proposes dechlorination by nucleophilic substitution, the mechanism by Takeshita et al. proposes dehydrochlorination. In this zipper mechanism by Takeshita, the dehydrochlorination of an adjacent monomer unit facilitates the loss of HCl from a two-carbon segment, leading to polyene. HCl is then converted to hydrochloric acid by dissolution in water. In subcritical water, chain scission occurred to generate low-molecular-weight compounds, mainly acetone, benzene, benzoic acid, benzaldehyde, phenol, and hydroquinone. It is proposed that intramolecular and intermolecular Diels–Alder reactions of polyenes lead to benzene, and the oxidation leads to acetone and benzene derivatives. More experiments are needed to confirm which mechanism actually operates. It may also be possible that different mechanisms occur under different conditions.
Condensation polymers
Polyethylene terephthalate (PET)
In general, PET decomposes mostly into a solid phase. At 240 °C in scw after 30 minutes all PET was depolymerized with the help of a zinc acetate catalyst, with a 90% solid yield of near-pure terephthalic acid (TA); the oil and gas yields were not given.26 The zinc acetate catalyst significantly accelerated the decomposition of PET as proved by the yield of 45% with catalyst vs. 2% without catalyst. In addition, the utilization of zinc acetate could decrease the required temperature by 60 °C. At 300 °C in scw after 90 minutes, 20% of the carbon was distributed into the oil phase, with a 75% solid yield of near-pure TA.27 Oil yield again was not explicitly given. At 350 °C in scw, a 10 wt% oil yield was reported by Passos et al., with a 70% solid yield of pure TA after 30 minutes.10 Under similar conditions after 20 minutes however, Seshasayee et al. reported a 5 wt% oil yield, with a higher 82 wt% solid yield.17 The main differences here were the reactor coating, being Teflon for the higher oil recovery and stainless steel for the higher solid TA recovery, and the heating rate, which was double in Seshasayee et al.'s study. Under supercritical conditions at 400 °C, Pedersen et al. reported no oil yield with medium size pellets after 15 minutes, but a 68.5% TA recovery in solid,28 while Seshasayee et al. reported a higher 72 wt% solid yield and 5 wt% oil yield after 30 minutes, with no stirring giving a higher TA yield.17 After 30 minutes at 425 °C and 450 °C the oil yield increased to 14 wt% and 16 wt%, respectively, again with a 70 wt% solid TA recovery. Passos et al. also showed that employing a base like potassium hydroxide would pull most of the solid TA into the aqueous phase as a salt, with the aqueous phase increasing from 18 wt% to 50 wt%, while the gas phase increased from 4 to 17 wt% and the solid phase decreased from 70 wt% to 25 wt%. The main reason why not all the solid TA was converted was due to an excess, as the potassium hydroxide was used as a catalyst.10 Furthermore, other alkaline catalysts such as sodium hydroxide and ammonia could also promote the depolymerization.In general, PET fully decomposes into its substituent monomers at both sub- and supercritical temperatures, although the solid TA yield decreased with higher temperatures. Given that at 350, 425, and 450 °C oil was formed, the lack of oil in Pedersen et al.'s study at 400 °C is unexpected.28 The notable, reported differences from Pedersen et al. are shorter reaction times and purging their reactor with nitrogen gas beforehand as well as mechanical stirring during the HTL process. Both the HTL oil and TA yields from the literature are summarized in Fig. 5.
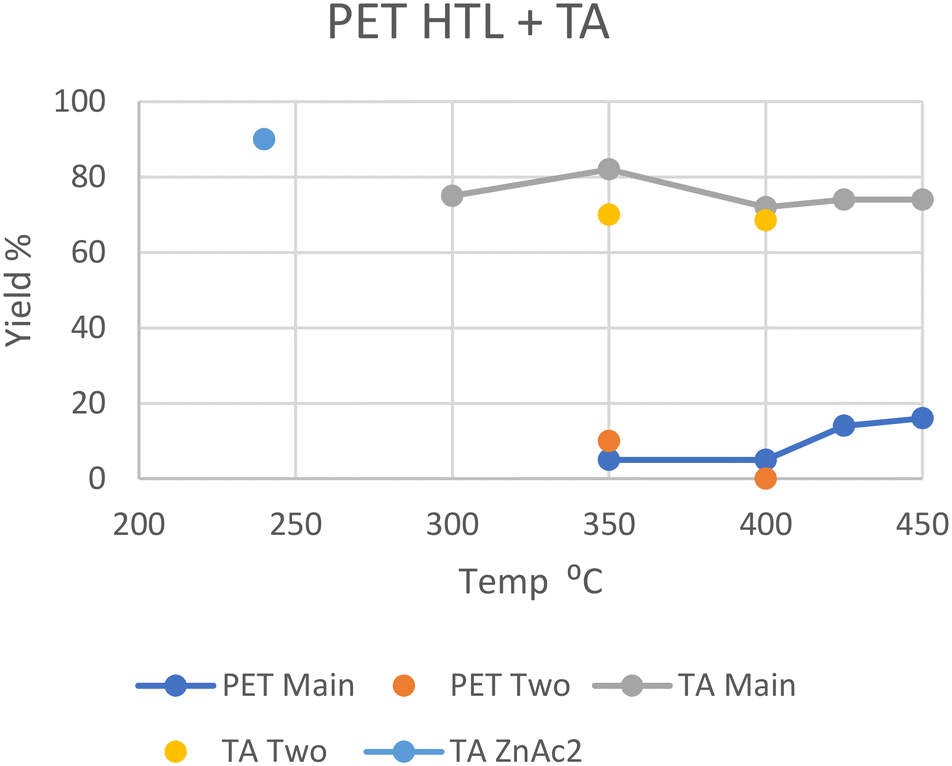 | ||
| Fig. 5 Summary of literature yields of HTL of PET (data provided in Table S4†). | ||
Thanks to the benzene groups, which are not prone to degradation, and the ester bonds present in PET, the polymer primarily cracks at the heteroatoms, shown in Scheme 7. The polymer first split into TA, which quickly formed a solid phase, as it does not dissolve well in water or organic solvent, and ethylene glycol monomers, which dissolved in the aqueous phase.27 Ethylene glycol further decomposed into acetaldehyde gas and small alkyls and alkenes through dehydration, which could then form long-chain fatty acids and diols through small-scale oligomerization, while TA formed benzoic acid at higher temperatures due to decarboxylation.
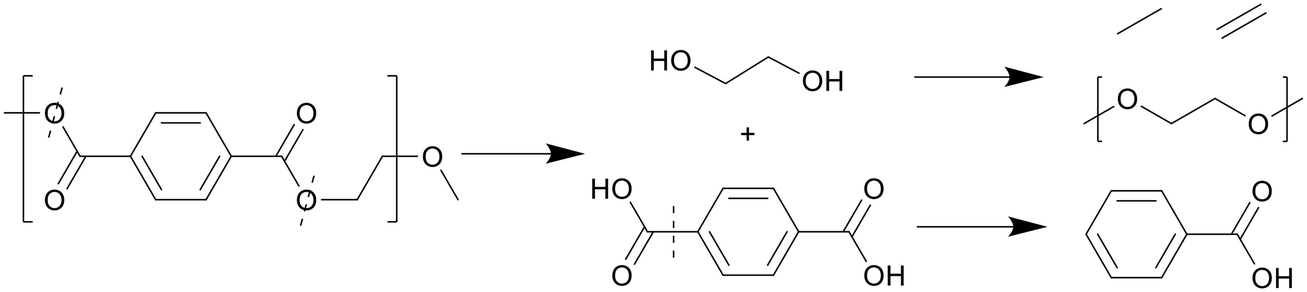 | ||
| Scheme 7 Proposed decomposition pathway of PET.27 | ||
Passos et al. added the alkali catalyst KOH to the HTL feedstock and reported that TA was converted into salt, which was then dissolved in the aqueous phase.10 This, however, also opened up the further decarboxylation of TA into benzoic acid, which proceeded at a higher rate compared to normal HTL, as the gaseous phase increased from 4 to 17 wt%. This may still be useful for recycling however, as dealing with the aqueous phase would be less difficult than separating the solid product stream in more complicated feedstock mixtures.
Liu et al. also used a catalyst in the HTL of PET in scw, namely, zinc acetate at 1.5 wt%, which provided a significant TA monomer recovery (90%) at a much lower temperature of 240 °C after only 30 minutes, which also allowed for a higher 60% ethylene glycol monomer recovery in the aqueous phase due to lower degradation.26 The yield is also shown in Fig. 5. Apparently, both acidic catalysts such as zinc acetate and alkaline catalysts such as KOH could accelerate the decomposition of PET by HTL. The application of an effective catalyst could increase the ion concentration in water, either the H+ or OH−, which can favor the cracking of polymers, lower the required temperature and improve product selectivity.
Polycarbonate (PC)
Like PET, polycarbonate also has heteroatoms in its backbone which water can interact with to split the polymer. Zhao et al. performed HTL at subcritical 250 °C, yielding 82.1 wt% oil from the solid after an hour, although the concentration of bisphenol A (BPA), the main monomer of PC, in the oil was not determined.20 Huang et al. performed HTL at 300 °C in scw, where it took 45 minutes for all the PC to depolymerize into oil and gas, giving a 37% yield of BPA.29 A similar reaction by Seshasayee et al. gave 65 wt% oil after an hour, with a slightly higher 47 wt% BPA recovery.17 Similar conditions employed by Zhao et al. showed a 91 wt% oil yield, although this increase may be due to the employment of a quartz reactor instead of a standard steel type.20At 350 °C, Passos et al. report around a 75 wt% oil yield after 20 minutes, 45% of which consists of BPA (33.8%), with no remaining solid.10 However, Zhao et al. again report a higher oil yield of 93 wt% after 60 minutes, of which only 8.2% is still BPA (7.7%), and no solid remains.20 Zhao et al. used a quartz reactor instead of steel, which allows for a higher heating rate. This higher oil yield and further degradation may also be explained by the larger water-to-plastic ratio and longer reaction times, respectively.
In SCW at 400 °C, Pedersen et al. reported a maximal 99.8 wt% oil yield after 15 minutes, with ∼15% being BPA.28 At a higher temperature of 425 °C, Seshasayee et al. reported a 60 wt% oil yield after 30 minutes with no remaining solid, where 12.5% was BPA (7.5%).17
The general yields are summarized in Fig. 6, and the series of higher yields from Zhao et al. in their quartz reactor can easily be seen, although an experiment of HTL in a steel reactor at 250 °C would be needed to prove this difference as the cause. Furthermore, the lower BPA yield at 350 °C from Zhao et al. compared to that of Passos et al. may also be explained by this, as BPA would decompose further in a reactor with a higher heating rate.
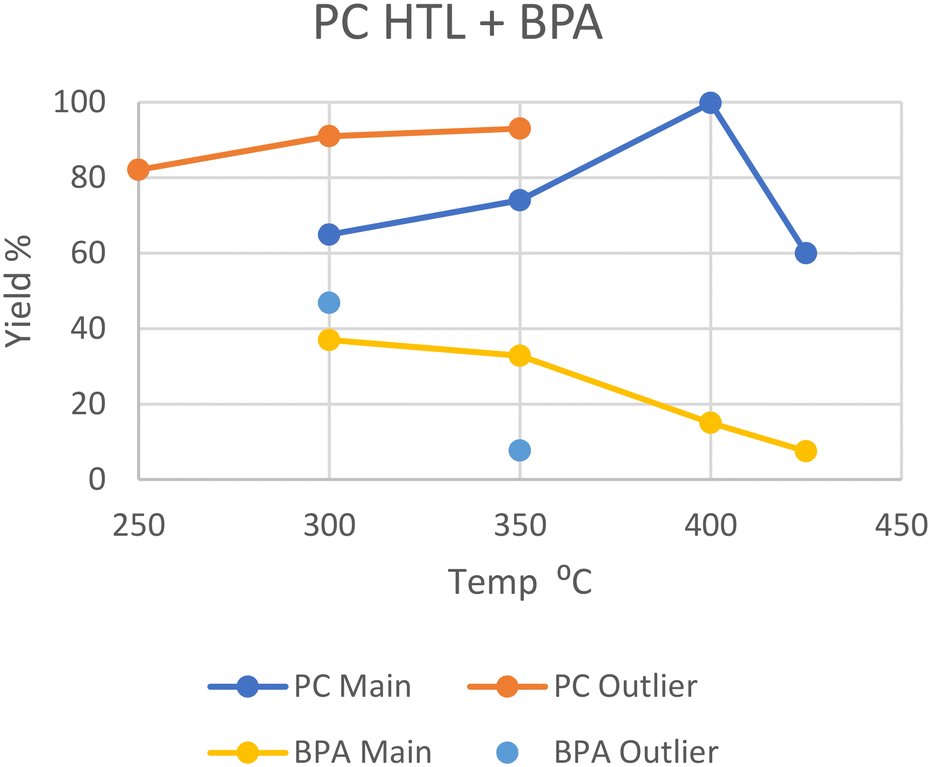 | ||
| Fig. 6 Summary of literature yields of HTL of PC (data provided in Table S5†). | ||
In general, PC completely depolymerizes into oil and gas under scw and SCW conditions, though the amount of BPA decreased through further degradation with increasing temperature and longer reaction times. It should also be noted however that BPA has been shown to have toxic effects on humans.
Like with PET, the presence of benzene rings and ester bonds in PC causes the polymer to break at the ester bonds first, forming BPA and C1 compounds, as shown in Scheme 8. The BPA quickly further decomposed by cracking at the isopropyl bridge between the benzenes, breaking the BPA up into phenol and p-isopropenylphenol, as shown by Jin et al., which then further decomposed into products like p-isopropyl phenol.30 There is also a small amount of p-tert-butylphenol present as oil within the first few minutes of HTL before any other decomposition products appeared, though this molecule appears to be a structural regulator present in PC, only appearing as a monomeric unit at the caps of the polymer molecules, and thus the easiest to be removed in liquefaction. At supercritical temperatures, p-ethyl phenol and p-methyl phenol were also seen, converted from the further decomposition of p-isopropyl phenol. Seshasayee et al. however also reported other methylated products of the previously mentioned compounds, like methoxy-2,4-dimethyl benzene and 3,4-dimethyl phenol at 450 °C.17
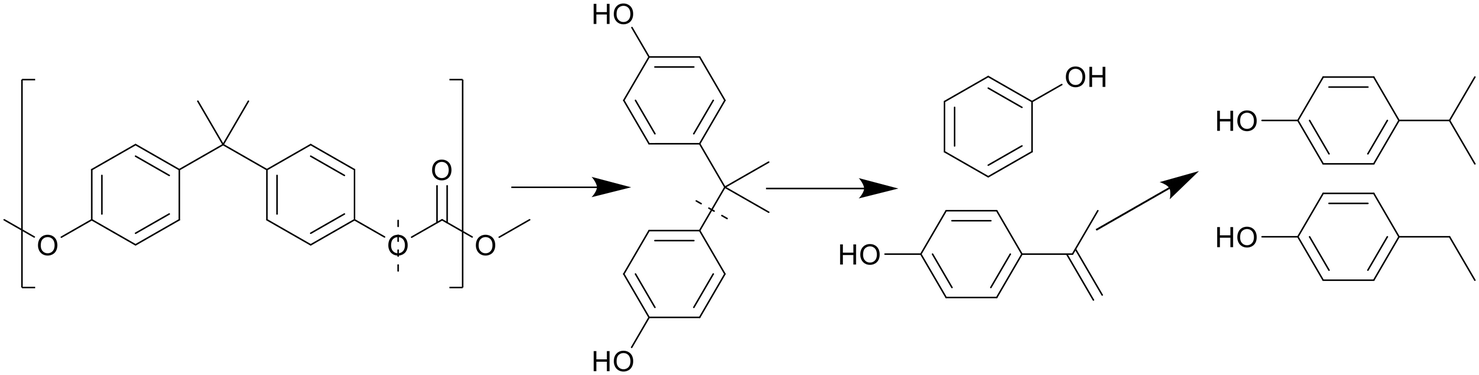 | ||
| Scheme 8 Proposed decomposition pathway of PC.30 | ||
Co-liquefaction
HTL of plastics has been shown to produce significant oil yields from the most common types of plastics, given the right temperatures and pressures, more so than most typical biomolecules present in biomass.31 However, for an efficient and practical implementation, mixtures of plastic would have to be processed together. As the different synthetic polymers require different conditions, and liquefaction products may interact with the degradation mechanisms of other plastic types, it is expected for plastic mixtures to require different conditions for optimal oil yields and might give higher or lower yields than expected if the plastic types were processed under said conditions separately.To this end, Seshasayee et al. performed an array of tests to identify which plastic types may interact synergistically or antagonistically at different temperatures. For instance, at 300 °C, binary plastic mixtures of PP, PET, and PC with PS gained a statistically highly significant synergy, going from 20 to 40 wt%, from 20 to 50 wt%, and from 50 to 70 wt%, respectively, as shown in Fig. 7.32 The other combinations showed no significant synergy or antagonism. In previous studies, PS was shown to produce significant oil starting at 350 °C when sufficiently powdered.11 The authors here speculate that the degradation products from PP, PET, or PC, presumably methyl radicals for the first, decomposition products from ethylene glycol for the second, and alkyl phenol radicals for the latter, as they are formed in oil at these low temperatures, promote the degradation of PS at 300 °C.
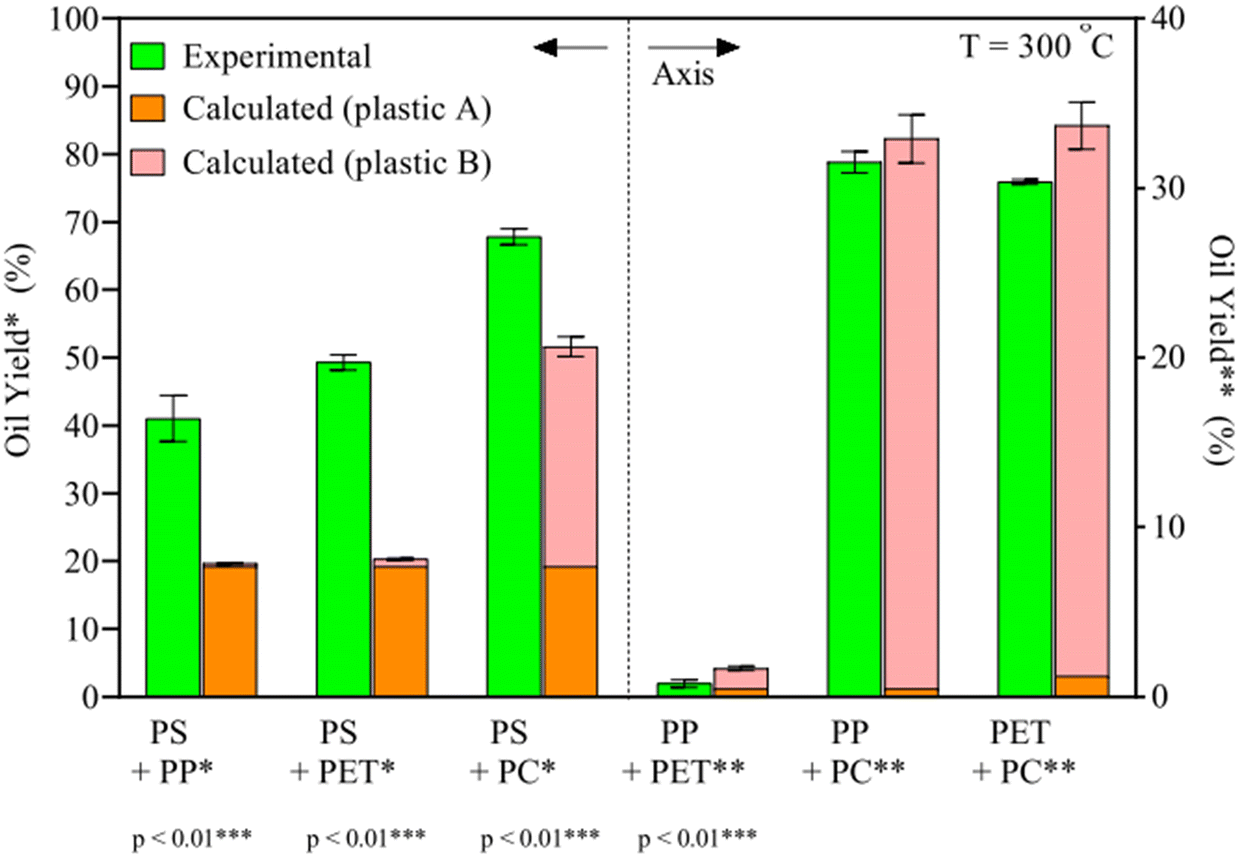 | ||
| Fig. 7 Synergy of binary plastic mixes.32 The binary plastic mixtures of PP, PET, and PC with PS show synergy and result in higher oil yields than individual components. | ||
A similar synergistic effect is seen at 425 °C for PP with PET and PC, showing a statistically high significant synergy, going from 2 to 18 wt% and from 34 to 60 wt% resprectively.31 The other combinations showed no significant synergy or antagonism. As PP normally produces the highest oil yields at 450 °C, the decomposition products of PET and PC must encourage the degradation of PP at lower temperatures. In general, it seems that the more easily liquefied plastics PC and PET encourage the decomposition of olefin-based plastics. How exactly PP, an olefin, is able to encourage the breakdown of PS far below its own liquefaction temperature is unclear. However, it may be the other way around in this case, with some small amounts of PS degradation products at 300 °C encouraging the breakdown of PP, which further encourages the breakdown of PS in return.
Jin et al. and Zhao et al. investigated the product composition from the liquefaction of several mixtures of HDPE with PP and LDPE with PP, respectively. The mixtures of HDPE and PP at ratios of 3:1, 1:1, and 1:3 at 450 °C did not have a change in the distribution of solid, liquid, and gas products.8 The composition of the oil however did change with the ratios of feedstock, as depicted in Fig. 8. Comparing PP product distribution to HDPE product distribution, the cyclics and olefins increased by only 4%, the aromatics and iso-paraffins increased by 10% and 12%, respectively, and the paraffins decreased by 24%. The iso-paraffins and aromatics logically increased, as PP produces far more iso-paraffins and olefins due to its methyl branches, the latter of which then converts further into cyclics and aromatics. Jin et al. did not report the olefin and cyclic percentages separately, so it is unclear whether the two rose similarly to their combined effect, or if they changed divergently and the changes balanced each other out. However, the former scenario is more likely because at 450 °C after 45 minutes, any additional olefins generated from PP would have reacted to form either cyclics, not changing the combined total, or further reacted into aromatics. As the decrease in paraffins would mostly account for the increase of iso-paraffins and olefins, the amount of olefins would have increased by 12%, 9% of which was converted into aromatics, with the remaining 3% only converting into cyclics. The individual compounds changed linearly with the change in product ratio from pure HDPE to pure PP however, indicating there were not any interactions between the two decomposition pathways, and the compound distributions are merely the distributions of the pure decomposition products added together at the corresponding ratios.
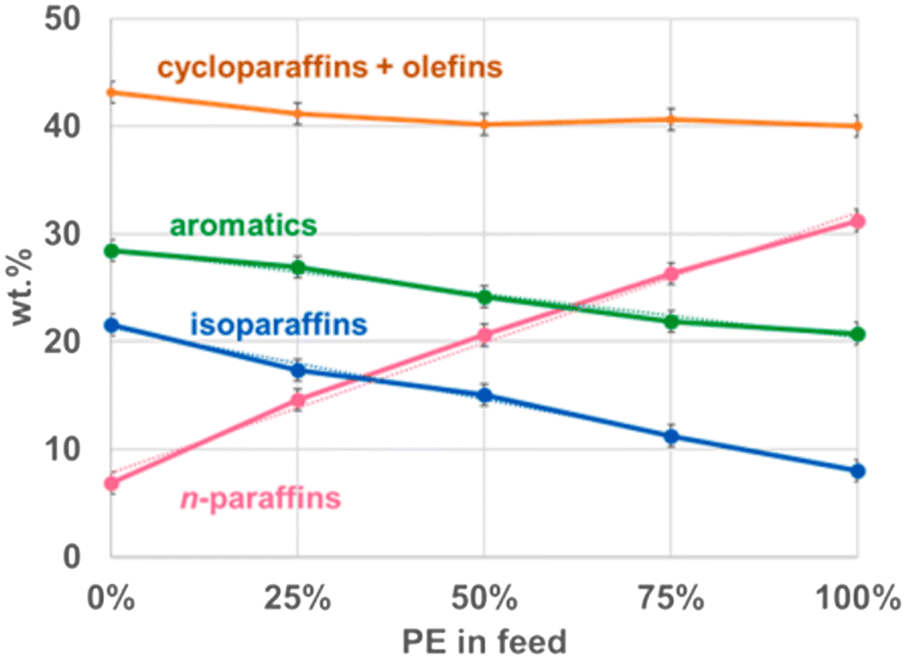 | ||
| Fig. 8 Oil composition of HDPE:PP mixtures.8 | ||
Zhao et al. reported a non-linear change with a variation in product ratio between LDPE and PP however, performed at a lower temperature of 400 °C, the results of which are shown in Fig. 9.13 When LDPE was half or more of the mixture, the cyclic and paraffin content in the oil was higher compared to that if the product composition had changed linearly according to the ratio of plastics, paraffin content sitting at 50% for the 1:1 mixture and at 56% for the 2:1 and 3:1 mixtures. The paraffin content decreased sharply to reach the content in pure PP once PP became the majority of the feedstock, going down to 28% and 8% for 1:2 and 1:3 mixtures, respectively. The cyclic content showed synergy for every ratio of the mixture, going from 12, 14, 19, 28, and 42 wt% for decreasing LDPE:PP ratio of 3:1, 2:1, 1:1, 1:2, and 1:3, respectively. The olefin content remained less than the linear combination scenario for every mixture of LDPE and PP, reaching the biggest disparity at a 1:1 ratio, and staying around 32, 30, 31, 44, and 50 for the same decreasing ratios of LDPE:PP.
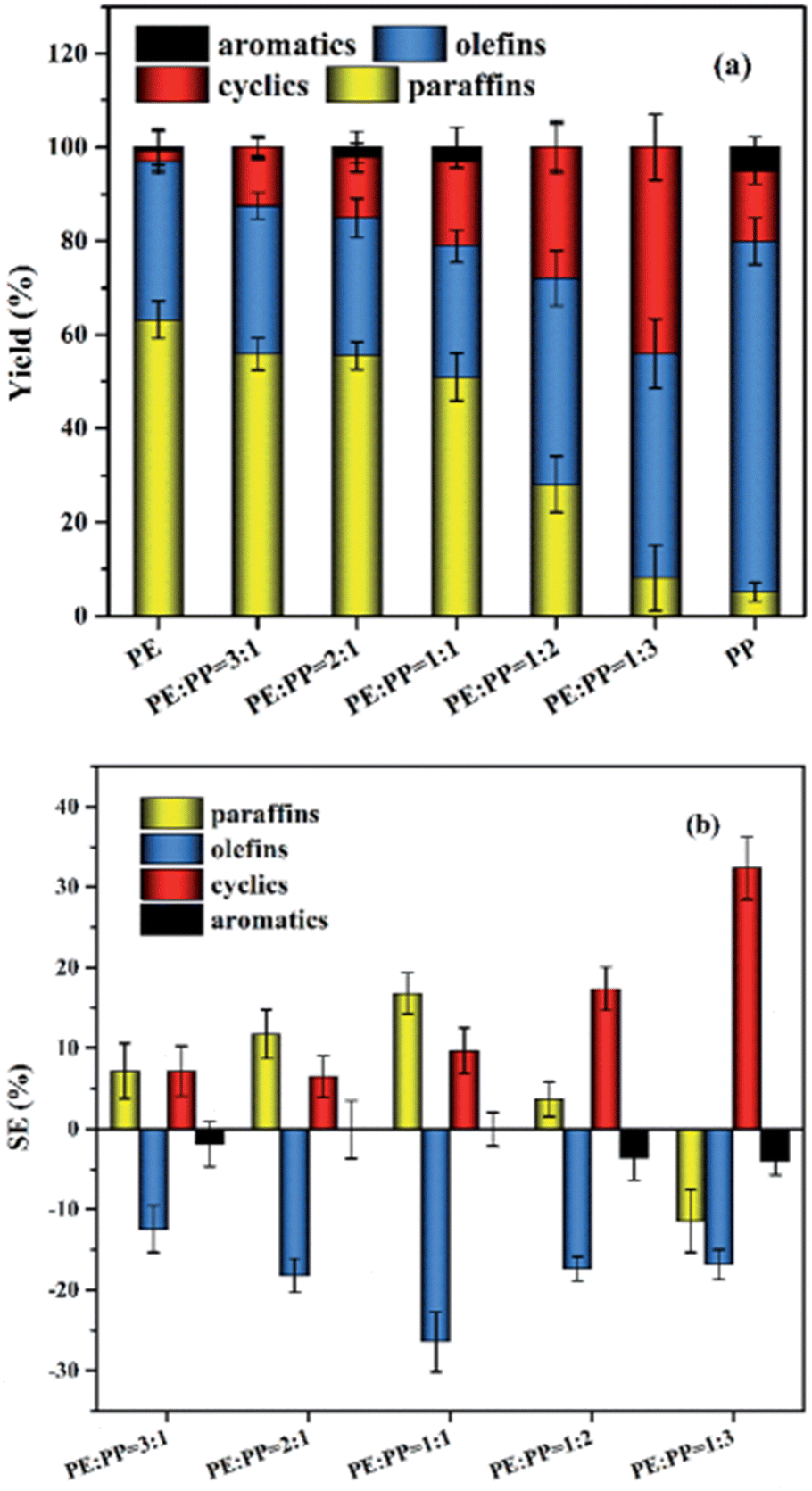 | ||
| Fig. 9 LDPE:PP product distribution (a) and synergy (b).13 | ||
The production of cyclics and paraffins seemed to be promoted by the increasing amount of olefin intermediates during degradation, enough to offset the theoretical decrease of paraffins as more PP is added compared to LDPE, which also would cause the constant lower olefin amounts compared to the theoretical amounts as it would be converted. However, at a 1:3 ratio the offset was not sufficient any longer to overcome the increase of olefin production, due to paraffin and olefin production being competitive within the degradation of a single plastic type and PP starting to dominate the mixture. The cyclics however did not lose this offset, as they are produced directly from olefins and their production is thus not competitive. The paraffin content at 1:3 was below the amount in the linear combination scenario however, indicating that the increased olefin content promoted the conversion of paraffin into cyclics, as the cyclic content sharply decreased once no LDPE was present in the feedstock at all.
Seshasayee et al. also performed an HTL on an equal mixture of PET, PS, PP, and PC, showing significant synergy at 300 °C of 9 wt%, going from 27 to 31.5 wt%, and at 400 °C, showing highly significant synergy of 26 wt%, going from 30 to 37 wt% yield, as shown in Fig. 10. The mixture showed antagonism at 350 °C and 425 °C, going from 35 to 30 wt% and from 38 to 34 wt%, respectively.31 The synergy at 300 °C most likely came from the previously mentioned breakdown of PS at lower temperatures. This would also explain the antagonism at 350 °C, as the oil that is normally produced at that temperature had already been produced at 300 °C, turning more into gas at the higher temperature. The same synergy/antagonism for 400 °C and 425 °C can similarly be explained by the earlier decomposition of PP, though in the binary plastic mixture experiments said synergy with PP was shown at 425 °C. That said, the experiment did not go over the effects at 400 °C, thus a full picture for comparison cannot be made.
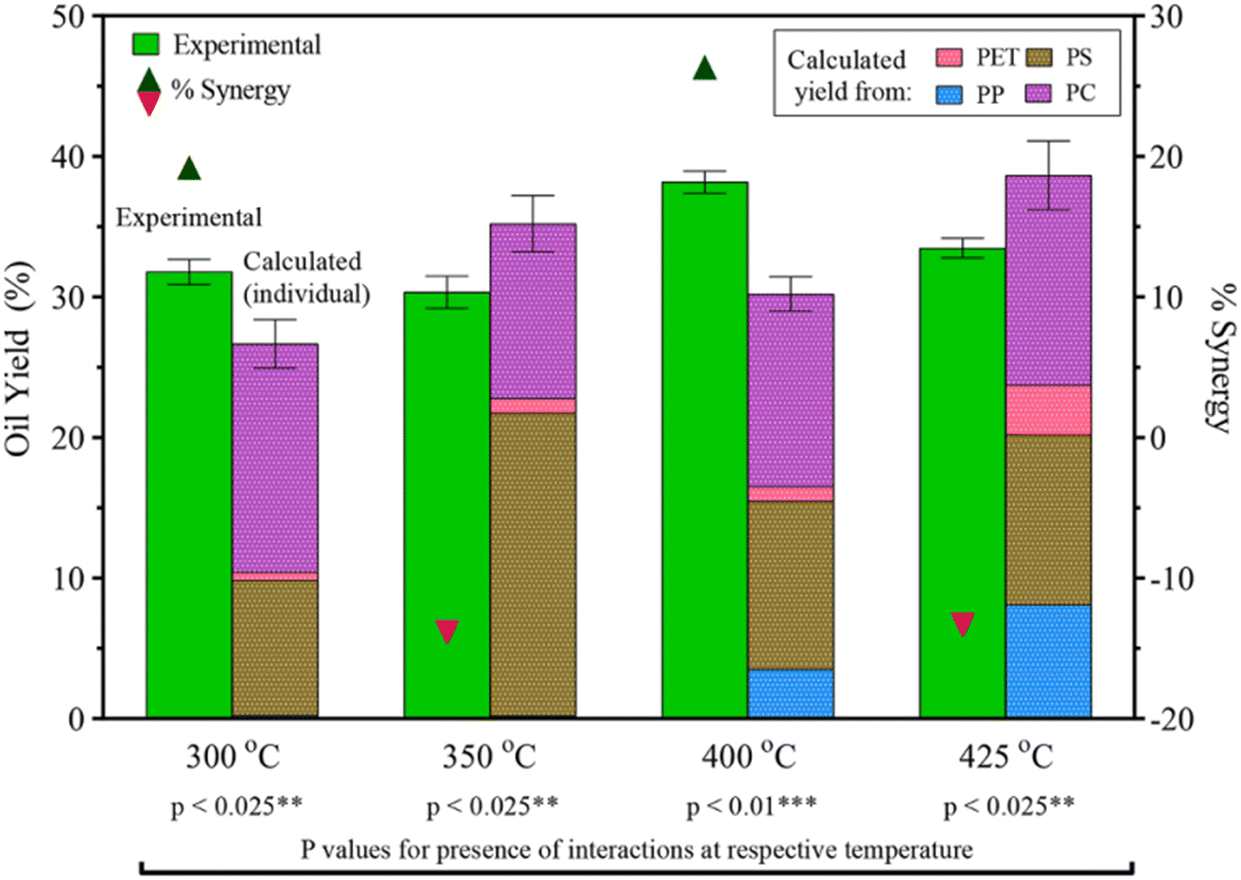 | ||
| Fig. 10 PET, PS, PP, PC mixture HTL yields and synergy.31 | ||
Jin et al. performed two studies with HDPE and PP consumer waste and found highly similar oil yields and composition compared to the breakdown of model HDPE and PP in HTL, except with an additional 6 wt% from inorganic contaminations.8
Sugano et al. also performed HTL on plastic waste in a batch and semi batch type reactor, using their steam explosion pretreatment, and showed decent oil yields of 54% and 66%, respectively, at 350 °C, though there was again more solid compared to model HTLs of PS and HDPE due to insoluble inorganic compounds.11
Chand et al. attempted an HTL of microplastics coming from sewage sludge produced by a wastewater treatment plant in a continuous reactor.33 At 400 °C in SCW the microplastics showed significant degradation. Analysis of the pre- and post-HTL products showed a 97 wt% loss of microplastics, though only a 25 wt% oil yield was found, with most of the decomposed microplastics being found in the solid residue. Analysis of samples at several points in the HTL process and product separation revealed the presence of PE, PP, PVC, PS, and PC as microplastics in the sewage sludge along with other plastics not covered here. The post-HTL samples showed that PE and PP were most resistant to degradation, although they still showed significant weight loss. Besides PE and PP, the other microplastics were all converted in the HTL process, and all the remaining microplastics could be isolated as solid product in the current procedure. Specific masses of plastics could not be determined. Additionally, the average size of the remaining microplastics decreased by the process. The identification process used by the authors was long and complicated, and complex mixtures may have produced unreliable results. As an example, some microplastics that were not detected in certain samples were detected in samples from further down the processing chain, showing that these microplastics were missed by the identification methods in the earlier samples. In general, however, Chand et al. have shown that HTL can be successfully used for the removal and simultaneous recovery of sewage sludge including the fraction of microplastics for bio-crude production of microplastics.
To understand how plastics would decompose in real-life applications of HTL, mixtures of plastics with biomass also need to be studied. Seshasayee et al. also performed several experiments between plastics and model biomolecules present in biomass in the previously mentioned study, said biomolecules being cellulose, starch, soy protein, (de)alkaline lignin, and stearic acid.31 As shown in Fig. 11, they reported a significant synergy with the model waste feedstock of the full plastic mix, consisting of PET, PS, PP, and PC, with the biomolecules at 300 °C, giving a yield of 14 wt% higher than the value calculated from individual plastic and model biomolecule yields, reaching 32 wt%. There is a 10.6 wt% difference with the calculated value from the full plastic mix and separate biomolecule mix added together, which also showed synergy compared to their individual mixes. Starting with 400 °C, there is no synergy present in the full model waste feedstock. In fact, supercritical temperatures show antagonism, while subcritical temperatures show synergy. HTL at 425 °C showed a 19 wt% antagonism, the yield going from a calculated 18 to 15 wt%.
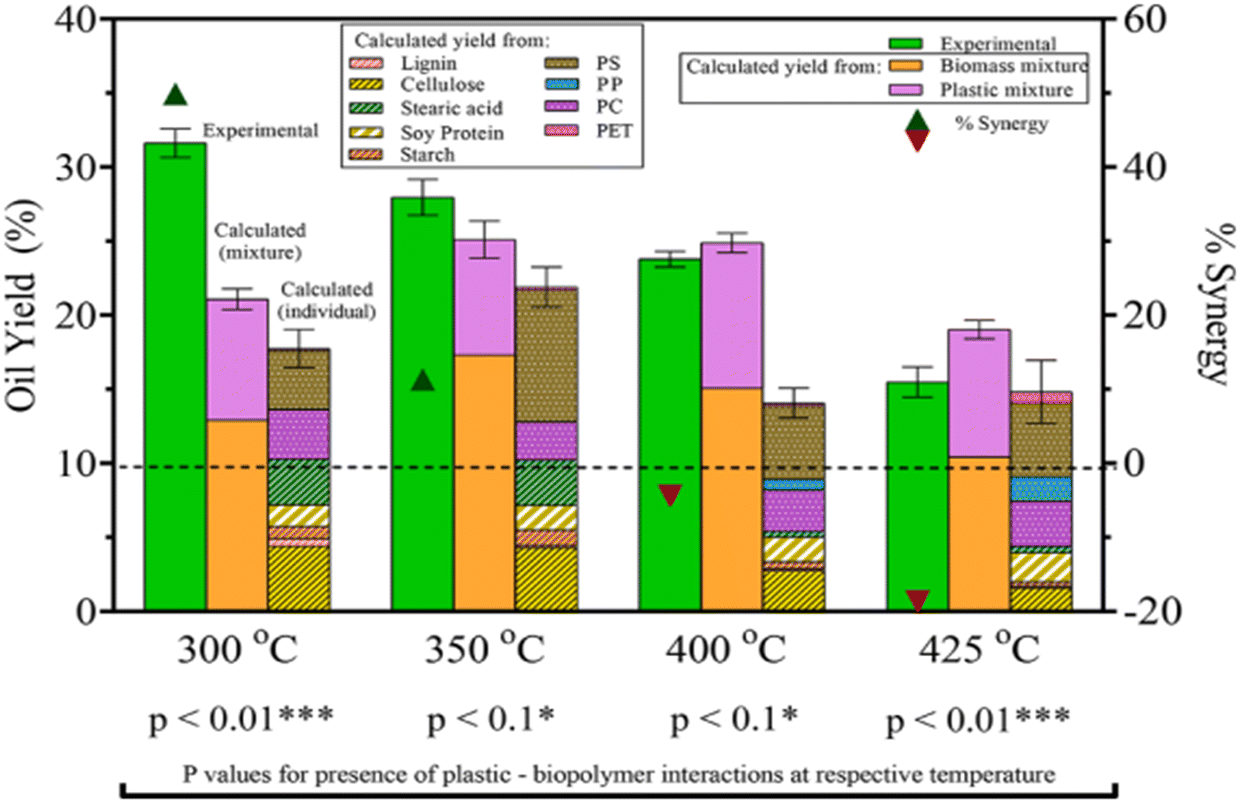 | ||
| Fig. 11 Biomass–plastic HTL yields and synergy. The combined HTL yields are higher than the calculated values for both mixtures and individual components.31 | ||
Seshasayee et al. concluded that the addition of both plastics that decompose at lower temperatures, like PC and PET, and biomass encourages the decomposition of polyolefins at much lower, subcritical temperatures, which then encourages further decomposition of other compounds. Further experiments between mixtures of individual biomolecules and plastic mix showed that all biomolecules except stearic acid contributed to this synergy at 300 °C, as shown in Fig. 12. For the mixtures of individual plastics (PET, PS, PP, and PC) with the biomass, all showed synergy, though PS gave the biggest effect, going from 22 to 46 wt%.
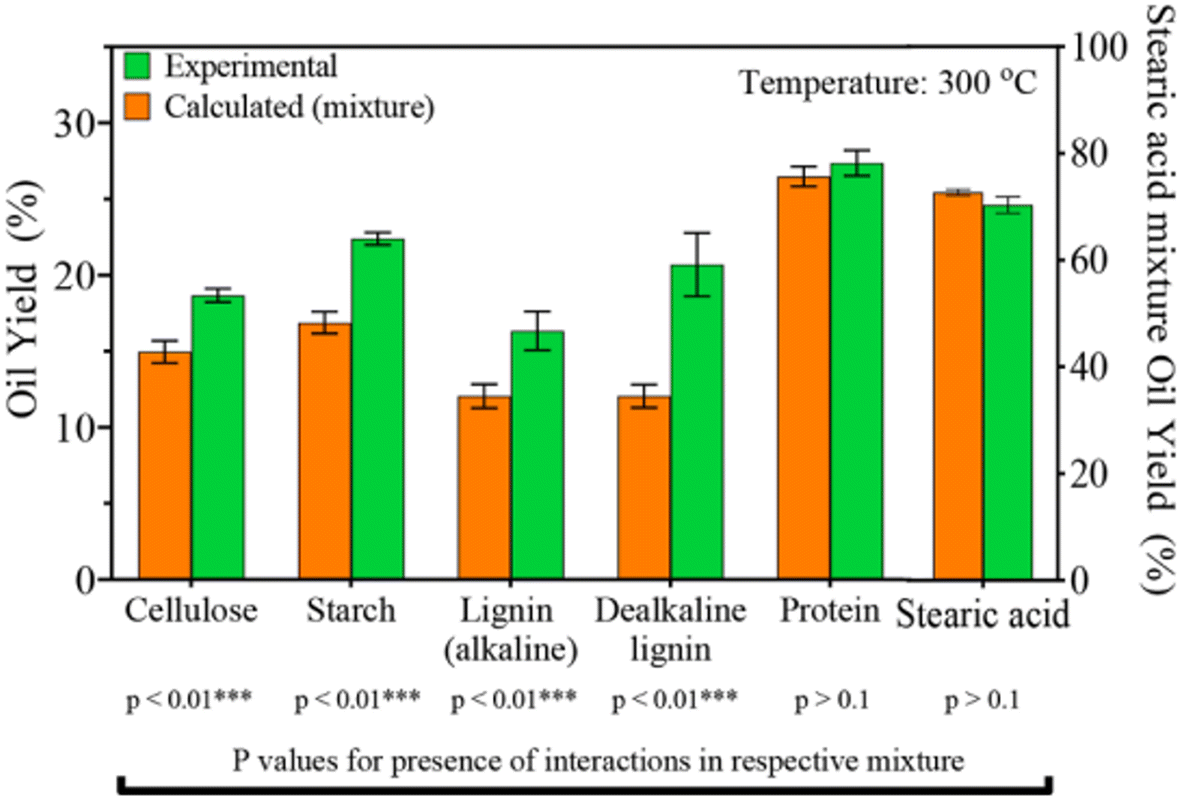 | ||
| Fig. 12 Synergy of plastic mix and biomolecules. Oil yields from HTL of mixtures of single biomolecule and plastics mixture. Anticipated yields are also shown.31 | ||
An HTL at 300 °C for 30 minutes with store-bought PS foam mixed with a blended mix of food waste, containing green beans, baked beans, chicken, potato salad, and parmesan cheese, was done to demonstrate a similar synergy with simulated food waste instead of model biomass. This gave a 76 wt% oil yield for a 4:1 PS to food waste mix, providing an additional 46 wt% synergy from a theoretical scenario where both components were liquefied separately.
Ciuffi et al. collected waste directly from a recycling plant, containing cellulose from paper, small amounts of metal, and plastics PET, PS, PVC, PE, and PP.34 HTL at 340 °C for 5 hours yielded a 7.7 wt% oil yield and an 11 wt% gas yield, with the remaining product being solid. Analysis of the oil showed significant PET and some PS degradation, but no decomposition products of the polyolefins PE and PP. No specific amounts were reported. Cellulose degradation was also seen. The aqueous phase also indicated a high dechlorination of the PVC present, though products of further PVC decomposition were not detected. That said, only a quarter of the oil products were successfully identified using GC-MS and GC-FID methods. The presence of a significant amount of chlorine atoms in the aqueous phase as well as the low oil yields might explain a significantly lower oil yield compared to the results of Seshasayee et al., as an acidic environment caused by PVC dechlorination may have encouraged charring and the production of solid products.
Previous research mainly studied the effect of temperatures and reaction times on the oil yields. Variation of other parameters such as pressure, water-to-plastic ratio, water fill rate, and heating rate were not given much attention. The pressures employed were around the vapor saturation curve or were not given in the papers. The heating rates were not varied within a study, though higher rates would allow for shorter reaction times prior to reaching the desired reaction temperature. The higher heat conducting coating may allow higher heating rates. Higher water fill rates may affect pressure deviations from the standard vapor saturation curve. The flowing water in both semi-batch and continuous reactors may quench primary radicals. The influence of mechanical stirring is not researched; however, Sugano et al. speculate that partial decomposition of plastics occurs through mechanochemical scission, as shown in their steam explosion pretreatment.
The polyolefins primarily decomposed into paraffins, olefins, cyclics, and aromatics, though olefins, cyclics, and aromatics formed an irreversible pathway. The ratio of paraffin to olefin hydrocarbons depended on the presence of side chains in the polymer, with PP and (HI)PS having a significantly larger olefin-to-paraffin ratio than HDPE and LDPE. PVC firstly dechlorinated, yielding a highly acidic aqueous phase of hydrochloric acid, and then decomposed into mostly olefins due to the dehydrochlorinations, which were converted into cyclics and aromatics mostly. The condensation polymers however were mainly converted into their original monomers thanks to the easily breakable oxygen bonds in the main chain and the more stable benzene rings. PET decomposed into TA at all temperatures, which became solid product, and ethylene glycol, which turned into small hydrocarbons. Most of the TA could be recovered through additional separation steps with the solid residue. PC yielded BPA, which in turn decomposed into a wide variety of phenols. A general overview of the decomposition pathways and lowest optimal temperature for the covered plastic types is shown in Scheme 9.
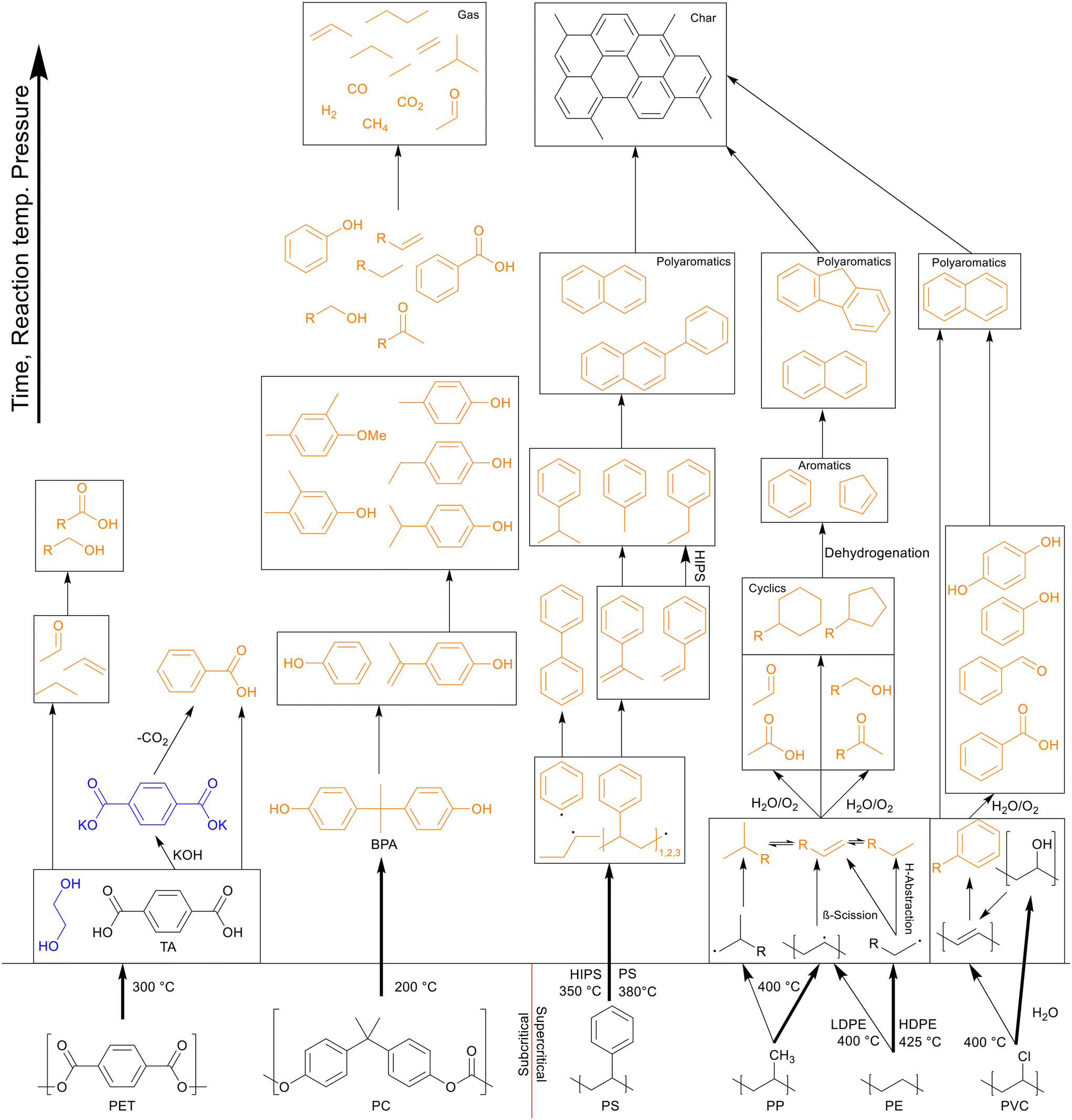 | ||
| Scheme 9 Overview of decomposition pathways and lowest optimal HTL temperatures for most common plastics. Black: solid, orange: liquid, blue: purely present in water. Reaction temperature, pressure and time increases from the bottom to the top of the figure; the decomposition pathways follow the same direction. | ||
Co-liquefaction of binary plastic mixtures revealed that polyolefins could be broken down at lower temperatures if more easily degradable plastics like PET and PC were present, with PS breaking down at 300 °C and PE and PP breaking down at 350 °C. The breakdown of the polyolefins would then further promote decomposition of the more easily degradable plastics. Co-liquefaction with PVC would result in unwanted charring through acidification of the solvent, which cannot be easily countered and should be avoided when possible. Liquefaction of an equal mass plastic mix containing PET, PC, PS, and PP with a model biomass mixture gave the highest synergy and yield at the low temperature of 300 °C, which is suitable for practical HTL application. Further experiments with plastic and biomass mixtures showed that PS was the largest contributor to the synergy.
Showing the versatility of liquefaction, the HTL of single plastic types from waste showed that decomposition and oil yields stayed mostly the same, though the inorganic impurities showed up as additional solid product. HTL of PS foam with actual food waste also showed similar high synergy to that observed in the HTL of plastics and model biomass. The application of HTL on microplastics from sewage sludge showed a reasonable conversion, with 25 wt% of the microplastics turning into oil and 97 wt% of the microplastics being converted into hydrocarbons in total, removing a significant amount of the microplastics from the waste. Several techniques are being developed for the chemical recycling of carbon-containing wastes and liquefaction is definitely a promising technique among them.35–37
Conclusions and outlook
In summary, we compared several studies to assess the optimal conditions for the chemical recycling of plastics by HTL. Even though studies on pure plastics are limited, a few general trends can be spotted, ignoring the occasional outlier. The polyolefins produced maximum oil yield in supercritical temperatures: HDPE and LDPE from 425 and 400 °C onwards, respectively, while PP performed best at 400 °C and up. PS, which is less thermally stable than PP and PE, peaked in a narrow range of 380 and 400 °C, while HIPS liquefied the most at a lower temperature of 350 °C. Since PP and PE have nonpolar bonds, the scw and SCW would be immiscible and the effect of HTL would be at the interface. Since condensation polymers such as PET and PC have heteroatoms in the main chain, the miscibility with scw or SCW is better and subcritical temperatures are sufficient to obtain oil. PC gives oil over a wide range of 200 to 400 °C, while PET broke down to TA monomers at high yields within the temperature range. PVC gave low oil yields at subcritical temperatures, and supercritical studies did not focus on oil yields. It should also be noted that unlike that of polyolefin plastics, the HTL of aromatic polymers such as PET, PS, and PC is rather focused on the production and recovery of their original monomers than the production of oily products.Overall, HTL of the most common plastics with typical biomass under milder conditions in subcritical water gave the highest oil yields. The plastics that degraded well under subcritical conditions promoted further degradation of those that normally only degrade well under supercritical conditions. Synergy occurs when mixing PP and PE with PET, which could play the role of phase transfer agent. PVC in the feedstock is an issue, as the optimal reaction temperature of 300 °C is enough for dechlorination and subsequent acidification, which encourages charring. The produced oils were typically energy rich and can be used both as a fuel source after further modification and as resources for resynthesizing virgin plastics.
The application of HTL towards the chemical recycling of plastics is only recent compared to HTL of biomass. As such, most studies that use plastics in HTL feedstock do so in conjunction with biomass. There should be more studies on the liquefaction of pure plastic or mixtures of plastic. The studies should also be comprehensive as not only temperatures and pressures but also other parameters such as water-to-feedstock ratio, reactor type, casing, purging, volume, stirring etc. could have an influence on the reaction output. Elemental and molecular analysis of oils is very important and missing in many cases. It defines the further applications of each oil. The gaseous components, char formation, and aqueous fraction and its treatments also need to be investigated further.
Abbreviations
| HTL | Hydrothermal liquefaction |
| HTC | Hydrothermal carbonization |
| HTG | Hydrothermal gasification |
| HDPE | High-density polyethylene |
| LDPE | Low-density polyethylene |
| PP | Polypropylene |
| PP&A | Polyester, polyamide, and acrylic polymers |
| PS | Polystyrene |
| HIPS | High-impact polystyrene |
| PVC | Polyvinyl chloride |
| PC | Polycarbonate |
| PET | Polyethylene terephthalate |
| TA | Terephthalic acid |
| EG | Ethylene glycol |
| BPA | Bisphenol A |
| scw | Subcritical water |
| SCW | Supercritical water |
| wt% | Weight percentage |
| HHV | Higher heating value |
| PE | Polyethylene |
| DCM | Dichloromethane |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon Europe Research and Innovation Programme under Grant Agreement No. 101058540 (PLASTICE Project, https://plastice.eu/).References
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- R. Yang, J. Kalsoom, C. Chen, W. Chen and K. C. Wu, Thermochemical Conversion of Plastic Waste into Fuels, Chemicals, and Value-Added Materials: A Critical Review and Outlooks, ChemSusChem, 2022, 15(11), e202200171 CrossRef CAS PubMed.
- D. Lachos-Perez, P. C. Torres-Mayanga, E. R. Abaide, G. L. Zabot and F. de Castilhos, Hydrothermal carbonization and Liquefaction: differences, progress, challenges, and opportunities, Bioresour. Technol., 2022, 343, e126084 CrossRef PubMed.
- K. P. Gopinath, V. M. Nagarajan, A. Krishnan and R. Malolan, A critical review on the influence of energy, environmental and economic factors on various processes used to handle and recycle plastic wastes: Development of a comprehensive index, J. Cleaner Prod., 2020, 274, e123031 CrossRef.
- O. V. Okoro and F. D. Faloye, Comparative Assessment of Thermo-Syngas Fermentative and Liquefaction Technologies as Waste Plastics Repurposing Strategies, AgriEngineering, 2020, 2(3), 378–392 CrossRef CAS.
- T. Moriya and H. Enomoto, Characteristics of polyethylene cracking in supercritical water compared to thermal cracking, Polym. Degrad. Stab., 1999, 65(3), 373–386 CrossRef CAS.
- K. Jin, P. Vozka, G. Kilaz, W. Chen and N. L. W. Wang, Conversion of polyethylene waste into clean fuels and waxes via hydrothermal processing (HTP), Fuel, 2020, 273, e117726 CrossRef.
- K. Jin, P. Vozka, C. Gentilcore, G. Kilaz and N. L. W. Wang, Low-pressure hydrothermal processing of mixed polyolefin wastes into clean fuels, Fuel, 2021, 294, e120505 CrossRef.
- X. Su, Y. Zhao, R. Zhang and J. Bi, Investigation on degradation of polyethylene to oils in supercritical water, Fuel Process. Technol., 2004, 85, 1249–1258 CrossRef CAS.
- J. S. dos Passos, M. Glasius and P. Biller, Screening of common synthetic polymers for depolymerization by subcritical hydrothermal liquefaction, Process Saf. Environ. Prot., 2020, 139, 371–379 CrossRef CAS.
- M. Sugano, A. Komatsu, M. Yamamoto, M. Kumagai, T. Shimizu, K. Hirano and K. Mashimo, Liquefaction process for a hydrothermally treated waste mixture containing plastics, J. Mater. Cycles Waste Manage., 2009, 11, 27–31 CrossRef CAS.
- H. Zhang, X. Su, D. Sun, R. Zhang and J. Bi, Investigation on degradation of polyethylene to oil in a continuous supercritical water reactor, J. Fuel Chem. Technol., 2007, 35(4), 487–491 CrossRef CAS.
- P. Zhao, Z. Yuan, J. Zhang, X. Song, C. Wang, Q. Guo and A. J. Ragauskas, Supercritical water co-liquefaction of LLDPE and PP into oil: properties and synergy, Sustainable Energy Fuels, 2021, 2, 575–583 RSC.
- M. Čolnik, P. Kotnik, Ž. Knez and M. Škerget, Hydrothermal decomposition of polyethylene waste to hydrocarbons rich oil, J. Supercrit. Fluids, 2021, 169, e105136 CrossRef.
- S. Wong, N. Ngadi, N. Amin, T. Abdullah and I. Inuwa, Pyrolysis of low density polyethylene waste in subcritical water optimized by response surface methodology, Environ. Technol., 2015, 37(2), 245–254 CrossRef.
- X. Zhao, L. Zhan, B. Xie and B. Gao, Products derived from waste plastics (PC, HIPS, ABS, PP and PA6) via hydrothermal treatment: Characterization and potential applications, Chemosphere, 2018, 207, 742–752 CrossRef CAS PubMed.
- M. S. Seshasayee and P. E. Savage, Oil from plastic via hydrothermal liquefaction: Production and characterization, Appl. Energy, 2020, 278, e115673 CrossRef.
- W. Chen, K. Jin and N. L. Wang, Use of Supercritical Water for the Liquefaction of Polypropylene into Oil, ACS Sustainable Chem. Eng., 2019, 7(4), 3749–3758 CrossRef CAS.
- H. Kwak, H. Shin, S. Bae and H. Kumazawa, Characteristics and kinetics of degradation of polystyrene in supercritical water, J. Appl. Polym. Sci., 2006, 101(1), 695–700 CrossRef CAS.
- X. Zhao, Y. Xia, L. Zhan, B. Xie, B. Gao and J. Wang, Hydrothermal Treatment of E-Waste Plastics for Tertiary Recycling: Product Slate and Decomposition Mechanisms, ACS Sustainable Chem. Eng., 2019, 7(1), 1464–1473 CrossRef CAS.
- B. Bai, H. Jin, C. Fan, C. Cao, W. Wei and W. Cao, Experimental investigation on liquefaction of plastic waste to oil in supercritical water, J. Waste Manage., 2019, 89, 247–253 CrossRef CAS.
- Y. Takeshita, K. Kato, K. Takahashi, Y. Sato and S. Nishi, Basic study on treatment of waste polyvinyl chloride plastics by hydrothermal decomposition in subcritical and supercritical regions, J. Supercrit. Fluids, 2004, 31, 185–193 CrossRef CAS.
- G. Gandon-Ros, A. Soler, I. Aracil and M. F. Gómez-Rico, Dechlorination of polyvinyl chloride electric wires by hydrothermal treatment using K2CO3 in subcritical water, Waste Manage., 2020, 102, 204–211 CrossRef CAS PubMed.
- Y. Sato, K. Kato, Y. Takeshita, K. Takahashi and S. Nishi, Decomposition of Polyvinylchloride using Supercritical Water, Jpn. J. Appl. Phys., 1998, 37, 6270–6271 CrossRef CAS.
- Y. Nagai, R. L. Smith Jr., H. Inomata and K. Arai, Direct Observation of Polyvinylchloride Degradation in Water at Temperatures up to 500 °C and at Pressures up to 700 MPa, J. Appl. Polym. Sci., 2007, 106, 1075–1086 CrossRef CAS.
- Y. Liu, M. Wang and Z. Pan, Catalytic depolymerization of polyethylene terephthalate in hot compressed water, J. Supercrit. Fluids, 2012, 62, 226–231 CrossRef CAS.
- R. Darzi, Y. Dubowski and R. Posmanik, Hydrothermal processing of polyethylene terephthalate and nylon-6 mixture as a plastic waste upcycling treatment: A comprehensive multi-phase analysis, J. Waste Manage., 2022, 143, 223–231 CrossRef CAS PubMed.
- T. H. Pedersen and F. Conti, Improving the circular economy via hydrothermal processing of high-density waste plastics, J. Waste Manage., 2017, 68, 24–31 CrossRef PubMed.
- Y. Huang, S. Liu and Z. Pan, Effects of plastic additives on depolymerization of polycarbonate in sub-critical water, Polym. Degrad. Stab., 2011, 96(8), 1405–1410 CrossRef CAS.
- H. Jin, B. Bai, W. Wei, Y. Chen, Z. Ge and J. Shi, Hydrothermal Liquefaction of Polycarbonate (PC) Plastics in Sub-/Supercritical Water and Reaction Pathway Exploration, ACS Sustainable Chem. Eng., 2020, 8(18), 7039–7050 CrossRef CAS.
- M. S. Seshasayee and P. E. Savage, Synergistic interactions during hydrothermal liquefaction of plastics and biomolecules, Chem. Eng. J., 2021, 417, e129268 CrossRef.
- M. S. Seshasayee, R. Stofanak and P. E. Savage, Component additivity model for plastics-biomass mixtures during hydrothermal liquefaction in sub-, near-, and supercritical water, iScience, 2021, 24(12), e103498 CrossRef PubMed.
- R. Chand, K. Kohansal, S. Toor, T. H. Pedersen and J. Vollertsen, J. Cleaner Prod., 2022, 335, e130383 CrossRef.
- B. Ciuffi, L. Rosi, E. Miliotti, G. Lotti, A. M. Rizzo and D. Chiaramonti, Batch Hydrothermal liquefaction of end-of-life plastic and oil characterization, E3S Web Conf., 2021, 238, e08004 Search PubMed.
- A. K. Panda, A. Alotaibi, I. V. Kozhevnikov and N. R. Shiju, Pyrolysis of Plastics to Liquid Fuel Using Sulphated Zirconium Hydroxide Catalyst, Waste Biomass Valorization, 2020, 11, 6337–6345 CrossRef CAS.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, Chemical Recycling of Used PET by Glycolysis Using Niobia-Based Catalysts, ACS Eng. Au, 2023, 3(1), 37–44 CrossRef CAS PubMed.
- J. Mors and N. R. Shiju, The synthesis of biooil using ambient pressure liquefaction of organic waste, Sustainable Chemistry for Climate Action, 2023, 2, 100013 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00510g |
| This journal is © The Royal Society of Chemistry 2024 |