
Polymer characterization by size-exclusion chromatography with multi-angle light scattering (SEC-MALS): a tutorial review
John B.
Matson
 *,
Anna Q.
Steele
,
Jonathan D.
Mase
and
Michael D.
Schulz
*
*,
Anna Q.
Steele
,
Jonathan D.
Mase
and
Michael D.
Schulz
*
Virginia Tech, Department of Chemistry and Macromolecules Innovation Institute, USA. E-mail: jbmatson@vt.edu; mdschulz@vt.edu
First published on 19th December 2023
Abstract
This tutorial review presents the theory and application of SEC-MALS with minimal equations and a focus on synthetic polymer characterization, serving as an entry point for polymer scientists who want to learn more about SEC-MALS. We discuss the principles of static light scattering, outline its capability to generate absolute weight-average molar mass values, and extend its application to SEC-MALS. Practical elements are emphasized, enabling researchers to appreciate how values for Mn, Mw, and Đ are determined in an SEC-MALS experiment and how experimental conditions and input values, such as the specific refractive index increment (dn/dc), influence the results. Several illustrative SEC-MALS experiments demonstrate the impact of separation quality on Mn (as opposed to Mw), the appearance of contaminants in SEC chromatograms from sample preparation, the influence of concentration on data quality, and how polymer topology affects molecular weight characterization in SEC. Finally, we address practical considerations, common issues, and persistent misconceptions.
Introduction
Molecular weight is among the most fundamental properties of a polymer. As such, polymer chemists have pursued numerous synthetic approaches to achieve control over this critical parameter. Synthetic methods now enable production of many types of polymers, often with precise control over molecular weight (aka, molar mass). Equally important is the molecular weight distribution, often described by the ratio of the weight-average molar mass (Mw) to the number-average molar mass (Mn), i.e., Mw/Mn, now commonly called dispersity (symbol Đ). Achieving a low Đ value (as close to 1.00 as possible) was a major focus in polymer synthesis over the past few decades,1–5 and tuning dispersity to a value of choice has recently become an active area of research.6–12 While modern synthetic methods offer ways to control both molecular weight and dispersity, determining the outcome of any polymer synthesis requires accurate characterization of these values to fully understand the advantages and limits of new synthetic methods.For decades, size-exclusion chromatography (SEC) has served as a polymer chemist's primary tool for characterizing molecular weight and molecular weight distribution. Historically, and even still in many labs, SEC experiments are run in comparison to several well-defined linear polymer standards, with molecular weights reported based on these chromatographic calibrations. While this approach has a long and generally successful history, it suffers from several limitations. Among these limitations is its inability to provide absolute molecular weight measurements for samples that differ in chemical structure from the standards or have a non-linear topology (architecture), producing instead only apparent molecular weight data (relative to linear standards) for the vast majority of samples. While corrections using Mark–Houwink–Sakurada (MHS) parameters (discussed below) can be applied in some cases, data accuracy depends on several factors, and MHS parameters are available only for limited homopolymers and a few copolymers.13 Consequently, other approaches for determining molecular weight have been integrated into SEC experiments over the years.
Static light scattering (SLS) has emerged as the most reliable and readily available approach to determining absolute molecular weights of polymers. This technique can be integrated into SEC experiments via in-line multi-angle light scattering (MALS) detectors. While traditional SEC experiments rely on accurate calibration and provide molecular weight data based on calibrated elution time values, SEC-MALS eliminates this requirement by measuring Mw directly. Information on dispersity and trace modality is still determined chromatographically. Thus, the combined SEC-MALS experiment is a powerful approach to fully and accurately characterize novel polymers in terms of molecular weight, molecular weight distribution, and other parameters. However, additional techniques are required to determine other key structural features, such as tacticity, topology, and comonomer content.
Characterization of polymers using SLS has a long and rich history, and the topic has been previously reviewed in considerable depth. For readers interested in the rigorous mathematical treatment of light scattering, excellent articles by Wyatt14 and Podzimek15 detail these elements of light scattering theory, as do Rubinstein and Colby in their textbook.16 In this tutorial review, we provide a conceptual basis with minimal equations to explain the theory and application of SEC-MALS to polymer characterization. We focus on practical elements with the goal of enabling researchers to understand how values for Mw, Mn, and Đ are derived in an SEC-MALS experiment and how input values (e.g., concentration, dn/dc) and experimental conditions (e.g., solvent, temperature) influence the results. Finally, we dispel some common misconceptions about light scattering and SEC-MALS. Many of these same principles also apply to protein characterization by SEC-MALS, but here we focus on synthetic polymers. Overall, we hope that this article will serve as a useful resource for polymer scientists who use SEC-MALS, on occasion or every day, and that it will illuminate the inner workings of this critical tool for polymer characterization.
Background & theory
We experience light scattering in our everyday lives when we look at the sky. As sunlight passes through the atmosphere, small particles (atmospheric gasses) scatter light with a wavelength dependence of 1/λ4, i.e., the shorter wavelengths scatter more. Because the solar spectrum has a greater contribution from blue light than violet light, and because our eyes are more sensitive to blue than violet, we perceive the sky as blue during the day. Sunrises and sunsets appear red because the light passes through more of the atmosphere than when the sun is overhead, so we observe more of the light that is not scattered, i.e., the longer wavelengths. Lord Rayleigh (aka, John William Strutt) was the first to explain this phenomenon in the late 19th century, from which he developed the field of light scattering, where some parameters bear his name (e.g., Rayleigh ratio, excess Rayleigh scattering).17–19 Various articles explain the phenomenon of atmospheric light scattering and the history of these discoveries in both lay terms and with considerable mathematical treatment.20,21Rayleigh's discoveries of the principles of light scattering in the atmosphere can also be applied to particles in solution, and these principles underpin the technologies that enable molecular weight characterization experiments by SLS, the type of solution light scattering used in SEC-MALS. MALS can be conducted either in batch-mode (e.g., on a solution of polymer in a vial) or in-line with an SEC instrument (SEC-MALS). In the context of MALS of polymer solutions, batch-mode experiments illustrate the basic concepts of the technique.
In a batch-mode MALS experiment, a glass vial containing a polymer solution is inserted into a MALS instrument with two or more SLS detectors. Critically, no chromatographic separation is applied to the polymer sample in a batch-mode MALS experiment so Mn and Đ are not determined, but batch-mode experiments can still yield valuable data. Specifically, these experiments provide the Mw of the polymer sample, without the need for routine calibration with standards of known Mw. With multiple detectors spaced at different angles, the angular dependence of light scattering can also provide information on the root mean-square radius of gyration (Rg) and the second virial coefficient (A2, a measure of polymer–solvent interactions). Molecules are in motion in all solutions, and in MALS experiments the measurement time is much longer than the rapid fluctuations of the particles. Time-dependent light-scattering measurements at faster timescales use these rapid fluctuations to measure diffusion coefficients and hydrodynamic radius—a technique called dynamic light scattering (DLS). We focus here on SLS because it is the method of light scattering used in SEC-MALS, but we refer the reader to several excellent reviews on DLS.22,23
Simplified for the sake of clarity, the basic light scattering equation for a polymer solution that scatters light equally in all directions (i.e., an isotropic scatterer, typically a polymer or particle of Rg < 10 nm) is as follows:
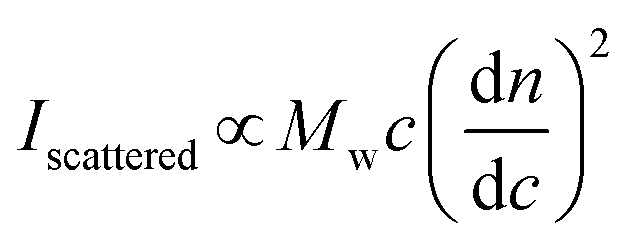 | (1) |
The terms Mn, Mw, and Đ are quite familiar to polymer chemists, but a brief reminder of the equations that define these terms is in order. These values are defined as described in eqn (2)–(4):
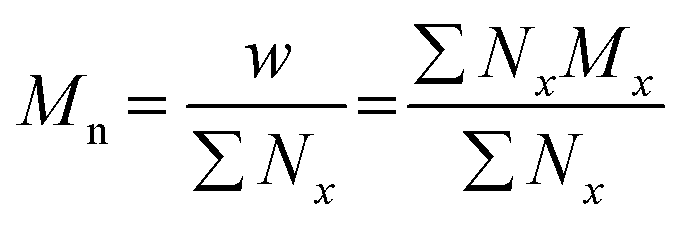 | (2) |
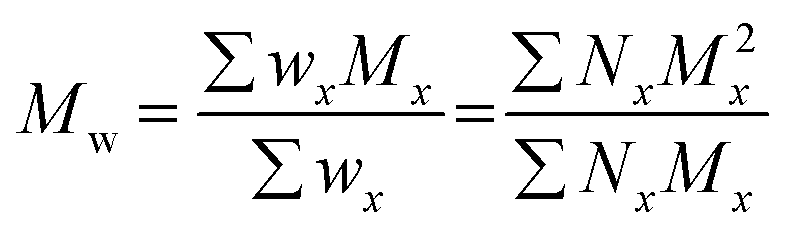 | (3) |
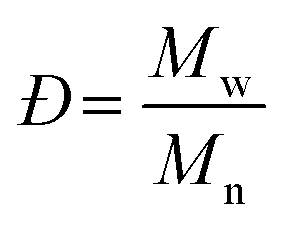 | (4) |
In eqn (2), w represents the total mass of a polymer sample (in units of g) and Nx is the number of macromolecules (in units of mol) of molar mass Mx. The sum of all values of Nx is simply the total moles in the polymer sample, and the units for Mn are g mol−1. Mn is commonly defined using either of the two summations shown in eqn (2). Mw also has units of g mol−1 and is defined as the fraction of two summations, where either of two forms is typically used (eqn (3)). Here, wx is the weight (in units of g) of macromolecules of molar mass Mx. Thus, Mn is a simple average of the molar masses of all polymer chains, whereas Mw gives greater weight to polymer chains with higher molar mass. The Mw/Mn ratio is dispersity (Đ), a measure of the breadth of the molar mass distribution.24 This parameter was formerly called the polydispersity index (PDI). We refer the reader to a classic textbook by Odian for a more detailed discussion on the derivation and physical meanings of Mn, Mw, and Đ.25
In a batch-mode MALS experiment, the Iscattered value is measured by the detectors, and the concentration of the polymer (c, in g mL−1) is determined by the experimenter when preparing the sample. The molecular weight value that a MALS detector measures is Mw, not Mn, because SLS depends on the mass concentration of particles, not the molar concentration. For a detailed explanation of the physics behind this phenomenon, we recommend Polymer Physics by Rubinstein and Colby (chapter 1),16 or a landmark 1948 paper on the topic by Zimm.26,27 Finally, the remaining key variable is the dn/dc value, which can be determined in several ways. The physical meaning of the dn/dc value, as well as how to measure it and its importance in the light scattering equation, is discussed in detail below.
The dn/dc value
When light travels from one medium into another, for example from air into water, the path of the light bends (refracts). The magnitude of this refraction depends on the specific media and the wavelength of light—this phenomenon explains why we see rainbows when light is refracted through water droplets in the air. The refractive index, n, describes both the degree to which the path of the light bends and the change in the speed of light as it travels through different media. For MALS of polymer solutions, the n value of the solution depends on the concentration of the solute, i.e., the polymer. The specific refractive index increment (the dn/dc value) describes the change in the refractive index (dn) of a solution with changing concentration of the solute (dc). In practice, the detector measures the differential refractive index (dRI)—the n value of a polymer solution divided by the n value of the solvent—which describes the deviation in n of a polymer solution from pure solvent. Put simply, the dn/dc of any solute dissolved in a solvent is the slope of the line generated when measuring the dRI of the solution at various concentrations of the solute (Fig. 1A).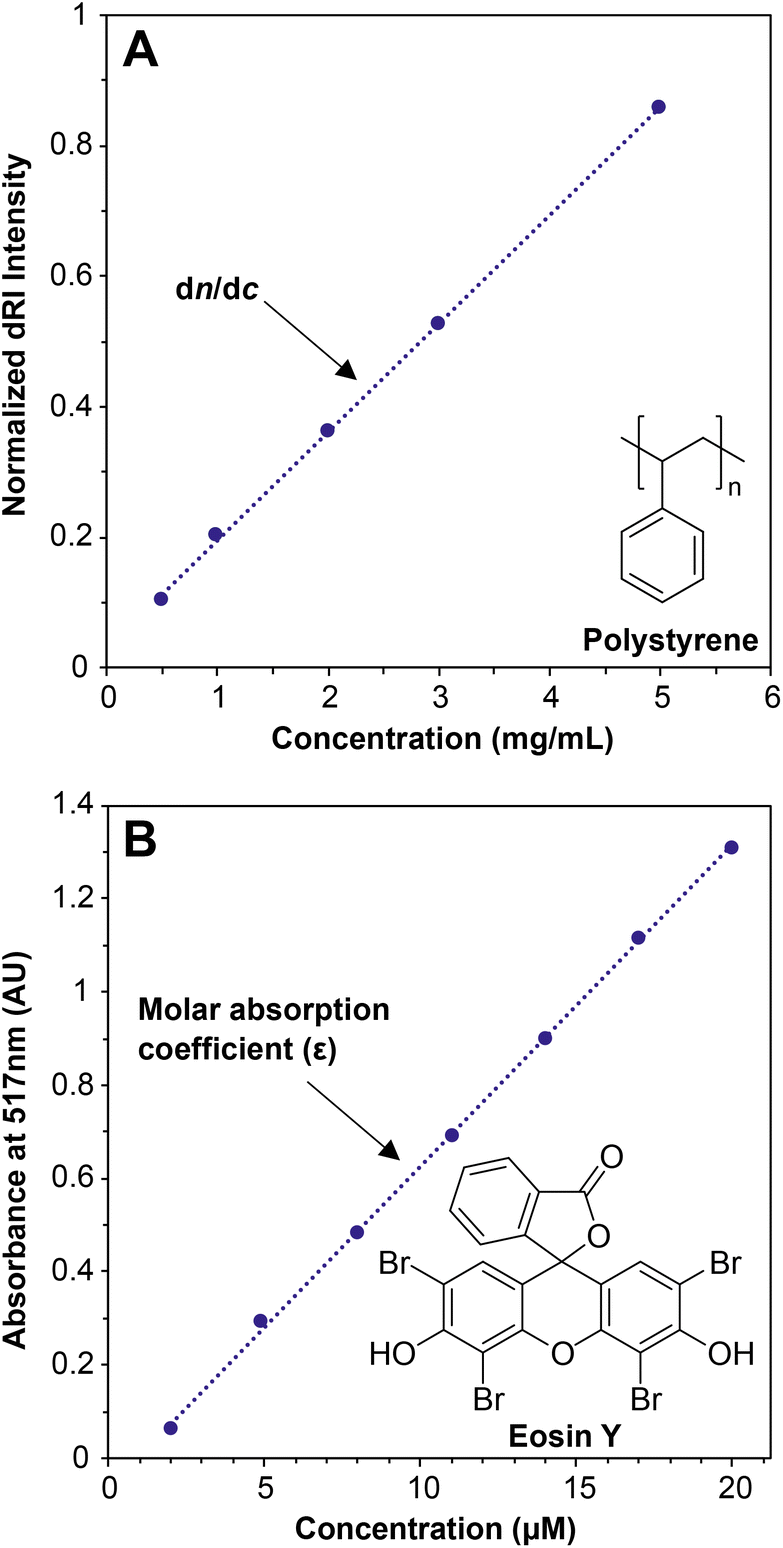 | ||
| Fig. 1 (A) Offline dn/dc experiment using various concentrations of a nominal 30 kg mol−1 polystyrene (PS) standard in THF, where the slope is equal to the dn/dc; (B) UV-vis experiment using various concentrations of eosin Y in water, where the slope is equal to the molar absorption coefficient (ε). | ||
The dn/dc value is quite similar conceptually to the molar absorption coefficient (previously called the molar extinction coefficient28) of a solute in UV-vis spectroscopy, which is determined by preparing a graph of absorption versus concentration at a given wavelength and measuring the slope (Fig. 1B). In a UV-vis experiment, more light is absorbed by the solute at a given wavelength as the concentration increases. At concentrations below which any inter-solute interactions occur, a line can be drawn through all of the points with a good fit, and the slope is the molar absorption coefficient. The dn/dc value of a polymer in a given solvent can be obtained in a similar experiment by measuring the dRI values of polymer solutions at various concentrations; the slope of the line connecting the points is the dn/dc, in units of mL g−1. Fortunately, dn/dc does not depend significantly on polymer molar mass once the polymer is longer than an oligomer, and it depends only slightly on laser wavelength. Therefore, accurate dn/dc values can be looked up for many common polymers in reference texts.13,29 Crucially, however, dn/dc inherently depends on the solvent (and temperature to a lesser degree), so the dn/dc must be measured in the same solvent as is used in the batch-mode SLS or SEC-MALS experiment.
Measuring a dn/dc value is accomplished in basically the same way as determining an absorption coefficient in a UV-vis experiment: Carefully prepare several (we recommend 5) known concentrations of the polymer in the solvent of choice, selecting a range that covers the concentration used in the batch-mode SLS or SEC-MALS experiment (0.5 to 10 mg mL−1 is a typical range). Next, measure the refractive index of each solution relative to a solvent blank using a dRI detector. Because nearly all SEC systems include a dRI detector, this experiment is relatively easy, and the instrument software may already be set up to do this analysis. Importantly, the polymer should not be fractionated in a column before analysis, so the columns must be disconnected from the SEC system before a dn/dc analysis is performed. In other words, this is an “offline” experiment. Before measuring dn/dc on a new polymer sample, we recommend measuring it first for a polymer with a known dn/dc value to confirm that the method works (e.g., polystyrene in THF has a dn/dc of 0.185 mL g−1 at 30 °C with a red laser).29 A potential downside of this method is that it can consume 20–50 mg of material to prepare enough of each solution for accurate measurements. An alternative but somewhat less accurate method to estimate dn/dc, called the 100% mass recovery method, is discussed below in the section The 100% mass recovery method to estimate dn/dc values. The 100% mass recovery method may be the only method available in cases where enough material cannot be obtained for offline dn/dc analysis. In most cases, dn/dc values fall in the range of 0.020–0.200 mL g−1, although they can be below 0 (in which case the plot in Fig. 1A would have a negative slope), with higher absolute values providing higher signal-to-noise ratios in MALS.
Batch-mode MALS
A batch-mode MALS experiment involves a single unfractionated sample; SEC-MALS, detailed below, is simply a series of batch-mode MALS experiments carried out in rapid succession on each fraction of the polymer after it exits the separation columns. To carry out a batch-mode MALS experiment, all that is needed is a solution of polymer of known concentration (in mg mL−1) and a known dn/dc value. The solution is analyzed either in a vial on an instrument specifically set up for batch-mode MALS or via continuous injection of the polymer solution directly into the flow cell of a MALS detector. The instrument measures (at multiple angles) the intensity of light scattered (Iscattered), which is proportional to the Mw value (eqn (1)). The Mw value is generated by solving a more complex form of eqn (1), which includes constants and additional components such as the form factor describing the angular dependence of light scattering.A batch-mode MALS experiment provides no information on Mn and therefore no information on Đ. However, we still find these experiments useful on occasion to determine Mw values for polymer samples that do not dissolve in available SEC solvents. Batch-mode MALS experiments can also be carried out on polymer samples that have undesired interactions with the stationary phase in the SEC columns. In any batch-mode MALS experiment, it is critical to rule out unintentional aggregation of the polymers in the chosen solvent, which will lead to an Mw value that is much higher than the actual value (see section Common errors and practical considerations for more on this topic). Finally, samples and solvent must be filtered to avoid dust, which also scatters light.
Beyond soluble polymer samples, batch-mode MALS can also be applied to estimating aggregation numbers in polymer assemblies such as block copolymer micelles, which cannot be measured by SEC because block copolymer aggregates tend to break up on the columns due to shear forces.30,31 The aggregation number can be estimated by dividing the Mw of the aggregates by the Mw of the individual block copolymer. As in all MALS experiments, the dn/dc of the block copolymer must be known accurately.
Somewhat beyond the scope of this tutorial review, but still worth mentioning, are the particular formalisms used to fit the data in a batch-mode MALS experiment. The Zimm, Debye, and Berry methods simply use different means to graph and interpret the same MALS data. Fortunately, the Zimm formalism can be used for most samples, and it is embedded into the software in modern MALS instruments. Podzimek provides a useful discussion of the relative strengths and weaknesses of these different formalisms, but his first point on this topic is most useful: “All formalisms provide similar results.”15 We recommend that users of SEC-MALS read this review and another by Podzimek32 to gain an understanding of the Zimm plot and related formalisms, and the second virial coefficient (A2), which are beyond the scope of this tutorial review.
Basic setup of an SEC experiment
In a typical SEC experiment, the polymer sample is dissolved in the same solvent as the mobile phase on the instrument (i.e., the solvent running through the columns and detectors). The specific concentration may vary, but it is typically between 0.5 and 10 mg mL−1. The sample solution is then injected—either manually or with an automated sampling module—onto the columns, which separate the polymer chains by size. Larger macromolecules elute first because they are excluded from the smaller pores of the stationary phase (i.e., the resin packed in the columns themselves). Smaller macromolecules elute later because they interact with the stationary phase to a greater extent. Thus, SEC separates polymers by their size in solution (hydrodynamic volume), not strictly by molar mass. The stationary phase in most SEC columns is composed of polystyrene crosslinked with divinylbenzene, but specially coated silica particles are sometimes used, particularly for aqueous SEC. Different stationary phase resins have different ranges of pore sizes, which may be better suited to separating different molecular weight ranges. As a result, many systems employ multiple columns to ensure that a wide range of molecular weights may be separated. A short guard column is also typically employed to prevent improper samples (i.e., those containing dust, aggregating materials, or insoluble polymers) or other impurities from damaging (clogging) the more expensive SEC separation columns. We refer the reader to an extensive review by Berek highlighting the underlying principles and limitations of SEC.33The choice of mobile phase for an SEC experiment depends on the typical samples to be analyzed on the instrument. Good solubility is paramount, and consequently the mobile phase is generally selected based on polymer solubility. For example, THF is typical for many nonpolar polymers, as is toluene. N,N-Dimethylacetamide (DMAc) and N,N-dimethylformamide (DMF) may be appropriate for more polar polymers. Often LiCl or another electrolyte is added to DMAc or DMF mobile phases to facilitate dissolution and to limit unwanted electrostatic interactions, particularly in the case of polysaccharides.34 For polyolefins such as polyethylene, 1,2,4-trichlorobenzene at elevated temperatures is a common mobile phase. Water, typically as a buffer solution with a preservative (i.e., sodium azide) to prevent microbial growth, is used as the mobile phase for polymers that dissolve in aqueous solutions. Finally, hexafluoroisopropanol (HFIP) is an extremely powerful solvent and can be used in SEC, sometimes with added salts, but its high volatility, acute toxicity, corrosiveness, and cost limit its widespread use.
After exiting the columns, the fractionated macromolecules in the mobile phase pass through a series of detectors (Fig. 2). Usually, the final (and sometimes only) detector is a dRI detector. At each data point in an SEC chromatogram (sometimes called an elugram or trace), the dRI detector collects the n value of the polymer solution as it exits the columns and compares it to the n value for pure mobile phase, computing the differential in the n values (dRI). Fortunately, the dRI detector provides a signal for nearly every type of polymer as long as there is a difference in n values between solute (polymer) and solvent (mobile phase) and provided that the sample does not absorb light at the laser wavelength. In the absence of a MALS detector, the dRI detector chromatogram can be used—applying a method called conventional column calibration, potentially including MHS parameters—to estimate the Mn, Mw, and Đ of the sample. The data produced by the dRI detector can also be used to estimate the dn/dc of the sample. Conventional column calibration, MHS parameters, and dn/dc estimation methods are discussed in more detail below.
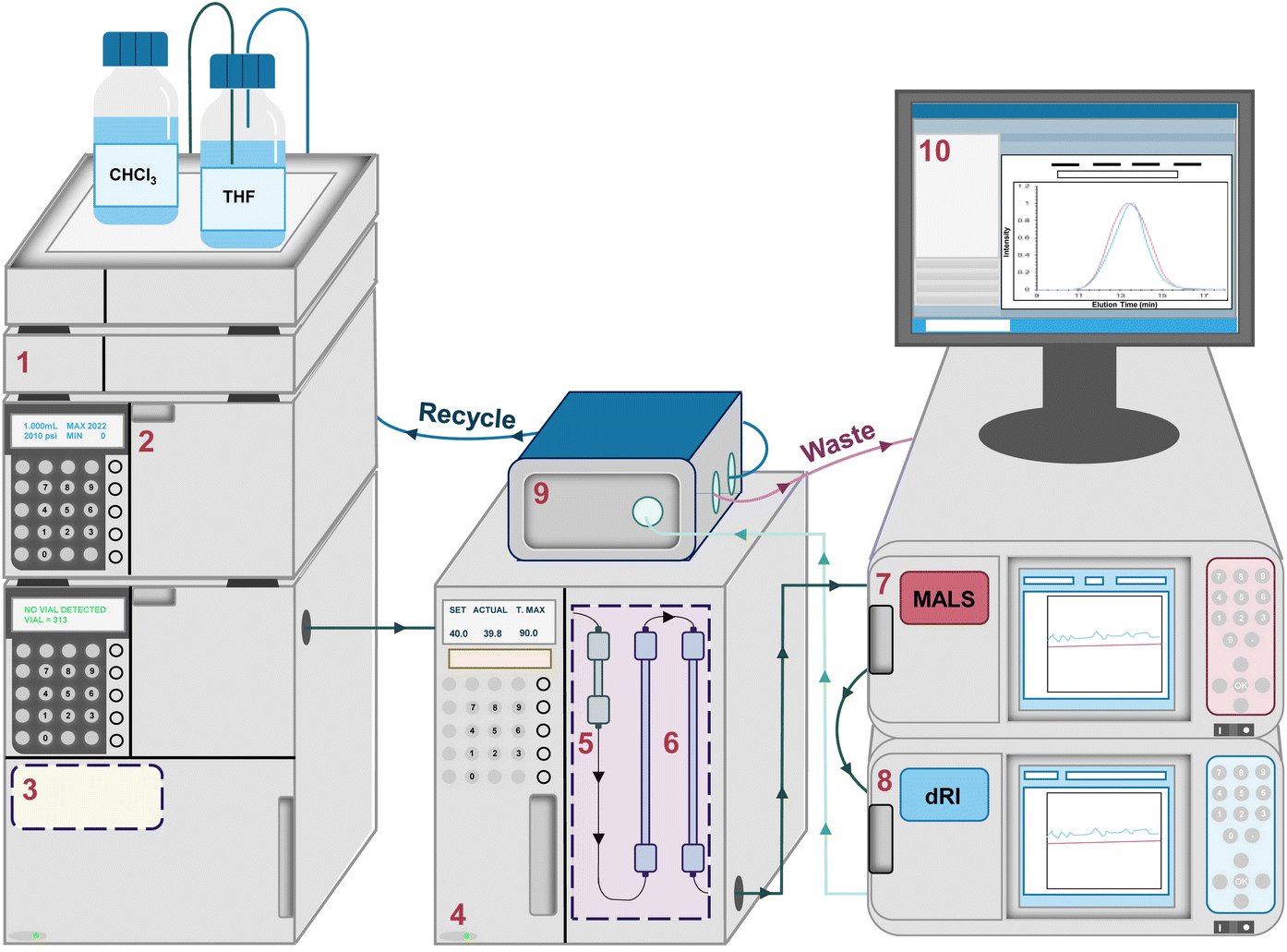 | ||
| Fig. 2 Schematic representation of a typical SEC-MALS system equipped with a (1) degassing unit, (2) solvent pump, (3) autosampler, (4) column oven, (5) guard column, (6) separation columns, (7) MALS detector, (8) dRI detector, (9) solvent recycling system, and (10) computer interface. | ||
Approaches to calibration in SEC without MALS
Many labs, past and present, rely on calibration methods for molecular weight determination by SEC alone, rather than absolute molecular weight determination using SEC-MALS. To place SEC-MALS in context with these other methods, we briefly discuss here the two most common forms of SEC that do not involve MALS: column calibration and universal calibration.In the column calibration method (sometimes called the conventional calibration method), a series of narrow dispersity polymers of known molar mass are used to calibrate the instrument. For example, commercially available, narrowly dispersed polystyrene samples of different molar masses are commonly used. Using these calibration data, elution time and molar mass can be correlated, and Đ can be inferred from the breadth of the peak in the chromatogram. While useful for qualitative comparisons among similar samples, this approach has significant limitations, most notably that it is only quantitatively accurate for polymers that have the same chemical and topological structure as the standards. For example, Mays and coworkers studied a set of star polymers and compared molecular weight values by SEC using either column calibration (dRI only) or using a dRI detector and a right-angle light scattering detector, revealing that Mn values were underestimated by as much as 2–3-fold when conventional column calibration was used.35 Additionally, Locock and coworkers studied a series of cationic poly(methacrylates) by SEC and other characterization methods.36 Column calibration using poly(methyl methacrylate) standards afforded values that were overestimated by 2–4-fold compared to those obtained by other, more accurate methods for these relatively low molecular weight samples (1H NMR spectroscopy and MALDI-TOF mass spectrometry). The inaccurate results by SEC with column calibration were likely due to differences of solvent quality and persistence length among the standards and the polymers being analyzed. Consequently, molar masses determined by SEC using the column calibration method are often described as “relative molar masses”, or more accurately “apparent molar masses”, to indicate that the given Mn or Mw values were determined relative to some standard.37
For many commercial polymers, MHS parameters—which relate polymer molecular weight to intrinsic viscosity—can be applied to improve the accuracy of the data when the polymer samples being tested are not the same type as those used for column calibration. However, MHS parameters are unknown for new types of polymer backbones, complex topologies, or block(y) copolymers. In these cases, an in-line viscosity detector (viscometer) can be used to measure relative viscosity, which can be combined with concentration measurements from a dRI detector to give intrinsic viscosity. This approach is “universal” because the product of intrinsic viscosity and molecular weight relates directly to hydrodynamic volume—the parameter driving separation on the column. Consequently, universal calibration can provide accurate molecular weight data when the SEC is calibrated with standards of known molecular weight, regardless of the polymer structure (provided no unwanted enthalpic effects perturb the measurement).38,39 While universal calibration becomes inaccurate for low molecular weight polymers,40 as well as very high molecular weight polymers,41 it is generally much more accurate than column calibration.
Analysis of SEC-MALS data
An SEC-MALS experiment requires fractionation (the SEC component), as well as a MALS detector and a concentration detector. The concentration detector is typically a dRI detector, but in some cases a UV-vis detector may be used; here we focus on SEC-MALS setups that include a dRI detector. As the fractionated polymer sample exits the separation columns, data are collected at rapid time intervals (e.g., every 0.5 s) by the dRI and MALS detectors, with corrections applied for the distance the sample travels between each detector to “line up” the data points accurately. Each data point (i) can be considered a single batch-mode SEC experiment. The dRI detector provides the concentration (ci in g mL−1) at each ith data point based on a given dn/dc value, and the MALS detector measures the intensity of light scattered (Iscattered,i) at multiple angles. Together they afford a molar mass value for the macromolecule sample eluting at each data point, called Mi. Note that Mi is not treated as a molar mass average, but rather a specific molar mass at the ith data point; the macromolecule sample eluting at each ith data point is assumed to be monodisperse, a critical assumption in SEC-MALS and SEC in general. In reality, Mi is a weight-average molar mass of the macromolecules eluting at each ith data point.Fig. 3A demonstrates how the instrument determines the Mn, Mw, and Đ values for the injected sample. For this example, we synthesized a polydisperse PS sample following a suspension polymerization method and then ran SEC-MALS in THF on the isolated polymer. The chromatogram shows the dRI and MALS detector responses versus time, where the dRI signal reflects polymer concentration at each data point (ci). In brief, ci is proportional to the baseline-subtracted signal from the dRI detector divided by dn/dc. The MALS signal depends on both polymer concentration and molar mass at each data point (Mi). (Note that there is a MALS trace for each angle in the MALS detector, but we show only the trace from the 90° detector throughout this tutorial review as a representative example.) For a more detailed mathematical treatment of the equations underlying detector responses, we refer the reader to recent articles by Podzimek.42,43
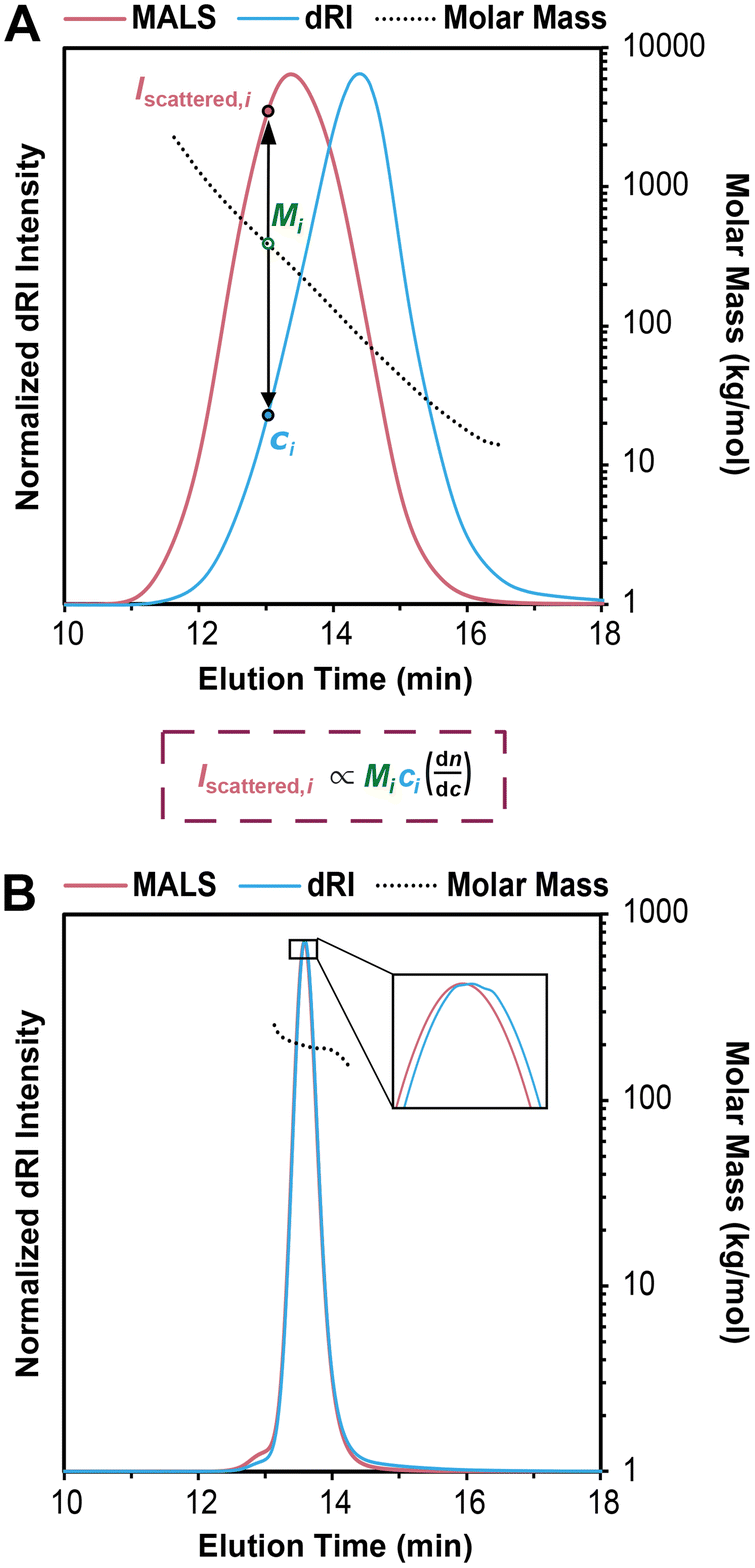 | ||
| Fig. 3 (A) SEC-MALS chromatogram overlaid with corresponding molar mass data for a polydisperse PS sample, where the intensity of light scattered (Iscattered,i) and concentration (ci) are used to determine the molecular weight (Mi) at each data point i, based on a more complex version of eqn (1) suited for SEC-MALS. Note that in SEC-MALS, Iscattered is proportional to dn/dc instead of (dn/dc)2 because the concentration term (ci) includes a dn/dc term in the denominator. (B) SEC-MALS chromatogram overlaid with corresponding molar mass data for a narrowly dispersed 200 kg mol−1 PS standard, where most of the data points in the middle of the peak have nearly the same Mi. | ||
In Fig. 3A, it is immediately clear that the two traces do not overlap, i.e., the MALS trace peak comes at 13 min while the peak for the dRI trace is at 14 min. This offset is not an error in the SEC experiment, but rather a result of the molecular weight dependence of light scattering. Macromolecules eluting earlier are larger than those eluting later, thus they scatter more light. Put another way, the macromolecules at an early elution time, 12 min for example, are low in concentration but high in molecular weight, affording a low intensity in the dRI trace but a high intensity in the MALS trace. At the other end of the chromatogram, for example at an elution time of 16 min, we see a moderate dRI signal, but a very low MALS signal, indicative of much lower molecular weight macromolecules.
Overlaid on top of the chromatogram in Fig. 3A is a series of points in black showing the Mi value at each data point. The instrument determines these Mi values by creating a partial Zimm plot (or a related plot called a Debye plot) for each data point. (Note that we show only every 12th data point here for sake of clarity.) To obtain each Mi value, the instrument software simply solves a more complex version of eqn (1) suited for SEC-MALS by using information on concentration from the dRI detector (ci) and information on scattered light intensity from the MALS detector (Iscattered,i). As noted above, the molar mass value measured by the MALS detector at each data point, Mi, is treated as a monodisperse macromolecule sample, but it is actually a weight-average molar mass value of the macromolecules eluting at each data point (because no separation is perfect). Calculating the Mw, Mn, and Đ values for the entire polymer sample becomes a straightforward summation problem that is simply a larger version of a type of question common in introductory polymer chemistry or polymer science classes: if you mix 1 g each of three monodisperse polymers with known Mn values x, y, and z, what are the values of Mn, Mw, and Đ for the final mixture? This problem is simply solved using the equations for Mn, Mw, and Đ (eqn (2)–(4)). In SEC-MALS, the same calculations are done but on a larger scale, where each data point (of typically 100–1000 total depending on the breadth of the peak) is treated as an individual monodisperse polymer.
In the case of this polydisperse PS sample in Fig. 3A, the Mi value for the data point at an elution time of 12 min is 1290 kg mol−1, and the value decreases steadily as elution time increases, dropping to 18 kg mol−1 for the data point at 16 min. Using the approximately 720 data points (the peak between ∼11 and ∼17 min elution time with data taken every 0.5 s), the instrument generates Mn, Mw, and Đ values for the entire sample. In this case, the values are Mn = 69 kg mol−1, Mw = 153 kg mol−1, and Đ = 2.22.
Fig. 3B shows an SEC-MALS experiment on a narrowly dispersed PS standard with a nominal Mn = 200 kg mol−1. In this case, the peak is quite sharp, and the dRI and MALS traces nearly overlap, consistent with a narrowly dispersed sample. For this standard, the SEC-MALS experiment shows that most of the data points in the middle of the peak have nearly the same Mi value (200 kg mol−1), while a few data points at earlier elution times have slightly larger molar masses and a few at late elution times have slightly lower molar masses. Again, this is typical of SEC standards with low Đ values. Note that high molecular weight PS standards typically contain a small amount of dimer, seen here as a small shoulder at 12.8 min. The very low concentration of these high molar mass species means that they do not substantially affect the results. In this case, we find that Mn = 194 kg mol−1, Mw = 198 kg mol−1, and Đ = 1.02.
The effect of separation quality on SEC-MALS
In an SEC-MALS experiment, assuming no aggregation and accurate values for dn/dc and other relevant constants, the Mw value is accurate, but the Mn value depends on the quality of the separation. In the theoretical case of a perfect separation, the assumption of a monodisperse macromolecule population at each data point is sound, and the Mn value for the sample is therefore perfectly accurate. In the case of good separation, the fraction eluting at each data point is nearly monodisperse, and the Mn value is reasonably accurate. In the case of poor separation, the assumption that each data point represents a monodisperse macromolecule sample is not sound. As a result, the Mn value is overestimated, and the Đ value (Mw/Mn) is therefore underestimated, i.e., the sample appears to be more narrowly dispersed than it actually is. Because no separation is perfect, Đ is underestimated in any SEC-MALS experiment, at least to some extent. This problem is discussed below in more detail (see section Dispersity in SEC and SEC-MALS).Fig. 4A illustrates this phenomenon using the same polydisperse PS sample synthesized in our labs as discussed above. In this series of SEC-MALS experiments, we demonstrate how the quality of the separation influences the Mw and apparent Mn values by examining the same polymer sample with different numbers of identical separation columns. In each SEC-MALS experiment, light scattering ensures that the Mw value is accurate, but the separation quality dramatically affects the apparent Mn value.
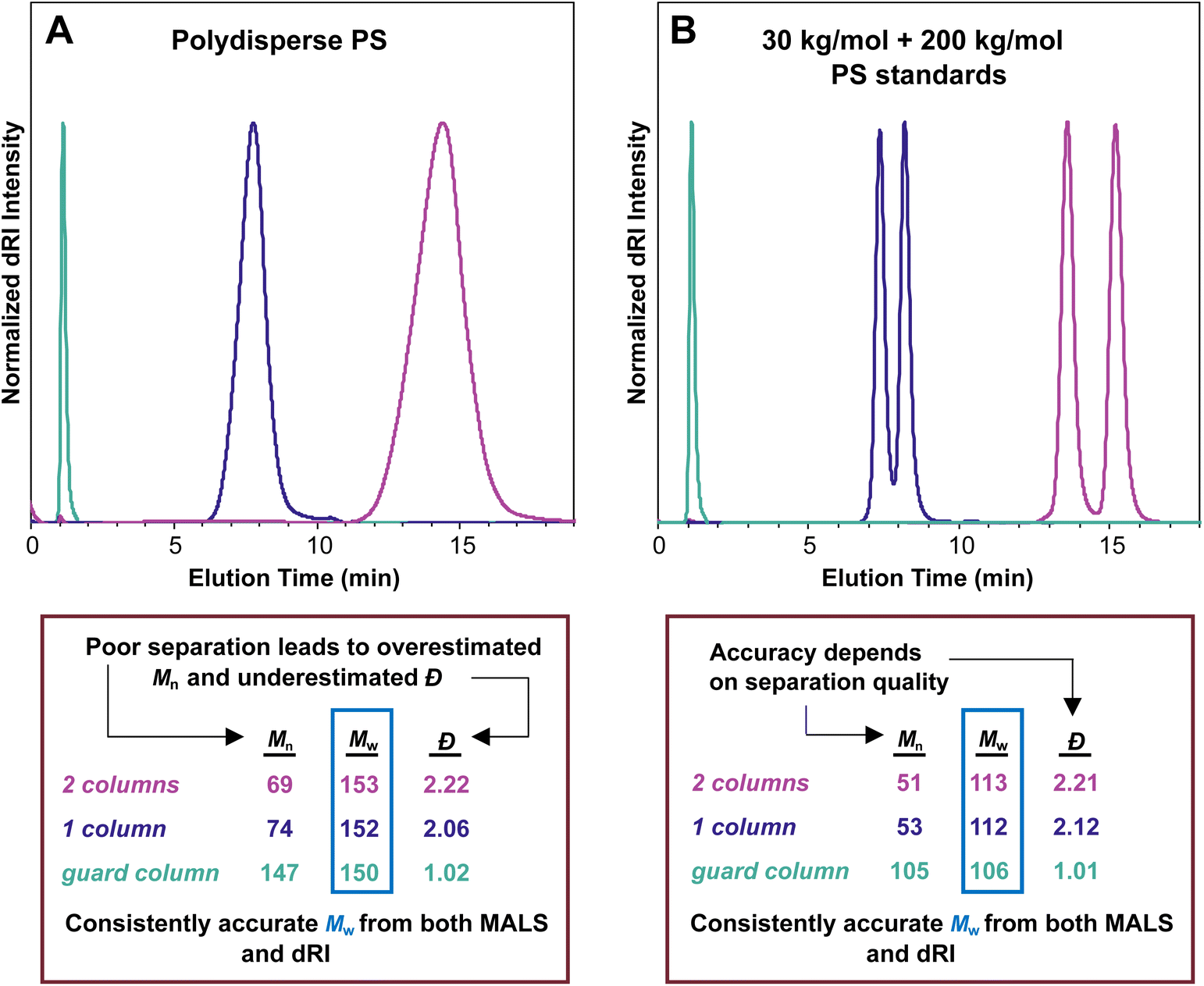 | ||
| Fig. 4 (A) SEC-MALS chromatograms of a polydisperse PS sample analyzed using two separation columns (pink trace), one separation column (purple trace), and only the guard column (teal trace); (B) SEC-MALS chromatograms of a 50/50 mixture by weight of 30 kg mol−1 and 200 kg mol−1 PS standards analyzed using two separation columns (pink trace), one separation column (purple trace), and only the guard column (teal trace). In both experiments, Mw remained largely constant, but decreasing separation quality led to increasingly overestimated Mn and increasingly underestimated Đ values. | ||
Using two separation columns appropriate for this size range and the literature dn/dc value for PS in THF (0.185 mL g−1), the molecular weight and Đ values were Mw = 153 kg mol−1, Mn = 69 kg mol−1, and Đ = 2.22 (pink trace). We assume good chromatographic separation under these conditions, so we accept that this Mn value is reasonably accurate. The situation changed slightly when using only one separation column (purple trace). In this experiment, the molecular weight and Đ values derived by the instrument were Mw = 152 kg mol−1, Mn = 74 kg mol−1, and Đ = 2.06. Compared with the previous run using two separation columns, the Mw value stayed virtually constant, but the Mn value increased somewhat, which decreased the Đ value. The elution time was reduced from around 14 min to around 8 min because a column was removed. A more dramatic change occurred when we removed both separation columns, running the experiment with only a guard column (teal trace), which provides very little separation. This experiment showed a very sharp peak at 1 min elution time with corresponding Mw = 150 kg mol−1, Mn = 147 kg mol−1, and Đ = 1.02. The Mw remained close to the value of 153 kg mol−1 determined with two columns, but the apparent Mn value was about twice as high as in the other runs, dropping the Đ value to afford what appeared to be a nearly monodisperse sample.
Taken together, this series of three SEC-MALS experiments on the same polydisperse polymer sample shows that Mw is consistently accurate, but Mn is heavily affected by the separation quality. Poor separation leads to an overestimated Mn value and an underestimated Đ value. In other words, Mn depends on the quality of the chromatography, but Mw does not.
A related set of experiments is shown in Fig. 4B. In this series of SEC-MALS runs, we mixed equal masses of two narrowly dispersed PS standards, one with nominal Mn = 30 kg mol−1 (actual Mn measured on our system with two separation columns = 28.8 kg mol−1), and the other with nominal Mn = 200 kg mol−1 (actual Mn measured on our system with two separation columns = 194 kg mol−1). We can use the definitions of Mn and Mw (eqn (2) and (3)) to calculate the expected Mn and Mw values (and therefore also the Đ value, eqn (4)) for this mixture: Mw = 111 kg mol−1, Mn = 50.2 kg mol−1, Đ = 2.21 (note that this calculation assumes that each standard is monodisperse). Using two separation columns and dn/dc = 0.185 mL g−1, we observed two peaks that were nearly baseline separated (pink trace). The molar mass and Đ values were Mw = 113 kg mol−1, Mn = 51 kg mol−1, and Đ = 2.21, lining up closely with expected values based on the calculations above. Removing one of the separation columns afforded peaks that were not as well separated but still clearly came from two distinct populations of macromolecules (purple trace). The corresponding molar mass and Đ values changed to Mw = 112 kg mol−1, Mn = 53 kg mol−1, and Đ = 2.12. This result reveals that just a single column separates these two polymers reasonably well, with only a small increase in Mn and a slightly reduced Đ. Removing both separation columns and retaining only the guard column eliminated the separation of the two populations of macromolecules (teal trace), and the corresponding values were Mw = 106 kg mol−1, Mn = 105 kg mol−1, and Đ = 1.01. These three SEC-MALS experiments on this pair of PS standards reveal similar results to those on the polydisperse PS sample: the measured Mw value for this mixture is quite consistent regardless of the number of columns, varying only by around 5% (113 to 106 kg mol−1), but the accuracy of the Mn value depends on the quality of the separation (105 to 51 kg mol−1).
Dispersity in SEC and SEC-MALS
Regardless of the type of SEC, Đ values are prone to error and should be viewed with some reservation. SEC-MALS underestimates Đ values because the macromolecule sample is assumed to be monodisperse at each data point in the chromatogram (Mi). Even with outstanding separation, some heterogeneity in the polymer population at each data point is inevitable. This underestimation of Đ values in SEC-MALS is often not addressed explicitly in the current literature, and the magnitude of the effect depends on the quality of the separation—with good separation the underestimation is small, but the underestimation increases as separation efficacy decreases.SEC with conventional calibration has been periodically suggested as the best method to obtain an accurate Đ value. In fact, historically it has been common practice to measure Mw with an SLS technique (batch-mode or SEC-MALS) and report Đ based on SEC with conventional calibration. Indeed, column calibration often affords a higher Đ value than SEC-MALS, but this Đ value is also prone to error. Guillaneuf and Castignolles showed in 2007 that solvent quality, as measured by the MHS alpha parameter, can dramatically affect Đ values determined using SEC with conventional calibration.44 In brief, if the eluent is a better solvent for the standard than for the studied polymers, then the Đ value is underestimated. If the eluent is a better solvent for the studied polymer than for the standards, then the Đ value is overestimated. Additionally, band broadening—the phenomenon where chains of the same molecular weight elute over a range of elution times due to molecular diffusion—must be taken into account to determine Đ accurately using SEC with conventional calibration.45,46
Accurate and precise measurement of the Đ value of a polymer sample is difficult. The best methods involve MALS to determine Mw and another method to accurately and precisely measure Mn, such as osmometry. As a word of caution, we note that Mn measured by end-group analysis using 1H NMR spectroscopy can be accurate and precise, but generating an Mn value of sufficient precision for an accurate Đ measurement requires attention to several details: (1) end-group fidelity must be confirmed using other techniques because any macromolecules that lack the diagnostic chain end proton(s) are not counted in this technique; (2) the NMR experiment must be designed with appropriate relaxation delays for accurate integral measurements; (3) the end-group protons must be in a region of the spectrum without overlap from the polymer backbone, solvent, or other impurities; (4) the spectrum must be worked up with careful attention when setting the baseline to obtain accurate integrations.
Finally, for the interested reader, we note that Harrisson suggests standard deviation as an alternative to Đ, discussing many of the issues in using this parameter as a measure of polymer molecular weight distribution.47 Note that measurements of standard deviation in molecular weight are subject to the same factors that affect measurements of Đ, including the SEC method, separation efficacy, band broadening, and solvent quality. Ultimately, we recommend not overinterpreting Đ values, regardless of how they are measured. Đ values determined by SEC-MALS are underestimated to varying degrees, and Đ values determined by conventional or universal calibration may be over- or underestimated. In cases where accurate and precise measurements of Đ values are critical, multiple techniques are needed.
The 100% mass recovery method to estimate dn/dc values
In an SEC-MALS experiment, the 100% mass recovery method is a simple way to estimate dn/dc that avoids the preparation of several samples. In short, the 100% mass recovery method assumes that all of the injected polymer mass (i.e., 100%) in an SEC experiment is accounted for within the selected peak in the dRI trace. In some instruments, the 100% mass recovery method can be set up as the default method for dn/dc determination in the software.The 100% mass recovery method can introduce a bit more error than an offline measurement, but it is typically accurate to within ∼20% with proper sample preparation. The advantage is that the 100% mass recovery method uses the data generated from the dRI detector in a single SEC experiment to estimate a dn/dc value based on a known amount of injected sample. It is analogous to estimating a molar absorption coefficient by UV-vis using a single concentration of the sample and measuring the slope of the line from that data point to the origin in a graph of absorption versus concentration. If the dRI trace exhibits a tail toward higher elution time, or if there are other reasons that the sample might “stick” to the columns, then this assumption is not valid. Sometimes this “sticking” issue can be solved by changing the sample concentration. For example, Gomez and coworkers found that poly(3-hexylthiophene-2,5-diyl) (P3HT) with an Mw value exceeding 30 kg mol−1 did not fully elute from the columns at an injected concentration of 1.0 mg mL−1, but it did fully elute at 0.5 mg mL−1.48
The 100% mass recovery method also relies heavily on an accurate value for the polymer mass injected onto the columns. This injected mass value requires that a sufficient amount of polymer was used to obtain an accurate mass measured on a balance when preparing the solution, and that the polymer is pure, i.e., free of solvent, water, residual monomer, and other potential contaminants. A 1H NMR spectrum can confirm purity or be used to estimate the amount of residual solvent, which can be factored into the polymer concentration value or injected mass value used in the experiment. Accurate injected mass values also require accurate injection volumes for the instrument. A persistent and consistent deviation in the dn/dc value obtained using the 100% mass recovery method versus a known dn/dc value may indicate an error in the injection volume. Methods to assess injection volume accuracy can be recommended by the manufacturer of the autosampler or injector/pump system. In summary, the 100% mass recovery method can be a reasonably accurate way to estimate dn/dc, but it depends heavily on the purity of the polymer sample, the accuracy in preparing the sample for the SEC experiment, and good calibration of the autosampler. Again, a good way to analyze the accuracy of this method is to test it on a sample with a well-established dn/dc value.
Determining dn/dc values for copolymers
As we have already seen, accurate dn/dc values are vital in SEC-MALS. For homopolymers, this value can be either looked up in reference texts, measured offline, or estimated using the 100% mass recovery method. However, the situation becomes more complicated for copolymers of all kinds, including random/statistical copolymers, block(y) copolymers, and graft (co)polymers.42 The challenges associated with accurately measuring block copolymer molecular weight have been recently reviewed, primarily in the context of universal calibration.49 SEC-MALS alleviates some of these challenges, but others arise, particularly with the dn/dc value. When several batches of copolymers are made and analyzed with different weight fractions of the two repeat units, each one will almost certainly have a different average dn/dc value. Fortunately, multiple methods can be used to measure or estimate the dn/dc values for a series of copolymer samples.The most accurate method to determine dn/dc values for a series of copolymers is to measure the average dn/dc value of each copolymer sample independently. As with homopolymers, this approach is most accurate when carried out using an offline dRI detector to measure the dRI of the copolymer sample at different concentrations and determining the average dn/dc value from the slope. However, this method is less accurate for copolymers than for homopolymers. This decrease in accuracy occurs because no copolymer sample is homogeneous, so the macromolecule population at each data point in an SEC-MALS chromatogram has a different dn/dc value reflecting the weight fractions of the two repeating units. Determining dn/dc at each data point is obviously impractical, so an average value for the entire sample must be assumed. This use of an average dn/dc value adds additional error to the measurement that is not present with homopolymers.
A less accurate but faster way to estimate the dn/dc values for a series of copolymers is to use the 100% mass recovery method. As discussed above in the context of homopolymers, this method relies on pure materials (no residual solvent or other small molecules), accurate mass and volume determination, and accurate injection volume. When these criteria are met, this method can be accurate to within 10–20%, but accuracy drops somewhat in the case of copolymers versus homopolymers for the same reason as offline measurements—the dn/dc value varies at each data point in a chromatogram, so an average dn/dc value must be assumed.
Finally, in many cases dn/dc values for copolymers can be estimated using the known dn/dc values of the homopolymers and their relative weight fractions in a given copolymer using the equation below:
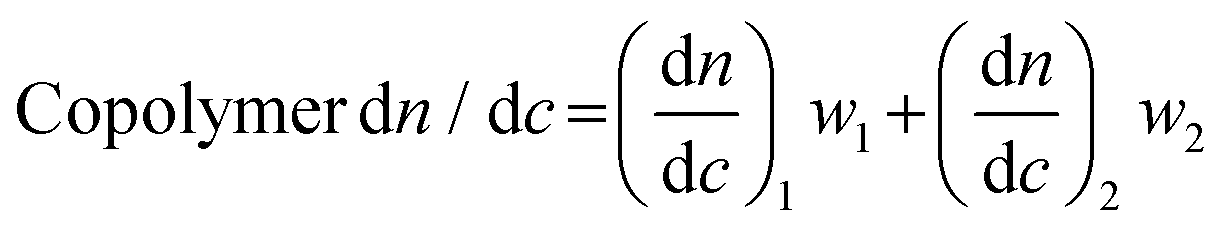 | (5) |
We note that eqn (5) provides a good estimate of dn/dc, but some reports show a lack of linearity in a plot of copolymer dn/dc versus wt% of one monomer, suggesting that its accuracy may depend on the exact copolymer system. For interested readers, several studies describe precise dn/dc measurements of various copolymers. For example, Coto and coworkers conducted a thorough evaluation of ethylene–propylene copolymers;50 Penlidis and coworkers determined dn/dc values in copolymers of alpha-methyl-styrene and methyl methacrylate;51 Rudin and coworkers measured dn/dc values of ethylene-vinyl acetate copolymers;52 and Chen et al. studied the effects of polybutadiene microstructure on dn/dc values.53
The method involving eqn (5) relies on known dn/dc values for each homopolymer and known weight fractions of each repeating unit. In many cases, the homopolymer dn/dc values can be looked up in reference texts, and the weight fraction of each repeating unit can be accurately determined using NMR spectroscopy or other spectroscopic techniques. While this method still suffers from the lack of homogeneity in copolymer samples, it is likely more accurate than the 100% mass recovery method for most copolymer systems.
Which method is best depends on the accuracy needed and the amount of material available. For the most accurate measurements, an offline dn/dc measurement for each copolymer sample is best. We recommend this method in a situation where one or a few copolymer compositions will be thoroughly studied. Alternatively, the 100% mass recovery method can yield moderately accurate results in SEC-MALS of copolymers with precise mass and volume measurements, high sample purity, and good autosampler calibration. Finally, the method using eqn (5) is often the simplest and in some cases can yield very accurate results, especially if the dn/dc values of the two homopolymers are close and the weight ratios of the monomer units can be accurately determined. When using the 100% mass recovery method, we recommend also estimating dn/dc using eqn (5) and comparing the two results.
Lastly, other methods beyond SEC-MALS can provide values for Mn and in some cases Mw for copolymers. These include two-dimensional chromatographic methods, NMR methods such as diffusion-ordered spectroscopy (DOSY), mass spectrometry, and in some cases elemental analysis. These methods were recently discussed by Michels and coauthors.49 However, we note that despite its complexities, SEC-MALS remains the most established method for measuring the molecular weight and molecular weight distribution of copolymers.
Contaminants, dissolved gasses, and small molecules in SEC-MALS experiments
An important consideration when preparing samples for SEC-MALS is the ability of the mobile phase to dissolve or extract compounds from any consumables used to prepare or transport the sample. Many commonly used plastic laboratory consumable products can leach significant amounts of small-molecule contaminants, such as plasticizers or other additives (Fig. 5). While this type of small-molecule contamination does not exhibit a significant light scattering signal, the dRI signal can overlap with low molecular weight polymer peaks. In cases where these signals may interfere with polymer signals, we recommend using only glass syringes and vials and filtering the sample quickly to avoid prolonged exposure to the plastic filter housing.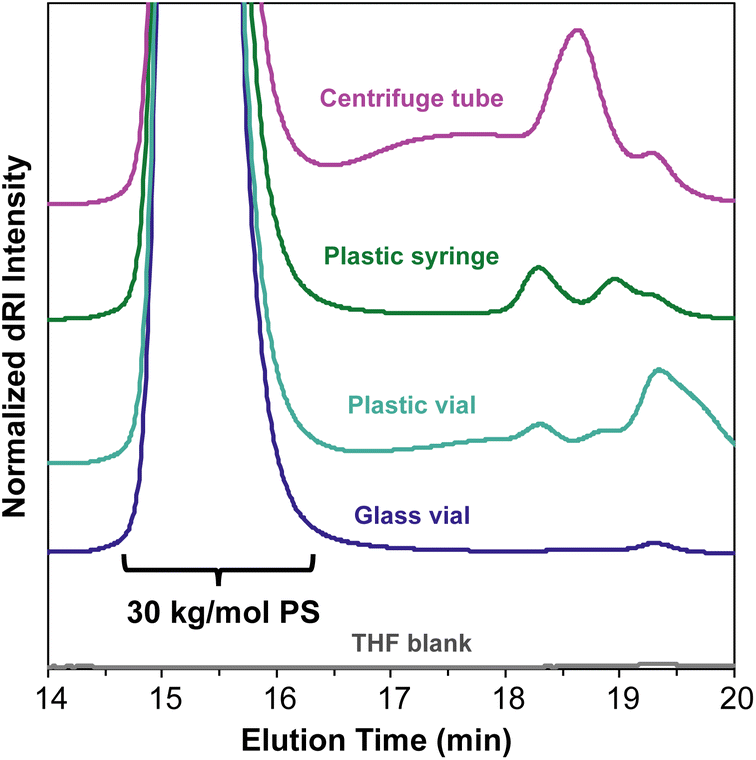 | ||
| Fig. 5 SEC-MALS chromatograms of a 30 kg mol−1 PS standard prepared with commonly used plastic laboratory consumable products. All samples were quickly filtered through a 0.22 μm polytetrafluoroethylene (PTFE) syringe filter and added to glass autosampler vials. | ||
In most published SEC-MALS chromatograms, only the region containing the polymer peak(s) of interest is shown. However, small-molecule, residual solvent, and/or dissolved gas peaks are commonly observed in a full SEC-MALS chromatogram. Small-molecule, salt, and solvent peaks typically appear at later elution times compared to polymer peaks, and they can have either a positive or negative inflection depending on their dRI in the selected mobile phase (Fig. 6). Low molecular weight polymer peaks may be distinguished from small-molecule and/or solvent peaks by examining the light scattering trace. Small molecules and solvents exhibit little to no light scattering signal, but depending on their concentration, can exhibit significant dRI signals. However, even low molecular weight oligomers often exhibit observable MALS and dRI signals.
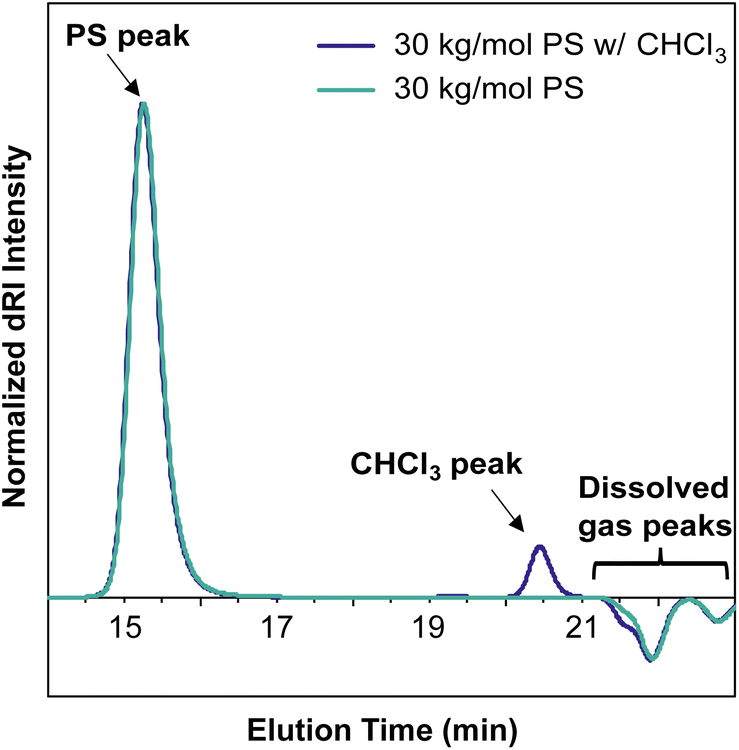 | ||
| Fig. 6 Overlaid SEC-MALS chromatograms of a 30 kg mol−1 PS standard in THF (teal trace) and a 30 kg mol−1 PS standard in THF doped with 1% CHCl3 as a representative small molecule contaminant (purple trace), which appears at an elution time of 20.5 min. Dissolved gas peaks have a negative inflection and appear at 21–23 min elution time. | ||
Other practical considerations and common errors
SEC-MALS data quality may be compromised by a number of factors, and an awareness of potential issues will allow a user to avoid misinterpretations. For example, certain polymers can interact with the column material, thereby complicating the separation. Poly(ethylene glycol) (PEG) of at least a moderate molecular weight, for instance, notoriously “drags” when eluting in THF on many SEC systems, which can result in the polymer peak partially overlapping with the low molecular weight region, thereby compromising the data. This challenge can often be circumvented by including specific additives in the mobile phase or changing to a different mobile phase (e.g., DMF in the case of PEG).Similarly, polymers may be poorly solubilized in the mobile phase resulting in aggregation, which is particularly prevalent with block copolymers. Aggregation can be identified by the presence of a low elution time peak with a molecular weight several times that of the main polymer peak, coming at a higher elution time. Although uncommon, when aggregates are relatively small they may appear as a high molecular weight shoulder on the main polymer peak that is more prominent in the MALS signal than in the dRI. A high molecular weight shoulder can also, however, indicate unaggregated high molecular weight material in the polymer sample, for example, due to coupling in a radical polymerization. The key to identifying aggregates is to recognize their large MALS signal (due to their high molar mass) combined with a small, sometimes negligible, dRI signal (due to the low concentration of the aggregates). Striegel demonstrates the use of SEC-MALS in identifying aggregation in a review on this topic.54 Aggregation can typically be avoided by judicious choice of mobile phase or, in certain cases, using mobile phase additives such as salts.
An accurate baseline is similarly crucial to producing high quality SEC data, so baselines should be examined for each trace (dRI and MALS traces at all angles) in each SEC-MALS experiment. While a gradually sloping baseline is typically not a problem, a wavy baseline may indicate a problem with the solvent pumps. More often, an automatic baseline setting that goes unchecked by the user may lead to the baseline ending on a dissolved gas or impurity peak, artificially distorting the baseline and compromising the data. Once the baseline is accurately set, peaks can then be selected, typically cut where the dRI signal drops below 5% of the signal maximum.
Polymers that fluoresce at the MALS detector laser wavelength present a particular problem for SEC-MALS because emitted photons can be erroneously counted as scattered photons. The result is an overestimated Mw value, sometimes several times higher than the true value.55 This problem can be addressed by using an incident laser wavelength that does not lead to fluorescence,56 applying emission filters that suppress detection of fluorescence,57 or adjusting the MALS detector signal using a mathematical correction based on sample-specific fluorescence data.58
Finally, we emphasize that synchronizing the dRI and MALS data points, a process often referred to as detector alignment, is a critical but sometimes overlooked setting. Both detectors collect data at each time point, but the sample reaches each detector at different times. Therefore, the “lag” from one instrument to the next, often called the inter-detector volume, must be accurately measured during the initial SEC-MALS system setup and testing of standards, and then applied in each SEC-MALS experiment. Sometimes the inter-detector volume is set improperly (e.g., left undefined or set to zero), which results in misalignment between the MALS and dRI detector signals. Misalignment leads to inaccurate values for both Mw and Mn, so an accurate value for inter-detector volume must be applied in the experimental template. We note that this problem regularly arises with newer users of the instrument who may be unaware of this critical setting. Fortunately, this error can usually be easily corrected in the instrument software even after samples have been run. However, we stress that regularly running low-dispersity standards can help identify issues like incorrect inter-detector volume before they become prevalent in the lab.
Common misconceptions
Misconception: the dRI trace provides the Mn, the MALS trace provides the Mw
Together they give this information, but neither one alone provides Mn or Mw in SEC-MALS. The dRI trace provides information on concentration at each data point (in mg mL−1), while the MALS detector response depends on both concentration and molar mass. Both chromatograms are required to generate values for Mn, Mw, and Đ. How the dRI and MALS detectors work together to provide values for Mn and Mw in SEC-MALS is discussed in greater detail above (see section Analysis of SEC-MALS data).Misconception: the polymer needs to be above a certain molar mass to generate a reliable MALS signal
The intensity of the MALS signal depends largely on three factors: sample concentration (c), dn/dc, and Mw (eqn (1)). Therefore, the detection limit and signal-to-noise ratio depend on all three factors, not just Mw. There has historically been some confusion on the ability of SEC-MALS to accurately characterize the molar mass of very small polymers, likely due to the differing capabilities of MALS in characterizing Mwversus Rg. While characterizing Rg for polymers with Rg < 10 nm is inaccurate with MALS, there is no hard lower limit for Mw. Mays and coworkers highlighted these differences in Mw and Rg characterization in a 1996 report,59 which at times has been misconstrued to suggest that Mw values for polymers with Rg < 10 nm are not accurate by SEC-MALS. This is not the case. Provided that there is sufficient MALS signal, SEC-MALS provides reliable and accurate Mw values even at molar masses of 1 kg mol−1 and below. To demonstrate this capability, we analyzed a 1.3 kg mol−1 PS standard at various concentrations and found that SEC-MALS can accurately measure Mw even at concentrations as low as 0.1 mg mL−1 due to the relatively high dn/dc value of PS in THF (Fig. 7). These chromatograms and molar mass results demonstrate that oligomers with at least a moderate dn/dc value can indeed be accurately characterized by SEC-MALS, even at quite low concentrations.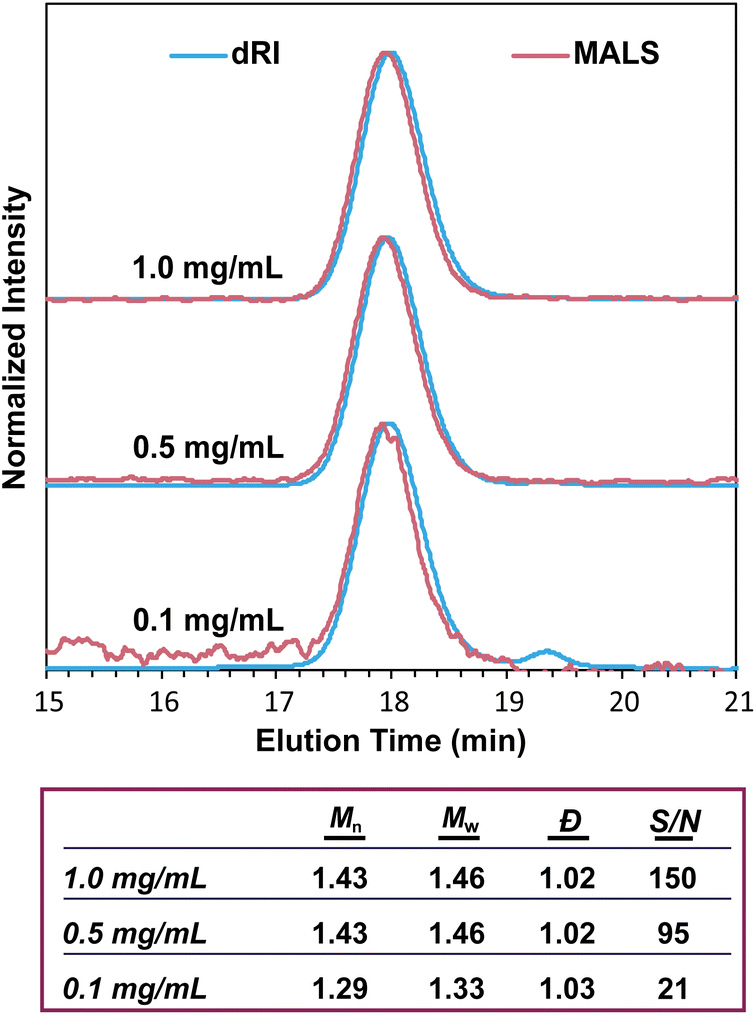 | ||
| Fig. 7 SEC-MALS chromatograms of a 1.3 kg mol−1 PS standard at various concentrations, showing the ability for SEC-MALS to accurately characterize this oligomer even at low concentrations. S/N denotes signal-to-noise ratio. | ||
Misconception: SEC-MALS instruments require periodic calibration with a series of standards
Unlike traditional SEC experiments, where molecular weight is determined based on a calibration plot of molar mass versus elution time derived from narrowly dispersed standards, SEC-MALS measures the Mw directly based on eqn (1). Consequently, column calibration is not required to get accurate, absolute Mw data by this technique, but values for Mn and Đ depend on separation quality (see sections The effect of separation quality on SEC-MALS and Dispersity in SEC and SEC-MALS). However, instrument calibration of the MALS detector is still needed on a routine basis. In our case, the manufacturer recommends annual calibration using toluene filtered through a 0.02 μm syringe filter.Misconception: SEC-MALS provides absolute Mw, Mn, and Đ values
In fact, the only absolute value that SEC-MALS provides is the Mw value for the injected sample. Values for Mn, and therefore Đ, both depend on the quality of the separation. See the sections The effect of separation quality on SEC-MALS and Dispersity in SEC and SEC-MALS for more discussion on this topic, including how separation quality influences Mn values but not Mw values.Misconception: SEC-MALS does not accurately measure branched polymers or polymers of other non-linear topologies
SEC-MALS determines the Mw of a sample regardless of topology, even in the case of highly branched polymers. As in linear polymers, the apparent Mn value increases with decreasing separation quality (see section The effect of separation quality on SEC-MALS). Branched polymers present challenges in separation that do not exist in linear polymers—for example, branched polymers have smaller hydrodynamic volumes than analogous linear polymers of equivalent molecular weight, so differences in branching structures among macromolecules in a polymer sample can lead to poor separation. Gaborieau and Castignolles discussed these differences in detail in a 2011 review.60 Regardless of separation quality, which influences the Mn value, SEC-MALS can accurately determine Mw for non-linear topologies. We demonstrate this fact in Fig. 8, which compares the SEC traces of linear and bottlebrush PS samples, where the molar mass values are very similar despite the different elution times of the two samples. When applying column calibration using narrowly dispersed PS standards to the dRI chromatogram from this bottlebrush polymer sample, an erroneous Mn value of 69 kg mol−1 was determined.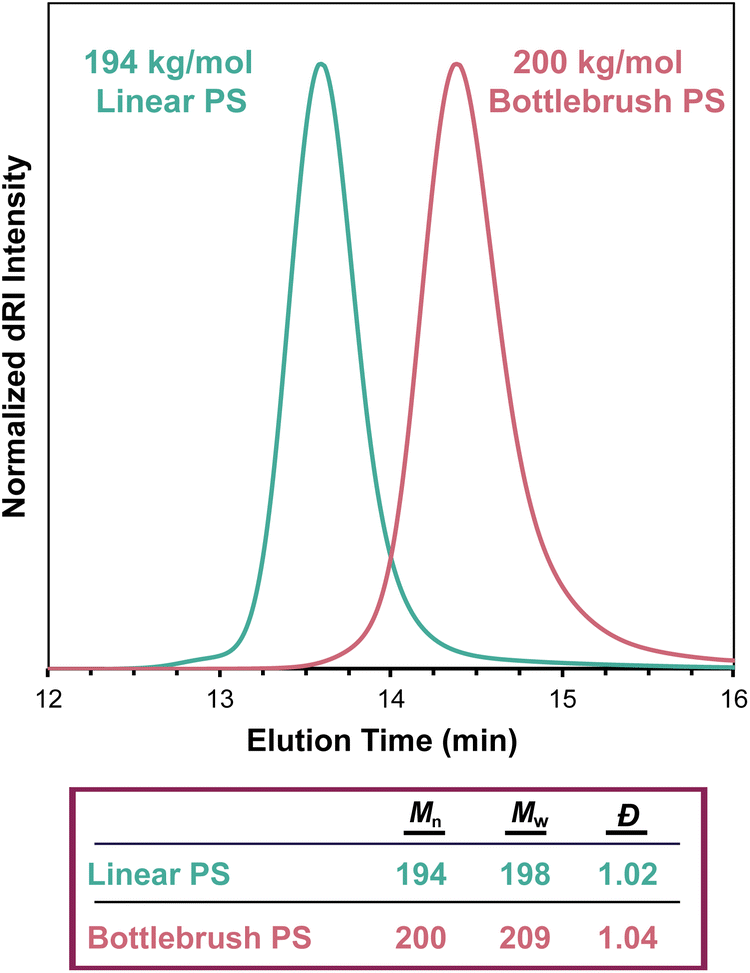 | ||
| Fig. 8 SEC-MALS chromatograms of a 194 kg mol−1 linear PS sample (teal trace, nominal 200 kg mol−1 standard) and a 200 kg mol−1 bottlebrush PS sample (pink trace), showing the difference in elution time as a result of polymer topology. | ||
Misconception: static light scattering and dynamic light scattering provide the same information
MALS uses static light scattering to determine the Rg (radius of gyration). In contrast, dynamic light scattering measures the time-dependent fluctuations in scattering intensity to determine a diffusion coefficient, from which Rh (hydrodynamic radius) is determined. Consequently, DLS requires accurate knowledge of the solution viscosity and temperature, whereas MALS requires accurate knowledge of the solution concentration and dn/dc value. Thus, the techniques provide related but distinct information. We refer the reader to separate reviews on DLS analysis.22,23Conclusions
SLS and SEC-MALS have emerged as powerful tools in polymer science, enabling precise absolute molecular weight characterization of polymers and proteins. Familiarity with the concepts underlying these measurements allows researchers to produce key insights into the structure of newly synthesized materials. We hope that this tutorial will clarify persistent misconceptions in this area and inspire young researchers to confidently use these techniques in their own work.Experimental section
Materials
All reagents were obtained from commercial vendors and used as received unless otherwise stated. The 30 kg mol−1 (listed Mw = 28.5 kg mol−1, Đ = 1.01) and 200 kg mol−1 (listed Mw = 206 kg mol−1, Đ = 1.01) PS standards were manufactured by Pressure Chemical Co. The narrowly dispersed PS standards used for SEC with conventional calibration were manufactured by Shodex (listed Mp values = 1.31, 3.95, 13.9, 55.1, 197, 591, and 3640 kg mol−1). HPLC-grade THF used for SEC and SEC-MALS was stabilized with 0.025 wt% butylated hydroxytoluene (BHT) as an inhibitor. Styrene was passed through a plug of basic alumina to remove radical inhibitors before use. Grubbs’ 3rd generation catalyst (G3) was prepared as previously described.61,62Synthesis and characterization methods
The polydisperse PS sample used in the SEC-MALS experiments presented here was synthesized using a suspension polymerization method. In brief, polyvinyl alcohol (PVA, manufacturer stated average Mw = 31–51 kg mol−1, 1.8 g) and NaCl (7.9 g, 130 mmol) were dissolved in DI water (240 mL) and added to a round-bottom flask equipped with a mechanical stirrer. Next, a solution of 2,2′-azobis(2-methylpropionitrile) (AIBN, 95 mg, 580 μmol) in styrene (22 mL, 190 mmol) was added, and the reaction mixture was heated to 80 °C while stirring. After 6 h, the reaction mixture was filtered to isolate the crude polymer product. The crude product was dissolved in CHCl3 (∼10 mL), then precipitated into hot DI water (90 °C, 1 L) while stirring to remove residual PVA, then filtered after 45 min and dried under vacuum.The 200 kg mol−1 bottlebrush PS sample was synthesized via grafting-through ring-opening metathesis polymerization (ROMP) of a norbornene macromonomer (50 mg, 8.6 μmol, Mn = 6.1 kg mol−1, Đ = 1.02) initiated by G3 (0.18 mg from a stock solution, 0.25 μmol) in EtOAc (0.43 mL), based on a previously reported procedure.63 The norbornene macromonomer was prepared by atom-transfer radical polymerization (ATRP) of styrene (30 mL, 262 mmol) with a norbornene-derived initiator (0.13 g, 0.48 mmol), CuBr (34 mg, 0.24 mmol), CuBr2 (53 mg, 0.24 mmol), and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 0.1 mL, 0.48 mmol) in DMF (9 mL) for 15 h at 90 °C, similar to previous reports.63,64
SEC-MALS characterization was carried out in THF (stabilized with 0.025 wt% BHT) at 1 mL min−1 at 30 °C using a Shimadzu LC-20AD HPLC equipped with an Agilent PLgel MIXED 10 μm guard column, two Agilent PLgel 10 μm MIXED-B columns (except where noted), a Wyatt DAWN HELEOS-II MALS detector, and a Wyatt Optilab T-rEx dRI detector. Although the MALS detector is calibrated annually, no polymeric calibration standards were used, and the known dn/dc value for polystyrene in THF of 0.185 mL g−1 was used to obtain absolute Mw values, except where noted.29
SEC with conventional calibration was performed on the same SEC system described above, using only the dRI chromatogram. A mixture of six narrowly dispersed PS standards (described in the materials section) was dissolved in the mobile phase and injected as a single sample. Analysis was performed using Wyatt's ASTRA software to generate a column calibration plot relating Mn to elution time.
All samples for SEC and SEC-MALS characterization were weighed into a glass vial, dissolved in the mobile phase (THF with 0.025 wt% BHT), then filtered through a 0.22 μm PTFE syringe filter using a glass syringe, unless otherwise specified. For the consumables contamination experiment (Fig. 5), 30 kg mol−1 PS standards were dissolved in the mobile phase and allowed to sit overnight at room temperature in their respective vials, syringes, or tubes before being filtered through a 0.22 μm PTFE syringe filter into an autosampler vial.
Offline experiments to determine dn/dc values were performed using a Wyatt Optilab T-rEx dRI detector equipped with a New Era InfusionOne syringe pump. THF was injected at the beginning and end of the experiment to establish a baseline. A series of solutions of known concentrations (0.5, 1, 2, 3, and 5 mg mol−1 PS in THF) was injected at 0.5 mL min−1 to determine the dRI value at each concentration (see Fig. 1A).
UV-vis measurements were carried out on a Cary 60 UV-Vis spectrophotometer. A stock solution of dye was made by adding 0.023 g of eosin Y (0.033 mmol) to a 200 mL volumetric flask, then dissolving in H2O. A series of solutions ranging from 2 to 20 μM were made from the stock solution using 20 mL volumetric flasks. Absorbance was measured from 400 to 600 nm at 1 nm intervals. The graph in Fig. 1B was then constructed by plotting the baseline-subtracted absorbance at 517 nm versus concentration for each solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Mohammed Alaboalirat and Dr Samantha J. Scannelli for synthesizing the macromonomer and bottlebrush polymer used in this work, as well as several employees of Wyatt Technology for their helpful suggestions on SEC-MALS experiments in our laboratories over the years. We also thank Professor C. Adrian Figg, Dr Brady A. Hall, and Jared G. Baker for critical reading and helpful suggestions on this manuscript. We appreciate the US National Science Foundation (CHE-2003662, DMR-2104602, CHE-2237487), the US Department of Energy (Office of Basic Energy Sciences under award number DE-SC0023035), the US National Institutes of Health (R01GM123508), and GlycoMIP (Cooperative Agreement DMR-1933525) for financial support of our research efforts in this area.References
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS PubMed.
- A. Plichta, M. Zhong, W. Li, A. M. Elsen and K. Matyjaszewski, Marcomol. Chem. Phys., 2012, 213, 2659–2668 CAS.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- C. W. Pester and E. M. Benetti, Adv. Mater. Interfaces, 2022, 9, 2201439 CrossRef CAS.
- D. T. Gentekos, L. N. Dupuis and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 1848–1851 CrossRef CAS PubMed.
- M. Rubens and T. Junkers, Polym. Chem., 2019, 10, 5721–5725 RSC.
- M. Rubens and T. Junkers, Polym. Chem., 2019, 10, 6315–6323 RSC.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley-Interscience, New York, 4th edn, 2003 Search PubMed.
- P. J. Wyatt, Anal. Chim. Acta, 1993, 272, 1–40 CrossRef CAS.
- S. Podzimek, in Multiple Detection in Size-Exclusion Chromatography, American Chemical Society, 2004, ch. 5, vol. 893, pp. 94–112 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- J. W. Strutt, London, Edinburgh Dublin Philos. Mag. J. Sci., 1871, 41, 107–120 CrossRef.
- L. Rayleigh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1881, 12, 81–101 CrossRef.
- L. Rayleigh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1899, 47, 375–384 CrossRef.
- A. D. Del Genio, Scientific American, 2003 Search PubMed.
- J. H. Williams, Why is the sky blue?, IOP Publishing, 2021 Search PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS PubMed.
- R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351–353 CrossRef.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., 2004 Search PubMed.
- B. H. Zimm, J. Chem. Phys., 1948, 16, 1093–1099 CrossRef CAS.
- B. H. Zimm, J. Chem. Phys., 2004, 16, 1093–1099 CrossRef.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- S. Mori and H. G. Barth, Size Exclusion Chromatography, Springer Berlin, Hiedelberg, 1999 Search PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- R. J. Carrazzone, X. Li, J. C. Foster, V. V. S. Uppala, C. E. Wall, A. R. Esker, L. A. Madsen and J. B. Matson, Macromolecules, 2021, 54, 6975–6981 CrossRef CAS PubMed.
- S. Podzimek, J. Appl. Polym. Sci., 1994, 54, 91–103 CrossRef CAS.
- D. Berek, J. Sep. Sci., 2010, 33, 315–335 CrossRef CAS PubMed.
- A. Striegel, Carbohydr. Polym., 1997, 34, 267–274 CrossRef CAS.
- Y. Huang, L. Bu, D. Zhang, C. Su, Z. Xu, L. Bu and J. W. Mays, Polym. Bull., 2000, 44, 301–307 CrossRef CAS.
- K. E. S. Locock, L. Meagher and M. Haeussler, Anal. Chem., 2014, 86, 2131–2137 CrossRef CAS PubMed.
- R. Stepto, T. Chang, P. Kratochvil, M. Hess, K. Horie, T. Sato and J. Vohlidal, Pure Appl. Chem., 2015, 87, 71–120 CrossRef CAS.
- A. Rudin and H. L. W. Hoegy, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 217–235 CrossRef CAS.
- H. C. Benoit, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1703–1704 CrossRef CAS.
- R. R. Chance, S. P. Baniukiewicz, D. Mintz, G. V. Strate and N. Hadjichristidis, Int. J. Polym. Anal. Charact., 1995, 1, 3–34 CrossRef CAS.
- F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738–6745 CrossRef CAS.
- S. Podzimek, J. Appl. Polym. Sci., 2014, 131, 40111 CrossRef.
- S. Podzimek, J. Appl. Polym. Sci., 2019, 136, 47561 CrossRef.
- Y. Guillaneuf and P. Castignolles, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 897–911 CrossRef CAS.
- J. L. Baumgarten, J. P. Busnel and G. R. Meira, J. Liq. Chromatogr. Relat. Technol., 2002, 25, 1967–2001 CrossRef CAS.
- D. Konkolewicz, J. W. Taylor, P. Castignolles, A. Gray-Weale and R. G. Gilbert, Macromolecules, 2007, 40, 3477–3487 CrossRef CAS.
- S. Harrisson, Polym. Chem., 2018, 9, 1366–1370 RSC.
- R. A. Fair, R. Xie, Y. Lee, R. H. Colby and E. D. Gomez, ACS Appl. Polym. Mater., 2021, 3, 4572–4578 CrossRef CAS.
- K. Philipps, T. Junkers and J. J. Michels, Polym. Chem., 2021, 12, 2522–2531 RSC.
- B. Coto, J. M. Escola, I. Suárez and M. J. Caballero, Polym. Test., 2007, 26, 568–575 CrossRef CAS.
- M. J. Leamen, N. T. McManus and A. Penlidis, J. Appl. Polym. Sci., 2004, 94, 2545–2547 CrossRef CAS.
- D. C. Bugada, R. Gagnon and A. Rudin, J. Appl. Polym. Sci., 1987, 34, 501–505 CrossRef CAS.
- X. Chen, Z. Xu, N. Hadjichristidis, L. J. Fetters, J. Carella and W. W. Graessley, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 777–779 CrossRef CAS.
- A. Striegel, Anal. Chem., 2005, 77, 104 A–113 A CrossRef CAS.
- D. Some, WP9002: Fluorescent macromolecules and nanoparticles: characterization of molar mass, size and charge, https://wyattfiles.s3-us-west-2.amazonaws.com/literature/white-papers/WP9002-Characterization-of-fluorescent-macromolecules-and-nanoparticles.pdf.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- D. Dong and A. L. Fricke, J. Appl. Polym. Sci., 1993, 50, 1131–1140 CrossRef CAS.
- Z. A. Pittman, M. E. McCarthy, M. R. Birtwistle and C. L. Kitchens, Biomacromolecules, 2022, 23, 3743–3751 CrossRef CAS PubMed.
- C. Jackson, Y.-J. Chen and J. W. Mays, J. Appl. Polym. Sci., 1996, 61, 865–874 CrossRef CAS.
- M. Gaborieau and P. Castignolles, Anal. Bioanal. Chem., 2011, 399, 1413–1423 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS PubMed.
- J. Liu, A. X. Gao and J. A. Johnson, J. Visualized Exp., 2013, e50874 Search PubMed.
- S. J. Scannelli, M. Alaboalirat, D. Troya and J. B. Matson, Macromolecules, 2023, 56, 3838–3847 CrossRef CAS.
- M. Alaboalirat, L. Qi, K. J. Arrington, S. Qian, J. K. Keum, H. Mei, K. C. Littrell, B. G. Sumpter, J.-M. Y. Carrillo, R. Verduzco and J. B. Matson, Macromolecules, 2019, 52, 465–476 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |