
Genetically engineered cell-derived nanovesicles for cancer immunotherapy
Shan
He
a and
Zongmin
Zhao
 *ab
*ab
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Illinois Chicago, Chicago, IL 60612, USA. E-mail: zhaozm@uic.edu
bTranslational Oncology Program, University of Illinois Cancer Center, Chicago, IL 60612, USA
First published on 25th March 2024
Abstract
The emergence of immunotherapy has marked a new epoch in cancer treatment, presenting substantial clinical benefits. Extracellular vesicles (EVs), as natural nanocarriers, can deliver biologically active agents in cancer therapy with their inherent biocompatibility and negligible immunogenicity. However, natural EVs have limitations such as inadequate targeting capability, low loading efficacy, and unpredictable side effects. Through progress in genetic engineering, EVs have been modified for enhanced delivery of immunomodulatory agents and antigen presentation with specific cancer targeting ability, deepening the role of EVs in cancer immunotherapy. This review briefly describes typical EV sources, isolation methods, and adjustable targeting of EVs. Furthermore, this review highlights the genetic engineering strategies developed for delivering immunomodulatory agents and antigen presentation in EV-based systems. The prospects and challenges of genetically engineered EVs as cancer immunotherapy in clinical translation are also discussed.
 Shan He | Dr Shan He is currently a postdoc fellow in the Department of Pharmaceutical Sciences at the University of Illinois Chicago. Dr He received her Ph.D. degree from the University of Hong Kong. Her current research focuses on the design and construction of bioinspired drug/gene delivery systems for cancer immunotherapy. |
 Zongmin Zhao | Dr Zongmin Zhao is an Assistant Professor in the Department of Pharmaceutical Sciences at the University of Illinois Chicago. Dr Zhao received his Ph.D. degree from Virginia Tech and was a postdoctoral associate at Harvard University. His research group focuses on developing cell-inspired technologies to improve the treatment of cancer, autoimmune diseases, and acute injuries. Dr Zhao has published over 60 articles in journals including Cell, Nature Biomedical Engineering, PNAS, Science Advances, Advanced Materials, etc. Dr Zhao is currently an Associate Editor of Bioengineering & Translational Medicine (Wiley). |
1. Introduction
Naturally derived nanomaterials like cell-based nanovesicles are gaining dramatic attention owing to their favorable biocompatibility, diminished immunogenicity, extended blood circulation time, and appropriate nano-scale dimensions, positioning them as attractive candidates for drug delivery. Extracellular vesicles (EVs), which encompass different types of membrane-enclosed particles secreted by cells into the extracellular milieu, play an important role in cell-to-cell communication.1,2 EVs can be classified into three major subclasses based on their size: small EVs (sEVs with <200 nm), large EVs (lEVs with 200–1000 nm), and apoptotic bodies (ABs with 100 nm–5 μm).3 Large EVs, known as microvesicles, are generated through direct outward budding of the plasma membrane. Apoptotic bodies are produced by cells undergoing apoptosis. Exosomes, a subset of small EVs, exhibit sizes between 50 and 150 nm. Their biogenesis commences with inward budding of the plasma membrane, followed by the formation of intraluminal vesicles during endosomal processing and release into the extracellular space (Fig. 1). As such, exosomes encapsulate both membranous and cytosolic molecules, making them captivating subjects in diverse biomedical domains in recent decades.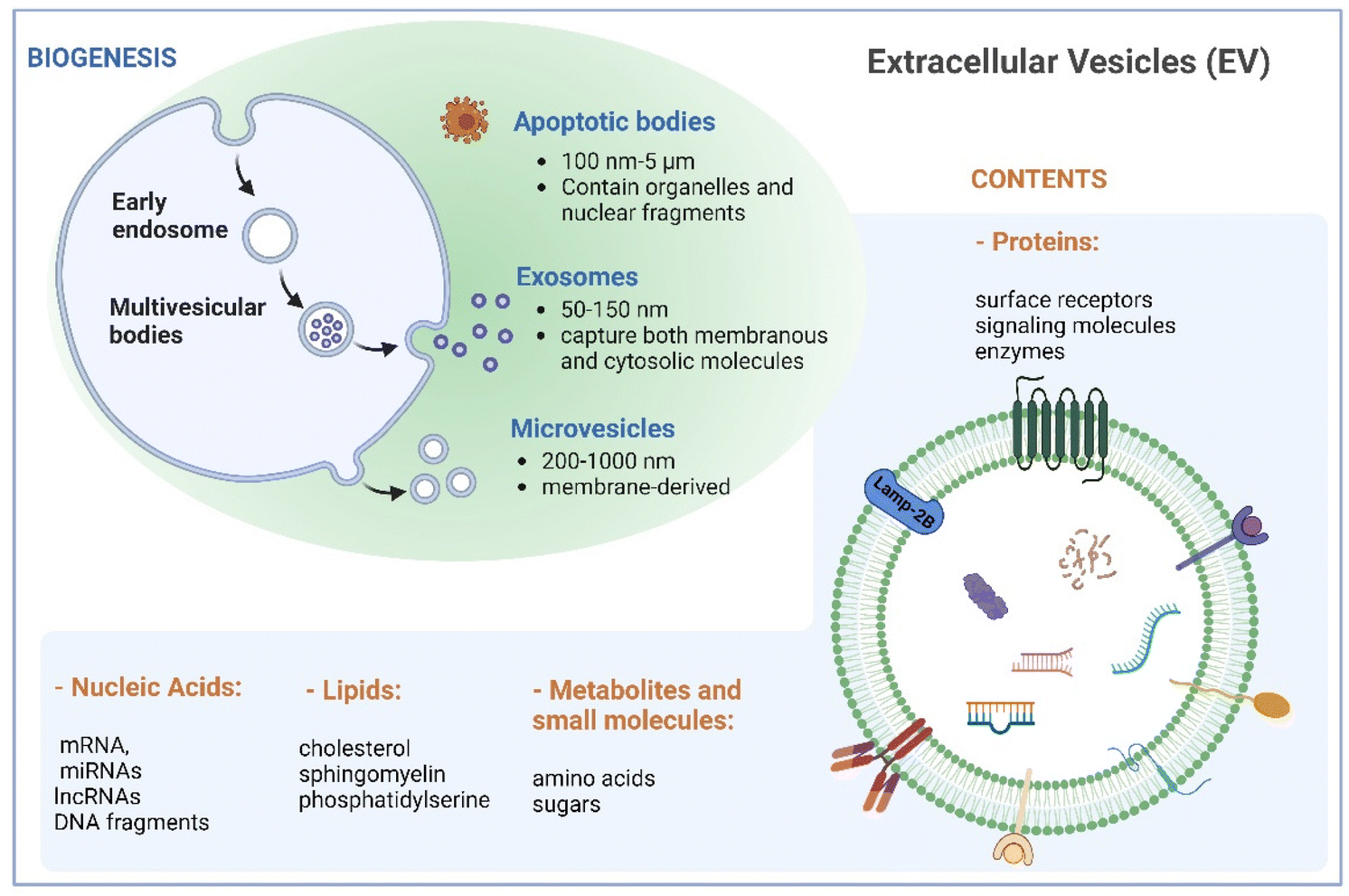 | ||
| Fig. 1 Biogenesis and characteristics of extracellular vesicles. Created with BioRender. | ||
Cancer immunotherapy, aiming to improve the ability of the immune system to recognize and eliminate tumors, represents one of the most rapidly advancing domains in clinical medicine. Steady development of cancer immunotherapy involves leveraging various elements of the immune system to treat tumors, including antibodies, cells, and proteins.3,4 Notable advancements have been made through approaches such as adoptive cell therapy based on the injection of tumor-activated T cells and checkpoint blockade inhibitors, which prevent the deactivation of cytotoxic T cells by blocking specific inhibitory receptor interactions. These strategies have significantly improved the long-term survival rates of patients with aggressive cancers.5,6
With recent advancements introducing EVs as promising candidates for various therapeutic and diagnostic applications, their utilization in cancer immunotherapy has garnered advantages. In comparison with conventional cell-based immunotherapy that requires meticulous and time-sensitive preparation, EV-based therapy is cell-free and thereby offers a more practical approach by exhibiting enhanced stability for extended storage without compromising efficacy.7–9 In addition, EVs outperform synthetic nanocarriers like liposomes or polymeric nanoparticles in terms of lower toxicity to healthy cells and environmentally friendly isolation, as they do not require organic solvents and various chemical materials for isolation.10 EVs originating from a specific source inherently carry biologically active molecules such as nucleic acids, proteins, and glycans, establishing their nature as connate carriers. Moreover, they can be modified via different methods to deliver various functional cargoes and target specific signaling pathways. However, although EVs have shown promise as versatile therapeutics, the optimal cargo composition and loading strategies for maximizing their efficacy in cancer immunotherapy remain to be determined. Additionally, strategies to enhance the targeting abilities of EVs while minimizing off-target effects need further investigation. Different aspects of EV-based systems for therapeutic applications have been discussed in many recent reviews.11–13 In this review, we specifically focus on and discuss recent advances in developing genetic engineering strategies for boosting EV-mediated cancer immunotherapy. Our discussions will help to identify key knowledge gaps in more effectively engineering EVs via genetic engineering as well as provide useful perspectives in introducing new functionalities to EVs as a powerful tool for cell-free cancer immunotherapy.
2. Extracellular vesicles for cancer therapy
2.1 Cell sources
Several studies have demonstrated that miRNAs encapsulated in native EVs derived from MSCs, such as miR-7-5p, miR-146b, miR-142-3p, and miR-let-7c, are capable of suppressing the development and metastasis of cancer.16–18 For example, a recent study19 by Wang et al. elucidated that miR-30b-5p shuttled by bone marrow MSC-derived exosomes could promote cell apoptosis and prevent tumorigenesis in non-small cell lung cancer (NSCLC) via the EZH2/PI3K/AKT axis. Conversely, a number of studies indicated the tumor-promoting effect of MSC-derived EVs,20–23 likely due to the enriched presence of tumor-supportive miRNAs (e.g., miRNA-21 and 34a) and proteins (e.g., PDGFR-β, TIMP-1, and TIMP-2).24 Ren et al.20 reported that EVs isolated from human bone marrow-derived MSCs significantly increased tumor growth, intra-tumoral angiogenesis as well as macrophage M2 polarization via miR-21-5p delivery. In another study, Biswas et al.21 showed that exosomes secreted by tumor-educated MSCs induced differentiation of monocytic myeloid-derived suppressor cells (MDSCs) into highly immunosuppressive M2 macrophages at tumor sites and thereby promoted breast cancer progression. Apart from regulating macrophage phenotypes, MSC-derived EVs have also been demonstrated to play a role in suppressing the proliferation and function of other immune cells, including natural killer (NK) cells, dendritic cells (DCs), T cells and B cells.25,26 In some cases, they could likely induce apoptosis in activated immune cells, leading to inhibited immune responses.
Apart from utilizing the intrinsic payload, MSC-derived EVs have been used as a carrier for anti-tumor immunotherapy. Deng et al.27 recently demonstrated the potential of EV-mediated delivery of CD38 siRNA to overcome the resistance of tumors to PD-1/PD-L1 inhibitors by promoting macrophage repolarization to the M1 phenotype.
In addition to employing naturally secreted EVs, parental cells or modified EVs post-isolation can be engineered to endow EVs with specific characteristics or cargoes to enhance their immunotherapeutic potential. Genetic engineering represents one versatile and effective strategy with bare limitations in cell sources. The HEK 293T cell line has been a popular candidate for its great transfectability to be a universal donor of EVs with customized properties. Of note, another source receiving great attention is red blood cells (RBCs) as they are readily available from the blood bank, thereby allowing large-scale EV production; additionally, they are enucleated cells devoid of DNA, causing no safety concern of horizontal gene transfer.66 Le and coworkers has developed a strategy to generate scalable amounts of RBC-derived EVs for the delivery of therapeutic RNA drugs such as antisense oligonucleotides (ASOs),67 siRNA,68 and Cas9 messenger RNA66 for cancer therapy with low cytotoxicity. The discussed cell sources and their characteristics are briefly summarized in Fig. 2.
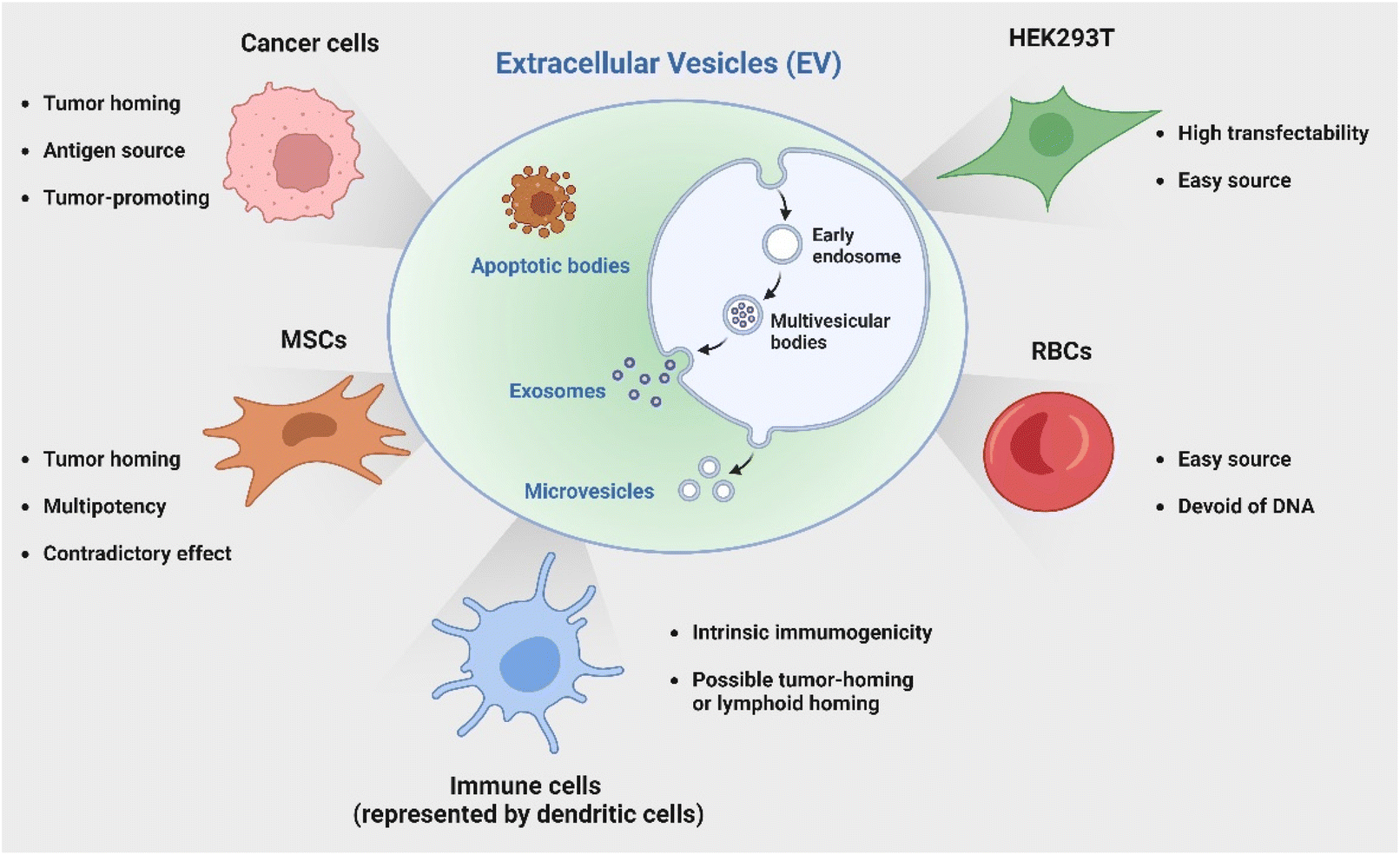 | ||
| Fig. 2 Various cell sources for the production of EVs for cancer immunotherapy. Created with BioRender. | ||
2.2 Isolation approaches
Although EVs have many appealing merits for cancer immunotherapy, a standardized isolation and purification procedure with high efficiency and simplicity remains one of the major challenges hindering their clinical translation.The most common approach to isolate EVs involves ultracentrifugation (UC) to process cell culture supernatants containing released EVs.69–71 With a series of centrifugation steps at progressively higher g-force, cell debris, large particles and some other unwanted particles can be removed, and EV pellets can be obtained. Disadvantages of this method include incomplete isolation, possible protein aggregates and damage with the ultra-high shear stress.72 Another established method, density-gradient centrifugation (DGC), based on ultracentrifugation utilizes density gradients induced by sucrose or iodixanol to separate components of different buoyant densities, thereby enabling efficient purification of EVs from protein aggregates,33,67 whereas this method requires a long processing time and expensive equipment, limiting its broad employment for scale-up applications.
Size exclusion chromatography (SEC), also known as spatial or molecular exclusion chromatography, uses a column with porous beads to allow small EVs to enter and entrap in the pores.73 This method is frequently used in combination with ultracentrifugation. It gives rise to higher yields and more intact physiological conditions of EVs. Similarly, ultrafiltration by using membrane filters with defined molecular weight cutoffs to concentrate and isolate EVs based on their size is an easy-to-use method but may cause EV deformation upon filtration. The tangential flow filtration (TFF) method utilizes a cutoff filter similar to ultrafiltration but is specifically designed to permit the discharge of waste in a direction orthogonal to the sample flow.74 This design prevents the formation of a filter cake and yields relatively high-purity EVs while continuously removing smaller molecules and contaminants.
Besides physiochemical properties, biological markers on the surface of EVs have been taken into consideration to develop isolation and purification methods. Immunoaffinity capture is a method that allows the selective capture of EVs expressing specific markers by antibodies that are generally prefixed to a particular carrier, such as magnetic particles or chromatography matrices. Currently, the most commonly used carrier is immunomagnetic beads. Clayton et al.75 have isolated EVs derived from APCs using magnetic beads coated with antibodies specific for MHC class II. Similarly based on immunoaffinity, a strategy using microfluidic devices with antibody-coated chip surfaces for EV isolation has been described in recent years.76–78 Kanwar et al. developed a platform called ExoChip to specifically capture CD63-specific exosomes from serum in an hour.77 With narrow channels interconnected with circular chambers, an extended retention time of exosomes in the channels led to more efficient capture. Moreover, the device could simultaneously quantify the isolated exosomes. However, EVs captured by specific antibodies can only induce a very small yield since they cannot be easily eluted as intact vesicles.
2.3 Specific target
It is widely acknowledged that EVs can inherit targeting ability from their parental cells, exemplified by the tumor-homing ability of M1 macrophage-derived EVs45 and the lymphoid homing capability of EVs derived from the mature DCs.79 Despite this, a comparative study has revealed that systemic administration of EVs derived from most cell lines induces preferential accumulation in the liver, lungs, spleen and gastrointestinal tract.80 To confer the targeting capabilities of EVs to other tissues or specific cell types, introducing specific targeting moieties to the EV surface is necessary. It has been validated that 80% of the proteins in exosomes are highly conserved among different cell types, some of which are used as the biomarkers of exosomes, including lysosome-associated membrane glycoprotein 2b (Lamp2b), TSG101, heat shock proteins, and the tetraspanins CD63, CD9, and CD81. Inspired by this concept, a well-studied strategy to endow EVs with targeting properties is to fuse targeting moieties with EV membrane proteins. This approach was used by Alvarez-Ervit et al., who engineered DCs to express a fusion protein of Lamp2b and brain-specific rabies viral glycoprotein (RVG), a peptide that binds the acetylcholine receptor.81 EVs isolated from these cells demonstrated increased accumulation in the brains of mice and successfully delivered the siRNA of interest, manifesting the potential of EVs as targeted drug delivery systems. The same approach was used by Tian et al. to achieve targeted delivery of doxorubicin to integrin-positive breast cancer cells in vitro and in vivo by exosomes that overexpress the αγ integrin-specific RGD peptide fused to Lamp2b.82 When using such a strategy to introduce target moieties, the choice of EV sorting domains should be considered, i.e., the surface protein that is known to be enriched on EVs, according to the cell source. For instance, in HEK 293T cells, fusing moieties to the tetraspanin CD9, CD63 or CD81 would result in a significantly higher expression on EVs as compared to fusing to Lamp2b or the C1C2 domain of lactadherin.83 Apart from utilizing exosomal surface proteins, foreign membrane proteins can be employed for the conjugation of targeting moieties. For example, Wang et al. designed a plasmid encoding fusion proteins of VSV-G fusogen and anti-GPC3 scFv, which can efficiently bind HCC cells.84 After lentiviral transduction, the obtained vesicles presented anti-GPC3 on their surface and showed significantly higher accumulation in HepG2 cells both in vitro and in vivo, thus facilitating anti-cancer targeted drug delivery.In the context of cancer immunotherapy, exosomes have been utilized for antibody display to activate T cells. A method of crosslinking T cells and cancer cells has been described by co-displaying anti-CD3 and anti-EGFR single-chain variable fragments (scFvs) on a construct of the transmembrane domain of human platelet-derived growth factor receptor (PDGFR), commercialized as pDisplay.85 The transfected cells expressing the fusion construct produced EVs containing both antibodies against CD3 and EGFR. In such a way, the engineered EVs could redirect and activate cytotoxic T cells against triple-negative breast cancer (TNBC) cells, eliciting potent antitumor immunity both in vitro and in vivo. Likewise, in a more recent study, Stranford et al. utilized the pDisplay system to passively load high-affinity T cell-targeting domains on the EV surface, enabling the targeted delivery of biomolecules to T cells.86 Specifically, the engineered EVs carrying substantially expressed anti-CD2 scFvs showed a 100-fold increase in binding efficiency for CD2+ Jurkat T cells than the non-targeted EVs (Fig. 3).
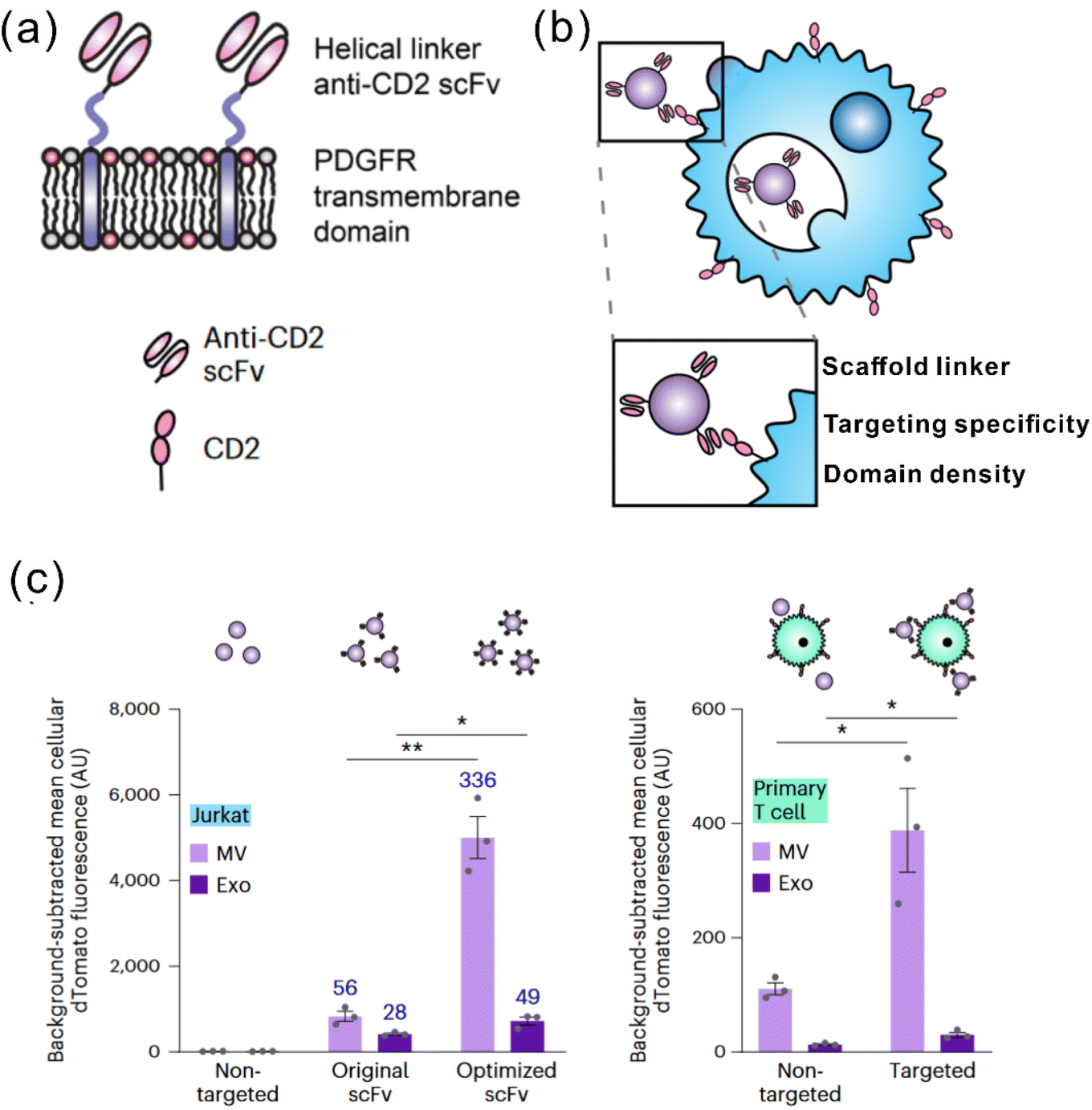 | ||
| Fig. 3 A representative study employing pDisplay to achieve the targeting ability of EVs to T cells. (a) Illustration of the structures of the PDGFR transmembrane domain for anti-CD2 scFv display. (b) Illustration of EVs achieving targeting to T cells through antibody–receptor interactions. (c) Targeting EVs can efficiently bind to both Jurkat T cells and primary human CD4+ T cells. Adapted from ref. 86 with permission from Springer Nature, copyright 2023. | ||
A more straightforward strategy to achieve the targeting ability of EVs involves an anchor peptide of CD63. Gao et al. reported a particular peptide, CP05, that shows a strong affinity towards the second extravesicular loop of CD63.87 Direct painting of exosomes with different moieties conjugated to CP05 redirected systemic distribution of exosomes to specific tissues. As a proof of concept, loading of a phosphorodiamidate morpholino oligomer on exosomes via CP05 at extremely low doses achieved elevated dystrophin expression in muscle with functional improvement without any detectable toxicity. This strategy provides a new avenue for exosome engineering, effective conjugation of targeting moieties,88 cargo delivery33 and surface modification30 with great simplicity, opening a new avenue for exosome engineering.
2.4 Biomimetic nanovesicles
Despite substantial advancements in the field of naturally-derived EVs, their relatively low yield remains a substantial impediment to widespread applications. Accordingly, various strategies have been developed to address the technical issues. Among them, biomimetic nanovesicles, also known as cell-derived nanovesicles (CNVs), have emerged as a promising alternative to EVs. CNVs are typically generated through mechanical extrusion of donor cells using nanosized filters or microchannels within microfluidic devices, resulting in a substantially higher yield.46,84,89–91 Notably, CNVs closely resemble EVs in terms of size and protein components. Studies have revealed that CNVs share more than 70% of the same membrane proteins found in naturally occurring EVs and their batch-to-batch variation can be well-controlled and minimized to 10%.90 In subsequent sections of this review, CNVs will be discussed within the broader context of EVs.A study by Jang et al. reported a 100-fold increase in the yield of CNVs compared to that of exosomes released from the same number of cells.89 This method relies on the serial extrusion of cells through filters with increasingly smaller pore sizes to generate CNVs. To validate the therapeutic efficacy, CNVs produced from macrophages were loaded with doxorubicin to treat immunocompetent CT26 mouse tumors. Encouragingly, CNVs showed similar anti-tumor effects to exosomes harvested from the same cells and caused minimal systemic side effects even with a 20-fold higher dose administration of the free drug. Employing this technique, Yang et al. generated CNVs from non-tumorigenic epithelial MCF-10A cells for siRNA delivery.91 The yield of the nanovesicles reached around 150-fold higher that of exosomes from the same cell source. Several exosomal biomarkers, including CD63, CD9, CD81and TSG101, were detected on the surface of both vesicle types, but at higher levels on secreted exosomes as anticipated. With a siRNA against cyclin-dependent kinase 4 (CDK4) loaded via electroporation, CNVs could significantly downregulate the CDK4 expression in vitro and in vivo, validating their potential as effective EV substitutes for drug delivery systems.
A recent study by Wang et al. further equipped CNVs with efficient cytosolic delivery and target specificity by genetically engineering the producer cells, i.e., HEK 293, to drive the co-expression of a viral fusogen, vesicular stomatitis virus G protein (VSV-G), and a cancer cell targeting moiety GPC3.84 The all-in-one CNV platform demonstrated effective and selective binding to tumor cells, fulfilling endosomal escape and cytosolic delivery of different therapeutics, such as siRNA, gelonin, and paclitaxel. Ultimately, drug-loaded CNVs resulted in an enhanced tumor treatment efficacy compared to the free drug as well as nanovesicles without targeting moieties or fusogens.
3. Genetically engineered EVs for cancer immunotherapy
Genetic engineering has undergone a revolutionary advancement in its fundamental technology, positioning it as a versatile tool across a broad spectrum of biomedical applications in recent decades. To broaden and improve the functionalities of EVs, genetic engineering has been substantially employed for EV modification. In addition to the previously discussed targeting capabilities, EVs have been genetically engineered to modulate immunity within the context of cancer immunotherapy (Fig. 4). This review delves into two primary engineering directions: immunomodulatory drug delivery and antigen presentation.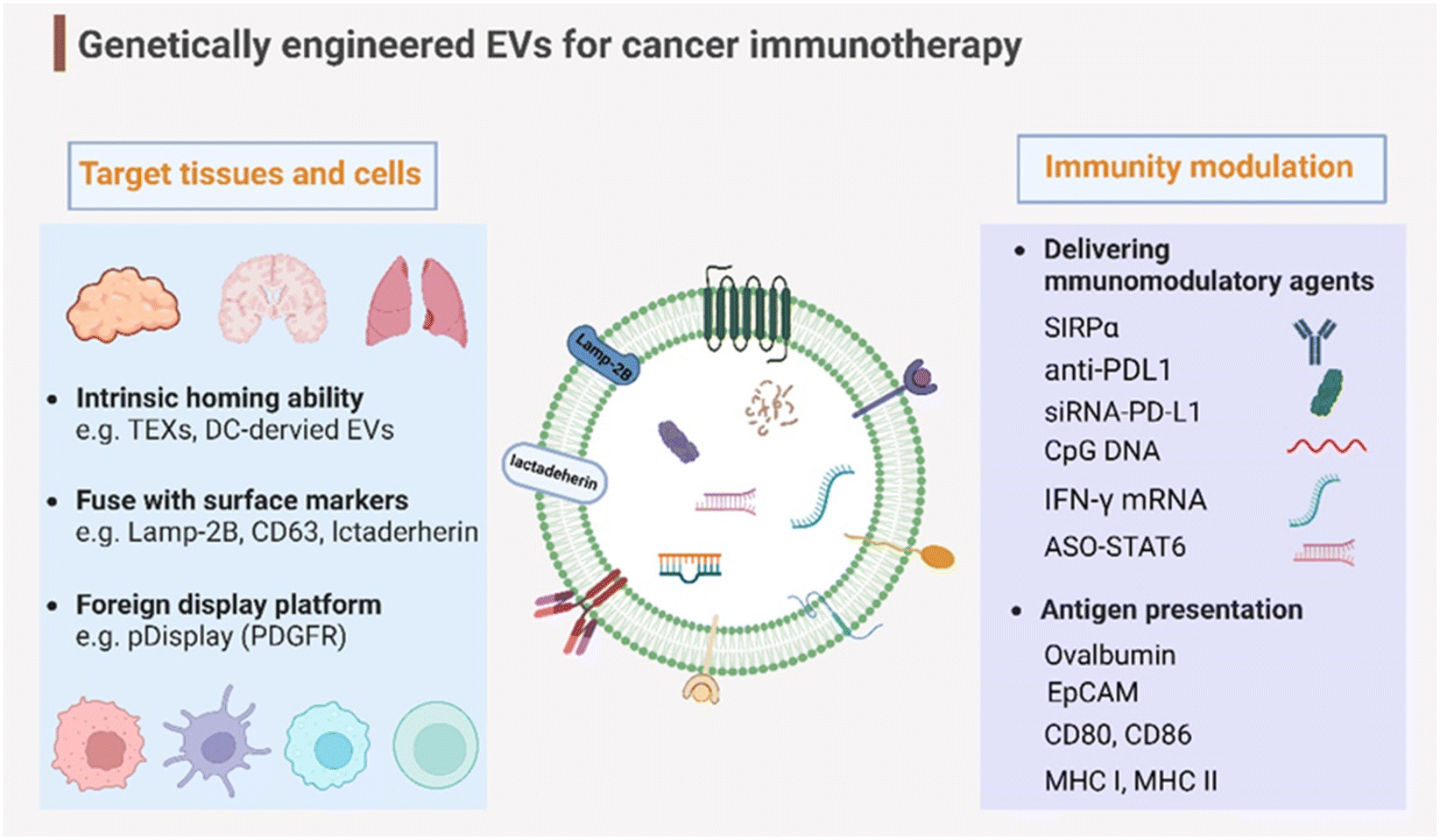 | ||
| Fig. 4 Genetic engineering endows EVs with targeting capabilities to specific tissues and organs and immunity modulation for cancer immunotherapy. Created with BioRender. | ||
3.1 Drug delivery
One hallmark of EVs is that they are naturally derived, endowing them with high biocompatibility. Moreover, the lipid bilayer membrane can be a natural barrier to prevent therapeutic cargoes from possible degradation by serum protease.92 Therefore, EVs have become increasingly appealing as delivery carriers when compared to synthetic carriers. Exosomes, in particular, have been considered a natural alternative to liposomes as delivery systems because of their low immunogenicity, evasion from phagocytosis by monocytes and macrophages, and enhanced retention in circulation.93 Here, we provide an overview of two different loading strategies. One is based on modulating the producer cells, commonly by genetic engineering approaches, to favor the endogenous loading of cargoes of interest during EV biogenesis. Alternatively, the cargoes can be loaded in EVs after their isolation by physical or chemical approaches. Examples of these loading techniques are shown in Table 1.| EV type | Loading strategy | Loading method | Isolation method | Donor cell | Genetic vector | Immunomodulatory agents | Cancer model | Ref. |
|---|---|---|---|---|---|---|---|---|
| Abbreviations: CNV (cell-derived nanovesicle), TACE (Transwell®-based asymmetric cell electroporation), CNP (cellular nanoporation), UC (ultracentrifugation), DGC (density gradient centrifugation), SEC (size exclusion chromatography), TFF (tangential flow filtration), MSC (mesenchymal stem cell), BM-MSC (bone marrow-derived MSC), RBC (red blood cell), DCs (dendritic cells), NPCC (nasopharyngeal carcinoma cell), MEF (mouse embryonic fibroblasts), CML (chronic myeloid leukemia), GBM (glioblastoma multiforme), TNBC (triple-negative breast cancer). | ||||||||
| Exosome | Exogenous | Electroporation | UC | BM-MSC | Lentiviral | CD38 siRNA | Hepa1-6 xenograft | 27 |
| Exosome | Exogenous | Streptavidin–biotin | UC | B16BL6 | Synthetic ODN | CpG DNA | B16BL6 xenograft | 31 |
| Exosome | Exogenous | Electroporation | DGC | PANC-02 | Synthetic siRNA | CCL22 siRNA | PANC-02 orthotopic model | 33 |
| Exosome | Exogenous | Transfection | DGC | RBC | Synthetic siRNA | ImmRNA 3p10la9 | 4T1 and CA1a xenograft | 67 |
| Exosome | Exogenous | Electroporation | UC | MSC | Synthetic siRNA | siRNA-KrasG12d | Panc-1 and KPC689 orthotopic models | 70 |
| Exosome | Exogenous | Scaffold protein binding | UC | HEK 293T | Synthetic ASO | ASO-STAT6 | CT26 xenograft | 71 |
| CNV | Endogenous | Viral transduction and lipid transfection | UC, DGC, and extrusion | DCs | Adenoviral and mammalian expression plasmid | GFP/OVA, M27-M30-TRP2, and anti-PD1 | B16F10, MC-38, and LLC xenograft | 79 |
| Exosome | Exogenous and endogenous | Electroporation and transfection | UC | M1 macrophage | Mammalian expression plasmid | PD-L1 siRNA and VSV-G | CT26 xenograft | 95 |
| CNV | Endogenous | Viral transduction | UC and extrusion | HEK 293T | Lentiviral | aPD-L1 | LLC xenograft | 100 |
| Exosome | Exogenous and endogenous | FCR–IGG binding and nanosecond pulse electroporation | UC | MEF and HEK 293T | Mammalian expression plasmid | Anti-CD71, anti-PDL1 and IFN-γ mRNA | GL261 and SB28 GBM | 106 |
| Exosome | Endogenous | Lipid transfection | UC | HEK 293T | Mammalian expression plasmid | IL-3 and BCR-ABL siRNA | LAMA84 or K562R CML xenograft | 108 |
| Exosome | Endogenous | Viral transduction | UC | NPCC | Lentiviral | miR-142-5p | 5-8FR NPCC | 103 |
| Exosome | Endogenous | TACE | UC and TFF | MEF and hBMSC | Synthetic mRNA/siRNA | TP53 mRNA and siKrasg12d | Orthotopic PANC-1 and patient-derived xenograft | 109 |
| Exosome | Endogenous | CNP | UC and DGC | MEF and DCs | Synthesized mRNA | PTEN mRNA | U87 and GL261 GBM xenograft | 105 |
| Exosome | Exogenous | Electroporation | UC | NK-92MI | — | PLK1 siRNA and PD-L1 siRNA | CT26 and hepg2 xenograft | 59 |
| Exosome | Endogenous | Transduction | UC | NK-92 | Lentiviral | CAR-Her2 | HER2+ breast tumor brain metastasis | 60 |
Despite the common use, a major concern of the electroporation method is the generation of siRNA nanoaggregates, potentially interfering therapeutic efficacy.67,96 Of note, some commercial non-viral transfection reagents showed higher loading efficiency than electroporation for the loading of siRNA into RBCEVs.67 Additionally, electroporation shows a limitation in loading large cargoes, such as mRNA and plasmid DNA. An early example was the delivery of CRISPR/Cas9 gene editing tool by RBC-derived EVs.66 Specifically, electroporation resulted in a loading efficiency of 18% for the Cas9 mRNA, and the observed gene knockout efficiency was only ∼10% when the EVs were tested to deliver the DNA plasmids of Cas9 and GFP gRNA to 293T-EGFP cells.
Some studies demonstrated the feasibility of simply incubating the EVs with the cargo of interest to allow surface encapsulation. This method is generally used for hydrophobic drugs for their ability to interact with and cross the lipid bilayer EV membrane.97 However, genetic cargoes, including RNA and DNA, are hydrophilic and thus are difficult to load passively into EVs by this method. To enhance the loading efficiency, the EV membrane can be genetically modified with some functional moieties as binding sites (anchor). For example, the streptavidin plasmid was transferred to the cells to produce SAC-EVs, which can specifically bind to biotinylated molecules to enable efficient loading of immunostimulatory CpG DNA.31 Dooley et al. generated exosomes overexpressing a surface glycoprotein, prostaglandin F2 receptor negative regulator (PTGFRN), via virally transfecting HEK 293 cells.98 PTGFRN+ exosomes allowed efficient tethering of a variety of cargoes, including cytokines, antibody fragments, RNA binding proteins, vaccine antigens and Cas9 (Fig. 5a). A subsequent study utilized the PTGFRN+ exosome platform for efficient exogenous loading of ASOs by co-incubation for 30 minutes.71 Compared with free ASOs, enhanced delivery mediated by the PTGFRN+ exosomes has been achieved to macrophages, monocytes, and MDSCs in the liver, peripheral blood, bone marrow, and tumor microenvironment (TME). As a proof of concept, ASO-STAT6 was delivered by the PTGFRN+ exosomes, showing effective reprogramming of immunosuppressive M2 macrophages to proinflammatory M1 macrophages both in vitro and in vivo. In syngeneic models of CRC and HCC, the exosome-mediated ASO-STAT6 monotherapy resulted in >90% tumor growth inhibition and 50–80% complete elimination. This study provides one of the first descriptions of an engineered exosome therapeutic candidate delivering an ASO targeting a transcription factor that controls the macrophage phenotype for cancer immunotherapy.
 | ||
| Fig. 5 Representative approaches of exogenous loading and endogenous loading for genetically engineered EVs. (a) Overexpressed PTGFRN+ on the exosome surface serves as a scaffold protein and achieves efficient loading of ASO-STAT6 with >2000 copies per exosome. Adapted from ref. 71 and 98 with permission from American Association for the Advancement of Science (copyright 2022) and from Cell Press (copyright 2021), respectively. (b) A CNP strategy achieved a high efficiency of large nucleic acid mRNA loading with a 100-fold higher level than the conventional electroporation method. Adapted from ref. 105 with permission from Springer Nature, copyright 2020. | ||
Apart from proteins, RNA cargoes can also be loaded into EVs in an endogenous manner. Both small RNAs and mRNAs encapsulated in exosomes can be transferred to another cell and be functional in the new location.101 Ohno et al. introduced miRNA let-7a into exosomes by transfecting HEK 293T cells using a lipofection method, which induced tumor inhibition in breast cancer mouse models.102 Zhu et al. achieved endogenous loading of microRNA 142-5p within TEXs for gene silencing of epidermal growth factor (EGF) and hepatocyte growth factor (HGF), thereby accelerating the radiosensitivity of nasopharyngeal carcinoma cells.103
Aiming at mRNA loading, Wang et al. developed an approach for selective recruitment of mRNA within microvesicles by taking advantage of the binding ability of the transactivator of transcription (Tat) protein to the stem loop-containing trans-activating response (TAR) element RNA.104 Specifically, two expression constructs were transferred into HEK 293 cells: one being a short Tat peptide fused to the C-terminus of arrestin domain containing protein 1 (ARRDC1) and the second being TAR fused to the 5′ end of a cargo mRNA. After binding occurred, the Tat-TAR system selectively packaged TAR-labeled mRNAs into the ARRDC1-mediated microvesicles (ARMMs) during EV biogenesis. A large macromolecular CRISPR/Cas9 complex was also successfully packaged into ARMMs via the interaction of ARRDC1 with the WW domains in WW-Cas9 fusion proteins. The cargo-loaded ARMMs demonstrated effective translation of mRNAs into functional proteins or CRISPR/Cas9-mediated gene silencing in recipient cells.
As previously mentioned, conventional electroporation can effectively insert small nucleic acids into exosomes but has poor performance in delivering large mRNA molecules. To address this problem, Yang et al. designed a cellular nanoporation (CNP) device to shuttle nucleic acid cargoes from the buffer into the cells attached on the chip surface under transient electrical pulses.105 This method allowed the production of large quantities of exosomes and efficient loading of miRNA with a higher concentration into the exosomes when compared with the conventional electroporation-based approach. More importantly, CNP achieved a high efficiency of large nucleic acid mRNA loading with a 100-fold higher level than bulk electroporation (Fig. 5b). Similarly, a recent report by Dong et al. developed a modified electroporation platform for higher yields of EVs (up to a 45-fold increase relative to the control group) and efficient mRNA loading.106 It is achieved by using nano- and millisecond pulse stimulation combined with a microfluidic setup. It is demonstrated that the level of mRNAs complementary to the plasmid DNA that had been encapsulated within the EVs was 103-fold higher than that in the control group. As a proof of concept, a mouse embryonic fibroblast line that stably expressed CD64 was engineered by the platform to produce IFN-γ mRNA loading EVs, which enabled the immobilization of anti-CD71 and anti-PD-L1 for preferential targeting to glioblastoma cells. Treatment with the immune sEVs induced increased infiltration of effector T cells, upregulation of MHC-I on GBM cells, and re-polarization of suppressive myeloid cells, and ultimately elicited potent anti-tumor efficacy in preclinical GBM models, including models that are intrinsically resistant to immunotherapy. Of note, endogenous loading strategies with an expression cassette for a therapeutic gene may lead to parallel enrichment of mRNA and protein in the extracellular vesicles generated by those cells,107 making it difficult to distinguish the newly translated protein from the mRNA in recipient cells.
A recent study has developed a strategy, termed GEMINI (genetically encoded multifunctional integrated nanovesicles), for achieving Cas9 delivery to T cells. EVs from Cas9 and sgRNA-expressing cells showed nuclease function.86 With the equipment of anti-CD2 scFvs for T cell targeting and VSV-G display for increased EV uptake, GEMINI validated its effectiveness in delivering recombinant Cas9 RNP by the complexing of Cas9 and single-guide RNA to cut the gene encoding C–X–C chemokine co-receptor type 4 in primary human CD4+ T cells. Although not in a cancer model, this work demonstrated the large therapeutic cargo delivery of EVs to defined cell populations, as well as providing integrated tools with elaborate designs for targeting, cargo loading and vesicle fusion, which can be widely applicable for a range of applications in cancer immunotherapy.
Together, these studies shed light on the great potential of EVs as carriers for therapeutic drugs to modulate immune activity in vitro and in vivo through a variety of approaches. Still, the selection of the most suitable method for clinical application remains to be determined. Beyond developing optimal EV-based delivery systems, thorough considerations should encompass the identification of the optimal payload and the evaluation of desired on-target effects, unwanted off-target effects and potential effects on non-target cells.
3.2 Antigen presentation
To increase the responsiveness to current cancer immunotherapy, researchers have made efforts to increase antigen presentation to induce activation of T cells. The ability to express membrane proteins is a unique feature of EVs as compared to synthetic carriers, providing new opportunities for developing antitumor vaccines. Compared to the classical vaccine administration containing soluble antigens, antigens presented by EVs showed a more potent antigen-specific antitumor immune response, highlighting the potential of EVs for vaccine development.Tumor-derived EVs, especially TEXs, naturally carry tumor-associated antigens, thereby holding the potential as cancer vaccines. Yet, the application of TEXs alone cannot produce satisfying antitumor immunity in vivo due to TEX-induced immunosuppression and limited immunogenicity. Modification to TEXs is necessary to enhance specific antigenicity. For example, Li et al. generated leukemia cell-derived exosomes (LEXs) harboring overexpressed CD80 and CD86 through lentiviral transduction, which promoted CD4+ T cell proliferation and Th1 cytokine secretion more efficiently than unmodified LEXs.110 Another study utilizing murine melanoma B16BL6 as producer cells developed a strategy to enhance the antigen capacity of TEXs with endogenous loading of immunostimulatory CpG DNA via streptavidin–biotin interactions.31 These CpG DNA-modified exosomes efficiently and simultaneously delivered tumor antigens together with the adjuvant to the same APC, inducing more potent antigen-specific immune response when compared with the simple co-administration of exosomes and CpG DNA.
The ability of DC-derived EVs to sustain similar immunostimulatory properties of their parent cells makes them among the highly auspicious candidates for anticancer vaccine development. A study has illustrated stronger in vivo immunostimulatory effects associated with smaller exosomes derived from DCs as compared with larger microvesicles, which is possibly due to the higher payload loading within exosomes.111 Liu et al. developed a strategy to deliver an antigen to lymph nodes (LNs) for activation of broad-spectrum T-cell responses using exosomes derived from DCs virally transduced with a plasmid encoding ovalbumin.79 A variety of functional molecules were assessed on the surface of the vesicles, including some co-stimulatory molecules that are involved in the process of antigen presentation (CD80, CD86, CD40 and so on) and various chemokine receptors that can enhance the migration to LN (CCR2, CCR5, CCR7). The engineered exosomes showed efficient accumulation in the peripheral LNs within 12 h and elicited robust CD8+ T-cell responses in vivo. Another interesting study for the development of cell-free DC-EV vaccines investigated the possibility of exploiting transcription-induced chimeric RNAs (chiRNAs) as neoantigens.112 Transcription-induced chiRNAs consist of fused mRNA transcripts originating from two or more unrelated genes. This study used chiRNAs encoding for astrotactin 2, an integral membrane protein involved in neural development, and pregnancy-associated plasma protein-A, a secreted metalloproteinase that cleaves the insulin-like growth factor binding proteins. The chiRNAs were introduced into DC-EVs by lentiviral transduction, forming an anticancer vaccine termed DEXA–P, which demonstrated antigen cross-presentation capability evidenced by high levels of MHC-I, MHC-II, ICAM, CD80, and CD86. Moreover, DEXA–P vaccination resulted in potent anti-tumor immunity and prolonged survival in a syngeneic immunocompetent esophageal cancer model, which is a difficult-to-treat ‘cold’ solid tumor.
Macrophage is another APC type and as well has been engineered to enhance antigen presentation with their EVs. Wang et al. recently described a strategy to equip M1 macrophage-derived vesicles with antigens by incorporating tumor cell nuclei, generating chimeric exosomes (aMT-exos).113 Tumor antigens, represented by ovalbumin, as well as various immune activation indicators represented by CD86, MHC I, and MHC II were observed in the aMT-exos. As a result, they showed potent efficacy in suppressing tumor growth and prolonging survival in multiple murine tumor models including lymphoma, TNBC, and melanoma. In terms of the underlying mechanisms, aMT-exos could enter LNs and activate prime T cells in both the classical antigen presentation-induced immunostimulatory manner (i.e. uptake by DCs or macrophages followed by downstream T cell activation) and a unique “direct exosome interaction” manner (i.e. direct activation of T cells with encapsulated factors). Engineered TEXs potentially can exert their functions in similar manners. For DC-derived EVs, they are more likely to only have the direct manner to interact with T cells, similar to their parental cells (Fig. 6).
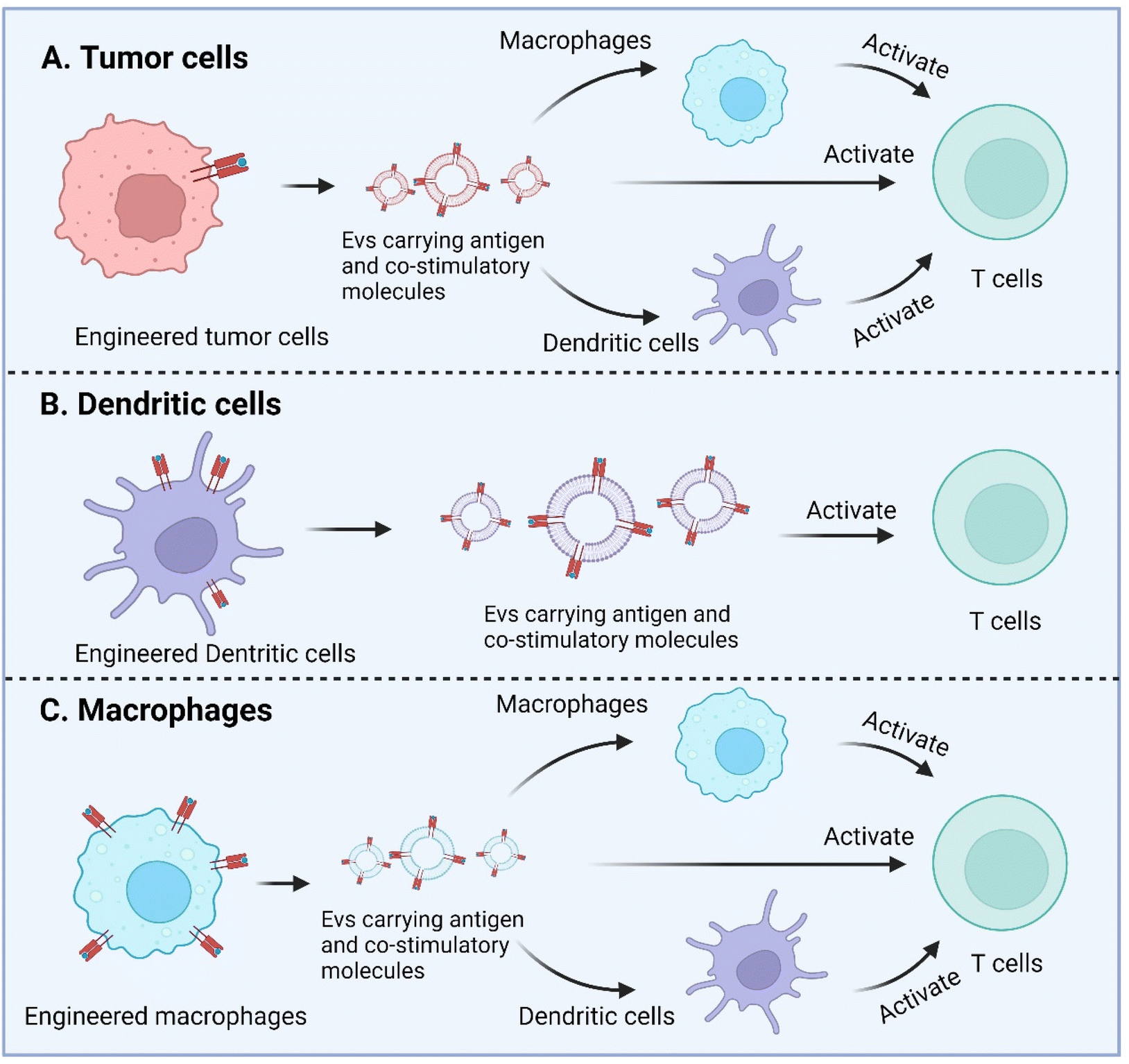 | ||
| Fig. 6 Enhanced antigen presentation by genetically engineered EVs derived from tumor cells, dendritic cells, and macrophages. Created with BioRender. | ||
Another strategy for enhanced antigen presentation to elicit immune response involves membrane engineering of tumor cells with foreign antigens. This may be achieved by exploiting the fusion ability of fusogenic liposomes or viruses. Inspired by this concept, Yang et al. developed a fusogenic exosome platform, mVSVG-Exo, by genetically introducing a viral fusion protein, VSV-G, to the surface of exosomes, thereby enabling transfer of active membrane proteins into the target cell surface.114 Subsequently, the platform was utilized to achieve xenogenization of tumor cells to either enhance the antigenicity or generate danger signals for increased recognition by the immune system.115 mVSVG-Exo mediated DC maturation by stimulating TLR4 signaling and induced cross-presentation of tumor antigens to CD8+ T cells. When combined with anti-PD-L1, the combinational treatment induced a specific and long-lasting systemic adaptive immune response that can eradicate the primary tumor and inhibit recurrence (Fig. 7). Imparting exosomes with fusion capabilities additionally facilitate delivery, in particular cytosolic delivery, of therapeutic agents. Relevant reports in this regard have been mentioned in previous sections.84,86,95
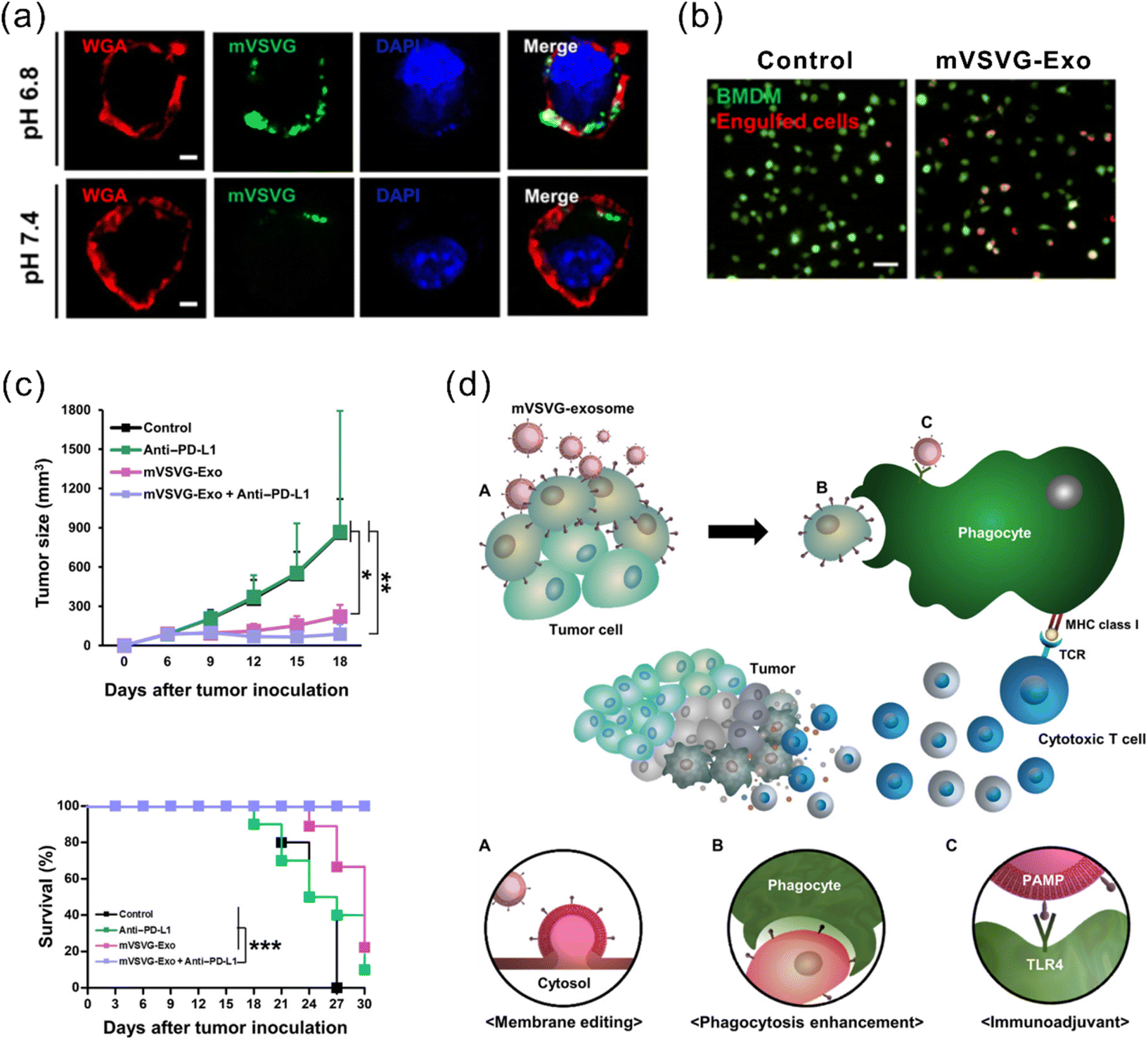 | ||
| Fig. 7 A fusogenic exosome platform, mVSVG-Exo, developed for the antigen xenogenization of tumor cells to enhance anti-tumor immunity. (a) mVSVG-Exo (green) fuses with cancer cell membranes (red) at pH 6.8. (b) Cancer cells fused with mVSVG-Exo can be readily engulfed by macrophages. (c) Combined treatment with mVSVG-Exo plus anti-PD-L1 effectively induces antitumor immunity evidenced by significantly inhibited tumor growth and increased survival rates. (d) Schematic illustration of the mVSVG-Exo-mediated tumor xenogenization strategy. Adapted from ref. 115 with permission from American Association for the Advancement of Science, copyright 2020. | ||
These studies open new avenues for engineering EVs for antigen-specific vaccines in cancer immunotherapy. Although it is yet far from the clinical translation of engineered EV-based vaccines, it is possible that an ‘all-in-one’ approach for generating therapeutically active EVs with antigen presentation, cytosolic delivery and minimal toxicity may be realized. The implementation of such a comprehensive approach holds promise for potential tangible clinical benefits in the future.
4. Conclusion and outlook
To sum up, the prospects of EV-based strategies in cancer immunotherapy are multifaceted. Firstly, being cell-free entities, EVs show enhanced stability and allow extended storage, which is particularly favorable for clinical applications. Secondly, their intrinsic biocompatibility, low immunogenicity, inherited surface protein presentation and encapsulation of bioactive agents make them outperformed synthetic nanocarriers. Besides, EVs can be tailored using various techniques, like genetic engineering, for targeted and efficient delivery of therapeutic agents, enhancing the precision of cancer treatment.Despite these promising aspects, unleashing the full potential of EV-based drug delivery in cancer immunotherapy as well as their clinical translation face several challenges.116 Owing to their inherent complexity, size heterogeneity, and batch-to-batch variations encountered both during biological processes inside the cells and during the manufacturing process, the product heterogeneity of EVs is higher than those of purely synthetic systems, potentially affecting the effectiveness of treatment. Therefore, standardization of the isolation, purification, and characterization methods for EVs is crucial for ensuring reproducibility. Of note is that large-scale production with cost-effectiveness for the clinical setting is in demand. While we mentioned in this review that some studies developed novel strategies for significantly increased yields of EVs, the required equipment or condition could be too complicated and costly for clinical-level production. To move the field forward, these pressing questions emerging from the initial stages need to be addressed.
From the translational standpoint, ensuring the safety profiles and efficacy reliability of EV-based therapy necessitates a comprehensive understanding of underlying mechanisms and potential consequences. Several pivotal questions emerge: to what extent do the functions of EV-based carriers rely on their cellular origin? What are the implications of EVs’ intrinsic cargo –nucleic acids, proteins and various factors, which will be co-delivered with the EV-based delivery systems to the target cells and may exert physiological effects? How to selectively isolate functional relevant EVs while discriminating against those deemed “useless” or “trash” EVs given the potential heterogenicity in cargo content among EVs? How do EVs dynamically interact with the complex TME? What happens to EV-based carriers once administered in vivo in terms of biodistribution, clearance, and persistence? Addressing these questions is essential for harnessing their merits in cancer immunotherapy and accelerating the clinical translation of EV-based delivery platforms. From the genetic engineering perspective, the specific focus in this manuscript, important questions include: what safety concerns arise from genetically engineered EVs? How can we overcome limitations in loading large therapeutic payloads (e.g., mRNA) without compromising EV stability? How do different encapsulation strategies influence the release of genetic cargo from EVs? How to achieve efficient endosomal escape to allow the genetic cargo to reach their intracellular target?
While understanding the essentials of EV biology remains pivotal, strategic combinations with other therapeutic modalities hold immense promise. Given the context of the volatility and complexity of tumor immune systems, achieving significant efficacy with individual therapy is challenging. Combining EV-based immunotherapy with other treatments, such as cell therapy and bio-scaffold-based strategies, holds the potential to boost the anti-tumor efficacy. Efforts can be directed towards optimizing synergistic effects and dosing schedules.
Moreover, if multi-disciplinary technologies can be combined as much as possible, such as optics, physics, clinical science, and chemistry, it would likely help to overcome existing barriers as well as expand the potential of EV-based therapy. The recent decades have witnessed rapid advancements in all these technologies. It is expected that the convergence of multidisciplinary research and strategic combinations will shape the clinical impact of EVs revolutionizing cancer immunotherapy and advancing precision medicine.
Abbreviations
| APCs | Antigen-presenting cells |
| ASOs | Antisense oligonucleotides |
| CNVs | Cell-derived nanovesicles |
| CNP | Cellular nanoporation |
| CML | Chronic myeloid leukemia |
| CRC | Colorectal cancer |
| DCs | Dendritic cells |
| DGC | Density gradient centrifugation |
| EVs | Extracellular vesicles |
| GBM | Glioblastoma multiforme |
| HCC | Hepatocellular carcinoma |
| HEK293 | Human embryonic kidney cells |
| IFN-γ | Interferon gamma |
| LNs | Lymph nodes |
| Lamp2b | Lysosome-associated membrane glycoprotein 2b |
| MHC | Major histocompatibility complex |
| MSCs | Mesenchymal stem cells |
| mRNA | Messenger RNA |
| miRNA | MicroRNA |
| MEF | Mouse embryonic fibroblasts |
| MDSCs | Myeloid-derived suppressor cells |
| NPCC | Nasopharyngeal carcinoma cells |
| NK | Natural killer |
| NSCLC | Non-small cell lung cancer |
| PC | Pancreatic cancer |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDGFR-β | Platelet-derived growth factor receptor beta |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death ligand 1 |
| RVG | Rabies viral glycoprotein |
| RBCs | Red blood cells |
| SIRPα | Signal regulatory protein α |
| scFvs | Single-chain variable fragments |
| SEC | Size exclusion chromatography |
| siRNA | Small interference RNA |
| TFF | Tangential flow filtration |
| TIMP-1 | Tissue inhibitor of metalloproteinases-1 |
| TLR | Toll-like receptor |
| TACE | Transwell®-based asymmetric cell electroporation |
| TNBC | Triple-negative breast cancer |
| TME | Tumor microenvironment |
| TAMs | Tumor-associated macrophages |
| TEXs | Tumor-derived exosomes |
| UC | Ultracentrifugation |
| VSV-G | Vesicular stomatitis virus G protein |
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We acknowledge financial support from the National Institute of General Medical Sciences of the National Institutes of Health under grant number R35GM150507 and Vahlteich Award from the College of Pharmacy at University of Illinois Chicago.References
- M. Tkach and C. Théry, Communication by Extracellular Vesicles: Where We Are and Where We Need to Go, Cell, 2016, 164, 1226–1232 CrossRef CAS PubMed.
- P. D. Robbins and A. E. Morelli, Regulation of immune responses by extracellular vesicles, Nat. Rev. Immunol., 2014, 14, 195–208 CrossRef CAS PubMed.
- T. Skotland and K. Sagini, et al., An emerging focus on lipids in extracellular vesicles, Adv. Drug Delivery Rev., 2020, 159, 308–321 CrossRef CAS PubMed.
- S. Araujo-Abad and M. Saceda, et al., Biomedical application of small extracellular vesicles in cancer treatment, Adv. Drug Delivery Rev., 2022, 182, 114–117 CrossRef PubMed.
- R. J. Motzer and N. M. Tannir, et al., Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma, N. Engl. J. Med., 2018, 378, 1277–1290 CrossRef CAS PubMed.
- M. D. Hellmann and N. A. Rizvi, et al., Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study, Lancet Oncol., 2017, 18, 31–41 CrossRef CAS PubMed.
- N. R. Anderson and N. G. Minutolo, et al., Macrophage-Based Approaches for Cancer Immunotherapy, Cancer Res., 2021, 81, 1201–1208 CrossRef CAS PubMed.
- Y. Zhang and J. Bi, et al., Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications, Int. J. Nanomed., 2020, 15, 6917–6934 CrossRef CAS PubMed.
- J. Frank and M. Richter, et al., Extracellular vesicles protect glucuronidase model enzymes during freeze-drying, Sci. Rep., 2018, 8, 12377 CrossRef PubMed.
- J. P. K. Armstrong and M. M. Stevens, Strategic design of extracellular vesicle drug delivery systems, Adv. Drug Delivery Rev., 2018, 130, 12–16 CrossRef CAS PubMed.
- I. K. Herrmann and M. J. A. Wood, et al., Extracellular vesicles as a next-generation drug delivery platform, Nat. Nanotechnol., 2021, 16, 748–759 CrossRef CAS PubMed.
- L. Wang and D. Wang, et al., Engineering Extracellular Vesicles as Delivery Systems in Therapeutic Applications, Adv. Sci., 2023, 10, 2300552 CrossRef CAS PubMed.
- H. Chen and T. Sun, et al., Extracellular vesicle-based macromolecule delivery systems in cancer immunotherapy, J. Controlled Release, 2022, 348, 572–589 CrossRef CAS PubMed.
- J. Xiao and L. Zeng, et al., Tumor-Tropic Adipose-Derived Mesenchymal Stromal Cell Mediated Bi2Se3 Nano-Radiosensitizers Delivery for Targeted Radiotherapy of Non-Small Cell Lung Cancer, Adv. Healthc. Mater., 2022, 11, 2200143 CrossRef CAS PubMed.
- H. Y. Lee and I. S. Hong, Double-edged sword of mesenchymal stem cells: Cancer-promoting versus therapeutic potential, Cancer Sci., 2017, 108, 1939–1946 CrossRef CAS PubMed.
- M. Ono and N. Kosaka, et al., Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells, Sci. Signaling, 2014, 7, ra63 CrossRef PubMed.
- L. Yu and S. Gui, et al., Exosomes derived from microRNA-199a-overexpressing mesenchymal stem cells inhibit glioma progression by down-regulating AGAP2, Aging, 2019, 11, 5300–5318 CrossRef CAS PubMed.
- D. Jiang and X. Wu, et al., Bone mesenchymal stem cell-derived exosomal microRNA-7-5p inhibits progression of acute myeloid leukemia by targeting OSBPL11, J. Nanobiotechnol., 2022, 20, 29 CrossRef CAS PubMed.
- T. Wu and Q. Tian, et al., Inhibitory role of bone marrow mesenchymal stem cells-derived exosome in non-small-cell lung cancer: microRNA-30b-5p, EZH2 and PI3K/AKT pathway, J. Cell. Mol. Med., 2023, 27, 3526–3538 CrossRef CAS PubMed.
- W. Ren and J. Hou, et al., Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery, J. Exp. Clin. Cancer Res., 2019, 38, 62 CrossRef PubMed.
- S. Biswas and G. Mandal, et al., Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer, J. Immunol., 2019, 203, 3447–3460 CrossRef CAS PubMed.
- W. Zhu and L. Huang, et al., Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo, Cancer Lett., 2012, 315, 28–37 CrossRef CAS PubMed.
- L. Sun and D. Li, et al., Exosomes derived from human umbilical cord mesenchymal stem cells protect against cisplatin-induced ovarian granulosa cell stress and apoptosis in vitro, Sci. Rep., 2017, 7, 2552 CrossRef PubMed.
- K. C. Vallabhaneni and P. Penfornis, et al., Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites, Oncotarget, 2015, 6, 4953–4967 CrossRef PubMed.
- M. Reis and E. Mavin, et al., Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Dendritic Cell Maturation and Function, Front. Immunol., 2018, 9, 2538 CrossRef PubMed.
- L. Carreras-Planella and M. Monguió-Tortajada, et al., Immunomodulatory Effect of MSC on B Cells Is Independent of Secreted Extracellular Vesicles, Front. Immunol., 2019, 10, 1288 CrossRef CAS PubMed.
- J. Deng and H. Ke, Overcoming the resistance of hepatocellular carcinoma to PD-1/PD-L1 inhibitor and the resultant immunosuppression by CD38 siRNA-loaded extracellular vesicles, OncoImmunology, 2023, 12, 2152635 CrossRef PubMed.
- R. L. G. Maus and J. W. Jakub, et al., Human Melanoma-Derived Extracellular Vesicles Regulate Dendritic Cell Maturation, Front. Immunol., 2017, 8, 358 Search PubMed.
- S. Yu and C. Liu, et al., Tumor Exosomes Inhibit Differentiation of Bone Marrow Dendritic Cells1, J. Immunol., 2007, 178, 6867–6875 CrossRef CAS PubMed.
- B. Zuo and H. Qi, et al., Alarmin-painted exosomes elicit persistent antitumor immunity in large established tumors in mice, Nat. Commun., 2020, 11, 1790 CrossRef CAS PubMed.
- M. Morishita and Y. Takahashi, et al., Exosome-based tumor antigens–adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA, Biomaterials, 2016, 111, 55–65 CrossRef CAS PubMed.
- Z. Tu and Y. Zhu, et al., Scavenging Tumor-Derived Small Extracellular Vesicles by Functionalized 2D Materials to Inhibit Tumor Regrowth and Metastasis Following Radiotherapy, Adv. Funct. Mater., 2022, 32, 2205663 CrossRef CAS.
- W. Zhou and X. Chen, et al., Exosomes derived from immunogenically dying tumor cells as a versatile tool for vaccination against pancreatic cancer, Biomaterials, 2022, 280, 121306 CrossRef CAS PubMed.
- J. Rossowska and N. Anger, et al., Antitumor Potential of Extracellular Vesicles Released by Genetically Modified Murine Colon Carcinoma Cells With Overexpression of Interleukin-12 and shRNA for TGF-β1, Front. Immunol., 2019, 10, 211 CrossRef CAS PubMed.
- M. Merad and P. Sathe, et al., The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting, Annu. Rev. Immunol., 2013, 31, 563–604 CrossRef CAS PubMed.
- B. Besse and M. Charrier, et al., Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC, OncoImmunology, 2016, 5, e1071008 CrossRef PubMed.
- J. Kowal and G. Arras, et al., Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E968–E977 CrossRef CAS PubMed.
- S. Chen and M. Lv, et al., Poly(I:C) enhanced anti-cervical cancer immunities induced by dendritic cells-derived exosomes, Int. J. Biol. Macromol., 2018, 113, 1182–1187 CrossRef CAS PubMed.
- A. Matsumoto and M. Asuka, et al., Antitumor immunity by small extracellular vesicles collected from activated dendritic cells through effective induction of cellular and humoral immune responses, Biomaterials, 2020, 252, 120112 CrossRef CAS PubMed.
- B. Besse and M. Charrier, et al., Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC, OncoImmunology, 2016, 5, e1071008 CrossRef PubMed.
- Z. Lu and B. Zuo, et al., Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models, J. Hepatol., 2017, 67, 739–748 CrossRef CAS PubMed.
- F. Xu and D. Jiang, et al., Engineering of dendritic cell bispecific extracellular vesicles for tumor-targeting immunotherapy, Cell Rep., 2023, 42, 113138 CrossRef CAS PubMed.
- L. Tang and Y. Yin, et al., Extracellular Vesicles-Derived Hybrid Nanoplatforms for Amplified CD47 Blockade-Based Cancer Immunotherapy, Adv. Mater., 2023, 35, 2303835 CrossRef CAS PubMed.
- Z. Xu and H. Huang, et al., A near-infrared light-responsive extracellular vesicle as a “Trojan horse” for tumor deep penetration and imaging-guided therapy, Biomaterials, 2021, 269, 120647 CrossRef CAS PubMed.
- M. Lu and H. Xing, et al., Photoactivatable Silencing Extracellular Vesicle (PASEV) Sensitizes Cancer Immunotherapy, Adv. Mater., 2022, 34, 2204765 CrossRef CAS PubMed.
- Y. W. Choo and M. Kang, et al., M1 Macrophage-Derived Nanovesicles Potentiate the Anticancer Efficacy of Immune Checkpoint Inhibitors, ACS Nano, 2018, 12, 8977–8993 CrossRef CAS PubMed.
- W. Nie and G. Wu, et al., Responsive Exosome Nano-bioconjugates for Synergistic Cancer Therapy, Angew. Chem., Int. Ed., 2020, 59, 2018–2022 CrossRef CAS PubMed.
- N. Seo and Y. Shirakura, et al., Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells, Nat. Commun., 2018, 9, 435 CrossRef PubMed.
- S. Shin and I. Jung, et al., Novel antitumor therapeutic strategy using CD4(+) T cell-derived extracellular vesicles, Biomaterials, 2022, 289, 121765 CrossRef CAS PubMed.
- J. Wahlgren and L. Karlson Tde, et al., Activated human T cells secrete exosomes that participate in IL-2 mediated immune response signaling, PLoS One, 2012, 7, e49723 CrossRef CAS PubMed.
- W. Fu and C. Lei, et al., CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity, Nat. Commun., 2019, 10, 4355 CrossRef PubMed.
- D. Jung and S. Shin, et al., Reprogramming of T cell-derived small extracellular vesicles using IL2 surface engineering induces potent anti-cancer effects through miRNA delivery, J. Extracell. Vesicles, 2022, 11, 12287 CrossRef CAS PubMed.
- A. Y. Jong and C. H. Wu, et al., Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells, J. Extracell. Vesicles, 2017, 6, 1294368 CrossRef PubMed.
- C.-H. Wu and J. Li, et al., Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells, J. Extracell. Vesicles, 2019, 8, 1588538 CrossRef CAS PubMed.
- Y. Qi and X. Zhao, et al., Opportunities and challenges of natural killer cell-derived extracellular vesicles, Front. Bioeng. Biotechnol., 2023, 11, 1122585 CrossRef PubMed.
- C. Federici and E. Shahaj, et al., Natural-Killer-Derived Extracellular Vesicles: Immune Sensors and Interactors, Front. Immunol., 2020, 11, 262 CrossRef CAS PubMed.
- L. Zhu and S. Kalimuthu, et al., Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by IL-15 priming, Biomaterials, 2019, 190, 38–50 CrossRef PubMed.
- M. Aarsund and F. M. Segers, et al., Comparison of characteristics and tumor targeting properties of extracellular vesicles derived from primary NK cells or NK-cell lines stimulated with IL-15 or IL-12/15/18, Cancer Immunol. Immunother., 2022, 71, 2227–2238 CrossRef CAS PubMed.
- M. Zhang and W. Shao, et al., Conscription of Immune Cells by Light-Activatable Silencing NK-Derived Exosome (LASNEO) for Synergetic Tumor Eradication, Adv. Sci., 2022, 9, 2201135 CrossRef CAS PubMed.
- B. L. Tao and R. X. Du, et al., Engineering CAR-NK cell derived exosome disguised nano-bombs for enhanced HER2 positive breast cancer brain metastasis therapy, J. Controlled Release, 2023, 363, 692–706 CrossRef CAS PubMed.
- J. Zhang and C. Ji, et al., Engineered neutrophil-derived exosome-like vesicles for targeted cancer therapy, Sci. Adv., 2022, 8, eabj8207 CrossRef CAS PubMed.
- S. Oggero and M. de Gaetano, et al., Extracellular vesicles from monocyte/platelet aggregates modulate human atherosclerotic plaque reactivity, J. Extracell. Vesicles, 2021, 10, 12084 CrossRef PubMed.
- J. Xiong and H. Chi, et al., Revolutionizing anti-tumor therapy: unleashing the potential of B cell-derived exosomes, Front. Immunol., 2023, 14, 1188760 CrossRef CAS PubMed.
- Q. Feng and X. Ma, et al., Engineered Bacterial Outer Membrane Vesicles as Controllable Two-Way Adaptors to Activate Macrophage Phagocytosis for Improved Tumor Immunotherapy, Adv. Mater., 2022, 34, 2206200 CrossRef CAS PubMed.
- J. Liang and F. Zhu, et al., Outer Membrane Vesicle-Based Nanohybrids Target Tumor-Associated Macrophages to Enhance Trained Immunity-Related Vaccine-Generated Antitumor Activity, Adv. Mater., 2023, 35, 2306158 CrossRef CAS PubMed.
- W. M. Usman and T. C. Pham, et al., Efficient RNA drug delivery using red blood cell extracellular vesicles, Nat. Commun., 2018, 9, 2359 CrossRef PubMed.
- B. Peng and T. M. Nguyen, et al., Robust delivery of RIG-I agonists using extracellular vesicles for anti-cancer immunotherapy, J. Extracell. Vesicles, 2022, 11, e12187 CrossRef CAS PubMed.
- B. Peng and Y. Yang, et al., Red blood cell extracellular vesicles deliver therapeutic siRNAs to skeletal muscles for treatment of cancer cachexia, Mol. Ther., 2023, 31, 1418–1436 CrossRef CAS PubMed.
- M. A. Livshits and E. Khomyakova, et al., Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol, Sci. Rep., 2015, 5, 17319 CrossRef PubMed.
- M. Mendt and S. Kamerkar, et al., Generation and testing of clinical-grade exosomes for pancreatic cancer, JCI Insight, 2018, 3, e99263 CrossRef PubMed.
- S. Kamerkar and C. Leng, et al., Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoASO-STAT6 leads to potent monotherapy antitumor activity, Sci. Adv., 2022, 8, eabj7002 CrossRef CAS PubMed.
- R. Linares and S. Tan, et al., High-speed centrifugation induces aggregation of extracellular vesicles, J. Extracell. Vesicles, 2015, 4, 29509 CrossRef PubMed.
- E. A. Mol and M. J. Goumans, et al., Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation, Nanomedicine, 2017, 13, 2061–2065 CrossRef CAS PubMed.
- S. Busatto and G. Vilanilam, et al., Tangential Flow Filtration for Highly Efficient Concentration of Extracellular Vesicles from Large Volumes of Fluid, Cells, 2018, 7, 273 CrossRef CAS PubMed.
- A. Clayton and J. Court, et al., Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry, J. Immunol. Methods, 2001, 247, 163–174 CrossRef CAS PubMed.
- J. Wang and W. Li, et al., Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells, ACS Appl. Mater. Interfaces, 2017, 9, 27441–27452 CrossRef CAS PubMed.
- S. S. Kanwar and C. J. Dunlay, et al., Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes, Lab Chip, 2014, 14, 1891–1900 RSC.
- M. Tayebi and Y. Zhou, et al., Exosome Purification and Analysis Using a Facile Microfluidic Hydrodynamic Trapping Device, Anal. Chem., 2020, 92, 10733–10742 CrossRef CAS PubMed.
- C. Liu and X. Liu, et al., A nanovaccine for antigen self-presentation and immunosuppression reversal as a personalized cancer immunotherapy strategy, Nat. Nanotechnol., 2022, 17, 531–540 CrossRef CAS PubMed.
- O. P. Wiklander and J. Z. Nordin, et al., Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting, J. Extracell. Vesicles, 2015, 4, 26316 CrossRef PubMed.
- L. Alvarez-Erviti and Y. Seow, et al., Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS PubMed.
- Y. Tian and S. Li, et al., A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy, Biomaterials, 2014, 35, 2383–2390 CrossRef CAS PubMed.
- G. Corso and W. Heusermann, et al., Systematic characterization of extracellular vesicles sorting domains and quantification at the single molecule–single vesicle level by fluorescence correlation spectroscopy and single particle imaging, J. Extracell. Vesicles, 2019, 8, 1663043 CrossRef CAS PubMed.
- L. Wang and G. Wang, et al., Bioinspired engineering of fusogen and targeting moiety equipped nanovesicles, Nat. Commun., 2023, 14, 3366 CrossRef CAS PubMed.
- Q. Cheng and X. Shi, et al., Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity, J. Am. Chem. Soc., 2018, 140, 16413–16417 CrossRef CAS PubMed.
- D. M. Stranford and L. M. Simons, et al., Genetically encoding multiple functionalities into extracellular vesicles for the targeted delivery of biologics to T cells, Nat. Biomed. Eng., 2023 DOI:10.1038/s41551-023-01142-x.
- X. Gao and N. Ran, et al., Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy, Sci. Transl. Med., 2018, 10, eaat0195 CrossRef PubMed.
- N. Ran and W. Li, et al., Autologous exosome facilitates load and target delivery of bioactive peptides to repair spinal cord injury, "Bioact. Mater., 2023, 25, 766–782 CAS.
- S. C. Jang and O. Y. Kim, et al., Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors, ACS Nano, 2013, 7, 7698–7710 CrossRef CAS PubMed.
- Y. Wen and Q. Fu, et al., Cell-derived nanovesicles prepared by membrane extrusion are good substitutes for natural extracellular vesicles, Extracell. Vesicles, 2022, 1, 100004 CrossRef PubMed.
- Z. Yang and J. Xie, et al., Functional exosome-mimic for delivery of siRNA to cancer: in vitro and in vivo evaluation, J. Controlled Release, 2016, 243, 160–171 CrossRef CAS PubMed.
- M. Colombo and G. Raposo, et al., Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- R. van der Meel and M. H. Fens, et al., Extracellular vesicles as drug delivery systems: lessons from the liposome field, J. Controlled Release, 2014, 195, 72–85 CrossRef CAS PubMed.
- S. Kamerkar and V. S. LeBleu, et al., Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer, Nature, 2017, 546, 498–503 CrossRef CAS PubMed.
- H. Liu and L. Huang, et al., Viral Protein-Pseudotyped and siRNA-Electroporated Extracellular Vesicles for Cancer Immunotherapy, Adv. Funct. Mater., 2020, 30, 2006515 CrossRef CAS.
- S. A. A. Kooijmans and S. Stremersch, et al., Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles, J. Controlled Release, 2013, 172, 229–238 CrossRef CAS PubMed.
- G. Fuhrmann and A. Serio, et al., Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins, J. Controlled Release, 2015, 205, 35–44 CrossRef CAS PubMed.
- K. Dooley and R. E. McConnell, et al., A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties, Mol. Ther., 2021, 29, 1729–1743 CrossRef CAS PubMed.
- E. Koh and E. J. Lee, et al., Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis, Biomaterials, 2017, 121, 121–129 CrossRef CAS PubMed.
- X. Li and T. Zhu, et al., Genetically Programmable Vesicles for Enhancing CAR-T Therapy against Solid Tumors, Adv. Mater., 2023, 35, e2211138 CrossRef PubMed.
- H. Valadi and K. Ekström, et al., Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- S.-i. Ohno and M. Takanashi, et al., Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells, Mol. Ther., 2013, 21, 185–191 CrossRef CAS PubMed.
- C. Zhu and X. Jiang, et al., Tumor-derived extracellular vesicles inhibit HGF/c-Met and EGF/EGFR pathways to accelerate the radiosensitivity of nasopharyngeal carcinoma cells via microRNA-142–5p delivery, Cell Death Discovery, 2022, 8, 17 CrossRef CAS PubMed.
- Q. Wang and J. Yu, et al., ARMMs as a versatile platform for intracellular delivery of macromolecules, Nat. Commun., 2018, 9, 960 CrossRef PubMed.
- Z. Yang and J. Shi, et al., Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation, Nat. Biomed. Eng., 2020, 4, 69–83 CrossRef CAS PubMed.
- S. Dong and X. Liu, et al., Adaptive design of mRNA-loaded extracellular vesicles for targeted immunotherapy of cancer, Nat. Commun., 2023, 14, 6610 CrossRef CAS PubMed.
- A. Mizrak and M. F. Bolukbasi, et al., Genetically Engineered Microvesicles Carrying Suicide mRNA/Protein Inhibit Schwannoma Tumor Growth, Mol. Ther., 2013, 21, 101–108 CrossRef CAS PubMed.
- D. Bellavia and S. Raimondo, et al., Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo chronic myelogenous Leukemia cell growth, Theranostics, 2017, 7, 1333–1345 CrossRef CAS PubMed.
- C.-L. Chiang and Y. Ma, et al., Dual targeted extracellular vesicles regulate oncogenic genes in advanced pancreatic cancer, Nat. Commun., 2023, 14, 6692 CrossRef CAS PubMed.
- J. Li and F. Huang, et al., A novel costimulatory molecule gene-modified leukemia cell-derived exosome-targeted CD4+ T cell vaccine efficiently enhances anti-leukemia immunity, Front. Immunol., 2022, 13, 1043484 CrossRef CAS PubMed.
- C. J. E. Wahlund and G. Güclüler, et al., Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo, Sci. Rep., 2017, 7, 17095 CrossRef PubMed.
- X. Xiong and X. Ke, et al., Neoantigen-based cancer vaccination using chimeric RNA-loaded dendritic cell-derived extracellular vesicles, J. Extracell. Vesicles, 2022, 11, e12243 CrossRef CAS PubMed.
- S. Wang and F. Li, et al., Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment, Sci. Transl. Med., 2021, 13, eabb6981 CrossRef CAS PubMed.
- Y. Yang and Y. Hong, et al., Virus-Mimetic Fusogenic Exosomes for Direct Delivery of Integral Membrane Proteins to Target Cell Membranes, Adv. Mater., 2017, 29, 1605604 CrossRef PubMed.
- G. B. Kim and G.-H. Nam, et al., Xenogenization of tumor cells by fusogenic exosomes in tumor microenvironment ignites and propagates antitumor immunity, Sci. Adv., 2020, 6, eaaz2083 CrossRef CAS PubMed.
- H. Clemmens and D. W. Lambert, Extracellular vesicles: translational challenges and opportunities, Biochem. Soc. Trans., 2018, 46, 1073–1082 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |