
Nano-PROTACs: state of the art and perspectives
Jie
Zhong†
 ab,
Ruiqi
Zhao†
b,
Yuji
Wang
a,
Yu-xiong
Su
*b and
Xinmiao
Lan
*a
ab,
Ruiqi
Zhao†
b,
Yuji
Wang
a,
Yu-xiong
Su
*b and
Xinmiao
Lan
*a
aBeijing Area Major Laboratory of Peptide and Small Molecular Drugs, Engineering Research Center of Ministry of Education of China, Beijing Laboratory of Biomedical Materials, School of Pharmaceutical Sciences, Capital Medical University, Beijing 100069, China. E-mail: xmiao.lan@ccmu.edu.cn
bDiscipline of Oral and Maxillofacial Surgery, Faculty of Dentistry, The University of Hong Kong, Hong Kong SAR 999077, China. E-mail: richsu@hku.hk
First published on 22nd January 2024
Abstract
PROteolysis TArgeting Chimeras (PROTACs), as a recently identified technique in the field of new drug development, provide new concepts for disease treatment and are expected to revolutionize drug discovery. With high specificity and flexibility, PROTACs serve as an innovative research tool to target and degrade disease-relevant proteins that are not currently pharmaceutically vulnerable to eliminating their functions by hijacking the ubiquitin–proteasome system. To date, PROTACs still face the challenges of low solubility, poor permeability, off-target effects, and metabolic instability. The combination of nanotechnology and PROTACs has been explored to enhance the in vivo performance of PROTACs regarding overcoming these challenging hurdles. In this review, we summarize the latest advancements in the building-block design of PROTAC prodrug nanoparticles and provide an overview of existing/potential delivery systems and loading approaches for PROTAC drugs. Furthermore, we discuss the current status and prospects of the split-and-mix approach for PROTAC drug optimization. Additionally, the advantages and translational potentials of carrier-free nano-PROTACs and their combinational therapeutic effects are highlighted. This review aims to foster a deeper understanding of this rapidly evolving field and facilitate the progress of nano-PROTACs that will continue to push the boundaries of achieving selectivity and controlled release of PROTAC drugs.
 Jie Zhong | Jie Zhong received his Master of Science in Biophysics under the guidance of Prof. Xing-Jie Liang from the National Center for Nanoscience and Technology, Chinese Academy of Sciences. He is currently studying at the Faculty of Dentistry, The University of Hong Kong as a PhD candidate. The main aim of his activities is focused on the study of drug delivery, biomaterials, polymer self-assembly, and immunotherapy for the applications of cancer therapy and infection management. |
 Ruiqi Zhao | Ruiqi Zhao obtained her Master of Stomatological Medicine degree from Peking Union Medical College, Tsinghua University. Currently, she is a Ph.D. student at the Faculty of Dentistry, The University of Hong Kong, focusing on the diagnosis and treatment of oral and maxillofacial-head and neck tumors. |
 Yu-xiong Su | Yu-xiong Su received his Ph.D. degree from Sun-Yat Sen University, China. Currently, he works as a Professor and Division Chief in the Department of Oral and Maxillofacial Surgery, University of Hong Kong. Prof. Su is the lead surgeon for maxillofacial oncology and microsurgical reconstruction in the Department of Oral and Maxillofacial Surgery, Queen Mary Hospital, Hong Kong. He is actively involved in clinical and translational research. His main clinical and research interests include oral and maxillofacial oncology, computer-assisted surgery and 3D printing in head and neck reconstruction, salivary gland diseases and sialendoscopy. |
 Xinmiao Lan | Xinmiao Lan obtained a master degree from the Faculty of Dentistry, The University of Hong Kong in 2015 and a Ph.D. degree from the same faculty in September 2019. She finished her postdoctoral fellowship under the guidance of Prof. Richard Yu-xiong Su at the University of Hong Kong. Currently, she is an Associate Professor at Capital Medical University and an Honorary Associate Professor at the University of Hong Kong. Her research interests are nanoparticles, advanced nanomaterials, chemotherapy, immunotherapy, and cancer therapy. |
1. Introduction
PROteolysis TArgeting Chimeras (PROTACs), an epochal therapeutic strategy employed to precisely degrade the protein of interest (POI), hold enormous potential in the treatment of intractable diseases, bringing better healthcare to human beings.1–4 PROTAC is a small heterobifunctional molecule that incorporates a linker connecting two distinct ligands, one of which is responsible for the recruitment and binding of the POI, whereas the other facilitates the recruitment and binding of E3 ubiquitin ligases. By bridging the POI to E3 ligases, PROTAC leads to the ubiquitination of the POI, which is labeled with ubiquitin for further degradation. This ubiquitin-tagged POI is subsequently recognized by the proteasome and degraded through the ubiquitin–proteasome system (UPS). After that, PROTAC is liberated and can bind to another POI, thus inducing the degradation of the POI in a cyclical and catalytic way.3,4 This mechanism allows PROTACs to accomplish their mission of disrupting the function of the POI with very low doses while minimizing side effects, accompanied by a longer duration of effect.2,4,5 In addition, by utilizing the unique strategy of targeted protein degradation, PROTACs can bypass the need for direct binding to the active site of the target protein, so they have the potential to inhibit previously “undruggable” targets and overcome drug resistance that traditional small molecule inhibitors often encountered.1,3,6The advantages of PROTACs have generated enthusiasm among researchers in both chemical biology and drug discovery fields, providing novel treatment options for various diseases including cancers,7 neurodegenerative disorders,8 and metabolic syndromes.9 However, despite the revolutionary role of PROTACs in the drug discovery paradigm by enabling precise and effective degradation of the POI, there are still some limitations that hinder the clinical application of PROTACs. Firstly, due to their unique molecular composition and structural characteristics, PROTACs often exhibit suboptimal water solubility and tissue permeability.10,11 Due to these limitations, conventional PROTAC molecules face barriers in being developed into drugs and experience limitations in drug absorption, distribution, metabolism, and excretion, potentially leading to limited therapeutic efficacy with a higher dosage requirement. Secondly, the lack of target tissue specificity in conventional PROTACs poses a challenge, as PROTACs indiscriminately degrade proteins in both normal and abnormal cells, leading to systemic toxicity. Consequently, it is imperative to achieve target-tissue delivery and optimize the metabolic stability of PROTACs.10,12,13
In recent years, increasing studies have investigated the utilization of nanotechnology to conquer the limitations of traditional PROTACs. The fabrication of versatile nanostructures offers PROTACs advantages such as enhancing solubility and tissue penetration, increasing target specificity, and providing controllable release capability, thus having the potential to revolutionarily improve targeted protein degradation and realize more effective and precise therapy.2,10,17 Moreover, possible smart delivery systems will collaboratively help to improve their solubility, permeability, and targetability, thus decreasing the risk of off-target toxicities. To provide an overview of the field of nano-PROTACs, we aim to explore how the introduction of nanotechnology and the construction of nanostructures can optimize PROTAC drugs and guide the discovery of new PROTAC drugs. The ingenious design of nanoplatforms and advanced delivery systems for PROTAC delivery is summarized in this review (Fig. 1). Our findings in this review are expected to shed light on future PROTAC drug design and promote the translation of PROTAC drugs to clinical applications.
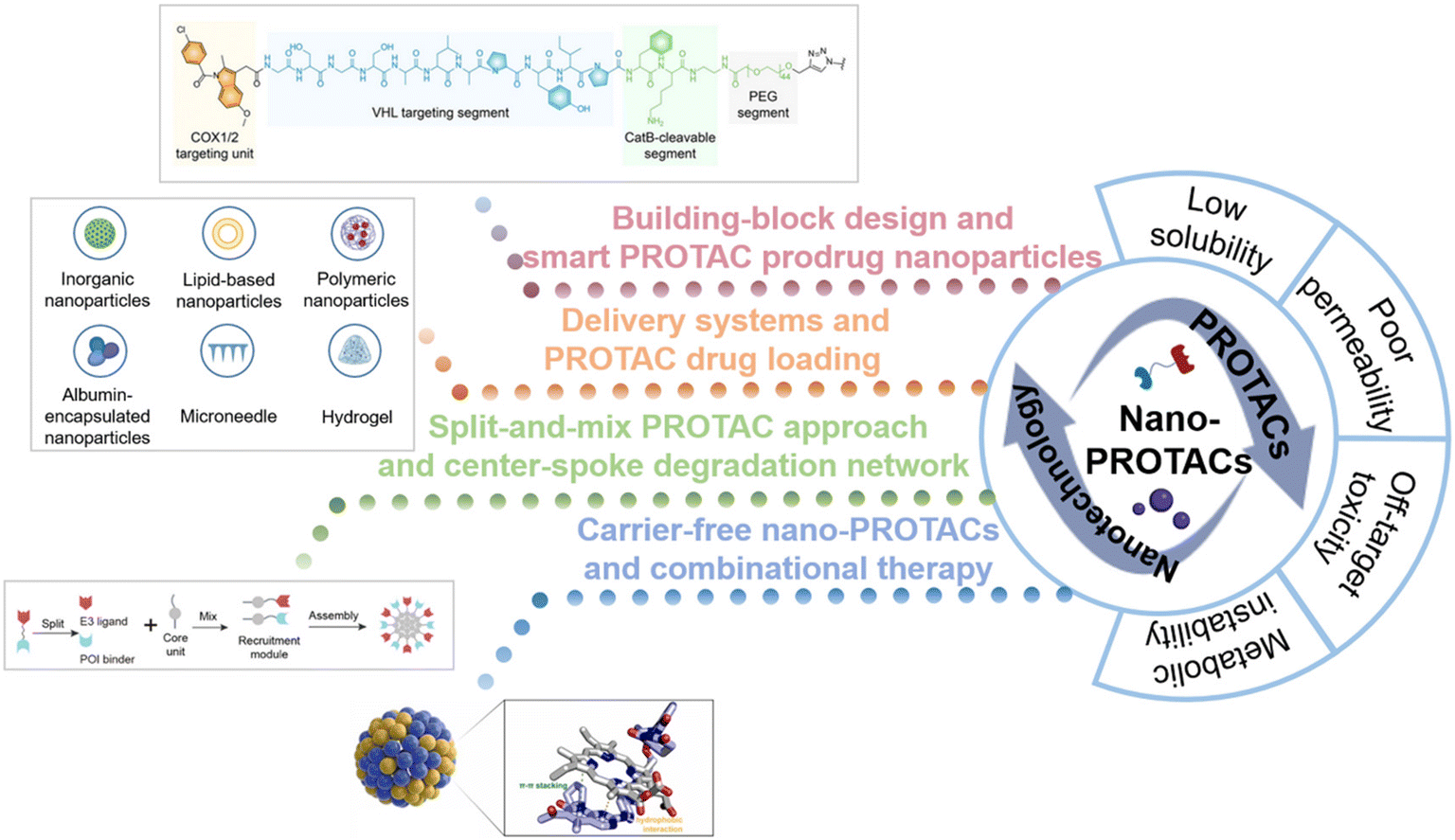 | ||
| Fig. 1 Schematic illustration of the combinational strategy of nanotechnology and PROTACs (nano-PROTACs): the typical shortcomings of traditional PROTACs and the nanotechnology-based strategies for PROTAC drug optimization. Reproduced from ref. 14 with permission. Copyright 2022, Wiley-VCH GmbH. Reproduced from ref. 15 with permission. Copyright 2023, American Chemical Society. Reproduced from ref. 16 with permission. Copyright 2023, Elsevier. | ||
2. Building-block design and smart PROTAC prodrug nanoparticles
For spatiotemporally controlled release of PROTACs and more efficient delivery, some stimuli-responsive linkers and functional groups were introduced to form advanced amphiphilic structures and further self-assemble to form PROTAC prodrug nanoparticles. Herein, we focus on the building-block design for smart PROTAC prodrug nanoparticles.To avoid the off-target tissue toxicity and improve the pharmacokinetic performances of conventional PROTACs, several chemical modifications on targeted groups have been explored. Based on the various membrane receptors on the cell surface in the targeted tissue, folate-caged,18,19 aptamer-conjugated,20,21 and antibody-conjugated22,23 PROTACs have been widely developed to facilitate PROTAC drug delivery by receptor-mediated endocytosis. To enable discrete control over PROTACs’ function, some labile protecting groups (such as photo-activated24–26 and hypoxia-activated27) have been installed on the POI ligands24,27 or E3 ligase ligands25,26,28 to form stimuli-responsive caged PROTACs. For example, after light exposure with a specific wavelength, the optical activation of small molecule-induced protein degradation was observed in terms of photo-caged PROTACs. By leveraging pathological cues and external triggers, stimuli-responsive caged PROTACs can provide spatiotemporal control of protein degradation, which enables more precise and efficient therapeutic effects.
Based on this strategy, light-controllable PROTAC prodrug nanoparticles to address the on-target off-tissue toxicity of PROTACs have recently been reported to spatiotemporally activate and release PROTACs.14,29–33 Briefly, PROTAC molecules are modified with responsive fragments with photo-caged groups to release PROTACs upon certain irradiation, accompanied by the de-caging and subsequent self-immolated cleavage of versatile responsive linkers (Fig. 2A). This strategy often simultaneously induces the self-assembly of the resultant PROTAC molecules into nanoscale particulate, both to achieve “nanoscale advantages”34,35 and to allow for further subsequent delivery system development. Zhang et al.14 introduced near-infrared (NIR) light-absorbing poly(cyclopentadithiophene-alt-benzothiadiazole) (PCB) groups and a cathepsin B (CatB)-cleavable segment, thus developing smart cyclooxygenase 1/2 (COX-1/2)-degrading PROTAC prodrug nanoparticles (SPNcox) (Fig. 2B). Specifically, PCB was reacted with sodium azide to obtain PCB-N3 and PROTACs were reacted with an alkyne-PEG-CatB cleavable segment. Then PCB-N3 was reacted with alkyne-PEG-PROTACs and alkyne-mPEG via a click reaction to obtain the final polymer. Due to the existence of the conjugated hydrophilic PEG chain and hydrophobic PCB backbone, this polymer showed self-assembly behavior in phosphate buffered saline (PBS) solutions. SPNcox can be activated by tumor-overexpressed CatB to induce the degradation of nanoparticles, followed by the release of COX-1/2-degrading PROTACs. In addition, in the presence of PCB, a photo-metabolic cancer immunotherapy effect was achieved, consequently improving tumor immunogenicity together with PROTACs. A similar building-block strategy has been reported recently in a number of PROTAC prodrug nanoparticles, with several PROTACs (such as COX-1/2,14 bromodomain-containing 4 (BRD4),29 indoleamine 2,3-dioxygenase (IDO),30 and Src homology 2 domain-containing phosphatase 2 (SHP2)31), stimuli-cleavable segments (such as CatB-cleavable,14,301O2-cleavable,29 and caspase 3-cleavable31), and photosensitizers (such as PCB,14,30 PA,29 and PplX31). These systems thus present a novel approach for spatiotemporal control over targeted protein degradation by PROTACs. Upon irradiation, embedded photosensitizers trigger the generation of 1O2 for the cleavage of responsive linkers and synergistic photodynamic therapy.
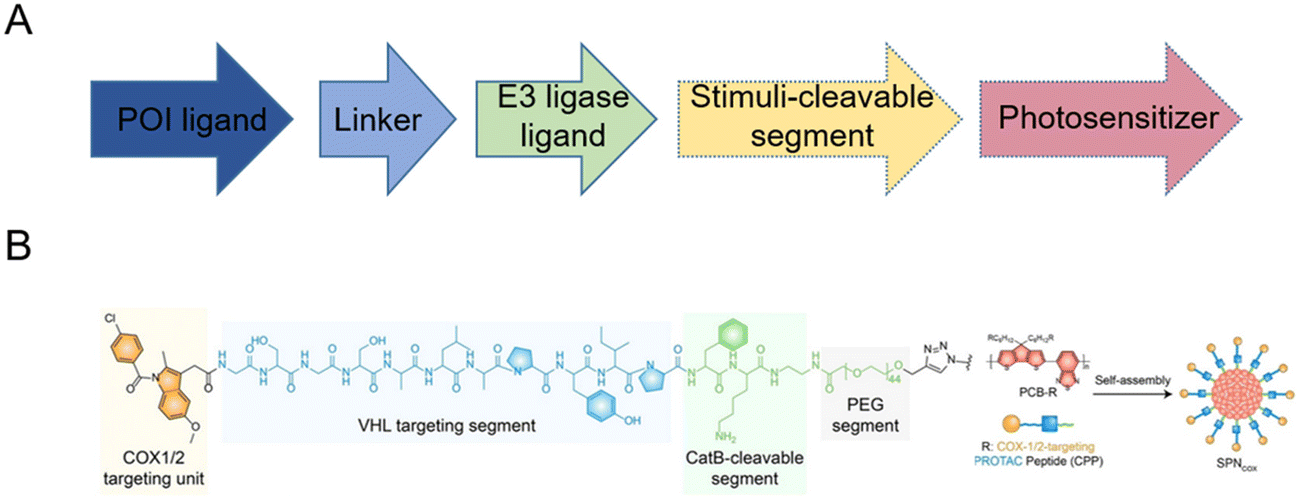 | ||
| Fig. 2 (A) Building-block design of light-controllable PROTAC prodrug nanoparticles. (B) Chemical structure and fabrication of NIR-activated COX-1/2-degrading PROTAC prodrug nanoparticles. Reproduced from ref. 14 with permission. Copyright 2022, Wiley-VCH GmbH. | ||
The above-mentioned strategies contribute to skillful on-target properties, leading to more efficient therapeutic effects for cancer therapy. Moreover, various treatment means, such as photodynamic therapy and immunotherapy, could be combined to control the targeted protein degradation and achieve a better curative effect.
3. Delivery systems and PROTAC drug loading
Except for remolded PROTAC structures, several delivery systems (Fig. 3) have been utilized to transport PROTACs to specific lesions. Conventional delivery systems consisting of inorganic nanoparticles, lipid nanoparticles, polymeric nanoparticles, and albumin-encapsulated nanoparticles have been concluded in this review. Besides these, local delivery systems such as microneedles and hydrogels were also utilized to make PROTACs more useful.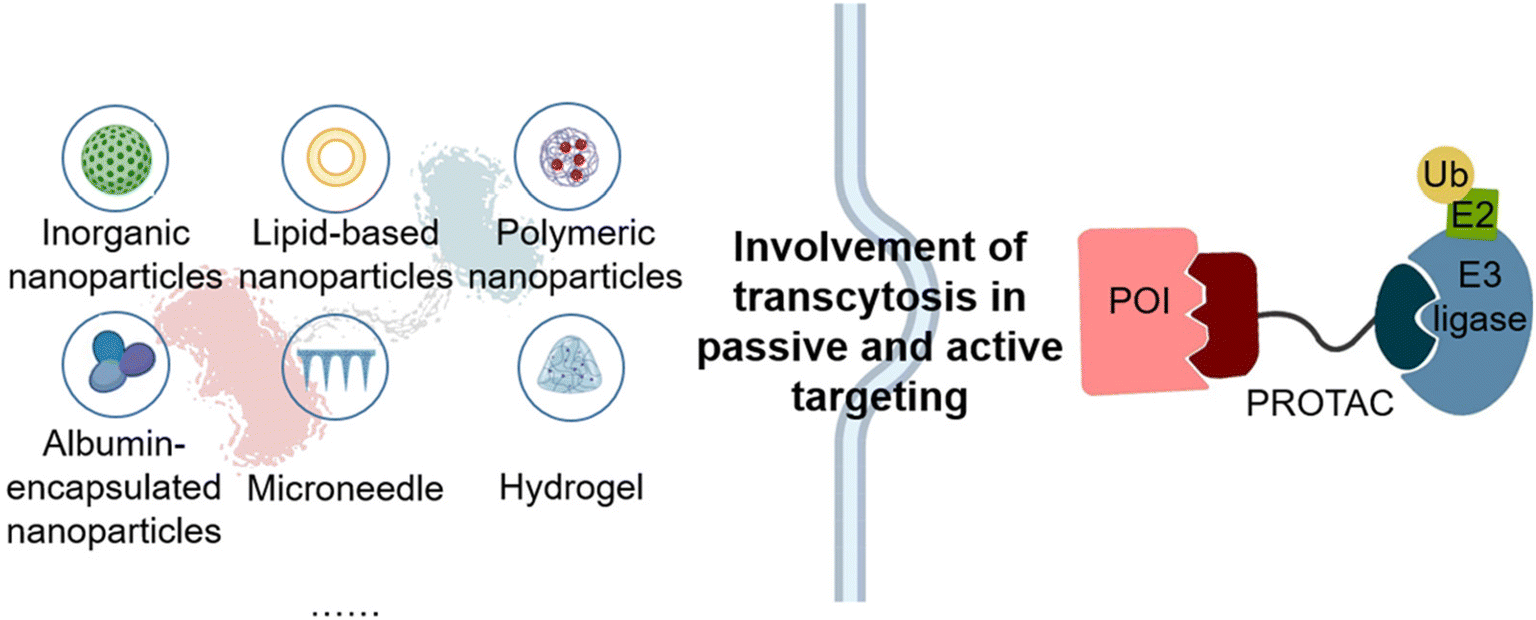 | ||
| Fig. 3 Delivery systems and PROTAC drug loading. | ||
3.1 Inorganic nanoparticles
Inorganic nanomaterials could serve as ideal PROTAC delivery systems due to their unique physicochemical properties, such as facile preparation and controlled shape regulation.36 The unique rigid structure of inorganic particles reduces the risk of drug leakage at off-target sites during circulation.37 Among them, gold nanoparticles have been widely developed for PROTAC drug delivery, especially peptide-based PROTACs due to the metal coordination between the peptide and the gold ion.38–42 For example, Ma et al.38 utilized ultrasmall gold nanoparticles to load peptide-based PROTAC drugs. Due to the presence of a cysteine in peptide-based PROTACs, the gold core could conjugate with PROTACs and branched poly(ethylenimine) (PEI) successfully (Fig. 4A). The resultant gold-PROTAC nanocomplexes showed a stable and uniform appearance and positive surface potentials, which could enable cell penetration. With high peptide PROTAC-loading efficiency and stability, more efficient androgen receptor degradation with potential clinical use in vivo was observed.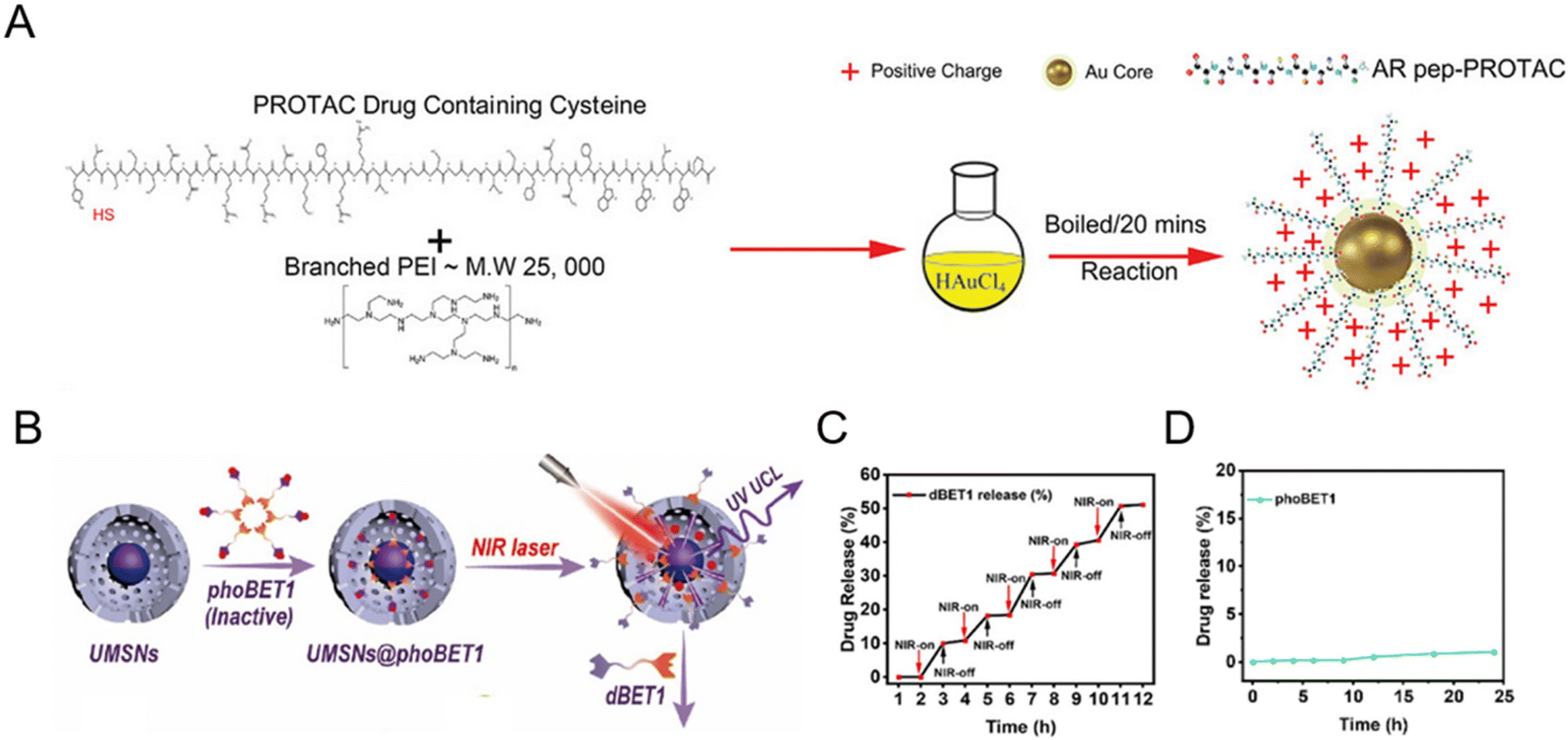 | ||
| Fig. 4 (A) Schematic depiction of the synthesis process of gold-PROTAC nanocomplexes with positive surfaces. Reproduced from ref. 38 with permission. Copyright 2022, Wiley-VCH GmbH. (B) Schematic illustration of the preparation of NIR-activated PROTACs loaded in mesoporous silica nanoparticles. (C) The release pattern of PROTAC dBET1 in mesoporous silica nanocages under 980 nm laser irradiation. (D) The release profile of phoBET1 in mesoporous silica nanoparticles in PBS buffer. Reproduced from ref. 32 with permission. Copyright 2023, American Chemical Society. | ||
In addition to gold nanoparticles, other inorganic nanomaterials such as mesoporous silica nanoparticles32,43 have also attracted considerable attention for PROTAC delivery with favorable pharmacokinetic properties. He and co-workers prepared photo-caged PROTAC-loaded mesoporous silica nanoparticles (phoBET1) for controllably targeted BRD4 degradation (Fig. 4B).32 As shown in Fig. 4C, the release of dBET1 could be controlled under alternating conditions of NIR light “on–off” irradiation, suggesting the controllable regulation and on-demand administration of the required PROTAC dosage by adjusting the irradiation time. Importantly, the released amount of phoBET1 in mesoporous silica nanoparticles was less than 1% in PBS buffer within 24 h (Fig. 4D), indicating that phoBET1 could be firmly encapsulated in mesoporous silica nanocages by physical adsorption to avoid premature leakage as mentioned before.37 Besides, as a kind of porous materials with uniform structures and adjustable pores, metal–organic frameworks (MOFs) are also worthy of further study for PROTAC delivery.44 Although inorganic nanoparticles are flexible for drug loading and surface modification, their potential non-biodegradability and toxicity are an impediment to sustainable translation. To fully develop inorganic nanoparticles for PROTAC delivery, more detailed safety evaluations and genomic/proteomic array tests should be conducted to access the sub-lethal cellular changes, both the short-term and long-term effects in vivo.45
3.2 Lipid-based and membrane-coated nanoparticles
With good biocompatibility, high payload, and surface modification flexibility, lipid-based nanoparticles have demonstrated the potential for PROTAC delivery. The most common lipid-based nanoparticles are spherical platforms that contain at least one lipid bilayer containing at least one internal aqueous compartment. The use of phospholipids and sterols is considered beneficial for PROTAC drug administration33,46–52 since these compounds are analogous to cellular membranes and facilitate cellular uptake.53 At the same time, the modification flexibility of basic liposome components endows PROTACs with the capability of tissue-targeting50 and controlled release. For example, by conjugating the folate group, PROTAC liposomal formulations showed folate-specific binding to folate receptors, where it rapidly released the drug to exert its POI-degrading effect.50 In a representative study,50 after folate conjugation, the mean fluorescence intensity of Cy5.5 in A549 cells (with overexpressed folate receptors) was 1.85-fold higher than that in L929 cells (Fig. 5A). In the meantime, in comparison with folate-unmodified liposomes, the Cy5.5 mean fluorescence intensity of liposomes containing folate groups was 2.50 times higher that in A549 cells (Fig. 5A). The enhanced cellular uptake with the modification of folate groups on PROTAC liposomes thus led to a significantly lower expression level of the abnormal protein NQO1 (Fig. 5B). To date, there are several lipid-based drugs that have been approved for clinical use by the FDA (US Food and Drug Administration), such as Doxil® (1995), DepoDur® (2004), and Exparel® (2011).54 In recent years, two liposomal vaccines were approved to combat COVID-19,53 showing the tremendous potential of lipid-based systems for PROTAC delivery in future clinical therapy. Despite the wide utility and proven efficacy of lipid-based nanoparticles for RNA delivery and COVID-19 vaccines, they still face some challenges. Firstly, the effect of lipid chemical structures and components on long-term storage stability and in-use stability is still poorly understood.55,56 Secondly, lipid-based formulations are not completely inert immunologically. For example, it has been found that pseudoallergic reactions related to complement activation are usually attributed to PEG-lipid components after COVID-19 vaccine administration.57,58 Moreover, anti-PEG antibodies detected in people vaccinated with the COVID-19 vaccine may lead to the rapid clearance of lipid-based formulations for regimens that require repeated administration, thus hindering the therapeutic effect.59,60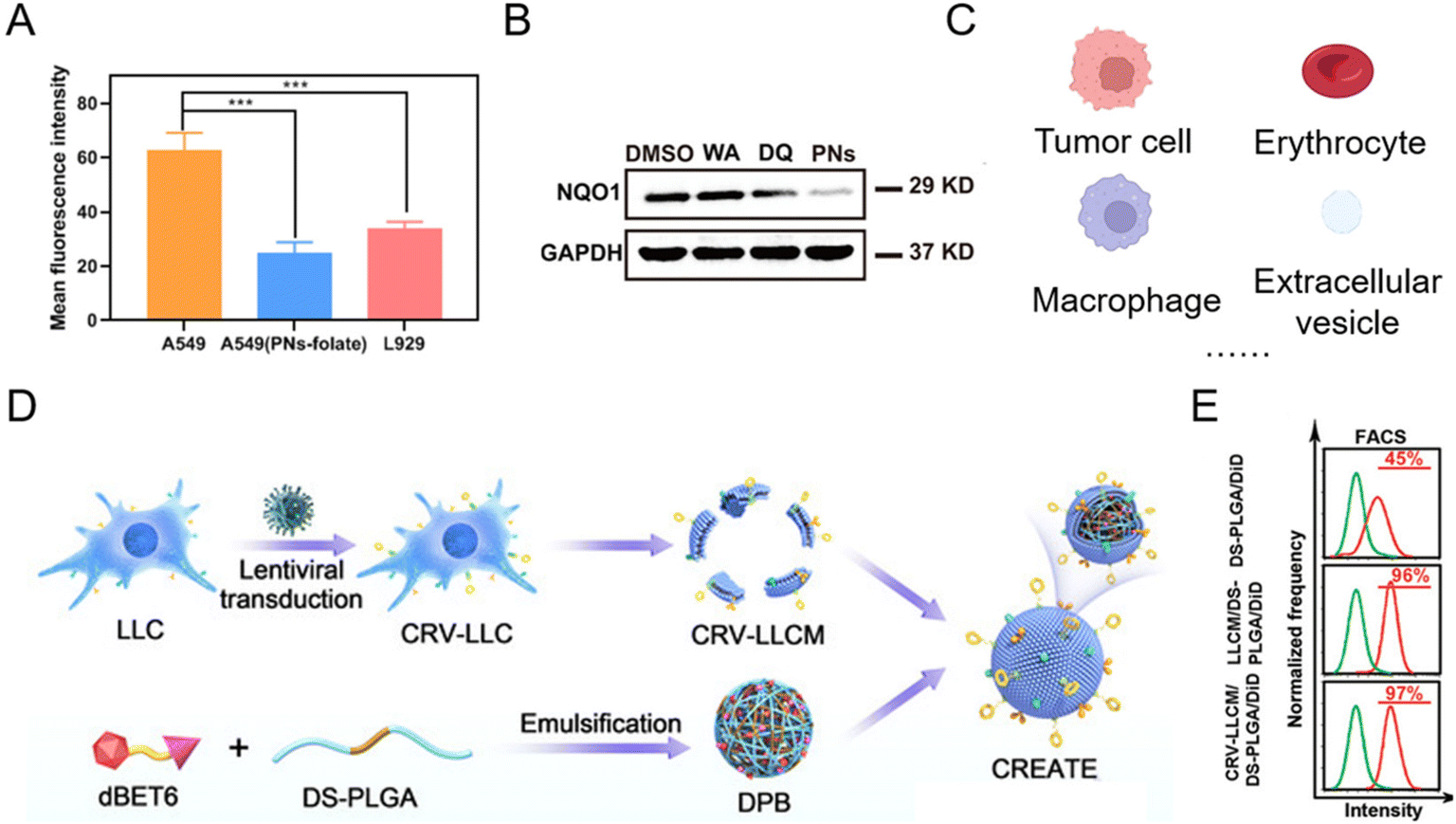 | ||
| Fig. 5 (A) Mean fluorescence intensities of Cy5.5 in A549 and L929 cells after being incubated with folate-modified PROTAC liposomes and PROTAC liposomes. (B) Abnormal protein NQO1 levels in A549 cells treated for 24 h with different groups. Reproduced from ref. 50 with permission. Copyright 2023, American Chemical Society. (C) Current natural membrane sources of biomimetic nanoparticles for PROTAC delivery. (D) Schematic illustration of engineering a lung cancer cell membrane-coated biomimetic BRD4-degrading PROTAC formulation. (E) The flow cytometry analysis of internalization of the membrane-coated BRD4-degrading PROTAC formulation in tumor cells. Reproduced from ref. 61 with permission. Copyright 2022, Wiley-VCH GmbH. | ||
In addition, some natural lipid sources have been leveraged as a practical strategy for drug loading (Fig. 5C).62–64 Since the low bioavailability and cell specificity of PROTAC reagents hinder their therapeutic applications, bioinspired membrane-coated technology is proposed as an ideal method to improve the performances of PROTACs, especially tumor cell membranes. Tumor cell membrane-coated systems are capable of homologous tumor targeting and have been widely used in targeted drug delivery systems for cancer therapy. Recently, more and more tumor cell membrane-coated systems have emerged for PROTAC delivery.43,61,65 For example, engineering lung cancer cell membrane in the biomimetic BRD4-degrading PROTACs prepared by Zhang et al.61 endowed the system with simultaneous targeting ability to lung cancer cells and tumor-associated macrophages (Fig. 5D). The flow cytometry analysis (Fig. 5E) revealed that the camouflage with the lung cancer membrane enhanced the internalization of PROTAC nanoparticles. In order to enhance targeting ability, prolong the circulation time of PROTAC drugs and meet other functional purposes, biomimetic nanoparticles for PROTAC delivery were observed in other membrane-coated systems (Fig. 5C), such as the erythrocyte membrane,41 the macrophage membrane,66 and extracellular vesicles.67
3.3 Polymeric nanoparticles
Polymeric nanoparticles consist of natural materials or synthetic polymers. Due to the inherent biocompatibility of the materials used, polymeric nanoparticles are widely used in the study of drug delivery vehicles. In addition, polymeric nanoparticles are simple to prepare, have adjustable physicochemical properties, and can be used to deliver a wide variety of drugs such as small molecules, biological macromolecules, proteins, and nucleic acids. Despite all the advantages mentioned above, studies on polymer nanoparticle-based PROTAC drugs61,68–75 are currently facing serious drawbacks for efficient encapsulation of PROTACs, smart delivery systems, and clinical translation. To solve these dilemmas, Liu et al.73 used two synthetic polymers, DSPE-PEG and PDSA, to prepare a biodegradable polymer nanoparticle for BRD4 degrader ARV-771 delivery (Fig. 6A). In the presence of disulfide bonds in PDSA, this polymer nanoparticle selectively released PROTACs in tumor cells in a glutathione (GSH)-responsive manner corresponding to the in vitro simulation (Fig. 6B). Moreover, this polymer nanoparticle showed enhanced bioavailability and accumulation of PROTACs in diseased cells and an in vivo anti-cancer effect with BRD4 degradation. In another study, He et al.74 proposed a combined treatment strategy for the co-delivery of doxorubicin and BRD4-degrading PROTAC ARV-825 (Fig. 6C). With the cRGD (αvβ3 integrin-targeted peptide)-modified polymer segment, polymeric micelles showed enhanced cellular uptake and targeting properties. Compared to free ARV-825 and doxorubicin, the resulting cRGD-modified polymer nanoparticles exhibited an apparent effect in vivo in overcoming acquired drug resistance due to the αvβ3 integrin-mediated cellular uptake and the improved polymeric nanoparticle-mediated controllability of PROTAC release. Yang and co-workers68 utilized a substance P peptide-modified polymer to form brain-targeted PROTAC polymeric nanoparticles (Fig. 6D). According to the molecular dynamics simulation in Fig. 6E, PROTAC molecule ARV-825 exhibited constant positional and conformational changes during the 10 ns of interaction with polymer mPEG-PDLLA, while ARV-825 is gradually encapsulated in the hydrophobic PDLLA chimeras of mPEG-PDLLA, ultimately forming stable drug-carrying micelles in both environments.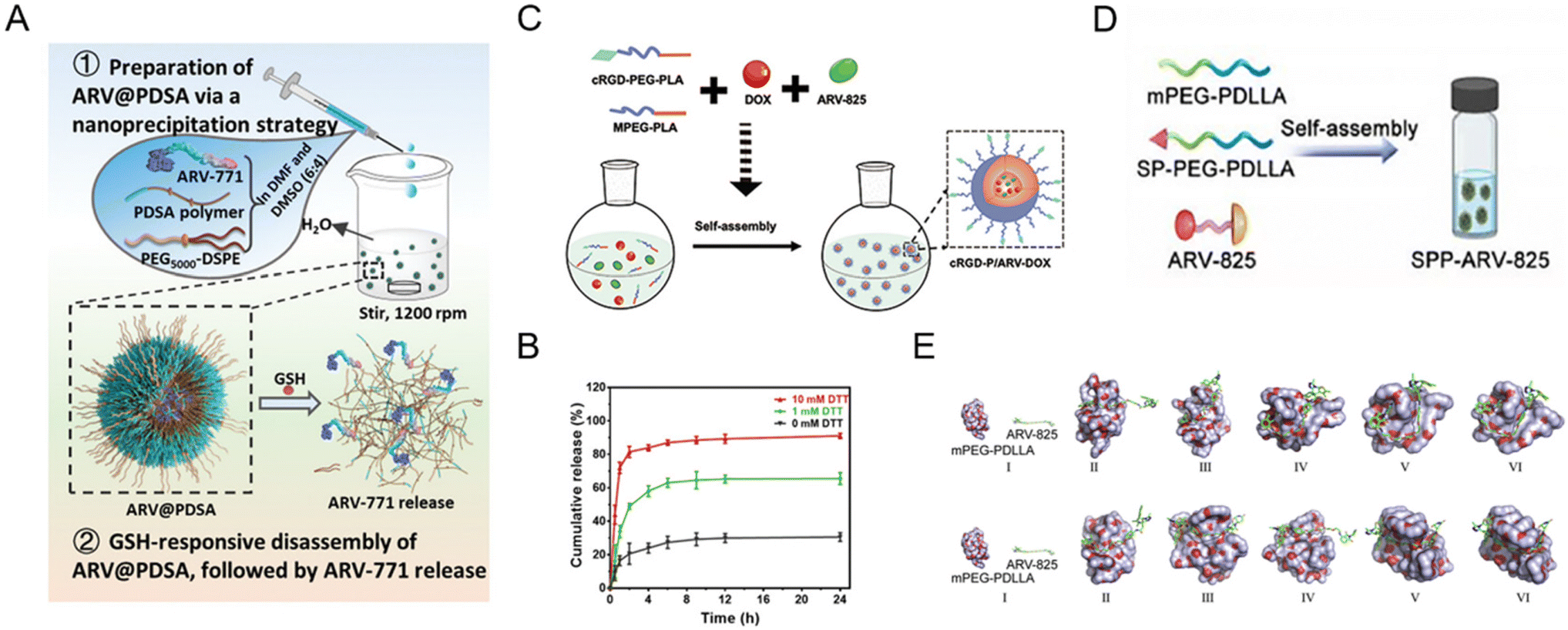 | ||
| Fig. 6 (A) Schematic illustration of the preparation of GSH-responsive BRD4-degrading PROTAC polymeric nanoparticles. (B) The release profiles of PROTACs from polymeric nanoparticles incubated with different dithiothreitol (DTT) concentrations. Reproduced from ref. 73 with permission. Copyright 2023, Wiley–VCH GmbH. (C) Schematic illustration of the preparation of cRGD-decorated polymeric nanoparticles for the co-delivery of doxorubicin and BRD4-degrading PROTACs. Reproduced from ref. 74 with permission. Copyright 2022, Elsevier. (D) Schematic illustration of the preparation of the substance P peptide-modified BRD4-degrading PROTAC polymeric nanoparticle formulation. (E) Molecular dynamics simulations of the interaction between mPEG-PDLLA and ARV-825 in a water environment and a tumor-mimicking environment. Reproduced from ref. 68 with permission. Copyright 2022, Elsevier. | ||
Polymeric nanoparticles can be synthesized using different techniques such as emulsification, nanoprecipitation, ionogelation, and microfluidization to prepare drug delivery systems with different properties. Thus, PROTAC molecules can be flexibly loaded in polymer nanoparticles, such as inside the hydrophobic core, bound to the particle surface, trapped in a polymer matrix, or chemically linked to the polymer structure to optimize the payload of PROTAC molecules.2 In addition, some polymeric materials with specific functionalities, such as targeting units and stimuli-responsive segments, can further optimize the pharmacokinetics to exert enhanced efficacy. However, the high immunogenicity of polymers remains a considerable limitation, and the high variability in mass production further limits their clinical translation.
3.4 Albumin-encapsulated nanoparticles
Albumin is a versatile carrier for drug delivery because it is non-toxic, non-immunogenic, biocompatible, and biodegradable. As an ideal carrier for delivering drugs, albumin nanoparticles have received widespread attention for their high binding capacity to a wide range of drugs76,77 without noticeable side effects. Due to the rapid growth of tumor tissues, most of them will bind to a large amount of albumin through glycoprotein 60 (GP60) receptor and secreted protein acidic and rich in cysteine (SPARC) protein uptake as their source of energy and amino acids. Benefiting from this, albumin nanoparticles or surface-modified albumin nanoparticles can be targeted to tumor tissues. To maximize PROTAC drugs’ therapeutic potential, lots of engineering albumin-based nanoplatforms were developed very recently (Fig. 7A).78–81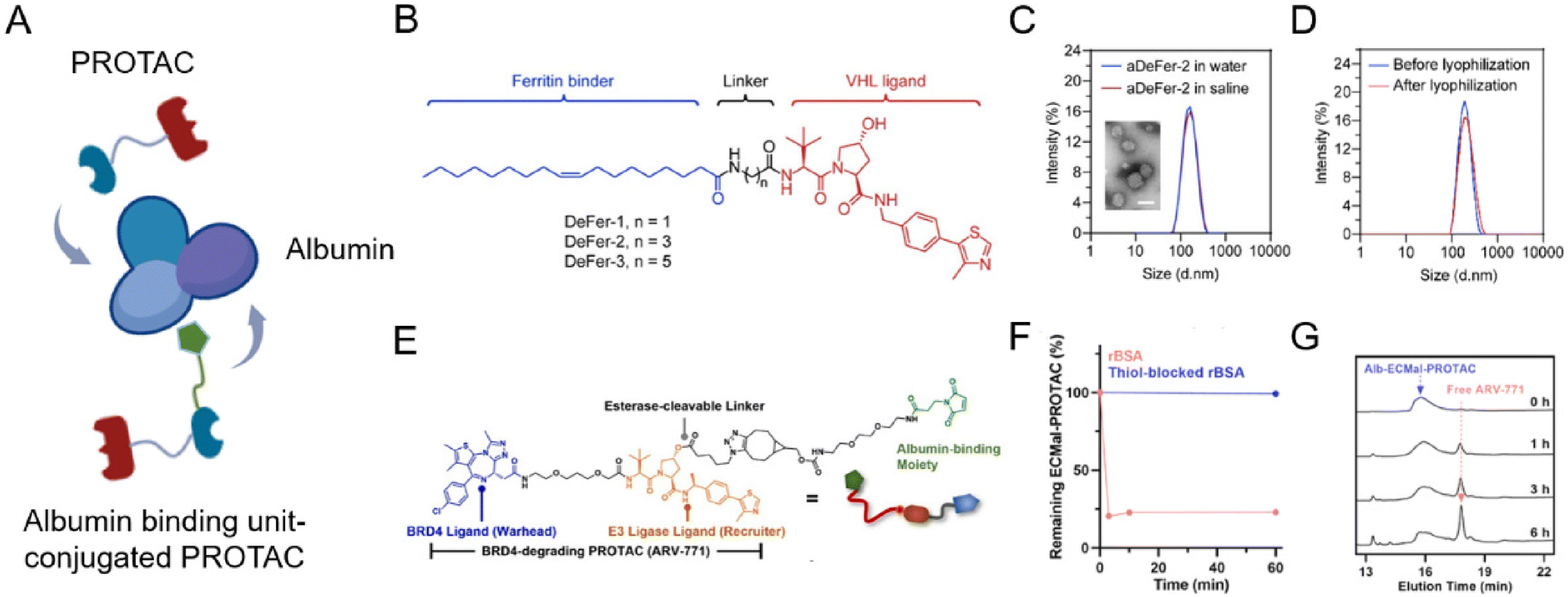 | ||
| Fig. 7 (A) Schematic depiction of the preparation of an albumin-encapsulated PROTAC delivery system. (B) Chemical structures of oleic acid-based VHL-recruiting PROTACs. (C) Size distribution of aDeFer-2 in water and saline and the morphology observed by TEM. Scale bar, 100 nm. (D) Size distribution of aDeFer-2 before and after lyophilization. Reproduced from ref. 78 with permission. Copyright 2023, American Chemical Society. (E) Chemical structures of albumin-binding moiety-modified BRD4-degrading PROTACs. (F) The HPLC peak of Alb-ECMal after incubation with BSA and thiol-blocked BSA. (G) The HPLC peak of Alb-ECMal PROTAC with an esterase solution (30 U mL−1). Reproduced from ref. 81 with permission. Copyright 2023, Elsevier. | ||
Considering this, Chen et al.78 conjugated oleic acid to the ligand of E3 ligase through different alkyl linkers to screen the most effective chimera (Fig. 7B). Based on the involvement of unsaturated long-chain fatty acids in the assembly of these identical iron-storage proteins, oleic acid was used as a binding agent for ferritin dimers. The screened ferritin-degrading PROTAC DeFer-2 was further bound with albumin (aDeFer-2) for favorable anticancer treatment efficacy. As shown in Fig. 7C, the obtained aDeFer-2 displayed a uniform spherical structure under a TEM (transmission electron microscope) and had a narrow size distribution. Moreover, no obvious changes were observed in size distribution before and after lyophilization (Fig. 7D), indicating the excellent stability of the albumin-encapsulated PROTAC delivery systems. The resultant aDeFer-2 nanoparticles demonstrated consistent and excellent ferritin degradation potency with DeFer-2, resulting in substantial tumor growth inhibition. In addition, a favorable systemic biosafety profile was observed. To exploit albumin as a more effective drug carrier, the in situ covalent binding of drugs to the unpaired thiol of the 34th cysteine residue in albumin was widely applied. Since the Thiol-Mal click reaction is feasible even under physiological conditions, drug molecules can be simply conjugated to native albumin in a hitchhiking way without destroying its structure. Recently, Cho et al.81 newly prepared a BRD4-degrading PROTAC (ARV-771) with an esterase-cleavable maleimide linker (Alb-ECMal PROTAC). The resulting precursor PROTACs were considered to hitchhike albumin in the blood plasma via a conjugated maleimide moiety. At first, in the presence of the conjugated maleimide moiety, PROTACs conjugated to bovine serum albumin (BSA) in a quick and efficient way (Fig. 7F), demonstrating the potential of sufficient albumin conjugation in blood. The HPLC (high performance liquid chromatography) peak of the released PROTAC showed the release of free ARV-771 with the enzymatic activity of esterase in Fig. 7G, indicating more active release of free ARV-771 after being delivered to esterase-overexpressed tumors. After being internalized to targeted tumor cells, they can be cleaved to release free BRD4-degrading PROTACs by the overexpressed esterase, resulting in actively intracellular BRD4 degradation and further anticancer efficacy.
Collectively, encapsulation with albumin not only enhances the stability and aqueous solubility of PROTACs, but also greatly enhances tumor-specific accumulation, even helping to clearly identify blood vessels, tumors, and adjacent boundaries. Nevertheless, this delivery system can still continue to be optimized. The introduction of the maleimide moiety provides ideas for the better binding of albumin to PROTACs. In addition, albumin has various functional groups, which can be modified with various ligands to further target tumor tissues or to produce other functionalities such as enhancing the permeability of tissues. For example, folate-conjugated albumin will help albumin-based PROTAC drugs actively target lesions.82 It is worth mentioning that receptors such as GP60 are mainly responsible for natural human serum albumin (HSA) transport in vivo, and it remains doubtful that drug-carrying albumin can be taken up by tumors without competitive inhibition in the presence of high concentrations of HSA in vivo. This, together with the enhanced permeability and retention (EPR) effect, often mentioned in terms of passive targeting,83 needs to be further investigated clearly in the future. The above issues are important prerequisites for the further development of an albumin-based nanoplatform for the delivery of PROTACs and should be the focus of future research.
3.5 Local delivery systems
Microneedle patches are powerful transdermal drug delivery systems that enable direct delivery of encapsulated drugs to the epidermis and dermis layers by physically overcoming the stratum corneum. With a minimally invasive approach, microneedle patch-assisted drug delivery systems have attracted soaring attention in several multifunctional therapies, such as cancers,84–86 hair follicle regeneration,87,88 wound healing,89 and rheumatoid arthritis.90 In January 2023, Wang et al.87 reported an androgen receptor-degrading PROTAC loaded dissolving microneedle patch for androgenetic alopecia and recrudescence treatment via single topical administration. This work first demonstrated the PROTAC drug delivery potential of a microneedle patch.A microneedle patch endows drugs with the capability of direct and sustained delivery to tissues, but at the same time, some necessary nanotechnology-based modifications are often used to fabricate PROTAC prodrugs to achieve more precise treatment.72,86 For example, Cheng et al.86 fabricated an estrogen receptor alpha (ERα)-degrading PROTAC loaded microneedle patch (Fig. 8A and B) for efficient breast cancer treatment. Before loading into biodegradable microneedle patches, PROTACs and palbociclib were encapsulated in a pH-sensitive micelle to form PROTAC prodrugs for deep tumor penetration and smart stimuli-responsive release. In another microneedle patch-assisted PROTAC delivery study by Huang et al.,72 to enhance the local intracellular targeted delivery, the RGD sequence was decorated onto the surface of PROTAC polymeric nanoparticles to target the integrin αVβ3 of cancer cells.
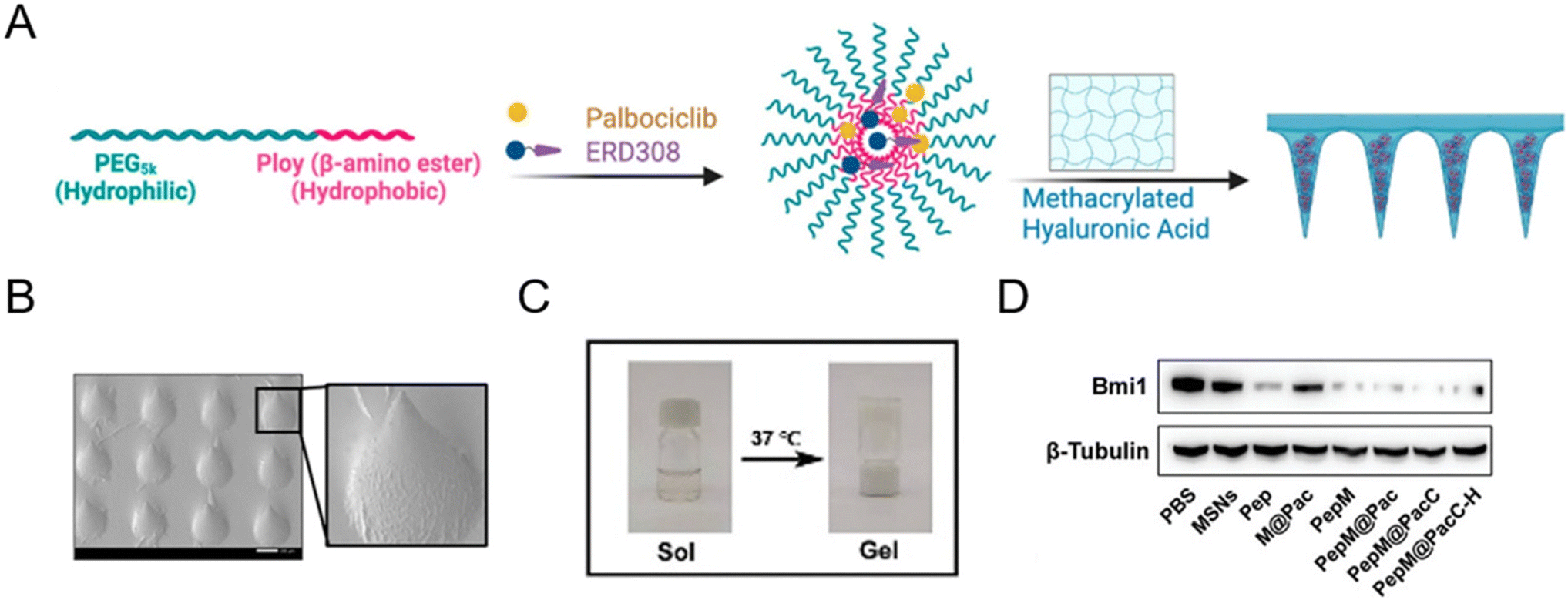 | ||
| Fig. 8 (A) Fabrication and characterization of PROTAC-encapsulated micelles and micelle-loaded microneedle patches. (B) SEM (scanning electron microscopy) image of PROTAC-loaded microneedle patches. Scale bar, 200 μm. Reproduced from ref. 86 with permission. Copyright 2023 American Chemical Society. (C) Gel–sol transition of a PROTAC-loaded hydrogel. (D) Immunoblotting for Bmi1 protein abundance after exposure to the indicated groups. Reproduced from ref. 43 with permission. Copyright 2023, Wiley-VCH GmbH. | ||
Aside from the afore-mentioned nanoparticles, some other delivery systems also hold potential to deliver PROTAC drugs. For example, hydrogel-based PROTAC delivery systems help PROTAC drugs circumvent their own unfavorable hydrophobicity and can be an important research direction for future PROTAC delivery technologies.43 Wu et al.43 demonstrated that PLGA–PEG–PLGA can serve as a favorable nanocomposite hydrogel for effective peptide PROTAC drug delivery with a sol–gel transition at 37 °C (Fig. 8C). According to Fig. 8D, the peptide PROTAC in the hydrogel maintained its original activity, inducing effective targeted protein Bmi1 degradation. Given the low toxicity, biodegradability, high delivery efficiency, and modifiability of these advanced materials, more delivery vectors are needed to deliver PROTAC drugs in the future.
4. Split-and-mix PROTAC approach and center-spoke degradation network
The split-and-mix PROTAC approach is employed as a special nanoplatform capable of facile screening and self-optimized biomolecule regulation.15,47,91–95 Specifically, this system consists of independent hydrophilic segments with recruiting targets of POIs and E3 ligases respectively. This strategy (Fig. 9A) will no longer focus on conjugating new groups to PROTAC ternary complexes, but will take the POI binder part and E3 ligand part as separate research objects (split) to study the self-assembled performance (mix) of the two modified parts in vitro and in vivo. The multivalent nature of this system not only ensures the protein degradation efficiency, but also helps to screen a facilely programmable ratio of E3 recruiters and POI recruiters. Due to this, multi-headed recruiters are exposed to multiple proximal E3 ligases with a center-spoke degradation network.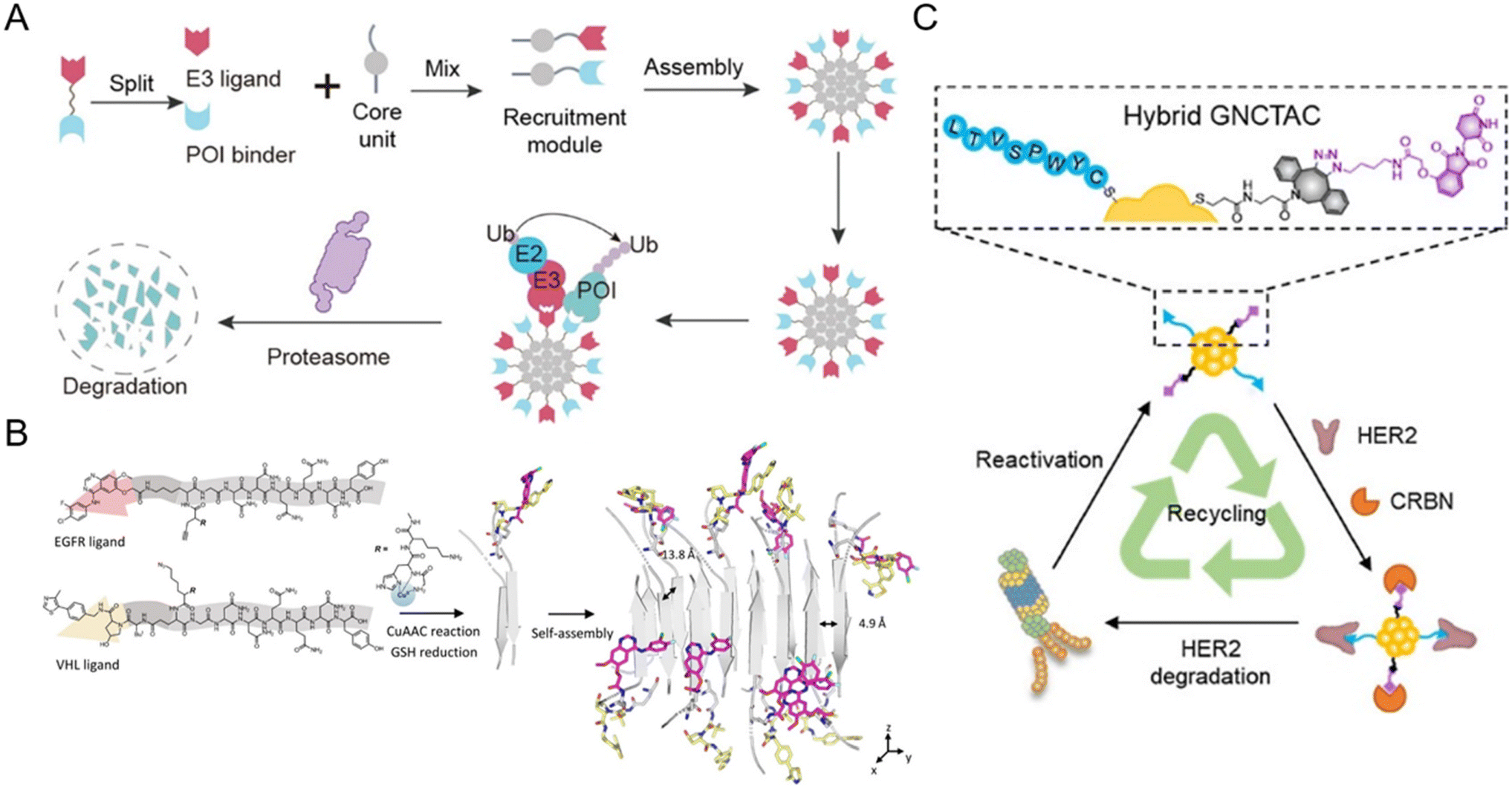 | ||
| Fig. 9 (A) Schematic illustration of the split-and-mix PROTAC approach and the center-spoke degradation network. Reproduced from ref. 15 with permission. Copyright 2023, American Chemical Society. (B) Chemical structure of the POI binder part and the E3 ligand part and the in situ self-assembly of EGFR-degrading split-and-mix PROTACs. Reproduced from ref. 91 with permission. Copyright 2023, Wiley-VCH GmbH. (C) Gold nanocluster-based HER2-degrading split-and-mix PROTACs. Reproduced from ref. 92 with permission. Copyright 2023, American Chemical Society. | ||
For example, Yang et al.15 selected a suitable and modifiable self-assembled monomer (diphenylglycine) as the core unit of the split-and-mix PROTAC system. The results clearly revealed that this model system can effectively degrade several proteins. Cellular nanofibrils built upon the peptide assembly can serve as degradation centers due to structurally diverse amino acid molecules. By some chemical methodologies such as click chemistry, the POI and E3 ligase can be recruited by a self-assembled peptide. The fact that in this strategy, the POI ligand and E3 ligase ligand molecules do not have to fuse into a ternary complex greatly improves the efficiency of protein degradation.91,95 Taking advantage of the in situ self-assembly strategy,96 Zhang et al.91 introduced a GHK-CuII moiety, an azido or alkyne group, and an assembly-driving peptide into the split-and-mix PROTAC system to realize dose-dependent and long-acting epidermal growth factor receptor (EGFR) degradation potency in vitro and in vivo (Fig. 9B). Besides peptide-based precursors, some studies utilized gold nanoclusters92,93 and carbon dots94 as the core units for the split-and-mix PROTAC approach. Wang et al.92 reported a gold nanocluster-hybrid PROTAC (GNCTAC) that perpetuated human epidermal growth factor receptor 2 (HER2) degradation (Fig. 9C). In this work, gold nanoclusters serve as a linker to provide the platform to connect targeting ligands. By recruiting cereblon (CRBN) in tumor cells through a small-molecule CRBN targeting ligand, more than 95% of HER2 in the human breast cancer cell line SKBR3 was degraded by GNCTACs. Further results demonstrated that the degradation duration was as long as 72 h, showing a catalytic-like reaction.
5. Carrier-free nano-PROTACs and combinational therapy
The principle of carrier-free nano-PROTACs is based on the self-assembly of PROTACs and other functional small molecules (Fig. 10A), attracting considerable interest in recent years. PROTACs generally have a conjugated structure and a planar structure based on double bonds, and these structures can form assemblies through hydrophobic interaction and π–π stacking interaction with other molecules, such as the widely used photosensitizer chlorin e6 (Ce6). The resultant nano-PROTAC particles generally have favorable dispersibility and a uniform nanosize distribution. With “nanoscale advantages”,34,35 it is generally believed that they have the potential to improve the tissue accumulation and blood circulation of PROTACs. Since co-assembled units are often therapeutic molecules, this co-delivery strategy can be a powerful tool for combinational therapy.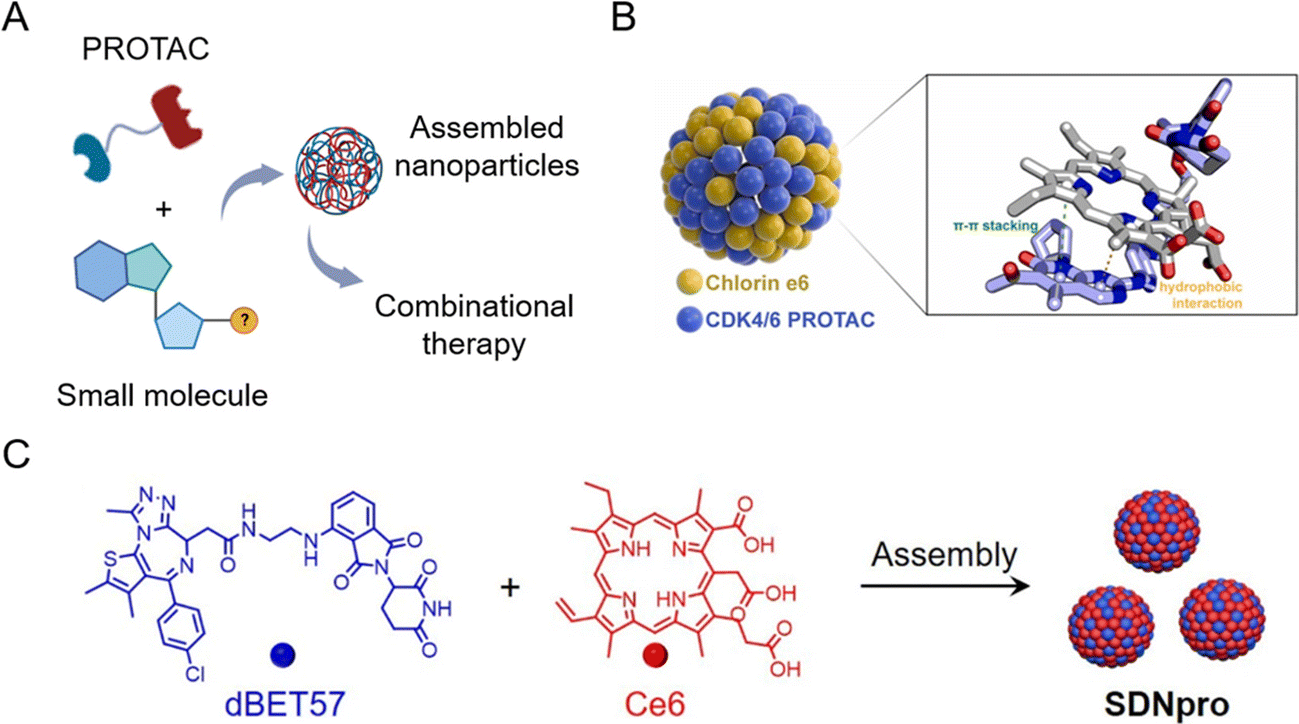 | ||
| Fig. 10 (A) Schematic illustration of carrier-free nano-PROTACs for combinational therapy. (B) Nano-PROTACs based on Ce6 and CDK4/6 PROTACs. Reproduced from ref. 16 with permission. Copyright 2023, Elsevier. (C) Nano-PROTACs (SDNpro) based on Ce6 and dBET57. Reproduced from ref. 97 with permission. Copyright 2023, American Chemical Society. | ||
Wang et al.16 used Ce6 to prepare self-assembled nanoparticles with cyclin-dependent kinase (CDK) 4/6 PROTACs (Fig. 10B). The resultant nanoparticles induced higher mitochondrial accumulation and activation, leading to an increased generation of reactive oxygen species and cell apoptosis. At the same time, photodynamic therapy induced by Ce6 evoked immunogenic cell death and the chemotaxis of immune cells cooperatively. This study demonstrated that carrier-free nano-PROTACs hold great potential for synergistic cancer photo-immunotherapy. A similar strategy was applied in another study by Zhao et al.97 To enhance photodynamic therapy by blocking DNA damage repair through BRD4 degradation, dBET57-Ce6 nano-PROTACs (SDNpro) have been developed (Fig. 10C). These reported carrier-free nano-PROTACs aimed at overcoming the off-target effect of PROTACs and enhancing the antitumor effect by combinational therapy strategies.
In carrier-free nanoplatforms, remarkable drug loading (could reach 100%) and excellent drug pharmacokinetics can always be observed since all the components used are functional. The simplicity of related preparation methods (e.g., nanoprecipitation) makes large-scale production feasible. Additionally, the insertion of stimuli-responsive linkers and tumor targeting ligands endows carrier-free nano-PROTACs with additional characteristics like on-target delivery and controllable release. After fully understanding the self-assembly behavior of PROTAC molecules with other drugs (such as photosensitizers, chemotherapeutic agents, and radiotherapeutic agents), more and more carrier-free nano-PROTACs will emerge with the overarching goal of achieving prolonged circulation times, deep tissue penetration, and high-efficiency synergistic treatment.
6. Conclusion and perspectives
As this review attempts to show, the combination of nanotechnology and PROTACs may become more commonplace in the future as a strategy to spatiotemporally control the release of PROTACs, which holds potential to improve efficiency and selectivity whilst mitigating the “hook effect” of PROTACs. At the same time, nanotechnology could be employed to greatly improve the druggability of PROTAC molecules. Additionally, thanks to the excellent loading capacity of various delivery systems and the construction of carrier-free nano-PROTACs, PROTACs can be co-delivered with other drugs for the purpose of combinational therapy. On the basis of the improvements and limitations discussed above, Table 1 systematically introduces a set of nano-PROTAC approaches for future rational design of PROTACs and their rapid translation to the clinic.| Nano-PROTAC approaches | Improvements | Limitations | Ref. |
|---|---|---|---|
| Building-block nano-PROTACs | On-demand release, combinational therapy, prolonged circulation | Potential immunogenicity, compromised in vivo stability, significant synthetic effort | 14 and 29–33 |
| Diverse delivery systems | Targeting effect, prolonged circulation, on-demand release | Potential immunogenicity, compromised in vivo stability, restricted encapsulation efficiency, limited application (e.g., hydrogel/microneedle, limited to a superficial lesion) | 2, 98 and 99 |
| Split-and-mix nano-PROTACs | Center-spoke degradation, facile screening of ligands | Potential immunogenicity | 15, 47 and 91–95 |
| Carrier-free nano-PROTACs | Combinational therapy, excellent drug loading capacity (could reach 100%) and drug pharmacokinetics, on-demand release | Intrinsic instability, insufficient targeting efficiency, biological barrier limitation | 16, 97 and 100 |
In addition to the urgent need to develop more POIs and E3 ligases for the construction of novel PROTACs, there are many reasons that hinder the translational process of nano-PROTACs from bench to bedside. The lack of reliable methods to study the structure of PROTACs and the assemblies of nano-PROTACs can be an important impediment. In addition to this, the safety of most of the additional ingredients used to construct nano-PROTACs has not been fully evaluated and needs to be studied in more detail. Another important pinch-point is manufacturing cost. Additional pharmaceutical excipients will increase the manufacturing cost of PROTAC drugs, thus hindering their globalization. In any case, with the development of biology, pharmacology, materials science, nanotechnology, industrial technology, and clinical science, new evaluation methods and new drug systems will be gradually established to promote the widespread clinical translation of PROTAC drugs and provide sufficient clinical benefit in the future. This comprehensive review and perspective can serve as a blueprint for both PROTAC researchers and nanomedicine researchers during their future ventures into the field of smart PROTAC drugs.
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (NSFC) project (grant No. 32301133), the University of Hong Kong Seed Fund for Basic Research (grant No. 104006533 and 104005845), the R&D Program of Beijing Municipal Education Commission (grant No. KM202310025022) and the Beijing Hospitals Authority Clinical Medicine Development of Special Funding Support (grant No. ZLRK202308 and CIT&TCD 20180332).References
- K. Li and C. M. Crews, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC.
- Y. Chen, I. Tandon, W. Heelan, Y. Wang, W. Tang and Q. Hu, Chem. Soc. Rev., 2022, 51, 5330–5350 RSC.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- B. Dale, M. Cheng, K. S. Park, H. Kaniskan, Y. Xiong and J. Jin, Nat. Rev. Cancer, 2021, 21, 638–654 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Signal Transduction Targeted Ther., 2022, 7, 113 CrossRef CAS PubMed.
- X. Li and Y. Song, J. Hematol. Oncol., 2020, 13, 50 CrossRef CAS PubMed.
- S. Tomoshige and M. Ishikawa, Angew. Chem., Int. Ed., 2021, 60, 3346–3354 CrossRef CAS PubMed.
- Q. Q. Zhou, H. T. Xiao, F. Yang, Y. D. Wang, P. Li and Z. G. Zheng, Pharmacol. Res., 2023, 188, 106627 CrossRef CAS PubMed.
- J. Gao, L. Yang, S. Lei, F. Zhou, H. Nie, B. Peng, T. Xu, X. Chen, X. Yang, C. Sheng, Y. Rao, K. Pu, J. Jin, Z. Xu and H. Yu, Sci. Bull., 2023, 68, 1069–1085 CrossRef CAS PubMed.
- C. Wang, C. Zheng, H. Wang, L. Zhang, Z. Liu and P. Xu, Eur. J. Med. Chem., 2022, 235, 114290 CrossRef CAS PubMed.
- R. G. Guenette, S. W. Yang, J. Min, B. Pei and P. R. Potts, Chem. Soc. Rev., 2022, 51, 5740–5756 RSC.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- C. Zhang, S. He, Z. Zeng, P. Cheng and K. Pu, Angew. Chem., Int. Ed., 2022, 61, e202114957 CrossRef CAS PubMed.
- F. Yang, Q. Luo, Y. Wang, H. Liang, Y. Wang, Z. Hou, C. Wan, Y. Wang, Z. Liu, Y. Ye, L. Zhu, J. Wu, F. Yin and Z. Li, J. Am. Chem. Soc., 2023, 145, 7879–7887 CrossRef CAS PubMed.
- T. Wang, Y. Zhang, K. Chen, Y. Huang, Y. Liu, S. Xu and W. Wang, Nano Today, 2023, 50, 101890 CrossRef CAS.
- J. Y. Lin, H. J. Liu, Y. Wu, J. M. Jin, Y. D. Zhou, H. Zhang, D. G. Nagle, H. Z. Chen, W. D. Zhang and X. Luan, Small, 2023, 19, e2207778 CrossRef PubMed.
- J. Liu, H. Chen, Y. Liu, Y. Shen, F. Meng, H. Kaniskan, J. Jin and W. Wei, J. Am. Chem. Soc., 2021, 143, 7380–7387 CrossRef CAS PubMed.
- H. Chen, J. Liu, H. Kaniskan, W. Wei and J. Jin, J. Med. Chem., 2021, 64, 12273–12285 CrossRef CAS PubMed.
- S. He, F. Gao, J. Ma, H. Ma, G. Dong and C. Sheng, Angew. Chem., Int. Ed., 2021, 60, 23299–23305 CrossRef CAS PubMed.
- Y. Liu, X. Qian, C. Ran, L. Li, T. Fu, D. Su, S. Xie and W. Tan, ACS Nano, 2023, 17, 6150–6164 CrossRef CAS PubMed.
- M. A. Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306–1312 CrossRef CAS PubMed.
- P. S. Dragovich, Chem. Soc. Rev., 2022, 51, 3886–3897 RSC.
- G. Xue, K. Wang, D. Zhou, H. Zhong and Z. Pan, J. Am. Chem. Soc., 2019, 141, 18370–18374 CrossRef CAS PubMed.
- Y. Naro, K. Darrah and A. Deiters, J. Am. Chem. Soc., 2020, 142, 2193–2197 CrossRef CAS PubMed.
- P. Pfaff, K. T. G. Samarasinghe, C. M. Crews and E. M. Carreira, ACS Cent. Sci., 2019, 5, 1682–1690 CrossRef CAS PubMed.
- W. Cheng, S. Li, X. Wen, S. Han, S. Wang, H. Wei, Z. Song, Y. Wang, X. Tian and X. Zhang, Chem. Commun., 2021, 57, 12852–12855 RSC.
- K. An, X. Deng, H. Chi, Y. Zhang, Y. Li, M. Cheng, Z. Ni, Z. Yang, C. Wang, J. Chen, J. Bai, C. Ran, Y. Wei, J. Li, P. Zhang, F. Xu and W. Tan, Angew. Chem., Int. Ed., 2023, 62, e202306824 CrossRef CAS PubMed.
- W. Wang, C. Zhu, B. Zhang, Y. Feng, Y. Zhang and J. Li, J. Am. Chem. Soc., 2023, 145, 16642–16649 CrossRef CAS PubMed.
- C. Zhang, Z. Zeng, D. Cui, S. He, Y. Jiang, J. Li, J. Huang and K. Pu, Nat. Commun., 2021, 12, 2934 CrossRef CAS PubMed.
- C. Zhang, M. Xu, S. He, J. Huang, C. Xu and K. Pu, Adv. Mater., 2023, 35, e2208553 CrossRef PubMed.
- Q. He, L. Zhou, D. Yu, R. Zhu, Y. Chen, M. Song, X. Liu, Y. Liao, T. Ding, W. Fan and W. Yu, J. Med. Chem., 2023, 66, 10458–10472 CrossRef CAS PubMed.
- L. Yao, N. Yang, W. Zhou, M. H. Akhtar, W. Zhou, C. Liu, S. Song, Y. Li, W. Han and C. Yu, Adv. Healthc. Mater., 2023, 12, e2300871 CrossRef PubMed.
- A. Kumar, F. Chen, A. Mozhi, X. Zhang, Y. Zhao, X. Xue, Y. Hao, X. Zhang, P. C. Wang and X. J. Liang, Nanoscale, 2013, 5, 8307–8325 RSC.
- Y. Tao, X. Lan, Y. Zhang, G. Qing, J. Wang, Y. Xiao, H. Chen, L. Liu, X. J. Liang and W. Guo, Expert Opin. Drug Delivery, 2022, 19, 985–996 CrossRef CAS PubMed.
- H. C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344–357 CrossRef CAS PubMed.
- K. Ariga, Q. Ji, M. J. McShane, Y. M. Lvov, A. Vinu and J. P. Hill, Chem. Mater., 2012, 24, 728–737 CrossRef CAS.
- B. Ma, Y. Fan, D. Zhang, Y. Wei, Y. Jian, D. Liu, Z. Wang, Y. Gao, J. Ma, Y. Chen, S. Xu and L. Li, Adv. Sci., 2022, 9, e2201859 CrossRef PubMed.
- S. Yan, J. Yan, D. Liu, X. Li, Q. Kang, W. You, J. Zhang, L. Wang, Z. Tian, W. Lu, W. Liu and W. He, Theranostics, 2021, 11, 6833–6846 CrossRef CAS PubMed.
- F. Niu, R. Yang, H. Feng, Y. Liu, R. Liu and B. Ma, Biochem. Biophys. Res. Commun., 2023, 684, 149125 CrossRef CAS PubMed.
- X. Zheng, J. Yan, W. You, F. Li, J. Diao, W. He and Y. Yao, Small, 2021, 17, 2100394 CrossRef CAS PubMed.
- W. Yang, W. Liu, X. Li, J. Yan and W. He, J. Adv. Res., 2023, 45, 59–71 CrossRef CAS PubMed.
- Y. Wu, X. Chang, G. Yang, L. Chen, Q. Wu, J. Gao, R. Tian, W. Mu, J. J. Gooding, X. Chen and S. Sun, Adv. Mater., 2023, 35, e2210787 CrossRef PubMed.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, e201606134 Search PubMed.
- L. Zhang, Y. Li and J. C. Yu, J. Mater. Chem. B, 2014, 2, 452–470 RSC.
- Y. Fu, D. Rathod and K. Patel, Exp. Cell Res., 2020, 396, 112275 CrossRef CAS PubMed.
- C. Song, Z. Jiao, Z. Hou, R. Wang, C. Lian, Y. Xing, Q. Luo, Y. An, F. Yang, Y. Wang, X. Sha, Z. Ruan, Y. Ye, Z. Liu, Z. Li and F. Yin, J. Am. Chem. Soc., 2023, 145, 21860–21870 CrossRef CAS PubMed.
- Y. Fu, A. Saraswat, Z. Wei, M. Y. Agrawal, V. V. Dukhande, S. E. Reznik and K. Patel, Pharmaceutics, 2021, 13 DOI:10.3390/pharmaceutics14102019.
- A. Saraswat, H. P. Vemana, V. V. Dukhande and K. Patel, Heliyon, 2022, 8, e08702 CrossRef CAS PubMed.
- H. Zhang, X. Xu, D. Yan, C. Ren, J. Zhang, M. Gu, Y. Wang, P. Wu, Z. Li, L. Kong and C. Han, ACS Appl. Mater. Interfaces, 2023, 15, 8946–8957 CrossRef CAS PubMed.
- J. Chen, M. Qiu, F. Ma, L. Yang, Z. Glass and Q. Xu, J. Controlled Release, 2021, 330, 1244–1249 CrossRef CAS PubMed.
- R. Vartak, A. Saraswat, Y. Yang, Z. S. Chen and K. Patel, Pharm. Res., 2022, 39, 2745–2759 CrossRef CAS PubMed.
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Vaccines, 2021, 9 DOI:10.3390/vaccines9040359.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9 DOI:10.3390/pharmaceutics9020012.
- M. N. Uddin and M. A. Roni, Vaccines, 2021, 9 DOI:10.3390/vaccines9091033.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- B. Cabanillas, C. A. Akdis and N. Novak, Allergy, 2021, 76, 1938–1940 CrossRef CAS PubMed.
- B. Cabanillas and N. Novak, Allergol. Int., 2021, 70, 313–318 CrossRef CAS PubMed.
- Y. Ju, J. M. Carreño, V. Simon, K. Dawson, F. Krammer and S. J. Kent, Nat. Rev. Immunol., 2023, 23, 135–136 CrossRef CAS PubMed.
- Y. Ju, W. S. Lee, E. H. Pilkington, H. G. Kelly, S. Li, K. J. Selva, K. M. Wragg, K. Subbarao, T. H. O. Nguyen, L. C. Rowntree, L. F. Allen, K. Bond, D. A. Williamson, N. P. Truong, M. Plebanski, K. Kedzierska, S. Mahanty, A. W. Chung, F. Caruso, A. K. Wheatley, J. A. Juno and S. J. Kent, ACS Nano, 2022, 16, 11769–11780 CrossRef CAS PubMed.
- H. T. Zhang, R. Peng, S. Chen, A. Shen, L. Zhao, W. Tang, X. H. Wang, Z. Y. Li, Z. G. Zha, M. Yi and L. Zhang, Adv. Sci., 2022, 9, e2202039 CrossRef PubMed.
- P. Dash, A. M. Piras and M. Dash, J. Controlled Release, 2020, 327, 546–570 CrossRef CAS PubMed.
- B. T. Luk and L. Zhang, J. Controlled Release, 2015, 220, 600–607 CrossRef CAS PubMed.
- J. Zhong, B. Xia, S. Shan, A. Zheng, S. Zhang, J. Chen and X. J. Liang, Biomaterials, 2021, 277, 121126 CrossRef CAS PubMed.
- R. Fan, S. He, Y. Wang, J. Qiao, H. Liu, L. Galstyan, A. Ghazaryan, H. Cai, S. Feng, P. Ni, G. Dong and H. Li, Am. J. Cancer Res., 2022, 12, 1027–1041 CAS.
- J.-H. Huang, C.-J. Huang, L.-N. Yu, X.-L. Guan, S.-W. Liang, J.-H. Li, L. Liang, M.-Y. Wei and L.-M. Zhang, Acta Pharmacol. Sin., 2023, 44, 1962–1976 CrossRef CAS PubMed.
- T. Wang, L. Sun, T. Ren, M. Hou, Y. Long, J.-H. Jiang and J. He, Nano Lett., 2023, 23, 9571–9578 CrossRef PubMed.
- T. Yang, Y. Hu, J. Miao, J. Chen, J. Liu, Y. Cheng and X. Gao, Acta Pharm. Sin. B, 2022, 12, 2658–2671 CrossRef CAS PubMed.
- J. Gao, B. Hou, Q. Zhu, L. Yang, X. Jiang, Z. Zou, X. Li, T. Xu, M. Zheng, Y. H. Chen, Z. Xu, H. Xu and H. Yu, Nat. Commun., 2022, 13, 4318 CrossRef CAS PubMed.
- O. Koshkina, T. Rheinberger, V. Flocke, A. Windfelder, P. Bouvain, N. M. Hamelmann, J. M. J. Paulusse, H. Gojzewski, U. Flögel and F. R. Wurm, Nat. Commun., 2023, 14, 4351 CrossRef CAS PubMed.
- A. Saraswat, M. Patki, Y. Fu, S. Barot, V. V. Dukhande and K. Patel, Nanomedicine, 2020, 15, 1761–1777 CrossRef CAS PubMed.
- J. Huang, Z. Yao, B. Li and Y. Ping, J. Controlled Release, 2023, 361, 270–279 CrossRef CAS PubMed.
- H. J. Liu, W. Chen, G. Wu, J. Zhou, C. Liu, Z. Tang, X. Huang, J. Gao, Y. Xiao, N. Kong, N. Joshi, Y. Cao, R. Abdi and W. Tao, Adv. Sci., 2023, 10, e2207439 CrossRef PubMed.
- Y. He, X. Zan, J. Miao, B. Wang, Y. Wu, Y. Shen, X. Chen, H. Gou, S. Zheng, N. Huang, Y. Cheng, Y. Ju, X. Fu, Z. Qian, P. Zhou, J. Liu and X. Gao, Mater. Today Bio, 2022, 16, 100423 CrossRef CAS PubMed.
- F. Wang, G. Dong, M. Ding, N. Yu, C. Sheng and J. Li, Small, 2023, e2306378 CrossRef PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 157, 168–182 CrossRef CAS PubMed.
- C. Lei, X. R. Liu, Q. B. Chen, Y. Li, J. L. Zhou, L. Y. Zhou and T. Zou, J. Controlled Release, 2021, 331, 416–433 CrossRef CAS PubMed.
- Y. Chen, W. Li, S. Kwon, Y. Wang, Z. Li and Q. Hu, J. Am. Chem. Soc., 2023, 145, 9815–9824 CrossRef CAS PubMed.
- Z. Hu, R. Li, X. Cui, C. Hu and Z. Chen, Chem. Eng. J., 2023, 469, 143883 CrossRef CAS.
- Q. Jiang, Y. Hu, Q. Liu, Y. Tang, X. Wu, J. Liu, G. Tu, G. Li, X. Lin, M. Qu, Y. Cai, X. Huang, J. Xu, Y. Deng, Z. Chen and L. Wu, J. Drug Targeting, 2023, 31, 411–420 CrossRef CAS PubMed.
- H. Cho, S. I. Jeon, M. K. Shim, C. H. Ahn and K. Kim, Biomaterials, 2023, 295, 122038 CrossRef CAS PubMed.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CrossRef CAS PubMed.
- X. Lan, J. She, D. A. Lin, Y. Xu, X. Li, W. F. Yang, V. W. Y. Lui, L. Jin, X. Xie and Y. X. Su, ACS Appl. Mater. Interfaces, 2018, 10, 33060–33069 CrossRef CAS PubMed.
- X. Lan, W. Zhu, X. Huang, Y. Yu, H. Xiao, L. Jin, J. J. Pu, X. Xie, J. She, V. W. Y. Lui, H. J. Chen and Y. X. Su, Nanoscale, 2020, 12, 18885–18898 RSC.
- X. Cheng, S. Hu and K. Cheng, ACS Nano, 2023, 17, 11855–11868 CrossRef CAS PubMed.
- R. Wang, T. Zhong, Q. Bian, S. Zhang, X. Ma, L. Li, Y. Xu, Y. Gu, A. Yuan, W. Hu, C. Qin and J. Gao, Small Methods, 2023, 7, e2201293 CrossRef PubMed.
- Y. Shi, J. Zhao, H. Li, M. Yu, W. Zhang, D. Qin, K. Qiu, X. Chen and M. Kong, Adv. Healthc. Mater., 2022, 11, e2200908 CrossRef PubMed.
- J. Chi, X. Zhang, C. Chen, C. Shao, Y. Zhao and Y. Wang, Bioact. Mater., 2020, 5, 253–259 Search PubMed.
- G. Du, P. He, J. Zhao, C. He, M. Jiang, Z. Zhang, Z. Zhang and X. Sun, J. Controlled Release, 2021, 336, 537–548 CrossRef CAS PubMed.
- N.-Y. Zhang, D.-Y. Hou, X.-J. Hu, J.-X. Liang, M.-D. Wang, Z.-Z. Song, L. Yi, Z.-J. Wang, H.-W. An, W. Xu and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202308049 CrossRef CAS PubMed.
- Z. Wang, M. Tan, W. Su, W. Huang, J. Zhang, F. Jia, G. Cao, X. Liu, H. Song, H. Ran, G. Nie and H. Wang, J. Med. Chem., 2023, 66, 6263–6273 CrossRef CAS PubMed.
- Y. Wang, L. Han, F. Liu, F. Yang, X. Jiang, H. Sun, F. Feng, J. Xue and W. Liu, Colloids Surf., B, 2020, 188, 110795 CrossRef CAS PubMed.
- W. Su, M. Tan, Z. Wang, J. Zhang, W. Huang, H. Song, X. Wang, H. Ran, Y. Gao, G. Nie and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202218128 CrossRef CAS PubMed.
- Z. Lin, B. A. Garcia and D. Lv, Angew. Chem., Int. Ed., 2023, e202316581 Search PubMed.
- J. Kim, S. Lee, Y. Kim, M. Choi, I. Lee, E. Kim, C. G. Yoon, K. Pu, H. Kang and J. S. Kim, Nat. Rev. Mater., 2023, 8, 710–725 CrossRef.
- L.-P. Zhao, X.-N. Rao, R.-R. Zheng, C.-Y. Huang, R.-J. Kong, H. Cheng, B. Li and S.-Y. Li, Nano Lett., 2023, 23, 6193–6201 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2014, 190, 15–28 CrossRef CAS PubMed.
- W. Poon, B. R. Kingston, B. Ouyang, W. Ngo and W. C. W. Chan, Nat. Nanotechnol., 2020, 15, 819–829 CrossRef CAS PubMed.
- S. Karaosmanoglu, M. Zhou, B. Shi, X. Zhang, G. R. Williams and X. Chen, J. Controlled Release, 2021, 329, 805–832 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |