DOI:
10.1039/D3MH01473H
(Communication)
Mater. Horiz., 2024,
11, 566-577
3D nitrogen-doped carbon frameworks with hierarchical pores and graphitic carbon channels for high-performance hybrid energy storages†
Received
13th September 2023
, Accepted 7th November 2023
First published on 7th November 2023
Abstract
In principle, hybrid energy storages can utilize the advantages of capacitor-type cathodes and battery-type anodes, but their cathode and anode materials still cannot realize a high energy density, fast rechargeable capability, and long-cycle stability. Herein, we report a strategy to synthesize cathode and anode materials as a solution to overcome this challenge. Firstly, 3D nitrogen-doped hierarchical porous graphitic carbon (NHPGC) frameworks were synthesized as cathode materials using Co–Zn mixed metal–organic frameworks (MOFs). A high capacity is achieved due to the abundant nitrogen and micropores produced by the MOF nanocages and evaporation of Zn. Also, fast ion/electron transport channels were derived through the Co-catalyzed hierarchical porosity control and graphitization. Moreover, tin oxide precursors were introduced in NHPGC to form the SnO2@NHPGC anode. Operando X-ray diffraction revealed that the rescaled subnanoparticles as anodic units facilitated the high capacity during ion insertion-induced rescaling. Besides, the Sn–N bonds endowed the anode with a cycling stability. Furthermore, the NHPGC cathode and SnO2@NHPGC achieved an ultrahigh energy density (up to 244.5 W h kg−1 for Li and 146.1 W h kg−1 for Na), fast rechargeable capability (up to 93C-rate for Li and 147C-rate for Na) as exhibited by photovoltaic recharge within a minute and a long-cycle stability with ∼100% coulombic efficiency over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
New concepts
The use of only a lithium-/sodium-ion battery and electrochemical capacitor cannot simultaneously realize a high energy density, fast rechargeable capability, and long-cycle stability. In this case, hybrid energy storages can theoretically be a solution to this challenge as the advantages of both batteries and capacitors can be exploited. Herein, we report a strategy to develop high-performance hybrid energy storages using 3D nitrogen-doped hierarchical porous graphitic carbon (NHPGC) frameworks derived from Co–Zn mixed MOFs as a cathode and a subnanometer tin oxide rescaled on NHPGC (SnO2@NHPGC) as an anode. These cathode and anode structures with a controlled hierarchical porosity for the rapid ion transport, graphitization for facile electron conduction, and rich redox-active sites for high capacity result in synergistic effects, delivering ultrahigh energy and fast-rechargeable power densities together with long-cycle stability. Consequently, this study provides a new route to develop high-capacity and high-rate cathode and anode structures for high-performance hybrid energy storage devices.
|
Introduction
Nowadays, electrochemical energy storage systems are essential in many applications ranging from portable electronic devices to electric vehicles (EVs) and large-scale grid systems.1,2 Meanwhile, the demand for high-performance energy storage materials that offer a high energy density for the prolonged operation in a single charge, fast rechargeable capability, and long cycle stability is rapidly growing. Presently, there are two types of electrochemical energy storage systems: batteries and electrochemical capacitors (ECs). Between them, lithium-ion batteries (LIBs) dominate the current energy storage market, while sodium-ion batteries (NIBs) are of great interest as alternatives to conventional LIBs, given that Na is ∼1000 times more abundant than Li.3–5 However, both LIBs and NIBs suffer from long diffusion lengths and phase transformations during repeated ion-insertion/desertion cycles, which lead to slow rechargeable capability and poor cycle stability. Similarly, electrochemical capacitors (ECs) suffer from surface-limited redox reactions, resulting in a low energy density. To overcome these drawbacks, hybrid energy storages (HESs), which refer to asymmetric full cells, is one of the great choices, since their operation voltage range can be extended using the different potential windows of capacitor-type cathode and battery-type anode electrodes.6,7 Therefore, a capacitor-type cathode capable of anion sorption reactions can be combined with a battery-type anode capable of cation insertion or conversion/alloying reactions to simultaneously attain a high energy density, fast rechargeable capability, and long-cycle stability.8–10 However, the low capacity of capacitor-type cathodes and the sluggish ion diffusion/poor electron conduction of battery-type anodes make it difficult to balance the charges and reaction kinetics between the cathode and anode over long cycles. Consequently, a high-capacity/high-rate cathode allowing rich anion adsorption/pseudocapacitive reaction sites as well as rapid anion transport and facile electron conduction channels and its counter anode possessing both rich active sites and fast cation transport and electron conduction channels are vital to achieve a high energy density and fast rechargeable capability over long cycles in hybrid energy storage devices.
In this case, capacitor-type cathode materials have been commonly fabricated using electrical double-layer capacitor (EDLC)-type carbon materials such as activated carbons and graphene,11–15 but EDLC-type carbon leads to a low capacitance, thereby limiting the capacitance in hybrid energy storage devices. Thus, a new capacitor-type cathode allowing higher capacitance than that of EDLC-type carbon electrodes is vital to realize high-performance hybrid energy storages. In addition, the incorporation of micropores and mesopores in a cathode material is required. The micropores can provide rich ion adsorption or reservoir sites for high capacity, while mesopores can offer diffusion channels for rapid ion movement.16–18 Alternatively, the electrical performances may be limited by a large amount of isolated and inaccessible micropores and high ion diffusion resistance due to the lack of mesoporous channels required for fast ion transport.19 In addition, graphitic carbons possess ordered sp2 carbons, contributing to high electrical conductivity.20 Hence, porous carbon having not only rich pore networks to facilitate ion transport, but also a high degree of graphitization to promote electron transfer should be designed to enable a high-rate performance in cathodes. In this case, metal–organic frameworks (MOFs) are promising materials to derive microporous ion adsorption sites as well as mesoporous ion transport channels.21 Also, the metal ions in their secondary building units (SBUs) and functionalized ligands can be utilized for catalytic graphitization and heteroatom doping.
In this work, we synthesized Co–Zn mixed MOF-derived cathode and anode materials to realize high performances in hybrid energy storage devices. Firstly, 3D nitrogen-doped hierarchical porous graphitic carbon (NHPGC) frameworks were synthesized as cathode materials from Co–Zn mixed metal–organic frameworks (MOFs). The porosity and graphitization in NHPGC were controlled by varying the proportion of Co and Zn atoms in the MOFs. The micropores were produced from the nanocages of the MOFs and vaporization of Zn. Co atoms were also utilized to offer catalytic sites for the formation of graphitic carbon layers and mesoporous channels were formed after the removal of the Co atoms. Additionally, a nitrogen-containing ligand (2-methylimidazole) was utilized to offer rich pseudocapacitive sites for high capacity and excellent electrolyte wettability. Besides, 3D anodic sites affording high capacity for lithium or sodium were conjugated in NHPGC to derive a high-capacity/high-rate anode. Subnanometer anodic sites were demonstrated to be generated via ion insertion-induced rescaling from larger crystals during cycling. We showed that the anodic subnanometer particles resulted in high capacity and fast kinetics. Also, they were shown to alleviate volume expansion/shrinkage without aggregation during ion insertion/extraction reactions. Besides, Sn–N bonds were demonstrated to allow long-term cycling stability. Furthermore, the cathode and anode electrodes were assembled into full-cell devices to attain high energy density, fast rechargeable capability, and long-cycle stability. Additionally, a charging kit using a solar cell was fabricated to demonstrate the fast charging capability of the full cell devices.
Results and discussion
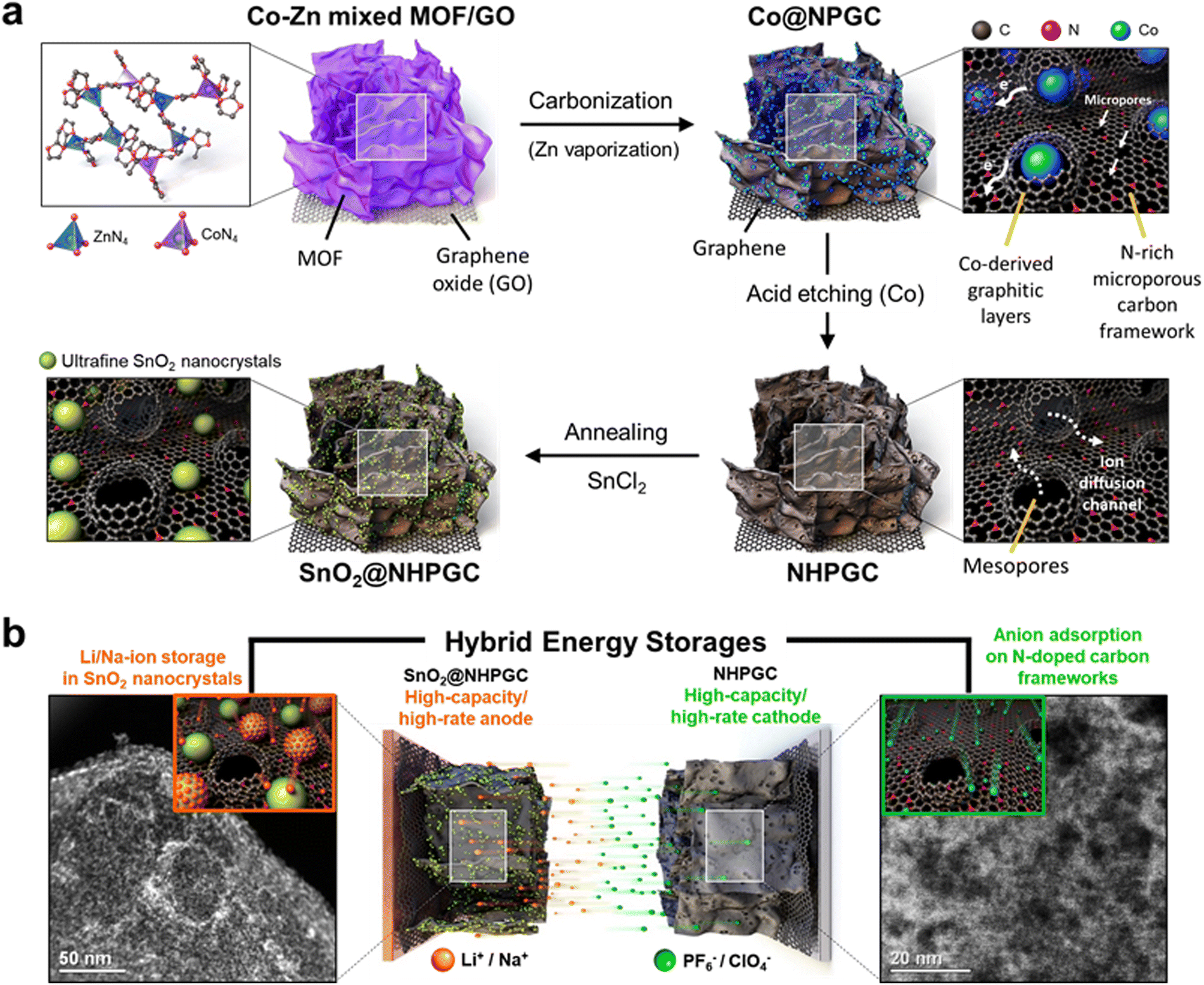
Fig. 1a illustrates the process for the synthesis of the NHPGC cathode and SnO2@NHPGC anode from Co–Zn mixed MOF/graphene oxide (GO). Firstly, we synthesized MOFs on GO, where Co and Zn ions were mixed in a ratio of 1:9 and coordinated by four nitrogen atoms (Co1–Zn9 MOF/GO). Then, a 3D Co-embedded nitrogen-doped porous graphitic carbon framework (Co@NPGC) was derived via carbonization in an Ar gas atmosphere at 900 °C. The graphitic layers were observed to be formed around Co particles. Subsequently, Co@NPGC was treated with nitric acid to etch the Co atoms. This led to the formation of NHPGC with mesoporous channels, which hereafter is denoted as Co1–Zn9 NHPGC. The thermogravimetric analysis (TGA) curves in Fig. S1 (ESI†) show the change in the mass of Co1–Zn9 MOF/GO during carbonization and the Co metal content in Co@NPGC. For the synthesis of SnO2@NHPGC, SnCl2 was added to NHPGC in water, and then annealed in an Ar/H2 atmosphere at 350 °C. This led to the formation of SnO2 crystals (∼3 nm) embedded in NHPGC. Fig. 1b illustrates the configuration of HES and the energy storage mechanism with NHPGC cathode storing anions on its pores/pseudocapacitive N sites and SnO2@NHPGC anode storing Li+ or Na+ cations by its rescaled subnanometer SnO2 sites.
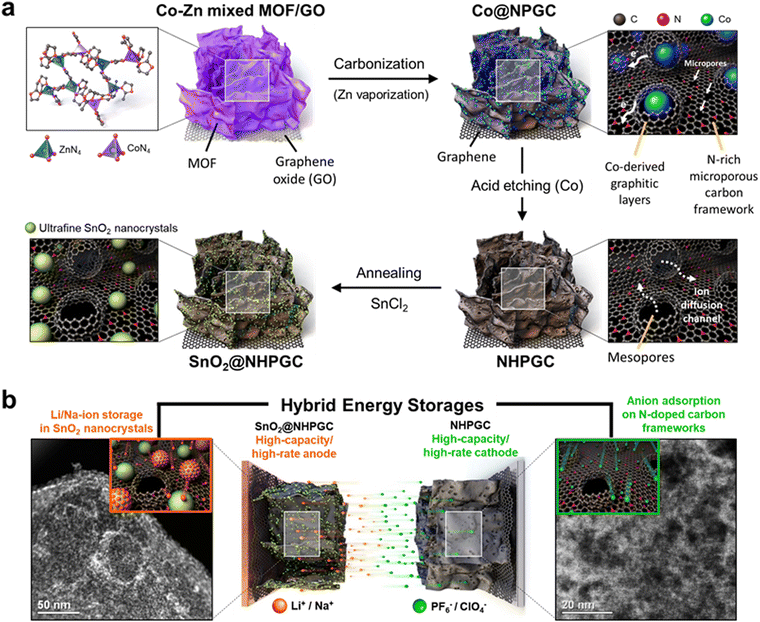 |
| | Fig. 1 A schematic illustration of synthesis processes and hybrid energy storage configuration. (a) Synthesis processes for NHPGC materials from Co–Zn mixed bimetallic MOFs on GO sheets and SnO2@NHPGC with ultrafine SnO2 nanocrystals integrated into NHPGC. (b) The hybrid energy storage configuration with the high-capacity/high-rate NHPGC cathode and SnO2@NHPGC anode. | |
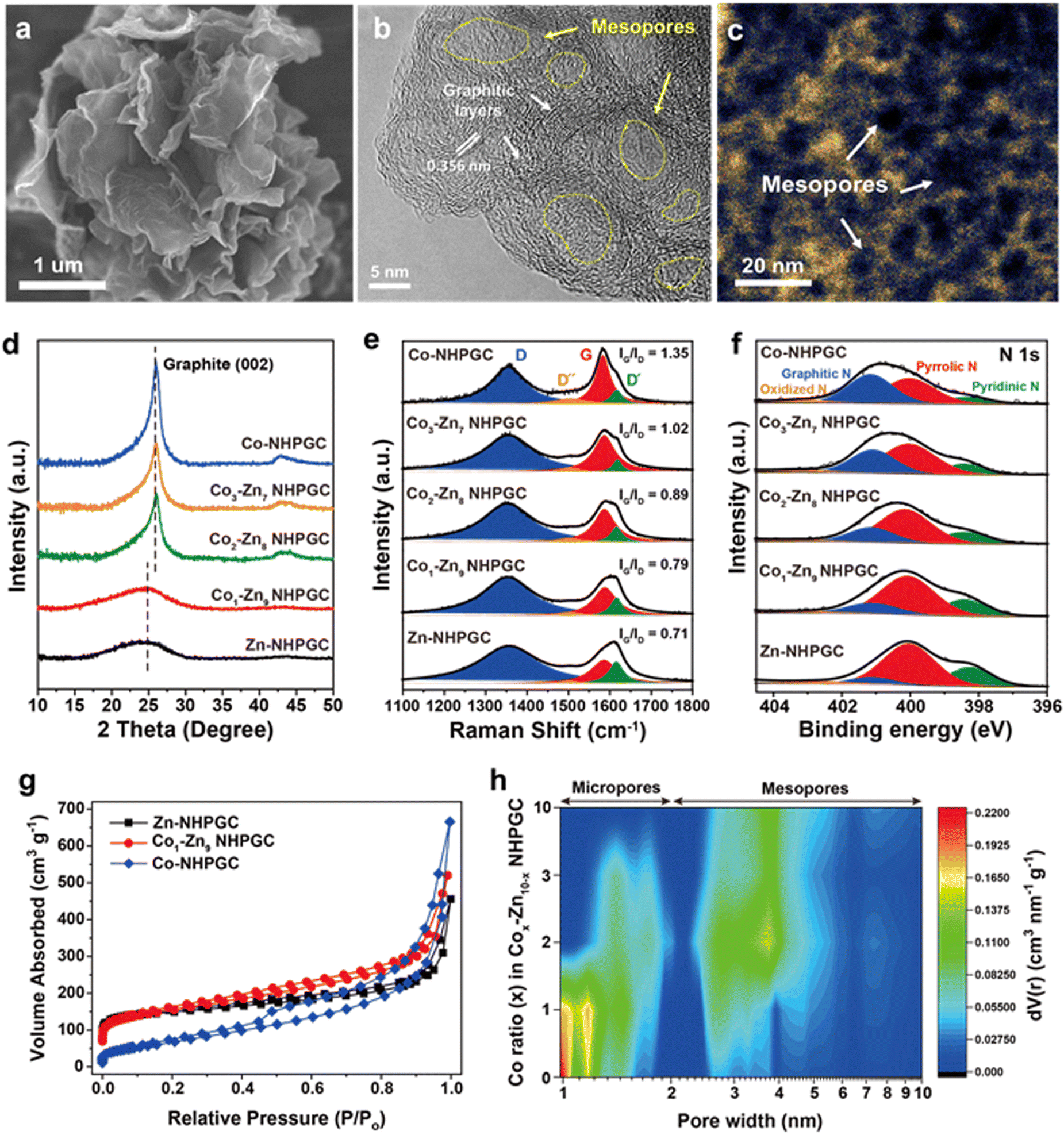
The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of Co1–Zn9 MOF/GO are shown in Fig. S2 (ESI†). Co–Zn mixed two-dimensional MOF sheets were grown on the GO substrate. After carbonization, it was observed that MOF/GO were pyrolyzed into carbon frameworks embedded with Co nanoparticles, and subsequently nitric acid treatment led to the total removal of the Co nanoparticles (Fig. S3, ESI†). The SEM image (Fig. 2a) indicates the well-preserved sheet shape of the MOF-derived carbon frameworks. In addition, the high-resolution TEM (HRTEM) image (Fig. 2b) reveals that 5 to 10 graphitic layers with a lattice spacing of 0.356 nm were formed on the porous carbon sheets via Co metal-catalyzed graphitization. The cavities surrounded by the graphitic layers serve as mesoporous channels. The mesoporous structures were further visualized through high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis (Fig. 2c), where the dark black regions in the temperature-mode image indicate pores with a size of less than 10 nm. Also, to elucidate the effect of the Co percentage on the structure of the carbon frameworks, we synthesized Cox–Zn10−x NHPGC with a Co ratio of x = 0, 2, 3, and 10 from Cox–Zn10−x MOF/GO. The Co and Zn ratios in the Cox–Zn10−x MOFs were revealed by inductively coupled plasma optical emission spectroscopy (ICP-OES), and the color of the powder changed from light brown to purple as the Co ratio increased from 0 to 10, as shown in Table S1 and Fig. S4 (ESI†). All the Cox–Zn10−x MOF/GO samples had a similar sheet-like shape, as shown in Fig. S5 (ESI†), but obvious morphological differences were observed in the carbon frameworks after carbonization (Fig. S6, ESI†). Notably, mesopores and graphitic layers were rarely detected in Zn-NHPGC (Fig. S6a and b, ESI†). Zn-NHPGC was mainly composed of micropores derived from the collapse of the MOF pores and vaporization of Zn during high-temperature annealing, while having an amorphous structure owing to the absence of Co metal. In contrast, a large number of mesopores and graphitic layers existed in Co2–Zn8, Co3–Zn7, and Co-NHPGCs (x = 2, 3, and 10), respectively. The pore size and number of graphitic layers were found to increase as the Co ratio increased, and thus even large pores of 50–100 nm were observed (Fig. S6c–e, ESI†), respectively. In addition, the structure of Cox–Zn10−x NHPGC was determined through powder X-ray diffraction (PXRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS) analyses. Fig. S7 (ESI†) shows the XRD patterns of Cox–Zn10−x MOF/GOs, and all the MOFs have the same diffraction patterns, corresponding to dia(Zn), a polymorph of ZIF-8.22 However, Fig. 2d shows that Co1–Zn9 and Zn-NHPGC give the weak and broad peaks at approximately 25°, whereas Co2–Zn8, Co3–Zn7, and Co-NHPGC showed a sharp peaks at 26°, which is attributed to the (002) reflection of graphite. This confirms that Co plays a role in the formation of the graphitic layers, as shown in the HRTEM image of Co-NHPGC (Fig. S8, ESI†).23 Besides, no diffraction peaks for Co metal was observed, indicating that the Co nanoparticles were completely removed via the nitric acid treatment, as also shown in the TEM and energy dispersive spectroscopy (EDS) mapping images in Fig. S9 (ESI†). The graphitization of NHPGC was analyzed by Raman spectroscopy (Fig. 2e). The peak at 1350 cm−1 is assigned to the D band, which his associated with structural defects. We observed that the peak at around 1600 cm−1 could be deconvoluted into two peaks at 1580 cm−1 (G band) and 1610 cm−1 (D′ band), corresponding to the in-plane stretching vibration mode of ideal and disordered graphitic lattices, respectively.24 The peak at 1500 cm−1 between the D and G band also corresponds to the D′′ band, which is associated with an amorphous phase.25 We determined the degree of graphitization using the intensity ratio between the G and D band (IG/ID). Co1–Zn9 NHPGC showed a higher IG/ID value (0.79) than Zn-NHPGC (0.71), indicating that Co1–Zn9 NHPGC has a higher degree of graphitization. Co-NHPGC with a highly graphitized structure had the highest value of 1.35, and IG/ID was found to increase as the amount of Co increased. This indicates that Co induced the graphitization of NHPGC. Moreover, the chemical state of Cox–Z10−x NHPGC was explored through X-ray photoelectron spectroscopy (XPS) analysis. Fig. 2f reveals the XPS N 1s spectra of NHPGCs. The N 1s peaks were deconvoluted into three components corresponding to pyridinic N (398.4 ± 0.2 eV), pyrrolic N (399.8 ± 0.2 eV), and graphitic N (400.8 ± 0.2 eV).26 Pyrrolic N and pyridinic N with unpaired electrons boost pseudocapacitive reactions for high capacity, while graphitic N leads to high electrical conductivity.27 Zn and Co1–Zn9 NHPGC predominantly consisted of pyrrolic N and pyridinic N, containing nitrogen concentrations of 7.49% and 5.03%, respectively (Fig. S10, ESI†). On the contrary, Co-NHPGC consisted mainly of graphitic N with a nitrogen concentration of 1.75%, supporting that a trade-off exists between the growth of graphitic layers and the concentration of nitrogen doping.28 The N2 adsorption–desorption isotherms (Fig. 2g and Fig. S11, ESI†) revealed the pore characteristics of the samples. Zn-NHPGC showed a type I isotherm, corresponding to a microporous structure, whereas NHPGCs derived from Co-containing MOFs exhibited isotherms gradually close to type IV as the Co ratio increased with a hysteresis loop, indicating a mesoporous structure. The pore size distribution and cumulative pore volume (Fig. 2h and Fig. S12, ESI†) indicate that Zn-NHPGC and Co1–Zn9 NHPGC possess abundant micropores and a high Co ratio induced the formation of mesopores in the carbon frameworks. In particular, Co1–Zn9 NHPGC possessed a micropore volume as large as that of Zn-NHPGC together with an increased volume of mesopores. The size of the mesopores ranged primarily from 3 to 5 nm, consistent with that observed in the HAADF-STEM image in Fig. 2c. Table S2 (ESI†) summarizes the pore structures of Cox–Zn10−x NHPGC, which were determined from the isotherms and pore size distribution. As the Co ratio increased, the Brunauer–Emmett–Teller (BET) specific surface area and volume ratio between the micropores and mesopores (Vmicro/Vmeso) were observed to decrease. In particular, Vmicro/Vmeso significantly decreased when x was higher than 1 in Cox–Zn10−x NHPGC. Co1–Zn9 NHPGC had the largest BET surface area of 554 m2 g−1 and hierarchical porosity with Vmicro/Vmeso of 33.5%. This verifies that the porosity can be optimized efficiently by adjusting the Co and Zn proportions in the synthesis of the MOFs, supporting that rich active sites with fast ion transport channels can be obtained without the addition of any pore activation agents.
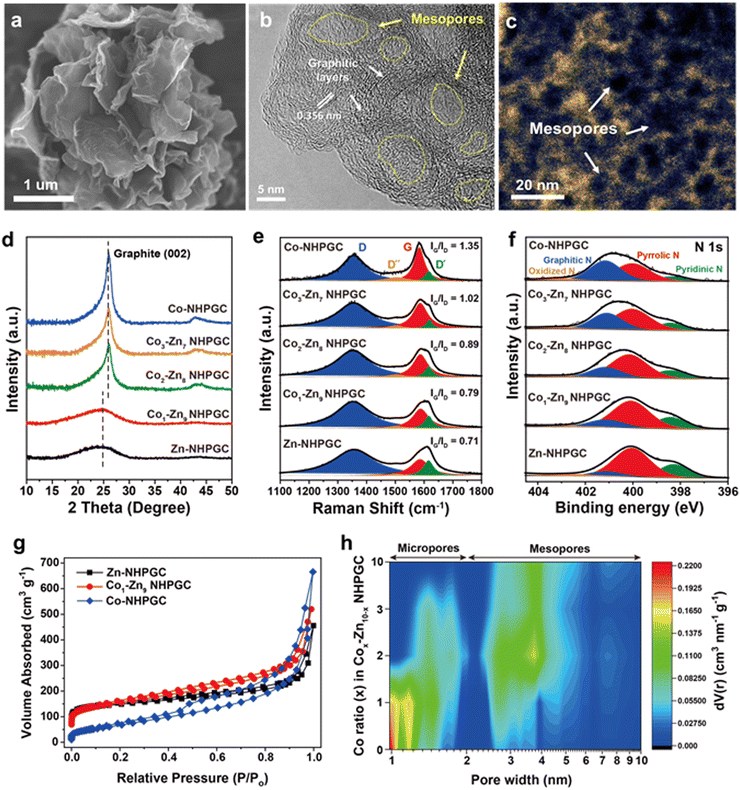 |
| | Fig. 2 The morphological and structural characterization of NHPGC cathode. (a) SEM and (b) HRTEM images of Co1–Zn9 NHPGC derived from Co1–Zn9 MOF/GO. (c) The HAADF-STEM image of Co1–Zn9 NHPGC in the temperature mode. (d) XRD, (e) Raman, and (f) XPS N 1s spectra of NHPGCs depending on the Co ratio, where the Raman spectra were normalized to the D band intensity for comparison. (g) The N2 adsorption isotherm and (h) contour plot of the pore size distribution for the NHPGCs synthesized with various Co ratios. | |
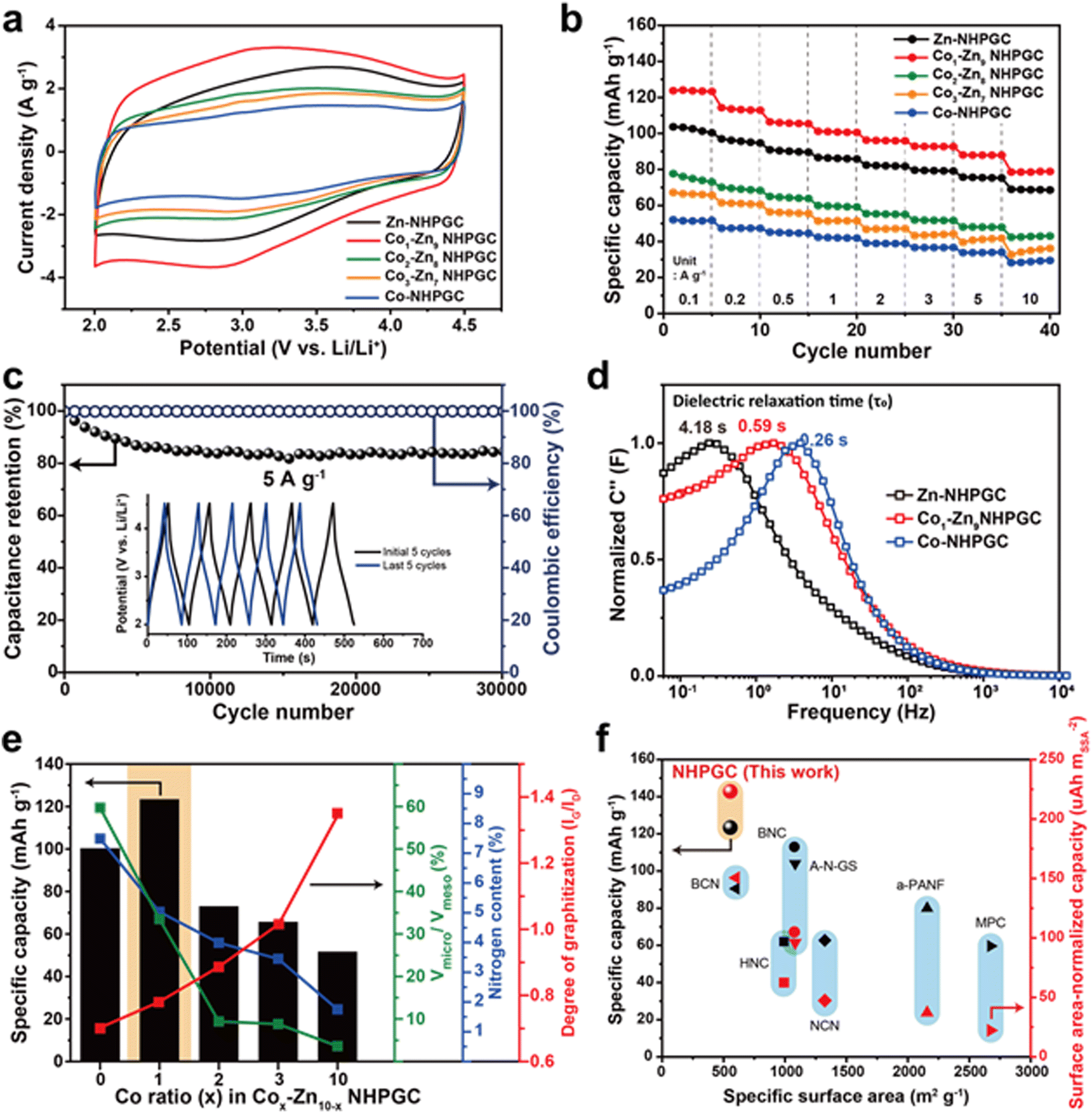
Fig. 3a reveals the cyclic voltammetry (CV) curves of the NHPGC cathode materials in the potential range of 2.0–4.5 V (vs. Li/Li+), in which their half cells were assembled using NHPGC as the working electrode and Li metal as the counter and a reference electrode in LiPF6 electrolyte. They present quasi-rectangular shapes, corresponding to electrical double-layer capacitance (EDLC)-type behaviors. Moreover, the humps at around 3.0 V indicate the pseudocapacitive reactions attributed to nitrogen species.29 The area under the CV curve of Co1–Zn9 NHPGC was the largest, whereas Co-NHPGC showed the smallest area owing to its insufficient volume of micropores serving as active sites. Fig. S13 (ESI†) demonstrates that all the Cox–Zn10−x NHPGC samples show analogous EDLC-type CV curves and the rectangular shapes are more distinct in Cox–Zn10−x NHPGC with higher Co proportions. The CV curve shape of Co1–Zn9 NHPGC was well preserved even at a high scan rate of 50 mV s−1, indicating its fast rate capability is attributed to enhanced electrical conductivity and ionic transport. Besides, the galvanostatic charge/discharge (GCD) curves for Cox–Zn10−x NHPGC (Fig. S14, ESI†) were found to have quasi-linear shapes, consistent with the CV curves. Co1–Zn9 NHPGC delivered the highest specific capacity of 123.4 mA h g−1 at 0.1 A g−1 and the high capacity retention of 78.5 mA h g−1 even at a high current density of 10 A g−1 (Fig. 3b), implying that the rich mesoporous ion transport channels boosted the ion adsorption on the micropores even at the 100-fold faster charging rate. Furthermore, Co1–Zn9 NHPGC exhibited superior cycle stability, as exhibited by high capacitance retention over 30000 cycles at 5 A g−1 together with voltage profiles in the initial and last 5 cycles (Fig. 3c). In addition, electrochemical impedance spectroscopy (EIS) measurements were carried out to elucidate the electrochemical behavior of Cox–Zn10−x NHPGCs with respect to the pore hierarchy, nitrogen-doping, and graphitization. Fig. S15 and Table S3 (ESI†) outline the Nyquist plots and summary of the resistances of Zn, Co1–Zn9 and Co-NHPGC, respectively. For a capacitor-type device, R1 (intercept on the X-axis) is the equivalent series resistance (ESR) and the diameter of the semicircle R2 corresponds to the charge transfer resistance at the electrode/electrolyte interface, and thus the sum of R1 and R2 can be interpreted as the internal resistance of the electrode.30 The R1 resistances for Co1–Zn9 and Co-NHPGCs were lower than that of Zn-NHPGC, indicating that the enhanced electrical conductivity is ascribed to the high degree of graphitization, while R2 is lower in Zn and Co1–Zn9 NHPGCs with high nitrogen contents. The diagonal line at the middle frequency between R2 and R3 is associated with the diffusion of ions. The shorter the length of this region, the faster the diffusion and adsorption process of ions in the porous structure.31 Given that the Co-induced mesoporous channels promote ionic transport in the electrolyte, Co-NHPGC had the highest diffusion rate, and Co1–Zn9 NHPGC also had higher diffusivity than Zn-NHPGC. This trend is also shown in the imaginary part of capacitance (C′′) versus frequency plot in Fig. 3d. The normalized capacitance is the maximum at a certain frequency (f0), and the corresponding time (1/f0) is called the dielectric relaxation time (τ0), which means how fast polarized electrolyte ions can be relaxed to equilibrium.17 The dielectric relaxation time for Zn-NHPGC was the largest (4.18 s), while Co1–Zn9 and Co-NHPGC showed shorter relaxation times of 0.59 and 0.26 s, respectively. It is noteworthy that including only 10 at% of cobalt in the Co–Zn mixed MOF significantly improved the ion diffusivity to a similar extent to that of the 100% Co-MOF-derived carbon. Fig. 3e summarizes the structural and electrochemical characteristics of the Cox–Zn10−x NHPGC cathodes. With an increase in the content of Co, the electrical conductivity improved by graphitization, and the ionic diffusion was enhanced due to the increased volume of mesoporous channels. However, the total nitrogen content including pyrrolic and pyridinic N as pseudocapacitive sites decreased. Also, the decrease in micropore volume led to the lack of adsorption sites. Consequently, Co1–Zn9 NHPGC exhibited the highest performance, which is attributed to the abundant micropores and nitrogen for copious ion adsorption reactions, rich mesopores for rapid ion transport, and high degree of graphitization for high electrical conductivity. The electrochemical performances of Co1–Zn9 NHPGC were observed to be superior to that of cathode materials reported to date (Fig. 3f and Table S4, ESI†). Although NHPGC showed the lowest specific surface area, it showed the highest specific capacity and surface area-normalized capacity, which is attributed to the maximized surface utilization efficiency enabled by the elaborate design of the carbon framework structure. To explore the effect of graphene on the cathode performance, we also synthesized Co1–Zn9 NHPGC without graphene by pyrolysis of Co1–Zn9 MOF without GO (Fig. S16, ESI†). It possessed a sheet shape and crater-like carbon layers, but its specific capacity was determined to be almost half that of Co1–Zn9 NHPGC. This demonstrates that the conductive graphene sheets promote the electrochemical performance in NHPGC.
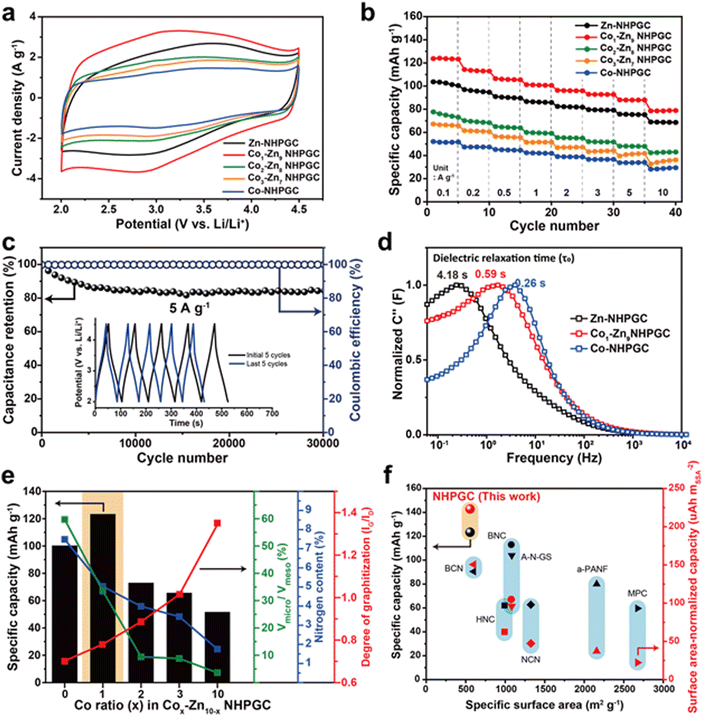 |
| | Fig. 3 Electrochemical performances of NHPGC cathode. (a) CV curves and (b) rate performances of NHPGCs. (c) The cycling stability and coulombic efficiency of Co1–Zn9 NHPGC at 5 A g−1. (d) The dielectric relaxation time of Zn, Co1–Zn9, and Co-NHPGC. (e) The specific capacity of NHPGCs in accordance with the volume ratio between micropores and mesopores, nitrogen content, and the degree of graphitization. (f) The specific capacity and surface-area normalized capacity of NHPGC compared with previously reported cathode materials. | |
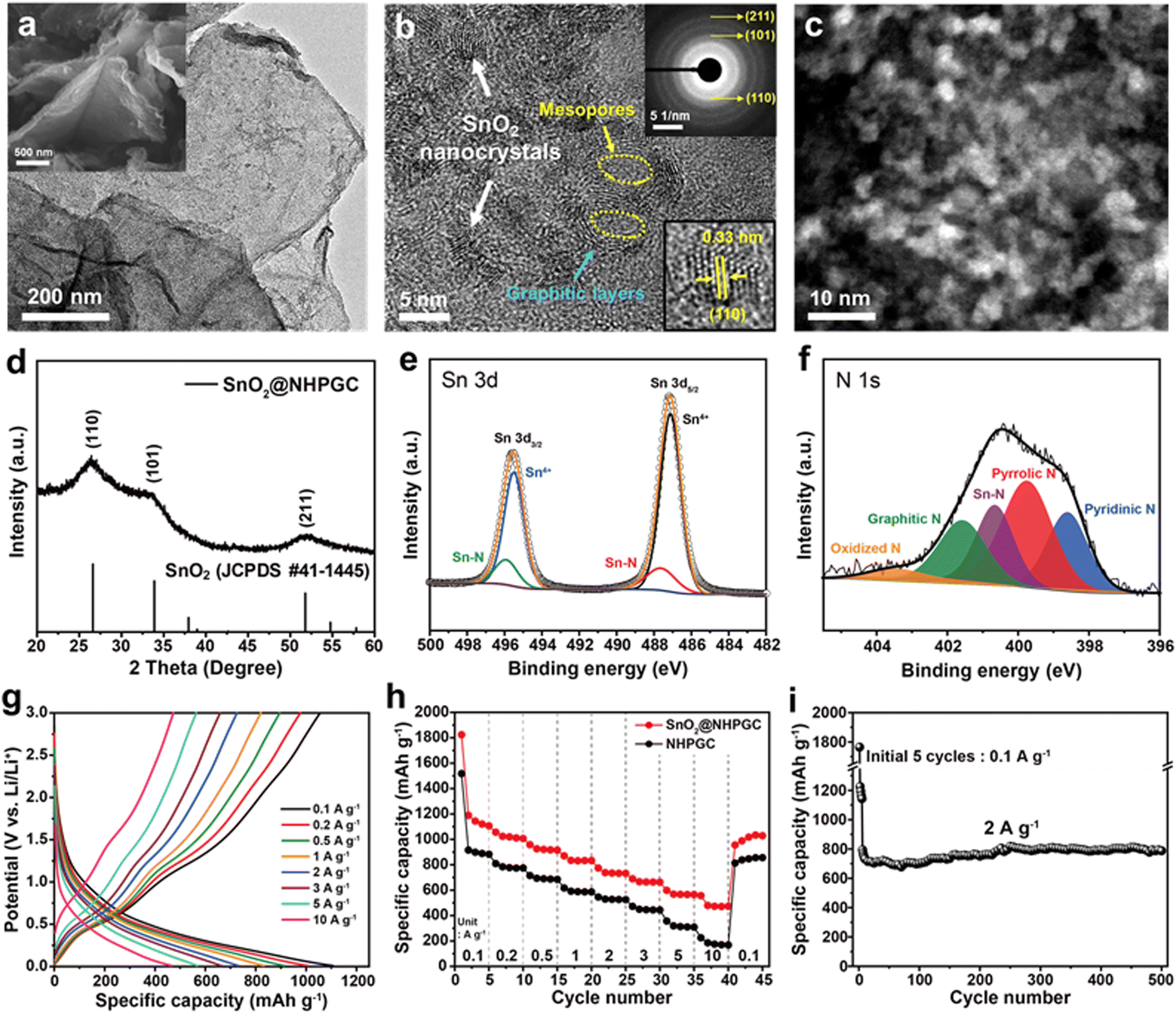
Subsequently, tin oxide nanocrystals were conjugated with Co1–Zn9 NHPGC to form a high-capacity/high-rate SnO2@NHPGC anode. NHPGC served as a matrix with an exquisite pore structure and rich nitrogen species. The SEM and TEM images of Fig. 4a and Fig. S17 (ESI†) reveal that tin oxide nanocrystals (3–5 nm) were uniformly grown without agglomeration on the nitrogen-doped carbon sheets. Also, Fig. 4b shows that graphite layers and mesopores coexist with nanocrystals. The selected area electron diffraction (SAED) patterns (upper right inset of Fig. 4b) reveal the (110), (101), and (211) reflections of SnO2, while the lower right inset shows the lattice spacing of 0.33 nm for the (110) plane. SnO2@NHPGC showed a broad XRD patterns due to the ultrafine size of the SnO2 nanocrystals (Fig. 4d). Besides, the deconvoluted Sn 3d spectra (Fig. 4e) show two sets of peaks. The peaks at 487.1 and 495.5 eV are associated with Sn4+ and the others at 487.7 and 495.9 eV correspond to Sn–N bonding.32 In the XPS N 1s spectra in Fig. 4f, the main peaks of pyridinic N, pyrrolic N, and graphitic N are observed together with the Sn–N bond at 400.7 eV,33 indicating the obvious interaction between Sn and NHPGC after the embedding of SnO2 nanocrystals. Also, TGA, as shown in Fig. S18 (ESI†), revealed the presence of 33.7 wt% SnO2 in SnO2@NHPGC. Fig. S19 (ESI†) shows the CV curves of the SnO2@NHPGC anode in the initial cycles between 0.01–3.0 V (vs. Li/Li+) at a scan rate of 0.2 mV s−1. The strong reduction peak at around 1 V in the first cycle corresponds to the conversion reaction of SnO2 to Sn, thereby resulting in the formation of solid–electrolyte interphase (SEI) layers.34 The peak at 0.1 V is attributed to the alloying reaction of Sn, forming LixSn, and Li intercalation in NHPGC.35 The oxidation peaks at 0.5 and 1.2 V are assigned to the dealloying of LixSn and the conversion reactions of Sn to SnO2, respectively. In the second and third cycles, the cathodic and anodic peaks in the CV curves almost overlapped, indicating the high reversibility of SnO2@NHPGC. Fig. 4g and h indicate the GCD curves of SnO2@NHPGC and its rate performance compared with Co1–Zn9 NHPGC at various current densities (Fig. S20, ESI†). The highest specific capacity of 1107 mA h g−1 was obtained at a current density of 0.1 A g−1. In addition, the high specific capacity of 472 mA h g−1 is achieved even at a 100-fold faster charging rate (10 A g−1). Although NHPGC also exhibited the high specific capacity of 883 mA h g−1 at 0.1 A g−1 due to its nitrogen species and defects serving as extra active sites, its specific capacity at fast charging rate of 10 A g−1 was only 174 mA h g−1. Fig. 4i presents the cycling performance of SnO2@NHPGC at 2 A g−1. After the initial 5 cycles at 0.1 A g−1, stable SEI layers were formed and SnO2@NHPGC showed excellent cycle stability, retaining a specific capacity of 787 mA h g−1 (99% retention in the 6th cycle) after 500 cycles.
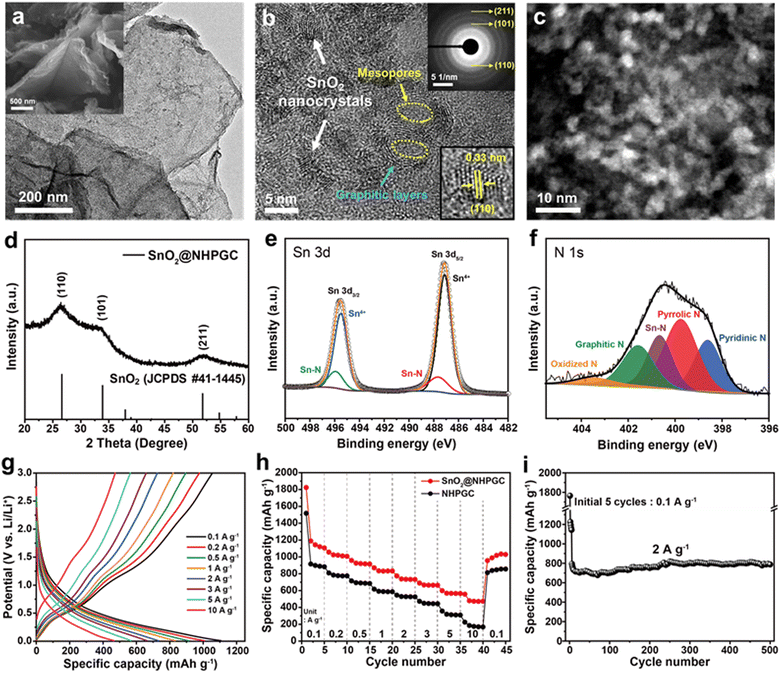 |
| | Fig. 4 The structural and electrochemical characterization of SnO2@NHPGC anode. (a) TEM and SEM (inset) images of SnO2@NHPGC. (b) HRTEM and (c) HAADF-STEM images of SnO2 nanocrystals grown on NHPGC. Inset images in (b) show the SAED patterns for the HRTEM image (upper right) and lattice spacing of SnO2 (lower right). (d) XRD patterns and (e) deconvoluted Sn 3d and (f) N 1s XPS spectra of SnO2@NHPGC. (g) GCD curves at different current densities, (h) rate performances compared to NHPGC, and (i) cycling performance of SnO2@NHPGC anode. | |
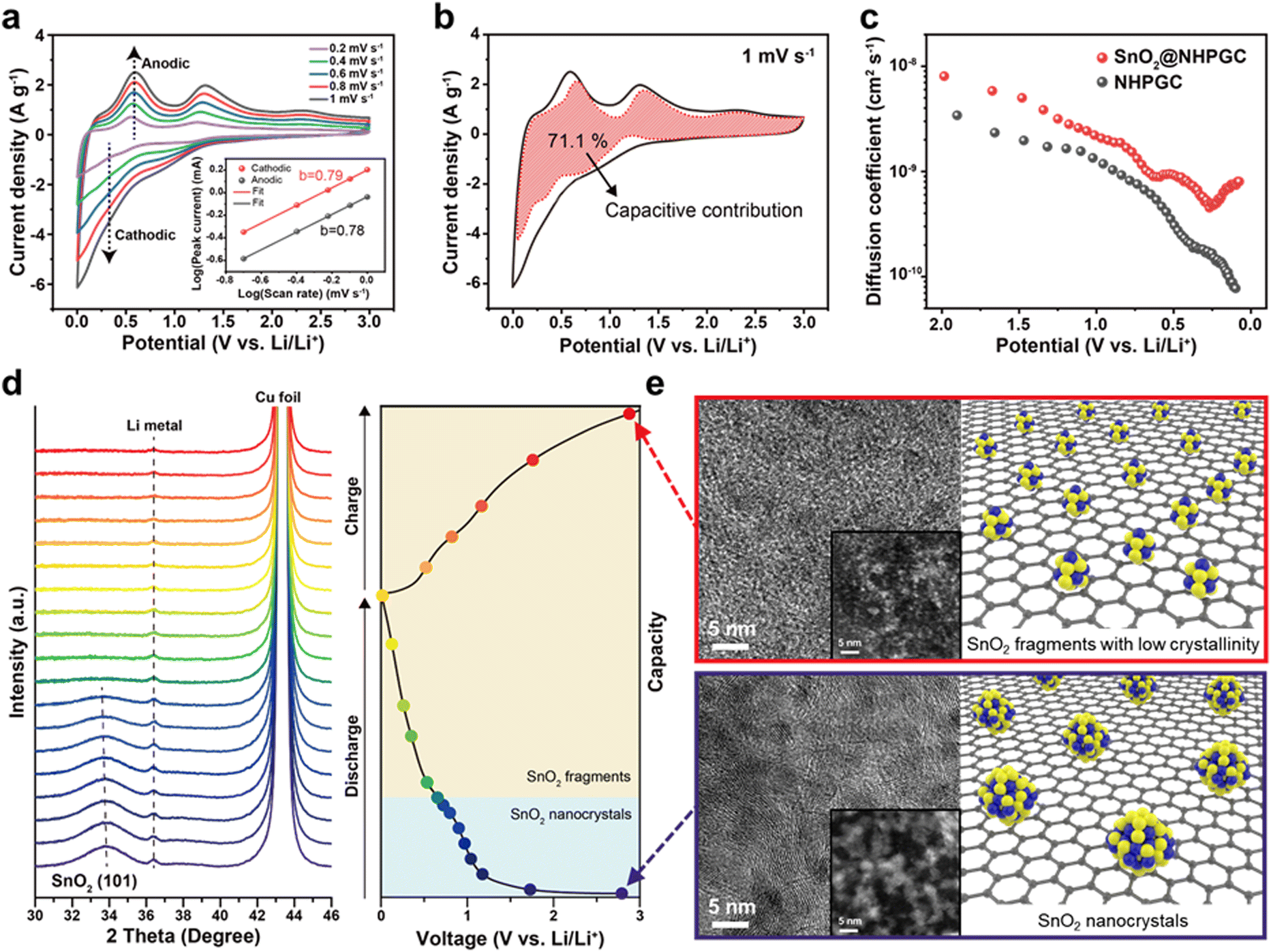
We also analyzed the reaction kinetics of the anode from the CV curves at various scan rates from 0.2 to 1 mV s−1 (Fig. 5a) using the power-law relationship, as follows:36
where
i is the peak current,
ν is the scan rate, and
a and
b are constants. The
b value can be calculated from the slope of the log(
i)
vs. log(
ν) plot, where a diffusion-controlled process is dominant for
b = 0.5, while the capacitive reaction is dominant for
b = 1. Meanwhile, the
b values of 0.79 for the cathodic peak and 0.78 for the anodic peak (inset of
Fig. 5a) show that both diffusion-controlled and surface-capacitive behaviors contribute to the lithium storage reactions. The capacitive contribution of SnO
2@NHPGC was also calculated using the current separation method
37 described by the following equation
where
i(
V) is the current at a given potential (
V),
ν is the scan rate, and
k1 and
k2 are constant values corresponding to the capacitive and diffusion-controlled reactions, respectively.
Fig. 5b shows that SnO
2@NHPGC gives a high capacitive contribution of 71.1% to the total capacity at 1 mV s
−1. Fig. S21 (ESI
†) further reveals that SnO
2@NHPGC led to about 20% higher capacitive contribution at the same scan rate than Co
1–Zn
9 NHPGC, thereby implying that the higher capacity and rate capability of SnO
2@NHPGC arise from the enhanced surface-capacitive reactions of the SnO
2 nanocrystals. To further analyze the reaction kinetics, the diffusion coefficient of Li ions (
DLi) was obtained by galvanostatic intermittent titration technique (GITT) measurements (
Fig. 5c and Fig. S22, ESI
†). The
DLi of SnO
2@NHPGC with a W-type curve by the phase transitions
38 is higher than that of NHPGC over the whole voltage range and determined to be 8.01 × 10
−10 cm
2 s
−1 in the full-discharge state (around 0.08 V), which is about 10-fold larger than that (7.74 × 10
−11 cm
2 s
−1) of NHPGC. Moreover, to determine the structural and morphological changes in the SnO
2 nanocrystals over the charge–discharge cycles,
operando XRD measurements were conducted at a current density of 50 mA g
−1 (
Fig. 5d). The SnO
2 (101) peak at 34° exhibited a shift toward lower angles and peak broadening, indicating the lattice expansion and fragmentation of the SnO
2 crystals during Li-ion insertion, respectively. The crystallinity of SnO
2 completely disappeared as the discharge voltage passed through the first plateau (∼0.7 V
vs. Li/Li
+), in which the conversion reaction occurred. Also, it showed no crystallinity even after being completely discharged to 0 V and charged to the initial voltage. The morphological transformations of the SnO
2 nanocrystals were also observed.
Fig. 5e displays the STEM-HAADF images of SnO
2@NHPGC before and after the discharge/charge cycle. The SnO
2 nanocrystals were rescaled into fragments of subnanometer sizes. The size-reduced particles were more favorable for the surface-capacitive reaction with fast kinetics, and the robust physical matrix enabled stable cycling. Fig. S23 (ESI
†) shows the galvanostatic cycling data at 100 mA g
−1 as well as HAADF-STEM image and XPS N 1s spectra of SnO
2@NHPGC collected after the cycling test. No aggregation of SnO
2 was observed and Sn–N bonding was preserved after long-term cycling. The fast kinetics of the fragmented SnO
2 and the presence of the Sn–N bond could effectively prevent coarsening, leading to long-term cycling stability. Subsequently, to verify the excellent performance and compatibility of SnO
2@NHPGC as a high-rate anode, its sodium-ion storage properties were also investigated (Fig. S24, ESI
†). A maximum specific capacity of 358 mA h g
−1 was achieved at 0.1 A g
−1 with a high specific capacity of 125 mA h g
−1 at 5 A g
−1. The surface-capacitive reaction contributed to 70.1%, and thus comparable to that for lithium storage. Considering that sodium ions have slower diffusion kinetics and lower reactivity due to their larger ionic radius than lithium ions,
39 it is notable that conjugating ultrafine SnO
2 in NHPGC enables fast and efficient surface-capacitive reactions. Fig. S24d (ESI
†) also shows the cycling performance of SnO
2@NHPGC at 2 A g
−1, and the specific capacity was maintained even after 2500 cycles.
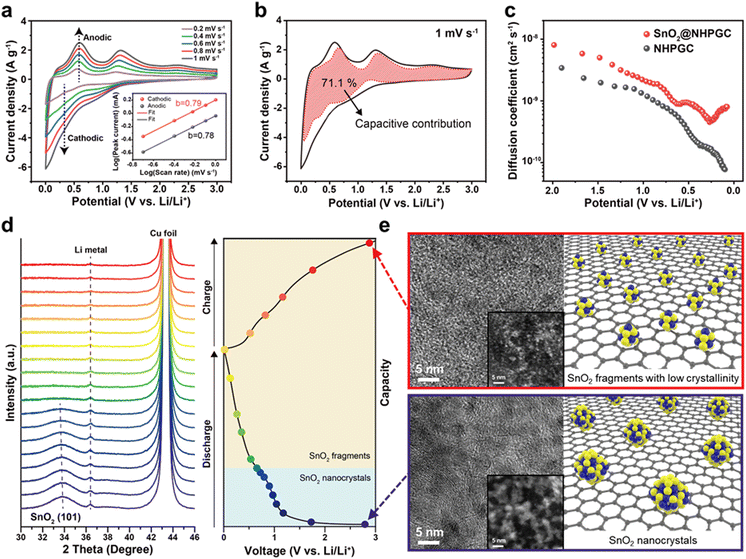 |
| | Fig. 5 The electrochemical energy storage mechanism of SnO2@NHPGC anode. (a) CV curves of SnO2@NHPGC at scan rates ranging from 0.2 to 1 mV s−1 and (b) the capacitive contribution at 1 mV s−1. The inset in (a) indicates log(i) vs. log(ν) plot at each anodic and cathodic peak. (c) The Li diffusion coefficient of SnO2@NHPGC and NHPGC. (d) Operando XRD data during the discharge/charge cycle at 50 mA g−1 and (e) corresponding TEM and STEM (inset) images with atomic structure of SnO2 nanocrystals (∼3 nm) and rescaled subnanoparticles (fragments less than 1 nm) before and after the discharge/charge cycle. | |
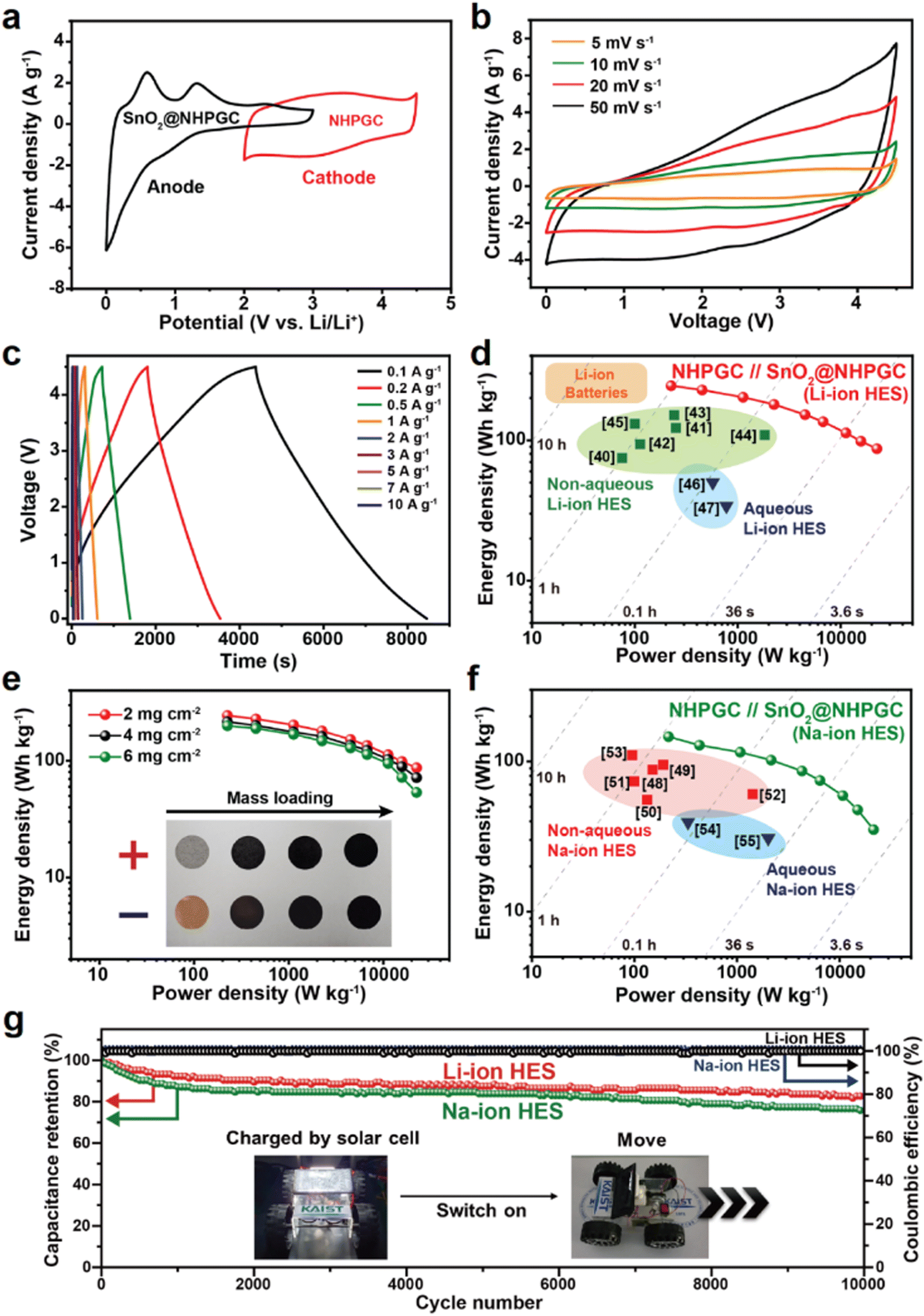
The NHPGC (Co1–Zn9 NHPGC) cathode and SnO2@NHPGC anode were also assembled into full cells for Li-ion HES and Na-ion HES. Fig. 6a shows the CV curves for the cathode and anode, where PF6− anions and Li+ cations are stored, respectively. To optimize the performance of the Li-ion HES, the energy density vs. power density of the HES was measured depending on the mass ratio of cathode to anode (Fig. S25, ESI†), and the optimal mass ratio of NHPGC to SnO2@NHPGC was determined to be 2.5:1. The Li-ion HES had a voltage window of 0–4.5 V together with a quasi-rectangular shape CV curve (Fig. 6b) and quasi-linear profiles in its GCD curves at various currents (Fig. 6c). As shown in the Ragone plot in Fig. 6d, the NHPGC//SnO2@NHPGC Li-ion HES achieved the maximum energy density of 244.5 W h kg−1 at a power density of 225 W kg−1 together with the high energy density of 87.1 W h kg−1 even at an extremely high power density of 22500 kW kg−1. This supports that the energy density of NHPGC//SnO2@NHPGC is superior to that of other non-aqueous Li-ion HES40–45 and aqueous Li-ion HES46,47 reported to date, as summarized in Table S5 (ESI†), and also comparable to that of a commercial LIB, while its power density outperforms that of a battery by about 100 fold. Besides, a high-mass loading test was performed by increasing the loading mass from 2 to 6 mg cm−2 (Fig. 6e). The maximum energy density was determined to be 215.87 and 201.06 W h kg−1 in 4 and 6 mg cm−2, respectively. It is notable that more than 80% of the energy density was maintained even at the three-fold higher mass loading. Also, NHPGC//SnO2@NHPGC Na-ion HES was fabricated with a 2:1 mass ratio for the cathode and anode. The CV and GCD curves in the range of 0–4.3 V show quasi-rectangular and quasi-linear shapes (Fig. S26, ESI†), respectively. The Ragone plot in Fig. 6f indicates that NHPGC//SnO2@NHPGC Na-ion HES achieved a high energy density of up to 146.1 W h kg−1 as well as high power density of up to 21500 W kg−1. The maximum energy density of NHPGC//SnO2@NHPGC Na-ion HES exceeds that of other non-aqueous Na-ion HES48–53 and aqueous Na-ion HES54,55 (Table S6, ESI†). Fig. 6g further shows that the Li-ion HES and Na-ion HES attained ∼100% coulombic efficiency over 10000 cycles. Additionally, the inset in Fig. 6g demonstrates that a mini electric car equipped with the series of NHPGC//SnO2@NHPGC Li-ion HES could run through ultrafast direct recharge from a photovoltaic module (Movie S1, ESI†).
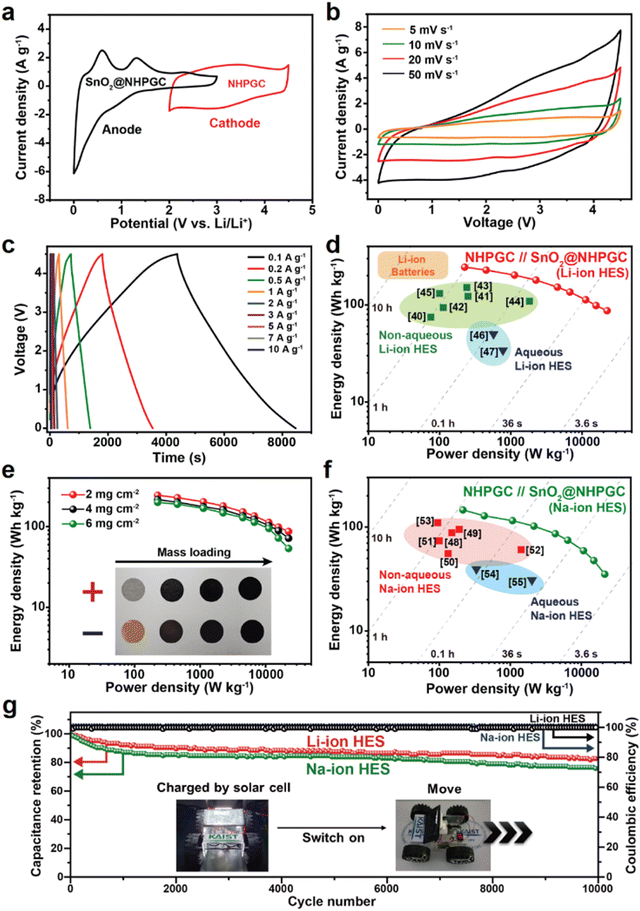 |
| | Fig. 6 Electrochemical performances of the NHPGC//SnO2@NHPGC hybrid energy storage (HES) full cell. (a) CV curves of NHPGC cathode and SnO2@NHPGC anode in the half-cell configuration. (b) CV curves at various scan rates and (c) GCD profiles of NHPGC//SnO2@NHPGC Li-ion HES at various current densities. (d) Ragone plots compared with other reported non-aqueous and aqueous Li-ion HES and commercial Li-ion batteries. (e) Ragone plot of Li-ion HES with different mass loadings. (f) The Ragone plot of Na-ion HES with previously reported non-aqueous and aqueous Na-ion HES. (g) The cycling stability and coulombic efficiencies for NHPGC//SnO2@NHPGC HES cells, where the inset image reveals a mini electric car with multiple full cells charged from a photovoltaic charging module. | |
Conclusions
In summary, we fabricated new cathode and anode materials using Co–Zn mixed bimetallic MOFs. Firstly, the Co to Zn ratio in the MOF was controlled to realize the optimal hierarchical porosity, nitrogen doping, and graphitization. This led to the synthesis of the NHPGC cathode with abundant mesopores as rapid ion transport channels, rich micropores/nitrogen atoms as ion adsorption/pseudocapacitive reaction sites for high capacity, and graphitic channels for facile electron conduction. Subsequently, embedding tin oxide precursors in NHPGC resulted in the formation of an anode material. Operando X-ray diffraction supported that the subnanoparticles rescaled during cycling and were available for high capacity. They were also proven to alleviate the volume expansion/shrinkage without aggregation and facilitate fast kinetics during the ion insertion/extraction reactions. In addition, the presence of Sn–N bonds was observed to result in cycling stability. Moreover, the NHPGC cathode and SnO2@NHPGC anode were assembled to fabricate HES devices. They were demonstrated to exploit the advantages of capacitive and battery-type reactions, as exhibited by their exceptionally high energy density (up to 244.5 W h kg−1 for Li and 146.1 W h kg−1 for Na) and ultrahigh power density (up to 22500 W kg−1 for ∼93 C-rate for Li and 21500 W kg−1 for ∼147 C-rate for Na), which are superior to that of a typical battery by about 100 fold. Consequently, a mini electric car equipped with multiple HES cells could run via fast charging from a photovoltaic module. Additionally, the HES exhibited ∼100% coulombic efficiency over 10000 cycles. Thus, this work supports that cathode and anode materials with rich active sites and high-rate transport networks are vital to develop high-performance energy storage systems for future technology.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was mainly supported by the National Research Foundation of Korea (2022M3H4A1A04096482, RS-2023-00229679) funded by the Ministry of Science and ICT.
Notes and references
- H. Sun, J. Zhu, D. Baumann, L. Peng, Y. Xu, I. Shakir, Y. Huang and X. Duan, Nat. Rev. Mater., 2019, 4, 45–60 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- T. Liu, Y. Zhang, Z. Jiang, X. Zeng, J. Ji, Z. Li, X. Gao, M. Sun, Z. Lin, M. Ling, J. Zheng and C. Liang, Energy Environ. Sci., 2019, 12, 1512–1533 RSC.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- L. Jin, C. Shen, A. Shellikeri, Q. Wu, J. Zheng, P. Andrei, J. G. Zhang and J. P. Zheng, Energy Environ. Sci., 2020, 13, 2341–2362 RSC.
- P. Han, G. Xu, X. Han, J. Zhao, X. Zhou and G. Cui, Adv. Energy Mater., 2018, 8, 1801243 CrossRef.
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng, R. Gong, J. P. Zheng and C. Zhang, Adv. Mater., 2018, 30, 1705670 CrossRef.
- J. Ding, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457–6498 CrossRef CAS.
- P. Jezowski, O. Crosnier, E. Deunf, P. Poizot, F. Béguin and T. Brousse, Nat. Mater., 2018, 17, 167–173 CrossRef CAS PubMed.
- Y. Zhang and S. J. Park, Carbon, 2017, 122, 287–297 CrossRef CAS.
- S. Ghosh, R. Santhosh, S. Jeniffer, V. Raghavan, G. Jacob, K. Nanaji, P. Kollu, S. K. Jeong and A. N. Grace, Sci. Rep., 2019, 9, 16315 CrossRef PubMed.
- F. Cheng, X. Yang, S. Zhang and W. Lu, J. Power Sources, 2020, 450, 227678 CrossRef CAS.
- S. Zhang, Y. Yu, M. Xie, C. Du, J. Chen, L. Wan and Y. Zhang, Appl. Surf. Sci., 2022, 589, 153011 CrossRef CAS.
- M. Athanasiou, S. N. Yannopoulos and T. Ioannides, Chem. Eng. J., 2022, 446, 137191 CrossRef CAS.
- F. Zhang, T. Liu, M. Li, M. Yu, Y. Luo, Y. Tong and Y. Li, Nano Lett., 2017, 17, 3097–3104 CrossRef CAS.
- L. Borchardt, D. Leistenschneider, J. Haase and M. Dvoyashkin, Adv. Energy Mater., 2018, 8, 1800892 CrossRef.
- R. Yan, M. Antonietti and M. Oschatz, Adv. Energy Mater., 2018, 8, 1800026 CrossRef.
- Q. Wang, J. Yan and Z. Fan, Energy Environ. Sci., 2016, 9, 729–762 RSC.
- S. Shi, X. Zhou, W. Chen, M. Chen, T. Nguyen, X. Wang and W. Zhang, RSC Adv., 2017, 7, 44632–44638 RSC.
- W. Yang, X. Li, Y. Li, R. Zhu and H. Pang, Adv. Mater., 2019, 31, 1804740 CrossRef.
- H. M. Titi, M. Arhangelskis, A. D. Katsenis, C. Mottillo, G. Ayoub, J.-L. Do, A. M. Fidelli, R. D. Rogers and T. Friščić, Chem. Mater., 2019, 31, 4882–4888 CrossRef CAS.
- S. Gadipelli, T. Zhao, S. A. Shevlin and Z. Guo, Energy Environ. Sci., 2016, 9, 1661–1667 RSC.
- M. Sharma, S. Rani, D. K. Pathak, R. Bhatia, R. Kumar and I. Sameera, Carbon, 2021, 184, 437–444 CrossRef CAS.
- S. Claramunt, A. Varea, D. López-Díaz, M. M. Velázquez, A. Cornet and A. Cirera, J. Phys. Chem. C, 2015, 119, 10123–10129 CrossRef CAS.
- L. F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS.
- J. Tang, R. R. Salunkhe, H. Zhang, V. Malgras, T. Ahamad, S. M. Alshehri, N. Kobayashi, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Sci. Rep., 2016, 6, 30295 CrossRef CAS PubMed.
- F. Sun, X. Liu, H. Bin Wu, L. Wang, J. Gao, H. Li and Y. Lu, Nano Lett., 2018, 18, 3368–3376 CrossRef CAS PubMed.
- B. A. Mei, O. Munteshari, J. Lau, B. Dunn and L. Pilon, J. Phys. Chem. C, 2018, 122, 194–206 CrossRef CAS.
- A. Eftekhari, ACS Sustainable Chem. Eng., 2019, 7, 3692–3701 CrossRef CAS.
- H. Guo Wang, Q. Wu, Y. Wang, X. Wang, L. Wu, S. Song and H. Zhang, Adv. Energy Mater., 2019, 9, 1802993 CrossRef.
- Z. Zhang, J. Huang, M. Zhang, Q. Yuan and B. Dong, Appl. Catal., B, 2015, 163, 298–305 CrossRef CAS.
- W. Ai, Z. Huang, L. Wu, Z. Du, C. Zou, Z. He, R. Shahbazian-Yassar, W. Huang and T. Yu, Energy Storage Mater., 2018, 14, 169–178 CrossRef.
- Y. Guo, X. Zeng, Y. Zhang, Z. Dai, H. Fan, Y. Huang, W. Zhang, H. Zhang, J. Lu, F. Huo and Q. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 17172–17177 CrossRef CAS.
- S. Wang, L. Li, W. He, Y. Shao, Y. Li, Y. Wu and X. Hao, Adv. Funct. Mater., 2020, 30, 2000350 CrossRef CAS.
- J. B. Cook, H. S. Kim, Y. Yan, J. S. Ko, S. Robbennolt, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2016, 6, 1501937 CrossRef.
- Y. Liu, C. Hu, L. Chen, Y. Hu, H. Jiang and C. Li, J. Energy Chem., 2022, 69, 450–455 CrossRef CAS.
- D. Dixon, M. Ávila, H. Ehrenberg and A. Bhaskar, ACS Omega, 2019, 4, 9731–9738 CrossRef CAS PubMed.
- C. Sun, X. Zhang, C. Li, K. Wang, X. Sun and Y. Ma, Energy Storage Mater., 2020, 24, 160–166 CrossRef.
- W. Yan, J. Su, Z. M. Yang, S. Lv, Z. Jin and J. L. Zuo, Small, 2021, 17, 2005209 CrossRef CAS.
- R. Bi, N. Xu, H. Ren, N. Yang, Y. Sun, A. Cao, R. Yu and D. Wang, Angew. Chem., 2020, 132, 4895–4898 CrossRef.
- Y. An, T. Liu, C. Li, X. Zhang, T. Hu, X. Sun, K. Wang, C. Wang and Y. Ma, J. Mater. Chem. A, 2021, 9, 15654–15664 RSC.
- G. Li, Z. Yin, H. Guo, Z. Wang, G. Yan, Z. Yang, Y. Liu, X. Ji and J. Wang, Adv. Energy Mater., 2019, 9, 1802878 CrossRef.
- S. Tao, R. Momen, Z. Luo, Y. Zhu, X. Xiao, Z. Cao, D. Xiong, W. Deng, Y. Liu, H. Hou, G. Zou and X. Ji, Small, 2023, 19, 2207975 CrossRef CAS.
- C. Li, W. Wu, S. Zhang, L. He, Y. Zhu, J. Wang, L. Fu, Y. Chen, Y. Wu and W. Huang, J. Mater. Chem. A, 2019, 7, 4110–4118 RSC.
- X. Li, H. Wu, A. M. Elshahawy, L. Wang, S. J. Pennycook, C. Guan and J. Wang, Adv. Funct. Mater., 2018, 28, 1800036 CrossRef.
- S. Li, J. Chen, X. Gong, J. Wang and P. S. Lee, Small, 2018, 14, 1804035 CrossRef.
- R. Yan, E. Josef, H. Huang, K. Leus, M. Niederberger, J. P. Hofmann, R. Walczak, M. Antonietti and M. Oschatz, Adv. Funct. Mater., 2019, 29, 1902858 CrossRef.
- M. L. Divya, S. Jayaraman, Y. S. Lee and V. Aravindan, Chem. Eng. J., 2021, 426, 130892 CrossRef CAS.
- X. Xiao, X. Duan, Z. Song, X. Deng, W. Deng, H. Hou, R. Zheng, G. Zou and X. Ji, Adv. Funct. Mater., 2022, 32, 2110476 CrossRef CAS.
- X. Liu, G. A. Elia, B. Qin, H. Zhang, P. Ruschhaupt, S. Fang, A. Varzi and S. Passerini, ACS Energy Lett., 2019, 4, 2675–2682 CrossRef CAS.
- Y. Song, X. Sun, L. Li, C. Zhang and F. Yin, Carbon, 2023, 204, 219–230 CrossRef CAS.
- K. Krishnamoorthy, P. Pazhamalai, S. Sahoo, J. H. Lim, K. H. Choi and S. J. Kim, ChemElectroChem, 2017, 4, 3302–3308 CrossRef CAS.
- E. B. T. H. Tanaya Das, T. Maiyalagan and N. Das, J. Alloys Compd., 2023, 931, 167501 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a