
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-site esterification: a tunable, reversible strategy to tailor therapeutic peptides for delivery†
Mark S.
Bannon
 a,
Jeffrey F.
Ellena
b,
Aditi S.
Gourishankar
a,
Spencer R.
Marsh
c,
Dilza
Trevisan-Silva
d,
Nicholas E.
Sherman
d,
L. Jane
Jourdan
c,
Robert G.
Gourdie
c and
Rachel A.
Letteri
*a
a,
Jeffrey F.
Ellena
b,
Aditi S.
Gourishankar
a,
Spencer R.
Marsh
c,
Dilza
Trevisan-Silva
d,
Nicholas E.
Sherman
d,
L. Jane
Jourdan
c,
Robert G.
Gourdie
c and
Rachel A.
Letteri
*a
aDepartment of Chemical Engineering, University of Virginia, Charlottesville, VA 22903, USA. E-mail: rl2qm@virginia.edu; Tel: +1 434 243 3628
bBiomolecular Magnetic Resonance Facility, School of Medicine, University of Virginia, Charlottesville, VA 22903, USA
cFralin Biomedical Institute, Virginia Tech Carillion School of Medicine, Roanoke, VA 24016, USA
dBiomolecular Analysis Facility, School of Medicine, University of Virginia, Charlottesville, VA 22903, USA
First published on 13th September 2024
Abstract
Peptides are naturally potent and selective therapeutics with massive potential; however, low cell membrane permeability limits their clinical implementation, particularly for hydrophilic, anionic peptides with intracellular targets. To overcome this limitation, esterification of anionic carboxylic acids on therapeutic peptides can simultaneously increase hydrophobicity and net charge to facilitate cell internalization, whereafter installed esters can be cleaved hydrolytically to restore activity. To date, however, most esterified therapeutics contain either a single esterification site or multiple esters randomly incorporated on multiple sites. This investigation provides molecular engineering insight into how the number and position of esters installed onto the therapeutic peptide α carboxyl terminus 11 (αCT11, RPRPDDLEI) with 4 esterification sites affect hydrophobicity and the hydrolysis process that reverts the peptide to its original form. After installing methyl esters onto αCT11 using Fischer esterification, we isolated 5 distinct products and used 2D nuclear magnetic resonance spectroscopy, reverse-phase high performance liquid chromatography, and mass spectrometry to determine which residues were esterified in each and the resulting increase in hydrophobicity. We found esterifying the C-terminal isoleucine to impart the largest increase in hydrophobicity. Monitoring ester hydrolysis showed the C-terminal isoleucine ester to be the most hydrolytically stable, followed by the glutamic acid, whereas esters on aspartic acids hydrolyze rapidly. LC-MS revealed the formation of transient intramolecular aspartimides prior to hydrolysis to carboxylic acids. In vitro proof-of-concept experiments showed esterifying αCT11 to increase cell migration into a scratch, highlighting the potential of multi-site esterification as a tunable, reversible strategy to enable the delivery of therapeutic peptides.
Design, System, ApplicationIn this manuscript, we report the molecular design of peptides with hydrolyzable ester groups to reversibly increase their hydrophobicity and net charge towards improving cell membrane permeability. As many peptides have multiple carboxylic acid esterification sites, there exist many combinations of numbers and positions of esters that can be installed within a single peptide. Thus, in this study, we provide insight into how many esters should be installed and where they should be installed to achieve the most optimal balance of enhancements in hydrophobicity and net charge conducive to increasing cell membrane permeability and the ability to eventually restore the original form of the therapeutic peptide. Proof-of-concept experiments show that delivering esterified peptides to scratched cell cultures improve cell migration into the scratch more than the unesterified analogs, indicating that esterifying the peptide enabled expected wound healing benefits. |
Introduction
Peptides are potent, versatile, and selective therapeutics that are more easily manufactured than larger biologics. Insulin, one of the most well-known and earliest therapeutic peptides, set the stage for the discovery and approval of over 80 others, including glucagon-like peptide-1 (GLP-1) receptor agonists to treat diabetes (e.g., dulaglutide and semaglutide), as well as the neuropeptide hormones oxytocin and vasopressin.1–3 Despite their benefits, therapeutic peptides only account for ∼5% of the $1.2 trillion global pharmaceutical market as of 2019.2 Clinical implementation of peptides is limited by a few key issues, one of which is low cell membrane permeability.4 Underscoring this point, less than 10% of the therapeutic peptides in active clinical development in 2018 had intracellular targets;5 yet, peptides with intracellular targets hold immense therapeutic promise. For example, α carboxyl terminus 11 (αCT11), a wound healing peptide with applications in dermal wound healing and cardioprotection, relies on interactions with the intracellular H2 domain of the transmembrane protein connexin 43 (Cx43) and/or the cytoplasmic PDZ-2 domain of the scaffolding protein ZO-1.6–8 To realize the full potential of peptides, such as αCT11, as viable therapeutics, it is imperative to increase their cell membrane permeability.Eukaryotic cell membrane permeability is determined by interactions between the drugs and the membrane itself, which is composed of a hydrophobic hydrocarbon layer flanked by intracellular and extracellular polar phospholipid headgroups.9 While cell membranes remain impermeable to many therapeutic peptides, cell-penetrating peptides (CPPs) are well documented to increase cell internalization of attached therapeutics.10,11 For example, appending αCT11 to the CPP antennapedia (RQPKIWFPNRRKPWKK),12 at the N-terminus (termed αCT1) enabled improvement in scar healing in a recent phase II clinical trial.8,13 CPPs often contain an excess of positive charge, usually from protonated lysine and/or arginine residues,14,15 conducive to electrostatic interactions with anionic intracellular phospholipid headgroups, which often have a high concentration of negatively charged phosphatidylserine.16–18 Further, as drug partitioning into the hydrophobic hydrocarbon region is also necessary for reaching targets both within the cell membrane and in the cytoplasm,19–21 CPP sequences often include hydrophobic amino acids (e.g., tryptophan and phenylalanine).22 Thus, for peptides with a high concentration of hydrophilic and/or anionic groups, such as αCT11 (56% hydrophilic residues, 3 cationic groups, 4 anionic groups), it will be essential to increase the hydrophobic character and/or to decrease the anionic character to enable access to targets within the cytoplasm and the membrane.
To date, methods to modify the hydrophobicity and/or net charge of peptide and protein therapeutics include stapling/cyclization, amide backbone methylation, supercharging, or ‘tagging’ a sequence with charged/hydrophobic amino acids (such as CPPs).23–32 Yet, such permanent modifications to molecular structure can alter activity, as observed with supercharged green fluorescent protein (GFP), which exhibited lower fluorescence relative to its unmodified form.25 Similarly, αCT11 alone restored 20% more left ventricular function in ex vivo ischemic cardiomyocytes than αCT1, suggesting that attaching the 16 amino acid CPP sequence decreased αCT11 wound healing activity.7 Further illustrating this point, the cardioprotective activity of αCT11 was found to be directly tied to its structure, where replacing the D5, D6, and E8 amino acids with alanine led to decreased interactions with Cx43 H2 and/or ZO-1 PDZ-2, and subsequent reduced activity in ex vivo ischemic mouse hearts.7 While it is likely that ischemic cardiomyocytes directly internalize αCT11 through open Cx43 hemichannels,7,33,34 antennapedia was required to enable skin wound healing.8 Therefore, while molecular modifications to peptide sequence are necessary to increase cell membrane permeability and access intracellular targets, we require reversible strategies to allow delivery of the native sequence to the intended target and maximize therapeutic activity.
To temporarily modify molecular structure while retaining activity, we can employ esterification (Scheme 1), a simple and widely used scheme where anionic, hydrophilic carboxylic acids (COOHs) are converted to esters (R–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OR) to simultaneously increase the hydrophobicity and reduce the net negative charge of therapeutics.35–38 After installation, esters can be hydrolyzed to restore the active, unesterified form of the therapeutic, a reaction that can be accelerated in the presence of endogenous esterases.39–42 Esterification has been used to increase the phospholipid membrane permeability of small molecules,43–45 proteins,46 and even peptides.47 While many of these small molecules and the therapeutic peptide thyrotropin-releasing hormone47 have just one COOH site, peptides can have multiple esterification sites.
O)OR) to simultaneously increase the hydrophobicity and reduce the net negative charge of therapeutics.35–38 After installation, esters can be hydrolyzed to restore the active, unesterified form of the therapeutic, a reaction that can be accelerated in the presence of endogenous esterases.39–42 Esterification has been used to increase the phospholipid membrane permeability of small molecules,43–45 proteins,46 and even peptides.47 While many of these small molecules and the therapeutic peptide thyrotropin-releasing hormone47 have just one COOH site, peptides can have multiple esterification sites.
 | ||
| Scheme 1 Fischer esterification of αCT11 (RPRPRDDLEI). | ||
αCT11 (RPRPDDLEI) has 4 possible esterification sites at the D5 (denoting the aspartic acid in the 5th position within the sequence), D6, E8, and C-terminal I9 residues, making esterification a suitable tool to reversibly increase cell membrane permeation. However, understanding how the number and position of installed esters affects hydrophobicity and hydrolysis/reversion into the therapeutically active form is critical to leveraging esterification as a reversible strategy to realize the therapeutic potential of peptides. Therefore, in this work, we install methyl esters onto αCT11 and assess how the number and position of installed esters affects the hydrophobicity and hydrolysis of esterified peptide formulations. Then, we test the viability of multi-site esterification with in vitro experiments designed to gauge peptide wound healing activity by cell migration into a scratch.
Experimental section
Materials
Fluorenylmethoxycarbonyl (Fmoc)-protected amino acids and 2-chlorotrityl chloride resin (0.6 mmol g−1) were purchased from Advanced ChemTech (Louisville, Kentucky). Diisopropyl carbodiimide (DIC, 99.8%), N,N′-dimethylformamide (DMF, 99%), Oxyma Pure (99%), piperidine (99%), N,N-diisopropylethylamine (DIPEA, 99%), trifluoroacetic acid (TFA, 99%), triisopropyl silane (TIPS, 98%), 2,2′-(ethylenedioxy)diethanethiol (DODT, 95%), diethyl ether (99%), methanol (MeOH, 99.8%), acetonitrile (ACN, HPLC-grade, 99.9%), hydrochloric acid (HCl, 37% in H2O), deuterated dimethyl sulfoxide (DMSO-d6, 99.5%), sodium bicarbonate (99.7%), sodium carbonate (99.5%), phosphate buffered saline tablets (PBS, 1 tablet per 200 mL for 1× concentration, pH 7.2–7.6), SpectraPor6 dialysis membranes (0.5–1 kDa), esterase from porcine liver (PLE, E3019, 19 units per mg solid), and Amicon ultra centrifugal filters (3 kDa MWCO) were purchased from Sigma Aldrich. Glacial acetic acid (99.7–100.5%) and potassium iodide (KI) were purchased from VWR. All chemicals were used as received. All water was purified by in-house reverse osmosis (RO). Ultrapure water refers to water purified by a thermo Scientific Barnstead Smart2Pure water purification system (18.2 mΩ cm).αCT11 synthesis
αCT11 was prepared using Fmoc-solid phase peptide synthesis with a CEM Liberty Blue automated microwave-assisted peptide synthesizer. To prepare the peptide with a COOH C-terminus, 2-chlorotrityl chloride resin (0.6 mmol g−1) was used, and the entire synthesis was conducted at 25 °C to prevent premature cleavage of the ester bond connecting the peptide to the resin, which was observed above 50 °C. DIC (1 M in DMF) and Oxyma Pure (1 M in DMF) were used to mediate amino acid coupling, except for the first amino acid, which was coupled to the resin using KI (0.125 M in DMF) and DIPEA (1 M in DMF). Piperidine (20% v/v in DMF) was used to deprotect Fmoc groups preceding amino acid additions. After synthesis, the peptide was cleaved from the resin using a deprotection cocktail composed of TFA, RO H2O, TIPS, and DODT (92.5/2.5/2.5/2.5 v/v) for 3 h at room temperature under constant stirring. Following deprotection, the peptide solution was separated from the resin by filtration, and the peptide was isolated by precipitation into diethyl ether and centrifugation (5 min, 2420 × g, 4 °C). The supernatant was decanted and the peptide was washed again with diethyl ether and isolated by centrifugation under the same conditions. The peptide pellet was dried under vacuum for 1 h, dissolved in 5% (v/v) ACN in H2O (both with 0.1% v/v TFA) and frozen in liquid N2 immediately prior to lyophilization for 48 h to produce a fluffy cake that was easy to manipulate. 60–70% yield was achieved for each synthesis.Fischer esterification of αCT11 with MeOH
For each esterification, between 20–150 mg of αCT11 was mixed at a 1600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of MeOH:αCT11 (400:1 mol MeOH:mol COOH) containing 5% HCl (v/v). The solution was stirred for 24 h, after which it was precipitated into diethyl ether and the peptide was isolated by centrifugation (5 min, 2420 × g, 4 °C). The supernatant was decanted, and the peptide was washed with diethyl ether, isolated by centrifugation under the same conditions, and dried under vacuum for 1 h. The peptide was dissolved in 5% (v/v) ACN in H2O (both with 0.1% TFA) and frozen with liquid N2 immediately prior to lyophilization for 48 h to produce a fluffy cake that was easy to manipulate. 50–70% yield was achieved for each esterification, assuming all 4 COOHs were esterified and peptides all contained TFA counterions.
:1 molar ratio of MeOH:αCT11 (400:1 mol MeOH:mol COOH) containing 5% HCl (v/v). The solution was stirred for 24 h, after which it was precipitated into diethyl ether and the peptide was isolated by centrifugation (5 min, 2420 × g, 4 °C). The supernatant was decanted, and the peptide was washed with diethyl ether, isolated by centrifugation under the same conditions, and dried under vacuum for 1 h. The peptide was dissolved in 5% (v/v) ACN in H2O (both with 0.1% TFA) and frozen with liquid N2 immediately prior to lyophilization for 48 h to produce a fluffy cake that was easy to manipulate. 50–70% yield was achieved for each esterification, assuming all 4 COOHs were esterified and peptides all contained TFA counterions.
Analytical reverse-phase high-performance liquid chromatography (RP-HPLC)
Analytical RP-HPLC was performed at 35 °C with a flow rate of 1 mL min−1 on a Waters e2695 Alliance Separations Module, equipped with a XBridge® C18 chromatographic separation column (4.6 mm × 50 mm, 3.5 μm beads) and a photodiode array detector (Waters 2489 UV/visible). The mobile phase consisted of ultrapurified water containing 0.1% v/v TFA (A) and ACN containing 0.1% v/v TFA (B). For a discussion of mobile phase gradient composition, see Table S1.† UV absorbance was monitored at 214 nm. For all chromatograms shown in Fig. 1, the retention time of each sample was normalized to the maximum absorbances of the unesterified αCT11 and fully esterified αCT11-4OMe peaks, which were set to normalized retention times (τ) of 0 and 1, respectively. Absorbance was also normalized to the minimum and maximum absorbances of each chromatogram, between τ = 0 and τ = 1. For the hydrolysis experiments, the retention time of each sample was reported relative to an αCT11 sample, which was set to a retention time of 0, and absorbance was not normalized. Peak area (%) for a given αCT11-related peak (αCT11) was calculated as the integration of the chromatogram within the respective bounds of that peak (ti,start and ti,end) relative to the sum of all integrals in the elution phase (between 5 and 15 min, Table S1†) of the chromatogram (eqn (1)). Since the solvent blanks (5% (v/v) ACN in H2O (both with 0.1% TFA), 1× PBS, or 100 mM carbonate buffer, when applicable) did not produce any features in the elution phase, we neglected solvent influence in the peak area calculations.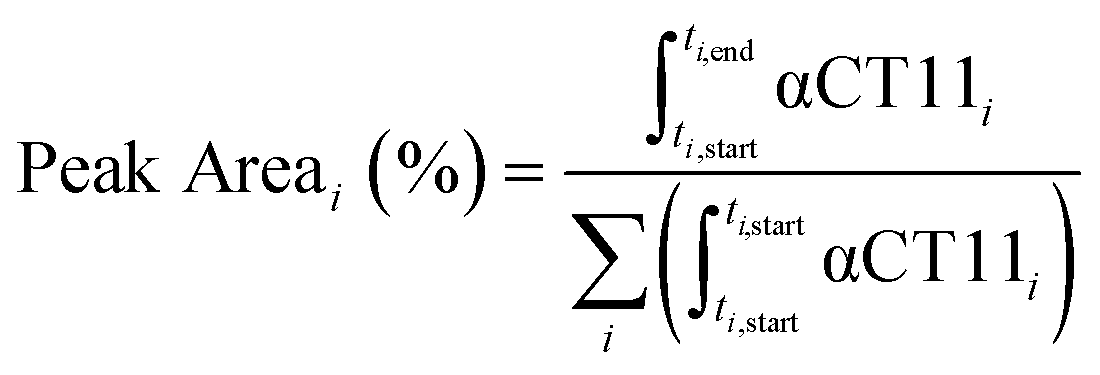 | (1) |
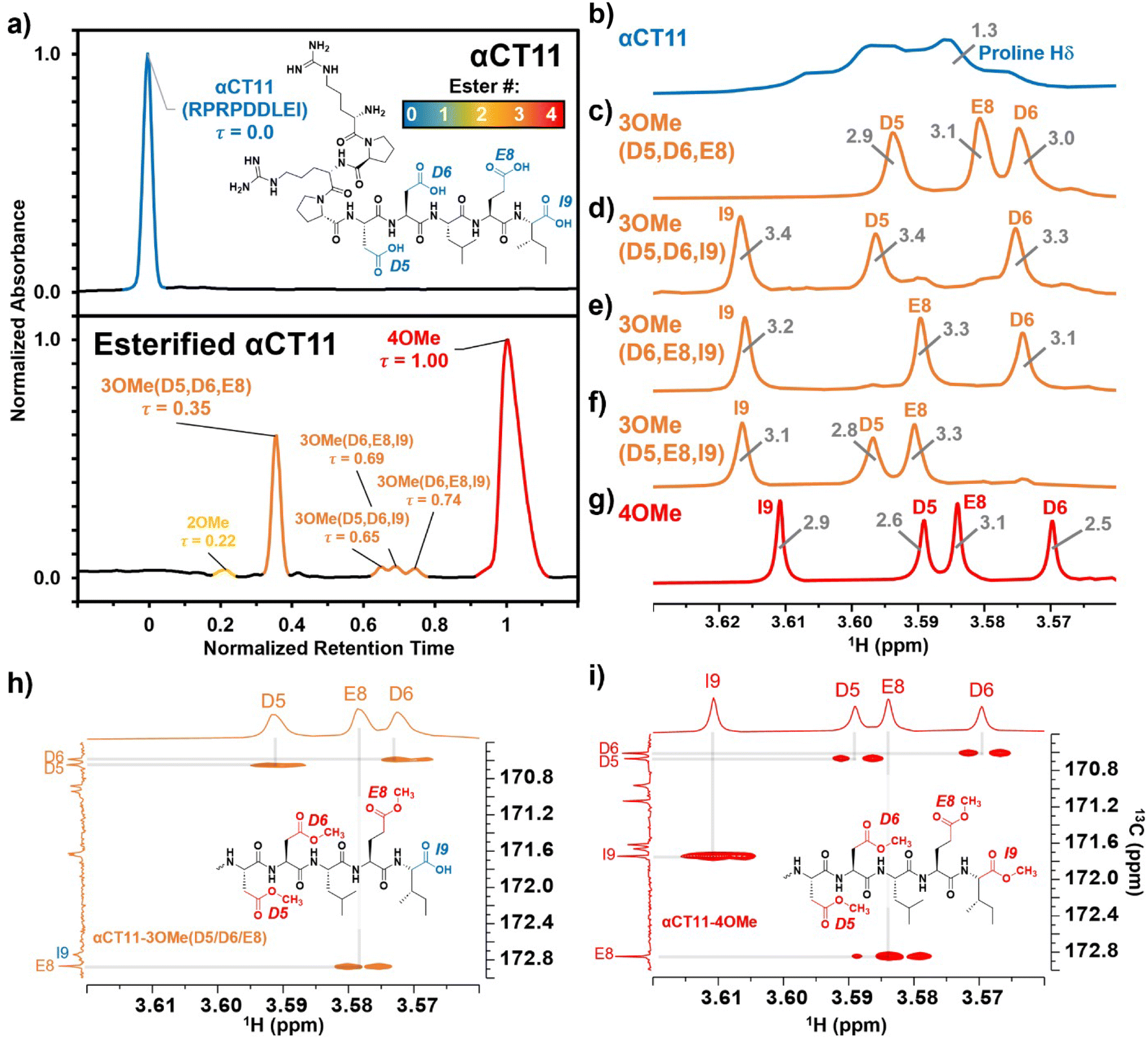 | ||
| Fig. 1 Identifying the numbers and positions of methyl esters installed onto αCT11 through Fischer esterification. a) RP-HPLC of αCT11 (peak is blue), αCT11 with 2 methyl esters (peak is yellow), 4 different conformations of αCT11 with 3 esterified esters in either the D5, D6, E8, and/or I9 residues (αCT11-3OMe(D5,D6,E8, or I9), peaks are orange), and fully esterified αCT11, with 4 methyl esters (αCT11-4OMe, peaks are red). Retention time, and thus hydrophobic character, increased with the number of esters installed onto αCT11. The mobile phase consisted of water and acetonitrile (ACN), each with 0.1% v/v TFA, and each sample eluted between 23 and 26% ACN. Retention times were normalized to those of the αCT11 and αCT11-4OMe samples. 1H NMR (800 MHz, DMSO-d6) spectra of b) αCT11 (blue), c–f) each of the 4 αCT11-3OMe samples (orange), and g) αCT11-4OMe (red) methyl ester proton singlets (3.56–3.63 ppm) show which amino acids are esterified in each formulation. Integrals for each peak are plotted in gray on the spectra. Selective heteronuclear multiple bond correlation (selHMBC) of h) αCT11-3OMe(D5,D6,E8) (orange) and i) αCT11-4OMe (red) shows the most abundant -3OMe peak (-3OMe(A)) has esters installed on the D5, D6, and E8 positions, but not on the C-terminal I9. | ||
Preparative RP-HPLC
To purify crude αCT11 variants and collect the isolated products, preparative RP-HPLC was performed at 25.52 mL min−1 at room temperature on a Waters Empower system, equipped with a XBridge® Prep C18 optimum bed density chromatographic separation column (30 mm × 150 mm, 5 μm beads) and a photodiode array detector (Waters 2489 UV/visible). UV absorbance was monitored at 214 nm. The mobile phase consisted of ultrapurified water containing 0.1% TFA (A) and ACN containing 0.1% TFA (B), where TFA was included to maintain pH. Mobile phase gradients can be found in Table S1.† For each purification, the eluent was collected at desired times and re-analyzed on analytical RP-HPLC using analogously scaled gradients to ensure purity, which was determined as the peak area fraction (eqn (1)) in the relevant retention time range (between τ = 0 and τ = 1). Like fractions were then combined and lyophilized to obtain solid, purified peptide. Between 30–80 mg of peptide were routinely injected, with 20–60% recovery achieved for each injection.Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS)
Molecular weights of each purified peptide sample were measured using a Shimadzu MALDI-8030 mass spectrometer with a 200 Hz solid-state laser (355 nm). The instrument was calibrated with a standard MALDI calibration kit (TOFMix and CHCA matrix, Shimadzu) within 0.5 Da, for which samples were dissolved at 670 femtomoles per μL in 70% v/v ACN with 0.1% v/v TFA. All samples were dissolved in 5% v/v ACN in ultrapure H2O, each with 0.1% v/v TFA (except for the acetate counterion-switched samples, which were dissolved in 5% v/v ACN in ultrapure H2O without any TFA), at approximately 1 mg mL−1 and co-crystallized in a 1:1 ratio (v/v) with a α-cyano-4-hydroxycinnamic acid (CHCA) matrix solution (5 mg mL−1 in 70% v/v ACN with 0.1% v/v TFA). Peaks with less than 2% intensity or those that could be attributed to a CHCA blank were not reported.
Nuclear magnetic resonance (NMR) spectroscopy
A Bruker Avance III 800 MHz spectrometer equipped with a 25 K cryoprobe was used to obtain NMR spectra. αCT11 formulations were dissolved in DMSO-d6 at 5 mg mL−1 when possible (i.e., αCT11, αCT11-3OMe(D5,D6,E8), αCT11-4OMe); however, as we were unable to obtain more than 1 mg of the minor αCT11-3OMe products (-3OMe(D5,D6,I9, D6,E8,I9, D5,E8,I9)), they were dissolved at less than 1 mg mL−1 in DMSO-d6. Chemical shifts were referenced to the DMSO-d6 residual peaks at 2.5 ppm or 39.5 ppm in 1H and 13C spectra, respectively. All integrals were referenced to the R1/R3 Hδ peak (3.11 ppm), which was set to 2 protons. 1D 1H and 13C, 2D H–H clean in-phase correlation spectroscopy (CLIPCOSY),48 H–H total correlation spectroscopy (TOCSY),49 H–H nuclear Overhauser spectroscopy (NOESY),50 H–C heteronuclear multiple bond correlation (HMBC),51 and selective H–C HMBC (selHMBC)52 spectra were obtained by using standard Bruker pulse programs. The TOCSY and NOESY mixing times were 100 ms and 250 ms, respectively. The center of the 13C dimension of the selective HMBC was set to 170.4 ppm and the 13C sweep width was 7.2 ppm; the selective 13C pulse had a Q3_surbop.1 shape and a 1470 Hz bandwidth. 50% nonuniform sampling was used for all 2D spectra except selHMBC, for which uniform sampling was used. Spectra were processed and analyzed with Mestrenova 14 and Topspin 3.Hydrolysis experiments
Stock solutions of αCT11, αCT11-3OMe(D5,D6,E8), and αCT11-4OMe in 5% (v/v) ACN in ultrapure H2O (0.1% v/v TFA) were prepared at 1 mg mL−1 using >10 mg peptide, for accuracy, and aliquots (0.6 mL) of each solution were lyophilized to minimize hydrolysis during storage at −20 °C. Each lyophilized sample was incubated in 2 mL of either 1× phosphate buffered saline (PBS, pH 7.4) or carbonate buffer (100 mM, pH 10), creating a final concentration of 0.1 mM for each sample. 1× PBS was prepared by dissolving a PBS tablet into 200 mL of distilled H2O. Carbonate buffer (100 mM) was prepared by combining 775 mg (9.23 mmol) of sodium bicarbonate and 1142 mg (10.77 mmol) of sodium carbonate in RO H2O (200 mL). Carbonate and PBS buffer concentrations were selected to exceed those of the protonatable/deprotonatable groups on the peptide samples (0.6 mM for αCT11, 0.4 mM for αCT11-3OMe(D5,D6,E8), and 0.3 mM for αCT11-4OMe) to provide adequate buffering. The pH of each buffer was measured using a Mettler Toledo Benchtop pH meter and was adjusted using 100 mM solutions of NaOH and HCl until the desired pH was achieved within a tenth of a unit. Solutions were stirred continuously at 37 °C and hydrolysis was monitored using RP-HPLC and MALDI-TOF MS. The percent of unesterified αCT11 recovered from hydrolysis was determined by integrating the chromatogram within the respective bounds (ti,start and ti,end) of the unmodified αCT11 peak (αCT11) at each timepoint relative to the sum of αCT11 and all esterified αCT11 (αCT11-OMei) peak integrals visible in the relevant retention time range (between the unmodified and most hydrophobic αCT11 peaks) of the RP-HPLC run (eqn (2)). As the buffered solvent alone produced no significant peaks within the elution phase (Table S1†) in their respective chromatograms, they were not considered in the recovery of unesterified αCT11 (%) calculations. Hydrolysis experiments for all αCT11-xOMe samples were repeated in triplicate. | (2) |
Porcine liver esterase (PLE) hydrolysis experiments
Lyophilized peptide samples were prepared as described for the hydrolysis experiments. A PLE (19 units per mg) stock solution (1 mg mL−1) was prepared in 1× PBS (pH 7.4) and was diluted to create 3 separate solutions: 0.526 mg mL−1 (20 units per mL), 0.053 mg mL−1 (2 units per mL), and 0.005 mg mL−1 (0.2 units per mL). Each solution (2 mL) was added to αCT11, αCT11-3OMe(D5,D6,E8), and αCT11-4OMe aliquots (final peptide concentration = 0.1 mM) to vary the PLE concentration between 1:1, 10:1, and 100:1 enzyme units:μmol peptide. Solutions were stirred continuously at 37 °C for 24 h and the peptide was isolated through centrifugal filtration (3 kDa membranes, 5 min, 2420 × g, 4 °C) prior to analysis. Isolated peptide samples were then filtered through a 0.45 μm filter and hydrolysis was monitored by RP-HPLC and MALDI-TOF MS.
Liquid chromatography-mass spectrometry (LC-MS)
LC-MS measurements were performed with a Thermo nanoEASY-LC 1200 coupled to a Thermo Orbitrap Exploris 480 mass spectrometer with an Easy Spray ion source. Hydrolysis of αCT11, αCT11-3OMe(D5,D6,E8) and -4OMe was halted by 0.1% (v/v) TFA, which lowered the pH to under 2.0, for less than 24 h prior to analysis. Samples (50 μL) were desalted using C-18 tips (Protocol: https://doi.org/10.17504/protocols.io.36wgqjzmyvk5/v1) suspended with 50 μL of 0.1% (v/v) formic acid (FA) and diluted 10× for LC-MS injection. Samples were injected into the mass spectrometer through an analytical PepMAP RSLC C-18 Easy Spray column (Thermo Scientific – 3 μm particle size, 100 Å pore size, 150 mm column length, 75 μm internal diameter) with a pre-column Acclaim PepMap 100 C-18 (Thermo Scientific – 3 μm particle size, 100 Å pore size, 20 mm column length, 75 μm internal diameter). The mobile phase consisted of water with 0.1% (v/v) FA (A) and 80% (v/v) ACN in ultrapure H2O with 0.1% v/v FA (B). Peptides were eluted from the column using an a ACN gradient in 0.1% (v/v) FA (5–60% B in 20 min, 60–95% B in 4 min, and hold at 95% B for 6 min). The mass spectrometer was operated in positive, data-dependent mode, in which one full MS scan as acquired in the m/z range of 375–1500 (1 μscan, resolution = 120 K, RF lens 40%, AGC = 300%, max inject = 60 ms) followed by MS/MS acquisition using higher energy collisional dissociation (HCD) of the 10 most intense ions (1 μscan, repeat count = 3, exclusion duration = 20 s, intensity threshold = 1 × 1006, 10 ppm tolerance, resolution = 30 K, AGC = 100%, NCE = 30) from the MS scan using window width of 2.0m/z. For each timepoint, a total ion chromatogram (m/z = 525.0–590.0 for +2 ions related to any αCT11 products) and a selective ion chromatogram (monoisotopic +2m/z, 10 ppm window) for each αCT11 species was measured, and the area of the selective ion chromatograms were corrected to account for different isotopes of each product by multiplying by a correction factor of 1.9 (Fig. S41, S42 and S45–S58†). Abundance (%) of different αCT11 species was calculated by integrating the area under the curve for each selective ion chromatogram, applying the correction factor, and dividing it by the area of the total ion chromatogram over the same timeframe.Trifluoroacetic acid counterion exchange
To replace TFA counterions that can be toxic with acetate counterions for the cell culture assay, we dissolved >10 mg of αCT11, αCT11-3OMe(D5,D6,E8), or αCT11-4OMe into distilled H2O (5–10 mL). We then placed the solution into a 0.5–1 kDa dialysis membrane. Each peptide sample was first dialyzed against 30% (v/v) acetic acid in RO H2O for 4 h to replace the counterions, followed by 3 additional dialysis solution changes (greater than 4 h each) against 0.1% (v/v) acetic acid in RO H2O to lower the overall concentration of acetic acid in the solution prior to lyophilization. After lyophilization, counterion-exchanged samples were weighed and dissolved in 5% v/v ACN in ultrapurified H2O (1 mg mL−1) and 1 mL aliquots were prepared and lyophilized, yielding 1 mg lyophilized samples of each αCT11 formulation.Cell-cultured scratch wound assays
Scratch wound assays were repeated as descripted in a prior publication.8 Cells used in this experiment were human dermal fibroblasts (huDF; ATCC, PCS-201-012). Medium used for huDF was Dulbecco's modified Eagle medium – high glucose (DMEM HG, 4.5 g L−1 glucose) with 2% normal calf serum (NCS; Thermo Fisher/Gibco, 16010-159, Lot 2490415) and 4% fetal bovine serum (FBS; Thermo Fisher/Gibco, 26140-079). Cells were expanded and stored in liquid N2 until plating on polypropylene culture dishes in culture medium (20 mL media per expansion). Cells were expanded to confluency, then passaged into 12-well plates and allowed to adhere and grow prior to removing serum from media to eliminate proliferation. Lyophilized counterion-exchanged peptide samples (αCT11, αCT11-3OMe(D5,D6,E8), αCT11-4OMe) were solubilized in the vehicle (DMSO in HEPES buffer) at 10 mM (100×). 10 μL of the 100× solution was then added to 1 mL of DMEM HG to dilute the concentration to ∼100 μM (1×). When serum was removed, either the 1× peptide solutions or the vehicle control were administered to the cells. The scratch wound assay was performed 24 h after the treatment, using a 200 μL sterile pipette tip to scratch the surface of the well, then cells were rinsed in 1× Dulbecco's phosphate buffered saline (dPBS) and provided fresh culture (1 mL per well) medium post-treatment. Cells were then imaged along the length of each scratch, and images were analyzed to determine initial scratch areas. After 6 h, cultures were rinsed in 1× dPBS, fixed in 2% paraformaldehyde, then rinsed 4 times in dPBS and stained in 1:20000 (v/v) Hoechst prior to imaging on a Leica SP8 laser scanning confocal microscope. The initial scratch area (Areax,0h) and final scratch area after 6 h (Areax,6h) were determined for each sample (x) using automated ImageJ Area Analysis software, then a “migration index” was calculated by dividing the difference between the initial area and the final area of the scratch by the initial area, with a larger migration index indicating greater movement towards a final scratch area of 0. Each migration index was reported relative to the average of that of the vehicle (v), creating a relative migration index (eqn (3)). The calculated relative migration indexes from 10 different images of 3 different cell cultures for each tested formulation were then averaged. Each formulation was statistically compared using a one-tailed paired 2 sample t-test assuming unequal variances. | (3) |
Results and discussion
Esterification of αCT11
To install methyl esters onto αCT11, we stirred the peptide in excess MeOH containing 5% (v/v) HCl. After 24 h, RP-HPLC revealed a mixture of peaks, each eluting later than unmodified αCT11, consistent with the expected increase in hydrophobicity upon esterification (Fig. 1a, S1 and S2, Table S2†). The product mixture was quite reproducible, as 3 reactions set up using independently prepared solutions led to nearly identical RP-HPLC chromatograms (Fig. S1†). With 4 esterification sites on αCT11(D5,D6,E8,I9), there are 15 possible esterified products; therefore, we next sought to separate the product mixture and, for the isolatable products, identify the number and position of the installed methyl esters. Then, to determine the effects of methyl ester number and position on peptide hydrophobicity, we compared the RP-HPLC retention times of each isolated product to gauge their relative hydrophobicity. For a more precise comparison of retention times for samples run in different batches of RP-HPLC mobile phase, we normalized each sample to an αCT11 and a fully esterified αCT11 sample set to normalized retention times τ = 0 and 1, respectively.We used preparative scale RP-HPLC to separate the product mixture, first focusing our attention on the most abundant (74% peak area) and latest eluting peak (τ = 1) (Fig. 1a, Table S2†). Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS) revealed this product to have a molecular weight of 1166.3 g mol−1, coinciding to αCT11 with all 4 sites esterified (αCT11-4OMe) (Fig. S3, Table S3†). That the fully esterified peptide is the most abundant product is consistent with the 400× molar excess of MeOH used in the reaction relative to αCT11 COOHs. The next most abundant peak (19% peak area) eluted at τ = 0.35 and had a molecular weight of 1151.6 g mol−1, corresponding to αCT11 with 3 methyl esters (αCT11-3OMe(A)) (Fig. 1a and S4, Tables S2 and S3†). Though the 3 peaks eluting at τ = 0.69, τ = 0.70, and τ = 0.75 accounted for only 4% of the total peak area, we were able to isolate them and determine that each of them also had 3 methyl esters (αCT11-3OMe(B–D)) (Fig. 1a and S5–S7, Tables S2 and S3†). Finally, we collected the product that eluted at τ = 0.22 and accounted for just 1% of the peak area, identifying it as αCT11 with 2 methyl esters (αCT11-2OMe) (Fig. 1a and S8, Table S2 and S3†). As the αCT11-2OMe and -3OMe products eluted before αCT11-4OMe, it is evident that hydrophobic character increases with the number of installed methyl esters. Yet, the 3 methyl ester products had a large range of retention times, spanning τ = 0.35–0.74, pointing to the substantial effect of ester position on αCT11 hydrophobicity.
To determine the positions of esters installed onto αCT11 in each isolated product, we used NMR spectroscopy. The 1H NMR spectrum of αCT11-4OMe (Fig. 1g and S9–S12, Table S4†) showed 4 distinct singlets at ∼3.6 ppm, each integrating to 3 protons, that were not present in the spectrum of unesterified αCT11 (Fig. 1b and S12a†). Ascribing these singlets to the methyl ester protons, we next used 2D NMR spectroscopy to assign these singlets to the D5, D6, E8, and I9 esterification sites. To do so, we first assigned the relevant carbonyl carbon resonances (i.e., the D5 and D6 γ carbons, the E8 δ carbon, and the C-terminal I9 carbonyl carbon) using a combination of 2D techniques, described in detail in Section S2 of the ESI† (Fig. S13–S24, Table S4†). Since the methyl ester protons are 3 bonds from these carbonyl carbons, we used H–C selective heteronuclear multiple bond correlation (selHMBC) spectroscopy, which shows correlations between protons and carbons separated by 2–4 bonds, to assign each methyl ester to a residue. We observed 4 clear correlations between the D5 and D6 γ carbons, the E8 δ carbon, and the C-terminal I9 carbonyl carbon and the 4 methyl esters (Fig. 1i). These correlations allowed us to assign the most downfield methyl ester singlet (3.61 ppm) as that on the C-terminal I9, followed by the D5 (3.59 ppm), E8 (3.58 ppm), and D6 (3.57 ppm) methyl ester singlets.
Similar analysis of the next most abundant product (αCT11-3OMe(A), τ = 0.35) showed esters installed on the D5, D6, and E8 positions (αCT11-3OMe(D5,D6,E8)) (Fig. 1c, h, S12c and S25–S28†). Since the chemical shift order of the methyl ester proton singlets was conserved in both the αCT11-3OMe(D5,D6,E8) and -4OMe 1H NMR spectra, we assumed that this order persisted across all esterified αCT11 formulations. With this assumption, we then used 1H NMR spectroscopy to identify the ester positions in the remaining -3OMe products. αCT11-3OMe(B) (τ = 0.65) had methyl esters on the D5, D6, and C-terminal I9 positions (αCT11-3OMe(D5,D6,I9)), -3OMe(C) (τ = 0.69) had esters on the D6, E8, and C-terminal I9 positions (αCT11-3OMe(D6,E8,I9)), and -3OMe(D) (τ = 0.74) had esters on the D5, E8, and C-terminal I9 positions (αCT11-3OMe(D5,E8,I9)) (Fig. 1d–f and S12†).
Having determined the positions of methyl esters installed onto the 4 αCT11-3OMe products, we compared their normalized RP-HPLC retention times to establish relationships between hydrophobicity and installed ester position (Fig. 1a). We note that the different pKas of the D5, D6, E8, and C-terminal I9 COOHs will result in different protonation states in physiological conditions, which will affect their relative hydrophobicities. However, as RP-HPLC comparisons were made in acidic conditions (0.1% (v/v) TFA), peptides should be fully protonated, allowing us to determine the relative increases in hydrophobicity upon ester installation at a constant protonation state. αCT11-3OMe(D5,D6,E8), the most abundant of the -3OMe products, lacked an ester on the C-terminal I9 site and eluted earliest (τ = 0.35). In contrast, the remaining 3 methyl ester products, all with the C-terminal I9 esterified, eluted later than αCT11-3OMe(D5,D6,E8) and close together with αCT11-3OMe(D5,D6,I9) at τ = 0.65, -3OMe(D6,E8,I9) at τ = 0.69, and -3OMe(D5,E8,I9) at τ = 0.74. Together, these data suggest that esterifying the C-terminal I9 imparts the largest increase in hydrophobic character.
Hydrolysis of esterified αCT11 formulations
After identifying the number and positions of methyl esters installed onto the various αCT11 products, we sought to determine the lability of the installed esters and subsequent recovery of the therapeutically active unesterified form of αCT11. We incubated the most abundant products αCT11-3OMe(D5,D6,E8) and -4OMe in 1× PBS (pH 7.4) at 37 °C and monitored ester hydrolysis as a function of time using RP-HPLC. Using these chromatograms, we then determined the recovery of unesterified αCT11 (%) as the integration of the αCT11 peak relative to the total integration between 5 and 15 min (Table S1,†eqn (2)). After 3 d, we only observed 6 ± 2% recovery of unesterified αCT11 from αCT11-3OMe(D5,D6,E8), and no unesterified αCT11 was recovered from -4OMe (Fig. 2 and S29–S31, Tables S5 and S6†). After 7 d, we observed 10.5 ± 2.3% recovery of unesterified αCT11 from αCT11-3OMe(D5,D6,E8), but only 1.1 ± 0.3% recovery from -4OMe was observed after 8 d. | ||
| Fig. 2 Recovery of unesterified αCT11 from esterified formulations in aqueous conditions. αCT11-3OMe(D5,D6,E8) (orange) and -4OMe (red) were incubated in 1× PBS (pH 7.4, squares) or 100 mM carbonate buffer (pH 10, triangles) and the amount of unmodified αCT11 present over ∼1 week was quantified using RP-HPLC. Basic conditions were required to fully hydrolyze installed esters, where αCT11-3OMe(D5,D6,E8) and -4OMe fully reverted to unesterified αCT11 after 3 d and 5 d in carbonate buffer (pH 10), respectively (denoted on the plot with an ‘x’). Full recovery of unesterified αCT11 was not achieved in 1× PBS. | ||
As 37 °C 1× PBS was insufficient to fully hydrolyze esterified αCT11 formulations, we attempted to catalyze ester hydrolysis with porcine liver esterase (PLE), which is often used to mimic native esterases.53,54 At high concentrations of PLE (10:1 and 100:1 PLE enzyme units:peptide μmol), we observed a loss of αCT11-related signal in both RP-HPLC and MALDI-TOF MS (Fig. S32–S37, Tables S7–S9†). Given that RP-HPLC and MALDI-TOF confirmed that unmodified αCT11 was stable for 7 d in 1× PBS (Fig. S31, Table S5†), we suspected that PLE was non-selectively catalyzing methyl ester and backbone amide hydrolysis, consistent with prior findings.55 Decreasing the PLE concentration to 1:1 PLE enzyme units:peptide μmol minimized loss of αCT11 signal but did not provide ester hydrolysis beyond that achieved in 1× PBS. Therefore, in our hands, PLE did not offer a viable route to selectively cleave these esters. A full discussion of the PLE hydrolysis experiments can be found in Section S3 of the ESI† (Fig. S32–S37, Tables S7–S9†).
To compare the time required to fully hydrolyze αCT11-3OMe(D5,D6,E8) and -4OMe, we used basic aqueous conditions (i.e., 100 mM carbonate buffer, pH 10) to accelerate ester hydrolysis. Having confirmed that αCT11 backbone amides were stable in 37 °C pH 10 carbonate buffer for 5 d, we monitored hydrolysis of the esterified products under these conditions (Fig. S38†). After only 6 h, we observed 54.7 ± 2.4% and 15.2 ± 0.4% recovery of unesterified αCT11 from αCT11-3OMe(D5,D6,E8) and -4OMe, respectively (Fig. 2, Table S6, Fig. S39 and S40†). We observed complete recovery of unesterified αCT11 from αCT11-3OMe(D5,D6,E8) and -4OMe after 3 d and 5 d, respectively, with liquid chromatography-mass spectrometry (LC-MS) showing both formulations to contain >95% αCT11 at these times (Fig. S41 and S42, Tables S10 and S11†).
Intermediates identified during esterified αCT11 hydrolysis
As early as 2 h after incubation, RP-HPLC chromatograms revealed the presence of several peaks eluting between the starting esterified product and unesterified αCT11 peaks in both physiological and basic conditions (Fig. S29 and S30†). MALDI-TOF MS showed both αCT11-3OMe(D5,D6,E8) and -4OMe samples to contain αCT11 with 1–3 esters after only 2 h (Fig. S43 and S44†). Further, we observed corresponding esterified products minus either 1 or 2 H2O molecules, suggesting intramolecular amide or imide formation. As a prior report involving peptides with side-chain esters and primary N-terminal amines showed that the primary amine was only involved in intramolecular ester cleavage when the ester was on the same N-terminal amino acid, we suspect that it is unlikely that the αCT11 N-terminal amine reacts with installed esters, and that this water loss is the result of intramolecular imide formation.56To identify the relative abundances of the different αCT11 hydrolysis intermediates, we used LC-MS. Specifically, we incubated αCT11-3OMe(D5,D6,E8) and -4OMe in 37 °C 1× PBS for 7 d. For each timepoint, we obtained a total ion chromatogram, which included all ions collected over the same m/z range (525–590 m/z region containing the most abundant +2 ions for all potential αCT11 species) inclusive of all expected hydrolysis intermediates identified by MALDI-TOF MS. We then collected selective ion chromatograms (SICs) specific to each intermediate and calculated the abundance (%) of each intermediate by dividing the integrated SIC signal by that of the total ion chromatogram (Fig. S45–S58, Tables S12 and S13†). Since LC-MS revealed both the αCT11-3OMe(D5,D6,E8) and -4OMe samples to contain just 6% and 4% of these starting products after only 2 h, respectively, it appears that 1 or more of the esters hydrolyze rapidly after exposure to aqueous solution (Fig. 3a and b, Table S12 and S13†). At this 2 h timepoint, the αCT11-3OMe(D5,D6,E8) formulation contained 40% αCT11 with 1 methyl ester missing 1 H2O molecule (i.e., -1OMe – 1 H2O), as well as 31% -1OMe – 2 H2O and 14% -2OMe (Fig. 3a, Table S12†). The αCT11-4OMe formulation hydrolyzed into -3OMe (16% abundance), 3OMe – 1 H2O (4% abundance), -2OMe (9% abundance), -2OMe – 1 H2O (47% abundance), and -2OMe – 2 H2O (19% abundance) (Fig. 3b, Table S13†). In all, more than 50% of each formulation contained imides after 2 h of hydrolysis, and these intermediates had higher LC-MS retention times than those with the same number of esters without imides, suggesting that these intramolecular reactions increase peptide hydrophobicity more than ester installation alone (Tables S12 and S13†). However, intermediates containing imides were transient, as less than 25% imide containing products remained after 6 h, less than 5% remained after 24 h, and less than 2% remained after 7 d in both esterified formulations. The transient nature of this water loss further supports that these intermediates are not intramolecular amides, but imides that are known to hydrolyze.57
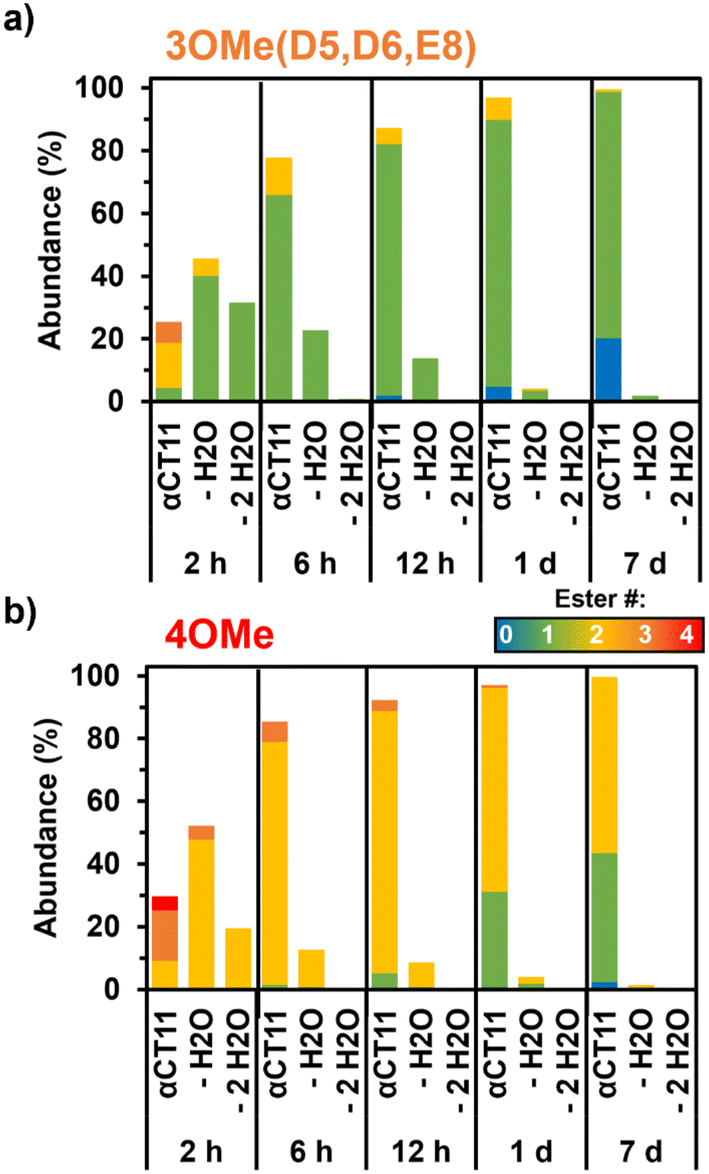 | ||
| Fig. 3 Relative abundance of intermediates observed during hydrolysis of esterified αCT11 formulations in 37 °C 1× PBS. LC-MS of a) αCT11-3OMe(D5,D6,E8) and b) -4OMe samples showed the presence of multiple esterified intermediates, including those containing intramolecular imides (– H2O, – 2 H2O), during hydrolysis. | ||
After 7 d, the αCT11-3OMe(D5,D6,E8) sample consisted of 79% -1OMe and 20% unmodified αCT11. αCT11-4OMe consisted of 56% -2OMe, 41% -1OMe, and 3% unmodified αCT11. As the αCT11-3OMe(D5,D6,E8) sample contained only a -1OMe intermediate after 7 d, yet the -4OMe sample contained both -1OMe and -2OMe intermediates, it stood to reason that an extra ester position was remaining on the -4OMe sample relative to -3OMe(D5,D6,E8). As αCT11-4OMe contained a C-terminal I9 methyl ester, which imbued the esterified formulations with the most hydrophobicity, we suspected it to be responsible for the increased hydrolytic stability of the -4OMe intermediates. To more directly gauge the hydrolytic stability of different ester positions, we sought to isolate the persistent αCT11-1OMe intermediates from each and determine which positions were esterified.
Hydrolytic stability of different ester positions on esterified αCT11 formulations
To determine the position of the most stable esters on αCT11-3OMe(D5,D6,E8) and -4OMe, we isolated the persistent αCT11-1OMe hydrolysis intermediates observed immediately prior to complete recovery of unesterified αCT11 in 37 °C 100 mM carbonate buffer (pH 10) (Fig. 4a, b, S39 and S40†). For these experiments, we used accelerated hydrolysis conditions to more rapidly obtain sufficient amounts of these persistent intermediates for 1H and 13C NMR spectroscopy, allowing αCT11-3OMe(D5,D6,E8) and -4OMe to hydrolyze for 6 and 24 h, respectively. After using preparative scale RP-HPLC to isolate the persistent hydrolysis intermediates and remove buffer salts, we used selHMBC to identify that the persistent hydrolysis intermediate of αCT11-3OMe(D5,D6,E8) (relative retention time (RRT) = 0.20) was αCT11 containing a single ester in the E8 position (-1OMe(E8)) (Fig. 4c). In the αCT11-4OMe formulation, the persistent hydrolysis intermediate (RRT = 0.62) was αCT11 containing a single ester in the I9 position (-1OMe(I9)) (Fig. 4d). As αCT11-3OMe(D5,D6,E8), which did not have an esterified C-terminal I9 position, fully hydrolyzed faster than αCT11-4OMe (Fig. 3), we concluded that the ester installed in the C-terminal I9 position was more hydrolytically stable than that in the E8 position. Additionally, the rapid appearance of the persistent intermediates in both samples under both physiological and basic conditions indicated that the D5 and D6 methyl esters hydrolyze quickly (Fig. S29, S30, S39 and S40†). Further, as the imide products are most abundant in samples collected at the earliest hydrolysis timepoints, it is likely that they involve the D5 and D6 methyl esters, suggesting that the transient intermediates we observe are aspartimides (Fig. 3, Tables S12 and S13†). That the aspartic acid (D5, D6) esters seem to hydrolyze fastest, followed by those on glutamic acid (E8) and finally C-terminal I9 also raises questions about the role of esterification site type vs. position in hydrolytic stability, which we plan to pursue in future studies. | ||
| Fig. 4 Hydrolytic stability of persistent hydrolysis intermediates of esterified αCT11. RP-HPLC chromatograms of a) αCT11-3OMe(D5,D6,E8) (orange) and b) αCT11-4OMe (red) in 37 °C 100 mM carbonate buffer (pH 10), showing each formulation after 6 and 24 h of hydrolysis, respectively. Persistent hydrolysis intermediates are highlighted in green, and αCT11 controls are in blue. Mobile phase composition (% B, gray) is plotted against relative retention time in each RP-HPLC chromatogram. 2D selHMBC of c) αCT11-3OMe(D5,D6,E8) (orange) and d) αCT11-4OMe (red) and their persistent hydrolysis intermediates (green) were used to show -3OMe(D5,D6,E8) hydrolyzes into -1OMe(E8) (green, left) and -4OMe hydrolyzes into -1OMe(I9) (green, right). | ||
Activity of esterified αCT11
Despite not observing full recovery of unesterified αCT11 from αCT11-3OMe(D5,D6,E8) or -4OMe in aqueous buffer at physiologically relevant conditions, we were interested to gauge the activity of the esterified αCT11 formulations in vitro. In prior work,8 we probed αCT11 wound healing activity by tracking cell migration into a scratch. In these experiments, we cultured human dermal fibroblasts (huDFs), with migratory behavior known to organize scar tissue,58–62 scratched them, and assessed their migration upon the addition of αCT1, which is αCT11 attached to the cell penetrating peptide antennapedia (RQPKIWFPNRRKPWKK).12 We found that αCT1 afforded higher cell migration into a scratch than the vehicle control, which was attributed to intracellular Cx43/ZO-1 binding-mediated wound healing. Therefore, we anticipated that installing methyl esters onto αCT11 would provide an alternative, reversible strategy towards realizing its intracellular localization. Thus, we performed in vitro cell culture scratch wound assays on huDFs (ATCC, PCS-201-012) treated with αCT11, αCT11-3OMe(D5,D6,E8), or -4OMe.Prior to their use in the scratch wound assays, each αCT11 peptide was dialyzed against 30% acetic acid to replace trifluoroacetate (TFA) counterions, known to skew physiological experiments,63,64 with acetate counterions. MALDI-TOF MS revealed that some hydrolysis occurred during the counterion exchange process, as the counterion switched αCT11-3OMe(D5,D6,E8) sample contained peaks commensurate with -3OMe, -2OMe, and -2OMe – 1 H2O (Fig. S59†). Similarly, the counterion switched αCT11-4OMe sample contained -4OMe, 3OMe, -3OMe – 1 H2O, -2OMe – 1 H2O, -2OMe – 2 H2O, and -1OMe (Fig. S60†). Despite some hydrolysis of the samples and the presence of intramolecular imide products, it was clear that both counterion-switched αCT11-3OMe(D5,D6,E8) and -4OMe still contained esters. Presumably, esters on the D5 and D6 aspartic acids hydrolyzed and some aspartimide formation occurred. Thus, we proceeded with these experiments to compare cell migration into the scratch resulting after addition of the esterified peptide formulations containing or lacking a C-terminal I9 methyl ester.
In the scratch assays, huDFs were cultured on polypropylene culture dishes for 24 h, after which the peptides or vehicle control were added, the cultures were scratched with a 200 μL pipette tip, and 10 images were taken at different positions along the scratch immediately and 6 h after scratching. We measured a “migration index” for the cells in each image, which was calculated by dividing the difference in the area of the scratch after 6 h by the initial area. We then divided each migration index by the average of those for cells treated with the vehicle control (DMSO in HEPES buffer) to calculate a “relative migration index” (eqn (3)) for each image. This was repeated 2 more times for a total of n = 3 scratched cell cultures, and we then calculated an overall average relative migration index for cells treated with each formulation. For cultures treated with αCT11-3OMe(D5,D6,E8) and -4OMe, we observed significantly higher relative migration indexes (-4OMe = 6.2 ± 2.1; -3OMe(D5,D6,E8) = 4.8 ± 1.5, with p < 0.05 for both formulations) than unmodified αCT11 (1.4 ± 0.4), which had a statistically similar relative migration index to the vehicle alone (1.0, p = 0.11) (Fig. 5, Tables S14 and S15†). Interestingly, despite the relative differences in hydrophobicity and hydrolytic stability observed between αCT11-3OMe(D5,D6,E8) and -4OMe, cultures treated with the 2 formulations did not have statistically significant relative migration indices (p = 0.19). Regardless, the higher relative migration indices observed in the cell cultures treated with esterified αCT11 suggest that esters offer benefit by facilitating cell internalization and/or increasing activity.
 | ||
| Fig. 5 Multi-site esterification of αCT11 increases cell migration into a scratch. a) Representative images of the initial scratch wounds (top) and the same wounds after 6 h (bottom) of cell culture. Prior to the scratch, samples were treated with either the vehicle control (DMSO in HEPES, pH 7.4, gray), unmodified αCT11 (blue), αCT11-3OMe(D5,D6,E8) (orange), or αCT11-4OMe (red). Dashed lines are added to guide the eye along the edges of the scratch. b) Relative migration indexes of scratched cells when exposed to the vehicle (gray), αCT11 (blue), -3OMe(D5,D6,E8) (orange), and -4OMe (red). Scratch assays were performed with a human adult dermal fibroblast line (ATCC, PCS-201-012). Plotted circular markers represent the average between n = 10 individual images of a single scratch, and their respected standard deviations are noted by the gray error bars. Plotted bars represent the average relative migration index across n = 3 different cell cultures, and the bolded black error bars represent the standard deviation of those averages. Statistics were determined from the average relative migration indexes of the n = 3 cell cultures using a one-tailed paired two-sample t-test assuming unequal variances, where # (gray) and * (blue) denote statistical significance relative to the vehicle and αCT11, respectively (p < 0.05) and “n.s.” represents no statistical significance (p > 0.05). | ||
Conclusions
Through these studies, we showed that varying the number of methyl esters installed onto the therapeutic peptide αCT11 is an effective way to reversibly modify the hydrophobicity and number of anionic COOHs; however, controlling the position of esters on the peptide is just as important. 2D NMR spectroscopy revealed which residues were esterified, a key development in establishing the role of ester position on the properties of esterified αCT11 formulations. Of the 4 sites on αCT11, the C-terminal I9 was the most influential in modifying αCT11 properties, as methyl esters on the C-terminal I9 imparted the highest increase in hydrophobicity and were the most hydrolytically stable. Considering that the D5/D6, E8, and C-terminal I9 methyl esters each affected αCT11 hydrophobicity and hydrolytic stability differently, these experiments raise important future questions about the relative roles of ester position (i.e., 5, 6, 8, C-terminus) vs. amino acid type (i.e., D, E, I) on modifying therapeutic peptide properties. During ester hydrolysis, we observed transient intramolecular imide intermediates that seemed to further increase hydrophobicity relative to products with just esters. Since aspartimide formation can lead to peptide racemization,57 further experiments will be needed to determine whether peptide stereochemistry remains intact; however, the increase in hydrophobicity is a promising alternative method for reversibly affecting therapeutic peptide properties. As αCT11 therapeutic activity is thought to rely on its interaction with the H2 domain of the transmembrane protein Cx43 and/or the PDZ2 domain of the scaffolding protein ZO-1,7 we were encouraged by the increased migration observed in in vitro scratch wound assays treated with both esterified αCT11 variants relative to those treated with unmodified αCT11. These results show that esterification provides a clear benefit to αCT11-mediated wound healing and raises exciting future questions about whether installed esters serve mainly to increase cell internalization of therapeutic peptides and/or to provide inherently higher activity themselves. Through reversible control over hydrophobicity and charge, multi-site esterification offers a compelling strategy to modify the hydrophobicity and net charge of hydrophilic peptides with multiple COOHs and tailor them for delivery, an imperative step towards transitioning intracellularly targeting therapeutic peptides into the clinic.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. G. G., L. J. J. and R. A. L. conceptualized the project. M. S. B. synthesized the materials, isolated them, and collected and analyzed the RP-HPLC and MALDI-TOF MS data for the esterification study. M. S. B. and J. F. E. collected and analyzed the NMR spectroscopy data. M. S. B. and A. S. G. collected and analyzed the RP-HPLC and MALDI-TOF MS data for the hydrolysis studies. N. E. S. and D. T. collected the LC-MS data for hydrolysis studies, and N. E. S., D. T., and M. S. B. analyzed the data. S. R. M. collected and analyzed the scratch wound assay data. M. S. B., A. S. G. and R. A. L. wrote the manuscript, and all authors contributed to revising the manuscript.Conflicts of interest
R. G. G. and S. R. M. are founders, shareholders and company officers at the Tiny Cargo Company Inc., which has licensed technology from Virginia Tech. L. J. J. is a shareholder in the Tiny Cargo Company Inc. R. G. G. is a founder and shareholder at FirstString Research/Xequel Bio Inc.Acknowledgements
This work has been supported by grants from the NIH (R. G. G. and R. A. L., R35HL161237-01 and R. A. L., R35GM147424), Seale Foundation (R. G. G.), CRCF (R. G. G. and R. A. L.), VBHRC catalyst (S. R. M., R. G. G., and R. A. L., Grant #1403), VTIP (R. G. G., S. R. M., CCF23-0035-PS), NSF (R. G. G., S. R. M., 2203330), and UVA School of Engineering Dean's Scholar Fellowship (M. S. B.).References
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- A. Meyer-Lindenberg, G. Domes, P. Kirsch and M. Heinrichs, Nat. Rev. Neurosci., 2011, 12, 524–538 CrossRef CAS PubMed.
- L. Di, AAPS J., 2015, 17, 134–143 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- J. M. Rhett, J. Jourdan and R. G. Gourdie, Mol. Biol. Cell, 2011, 22, 1516–1528 CrossRef CAS PubMed.
- J. Jiang, D. Hoagland, J. A. Palatinus, H. He, J. Iyyathurai, L. J. Jourdan, G. Bultynck, Z. Wang, Z. Zhang, K. Schey, S. Poelzing, F. X. McGowan and R. G. Gourdie, J. Am. Heart Assoc., 2019, 8, 16 Search PubMed.
- J. Montgomery, W. J. Richardson, S. Marsh, J. M. Rhett, F. Bustos, K. Degen, G. S. Ghatnekar, C. L. Grek, L. J. Jourdan, J. W. Holmes and R. G. Gourdie, FASEB J., 2021, 35(8), e21762 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Waller, in Molecular Biology of the Cell, Garland Science, New York, NY, 4th edn, 2002 Search PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed.
- A. K. Sato, M. Viswanathan, R. B. Kent and C. R. Wood, Curr. Opin. Biotechnol., 2006, 17, 638–642 CrossRef CAS PubMed.
- D. Derossi, G. Chassaing and A. Prochiantz, Trends Cell Biol., 1998, 8(2), 84–87 CrossRef CAS PubMed.
- C. L. Grek, J. Montgomery, M. Sharma, A. Ravi, J. S. Rajkumar, K. E. Moyer, R. G. Gourdie and G. S. Ghatnekar, J. Invest. Dermatol., 2017, 137, 620–630 CrossRef CAS PubMed.
- A. E. Cardenas, R. Shrestha, L. J. Webb and R. Elber, J. Phys. Chem. B, 2015, 119, 6412–6420 CrossRef CAS PubMed.
- A. Gräslund, F. Madani, S. Lindberg, Ü. Langel and S. Futaki, J. Biophys., 2011, 2011, 414729 Search PubMed.
- P. A. Leventis and S. Grinstein, Annu. Rev. Biophys., 2010, 39, 407–427 CrossRef CAS PubMed.
- A. Zachowski, Biochem. J., 1993, 294, 1–14 CrossRef CAS PubMed.
- S. C. Povilaitis and L. J. Webb, J. Phys. Chem. Lett., 2023, 14, 5841–5849 CrossRef CAS PubMed.
- S. J. Marrink and H. J. C. Berendsen, J. Phys. Chem., 1994, 98, 4155–4168 CrossRef CAS.
- R. A. Conradi, A. R. Hilgers, N. F. Ho and P. S. Burton, Pharm. Res., 1992, 9, 435–439 CrossRef CAS PubMed.
- Q. Al-Awqati, Nat. Cell Biol., 1999, 1, E201–E202 CrossRef CAS PubMed.
- D. Kalafatovic and E. Giralt, Molecules, 2017, 22(22), 1929 CrossRef PubMed.
- C. E. Mills, A. Obermeyer, X. Dong, J. Walker and B. D. Olsen, Langmuir, 2016, 32, 13367–13376 CrossRef CAS PubMed.
- R. A. Kapelner, R. S. Fisher, S. Elbaum-Garfinkle and A. C. Obermeyer, Chem. Sci., 2022, 13, 14346–14356 RSC.
- R. A. Kapelner and A. C. Obermeyer, Chem. Sci., 2019, 10, 2700–2707 RSC.
- X. Jia, Y. K. Y. Chin, A. H. Zhang, T. Crawford, Y. Zhu, N. L. Fletcher, Z. Zhou, B. R. Hamilton, M. Stroet, K. J. Thurecht and M. Mobli, Commun. Chem., 2023, 6, 1–10 CrossRef PubMed.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS PubMed.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed.
- S. Liras and K. F. McClure, ACS Med. Chem. Lett., 2019, 10, 1026–1032 CrossRef CAS PubMed.
- A. C. Rand, S. S. F. Leung, H. Eng, C. J. Rotter, R. Sharma, A. S. Kalgutkar, Y. Zhang, M. V. Varma, K. A. Farley, B. Khunte, C. Limberakis, D. A. Price, S. Liras, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, MedChemComm, 2012, 3, 1282–1289 RSC.
- T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510–2511 CrossRef CAS PubMed.
- C. K. Wang, S. E. Northfield, B. Colless, S. Chaousis, I. Hamernig, R. J. Lohman, D. S. Nielsen, C. I. Schroeder, S. Liras, D. A. Price, D. P. Fairlie and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17504–17509 CrossRef CAS PubMed.
- J. Neijssen, C. Herberts, J. W. Drijfhout, E. Reits, L. Janssen and J. Neefjes, Nature, 2005, 434, 83–88 CrossRef CAS PubMed.
- K. Shintani-Ishida, K. Uemura and K.-I. Yoshida, Am. J. Physiol., 2007, 293(3), H1714–H1720 CAS.
- K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750–771 CrossRef CAS PubMed.
- L. D. Lavis, ACS Chem. Biol., 2008, 3, 203–206 CrossRef CAS PubMed.
- O. H. Chan and B. H. Stewart, Drug Discovery Today, 1996, 1, 461–473 CrossRef CAS.
- K. Beaumont, R. Webster, I. Gardner and K. Dack, Curr. Drug Metab., 2005, 4, 461–485 CrossRef PubMed.
- B. M. Liederer and R. T. Borchardt, J. Pharm. Sci., 2006, 95, 1177–1195 CrossRef CAS PubMed.
- E. Afrimzon, A. Deutsch, Y. Shafran, N. Zurgil, J. Sandbank, I. Pappo and M. Deutsch, Clin. Exp. Metastasis, 2008, 25, 213–224 CrossRef CAS PubMed.
- O. Lockridge and D. M. Quinn, in Comprehensive Toxicology, ed. C. A. McQueen, Elsevier, Oxford, 2nd edn, 2010, pp. 243–273 Search PubMed.
- E. M. Larsen and R. J. Johnson, Drug Dev. Res., 2019, 80, 33 CrossRef CAS PubMed.
- D. Bratosin, L. Mitrofan, C. Palii, J. Estaquier and J. Montreuil, Cytometry, Part A, 2005, 66, 78–84 CrossRef PubMed.
- P. A. Burnouf, Y. L. Leu, Y. C. Su, K. Wu, W. C. Lin and S. R. Roffler, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- W. D. Gray, A. J. Mitchell and C. D. Searles, MethodsX, 2015, 2, 360–367 CrossRef PubMed.
- K. A. Mix, J. E. Lomax and R. T. Raines, J. Am. Chem. Soc., 2017, 139, 14396–14398 CrossRef CAS PubMed.
- K. Prokai-Tatrai, V. Nguyen, A. D. Zharikova, A. C. Braddy, S. M. Stevens and L. Prokai, Bioorg. Med. Chem. Lett., 2003, 13, 1011–1014 CrossRef CAS PubMed.
- M. R. M. Koos, G. Kummerlöwe, L. Kaltschnee, C. M. Thiele and B. Luy, Angew. Chem., Int. Ed., 2016, 55, 7655–7659 CrossRef CAS PubMed.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- R. Wagner and S. Berger, J. Magn. Reson., Ser. A, 1996, 123, 119–121 CrossRef CAS PubMed.
- W. Willker, D. Leibfritz, R. Kerssebaum and W. Bermel, Magn. Reson. Chem., 1993, 31, 287–292 CrossRef CAS.
- T. D. W. Claridge and I. Pérez-Victoria, Org. Biomol. Chem., 2003, 1, 3632–3634 RSC.
- J. V. Jun, Y. D. Petri, L. W. Erickson and R. T. Raines, J. Am. Chem. Soc., 2023, 145, 6615–6621 CrossRef CAS PubMed.
- L. Tian, Y. Yang, L. M. Wysocki, A. C. Arnold, A. Hu, B. Ravichandran, S. M. Sternson, L. L. Looger and L. D. Lavis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4756–4761 CrossRef CAS PubMed.
- Q. Zhou, Q. Xiao, Y. Zhang, X. Wang, Y. Xiao and D. Shi, Sci. Rep., 2019, 9, 1–12 CrossRef PubMed.
- Y. Zhu, Y. Shmidov, E. A. Harris, M. H. Theus, R. Bitton and J. B. Matson, Nat. Commun., 2023, 14, 3635 CrossRef CAS PubMed.
- K. Neumann, J. Farnung, S. Baldauf and J. W. Bode, Nat. Commun., 2020, 11, 982 CrossRef CAS PubMed.
- W. J. Richardson, B. Kegerreis, S. Thomopoulos and J. W. Holmes, Biomech. Model. Mechanobiol., 2018, 17, 1569–1580 CrossRef PubMed.
- M. Xue and C. J. Jackson, Adv. Wound Care, 2015, 4, 119–136 CrossRef PubMed.
- A. D. Rouillard and J. W. Holmes, J. Physiol., 2012, 590, 4585–4602 CrossRef CAS PubMed.
- S. McDougall, J. Dallon, J. Sherratt and P. Maini, Philos. Trans. R. Soc., A, 2006, 364, 1385–1405 CrossRef CAS PubMed.
- S. M. Karppinen, R. Heljasvaara, D. Gullberg, K. Tasanen and T. Pihlajaniemi, F1000Research, 2019, 8, F1000 Faculty Rev–787 Search PubMed.
- J. Cornish, K. E. Callon, C. Q. X. Lin, C. L. Xiao, T. B. Mulvey, G. J. S. Cooper and I. R. Reid, Am. J. Physiol., 1999, 277, 779–783 Search PubMed.
- M. E. Tipps, S. V. Iyer and S. John Mihic, Neuropharmacology, 2012, 63, 368–373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, characterization data and analysis, alternate plots, and ESI data (DOC). See DOI: https://doi.org/10.1039/d4me00072b |
| This journal is © The Royal Society of Chemistry 2024 |