
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing enzymatic activity with nanoparticle display – an updated compendium and engineering outlook
Shelby L.
Hooe
 ,
Joyce C.
Breger
and
Igor L.
Medintz
*
,
Joyce C.
Breger
and
Igor L.
Medintz
*
Center for Bio/Molecular Science and Engineering, Code 6900, U.S. Naval Research Laboratory, Washington, D.C., 20375, USA. E-mail: Igor.medintz@nrl.navy.mil
First published on 29th April 2024
Abstract
Almost all utilization of biocatalysis in the burgeoning field of synthetic biology requires not only enzymes but also that they function with peak efficiency, especially when paired with other enzymes in designer multistep cascades. This has driven concerted efforts into enhancing enzymatic performance by attaching them to macroscale scaffolding materials for display. Although providing for improved long-term stability, this attachment typically comes at the cost of decreased catalytic efficiency. However, an accumulating body of data has confirmed that attaching enzymes to various types of nanoparticle (NP) materials can often dramatically increase their catalytic efficiency. Many of the causative mechanisms that give rise to such enhancement remain mostly unknown but it is clear that the unique structured and interfacial environment that physically surrounds the NP material is a major contributor. In this review, we provide an updated and succinct overview of the current understanding and key factors that contribute to enzymatic enhancement by NP materials including the unique structured NP interfacial environment, NP surface chemistry and size, and the influence of bioconjugation chemistry along with enzyme mechanics. We then provide a detailed listing of examples where enzymes have displayed enhanced activity of some form when they are displayed on a NP as organized by material types such as semiconductor quantum dots, metallic NPs, DNA nanostructures, and other more non-specific and polymeric nanomaterials. This is followed by a description of what has been learned about enhancement from these examples. We conclude by discussing what more is needed for this phenomenon to be exploited and potentially translated in the design and engineering of far more complex molecular systems and downstream applications.
Design, System, ApplicationEnzymes are constantly finding utility in biotechnology and especially within the exponentially growing field of synthetic biology. Across this vast application space it is many times desirable to enhance and improve the kinetic activity of the enzymes and especially in configurations when they are paired with other enzymes in designer multistep cascades. Although mutational selection and enzyme evolution are clearly important ways to address the need for catalytic improvements, they still require significant expertise and resources to be properly undertaken. However, a growing body of literature confirms that displaying enzymes on nanoparticles (NPs) can provide for significant kinetic improvements in their performance. In this review, we provide a state of the art compendium of enzymatic enhancement by NP attachment as a function of major NP material classes. We further describe what is currently known about this enhancement phenomenon with a focus that includes contributions from the unique structured interfacial environment that surrounds NPs, NP surface chemistry and size, and the influence of bioconjugation chemistry along with enzyme mechanics. We then look towards the future and discuss what more is needed for this phenomenon to be fully exploited and translated in the design and engineering of far more complex molecular systems and downstream applications. |
Introduction
Enzymes are the key catalysts that speed up almost all biochemical reactions including many of those that would occur exceedingly slowly. This has made almost all attempts at biosynthesis without enzymes effectively a non-viable process. However, though a given enzyme may speed up a catalytic transformation quite significantly by several orders of magnitude, it can be even more desirable to enhance the kinetic activity of that enzyme even further than what can be initially achieved or apparent within the context of several different applications. Such enhancement above an enzyme's native rate of activity allows for increased throughput and yield in an assay or biocatalytic industrial application, decreasing substrate requirements and improving output in research and diagnostic assays or pharmaceutical candidate/activity screening, along with matching kinetic rates between jointly-coupled enzymes in designer multienzyme cascaded reactions amongst other potential utility.1–5 There are several different approaches to achieving such enhancement, some of which have been applied independently or even jointly, and these include looking for more permissive or active enzyme homologs from other source species, mutational selection and/or evolution for optimized versions of the enzyme itself, and parametric adjustment of overall components and reaction conditions in a multienzyme cascade within a design of experimental framework.6–16 Interestingly, attaching enzymes to macroscale scaffolds such as surfaces, beads, and resins can help increase an enzyme's structural stability and, in turn, it's viable lifetime providing for long-term application along with potential reuse by allowing for the attached enzymes to be removed with the scaffolding and added to another reaction.17,18 However, the latter is not normally pursued to enhance enzyme activity such as the catalytic rate or kcat, for example, primarily because chemical attachment to macroscale scaffolding materials is usually achieved with a concomitant decrease in that enzyme's kinetic properties.17–19 This is believed to arise as a result of the linkage chemistry decreasing the enzyme's overall freedom of movement, or by blocking or limiting access to its active site, along with the possibility of chemically modifying key residues needed for catalysis during attachment or association with the scaffold.20–22 Another aspect to appreciate is that, in many cases, enzyme structures are somewhat metastable, which allows them to better sample their substrates, and any structural impediment to this can be functionally deleterious.23–26Somewhat counterintuitively, a growing number of recent reports have confirmed that attaching or displaying enzymes on nanoparticles (NPs) or various other similarly-sized nanoscale scaffolding materials can increase enzymatic activity, sometimes to quite a significant extent, for example by increasing the kcat apparent by 50-fold or more (vide infra). These nanomaterials can range from those that are primarily inorganic such as metallic NPs to those that are predominantly biological such as dense DNA constructs. We note that there exist almost no current review articles or other sources that bring together a detailed listing of which enzymes have been enhanced and what NP materials this has occurred with. Although an excellent conceptual reference, the main source material in this vein originates from 2012 and this reference does not include anything that occurred subsequently in the past 12 years.27 The only other partial sources in this vein include ref. 28–30. Our focus in this review is to provide a brief discussion of some of the key factors associated with achieving NP enhancement of enzymatic activity based on what is understood about this phenomenon. See Fig. 1 for a schematic of many of the inter-related factors that are found at the NP–enzyme interface and which can influence enzymatic activity and especially kinetic enhancement. We then provide a state of the art compendium of examples where such an enhancement phenomenon has been observed across different nanoparticulate materials. We conclude with a discussion of how this phenomenon can be exploited and potentially translated during the design and engineering of more complex molecular systems. The latter can help drive unique enzymatic applications in synthetic biology and other related biotechnological fields.
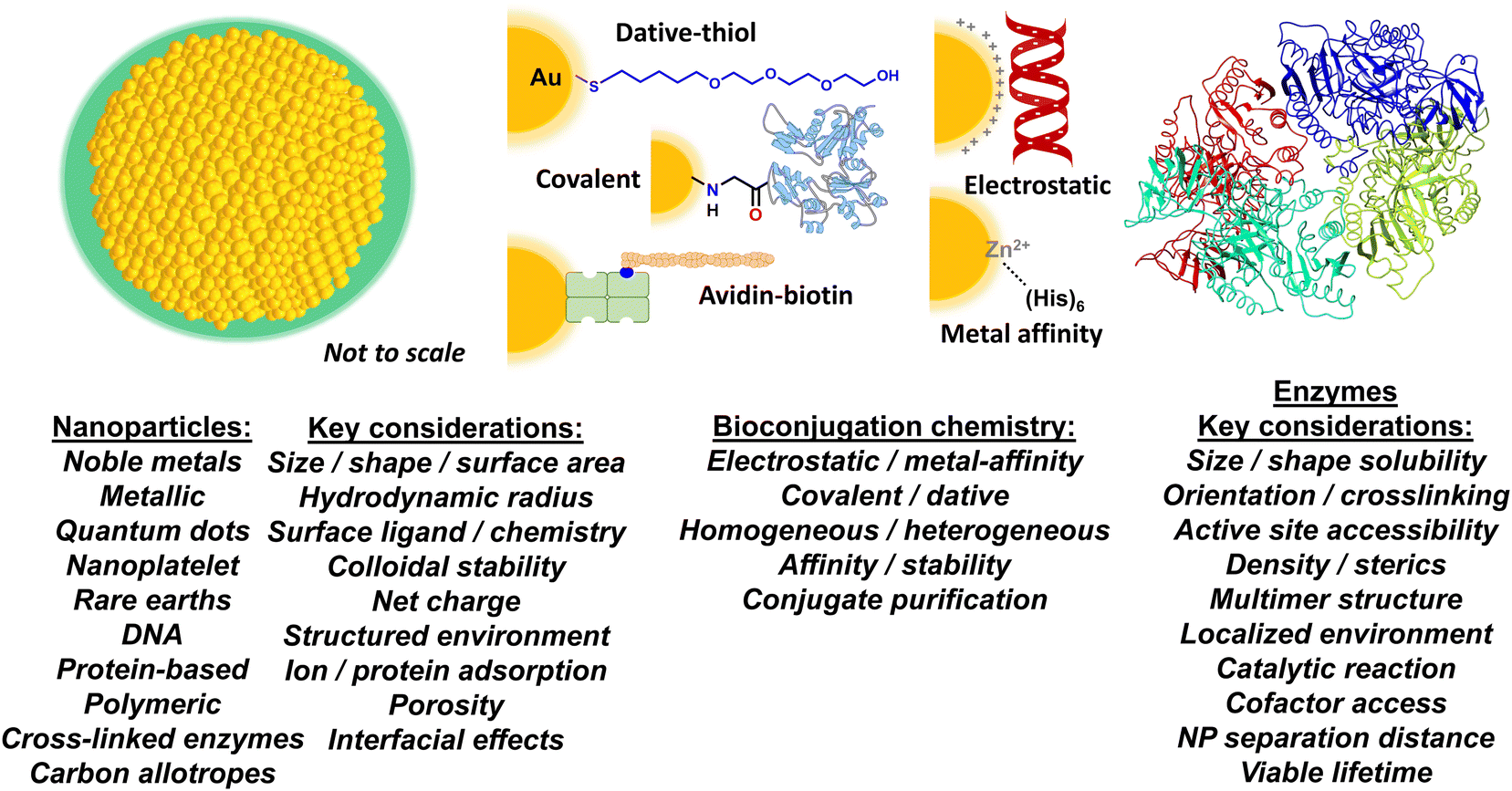 | ||
| Fig. 1 Nanoparticle–enzyme bioconjugation and key considerations for accessing enhanced enzymatic activity. Schematic depicting a prototypical NP structure (left), different bioconjugation chemistries (middle), and an enzyme (right). The NP (gold) is surrounded by a layer of surface ligands (green), which provide it with colloidal stability. Bioconjugation chemistries shown include: a poly(ethylene glycol) or PEG attached to a gold NP surface via dative thiol–gold interactions; covalent amide bond formation between an aminolated NP and a carboxyl group on a protein; binding of a biotinylated peptide to an avidin modified NP; electrostatic assembly of a negatively charged DNA to a positively charged NP surface; and MAC between the (His)6 motif and the surface ions of a Zn-overcoated quantum dot (QD). The listings underneath include some examples on NP material classes and bioconjugation chemistry approaches along with key considerations that can effect or are implicated in the generalized phenomenon of enzymatic acceleration when displayed on a NP surface. | ||
Colloidal nanoparticles and their unique interface
The European Commission defines a nanomaterial as “a natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range of 1 nm up to 100 nm”.31 There are, of course, many other definitions, depending upon the source and application.32 These definitions are generally extended to state that the material should not be exclusively naturally occurring; therefore a protein by itself would generally not be classified as a nanomaterial, however, an artificially engineered NP assembled from several naturally occurring proteins would. For these purposes, we define a NP as an engineered nanomaterial that has one of its dimensions <100 nm regardless of what it is constituted from. NPs can also be assembled from chemicals such as polymers or even constituted from carbon allotropes.32The current application under discussion, where NPs are interfaced with enzymes, requires that the NP materials be dispersible in aqueous media such as buffers. Since most NPs consisting of metals, semiconductors, and carbonaceous materials are synthesized within organic media or at high temperature, this typically means that the NPs must be further modified post-synthetically to display molecules on their surface, referred to as NP ‘surface ligands’ in the parlance of the field. These ligands impart colloidal stability to the NPs in aqueous media. Ligands can range chemically from small charged molecules to long poly(ethylene glycol) or PEG derivatives along with amphiphilic polymers and many other similarly functional materials such as polyelectrolytes (Fig. 1). The key concept to appreciate is that one part of the ligand interacts with the NP surface while another provides the NP with colloidal stability in the medium (i.e., the particles remain evenly and stably distributed throughout the solution). Obviously, the size and chemical properties of these ligands will all influence the activity of an enzyme that is attached to it or placed in or around it. In contrast to this, biological-based NPs assembled from proteins or DNA, for example, typically have intrinsic colloidal stability in aqueous media given that their constituent materials are naturally soluble in water. The interested reader is referred to several excellent reviews on this subject for more information.33–38
More pertinently, recent work has revealed that colloidal NPs universally structure their surrounding environment through the physicochemical influence of the dense ligand layer that provides the colloidal stability to the NPs.39,40 The characteristics of this structured environment remain mostly speculative as the requisite metrology to probe these nanoscale confines does not yet exist.41,42 However, it is believed that this structured environment includes pH, ionic, charge, and density gradients that may extend to twice the NP diameter in some cases. This nanoscale structuring of the surrounding environment and the immediate boundary layer that it influences between the NP–enzyme conjugate and the bulk environment is believed to be a major contributing factor to the enzymatic enhancement described below.2,43,44
Nanoparticles – selected methods for enzyme bioconjugation
How an enzyme is chemically attached to or otherwise displayed on a NP is obviously another major contributing factor to both accessing kinetic enhancement and the magnitude with which it is manifested (Fig. 1). From an idealized perspective, the chemistry used to bioconjugate an enzyme to a NP should allow for control over: i – the ratio of enzyme attached per NP; ii – the distance between the enzyme and NP; iii – the orientation of the enzyme on the NP; iv – the affinity of the enzyme attachment on the NP; v – provide for homogeneous attachment and a homogenous final bioconjugate structure; and, vi – all of these should be replicable with another unrelated NP and enzyme as desired.45 The reality is that for most NP–protein bioconjugation reactions currently utilized, the majority of these properties are not achievable in any method let alone even jointly approach a plurality. The breadth of NP–bioconjugation chemistries along with their considerations has been extensively discussed in several focused review articles.46–55The benefits and liabilities of each bioconjugation chemistry should be carefully considered in terms of the composite material's final application and whether that choice of chemistry is appropriate or compatible for that purpose or not. For example, protein assembly to noble metal NPs and especially gold NPs (AuNPs) often relies on dative bonding between thiols and the gold surface.56–58 Many times, this requires that cysteine residues with their thiol side group be recombinantly introduced into the protein in a surface available position to facilitate the interaction. If the enzyme already has a cysteine or a cysteine-based dithiol in its structure, introduction of further cysteine(s) can result in thiol scrambling and loss of activity. Moreover, if a single thiol is used, it can have a strong rate of desorption from the gold surface, which can impinge on a need for extended bioconjugate stability; this can be somewhat alleviated by having multiple thiols participate in the chemistry.59,60 In contrast to dative thiol bonding, other bioconjugation chemistries rely on covalent chemistries with use of the common carboxyl–amine linkage by amide bond formation using EDC-NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide – N-hydroxysuccinimide ester) reactants being perhaps the most popular.52 To facilitate this type of linkage, the NPs often display either the requisite carboxyl or amine group on their surface ligands with the other cognate chemical functionality originating from the enzyme. However, the ubiquitous presence of these groups on most proteins usually results in heterogeneous enzyme display along with cross-linked NP–enzyme clusters blocking active sites. If these same groups are part of the enzyme's active site, this chemistry can further inactivate the enzyme altogether. Additionally, the large concentrations of reactants used in these reactions lead to a need for extensive purification. Although ubiquitous and certainly powerful, biotin–avidin chemistry also comes with its own set of issues. Unless a labeling site is recombinantly introduced into a protein for site-specific biotinylation during expression,61 it is typically introduced into the protein using derivatives of the above heterogeneous covalent chemistry targeting amines, thiols or carboxyls. Most avidins are naturally tetravalent and attached to the NP via some type of covalent chemistry or electrostatically. This means that, in conjugation with heterogeneous biotinylation, the enzyme or other biomolecule will also be displayed in a variety of heterogeneous orientations that cannot be controlled and may even crosslink the NPs if it displays multiple biotin moieties.62
Our work assembling enzymes to semiconductor QDs has mostly relied on metal affinity coordination (MAC) between (His)6-motifs introduced at a protein's C- or N-termini and the Zn on a ZnS shell that overcoats the core/shell QD structure.63 The (His)6-motifs are usually introduced into the proteins recombinantly for subsequent purification using metal-affinity media and are thus already present on many expressed proteins.64 Extensive characterization has shown that the imidazole side chains of (His)6 will coordinate to the Zn on the QD surface with very high affinity (Kd ∼1 nM), as it is available in the appropriate 2+ valence state similar to the transition metals (e.g. Ni, Co, Cr) commonly found as chelates in metal affinity media such as nitrilotriacetic acid (NTA).50,65 Even when access to the QD's ZnS shell may be precluded by a large surface solubilizing ligand, as long as the QD's surface displays multiple carboxyls, the same type of polyhistidine binding can still be accomplished in practice.66,67 Exposing these carboxylated QDs to small amounts of transition metals allows the ions to be coordinated by the carboxyls and functionally mimic the NTA group. More pertinently, attaching enzymes to QDs in this manner occurs almost spontaneously and provides for control over enzyme orientation on the QD and the average enzyme ratio per QD, which follows a Poisson distribution and displays an upper limit dictated purely by steric fitting considerations.68 One potential issue here is that many enzymes are tetrameric as self-assembled from monomeric units and will thus display multiple (His)6 at their multiple termini; this, in turn, will crosslink the QDs and enzymes into clusters. As discussed below, this can actually be very desirable when creating channeled multienzyme cascades with QDs.
Methods for quantifying enzyme kinetics and changes thereof
For appropriate context towards understanding the enhancement phenomenon, it is helpful to first consider enzyme activity within the formalism of the simplest-generalized version of the Michaelis–Menten (MM) model of enzymatic activity as given in eqn (1):69 | (1) |
 | (2) |
A second order rate constant can also be derived from the ratio of kcat/KM; this is sometimes referred to as the specificity constant, and is also used as a metric for describing enzyme efficiency.69 The data shown in Fig. 2E and F represents a sample of how enzyme kinetic data is commonly plotted. Obviously, more complex enzymatic mechanisms will necessitate far more complex models than this.
 | ||
| Fig. 2 Enhanced maltase activity when attached to semiconductor quantum dots (QDs) or nanoplatelets (NPLs). (A) Native maltase catalytic reaction and its activity on the (B) colorimetric 4-nitrophenyl α-D-glucopyranoside substrate, which is used to monitor activity in the assays shown. At scale schematic depiction of maltase as assembled to (C) ∼4 nM diameter 520 nm emitting CdSe/CdS/ZnS core/shell/shell QDs224 and (D) ca. 19 × 17 × 2.6 nm (LWH) 585 nm emitting CdSe/ZnS core/shell NPL materials. Both the QDs and the NPLs were surface functionalized with the zwitterionic CL4 ligand (see Fig. 3).149 Protein structure coordinates obtained from the protein structure of human maltase-glucoamylase PDB # 3CTT, originally derived from ref. 225. (E) Enzymatic activity progress curves for a constant concentration of 25 picomoles of maltase as assembled at the indicated increasing ratios to 4 nm diameter QDs in the presence of 4000 μM colorimetric substrate. (F) Initial rate of 25 picomoles of maltase enzyme as assembled to the indicated increasing ratios of the NPL materials versus increasing concentrations of the colorimetric substrate. Maltase kcat apparently increased ∼50× and then ∼125× when displayed on the QDs and NPLs, respectively, as indicated by the arrows. Maltase concentration held constant while QD/NPL concentration varied to achieve the indicated ratios. Data in panels E and F drawn from ref. 2. | ||
Some additional methods for quantifying enhancement in the observed activity of an enzyme include monitoring the overall amount of product formed under a given set of conditions and specific activity. Specific activity provides a metric for the activity of an enzyme per milligram of total protein and is usually given in units of μmol min−1 mg−1. In its classical usage, specific activity provides a measurement or indirect reflection of enzyme purity in the mixture. Important to note is that in the current context where enzymes are displayed on NPs, a change in specific activity between free enzymes or as attached to NPs does not necessarily mean that it is now purer when attached to the NP in the strict sense of the definition, rather that it has become more active.
Compendium of enzymatic enhancement examples
In this section, we provide a somewhat detailed, but certainly not exhaustive listing, of examples where enzyme activity has been enhanced when attached to some type of NP material. To qualify, the NP material has to have at least one of its dimensions within a size ≤100 nm. We further separate the data by NP material types including semiconductor QDs and related nanoplatelet (NPL) materials in Table 1, AuNPs in Table 2, metallic NPs in Table 3, other NP materials in Table 4, and DNA-based NPs in Table 5. The activity itself also has to have some reported quantifiable metric of enhancement. Some pertinent and/or relevant details are also provided on the NP surface and bioconjugation chemistries used for each example along with how the enzyme activity was assayed.| Material (size-diameter) | Enzyme | NP-attachment chemistry | Reaction | Resultsa | Ref. |
|---|---|---|---|---|---|
| Notes: MAC = metal affinity coordination; NPLs = nanoplatelets (585 nm emitting CdSe/ZnS core/shell four monolayers CdSe); QDs = quantum dots; TMB = tetramethylbenzidine; LWH = length × width × height.a Reported relative to the freely diffusing enzyme (without immobilization). | |||||
| CdSe/ZnS core–shell 520 nm QDs (∼4.0 nm) | Horseradish peroxidase (HRP) | HRP-(His)6 – QD ZnS shell metal affinity coordination (MAC) | Oxidation of TMB | >2× kcat | 94 |
| CdSe/ZnS core–shell 525 nm QDs (∼4.2 nm) | Phosphotriesterase (PTE) | PTE-(His)6 – QD ZnS shell MAC | Paraoxon to p-nitrophenol | ∼4× initial rate; ∼2× kcat/KM | 72 |
| Benzaldehyde lyase (Bal) | Bal-(His)6 – QD ZnS shell MAC | Benzaldehyde + acetaldehyde to (R)-2-hydroxy-1-phenylpropan-1-one | ∼30% increase in kcat > 3× kcat/KM | 95 | |
| CdSe/ZnS core shell (∼4/9 nm) | Engineered PTE trimer | PTE-(His)6 – QD ZnS shell MAC | Paraoxon to p-nitrophenol | ∼2× kcat; ∼2× kcat/KM | 96 |
| CdSe/CdS/ZnS core/shell/shell 520 nm QDs (∼4.0 nm) | Lactate dehydrogenase (LDH) | LDH-(His)6 – QD ZnS shell MAC | Pyruvate to lactate | >50× in turnover on QD/∼10× in kcat/KM on QD and NPL | 97, 2 |
| NPLs (LWH of ∼19.2 × 17.3 × 2.6 nm) | Maltase (Mlt) | Mlt-(His)6 – QD/NPL ZnS MAC | Maltose to glucose | ∼50× kcat on QD | |
| ∼125× kcat on NPL | |||||
| Glucokinase (Glk) | Glk-(His)6 – QD/NPL ZnS MAC | Glucose to glucose-6-phosphate | ∼12× kcat on QD | ||
| ∼7× kcat on NPL | |||||
| Phosphofructokinase (PFK) | PFK-(His)6 – QD/NPL ZnS MAC | Fructose-6-phosphate to fructose-1,6-bisphosphate | ∼4× kcat on NPL | ||
| Glyceraldehyde-3-phos-phate dehydrogenase(GPD) | GPD-(His)6 – QD/NPL ZnS MAC | Glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate | ∼5× kcat on QD | ||
| ∼3× kcat on NPL | |||||
| Phosphoglycerate kinase (PGK) | PGK-(His)6 – QD/NPL ZnS MAC | 1,3-Bisphosphoglycerate to 3-phosphoglycerate | ∼3× kcat on QD | ||
| 2.3× kcat on NPL | |||||
| Phosphoglycerate mutase (PGM) | PGM-(His)6 – QD/NPL ZnS MAC | 3-Phosphoglycerate to 2-phosphoglycerate | ∼6× kcat on QD and | ||
| ∼4× kcat on NPL | |||||
| CdSe/ZnS core–shell 523 nm QDs (∼4.3 nm) | Glucose dehydrogenase (GDH) | GDH-(His)6 – QD ZnS shell MAC | D-Glucose to D-glucono-1,5-lactone | ∼5× kcat/KM | 98 |
| CdSe/ZnS core–shell 525 nm QDs (∼4.2 nm) | Beta-galactosidase (β-gal) | β-Gal-(His)6 – QD ZnS shell MAC | 2-Nitrophenyl β-D-galactopyranoside to 2-nitrophenol and galactose | ∼4× kcat | 70 |
| Alkaline phosphatase (AlkP) | AlkP-(His)6 – QD ZnS shell MAC | Conversion of 4-methylumbelliferyl phosphate to 4-methylumbelliferyl | 25% increase in kcat | 99 | |
| CdSe/ZnS core–shell 630 nm QDs (size not reported) | Lysozyme (Lyz) | Lyz-(His)6 – QD ZnS shell MAC | Hydrolysis of glycosidic bonds | ∼2–2.5× activity | 100 |
| CdSe/ZnS core–shell 545 and 605 nm QDs (∼5 and 10 nm) | Endogluconase and exogluconase | Enzyme (His)6 – QD ZnS shell MAC | Cellulose digestion | 4.3–4.9× initial rate | 101 |
| CdSe QDs (20 nm) | Cellulase catalytic domain | Biotinylated-enzyme to streptavidin QDs | Cellulose digestion | >7× sugar yield | 102 |
| Nanoparticle material (size-diameter) | Enzyme | NP-Attachment chemistry | Reaction | Resultsa | Ref. |
|---|---|---|---|---|---|
| Notes: ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt; BApNA = N-benzoyl-DL-arginine-4-nitroanilide hydrochloride; EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride; NHS = N-hydroxysuccinimide; NPs = nanoparticles; NTA = nitrilotriacetic acid. PTE = Phosphotriesterase.a Reported relative to the freely diffusing enzyme (without immobilization). | |||||
| AuNPs (20 nm) | Glucose oxidase (GOx) | Thiol bonding at the cysteine-rich side of GOx | Glucose conversion to gluconic acid and H2O2 | 3× specific activity | 103 |
| AuNPs (3 nm) | Laccase | Hybrid immobilization via adsorption or amide coordination | Oxidation of ABTS | 2× kcat/KM | 104 |
| AuNPs (50 nm) | Nitroreductase (NTR) | Amino groups of Cys-tagged NTR | Reduction of CB1954 prodrug | 22% decrease in KM | 105 |
| 112% increase in kcat | |||||
| 512% increase in kcat/KM | |||||
| AuNPs (30 nm) | Pepsin | Enzyme immobilization via adsorption | Hydrolysis of peptide bonds | 79% decrease KM | 106 |
| 118% increase in VMax | |||||
| 110% increase in kcat/KM | |||||
| AuNPs of varying size (1.5–100 nm) | PTE | PTE-(His)6 conjugation to Ni2+-NTA AuNP surface | Conversion of paraoxon to p-nitrophenol | 3 to 10× kcat | 107 |
| 2× kcat/KM | |||||
| HS-PEG7-COOH functionalized AuNPs (10–30 nm) | Pepsin | EDC cross-linking | Hydrolysis of peptide bonds | 73% decrease in KM and 107% increase in kcat/KM | 108 |
| Polyelectrolyte functionalized AuNPs (36.4 nm) | Papain | Amide coupling between amino groups on papain and the COOH AuNPs (EDC/NHS) | Conversion of BApNA to p-nitroaniline | 59% decrease in KM | 71 |
| 4211% increase in kcat | |||||
| 6667% increase in kcat/KM | |||||
| Cysteamine surface functionalized AuNPs (25 nm) | Lipase | Carboxyl group of enzyme and AuNPs EDC/NHS cross-linking | Conversion of p-nitrophenyl palmitate to p-nitrophenol | 41% decrease in KM | 109 |
| 181% increase in kcat/KM | |||||
| Citrate modified AuNPs of varying size (5–20 nm) | PTE | PTE-(His)6 conjugation to Ni2+-NTA AuNP surface | Conversion of paraoxon to p-nitrophenol | ∼17× Vmax | 110 |
| Citrate modified AuNPs (47 nm) | Rhamnulose-1-phosphate aldolase | Adsorption | Aldol addition | ∼4× initial rate | 111 |
| Au nanostars (37 and 83 nm Feret diameter) | Amylase | (His)6 conjugation to Ni2+-NTA AuNP surface | 4-Nitrophenol colorimetric substrate | 76% increase in kcat | 77 |
| Glucokinase | NADH via coupled enzyme assay | 156% increase in kcat | |||
| Material (size-diameter) | Enzyme | NP-Attachment chemistry | Reaction | Resultsa | Ref. |
|---|---|---|---|---|---|
| Notes: ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt; APTMS = (3-aminopropyl)-trimethoxysilane; APTES = 3-aminopropyltriethoxysilane; 2,6-DMP = 2,6-dimethoxy-phenol; EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride; CLEA = cross-linked enzyme aggregate; NPs = nanoparticles NTA = nitrilotriacetic acid.a Reported relative to the freely diffusing enzyme (without immobilization).b Full 3-D size, not NP size. | |||||
| CLEA – amino-functionalized iron oxide NPs (50–100 nm) | Pectinase (containing xylanases and cellulases) | Cross-linking via glutaraldehyde addition | Hydrolysis of wheat straw | 1.8× half-life | 112 |
| Iron oxide with polyacrylic acid – gallic acid (13.1 nm) | Laccase | EDC cross-linking | Oxidation of ABTS | 4.4-Fold increase in initial rate | 87 |
| Cu(OH)2 nanocages (170 nm) | Laccase | Enzyme amines to APTMS-functionalized nanocage surface | Oxidation of 2,6-DMP or ABTS | 14- to 18× kcat | 89 |
| 8- to 14× kcat/KM | |||||
| Nano-sized Fe3O4 modified with APTES (12 nm) | Lipase | Enzyme immobilization via the aldehyde activated magnetite | Hydrolysis of either p-nitrophenol butyrate or p-nitrophenol palmitate | 9.2× p-nitrophenol butyrate 32× p-nitrophenol palmitate | 113 |
| Ni-NTA/H2N-SiO2@Fe3O4 NPs (50 nm) | Bienzyme hydroxylase monooxygenase (HpaBC) | HpaB-(His)6 and HpaB-(His)6 to Ni-NTA | ortho-Hydroxylation of 4-hydroxyphenylacetate | 2.6× increase in product formation | 114 |
| Cu3(PO4)2 nanoflowers | Laccase | Immobilization via amide groups of enzyme and Cu2+ | Oxidation of epinephrine or ABTS | 1.5× and 3.6× initial rate/6.5× product | 115–117 |
| (3 μm/2 μm/5–8 μm)b | |||||
| (15–20 μm)b | HRP | Oxidation of o-phenylenediamine | 5.1× initial rate | 118 | |
| (20 μm)b | Lipase | Conversion of 4-nitrophenyl acetate | 4.6× Vmax | 119 | |
| (Size not reported) | Carbonic anhydrase | Conversion of paraoxon | 2.9× product formation | 120 | |
| (20–40 nm) | Lipase | Lauric acid 1-dodecanol esterification | 51× specific activity | 84 | |
| (100 nm) | Hydroxylase | Conversion of 2,4-dichlorophenol to 3,5-dichlorocatechol | 1.6× specific activity | 121 | |
| Cu2O NP (<350 nm) | Laccase | Immobilization via amide groups of enzyme and Cu2+ | Oxidation of syringaldazine | 4.0× increase in product formation | 122 |
| Co3(PO4)2·8H2O (1 μm × 450 nm) | Organophosphorus hydrolase | Co2+ binding to allosteric site of enzyme | Paraoxon conversion to p-nitrophenol | 3× increase in product formation | 123 |
| Co3(PO4)2 nanoflower (7 μm × 200 nm) | D-Psicose 3-epimerase | Enzyme binding to Co2+ | D-Fructose conversion to D-psicose | 7.2× specific activity | 124 |
| Cu2O nanowire mesocrystal (120 nm pore size, dia ∼90 nm) | Laccase | Enzyme binding to Cu2+ | Oxidation of syringaldazine | 10× specific activity | 88 |
| Fe3O4 nanoring (70 nm outer diameter height 50 nm) | Beta-galactosidase | Immobilization via enzyme binding to Fe2+ | o-Nitrophenyl-β-galactoside conversion to o-nitrophenol | 1.8× increase in product formation | 125 |
| Cu3(PO4)2·3H2O (30 nm) | Urease | Enzyme binding to Cu2+ | Ureas conversion of urea to CO2 NH3 | 40× specific activity | 126 |
| Membrane/Cu nanoflower (2 μm pore size, 4 μm diameter) | Laccase | Enzyme amide groups and Cu2+ | Oxidative coupling of phenol to 4-aminopyrine to form antipyrine dye | 2.0-fold increase in product formation | 127 |
| Material (size-diameter) | Enzyme | NP-Attachment chemistry | Reaction | Resultsa | Ref. |
|---|---|---|---|---|---|
| Notes: ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt; AOT = sodium bis-2-(ethylhexyl) sulfosuccinate; APTES = 3-aminopropyltriethoxysilane; BSA = bovine serum albumin; CLEA = cross-linked enzyme aggregate; EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride; MWCNT = multiwalled carbon nanotubes; NHS = N-hydroxysuccinimide; NPs = nanoparticles; PAA/PPEGA = block copolymer [poly(acrylic acid)/poly(poly(ethylene glycol) acrylate)]; PTE = phosphotriesterase; PVPP = polyvinyl pyrrolidone; SWCNT = single-walled carbon nanotube. TMB = tetramethylbenzidine.a Reported relative to the freely diffusing enzyme (without immobilization). | |||||
| Graphene oxide (GO) – MgNP (GO size 0.5–5 μm) | α-Amylase (amy) | Amy-Mg affinity GO cross-linking via glutaraldehyde | Cleavage of 1,4-α-D-glycosidic bond in linear amylose and amylopectin | 2.3×/4.3× (8 °C/90 °C) Vmax; 2.5× product formation (18 °C) | 128 |
| GO-GQDs (2.3 nm × 0.9 nm) | HRP | Covalent attachment | Oxidation of TMB | 1.9× KM | 129 |
| Magnetic Fe3O4 NP CLEAs (30–50 nm) | Lipase | AOT-activated CLEA lipase immobilized to NP with APTES | Transesterification of 2-phenylethanol and vinyl acetate | 20× increase in product formation | 130 |
| MWCNTs (size not reported) | Lipase | EDC/NHS cross-linking | Hydrolysis of p-nitrophenol palmitate | 10× specific activity | 131 |
| Polydopamine coated AgNP (20 nm) | Trypsin | Enzyme immobilization via catechol groups on AgNPs | Hydrolysis of proteins | 1.6× hydrolysis casein, 8–17% increase ovalbumin and BSA | 132 |
| Polycaprolactone (PCL) nanofibers (324 nm in diameter) | Lipase | Immobilization via PCL backbone | Hydrolysis/transesterification of p-nitrophenyl palmitate | 14× specific activity | 133 |
| Carbon dot (3 nm) | Laccase | Carbon dot phosphate backbone | Oxidation of ABTS | 1.9× specific activity | 134 |
| Cu2+-adsorbed pyrene-PAA/PPEGA (50 nm) | Laccase | Enzyme amide groups and Cu2+ | Oxidation of ABTS | 4.5× specific activity | 135 |
| 3× kcat | |||||
| SWCNT (diameter: 1–2 nm, length: 5–30 μm) | Laccase | Enzyme adsorption | Oxygen reduction | 6× increase in electrocatalytic current | 136 |
| Mesoporous silica NP (6 nm) | Laccase | Enzyme adsorption | Oxidation of ABTS | 1.2× half life | 137 |
| Carbon nanotube (diameter: 20–40 nm; length: 5–15 mm) | Lipase | Enzyme adsorption | Lauric acid 1-dodecanol esterification | 68× specific activity | 84 |
| ZIF-8 MOF (300 nm with pore sizes of 5–20 nm) | Cytochrome c | PVPP-modified enzyme assembly to Zn2+ of MOF | Amplex red conversion resorufin | 10× specific activity | 138 |
| Pluronic polymer (30 nm) | Lipase | Amphiphilic grafting | Hydrolysis of 4-nitrophenyl butyrate | 67× specific activity | 90 |
| Cytochrome c | Conversion of 2,2′-azinobis-(2-ethylbenzthiazoline-6-sulfonate) | 670× specific activity | |||
| Fe3O4 NP hydrogel (size not reported) | L-2-HADST dehalogenase | Conjugation to acrylic acid via the protein's lysines | Dehalogenation of L-2-haloalkanoates to D-2-hydroxyalkanoates | 2.0× kcat/KM | 139 |
| Siliceous mesocellular foam (mesoporous dia. ∼36 nm) | Lipase | Enzyme adsorption | Tributyrin conversion to butyric acid | 25× specific activity | 140 |
| DNA nanostructure (size or structure) | Enzyme | NP-Attachment chemistry | Reaction | Resultsa | Ref. |
|---|---|---|---|---|---|
| Notes: ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt; bp = base pair; NTA = nitrilotriacetic acid; Sulfo-EMCS N-ε-maleimidocaproyl-oxysulfosuccinimide ester-.a Reported relative to the freely diffusing enzyme (without immobilization). | |||||
| DNA triangle (120 nm length per side) | HRP | Covalent attachment to free lysine amine side chains | Oxidation of a series of phenol derivatives | >3× specific activity | 141 |
| DNA origami triangle (120 nm length per side) | β-Amylase | NTA-modified 22 bp-oligomer guide strand bound to enzyme's His6 in presence of Cu(II) ions | Hydrolysis of every second α-1,4 glycosidic linkage in starch | 4× kcat | 44 |
| 3× kcat/KM | |||||
| Maltase | Hydrolysis central α-1,4 glycosidic maltose bond yielding 2 glucose | >35× kcat | |||
| 2× kcat/KM | |||||
| Glucokinase | Phosphorylation of glucose to glucose-6-phosphate | >3× kcat | |||
| 11.5× kcat/KM | |||||
| 3D octahedral DNA scaffold (12 six-double helix bundles and 120 staples) | Glucose oxidase | Sulfo-EMCS treated enzymes assembled directly to activated oligonucleotides | Conversion of glucose to gluconic acid | 3.5× kcat | 142 |
| DNA nanocage (∼54 nm × 27 nm × 26 nm; inner cavity: 20 nm × 20 nm × 17 nm) | Glucose oxidase | Oligonucleotide-conjugated enzyme annealed directly to complementary DNA nanocage | Conversion of glucose to gluconic acid | 5.4× kcat | 143 |
| HRP | Oxidation of ABTS | 9.5× kcat | |||
| Rectangle (70 × 100 nm2) on microbeads | (R)-Selective alcohol dehydrogenase | Halotag based oligonucleotide binder protein. | Convert 5-nitrononae-2,8-dione 1 to (R)-syn/anti-hydroxyketones (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) :40) |
2.2× kcat | 144 |
| 2D DNA triangle 25 nm | HRP | Covalent chemistry with bifunctional linker | Various substrates including p-aminophenol | 100–300× activity | 141 |
| Square pyramidal DNA scaffold ∼35–45 nm | Xylose reductase | Leucine zipper/Halo-tag enzyme fused to DNA binding proteins | NADH cofactor production or consumption | 3–4× turnover frequency | 145 |
| Xylitol dehydrogenase | |||||
| 48.5 kbp lambda phage DNA | β-Lactamase | Streptavidin-biotin chemistry | Chromogenic nitrocefin substrate | ∼2× kcat 1.7× kcat/KM | 146 |
| 3.8× activity | |||||
In Table 1, we list representative results collected for different individual enzymes displayed on the surface of semiconductor QDs. Notably, all the examples mentioned using QDs rely exclusively on the MAC of the enzyme's pendant (His)6-motifs to the QD's ZnS shell for NP bioconjugation. The pendant coordination to one of the enzyme's end termini suggests that minimal to no perturbations were being contributed to these systems from heterogeneous enzyme assembly. The reported range of different QD sizes, which have been used to study the effect of immobilization on the enhancement of an individual enzyme's activity, is moderately narrow, ranging from ∼4.0 up to 10 nm in QD diameter. Although QD emission is known for being size-tunable, the inherent chemistry that gives rise to this quantum confined property limits the size of the final QD material to a relatively small range in contrast to other materials such as AuNPs, see below. Almost half of the examples (7 out of 15) in Table 1 originate from ref. 2, where QD-displayed enzymes were allowed to form self-assembled nanoclusters to engage in channeled activity. In that study it was important to profile the kinetic activity of each enzyme independently when displayed on the QD to better numerically simulate and match their relative activities during cascaded assays by controlling their ratios to each other when present in these clusters. Although not listed in the current table, this same reference also has data for two other QD sizes of 9.7 and 13.4 nm diameter, where the same 7 enzymes also displayed some enhancement but not of the same magnitude as when attached to the smallest ∼4.0 nm diameter QDs.
Assembly of individual enzymes onto semiconductor QDs led to improvements in the enzyme's catalytic rate (kcat) ranging from 2–50 times greater than what was obtained with the freely diffusing enzyme. Similarly, 2–10× improvements in enzyme efficiency (kcat/KM) were also reported. Of the 14 reported enzymes in Table 1, half have also been immobilized onto ∼585 nm emitting CdSe/ZnS core/shell NPLs with an average L × W × H of ∼19.2 × 17.3 × 2.6 nm. Utilizing these NPLs with their larger size and greater dimensionality instead of semiconductor QDs led to 2–125× improvements in kcat and ∼10× improvements in kcat/KM. It is worth highlighting that of all the different enzymes that have been immobilized onto either semiconductor QDs or NPLs, maltase (Mlt) showed the greatest improvement in activity when immobilized relative to the freely diffusing enzyme. As shown in Fig. 2, maltase was being studied for the conversion of maltose to glucose as part of a 10 enzyme joint saccharification and glycolytic cascade and displayed a remarkable 50× and 125× improvement in kcat when immobilized onto either QDs or NPLs, respectively.2 This result directly speaks to the improvements in kinetic activity that can be potentially accessed by this phenomenon.2 Studies with beta-galactosidase (β-gal) attachment to QDs revealed further and somewhat counterintuitive findings about the enhancement mechanism.70 Although expressed as a monomer (∼116.3 kDa) from a single gene during E. coli production, the functional form of this enzyme is an obligate tetramer of ca. 465 kDa displaying 4×(His)6, one at each of the four monomers' termini, and has a length that is slightly greater than 10 nm. This large size means that, in practice, this enzyme is not displayed at a QD surface but rather displays up to 4 QDs around its surface. Here, selective display of QDs around β-gal's surface resulted in a further 3× enhancement in kcat. Table 1, as well as the subsequent tables, also reveals a wide range of activity improvements reported across a variety of different enzymes from different sources and manifesting a variety of different catalytic processes suggesting that such enhancement is not limited to only certain enzymes or catalytic mechanisms.
Table 2 outlines results reported for the assembly of individual enzymes onto AuNPs. Here, a wide range of differentially-sized AuNPs, ranging from around ca. 1.5 up to 100 nm in NP diameter, were utilized to study the effect of NP assembly on enzyme activity. Additionally, a variety of surface-modifications for the reported AuNP systems were explored including HS-PEG7-COOH functionalized AuNPs, polyelectrolyte functionalized AuNPs, and cysteamine functionalized AuNPs with the rest assumed to be citrate modified AuNPs. The type of enzyme attachment strategy also varies across the AuNPs possessing these different surface functionalizations including, adsorption, thiol-bonding, MAC, and covalent chemistry. Assembly of individual enzymes onto AuNPs led to improvements in the enzyme's kcat parameter ranging from 300× improvement to a truly remarkable 4211% greater than what could be achieved with the freely diffusing enzyme. Similarly, 100–6667% improvements in the enzyme kcat/KM and 22–79% decrease in the enzyme KM were reported across the different AuNPs. That the KM values did not undergo similar changes in magnitude again suggests that some of these enhancements may arise from changes in the rate of EP release (vide infra). Notably, papain assembly onto polyelectrolyte functionalized AuNPs displayed the largest improvement in overall enzyme activity compared to the freely diffusing enzyme. For example, the protease papain was studied for the conversion of N-benzoyl-DL-arginine-4-nitroanilide hydrochloride (BApNA) to p-nitroaniline. When assembled onto polyelectrolyte functionalized AuNPs (36.4 nm in diameter), the papain enzyme displayed a 59% decrease in KM, a 4211% increase in kcat, and a 6667% increase in kcat/KM relative to the non-assembled enzyme. However, these results were attributed less to the AuNP aspect of the material and more to the charged polymer surface and high papain loading capacity achieved with the polymer chains of the polyelectrolyte surface.71 It may be that the high charge density of the polyelectrolyte surface also contributed in other ways such as providing for localized substrate sequestration. See also related discussion on DNA structures below.
The unique material chemistry available with AuNPs has also provided useful properties for these types of studies. The above-mentioned study where AuNPs ranging over almost 2 orders of magnitude in size (from 1.5 to ∼100 nm diameter) were used to ascertain the effect of NP size on PTE catalytic enhancement would probably have not been possible with other individual NP types.72 This is because the unique chemistry of AuNPs allows for this size range to be achieved, although synthesis of some of the larger sizes usually necessitates using smaller AuNP particulates as seeds for further growth. There are also a variety of other AuNP shapes synthetically available including triangles, nanostars, and nanoflowers for evaluation with enzymes.73–76 Díaz and coworkers assayed the activity of amylase, maltase, and glucokinase as assembled to both large and small gold nanostars as part of a study looking at their cascaded activity.77 Due to their shape, these NPs are characterized by their Feret diameters meaning that they were measured along a specific plane that could include the largest end-to-end distance between the stars tips. Díaz found that both amylase and glucokinase kcat increased between 76% and 156%, respectively. In contrast to what was seen with QDs above, maltase activity actually decreased when displayed on these NPs by almost 50%. It is not readily apparent why this enzyme would now decrease in activity after increasing by almost 150× when attached to NPLs above.77 This is a prime example of where differences among inherent properties such as NP size, shape, surface ligand/solubilization approach, and bioconjugation chemistry could all potentially contribute to this stark difference. We also note that considerable research has been undertaken using lasers to plasmonically heat AuNPs, which, in turn, can transduce the heat to the localized surroundings including an enzyme on its surface in a very rapid manner before the heat dissipates into the bulk.78–82 This may represent a possible tool for probing the narrow confines of the NP–enzyme interface by selectively activating or speeding up its activity in an effort to understand how enzymatic enhancement is manifest.
The data compiled in Table 3 summarizes results reported from the assembly of individual enzymes onto metallic NPs other than gold including those synthesized from iron (Fe), cobalt (Co), and copper (Cu). Unlike the AuNPs above, these NPs will exist mostly as a variety of complex oxides based upon how they were synthesized.83 The NPs in this table include a variety of NP shapes each with its own unique dimensionality including nanocages, nanowires, nanorings, and nanoflowers. They also vary widely in size ranging from 12 nm to 20 μm with the latter reflecting the overall dimensions of the materials and not their nanoscale components, which include those that are <100 nm in size. Enzyme assembly to this class of materials relies heavily on enzyme functionalization via the metal (M2+; M = Fe, Co, or Cu) center of the produced nanomaterial. Assembly of individual enzymes onto these metallic nanomaterials led to improvements in their specific activity with observed increases ranging from 1.6 up to 51× greater than what was obtained from the freely diffusing enzyme.
Similarly, a range of 2.6× up to 6.5× improvements in enzyme product formation and 1.5–5.1× increases in the enzyme initial rate were reported across the different metallic nanomaterials. Of these materials, the greatest enhancement was observed with a lipase enzyme assembled onto a Cu3(PO4)2 nanoflower (20–40 nm in size). This lipase was studied for the esterification of lauric acid 1-dodecanol and when assembled onto a Cu3(PO4)2 nanoflower demonstrated a 51× improvement in specific activity (i.e., improved activity) relative to the freely diffusing enzyme.84 Lipases are interesting enzymes, some of which are known to have hinges over their binding pockets and undergo enhancement or more correctly display a ‘turn on’ in activity at organic–inorganic interfaces and cell membranes.85,86 This is thought to help them better access substrates with partial solubility. Still not known is how this mechanism would be affected by NP display, although the unique environment around the NP is also commonly considered an interface. We also note that data on the activity of the enzyme laccase as attached across different NP materials was reported. Here, it is seen that attachment to iron oxide NPs (13.1 nm dia.) resulted in a >4-fold increase in specific activity with the same caveat given here about interpreting this metric as above.87 Similarly, 10× increases in specific activity are reported for attachment to Cu2O nanowires (90 nm) while ca. 14–18× improvements in kcat and kcat/KM are achieved for laccase display on Cu(OH)2 nanocages (170 nm).88,89 Along with some other examples of increased laccase activity on other NP materials given in Table 3, the similarity of the activity increases with their magnitude being almost all within 10-fold of each other suggests that a common enhancement mechanism is responsible for what is observed with this particular enzyme.
Table 4 summarizes results reported for the assembly of several different enzymes onto other NP and NP-like materials that do not fall into the same scaffolding material categories as the systems presented previously. This listing comprises a variety of different materials, including metal organic frameworks (MOFs), hydrogels, polymer materials, cross-linked enzyme aggregates (CLEAs), silica-based materials, and carbon/graphene or carbon allotrope-derived materials. The NP materials vary in overall size ranging from ∼3 nm to 5 μm with the caveat that the latter have some dimensional property in a size range of <100 nm. Additionally, there is a significant amount of variation in the enzyme to NP assembly strategies utilized for these materials including covalent attachment, adsorption, grafting, and direct chemical enzyme cross-linking. The latter are again generally undertaken with heterogeneous conjugation chemistries leading to functionally-mixed and heterogeneous ensemble materials. NP–enzyme assembly in this case led to improvements in the enzyme's product formation rates ranging from 2.5 up to 20× and increases in specific activity ranging from 1.9 up to 670× greater than what could be achieved with the freely diffusing enzyme. Of these materials, the greatest enhancement was observed with cytochrome c as assembled onto a pluronic polymer material (30 nm in size). The pluronic polymer was composed of a central hydrophobic polypropylene oxide block attached to two hydrophilic poly(ethylene) oxide side blocks. This enzyme was assayed with the conversion of 2,2′-azinobis-(2-ethylbenzthiazoline-6-sulfonate) and when conjugated to the pluronic polymer material demonstrated an impressive 670× improvement in specific activity relative to the freely diffusing enzyme.90 As above, the increase in specific activity probably does not arise from an improvement in purity per the strict definition, but rather should be interpreted as an increase in apparent activity when attached to the NP material.
It should be noted that CLEAs are a popular approach to exploiting the activity of multiple linked enzymes in the context of a multienzyme cascade within both research and industrial applications.91,92 They are also many-times formed using heterogeneous covalent crosslinking chemistries with reactive agents such as glutaraldehyde.93 This can and will lead to an exacerbation of the aforementioned covalent chemistry issues that affect the catalytic activity of a given enzyme across the resulting ensemble CLEA material. More pertinently, the CLEAs that are formed in this way are typically evaluated in the context of the intended final application, i.e. cascaded activity by a linked multienzyme assembly, along with product formation and not in terms of changes in individual enzyme rates. This makes collecting requisite kinetic data for each enzyme's individual performance in this configuration rather challenging.
Lastly, Table 5 summarizes results reported for the assembly of individual enzymes onto different DNA-based nanostructures including DNA origami triangles, octahedra, rectangles, and nanocages. These materials vary minimally in size, relative to some of the other materials, ranging from ∼20–120 nm in length for any side of the DNA nanostructure. Given that each of these nanostructures is assembled from multiple smaller DNA strands that are hybridized together into the final architecture, the definition for having at least one dimension <100 nm is not as rigidly constraining. The enzyme assembly strategies utilized for these DNA-based nanostructures include direct annealing of enzyme-labeled DNA to complementary DNA, (His)6-MAC, direct attachment by DNA binding proteins when fused to the enzyme, and other covalent attachment methods. Aside from horse radish peroxidase (HRP), which had much higher enhancement activity, assembly of individual enzymes onto DNA nanostructures led to improvements in enzyme kcat ranging from 3–35× greater than what could be achieved with the freely diffusing enzyme and 2–11.5× improvements in the enzyme kcat/KM. Of these materials, the greatest enhancement was observed with the maltase enzyme assembled onto a DNA origami triangle (120 nm in length per side). This maltase enzyme was implemented for the hydrolysis of a central α-1,4 glycosidic bond in maltose and related maltosidic sugars derived from amylose as part of a saccharification pathway yielding two glucose molecules when assembled onto the DNA triangle. In this context, maltase demonstrated a >35× improvement in kcat relative to the freely diffusing enzyme.5 Notably, this same maltase enzyme showed the greatest overall enhancement in activity relative to other reported systems for both QD and NPL-based scaffolding materials (Table 1).
Towards elucidating the mechanism of enzyme enhancement by nanoparticles
As originally suggested by Ansari and others, enzymes commonly display enhanced activity when they are displayed on a NP surface.27–29,147 Reports of this phenomenon have been steadily accumulating over the last 10-plus years and indeed many of the examples in this time period are listed in the tables included and further expounded upon in the corresponding discussion. A prototypical example of how such enhancement would manifest is provided in Fig. 2. Here, the enzyme maltase originating from Schizosaccharomyces pombe as expressed in laboratory E. coli was attached to both semiconductor QDs and nanoplatelets (NPLs) by metal affinity via its terminal (His)6 motif. NPLs are a new form of quasi 2-dimensional materials synthesized from the same constituents as QDs but having far larger length × width dimensions with a very shallow height.148 When the 74.5 kDa monomeric maltase protein was attached to ∼4 nm diameter QDs at a ratio of <1 per QD, kcat increased ca. 50×. When assembled to 19 × 17 × 2.6 nm (LWH) 585 nm emitting CdSe/ZnS core/shell NPL materials at the same ratio, maltase manifests a ∼125-fold increase in kcat.2Fig. 2B and C show at scale schematics of maltase as attached to these two different nanomaterials. When considered at face value, these depictions provide strong support for the unique NP environment serving to enhance maltase activity as the NPL clearly has far more of such an environment surrounding it and the relatively smaller maltase enzyme. Steric considerations alone suggest that a range of 4–8 and 21–31 maltase could fit around these two differentially shaped/sized QD and NPL materials, respectively.2Studies utilizing the enzyme phosphotriesterase (PTE), which originates from Brevundimonas diminuta, as assembled to QDs and AuNPs have yielded perhaps the most insight into the underlying mechanisms that give rise to NP-enzymatic enhancement.72,107 This 341 residue protein forms an obligate dimer and was expressed with a C-terminal (His)6 motif for assembly to the ZnS surface of QDs again by MAC. Since the C-termini in the dimer are in very close proximity to each other, it is assumed that this enzyme does not crosslink QDs when assembled to them as they both should bind to the same NP. Fig. 3A schematically depicts the components of this system including the enzymatic reaction and the structure of the paraoxon substrate, zwitterionic DHLA-CL4 ligand used to make the QDs colloidally stable, and a triethyl phosphate competitive inhibitor.72,149Fig. 3A compares the initial rates of PTE activity when consuming paraoxon as the PTE concentration incrementally increases and it remains freely diffusing or as assembled to a fixed concentration of 625 nm emitting CdSe/ZnS core/shell QDs with a corresponding increase in ratios per QD. PTE kcat more than doubled from ∼45 s−1 to >110 s−1 when displayed on the QDs at ratios of 6–8 per QD. The upper assembly limit of PTE to these 9.2 ± 0.8 nm diameter QDs was estimated at 28 confirming that the ratios utilized in the assays did not approach a steric limit with all enzymes present assumed to be attached to the QDs. Interestingly, PTE kcat increased by 4-fold when displayed on smaller 525 nm emitting QDs (diameter ∼4.2 ± 0.5 nm). PTE KM was concomitantly decreased (meaning a lowering of affinity with an increased value of KM) as kcat increased in these examples. This is not unexpected because kcat and KM are usually mechanistically-linked with one changing in the converse direction of the other unless the enzyme undergoes very complex changes in its mechanism of activity.69 One of the utilities or potential benefits of such enzymatic enhancement that still remains mostly unexplored is highlighted in Fig. 3C where it was revealed that PTE display on QDs allows it to remain far more active with a much higher concentration of triethyl phosphate competitive inhibitor present than the free enzyme. Indeed the Ki value tripled from ca. 8.8 up to 24.2 mM when the enzyme was attached to the surface of the QDs.
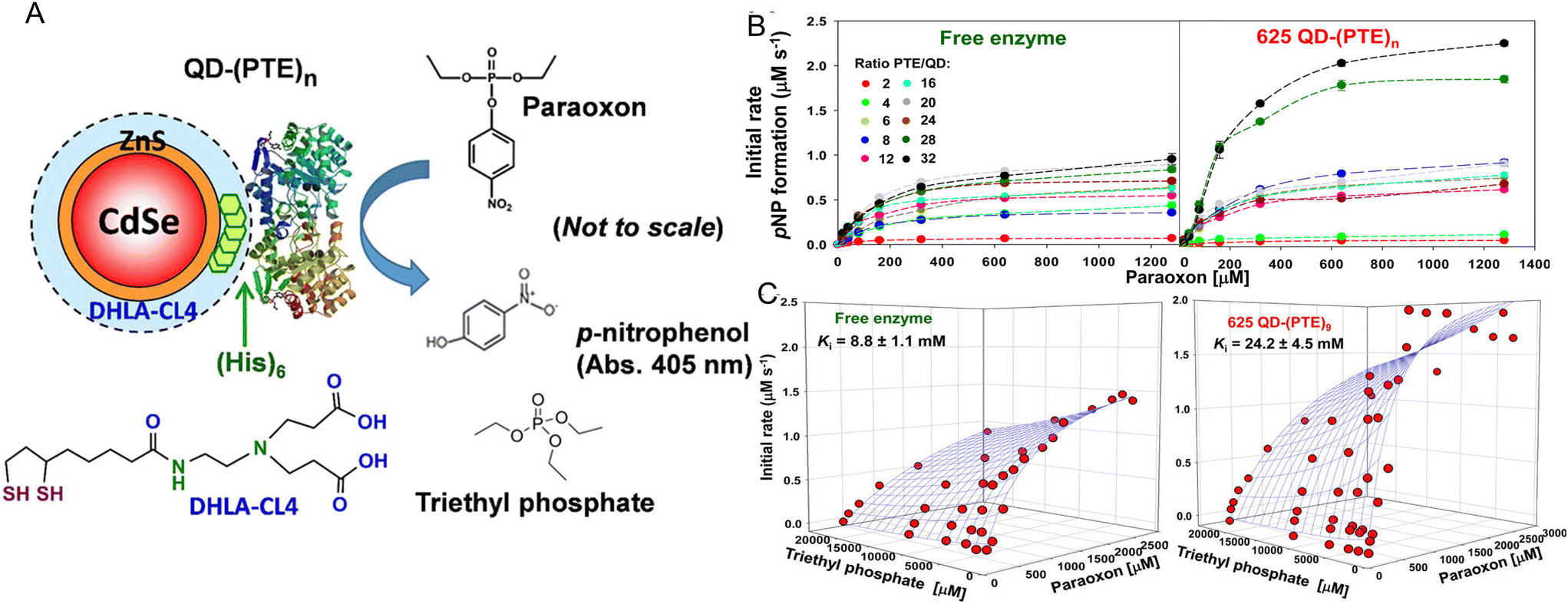 | ||
| Fig. 3 QD phosphotriesterase bioconjugate and paraoxon hydrolysis. (A) Schematic of a CdSe/ZnS core/shell QD surface-functionalized with the DHLA-CL4 ligand.149 PTE is ratiometrically self-assembled to the QD surface by its terminal hexahistidine (His)6 sequence. The average number or valency of PTE per QD is controlled through the molar stoichiometry added during assembly and the conjugates are directly utilized without subsequent purification. PTE hydrolysis of paraoxon substrate to p-nitrophenol product, which absorbs at 405 nm, is used to monitor enzyme activity. Structure of the PTE competitive inhibitor triethyl phosphate. Note, not to scale. (B) Initial rates of p-nitrophenol product formation for (left) free PTE enzyme and (right) 625 QD-(PTE)n bioconjugates assembled at the indicated ratios of n when exposed to an increasing concentration of paraoxon substrate. (C) 3D plots of PTE initial rates versus increasing paraoxon concentration in the presence of increasing triethyl phosphate inhibitor for (left) free enzyme and 625 QD-(PTE)9 (right); estimated Ki values are included with each. Reproduced with permission from ref. 72 Copyright 2015 American Chemical Society. | ||
Fig. 4 presents representative data providing insight into the mechanisms that underlie PTE enhancement on QDs. Fig. 4A shows results from assaying PTE both when freely diffusing and as assembled to the above 525 and 625 nm emitting QDs as the temperature is raised in the form of an Arrhenius plot.72 Thermally activated changes are seen in all cases with an activation energy of 37.9 kJ mol−1 estimated for the free enzyme. That the QD-displayed enzymes show the same slope over temperature change as the free enzyme confirms that their activation energies are the same. Thus, the enzymatic enhancement from QD attachment does not arise from a change or especially lowering of the enzyme's energy barrier, at least for PTE. The possibility of lowering an enzyme's energy of activation by NP display should not, however, be discounted. For example, Kouassi and colleagues reported that attachment of cholesterol oxidase to 10–56 nm diameter magnetic Fe3O4 NPs lowered the enzyme's activation energy by >30% from 13.6 to 9.3 kJ mol−1.150Fig. 4B shows results collected in a similar format of assaying free PTE enzyme activity versus that of when QD displayed in buffer with increasing concentration of sucrose present. Raushel previously used this same format to show that PTE experienced decreased turnover as the microviscosity of the localized environment increased with increasing sucrose presence in the buffer and concluded that this decreased activity arose from a decrease in k2, the rate of E-P release, which was postulated to be the enzyme's rate limiting step similar to that of many other enzymes.69,151,152
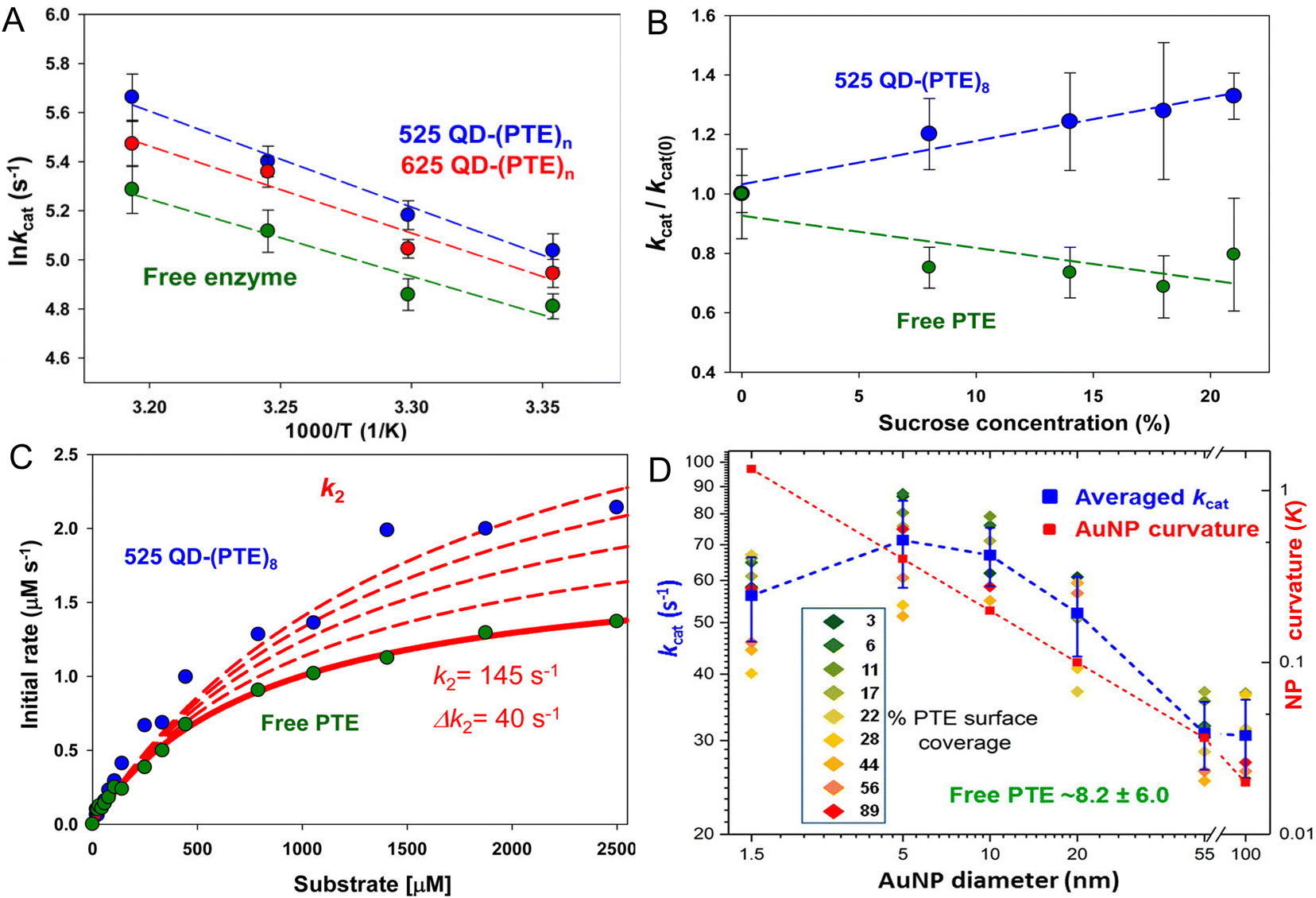 | ||
| Fig. 4 Analyses of QD phosphotriesterase bioconjugate activity. (A) Arrhenius plot of averaged lnkcat values versus inverse temperature (Kelvin) for QD–PTE assemblies and free enzyme. Slopes of the fitted data were −3.9 ± 0.4, −3.5 ± 0.5, and −3.1 ± 0.6 (average = −3.5 ± 0.4) for the 525 QD, 625 QD, and free enzyme, respectively. (B) Analysis of PTE kcat on and off QD versus increasing sucrose concentration. Plots of normalized PTE kcat values from 525 QD–(PTE)8 conjugates (blue) and equivalent amounts of free PTE versus increasing sucrose concentration. Linear fits added to each data series. (C) Plot comparing the effect of potential changes in k2 on initial PTE rates. The experimental rates of free PTE (green) and 525 QD–(PTE)8 (blue) versus substrate concentration are plotted. An initial k2 value of 145 s−1 was derived from the experimental data. The effect on initial rates of increasing the k2 value by increments of 40 s−1 is then estimated with the red dashed lines. Note the overall qualitatively good fit between free and on QD experimental formats. (D) Comparison of averaged PTE kcat values as AuNP surface coverage increases for the AuNP size series following the indicated color-coding (inset). The average value (of individual replicates) is shown in blue along with its standard deviation. The dashed blue line joins the average values together. Unassembled free PTE kcat value (8.2 ± 6.0 s−1) indicated in green. AuNP curvature (κ = 1/radius) values in log scale are plotted in red with a line of best fit. Percent PTE coverage was based on ratios of 2.5, 8, 32, 128, 969, and 3202 PTE being assembled to the 1.5-, 5-, 10-, 20-, 55-, and 100 nm-diameter NPs as the 100% coverage maxima. PTE concentration held constant while NP concentration varied to achieve the indicated ratios. Panels A–C reproduced with permission from ref. 72 Copyright 2015 American Chemical Society. Panel D reproduced with permission from ref. 107 Copyright 2019 American Chemical Society. | ||
In essence, as the viscosity increases it makes it harder for the enzyme to release its product. However, when PTE is assembled to QDs, the opposite effect is seen strongly suggesting that QD display somehow alleviates this rate-limiting step even though the localized viscosity should be just as high and the enzyme should encounter the same difficulty in releasing P. By being displayed on the QD surface, the PTE enzyme is now somehow protected from this viscosity increase – presumably by the localized environment around the QD, which is radically different from that of bulk solution. Supporting this notion, Fig. 4C shows simulations fitting the experimentally derived data for both free and QD attached PTE enzymes from Fig. 4B where the rate of k2 alone was modified while all other variables were kept constant. This excellent fit to the experimental data was not seen when attempting the same exercise while varying k1. This exercise suggests that by just increasing the product dissociation rate and effectively alleviating this rate-limiting step, one could potentially explain this effect of QD-enzyme enhancement.
Lastly, Fig. 4D draws from a study where a series of increasingly larger AuNPs were used to ascertain the effect of NP size on PTE catalytic enhancement.72 This study showed that PTE kcat increased with NP size from 1.5 nm up to the range of ∼5–10 nm AuNP diameter and then steadily decreases as the NP size increases and the NP curvature decreases although the catalytic rate at the largest 100 nm diameter size is still 3–4× that of the free PTE. PTE enhancement is also consistently greater with a lower percentage of surface coverage across all NP sizes tested. These PTE studies all utilized (His)6-MAC for NP bioconjugation, thus all the enzymes were in the same orientation. That the enhancement was highest with the lowest NP display ratio or lowest surface coverage argues that enzyme freedom of movement, even on a NP surface, is still important. Attaching this enzyme to a bulk flat surface using the same chemistry would orient it on the surface in the same manner, yet enzymatic enhancement is normally not seen in this configuration. It is believed that the reason behind this is that such bulk surfaces lead to stagnation layers near the enzyme where the local substrate is rapidly depleted.5,28,30 Cumulatively, the PTE data shows that enhancement is dependent on NP size and curvature with smaller NPs performing better, does not arise from a change to the enzyme's thermodynamics, and most likely arises from a putative change or alleviation of k2, the enzyme's rate limiting step. We postulate and some of the experiments discussed above support the notion that the latter arises in part from the unique and structured environment that is found around colloidal NPs (vide supra).
Results from displaying QDs around β-gal's surface, which provided for a further 3× enhancement in that enzyme's kcat, are also quite informative even if somewhat counterintuitive (Table 1). β-Gal is considered to be a diffusion limited enzyme meaning that it already functions at its optimal rate and should demonstrate a hard-ceiling towards increased activity arising purely from continuously increasing the concentration of the substrate. This suggested that either a rather unlikely super-diffusional rate of substrate accessibility was now contributing to activity in the presence of QDs or some other change in activity was taking place. Assuming a similar overall rate-limitation arising from k2, the rate of E–P release as discussed above, it seems likely that an alleviation of this step would again be responsible for the observed enhancement. Supporting this, the largest increase in kcat was noted for the β-gal enzyme assemblies displaying the maximum of 4 QDs rather than lower valencies as was seen above with maltase (Fig. 2). We hypothesize that the larger size of this enzyme now requires more QDs to be displayed around it to reap the full benefits of the unique NP interfacial environment. Alternatively, some evidence suggests that proton transfer during β-gal's catalysis is part of the rate-limiting step for kcat.153 This would then suggest that the unique QD environment acts to alleviate this limiting mechanism somehow by allowing for an increased rate of proton transfer.
We also note that aside from results reported with NPLs, enzyme enhancement appears to generally increase when smaller NP materials are utilized.2,70,72,95,98,99,107,110 Further support for the importance of smaller NP size on achieving enhancement comes from the work of Mukai et al.,154 who assembled the same 7–10 glycolytic enzymes (albeit cloned from mice testis) as Breger et al.,2via His6 MAC to 500 nm diameter Ni-NTA silica NPs in different cascaded combinations. In contrast to Breger's finding of kinetic enhancement with half of the enzymes in the cascade, they found that enzyme activity decreased in all cases when tethered to their significantly larger NPs. Cumulatively, this suggests that the enzyme activity should achieve its maximum enhancement for NP materials with zero dimensionality – or the no NP present control. That this also does not occur again argues for the unique NP environment contributing to this phenomenon.
Important to mention is that a frequent alternative explanation put forth is that of localized substrate sequestration around the NP–enzyme bioconjugate.155–161 This should not be discounted as it may actually be a contributor to some of the examples seen with DNA-based NP structures. Here, the high-localized concentration of DNA in conjunction with its high charge density may actually be able to build up significant localized concentrations of substrates especially if they are oppositely charged and this, in turn, can increase the rate of substrate binding and catalysis. For the specific case of horseradish peroxidase, HRP (Table 5), Lin and Wheeldon found that attachment of this enzyme to a 2-dimensional 25 nm DNA triangle led to 100–300× increases in activity depending upon what type of substrate was used in the subsequent assay.141 By performing kinetic assays using a library of different HRP chemical substrates, it was revealed that the DNA scaffolding itself could enhance the HRP activity in a manner that is akin to the so called Sabatier principle. This principle is drawn from the field of heterogeneous catalysis and posits that the binding between the substrate and the catalyst should be “just right” and should be neither too weak nor too strong. If substrates bind too weakly, they will fail to associate with the catalyst and result in no net reaction while strongly bound substrates will be conversely too slow to dissociate and prevent subsequent reactions via product inhibition by blocking of the active sites. Lin and Wheeldon concluded that here the substrates bound to the DNA scaffold and not the enzyme but the overarching trend expected for the Sabatier principle remained in place where the weakly and strongly binding substrates manifested no kinetic enhancement in their activity rates while that of the intermediately bound or ‘just right’ substrates resulted in the highest >300% enhancement in HRP activity. Presumably, this binding led to high-localized concentrations of given substrates around the DNA scaffold, which, in turn, effectively allowed for increased enzyme activity by promoting substrate binding. Hess has posited similar pH-driven changes being responsible for enhancement of HRP and other enzymes when attached to DNA scaffolding.162 There is also evidence for this effect being related to having high localized concentrations of DNA in a NP assembly. For example, Zhao reported that direct conjugation of the enzymes sarcosine oxidase and HRP to single oligonucleotide strands significantly reduced their activity.163 However, it should also be noted that the same report also confirms that inserting the DNA-tethered enzymes into a tetrahedral DNA structure significantly increased their activity to beyond that seen for the free unmodified enzyme.163
Wheeldon drew upon these results and some other similar studies that examined enzymatic rate changes in the context of localized physiochemical characteristics to suggest a set of design rules for enzyme–DNA nanostructures where enhanced catalysis is desired.156 A mixed approach of molecular simulations and kinetic analysis was able to confirm that interactions between enzyme substrates and DNA-scaffolded enzyme nanostructures could increase local substrate concentrations. The enhancement would manifest as a reduction in the apparent KM and increased enzyme efficiency. The primary determinants to accessing this effect reduce primarily to exerting control over the local physical and chemical environment to obtain the required increases in the local concentration of substrates. Although scaffolding of enzymes on DNA may allow them to manifest enhancements in their activity, the reasons that give rise to this may be amongst the most complex to elucidate mechanistically.141,144,164 As Wheeldon elegantly demonstrated above,156 the physicochemical characteristics of the enzyme, substrate, DNA scaffold itself, the buffering environment, and especially that of the buffer's pH will all interact in a complex manner that can be hard to predict. Given its strongly-charged nature and the way that this is displayed in an almost uniform manner, it would not be unexpected for DNA to universally structure its localized environment as well in a manner akin to that predicted for the colloidal NPs above.39,40 This would certainly add another confounding variable to the determinative mix. Fortunately, many of these properties can be isolated and changed independent from the others (e.g. buffer pH) and therefore may allow for parametric testing to elucidate some of the contributions and underlying roles of each. There have also been examples reported of mixed NP–DNA–enzyme systems that also yielded some form of enzymatic improvement.165–167 The material complexity here suggests that parsing out and understanding the enhancement mechanism may be incredibly complicated in this situation as localized substrate sequestration from DNA and NP interfacial structuring could both be simultaneously present and contribute to different extents.
An alternative approach to enhancement using substrate–NP attachment
It has long been known that assembling ligands around a NP could significantly improve the observed binding affinity for a receptor specific to that ligand presumably through localized density and avidity effects.168,169 Following from this belief in a conceptual manner, enzymatic enhancement at a NP interface was originally observed for a configuration opposite to that focused on above – where NPs were assembled with repeated copies of a substrate in somewhat high density and then exposed to a freely diffusing enzyme.170 Perhaps the best understanding of these systems is that of proteases acting upon NP-displayed peptidyl substrates and originates from the seminal studies of Algar.171 Using QD donors displaying different densities or valencies of dye-labeled acceptor peptidyl substrates and quantitative monitoring of reaction kinetics by changes to real-time QD-dye Förster resonance energy transfer (FRET) interactions as a prototypical system,172 Algar showed that trypsin's observed kcat/KM increased almost 5× when the peptide substrate was displayed on a QD.171 Analyzing these results in the context of several different mechanisms of enzymatic activity at a surface interface, Algar posited a scooting mode of trypsin activity where the enzyme interacts with a given QD-peptide(n) substrate due to augmented avidity, rapidly consumes all the substrate on that QD, and then diffuses away to another encounter. The Algar group has extensively characterized the activity of different proteases in this configuration and has gone on to develop several multiplexed sensor configurations including portable versions for monitoring simultaneous protease activity.173,100,174–180 They, and others, have also looked at the influence of different NP surface ligands and enzyme net charge on this type of activity.181–183 Although mechanistically different from the enhancement seen with NP display of enzymes, it is quite possible that the unique structured NP environment could also contribute to some of the kinetic improvements seen here as well. In this scenario, it may be interesting to test the role of E–P dissociation by performing experiments with increasing viscosities to see if it is a contributor.97Future outlook – systems engineering
The depth and breadth of examples listed above, which include 40 different enzymes and >10 types of NP material families, make it clear that enhancement of enzymatic activity by NP display is a real phenomenon and not just a curiosity or misinterpretation of complex results occurring between different experimental configurations. It is also clear that before engaging in designer engineering of molecular systems geared towards exploiting such enhancements, far more fundamental studies are still needed. Such studies need to be performed across many different NP materials/types and sizes along with different bioconjugation chemistries. More importantly, these studies need to be performed in a systematic and parametrized manner so that both qualitative and quantitative conclusions can be drawn from an analysis of the results. The enzyme maltase presents an interesting example in this vein. Maltase activity in the form of kcat was enhanced by ca. 50 fold when displayed on 4.0 nm dia. QDs, by almost 150× when attached to 19.2 × 17.3 × 2.6 nm NPLs, 35× on a 120 nm per side DNA origami triangle, but then decreased by almost 50% when attached to Au nanostars of 37 and 83 nm Feret dia.5,77,107 Pertinently, the enzyme utilized in these examples is the exact same monomeric version and was assembled to the materials using the same approach – namely that of MAC by its terminal (His)6 precluding confounding effects from any potential NP crosslinking or heterogeneous orientation. This suggests that differences arise not from the enzyme itself but rather from the effect of the materials in the form of the NP dimensionality along with the surface ligand chemistry used to make the NPs colloidally stable. Another important factor to consider if undertaking these types of parametric studies would be to factor in not only many different enzyme types, i.e. different catalytic mechanisms, but also enzymes that have different rate-limiting steps beyond the more common EP release. Of course, more complex enzymatic mechanisms including those that have multiple substrates and multiple reaction steps will necessitate more complex studies. It will also be important to gather data on any changes to a given enzyme's energy of activation when attached to a NP. Work with PTE showed that this was not a factor for that enzyme's attachment to QDs,72 however, alternate work did show a reduction when cholesterol oxidase was attached to Fe3O4 magnetic particles.150 Given the dearth of information currently available about this, the role of changes in enzymatic activation barriers in this context remains an open question. There is also the possibility that these changes or even increases in enzyme activation energy when attached to NP can be correlated to different types of catalytic transformations based on the enzyme type and mechanism of action.One fascinating possibility towards performing the type of parametric analysis intimated above without having to engage in exhaustive high-density mechanistic studies is that of drawing data from the literature and subjecting it to artificial intelligence (AI) or machine learning (ML) based analysis.184,185 This approach would draw from the existing body of data in the literature and compiles as many quantitative (e.g. NP size, kcat enhancement, enzyme molecular weight, etc.) and categorical (e.g. NP surface ligand type, substrate type, bioconjugation chemistry used, NP component materials, etc.) descriptors or attributes as possible for subsequent analysis. Moreover, negative data in the form of decreases to enzyme activity would be extremely useful in this context and do not have to remain in the background as so-called ‘dark data.’186 Indeed, the latter type of data may be especially useful as it can allow for parsing of factors that correlate negatively with enhancement and not just those that contribute positively to give a more balanced analysis and deeper insight into the underlying mechanisms. This type of AI-based approach has so far proven to be quite useful for analyzing NP toxicity across hundreds of different material preparations and cell-lines along with design of new electrocatalytic NP materials.187–189 An important caveat with this type of approach is that to be really useful it needs extremely large data sets that have huge numbers of examples and that are very well curated in terms of attribute type and number.190 This suggests that the above compendium is not anywhere near meeting a critical baseline requirement for such an analysis.
Turning towards incorporating NP-enzyme enhancement within engineered molecular systems, several applications currently in use can potentially benefit from this phenomenon in the near term. For example, many enzymatic assays in both research and industrial use require key cofactors for use as electron donors or acceptors in a given reaction.191–193 The vast majority of these cofactors, including especially the redox-active nicotinamide adenine dinucleotide cofactor(s) NAD(P)+/NAD(P)H, for example, are exceedingly expensive to purchase directly and, in many cases, become the most expensive reagent utilized in these assays. Moreover, buildup of the reduced or oxidized form of the cofactor during the reaction can also poison it. This has led towards sustained efforts to recycle these cofactors in a given reaction using recycling enzymes that are driven by some far cheaper substrates such as glucose towards improving economic viability.191–194 In a recent report, Breger and co. showed that glucose dehydrogenase driven reduction of NAD+ → NADH (and similarly NAD(P)+) could be enhanced by up to 10× simply by displaying it on QDs.98 This could certainly increase turnover in these reactions along with even further reducing costs by allowing for even less substrate to be used. Following from this example, many diagnostic assays are based on some form of enzymatic readout such as, for example, the ubiquitous enzyme-linked immunosorbent assay (ELISA) and reverse dot-blot hybridization assays.195–197 Increasing the apparent catalytic activity of the enzymes utilized in such formats may represent another cost-effective and powerful way to decrease their reaction time along with the amount of enzyme required. More generally, increasing the activity of enzymes used commercially in all manner of stand-alone industrial applications may help increase their cost-effectiveness along with decreasing the requisite reaction times.3 This would be especially beneficial for enzymes sourced from complex eukaryotes, many of which display low endogenous catalytic rates when applied extracellularly in non-native reaction envirnoments.198,199
The potential application where we believe that NP-enzyme enhancement may play an outsized role is within designer minimalistic cell-free multienzyme cascaded reactions.1–6,200 Rather than engineering and maintaining a cellular system to produce a desired enzymatic product, as is the current major focus in the growing field of synthetic biology, minimizing the required reactions to where all that is present are the requisite enzymes, substrates, and cofactors can allow for production of target molecules that are normally toxic to cells or otherwise not favored due to competing intracellular pathways and cross-inhibiting reactions.201–205 Moreover, such a format can allow for potential incorporation of xenobiotic or non-natural substrates towards making new molecules enzymatically.6,200 The primary issue faced by these types of reaction formats is that of diffusion limitations and enzyme stability and it is here where NP display of enzymes can contribute. Although not focused on here, attaching enzymes to NPs can and, in many cases, does increase their long-term stability and viable lifetime along with increasing their catalytic activity.2,72,97,98 More importantly, assembly of multiple linked enzymes that constitute a biomolecular cascade with NPs into cross-linked nanoclusters can allow them to access intermediary or probabilistic channeling. Channeling is a process that occurs when enzymes are brought into close proximity of each other and where the diffusion of an intermediary from one upstream enzyme into the bulk is decreased and it instead encounters the next downstream enzyme in a cascade so as to produce a net increase in flux through the multistep system. This nanoscale diffusion-limited process is the most efficient form of multienzyme catalysis and occurs when the apparent catalytic rate of flux ≫ diffusion rate.147,161,206,207 Vranish, Breger and others have shown that QDs can crosslink with multimeric enzymes present in a cascade to form QD–enzyme nanoclusters that speed up overall catalytic flux by orders of magnitude via channeling versus the same concentration of enzyme freely diffusing in solution.2,95,97,98 Within these clusters, enzymatic intermediaries have a high probability of finding the next enzyme in the cascade due to the high localized density of enzymes present. Not only could NP display of enzymes enhance the activity of many of the participants in these cascades, but it could also enhance the rate or efficiency of the channeling process. Moreover, by initially measuring the apparent catalytic rates and affinity of each enzyme when present on a NP and enhanced, one can undertake numerical simulations to optimize the relative ratio of each enzyme present in the cluster to the others so as to maximize the rate of channeled flux.2 Overall, this represents a promising approach to increasing the productivity of multienzyme cascades while also potentially requiring a lot less enzyme material. A complementary and mostly unmet need in the field of synthetic biology is that of sensors to monitor and report on the pathways being utilized and the concentrations of products or intermediaries in a reaction regardless of whether it is intracellular or in a cell-free format.208,209 NP–enzyme and NP–substrate sensors may also find utility here where they would directly benefit from improved performance.210 Combining enzymes with QD's intrinsic photoluminescence and FRET or other energy transfer modalities can allow for sensor assemblies that are capable of extremely complex and concatenated Boolean logic functions.211–213 This can allow such sensors to perform rudimentary computational analysis and provide a more complex and nuanced data output on the underlying processes being monitored.
Although our focus here has been on enzymatic enhancement when displayed on a NP surface, we are cognizant that such attachment can also lead to changes in enzyme stability and as mentioned before this may not necessarily be in a detrimental manner. Examining enzymatic channeling in a coupled pyruvate kinase–lactate dehydrogenase (LDH) enzymatic system assembled on QDs, Vranish noted that assembly of the latter LDH significantly increased that enzyme's stability.97 LDH is an obligate tetramer that normally dissociates and ceases functioning at around 10 nM concentration, however, displaying it on QDs allowed it to continue catalyzing turnover even when diluted by a further 1.5 orders of magnitude. In this case, the tetrameric enzyme's 4-pendant terminal (His)6 moieties cross-linked with the QDs into clusters that presumably helped facilitate structural stabilization. This is certainly a promising result and suggests that not only should enzymes be tested for catalytic enhancement when attached to NPs, but they should undergo further experimental evaluation to determine if they are also stabilized in this context. Such parametric analysis could help provide further information on which types of enzyme structures are more amenable to being incorporated into such configurations. The interested reader is directed towards further reviews describing enzymatic stabilization by NP structures.214–217
The long-term outlook also remains promising especially given how quickly the fields of synthetic biology and biotechnology in general are moving. Enzyme evolution coupled with AI offers the prospect of creating new enzymes capable of new transformations and these too should be amenable to enhancement by NP display.218–220 There is also the possibility of accessing the same enhancement phenomenon but without the need for an actual NP material to be present. For example, protein–protein binding motifs and fusion/ligase sequences such as the SpyTag-SpyCatcher system and its myriad derivatives have now matured to the point that they can be designed to allow designer multiprotein clusters to form while still providing stoichiometric control over the participants.221–223 This could potentially allow self-assembly or formation of designer CLEAs where component enzymes manifest some enhancement due to the structure's NP-like properties. The ability of NP-enzyme enhancement to allow PTE to tolerate and even work better than the free enzyme in the presence of a triethyl phosphate competitive inhibitor is also quite intriguing (Fig. 3C).72 This suggests possible utility in drug screening assays where a given chemical compound may inhibit more than one target enzyme. This would also allow for larger amounts of an inhibitor to be used in an assay. Lastly, it is well worth considering that the actual implementation of NP-enzyme enhancement does not have to be perfect or fully optimized to generate a benefit from its effects – any enhancement can be helpful in many cases. In summary, although a significant amount of work still needs to be undertaken to fully utilize NP-enhancement of enzyme activity in a reliable and engineered manner, the significant benefits from achieving this suggest that much will be gained by this pursuit in the near and long term.
Author contributions
SLH, JCB, and ILM all conceived and prepared the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge the Office of Naval Research (ONR), the U.S. Naval Research Laboratory (NRL), and the NRL Nanoscience Institute for funding support. ILM acknowledges the National Institute of Food and Agriculture, U.S. Department of Agriculture, under Award #2020-67021-31254, and the Strategic Environmental Research and Development Program (SERDP), under Award #WP21-1073 New Start Project (W74RDV03497375).References
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- J. C. Breger, J. N. Vranish, E. Oh, M. H. Stewart, K. Susumu, G. Lasarte-Aragonés, G. A. Ellis, S. A. Walper, S. A. Díaz, S. L. Hooe, W. P. Klein, M. Thakur, M. G. Ancona and I. L. Medintz, Self Assembling Nanoparticle Enzyme Clusters Provide Access to Substrate Channeling in Multienzymatic Cascades, Nat. Commun., 2023, 14, 1757 CrossRef CAS PubMed.
- A. Basso and S. Serban, Industrial Applications of Immobilized Enzymes-A Review, Mol. Catal., 2019, 479, 35–54 Search PubMed.
- J. M. Choi, S. S. Han and H. S. Kim, Industrial Applications of Enzyme Biocatalysis: Current Status and Future Aspects, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- G. A. Ellis, W. P. Klein, G. Lasarte-Aragones, M. Thakur, S. A. Walper and I. L. Medintz, Artificial Multienzyme Scaffolds: Pursuing in Vitro Substrate Channeling with an Overview of Current Progress, ACS Catal., 2019, 9, 10812–10869 CrossRef CAS.
- S. L. Hooe, A. D. Smith, S. N. Dean, J. C. Breger, G. A. Ellis and I. L. Medintz, Multienzymatic Cascades and Nanomaterial Scaffolding-A Potential Way Forward for the Efficient Biosynthesis of Novel Chemical Products, Adv. Mater., 2023, 36, 2309963 CrossRef PubMed.
- M. Ali, H. M. Ishqi and Q. Husain, Enzyme Engineering: Reshaping the Biocatalytic Functions, Biotechnol. Bioeng., 2020, 117, 1877–1894 CrossRef CAS PubMed.
- R. Chowdhury and C. D. Maranas, From Directed Evolution to Computational Enzyme Engineering-A Review, AIChE J., 2020, 66, e16847 CrossRef CAS.
- R. Feehan, D. Montezano and J. S. G. Slusky, Machine Learning for Enzyme Engineering, Selection and Design, Protein Eng., Des. Sel., 2021, 34, 1–10 CAS.
- W. Q. Li, M. Bilal, A. K. Singh, F. Sher, S. S. Ashraf, M. Franco, J. H. P. Américo-Pinheiro and H. M. N. Iqbal, Broadening the Scope of Biocatalysis Engineering by Tailoring Enzyme Microenvironment: A Review, Catal. Lett., 2023, 153, 1227–1239 CrossRef CAS.
- E. Radley, J. Davidson, J. Foster, R. Obexer, E. L. Bell and A. P. Green, Engineering Enzymes for Environmental Sustainability, Angew. Chem., 2023, 62, e202309305 CrossRef CAS PubMed.
- J. Yang, F.-Z. Li and F. H. Arnold, Opportunities and Challenges for Machine Learning-Assisted Enzyme Engineering, ACS Cent. Sci., 2024, 10, 226–241 CrossRef CAS PubMed.
- M. T. Reetz, G. Qu and Z. Sun, Engineered enzymes for the synthesis of pharmaceuticals and other high-value products, J. Mol. Graphics, 2024, 3, 19–32 Search PubMed.
- M. Reetz, Making Enzymes Suitable for Organic Chemistry by Rational Protein Design, ChemBioChem, 2022, 23, e202200049 CrossRef CAS PubMed.
- W. Finnigan, M. Lubberink, L. J. Hepworth, J. Citoler, A. P. Mattey, G. J. Ford, J. Sangster, S. C. Cosgrove, B. Z. da Costa, R. S. Heath, T. W. Thorpe, Y. Yu, S. L. Flitsch and N. J. Turner, RetroBioCat Database: A Platform for Collaborative Curation and Automated Meta-Analysis of Biocatalysis Data, ACS Catal., 2023, 13, 11771–11780 CrossRef CAS PubMed.
- R. J. Young, S. L. Flitsch, M. Grigalunas, P. D. Leeson, R. J. Quinn, N. J. Turner and H. Waldmann, The Time and Place for Nature in Drug Discovery, JACS Au, 2022, 2, 2400–2416 CrossRef CAS PubMed.
- Z. Al-Qodah, M. Al-Shannag, M. Ai-Busoul, I. Penchev and W. Orfali, Immobilized Enzymes Bioreactors Utilizing a Magnetic Field: A Review, Biochem. Eng. J., 2017, 121, 94–106 CrossRef CAS.
- Y. Xie, J. An, G. Y. Yang, G. Wu, Y. Zhang, L. Cui and Y. Feng, Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site, J. Biol. Chem., 2014, 289, 7994–8006 CrossRef CAS PubMed.
- I. Eş, J. D. G. Vieira and A. C. Amaral, Principles, Techniques, and Applications of Biocatalyst Immobilization for Industrial Application, Appl. Microbiol. Biotechnol., 2015, 99, 2065–2082 CrossRef PubMed.
- D. Gurgel, Y. A. Vieira, R. O. Henriques, R. Machado, B. F. Oechsler, A. Furigo and D. de Oliveira, A Comprehensive Review on Core-Shell Polymeric Particles for Enzyme Immobilization, ChemistrySelect, 2022, 7, e202202285 CrossRef CAS.
- T. Jesionowski, J. Zdarta and B. Krajewska, Enzyme Immobilization by Adsorption: A Review, Adsorption, 2014, 20, 801–821 CrossRef CAS.
- A. Valls-Chivas, J. Gómez, J. I. Garcia-Peiro, F. Hornos and J. L. Hueso, Enzyme-Iron Oxide Nanoassemblies: A Review of Immobilization and Biocatalytic Applications, Catalysts, 2023, 13, 980 CrossRef CAS.
- D. K. Ghosh and A. Ranjan, The Metastable States of Proteins, Protein Sci., 2020, 29, 1559–1568 CrossRef CAS PubMed.
- T. Sivaraman and T. Richa, Cryptic Intermediates and Metastable States of Proteins as Predicted by OneG Computational Method, J. Biomol. Struct. Dyn., 2022, 40, 7899–7914 CrossRef CAS PubMed.
- B. Turk, I. Dolenc, E. Zerovnik, D. Turk, F. Gubensek and V. Turk, Human Cathepsin B Is a Metastable Enzyme Stabilized by Specific Ionic Interactions Associated with the Active Site, Biochemistry, 1994, 33, 14800–14806 CrossRef CAS PubMed.
- T. H. Kim, P. Mehrabi, Z. Ren, A. Sljoka, C. Ing, A. Bezginov, L. Ye, R. Pomès, R. S. Prosser and E. F. Pai, The Role of Dimer Asymmetry and Protomer Dynamics in Enzyme Catalysis, Science, 2017, 355, eaag2355 CrossRef PubMed.
- S. A. Ansari and Q. Husain, Potential Applications of Enzymes Immobilized On/In Nano Materials: A Review, Biotechnol. Adv., 2012, 30, 512–523 CrossRef CAS PubMed.
- B. J. Johnson, W. R. Algar, A. P. Malanoski, M. G. Ancona and I. L. Medintz, Understanding Enzymatic Acceleration at Nanoparticle Interfaces: Approaches and Challenges, Nano Today, 2014, 9, 102–131 CrossRef CAS.
- S. W. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Increasing the Activity of Immobilized Enzymes with Nanoparticle Conjugation, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- J. N. Vranish, M. G. Ancona, S. A. Walper and I. L. Medintz, Pursuing the Promise of Enzymatic Enhancement with Nanoparticle Assemblies, Langmuir, 2018, 34, 2901–2925 CrossRef CAS PubMed.
- ISO 80004-1:2023 Nanotechnologies – Vocabulary; Part 1: Core vocabulary. https://www.iso.org/standard/79525.html.
- M. Vert, Y. Doi, K. H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Terminology for Biorelated Polymers and Applications (IUPAC Recommendations 2012), Pure Appl. Chem., 2012, 84, 377–408 CrossRef CAS.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, The Role of Ligands in the Chemical Synthesis and Applications of Inorganic Nanoparticles, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- R. Huang, D. C. Luther, X. Z. Zhang, A. Gupta, S. A. Tufts and V. M. Rotello, Engineering the Interface between Inorganic Nanoparticles and Biological Systems through Ligand Design, Nanomaterials, 2021, 11, 1001 CrossRef CAS PubMed.
- M. Iijima and H. Kamiya, Surface Modification for Improving the Stability of Nanoparticles in Liquid Media, Kona Powder Part. J., 2009, 27, 119–129 CrossRef CAS.
- M. Rambukwella, N. A. Sakthivel, J. H. Delcamp, L. Sementa, A. Fortunelli and A. Dass, Ligand Structure Determines Nanoparticles' Atomic Structure, Metal-Ligand Interface and Properties, Front. Chem., 2018, 6, 330 CrossRef PubMed.
- S. Sarkar, E. Guibal, F. Quignard and A. K. SenGupta, Polymer-Supported Metals and Metal Oxide Nanoparticles: Synthesis, Characterization, and Applications, J. Nanopart. Res., 2012, 14, 715 CrossRef.
- Y. L. Wang, Z. Li and Z. H. Liu, Water Solubilization of Upconversion Nanoparticles, Prog. Chem., 2016, 28, 617–627 CAS.
- S. L. J. Thomä, S. W. Krauss, M. Eckardt, P. Chater and M. Zobel, Atomic Insight into Hydration Shells Around Facetted Nanoparticles, Nat. Commun., 2019, 10, 995 CrossRef PubMed.
- M. Zobel, R. B. Neder and S. A. J. Kimber, Universal Solvent Restructuring Induced by Colloidal Nanoparticles, Science, 2015, 347, 292–294 CrossRef CAS PubMed.
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Analyzing Nanomaterial Bioconjugates: A Review of Current and Emerging Purification and Characterization Techniques, Anal. Chem., 2011, 83, 4453–4488 CrossRef CAS PubMed.
- D. Mathur and I. L. Medintz, Analyzing DNA Nanotechnology: A Call to Arms For The Analytical Chemistry Community, Anal. Chem., 2017, 89, 2646–2663 CrossRef CAS PubMed.
- J. C. Breger, E. Oh, K. Susumu, W. P. Klein, S. A. Walper, M. G. Ancona and I. L. Medintz, Nanoparticle Size Influences Localized Enzymatic Enhancement-A Case Study with Phosphotriesterase, Bioconjugate Chem., 2019, 30, 2060–2074 CrossRef CAS PubMed.
- W. P. Klein, R. P. Thomsen, K. B. Turner, S. A. Walper, J. Vranish, J. Kjems, M. G. Ancona and I. L. Medintz, Enhanced Catalysis from Multienzyme Cascades Assembled on a DNA Origami Triangle, ACS Nano, 2019, 13, 13677–13689 CrossRef CAS PubMed.
- I. Medintz, Universal Tools for Biomolecular Attachment to Surfaces, Nat. Mater., 2006, 5, 842 CrossRef CAS PubMed.
- S. Avvakumova, M. Colombo, P. Tortora and D. Prosperi, Biotechnological Approaches Toward Nanoparticle Biofunctionalization, Trends Biotechnol., 2014, 32, 11–20 CrossRef CAS PubMed.
- M. Di Marco, S. Shamsuddin, K. A. Razak, A. A. Aziz, C. Devaux, E. Borghi, L. Levy and C. Sadun, Overview of the Main Methods Used to Combine Proteins with Nanosystems: Absorption, Bioconjugation, and Encapsulation, Int. J. Nanomed., 2010, 5, 37–49 CAS.
- G. A. Ellis, S. N. Dean, S. A. Walper and I. L. Medintz, Quantum Dots and Gold Nanoparticles as Scaffolds for Enzymatic Enhancement: Recent Advances and the Influence of Nanoparticle Size, Catalysts, 2020, 10, 83 CrossRef CAS.
- A. J. Sivaram, A. Wardiana, C. B. Howard, S. M. Mahler and K. J. Thurecht, Recent Advances in the Generation of Antibody-Nanomaterial Conjugates, Adv. Healthcare Mater., 2018, 7, 1700607 CrossRef PubMed.
- J. B. Blanco-Canosa, M. Wu, K. Susumu, E. Petryayeva, T. L. Jennings, P. E. Dawson, W. R. Algar and I. L. Medintz, Recent Progress in the Bioconjugation of Quantum Dots, Coord. Chem. Rev., 2014, 263, 101–137 CrossRef.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, The Controlled Display of Biomolecules on Nanoparticles: A Challenge Suited to Bioorthogonal Chemistry, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Functionalizing Nanoparticles with Biological Molecules: Developing Chemistries that Facilitate Nanotechnology, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- S. A. Walper, K. B. Turner and I. L. Medintz, Enzymatic Bioconjugation of Nanoparticles: Developing Specificity and Control, Curr. Opin. Biotechnol., 2015, 34, 232–241 CrossRef CAS PubMed.
- T. L. Jennings, S. G. Becker-Catania, R. C. Triulzi, G. L. Tao, B. Scott, K. E. Sapsford, S. Spindel, E. Oh, V. Jain, J. B. Delehanty, D. E. Prasuhn, K. Boeneman, W. R. Algar and I. L. Medintz, Reactive Semiconductor Nanocrystals for Chemoselective Biolabeling and Multiplexed Analysis, ACS Nano, 2011, 5, 5579–5593 CrossRef CAS PubMed.
- D. E. Prasuhn, J. B. Blanco-Canosa, G. J. Vora, J. B. Delehanty, K. Susumu, B. C. Mei, P. E. Dawson and I. L. Medintz, Combining Chemoselective Ligation with Polyhistidine-Driven Self-Assembly for the Modular Display of Biomolecules on Quantum Dots, ACS Nano, 2010, 4, 267–278 CrossRef CAS PubMed.
- N. Bhatt, P. J. J. Huang, N. Dave and J. W. Liu, Dissociation and Degradation of Thiol-Modified DNA on Gold Nanoparticles in Aqueous and Organic Solvents, Langmuir, 2011, 27, 6132–6137 CrossRef CAS PubMed.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, The Chemistry of the Sulfur-Gold Interface: In Search of a Unified Model, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- P. X. Zhao, N. Li and D. Astruc, State of the Art in Gold Nanoparticle Synthesis, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- E. Oh, K. Susumu, A. J. Mäkinen, J. R. Deschamps, A. L. Huston and I. L. Medintz, Colloidal Stability of Gold Nanoparticles Coated with Multithiol-Poly(ethylene glycol) Ligands: Importance of Structural Constraints of the Sulfur Anchoring Groups, J. Phys. Chem. C, 2013, 117, 18947–18956 CrossRef CAS.
- C. E. Bradburne, J. B. Delehanty, K. Boeneman Gemmill, B. C. Mei, H. Mattoussi, K. Susumu, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Cytotoxicity of quantum dots used for in vitro cellular labeling: Role of QD surface ligand, delivery modality, cell type and direct comparison to organic fluorophores, Bioconjugate Chem., 2013, 24, 1570–1583 CrossRef CAS PubMed.
- L. P. Tan and S. Q. Yao, Intein-Mediated, in vitro and in vivo Protein Modifications with Small Molecules, Protein Pept. Lett., 2005, 12, 769–775 CrossRef CAS PubMed.
- K. Boeneman, J. R. Deschamps, S. Buckhout-White, D. E. Prasuhn, J. B. Blanco-Canosa, P. E. Dawson, M. H. Stewart, K. Susumu, E. R. Goldman, M. Ancona and I. L. Medintz, Quantum Dot DNA Bioconjugates: Attachment Chemistry Strongly Influences the Resulting Composite Architecture, ACS Nano, 2010, 4, 7253–7266 CrossRef CAS PubMed.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, Kinetics of Metal-Affinity Driven Self-Assembly Between Proteins or Peptides and CdSe-ZnS Quantum Dots, J. Phys. Chem. C, 2007, 111, 11528–11538 CrossRef CAS.
- J. J. Lichty, J. L. Malecki, H. D. Agnew, D. J. Michelson-Horowitz and S. Tan, Comparison of Affinity Tags for Protein Purification, Protein Expression Purif., 2005, 41, 98–105 CrossRef CAS PubMed.
- E. Hochuli, H. Döbeli and A. Schacher, New Metal Chelate Adsorbent Selective for Proteins and Peptides Containing Neighbouring Histidine Residues, J. Chromatogr. A, 1987, 411, 177–184 CrossRef CAS PubMed.
- K. Boeneman, J. B. Delehanty, K. Susumu, M. H. Stewart and I. L. Medintz, Intracellular Bioconjugation of Targeted Proteins with Semiconductor Quantum Dots, J. Am. Chem. Soc., 2010, 132, 5975 CrossRef CAS PubMed.
- A. M. Dennis, D. C. Sotto, B. C. Mei, I. L. Medintz, H. Mattoussi and G. Bao, Surface Ligand Effects on Metal-Affinity Coordination to Quantum Dots: Implications for Nanoprobe Self-Assembly, Bioconjugate Chem., 2010, 21, 1160–1170 CrossRef CAS PubMed.
- T. Pons, I. L. Medintz, X. Wang, D. S. English and H. Mattoussi, Solution-Phase Single Quantum Dot Fluorescence Resonance Energy Transfer, J. Am. Chem. Soc., 2006, 128, 15324–15331 CrossRef CAS PubMed.
- A. Cornish-Bowden, Fundamentals of Enzyme Kinetics, Wiley-Blackwell, Weinheim, Germany, 4th edn, 2012 Search PubMed.
- C. W. Brown Iii, E. Oh, D. A. Hastman, S. A. Walper, K. Susumu, M. H. Stewart, J. R. Deschamps and I. L. Medintz, Kinetic Enhancement of the Diffusion-Limited Enzyme Beta-Galactosidase when Displayed with Quantum Dots, RSC Adv., 2015, 5, 93089–93094 RSC.
- S. Liu, M. Höldrich, A. Sievers-Engler, J. Horak and M. Lämmerhofer, Papain-Functionalized Gold Nanoparticles as Heterogeneous Biocatalyst for Bioanalysis and Biopharmaceuticals Analysis, Anal. Chim. Acta, 2017, 963, 33–43 CrossRef CAS PubMed.
- J. C. Breger, M. G. Ancona, S. A. Walper, E. Oh, K. Susumu, M. H. Stewart, J. R. Deschamps and I. L. Medintz, Understanding How Nanoparticle Attachment Enhances Phosphotriesterase Kinetic Efficiency, ACS Nano, 2015, 9, 8491–8503 CrossRef CAS PubMed.
- L. L. Lu and G. P. Yin, Shape-Controlled Synthesis and Applications of Platinum Nanocrystals, Prog. Chem., 2010, 22, 338–344 CAS.
- M. Bilal, E. Oh, R. Liu, J. C. Breger, I. L. Medintz and Y. Cohen, Bayesian network resource for meta-analysis: cellular toxicity of quantum dots, Small, 2019, 15, 1900510 CrossRef PubMed.
- E. Oh, J. B. Delehanty, C. A. Klug, K. Susumu, W. R. Algar, R. Goswami and I. L. Medintz, Utility of PEGylated Dithiolane Ligands for Direct Synthesis of Water-Soluble Au, Ag, Pt, Pd, Cu and AuPt Nanoparticles, Chem. Commun., 2018, 54, 1956–1959 RSC.
- A. Sangtani, K. H. Lee, O. K. Nag, K. Susumu, R. J. Weiblen, M. Kim, I. Vurgaftman, S. C. Liou, J. B. Delehanty and E. Oh, Seedless Synthesis of Disulfide-Grafted Gold Nanoflowers with Size and Shape Control and Their Photothermally Mediated Cell Perforation, Chem. Mater., 2022, 35, 163–176 CrossRef.
- S. A. Díaz, P. Choo, E. Oh, K. Susumu, W. P. Klein, S. A. Walper, D. A. Hastman, T. W. Odom and I. L. Medintz, Gold Nanoparticle Templating Increases the Catalytic Rate of an Amylase, Maltase, and Glucokinase Multienzyme Cascade through Substrate Channeling Independent of Surface Curvature, ACS Catal., 2021, 11, 627–638 CrossRef.
- G. Baffou and H. Rigneault, Femtosecond-Pulsed Optical Heating of Gold Nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 035415 CrossRef.
- Z. P. Qin and J. C. Bischof, Thermophysical and Biological Responses of Gold Nanoparticle Laser Heating, Chem. Soc. Rev., 2012, 41, 1191–1217 RSC.
- D. A. Hastman, J. S. Melinger, G. L. Aragonés, P. D. Cunningham, M. Chiriboga, Z. J. Salvato, T. M. Salvato, C. W. Brown, D. Mathur, I. L. Medintz, E. Oh and S. A. Díaz, Femtosecond Laser Pulse Excitation of DNA-Labeled Gold Nanoparticles: Establishing a Quantitative Local Nanothermometer for Biological Applications, ACS Nano, 2020, 14, 8570–8583 CrossRef CAS PubMed.
- D. A. Hastman, E. Oh, J. S. Melinger, C. M. Green, A. J. P. Thielemann, I. L. Medintz and S. A. Díaz, Smaller Gold Nanoparticles Release DNA More Efficiently During fs Laser Pulsed Optical Heating, Small, 2023, 2303136 Search PubMed.
- R. Kozlowski, J. Zhao and R. B. Dyer, Acceleration of Catalysis in Dihydrofolate Reductase by Transient, Site-Specific Photothermal Excitation, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014592118 CrossRef CAS PubMed.
- A. M. Negrescu, M. S. Killian, S. N. V. Raghu, P. Schmuki, A. Mazare and A. Cimpean, Metal Oxide Nanoparticles: Review of Synthesis, Characterization and Biological Effects, J. Funct. Biomater., 2022, 13, 274 CrossRef CAS PubMed.
- K. Li, J. Wang, Y. He, M. A. Abdulrazaq and Y. Yan, Carbon Nanotube-Lipase Hybrid Nanoflowers with Enhanced Enzyme Activity and Enantioselectivity, J. Biotechnol., 2018, 281, 87–98 CrossRef CAS PubMed.
- P. Reis, K. Holmberg, H. Watzke, M. E. Leser and R. Miller, Lipases at Interfaces: A Review, Adv. Colloid Interface Sci., 2009, 147-148, 237–250 CrossRef CAS PubMed.
- M. L. Verma, W. Azmi and S. S. Kanwar, Microbial Lipases: At the Interface of Aqueous and Non-Aqueous Media - A Review, Acta Microbiol. Immunol. Hung., 2008, 55, 265–294 CrossRef CAS PubMed.
- C. Iriarte-Mesa, S. Díaz-Castañón and D. G. Abradelo, Facile Immobilization of Trametes versicolor Laccase on Highly Monodisperse Superparamagnetic Iron Oxide Nanoparticles, Colloids Surf., B, 2019, 181, 470–479 CrossRef CAS PubMed.
- G. Li, P. Ma, Y. He, Y. Zhang, Y. Luo, C. Zhang and H. Fan, Enzyme–Nanowire Mesocrystal Hybrid Materials with an Extremely High Biocatalytic Activity, Nano Lett., 2018, 18, 5919–5926 CrossRef CAS PubMed.
- O. Silva-Torres, L. Bojorquez-Vazquez, A. Simakov and R. Vazquez-Duhalt, Enhanced Laccase Activity of Biocatalytic Hybrid Copper Hydroxide Nanocages, Enzyme Microb. Technol., 2019, 128, 59–66 CrossRef CAS PubMed.
- J. Zhu, Y. Zhang, D. Lu, R. N. Zare, J. Ge and Z. Liu, Temperature-Responsive Enzyme–Polymer Nanoconjugates with Enhanced Catalytic Activities in Organic Media, Chem. Commun., 2013, 49, 6090–6092 RSC.
- S. Velasco-Lozano, F. López-Gallego, J. Mateos-Díaz and E. Favela-Torres, Cross-Linked Enzyme Aggregates (CLEA) in Enzyme Improvement – A Review, Biocatalysis, 2015, 1, 166–177 Search PubMed.
- R. A. Sheldon, Cross-Linked Enzyme Aggregates as Industrial Biocatalysts, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef CAS.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Glutaraldehyde in Bio-Catalysts Design: A Useful Crosslinker and a Versatile Tool in Enzyme Immobilization, RSC Adv., 2014, 4, 1583–1600 RSC.
- J. N. Vranish, M. G. Ancona, E. Oh, K. Susumu and I. L. Medintz, Enhancing Coupled Enzymatic Activity by Conjugating One Enzyme to a Nanoparticle, Nanoscale, 2017, 9, 5172–5187 RSC.
- S. Hooe, J. Breger, S. Dean, K. Susumu, E. Oh, S. Walper, G. A. Ellis and I. L. Medintz, Benzaldehyde Lyase Kinetic Improvements, Potential Channeling to Alcohol Dehydrogenase, and Substrate Scope when Immobilized on Semiconductor Quantum Dots, ACS Appl. Nano Mater., 2022, 5, 10900–10911 CrossRef CAS.
- J. C. Breger, S. A. Walper, E. Oh, K. Susumu, M. H. Stewart, J. R. Deschamps and I. L. Medintz, Quantum Dot Display Enhances Activity of a Phosphotriesterase Trimer, Chem. Commun., 2015, 51, 6403–6406 RSC.
- J. N. Vranish, M. G. Ancona, E. Oh, K. Susumu, G. Lasarte Aragonés, J. C. Breger, S. A. Walper and I. L. Medintz, Enhancing Coupled Enzymatic Activity by Colocalization on Nanoparticle Surfaces: Kinetic Evidence for Directed Channeling of Intermediates, ACS Nano, 2018, 12, 7911–7926 CrossRef CAS PubMed.
- J. C. Breger, E. R. Goldman, K. Susumu, E. Oh, C. M. Green, S. L. Hooe, M. Thakur, I. L. Medintz and G. A. Ellis, Enzyme Assembly on Nanoparticle Scaffolds Enhances Cofactor Recycling and Improves Coupled Reaction Kinetics, Nanoscale, 2023, 15, 10159–10175 RSC.
- J. C. Claussen, A. Malanoski, J. C. Breger, E. Oh, S. A. Walper, K. Susumu, R. Goswami, J. R. Deschamps and I. L. Medintz, Probing the Enzymatic Activity of Alkaline Phosphatase within Quantum Dot Bioconjugates, J. Phys. Chem. C, 2015, 119, 2208–2221 CrossRef CAS.
- E. Petryayeva, T. Jeen and W. R. Algar, Optimization and Changes in the Mode of Proteolytic Turnover of Quantum Dot–Peptide Substrate Conjugates through Moderation of Interfacial Adsorption, ACS Appl. Mater. Interfaces, 2017, 9, 30359–30372 CrossRef CAS PubMed.
- S.-L. Tsai, M. Park and W. Chen, Size-Modulated Synergy of Cellulase Clustering for Enhanced Cellulose Hydrolysis, Biotechnol. J., 2013, 8, 257–261 CrossRef CAS PubMed.
- D.-M. Kim, M. Umetsu, K. Takai, T. Matsuyama, N. Ishida, H. Takahashi, R. Asano and I. Kumagai, Enhancement of Cellulolytic Enzyme Activity by Clustering Cellulose Binding Domains on Nanoscaffolds, Small, 2011, 7, 656–664 CrossRef CAS PubMed.
- Y. Wang, R. Jonkute, H. Lindmark, J. D. Keighron and A.-S. Cans, Molecular Crowding and a Minimal Footprint at a Gold Nanoparticle Support Stabilize Glucose Oxidase and Boost Its Activity, Langmuir, 2020, 36, 37–46 CrossRef CAS PubMed.
- S. Guo, H. Li, J. Liu, Y. Yang, W. Kong, S. Qiao, H. Huang, Y. Liu and Z. Kang, Visible-Light-Induced Effects of Au Nanoparticle on Laccase Catalytic Activity, ACS Appl. Mater. Interfaces, 2015, 7, 20937–20944 CrossRef CAS PubMed.
- P. Ball, E. Thompson, S. Anderson, V. Gwenin and C. Gwenin, Time Dependent HPLC Analysis of the Product Ratio of Enzymatically Reduced Prodrug CB1954 by a Modified and Immobilised Nitroreductase, Eur. J. Pharm. Sci., 2019, 127, 217–224 CrossRef CAS PubMed.
- M. Höldrich, S. Liu, M. Epe and M. Lämmerhofer, Taylor Dispersion Analysis, Resonant Mass Measurement and Bioactivity of Pepsin-Coated Gold Nanoparticles, Talanta, 2017, 167, 67–74 CrossRef PubMed.
- J. C. Breger, E. Oh, K. Susumu, W. P. Klein, S. A. Walper, M. G. Ancona and I. L. Medintz, Nanoparticle Size Influences Localized Enzymatic Enhancement-A Case Study with Phosphotriesterase, Bioconjugate Chem., 2019, 30, 2060–2074 CrossRef CAS PubMed.
- M. Höldrich, A. Sievers-Engler and M. Lämmerhofer, Gold Nanoparticle-Conjugated Pepsin for Efficient Solution-Like Heterogeneous Biocatalysis in Analytical Sample Preparation Protocols, Anal. Bioanal. Chem., 2016, 408, 5415–5427 CrossRef PubMed.
- S. Shikha, K. G. Thakur and M. S. Bhattacharyya, Facile Fabrication of Lipase to Amine Functionalized Gold Nanoparticles to Enhance Stability and Activity, RSC Adv., 2017, 7, 42845–42855 RSC.
- J. A. Hondred, J. C. Breger, N. T. Garland, E. Oh, K. Susumu, S. A. Walper, I. L. Medintz and J. C. Claussen, Enhanced Enzymatic Activity from Phosphotriesterase Trimer Gold Nanoparticle Bioconjugates for Pesticide Detection, Analyst, 2017, 142, 3261–3271 RSC.
- I. Ardao, J. Comenge, M. D. Benaiges, G. Álvaro and V. F. Puntes, Rational Nanoconjugation Improves Biocatalytic Performance of Enzymes: Aldol Addition Catalyzed by Immobilized Rhamnulose-1-Phosphate Aldolase, Langmuir, 2012, 28, 6461–6467 CrossRef CAS PubMed.
- M. Perwez, J. Ahmed Mazumder and M. Sardar, Preparation and Characterization of Reusable Magnetic Combi-CLEA of Cellulase and Hemicellulase, Enzyme Microb. Technol., 2019, 131, 109389 CrossRef CAS PubMed.
- J.-F. Zhao, J.-P. Lin, L.-R. Yang and M.-B. Wu, Enhanced Performance of Rhizopus oryzae Lipase by Reasonable Immobilization on Magnetic Nanoparticles and Its Application in Synthesis 1,3-Diacyglycerol, Appl. Biochem. Biotechnol., 2019, 188, 677–689 CrossRef CAS PubMed.
- J. Liao, S. Han, X. Li, J. He, F. Secundo and H. Liang, Co-Immobilization of Two-Component Hydroxylase Monooxygenase by Functionalized Magnetic Nanoparticles for Preserving High Catalytic Activity and Enhancing Enzyme Stabilty, Int. J. Biol. Macromol., 2020, 164, 3163–3170 CrossRef CAS PubMed.
- J. Ge, J. Lei and R. N. Zare, Protein–Inorganic Hybrid Nanoflowers, Nat. Nanotechnol., 2012, 7, 428–432 CrossRef CAS PubMed.
- B. S. Batule, K. S. Park, M. I. Kim and H. G. Park, Ultrafast Sonochemical Synthesis of Protein-Inorganic Nanoflowers, Int. J. Nanomed., 2015, 10, 137–142 CAS.
- J. Rong, T. Zhang, F. Qiu and Y. Zhu, Preparation of Efficient, Stable, and Reusable Laccase–Cu3(PO4)2 Hybrid Microspheres Based on Copper Foil for Decoloration of Congo Red, ACS Sustainable Chem. Eng., 2017, 5, 4468–4477 CrossRef CAS.
- Z. Lin, Y. Xiao, Y. Yin, W. Hu, W. Liu and H. Yang, Facile Synthesis of Enzyme-Inorganic Hybrid Nanoflowers and Its Application as a Colorimetric Platform for Visual Detection of Hydrogen Peroxide and Phenol, ACS Appl. Mater. Interfaces, 2014, 6, 10775–10782 CrossRef CAS PubMed.
- J. Cui, Y. Zhao, R. Liu, C. Zhong and S. Jia, Surfactant-Activated Lipase Hybrid Nanoflowers with Enhanced Enzymatic Performance, Sci. Rep., 2016, 6, 27928 CrossRef CAS PubMed.
- L. Duan, H. Li and Y. Zhang, Synthesis of Hybrid Nanoflower-Based Carbonic Anhydrase for Enhanced Biocatalytic Activity and Stability, ACS Omega, 2018, 3, 18234–18241 CrossRef CAS.
- X. Fang, C. Zhang, X. Qian and D. Yu, Self-Assembled 2,4-Dichlorophenol Hydroxylase-Inorganic Hybrid Nanoflowers with Enhanced Activity and Stability, RSC Adv., 2018, 8, 20976–20981 RSC.
- A. Mukhopadhyay, A. K. Dasgupta and K. Chakrabarti, Thermostability, pH Stability and Dye Degrading Activity of a Bacterial Laccase are Enhanced in the Presence of Cu2O Nanoparticles, Bioresour. Technol., 2013, 127, 25–36 CrossRef CAS PubMed.
- L. Han and A. Liu, Novel Cell–Inorganic Hybrid Catalytic Interfaces with Enhanced Enzymatic Activity and Stability for Sensitive Biosensing of Paraoxon, ACS Appl. Mater. Interfaces, 2017, 9, 6894–6901 CrossRef CAS PubMed.
- L. Zheng, Y. Sun, J. Wang, H. Huang, X. Geng, Y. Tong and Z. Wang, Preparation of a Flower-Like Immobilized D-Psicose 3-Epimerase with Enhanced Catalytic Performance, Catalysts, 2018, 8, 468 CrossRef.
- R. Xiong, W. Zhang, Y. Zhang, Y. Zhang, Y. Chen, Y. He and H. Fan, Remote and Real Time Control of an FVIO–Enzyme Hybrid Nanocatalyst Using Magnetic Stimulation, Nanoscale, 2019, 11, 18081–18089 RSC.
- B. Somturk, I. Yilmaz, C. Altinkaynak, A. Karatepe, N. Özdemir and I. Ocsoy, Synthesis of Urease Hybrid Nanoflowers and Their Enhanced Catalytic Properties, Enzyme Microb. Technol., 2016, 86, 134–142 CrossRef CAS PubMed.
- L. Zhu, L. Gong, Y. Zhang, R. Wang, J. Ge, Z. Liu and R. N. Zare, Rapid Detection of Phenol Using a Membrane Containing Laccase Nanoflowers, Chem. – Asian J., 2013, 8, 2358–2360 CrossRef CAS PubMed.
- N. Dutta, S. Biswas and M. K. Saha, Nano-Magnesium Aided Activity Enhancement and Biophysical Characterization of a Psychrophilic α-Amylase Immobilized on Graphene Oxide Nanosupport, J. Biosci. Bioeng., 2017, 124, 15–22 CrossRef CAS PubMed.
- Z. Xiaoyan, J. Yuanyuan, L. Zaijun, G. Zhiguo and W. Guangli, Improved Activity and Thermo-Stability of the Horse Radish Peroxidase with Graphene Quantum Dots and its Application in Fluorometric Detection of Hydrogen Peroxide, Spectrochim. Acta, Part A, 2016, 165, 106–113 CrossRef PubMed.
- W. Zhang, H. Yang, W. Liu, N. Wang and X. Yu, Improved Performance of Magnetic Cross-Linked Lipase Aggregates by Interfacial Activation: A Robust and Magnetically Recyclable Biocatalyst for Transesterification of Jatropha Oil, Molecules, 2017, 22, 2157 CrossRef PubMed.
- B. P. Dwivedee, J. Bhaumik, S. K. Rai, J. K. Laha and U. C. Banerjee, Development of Nanobiocatalysts Through the Immobilization of Pseudomonas fluorescens Lipase for Applications in Efficient Kinetic Resolution of Racemic Compounds, Bioresour. Technol., 2017, 239, 464–471 CrossRef CAS PubMed.
- I. Siddiqui and Q. Husain, Stabilization of Polydopamine Modified Silver Nanoparticles Bound Trypsin: Insights on Protein Hydrolysis, Colloids Surf., B, 2019, 173, 733–741 CrossRef CAS PubMed.
- J. Song, D. Kahveci, M. Chen, Z. Guo, E. Xie, X. Xu, F. Besenbacher and M. Dong, Enhanced Catalytic Activity of Lipase Encapsulated in PCL Nanofibers, Langmuir, 2012, 28, 6157–6162 CrossRef CAS PubMed.
- H. Li, S. Guo, C. Li, H. Huang, Y. Liu and Z. Kang, Tuning Laccase Catalytic Activity with Phosphate Functionalized Carbon Dots by Visible Light, ACS Appl. Mater. Interfaces, 2015, 7, 10004–10012 CrossRef CAS PubMed.
- T. Chen, Y. Xu, Z. Peng, A. Li and J. Liu, Simultaneous Enhancement of Bioactivity and Stability of Laccase by Cu2+/PAA/PPEGA Matrix for Efficient Biosensing and Recyclable Decontamination of Pyrocatechol, Anal. Chem., 2017, 89, 2065–2072 CrossRef CAS PubMed.
- F. Wu, L. Su, P. Yu and L. Mao, Role of Organic Solvents in Immobilizing Fungus Laccase on Single-Walled Carbon Nanotubes for Improved Current Response in Direct Bioelectrocatalysis, J. Am. Chem. Soc., 2017, 139, 1565–1574 CrossRef CAS PubMed.
- J. Forde, E. Tully, A. Vakurov, T. D. Gibson, P. Millner and C. Ó'Fágáin, Chemical Modification and Immobilisation of Laccase from Trametes hirsuta and from Myceliophthora thermophila, Enzyme Microb. Technol., 2010, 46, 430–437 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, One-Pot Synthesis of Protein-Embedded Metal–Organic Frameworks with Enhanced Biological Activities, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- L. D. Knecht, N. Ali, Y. Wei, J. Z. Hilt and S. Daunert, Nanoparticle-Mediated Remote Control of Enzymatic Activity, ACS Nano, 2012, 6, 9079–9086 CrossRef CAS PubMed.
- Q. Jin, G. Jia, Y. Zhang, Q. Yang and C. Li, Hydrophobic Surface Induced Activation of Pseudomonas cepacia Lipase Immobilized into Mesoporous Silica, Langmuir, 2011, 27, 12016–12024 CrossRef CAS PubMed.
- J. L. Lin and I. Wheeldon, Kinetic Enhancements in DNA-Enzyme Nanostructures Mimic the Sabatier Principle, ACS Catal., 2013, 3, 560–564 CrossRef CAS.
- Y. Xiong, J. Huang, S.-T. Wang, S. Zafar and O. Gang, Local Environment Affects the Activity of Enzymes on a 3D Molecular Scaffold, ACS Nano, 2020, 14, 14646–14654 CrossRef CAS PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nanocaged Enzymes with Enhanced Catalytic Activity and Increased Stability Against Protease Digestion, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- S. Kröll, T. Burgahn, K. S. Rabe, M. Franzreb and C. M. Niemeyer, Nano- and Microscale Confinements in DNA-Scaffolded Enzyme Cascade Reactions, Small, 2023, 20, 2304578 CrossRef PubMed.
- P. Lin, H. Dinh, Y. Morita, Z. X. Zhang, E. Nakata, M. Kinoshita and T. Morii, Evaluation of the Role of the DNA Surface for Enhancing the Activity of Scaffolded Enzymes, Chem. Commun., 2021, 57, 3925–3928 RSC.
- S. Rudiuk, A. Venancio-Marques and D. Baigl, Enhancement and Modulation of Enzymatic Activity through Higher-Order Structural Changes of Giant DNA-Protein Multibranch Conjugates, Angew. Chem., Int. Ed., 2012, 51, 12694–12698 CrossRef CAS PubMed.
- G. A. Ellis, S. A. Díaz and I. L. Medintz, Enhancing Enzymatic Performance with Nanoparticle Immobilization: Improved Analytical and Control Capability for Synthetic Biochemistry, Curr. Opin. Biotechnol., 2021, 71, 77–90 CrossRef CAS PubMed.
- B. T. Diroll, B. Guzelturk, H. Po, C. Dabard, N. Y. Fu, L. Makke, E. Lhuillier and S. Ithurria, 2D II-VI Semiconductor Nanoplatelets: From Material Synthesis to Optoelectronic Integration, Chem. Rev., 2023, 123, 3543–3624 CrossRef CAS PubMed.
- K. Susumu, E. Oh, J. B. Delehanty, J. B. Blanco-Canosa, B. J. Johnson, V. Jain, W. J. Hervey, W. R. Algar, K. Boeneman, P. E. Dawson and I. L. Medintz, Multifunctional Compact Zwitterionic Ligands for Preparing Robust Biocompatible Semiconductor Quantum Dots and Gold Nanoparticles, J. Am. Chem. Soc., 2011, 133, 9480–9496 CrossRef CAS PubMed.
- G. K. Kouassi, J. Irudayaraj and G. McCarty, Examination of Cholesterol Oxidase Attachment to Magnetic Nanoparticles, J. Nanobiotechnol., 2005, 3, 1 CrossRef PubMed.
- S. R. Caldwell, J. R. Newcomb, K. A. Schlecht and F. M. Raushel, Limits of Diffusion in the Hydrolysis of Substrates by the Phosphotriesterase from Pseudomonas-Diminuta, Biochemistry, 1991, 30, 7438–7444 CrossRef CAS PubMed.
- E. D. Watt, H. Shimada, E. L. Kovrigin and J. P. Loria, The Mechanism of Rate-Limiting Motions in Enzyme Function, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11981–11986 CrossRef CAS PubMed.
- D. H. Juers, B. W. Matthews and R. E. Huber, LacZ β-Galactosidase: Structure and Function of an Enzyme of Historical and Molecular Biological Importance, Protein Sci., 2012, 21, 1792–1807 CrossRef CAS PubMed.
- C. Mukai, L. Z. Gao, J. L. Nelson, J. P. Lata, R. Cohen, L. R. Wu, M. M. Hinchman, M. Bergkvist, R. W. Sherwood, S. Zhang and A. J. Travis, Biomimicry Promotes the Efficiency of a 10-Step Sequential Enzymatic Reaction on Nanoparticles, Converting Glucose to Lactate, Angew. Chem., Int. Ed., 2017, 56, 235–238 CrossRef CAS PubMed.
- W. Abdallah, X. Hong, S. Banta and I. Wheeldon, Microenvironmental Effects can Masquerade as Substrate Channelling in Cascade Biocatalysis, Curr. Opin. Biotechnol., 2022, 73, 233–239 CrossRef CAS PubMed.
- Y. N. Gao, C. C. Roberts, A. Toop, C. E. A. Chang and I. Wheeldon, Mechanisms of Enhanced Catalysis in Enzyme-DNA Nanostructures Revealed through Molecular Simulations and Experimental Analysis, ChemBioChem, 2016, 17, 1430–1436 CrossRef CAS PubMed.
- L. Lancaster, W. Abdallah, S. Banta and I. Wheeldon, Engineering Enzyme Microenvironments for Enhanced Biocatalysis, Chem. Soc. Rev., 2018, 47, 5177–5186 RSC.
- X. Y. Lang, X. Hong, C. A. Baker, T. C. Otto and I. Wheeldon, Molecular Binding Scaffolds Increase Local Substrate Concentration Enhancing the Enzymatic Hydrolysis of VX Nerve Agent, Biotechnol. Bioeng., 2020, 117, 1970–1978 CrossRef CAS PubMed.
- X. Y. Lang, L. L. Zhu, Y. N. Gao and I. Wheeldon, Enhancing Enzyme Activity and Immobilization in Nanostructured Inorganic-Enzyme Complexes, Langmuir, 2017, 33, 9073–9080 CrossRef CAS PubMed.
- J. L. Lin, L. Palomec and I. Wheeldon, Design and Analysis of Enhanced Catalysis in Scaffolded Multienzyme Cascade Reactions, ACS Catal., 2014, 4, 505–511 CrossRef CAS.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Substrate Channelling as an Approach to Cascade Reactions, Nat. Chem., 2016, 8, 299–309 CrossRef CAS PubMed.
- Y. F. Zhang, Q. Wang and H. Hess, Increasing Enzyme Cascade Throughput by pH-Engineering the Microenvironment of Individual Enzymes, ACS Catal., 2017, 7, 2047–2051 CrossRef CAS.
- H. P. Zhao, M. Q. Li, S. S. Lu, N. Cao, X. L. Zuo, S. P. Wang and M. Li, The Enhancement of Enzyme Cascading via Tetrahedral DNA Framework Modification, Analyst, 2023, 148, 906–911 RSC.
- S. Kröll and C. M. Niemeyer, Nucleic Acid-based Enzyme Cascades-Current Trends and Future Perspectives, Angew. Chem., Int. Ed., 2023, 63, e202314452 CrossRef PubMed.
- J. Breger, S. Buckhout-White, S. Walper, E. Oh, K. Susumu, M. Ancona and I. Medintz, Assembling High Activity Phosphotriesterase Composites Using Hybrid Nanoparticle Peptide-DNA Scaffolded Architectures, Nano Futures, 2017, 1, 011002 CrossRef.
- A. Samanta, S. Buckhout-White, E. Oh, K. Susumu and I. L. Medintz, Exploring Attachment Chemistry with FRET in Hybrid Quantum Dot Dye-Labeled DNA Dendrimer Composites, Mol. Syst. Des. Eng., 2018, 3, 314–327 RSC.
- C. L. Dwyer, S. A. Díaz, S. A. Walper, A. Samanta, K. Susumu, E. Oh, S. Buckhout-White and I. L. Medintz, Chemoenzymatic Sensitization of DNA Photonic Wires Mediated through Quantum Dot Energy Transfer Relays, Chem. Mater., 2015, 27, 6490–6494 CrossRef CAS.
- O. Zimmer and A. Goepferich, How Clathrin-Coated Pits Control Nanoparticle Avidity for Cells, Nanoscale Horiz., 2023, 8, 256–269 RSC.
- M. H. Li, H. Zong, P. R. Leroueil, S. K. Choi and J. R. Baker, Ligand Characteristics Important to Avidity Interactions of Multivalent Nanoparticles, Bioconjugate Chem., 2017, 28, 1649–1657 CrossRef CAS PubMed.
- A. E. Prigodich, A. H. Alhasan and C. A. Mirkin, Selective Enhancement of Nucleases by Polyvalent DNA-Functionalized Gold Nanoparticles, J. Am. Chem. Soc., 2011, 133, 2120–2123 CrossRef CAS PubMed.
- W. R. Algar, A. Malonoski, J. R. Deschamps, J. B. Banco-Canosa, K. Susumu, M. H. Stewart, B. J. Johnson, P. E. Dawson and I. L. Medintz, Proteolytic Activity at Quantum Dot-Conjugates: Kinetic Analysis Reveals Enhanced Enzyme Activity and Localized Interfacial “Hopping”, Nano Lett., 2012, 12, 3793–3802 CrossRef CAS PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, FRET as a Biomolecular Research Tool-Understanding its Potential While Avoiding Pitfalls, Nat. Methods, 2019, 16, 815–829 CrossRef CAS PubMed.
- Z. Xiao, G. H. Darwish, K. Susumu, I. L. Medintz and W. R. Algar, Prototype Smartphone-Based Device for Flow Cytometry with Immunolabeling via Supra-Nanoparticle Assemblies of Quantum Dots, ACS Meas. Sci. Au, 2021, 2, 57–66 CrossRef PubMed.
- W. R. Algar, T. Jeen, M. Massey, W. J. Peveler and J. Asselin, Small Surface, Big Effects, and Big Challenges: Toward Understanding Enzymatic Activity at the Inorganic Nanoparticle-Substrate Interface, Langmuir, 2019, 35, 7067–7091 CrossRef CAS PubMed.
- T. Jeen and W. R. Algar, Mimicking Cell Surface Enhancement of Protease Activity on the Surface of a Quantum Dot Nanoparticle, Bioconjugate Chem., 2018, 29, 3783–3792 CrossRef CAS PubMed.
- M. Massey, H. Kim, E. M. Conroy and W. R. Algar, Expanded Quantum Dot-Based Concentric Forster Resonance Energy Transfer: Adding and Characterizing Energy-Transfer Pathways for Triply Multiplexed Biosensing, J. Phys. Chem. C, 2017, 121, 13345–13356 CrossRef CAS.
- E. Petryayeva and W. R. Algar, Multiplexed Homogeneous Assays of Proteolytic Activity Using a Smartphone and Quantum Dots, Anal. Chem., 2014, 86, 3195–3202 CrossRef CAS PubMed.
- H. Y. Tsai, H. Kim, M. Massey, K. D. Krause and W. R. Algar, Concentric FRET: A Review of the Emerging Concept, Theory, and Applications, Methods Appl. Fluoresc., 2019, 7, 042001 CrossRef CAS PubMed.
- M. Wu and W. R. Algar, Acceleration of Proteolytic Activity Associated with Selection of Thiol Ligand Coatings on Quantum Dots, ACS Appl. Mater. Interfaces, 2015, 7, 2535–2545 CrossRef CAS PubMed.
- M. Wu, E. Petryayeva and W. R. Algar, Quantum Dot-Based Concentric FRET Configuration for the Parallel Detection of Protease Activity and Concentration, Anal. Chem., 2014, 86, 11181–11188 CrossRef CAS PubMed.
- S. A. Díaz, S. Sen, K. B. Gemmill, C. W. Brown, E. Oh, K. Susumu, M. H. Stewart, J. C. Breger, G. L. Aragonés, L. D. Field, J. R. Deschamps, P. Král and I. L. Medintz, Elucidating Surface Ligand-Dependent Kinetic Enhancement of Proteolytic Activity at Surface-Modified Quantum Dots, ACS Nano, 2017, 11, 5884–5896 CrossRef PubMed.
- K. D. Krause, K. Rees and W. R. Algar, Assessing the Steric Impact of Surface Ligands on the Proteolytic Turnover of Quantum Dot–Peptide Conjugates, ACS Appl. Mater. Interfaces, 2023, 15, 57799–57811 CAS.
- S. A. Díaz, A. P. Malonoski, K. Susumu, R. V. Hofele, E. Oh and I. L. Medintz, Probing the Kinetics of Quantum Dot-Based Proteolytic Sensors, Anal. Bioanal. Chem., 2015, 407, 7307–7318 CrossRef PubMed.
- P. Linardatos, V. Papastefanopoulos and S. Kotsiantis, Explainable AI: A Review of Machine Learning Interpretability Methods, Entropy, 2021, 23, 18 CrossRef PubMed.
- A. M. Rahmani, E. Yousefpoor, M. S. Yousefpoor, Z. Mehmood, A. Haider, M. Hosseinzadeh and R. A. Naqvi, Machine Learning (ML) in Medicine: Review, Applications, and Challenges, Mathematics, 2021, 9, 2970 CrossRef.
- M. Björnmalm, M. Faria and F. Caruso, Increasing the Impact of Materials in and beyond Bio-Nano Science, J. Am. Chem. Soc., 2016, 138, 13449–13456 CrossRef PubMed.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. L. Medintz, Meta-analysis of cellular toxicity for cadmium-containing quantum dots, Nat. Nanotechnol., 2016, 11, 479–486 CrossRef CAS PubMed.
- M. Bilal, E. Oh, R. Liu, J. C. Breger, I. L. Medintz and Y. Cohen, Bayesian Network Resource for Meta-Analysis: Cellular Toxicity of Quantum Dots, Small, 2019, 15, 1900510 CrossRef PubMed.
- M. Zhong, K. Tran, Y. M. Min, C. H. Wang, Z. Y. Wang, C. T. Dinh, P. De Luna, Z. Q. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. L. Che, M. Liu, A. Seifitokaldani, Y. J. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Accelerated Discovery of CO2 Electrocatalysts Using Active Machine Learning, Nature, 2020, 581, 178 CrossRef CAS PubMed.
- L. H. Wu, S. Chen, L. Guo, S. Shpyleva, K. Harris, T. Fahmi, T. Flanigan, W. D. Tong, J. S. Xu and Z. Ren, Development of Benchmark Datasets for Text Mining and Sentiment Analysis to Accelerate Regulatory Literature Review, Regul. Toxicol. Pharmacol., 2023, 137, 105287 CrossRef CAS PubMed.
- C. S. Morrison, W. B. Armiger, D. R. Dodds, J. S. Dordick and M. A. G. Koffas, Improved Strategies for Electrochemical 1,4-NAD(P)H2 Regeneration: A New Era of Bioreactors for Industrial Biocatalysis, Biotechnol. Adv., 2018, 36, 120–131 CrossRef CAS PubMed.
- R. A. Rocha, A. J. North, R. E. Speight, C. C. Williams and C. Scott, Cofactor and Process Engineering for Nicotinamide Recycling and Retention in Intensified Biocatalysis, Catalysts, 2022, 12, 1454 CrossRef CAS.
- I. Zachos, C. Nowak and V. Sieber, Biomimetic Cofactors and Methods for Their Recycling, Curr. Opin. Chem. Biol., 2019, 49, 59–66 CrossRef CAS PubMed.
- P. H. Opgenorth, T. P. Korman and J. U. Bowie, A Synthetic Biochemistry Molecular Purge Valve Module that Maintains Redox Balance, Nat. Commun., 2014, 5, 4113 CrossRef CAS PubMed.
- I. Medintz, L. Chiriboga, L. McCurdy and L. Kobilinsky, Restriction Fragment Length Polymorphism and Polymerase Chain Reaction-HLA DQ Alpha Analysis of Casework Urine Specimens, J. Forensic Sci., 1994, 39, 1372–1380 CrossRef CAS PubMed.
- P. J. Tighe, R. R. Ryder, I. Todd and L. C. Fairclough, ELISA in the Multiplex Era: Potentials and Pitfalls, Proteomics: Clin. Appl., 2015, 9, 406–422 CAS.
- S. Aydin, A Short History, Principles, and Types of ELISA, and Our Laboratory Experience with Peptide/Protein Analyses Using ELISA, Peptides, 2015, 72, 4–15 CrossRef CAS PubMed.
- M. Asgher, M. Shahid, S. Kamal and H. M. N. Iqbal, Recent Trends and Valorization of Immobilization Strategies and Ligninolytic Enzymes by Industrial Biotechnology, J. Mol. Catal. B: Enzym., 2014, 101, 56–66 CrossRef CAS.
- Z. B. Liu, R. Weis and A. Glieder, Enzymes from Higher Eukaryotes for Industrial Biocatalysis, Food Technol. Biotechnol., 2004, 42, 237–249 CAS.
- S. L. Hooe, G. A. Ellis and I. L. Medintz, Alternative Design Strategies to Help Build the Enzymatic Retrosynthesis Toolbox, RSC Chem. Biol., 2022, 2, 1301–1313 RSC.
- M. El Karoui, M. Hoyos-Flight and L. Fletcher, Future Trends in Synthetic Biology—A Report, Front. Bioeng. Biotechnol., 2019, 7, 175 CrossRef PubMed.
- J. Garamella, D. Garenne and V. Noireaux, TXTL-Based Approach to Synthetic Cells, in Metabolons and Supramolecular Enzyme Assemblies, ed. C. SchmidtDannert and M. B. Quin, 2019, vol. 617, pp. 217–239 Search PubMed.
- D. Garenne and V. Noireaux, Cell-Free Transcription-Translation: Engineering Biology from the Nanometer to the Millimeter Scale, Curr. Opin. Biotechnol., 2019, 58, 19–27 CrossRef CAS PubMed.
- J. U. Bowie, S. Sherkhanov, T. P. Korman, M. A. Valliere, P. H. Opgenorth and H. Liu, Synthetic Biochemistry: The Bio-Inspired Cell-Free Approach to Commodity Chemical Production, Trends Biotechnol., 2020, 38, 766–778 CrossRef CAS PubMed.
- S. L. Hooe, M. Thakur, G. Lasarte-Aragonés, J. C. Breger, S. A. Walper, I. L. Medintz and G. A. Ellis, Exploration of the In Vitro Violacein Synthetic Pathway with Substrate Analogues, ACS Omega, 2024, 9, 3894–3904 CAS.
- J. S. Easterby, The Analysis of Metabolite Channelling in Multienzyme Complexes and Multifunctional Proteins, Biochem. J., 1989, 264, 605–607 CrossRef CAS PubMed.
- H. O. Spivey and J. Ovadi, Substrate Channeling, Methods, 1999, 19, 306–321 CrossRef CAS PubMed.
- I. Del Valle, E. M. Fulk, P. Kalvapalle, J. J. Silberg, C. A. Masiello and L. B. Stadler, Translating New Synthetic Biology Advances for Biosensing Into the Earth and Environmental Sciences, Front. Microbiol., 2021, 11, 618373 CrossRef PubMed.
- I. C. MacDonald and T. L. Deans, Tools and Applications in Synthetic Biology, Adv. Drug Delivery Rev., 2016, 105, 20–34 CrossRef CAS PubMed.
- J. C. Breger, K. Susumu, G. Lasarte-Aragonés, S. A. Díaz, J. Brask and I. L. Medintz, Quantum Dot Lipase Biosensor Utilizing a Custom-Synthesized Peptidyl-Ester Substrate, ACS Sens., 2020, 5, 1295–1304 CrossRef CAS PubMed.
- J. C. Claussen, W. R. Algar, N. Hildebrandt, K. Susumu, M. G. Ancona and I. L. Medintz, Biophotonic Logic Devices Based on Quantum Dots and Temporally-Staggered Forster Energy Transfer Relays, Nanoscale, 2013, 5, 12156–12170 RSC.
- J. C. Claussen, N. Hildebrandt, K. Susumu, M. G. Ancona and I. L. Medintz, Complex Logic Functions Implemented with Quantum Dot Bionanophotonic Circuits, ACS Appl. Mater. Interfaces, 2014, 6, 3771–3778 CrossRef CAS PubMed.
- D. A. Hastman, S. Hooe, M. Chiriboga, S. A. Díaz, K. Susumu, M. H. Stewart, C. M. Green, N. Hildebrandt and I. L. Medintz, Multiplexed DNA and Protease Detection with Orthogonal Energy Transfer on a Single Quantum Dot Scaffolded Biosensor, ACS Sens., 2024, 9, 157–170 CrossRef CAS PubMed.
- J. Kim, J. W. Grate and P. Wang, Nanostructures for enzyme stabilization, Chem. Eng. Sci., 2006, 61, 1017–1026 CrossRef CAS.
- R. S. Ahmad, Meryam, Enzyme Immobilization: An Overview on Nanoparticles as Immobilization Matrix, Biochem. Anal. Biochem., 2015, 4, 1000178 Search PubMed.
- P. Asuri, S. S. Karajanagi, A. A. Vertegel, J. S. Dordick and R. S. Kane, Enhanced Stability of Enzymes Adsorbed onto Nanoparticles, J. Nanosci. Nanotechnol., 2007, 7, 1675–1678 CrossRef CAS PubMed.
- E. T. Hwang and M. B. Gu, Enzyme stabilization by nano/microsized hybrid materials, Eng. Life Sci., 2013, 13, 49–61 CrossRef CAS.
- D. Davidi, L. M. Longo, J. Jablonska, R. Milo and D. S. Tawfik, A Bird's-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations, Chem. Rev., 2018, 118, 8786–8797 CrossRef CAS PubMed.
- W. D. Jang, G. B. Kim, Y. Kim and S. Y. Lee, Applications of Artificial Intelligence to Enzyme and Pathway Design for Metabolic Engineering, Curr. Opin. Biotechnol., 2022, 73, 101–107 CrossRef CAS PubMed.
- G. Y. Li, Y. J. Dong and M. T. Reetz, Can Machine Learning Revolutionize Directed Evolution of Selective Enzymes?, Adv. Synth. Catal., 2019, 361, 2377–2386 CrossRef CAS.
- A. H. Keeble and M. Howarth, Power to the Protein: Enhancing and Combining Activities Using the Spy Toolbox, Chem. Sci., 2020, 11, 7281–7291 RSC.
- A. R. Sutherland, M. K. Alam and C. R. Geyer, Post-Translational Assembly of Protein Parts into Complex Devices by Using SpyTag/SpyCatcher Protein Ligase, ChemBioChem, 2019, 20, 319–328 CrossRef CAS PubMed.
- N. J. Alves, K. B. Turner, M. A. Daniele, E. Oh, I. L. Medintz and S. A. Walper, Bacterial Nanobioreactors–Directing Enzyme Packaging into Bacterial Outer Membrane Vesicles, ACS Appl. Mater. Interfaces, 2015, 7, 24963–24972 CrossRef CAS PubMed.
- K. Susumu, L. D. Field, E. Oh, M. Hunt, J. B. Delehanty, V. Palomo, P. E. Dawson, A. L. Huston and I. L. Medintz, Purple-, Blue-, and Green-Emitting Multishell Alloyed Quantum Dots: Synthesis, Characterization, and Application for Ratiometric Extracellular pH Sensing, Chem. Mater., 2017, 29, 7330–7344 CrossRef CAS.
- F. Cardona, C. Parmeggiani, E. Faggi, C. Bonaccini, P. Gratteri, L. Sim, T. M. Gloster, S. Roberts, G. J. Davies, D. R. Rose and A. Goti, Total Syntheses of Casuarine and Its 6-O-α-Glucoside: Complementary Inhibition towards Glycoside Hydrolases of the GH31 and GH37 Families, Chem. – Eur. J., 2009, 15, 1627–1636 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |