
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: optimisation of drinking water biofilm cell detachment and sample homogenisation methods for rapid quantification via flow cytometry†
Frances C.
Pick
*a and
Katherine E.
Fish
 *ab
*ab
aSheffield Water Centre, Department of Civil and Structural Engineering, The University of Sheffield, UK. E-mail: k.fish@sheffield.ac.uk
bNERC Environmental Omics Facility, School of Biosciences, University of Sheffield, Sheffield, UK
First published on 8th February 2024
Abstract
Understanding biofilm microbial loads and viability within drinking water pipes is critical to inform sustainable management of ageing infrastructure to protect future water quality. This study establishes an optimised method for robustly harvesting and quantifying cells of biofilms sampled from drinking water systems. Extensive research was conducted to determine the best way to remove biofilms of diverse ages (3–9 months) from different sampling surfaces (pipe sections or coupons) and create homogenised samples for rapid cell enumeration using flow cytometry. Utilising a standardised brushing technique, the optimised approaches delivered the greatest yield of biofilm cells (nine times more cells removed than using sonication) and simultaneously homogenized samples without affecting integrity of intact cells. The optimal brushing strategy differed slightly between sampling surfaces (15 brush strokes for pipe sections, 30 for coupons). When applied to biofilms from a full-scale pipe system, the optimised sampling and flow cytometry methods consistently showed the same trends in biofilm cell concentrations as obtained via molecular analysis (qPCR), but more quickly and from a smaller sample area. Application of the optimised biofilm preparation approach to samples from operational DWDS will ensure that greater yield and more representative samples are collected and analysed, which is critical for any downstream biofilm characterisation or assessment of operational performance.
Water impactAppropriate and optimised drinking water biofilm removal/harvesting is critical in generating representative samples for any subsequent microbiome characterisation or assessment of operational system performance. As such, the optimised methods presented are of vital importance to biofilm research and the wider water sector as they will facilitate generation and translation of understanding to real-systems. The presented method, combined with advances in flow cytometry, has the potential to provide novel insights into biofilm fouling rates and interventions within DWDS environments. Ultimately, this will lead to proactive and sustainable management of water systems, protecting water quality. |
1 Introduction
Drinking water distribution systems (DWDS) are designed and managed to maintain the microbial quality, and thereby the biostability,1 of drinking water reaching the consumer. Monitoring microbial water quality is critical to assess DWDS performance and protect public health. Methods traditionally used to monitor the microbial quality of water are based on culture dependent techniques that detect microorganisms in bulk-water samples. Such methods are under representative of microbial loads in DWDS as they only consider culturable planktonic organisms and do not take into account attached microorganisms residing within biofilms in DWDS.2 More recently, cultivation independent methods, such as flow cytometry and adenosine triphosphate (ATP) have gained considerable interest as these methods are rapid and able to detect both culturable and non-culturable microorganisms.3 Robust flow cytometry protocols have been developed4,5 and applied to quantify the absolute number of cells in different water types or locations in the DWDS,6,7 and to assess treatment efficiency.8–10 These studies have comprehensively demonstrated the benefits of flow cytometry and insights this analysis can provide. However, the majority of these protocols and applications focus on enumerating microorganisms within the bulk-water and not within biofilms.The majority of microbial material within DWDS is found within drinking water biofilms,11 attached to the pipe wall via a matrix of extracellular polymeric substances (EPS). Biofilms have the potential to degrade drinking water quality via interactions with both the pipe wall and the bulk-water. Biofilm mobilisation into the bulk-water can cause discolouration and other aesthetic quality issues, potentially posing a risk to public health if pathogens are mobilised,12 hence there is a need to reliably characterise biofilms from drinking water systems to understand and control associated risks. DWDS cleaning programmes, such as mains conditioning,13 flushing,14 or pigging15 are used to remove or manage material from the pipe walls of the DWDS. Understanding and characterising biofilms or the biofouling rate within DWDS is critical to plan the type and frequency of network interventions.16
Until recently, biofilm sampling in situ within DWDS relied on either using cut-out sections of pipe or inserting devices, such as the Pennine Water Group (PWG) coupon17 or the ‘biofilm sampler’.18 Emerging technologies, such as the biofilm monitoring device (BMD),16 offer a simple way to sample biofilms within operational DWDS, and study biofilm formation rates in distribution systems with different water qualities. However, there is currently no universally accepted method for optimal biofilm removal/harvesting from sampled surfaces (required to ensure representative samples are analysed) or the subsequent homogenisation of the biofilm samples, which is critical for downstream analyses such as flow cytometry that require homogenised samples for accurate quantification. Although, not drinking water specific, Buckingham-Meyer, Miller19 note that vortexing, sonication (with a water bath) and scraping are common methods for separating biofilm from the sampled surface and demonstrate optimisation for silicon catheter tubing, polycarbonate CDC reactor coupons and glass surfaces. Note that herein the term “biofilm removal” will be used to refer to collecting all (or as much as possible) of a biofilm from a sampled surface, this process is also referred to as “harvesting” or “processing” in the literature (Buckingham-Meyer et al., 2022).19 Some drinking water studies also use (repeated) sonication via a water bath, with glass beads for removal of biofilm from drinking water pipe sections or from a shower hose, followed by homogenisation using a sonicating needle.20,21 Others remove biofilm by brushing the sampled surface such as PWG coupons that have been inserted into drinking water pipes in a full-scale facility or an operational network.16,17,22
Flow cytometry requires a homogenised sample of single cells/events to be most effective. The biofilm EPS (responsible for adhesion/cohesion) promotes clustering, and therefore in order to have homogenised, representative samples, an external disturbance must be applied. However, care must be taken during sample homogenisation to avoid cell damage.23 Gagnon and Slawson24 compared four drinking water biofilm homogenisation methods including the use a tissue blender, vortex, stomacher and a sonicator; and found that stomaching provided the highest enumeration (in terms of heterotrophic plate counts (HPCs) and total cell counts). However, intact cell counts were not assessed so impact of the method on cell viability cannot be evaluated. There is a need to understand which biofilm removal and homogenisation methodology is most appropriate to use when studying drinking water biofilms from full-scale and field systems. Moreover, it is essential to determine if these methods can be further optimised to transfer as many cells as possible from the sampled surface but with minimal impact on cell integrity, to ensure the most representative biofilm samples are analysed.
This research aimed to evaluate the impact of different drinking water biofilm removal and homogenisation methods on cell quantity and viability, to inform the most appropriate removal method to ensure representative drinking water biofilm cell quantification via flow cytometry. Existing methods used to remove drinking water biofilms from sampling devices were compared and optimised, and their advantages and limitations considered. Homogenisation methods, including vortexing and sonication with a sonicating needle, were assessed. The methods were trialled on developing and mature biofilms, and flow cytometry quantification trends were compared to those obtained using molecular analysis (qPCR), to provide reliable and useful methodological data for further DWDS biofilm research so that representative data is collected that is comparable across studies.
2 Methods
2.1 Experimental overview
A series of laboratory trails were conducted to: (i) determine the optimal brushing method to remove drinking water biofilm from a pipe surface (Biofilm Monitoring Device – BMD or coupons from a bioreactor); (ii) compare and evaluate biofilm removal methods (brushing vs. glass bead sonication); and (iii) determine the optimal method to homogenise drinking water biofilms for cell enumeration using flow cytometry. Subsequently, the optimised removal, homogenisation and flow cytometry methods were applied to enumerate the cells in biofilms sampled from a full-scale DWDS (using coupons), verifying the method via comparison to molecular based quantification (qPCR).2.2 Biofilm sampling devices
Unless specified, all biofilm cell removal and homogenisation trials were conducted using biofilms grown within biofilm monitoring devices (BMD) developed at The University of Sheffield (Fig. 1). The BMD consisted of a series of short and identical polyethylene pipe lengths (53 mm) suitable for biofilm sampling, which can be connected directly to an operational drinking water supply.16 A flow valve was attached to the outlet (Akro Valves Ltd, UK) to maintain a consistent flow rate of 1 L min−1 with a shear stress of 0.16 Pa.16 The biofilms used in the removal and homogenisation experiments were grown in a BMD for either 3 or 6 months.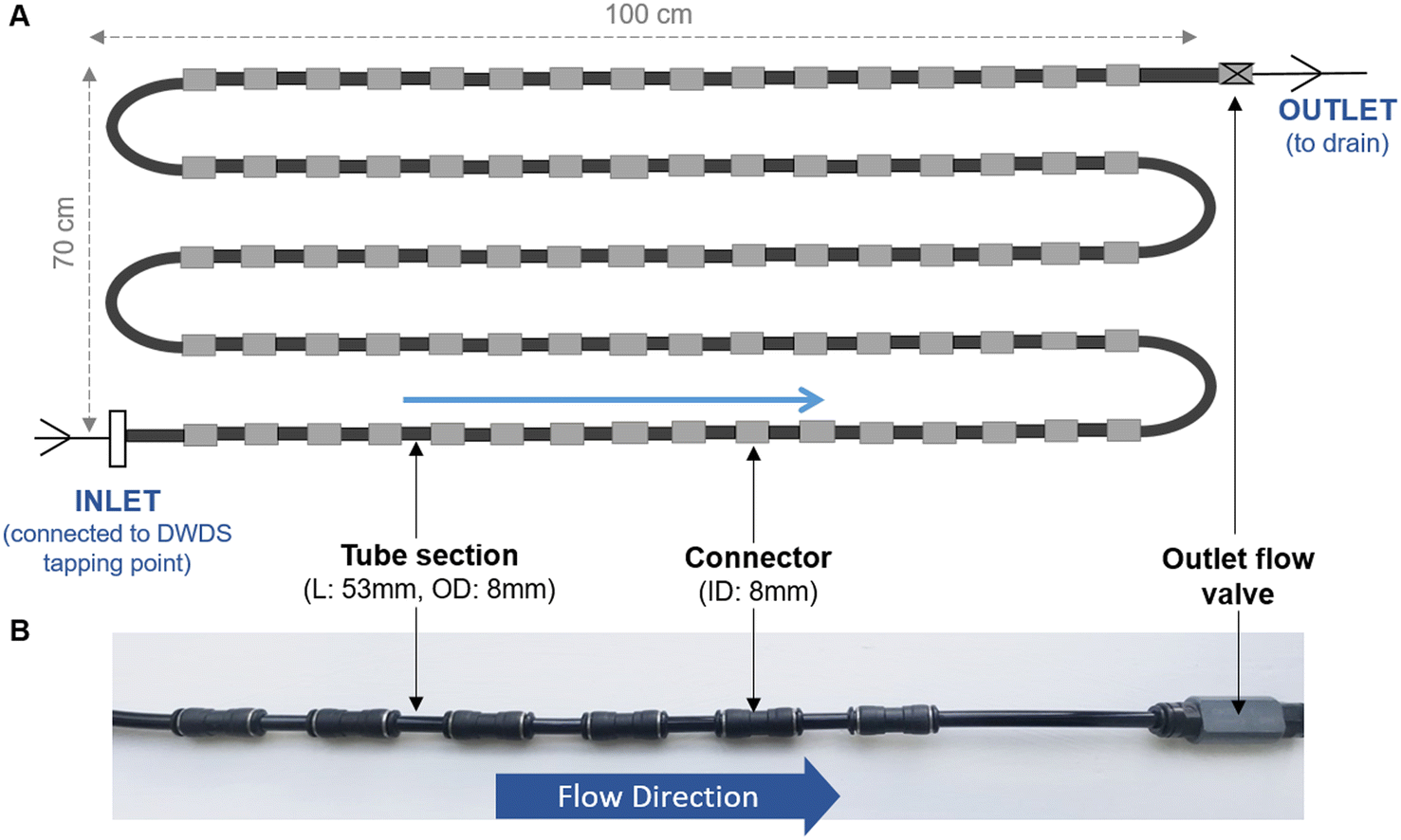 | ||
| Fig. 1 Biofilm monitoring device (BMD) schematic (A) and image (B). L = length; OD = outside diameter, ID = inner diameter, blue arrows indicate direction of water flow. (A) Schematic showing installation of five rows of the BMD (row L = 100 cm, height of all rows = 70 cm, total L ∼ 500 cm). Note schematic does not show the full number of pipe sections used in an installation. (B) Photo of short section of BMD in situ. | ||
In addition to the BMD, an unpressurised drinking water bioreactor, containing square (2 cm × 2 cm) high density polyethylene (HDPE) coupons (Fig. S1†) was used to develop biofilms for 9 months. The bioreactor was run at a steady state flow of 0.4 L s−1, replicating a shear stress (0.12 Pa), comparable to that experienced in UK operational DWDS, based on an average flow of 0.4 L s−1 in 75–100 mm diameter pipes.25 The use of HDPE coupons facilitated the optimisation of the brushing technique on an alternative surface shape and a material relevant to full-scale DWDS pipelines. Biofilm samples from the bioreactor were used during the optimisation of the brushing removal method.
2.3 Biofilm growth and sampling for removal and homogenisation tests
To establish the suitability of applying the removal and homogenisation methods across differently aged biofilms, trials were performed on developing (3 months) and mature (6–9 months) biofilms. It would be desirable for the optimised method to be suitable for application to mature and young biofilms alike to enable temporal studies of drinking water biofilm growth and dynamics with the same methods.All experiments were performed in a temperature-controlled (16 °C) laboratory at the University of Sheffield. Prior to installation, all pipe sections (or coupons) and brushes were cleaned via sonication with a 2% (w/v) sodium dodecyl sulphate (SDS) solution and autoclaved.16 Both the BMD and bioreactor were supplied with water from the local DWDS (surface water source), which is distributed via a cast iron trunk main direct into the building that houses the facility. No local dosing of organisms was used so the biofilms developed naturally. Throughout the biofilm growth periods, bulk-water quality was monitored every two weeks (n = 3), in all instances water quality was within the UK regulations with standard parameters being monitored (see Table S1†).
During sampling of individual pipe sections from the BMD (n = 3 or n = 5), the flow was temporarily switched off (<1 minute) for removal of a pipe section before the remaining pipe sections were reconnected and flow resumed. When sampling the bioreactor HDPE coupons (n = 5), flow was paused, clean coupons were used to replace sampled coupons, before the flow was resumed. In all experiments, control samples were also collected (n = 3 or n = 5), which included phosphate buffer solution (PBS) being poured over coupons or pipe sections (i.e. no brushing or sonication). An overview of the biofilm samples collected (age, sampling surface) and experiments conducted on those samples is presented in Table 1.
| Biofilm age | Sampling surface | Brushing optimisation | Removal comparisons | Homogenisation comparisons | |
|---|---|---|---|---|---|
| Description | Months | ||||
| BMD: biofilm monitoring device; HDPE: high density polyethylene. | |||||
| Developing | 3 | BMD | — | Brushing | Vortex |
| Sonicating needle | |||||
| Glass bead sonication | Vortex | ||||
| Sonicating needle | |||||
| Mature | 6 | BMD | BMD optimisation | Brushing | Vortex |
| Sonicating needle | |||||
| Mature | 9 | HDPE coupon | HDPE coupon optimisation | Brushing | — |
2.4 Biofilm removal
When brushing to remove biofilm from the BMD, suspensions were created using an adapted version of the technique described in Fish, Collins;26 using sterile nylon cylinder brushes (Lessmann, Germany; diameter 6 mm, length 80 mm) biofilm was removed from the interior surface of the BMDs into a 30 mL volume of PBS. To determine the optimum number of brush strokes, pipe sections (n = 5) were removed from a BMD and brushed five times using circular brush strokes (rinsing the brush into a 30 mL volume of PBS after each brush), the resulting biofilm suspensions was then enumerated using flow cytometry (section 3.6). Subsequently, the same BMD pipe sections were brushed a further five times (rinsing the brush in PBS after each brush) and the resulting suspension was enumerated using flow cytometry to assess any additional biofilm that had been removed. This process was repeated five times, up to a total of 25 brush strokes.
Biofilm was removed from HDPE coupons (n = 5) by brushing the “top” surface (i.e. the surface that was in contact with the bulk-water during growth) into 30 mL of sterile PBS, using a sterile toothbrush (nylon, bristle dimensions 30 mm × 10 mm × 12 mm, standard toothbrush of medium firmness) and brushing for a set number of brush strokes. The HDPE coupons were brushed horizontally and vertically for 0, 10, 20, 30, 40 and 50 brushes (in both directions), rinsing the toothbrush in the PBS after 10 strokes in one orientation. The biofilm suspensions were analysed using flow cytometry (section 3.6) after each set of 10 brush strokes in both orientations.
The optimal number of brush strokes was ascertained by using flow cytometry to quantify cell concentrations and viability after each set of brush strokes (five for BMDs, 10 for HDPE coupons) and determine the point at which no further cells were being removed from the BMD or HDPE surface.
Biofilm removal using sonication and glass beads was conducted using the protocol outlined in Proctor, Gächter.20 In summary, pipe sections (n = 5) were aseptically removed from the BMD, one end of the pipe section was sealed (using parafilm) and a mixture of glass beads (0.5 mL) and PBS (0.5 mL) was added, the open end of the pipe section was then also sealed (with parafilm). The sealed BMD pipe section was inverted five times and sonicated in a water bath (Advantage-Lab™, Belgium) for 5 minutes. The resulting biofilm suspension was collected into a 50 mL falcon pipe (the beads were retained) and the BMD was filled with fresh PBS and sonicated again. This process was repeated for five rounds of sonication and PBS replacement. After the final round, the glass beads were removed and discarded, and the pipe sections filled up with PBS and inverted 30 times. This final rinse water was added to the biofilm suspension, and the entire biofilm suspension was sonicated for 0.5 minutes.
Removal methods were compared using 3 month old biofilm samples (n = 10), sampled randomly from a BMD device (Table 1). Half of the BMD pipe sections (1–5) were brushed first and cell removal was quantified via flow cytometry, followed by water-bath sonication with glass beads and any additional cell removal assessed using flow cytometry. The other BMD sections (6–10) had the biofilm removal using water-bath sonication with glass beads applied first; cell removal was quantified via flow cytometry, followed by secondary removal using brushing and any additional cell removal was quantified via flow cytometry.
2.5 Biofilm homogenisation optimisation
To evaluate homogenisation efficiency two methods were compared, vortexing (gentle mixing) and sonication with a sonicating needle (harsher homogenisation method) using suspensions of developing (3-months) and mature (6-months) biofilms. The biofilm suspensions were generated using either the optimised brushing or sonication with glass bead protocols as indicated (Table 1) to assess any impact of upstream biofilm removal method on the efficiency of the homogenisation method.Each homogenisation method (vortexing or sonication-needle) was conducted for a series of increasing longevities to ascertain the optimal time and technique to ensure that biofilm samples were homogenised but no cells were damaged in the process. Samples were either vortexed (speed setting 6, Vortex-Genie 2, Scientific Industries, Inc. USA) or sonicated (20 kHz (20.000 cycles per second), Jencons High Intensity Ultrasonic Processor Model GE 50, Jencons. Scientific Ltd. UK) for 0.5, 1, 2, 4 and 8 minutes. All sonicating times were conducted in a series of 0.5 minutes with the sample on ice to ensure samples were not overheated. The biofilm samples were subsequently analysed using flow cytometry to ascertain cell concentrations, viability and homogenisation (via singlet–doublet analysis), using the flow cytometry gating strategy detailed in Fish, Reeves-McLaren.22 A singlet–doublet gate was added to the standard BD C6 Accuri template to distinguish singlets from doublets and evaluate the proportion of the collected data that was classified as single events. In other fields, such as medical applications, a threshold such as ≥98% is set (i.e. ≥98% of the data collected for each sample is singlet in nature) to ensure the purity of data.27 Analysis historic flow cytometry data of bulk-water samples (accepted as being homogenised samples) showed that for each sample 97–100% of the data points collected were singlets (98.5% on average), therefore a biofilm sample would be classed as homogenised if the singlet–doublet% was ≥98%.
2.6 Cell enumeration using flow cytometry
A 500 μl volume of each biofilm suspension was stained and analysed in accordance with the flow cytometry protocol detailed elsewhere.28 Briefly, 0.5 mL of the sample was stained with either SYBR Green (Invitrogen™ by Thermo Fisher Scientific, USA) to count total cells, or SYBR Green and propidium iodide (Invitrogen™ by Thermo Fisher Scientific, USA) to count intact cells. Samples were incubated (10 minutes, 37 °C) and analysed with a BD Accuri C6 flow cytometer (50 μL, medium flow rate). The flow cytometer template was edited to include singlet–doublet analysis, providing a quantitative assessment of sample homogenisation (Fish et al. 2020).22 In this study, samples for which ≥98% of the data collected were singlets, were classed as well homogenised. To convert the cell counts into cell concentrations (ICC/mm2 or TCC/mm2), eqn (1) was used: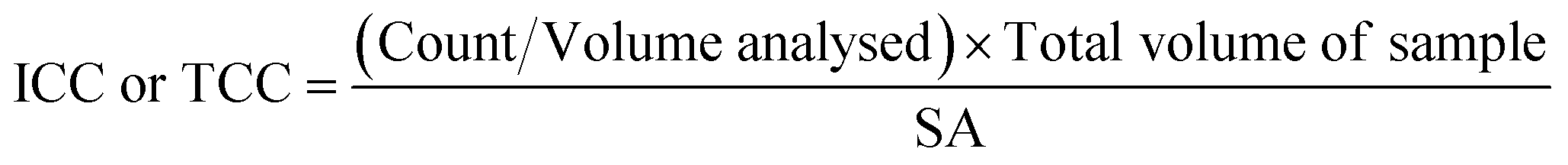 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μl) and SA is the surface area from which the biofilm was removed (915.78 mm2 for pipe sections within the BMD and 400 mm2 for coupons). Preliminary tests of technical replication showed no differences, so only biological replication samples were undertaken (n = 3 or n = 5 depending on the experiment).
000 μl) and SA is the surface area from which the biofilm was removed (915.78 mm2 for pipe sections within the BMD and 400 mm2 for coupons). Preliminary tests of technical replication showed no differences, so only biological replication samples were undertaken (n = 3 or n = 5 depending on the experiment).
2.7 Flow cytometry vs. qPCR using full-scale DWDS biofilms
To verify the application of flow cytometry as a rapid technique to quantify DWDS biofilm cell concentration, biofilms were developed within a full-scale DWDS experimental system under three different chlorine concentrations (Fig. S2†) and cell concentrations were enumerated using flow cytometry and qPCR. Results were expressed as concentrations per area and datasets from the two methods were compared to ascertain if flow cytometry showed the same trends as qPCR.The full-scale experimental system has been described in detail elsewhere29 as have the chlorine testing experimental design and other results relating to water quality and biofilm characterisation.22 In brief, three independent systems each comprising a tank, loop of HPDE pipe (200 m long, 79 mm internal diameter) and online water quality monitors (flow, pressure, turbidity and free chlorine) were supplied with water from the local DWDS. Drinking water was pumped around each system following a typical residential pattern of demand (double-peaked diurnal flow profile, peak 0.54 l s−1, low flow 0.23 l s−1). A trickle turnover of 24 hours was set to preserve baseline water qualities. Each of the three loops had HDPE Pennine Water Group coupons17 installed for biofilm sampling (these coupons comprise an outer coupon and removable insert enabling dual analysis of the same sample). Biofilms were grown under one of three chlorine-residual concentrations (low, medium, high) for a 28-day period, at 16 °C (representative of UK summer water temperatures). Subsequently, biofilms were exposed to a series of increasing flow rates (0.74, 3.58, 5.10 and 6.29 l s−1) to increase the shear stress at the pipe wall (0.09, 1.57, 3.05, 4.53 Pa) and ascertain biofilm stability under hydraulic changes representative of those in operational networks and used as a cleaning intervention (described in detail in Fish et al. 2020).22
Chlorine concentrations varied over the 28 days due to natural variations in the incoming supply water as captured in the medium regime (control). Chlorine concentration was boosted (dosing with a 1:15 v/v dilution of 12% sodium hypochlorite) or reduced (dosing with 1% sodium ascorbate) for the high and low regimes, respectively. Dosing solutions were added into the tank of the appropriate loop via a peristaltic pump (Watson and Marlow 505). On average, the free-chlorine concentrations were 0.05 mg L−1 (±0.06), 0.45 mg L−1 (±0.05) and 0.80 mg L−1 (±0.16) in the low, medium and high regimes, respectively. All other water quality parameters (and variation therein) were maintained across the three chlorine regimes by running the three systems co-currently.
Biofilm samples were collected at day 0 (sampled 90 minutes into each chlorine regime), day 28 and post-flush (after completion of the entire flushing phase), without draining the loops to limit the impact of sampling on the biofilms within the systems. Sterile coupons were used as negative controls for the biofilm removal stage. For flow cytometry analysis biofilms were taken in triplicate, and biofilm was removed from the insert of the PWG coupons (surface area 90 mm2). For qPCR analysis biofilm was removed from the outer coupon (n = 5, surface area 314.16 mm2), to provide more biomass for downstream DNA based analyses. In all instances, biofilm samples were obtained from the coupon surface using the optimised removal and homogenisation methodology. The cell concentrations within the resulting suspensions were then analysed using either the flow cytometry method described previously or the qPCR method detailed in Fish and Boxall.29 Briefly, qPCR entailed filtering biofilm suspensions (47 mm, 0.22 μm pore nitrocellulose membrane; Millipore, USA) and extracting DNA using the proteinase K chemical lysis method, with cetyltrimethyl ammonium bromide (CTAB) incubation.26,30 Copies of the bacterial 16S rRNA gene and fungal ITS region were quantified using a StepOne qPCR system (Applied Biosystems), including internal standard curves from environmental samples (R2 ≥ 0.984) and internal calibration standards to normalise/calibrate data and enable comparisons between plates. The primers used were Eub338 (5′-ACTCCTACGGGAGGCAGCAG-3′) and Eub518 (5′-ATTACCGCGGCTGCTGG-3′)29,31,32 for bacteria, and ITS1F (5′-TCCGTAGGTGAACCTGCGG-3′) and 5.8S (5′-CGCTGCGTTCTTCATCG-3′)29,33,34 for fungi. All qPCR reactions were undertaken in triplicate and amplified according to the Quantifast SYBR Green PCR kit (Qiagen, UK): 12.5 μL QuantiFast SYBR Green PCR MasterMix, 9 μL nuclease free water (Ambion, Warrington, UK), 1.25 μL of each primer (10 mM) and 1 μL of DNA template (or nuclease free water for the controls). The cycling conditions used for the qPCR were 95 °C for 5 min, then 35 cycles of 95 °C for 10 s and 60 °C for 30 s. The number of gene copies was determined using the StepOne software.
2.8 Data analysis
Due to different datasets being used in this research, a range of statistical tests were applied, with the p-values (significance level was <0.05) being reported along-side any other relevant values specific to each test. The normality of data was analysed using the Shapiro–Wilk test. Parameters were not normally distributed, therefore they were compared using non-parametric statistics, specifically Kruskal–Wallis (for comparison of >2 datasets, df = 2 in all cases, χ2 values presented in results) or Wilcoxon (for comparison of two datasets, W values reported). All statistical analysis and graphical plots were generated in R v4.1.0 with a significance level of p < 0.05. R packages used include: ggplot2, grid and beeswarm.3 Results and discussion
3.1 Biofilm removal: brushing optimisation
Optimisation of the brushing process to fully remove DWDS biofilm from the surface of a pipe section (in this case a BMD) was determined using mature biofilms (developed for 6 months), which would likely have a greater biomass than younger biofilms.35 Thus the optimised protocol would be suitable for use across a range of ages of biofilm samples. Assessment of the efficiency of biofilm removal using different iterations of brushing and rinsing is shown in Fig. 2, with respect to cell quantity (Fig. 2A) and viability (Fig. 2B) in the biofilm suspension generated. The largest proportion of total and intact cell were removed after five brushes, followed by a magnitude difference between subsequent sets of five brushes. The majority of cells (average of 95% TCC and 98% ICC) were removed after 5 brushes, with an additional 4% of TCC and 1% of ICC removed by increasing to 10 brushes. Very minimal amounts of additional cells (<1% of TCC and ICC) were removed after 15 brushes. No further cells were detected in the 20 or 25 brush conditions (Fig. 2A, B and S3† shows non-logged data), indicative that all cells had been removed from the BMD after 15 brushes.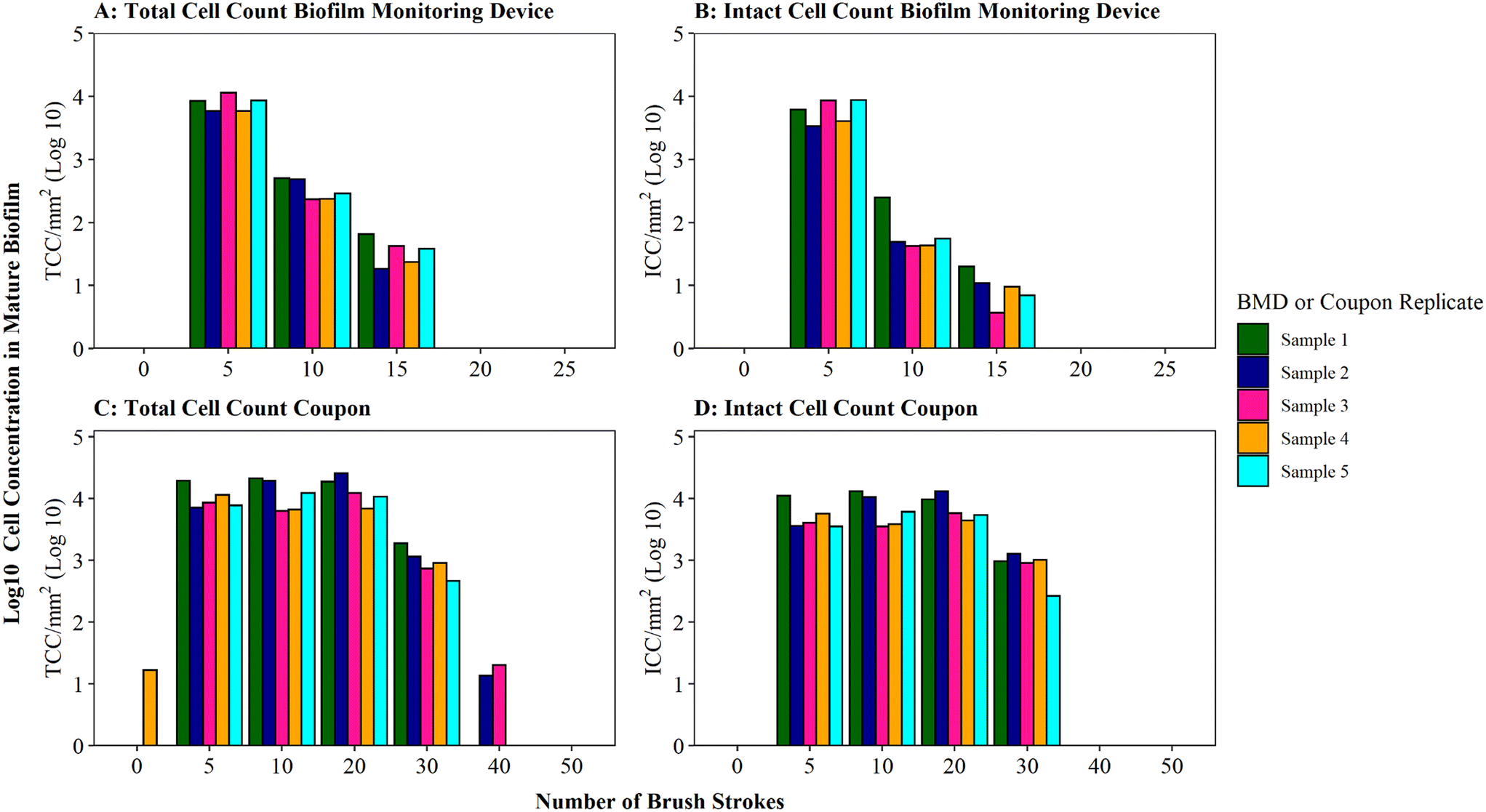 | ||
| Fig. 2 Brushing efficiency to remove biofilm from biofilm monitoring devices (A and B) and high-density polyethylene coupons (C and D). Total cell counts (TCC) and intact cell counts (ICC) obtained from each BMD or coupon (n = 5) are presented. Brushes refers to brush strokes. “0” brushes is defined as phosphate buffer solution (PBS) poured over the surface of the BMD or coupon to act as a control. Note data is logged so brush 0 count in panel C is only 17 cells compared to 11311 at brush 5 (see Fig. S5† for raw data). | ||
Coupons are a commonly used sampling surface when studying drinking water biofilms36–38 and offer an alternative to pipe sections. As BMD and coupon sampling surfaces require different brushing approaches, the impact of brushing longevities (in both horizontal and vertical orientations) on cell removal was tested specifically with mature drinking water biofilms developed on HDPE coupons (Fig. 2C and D). The majority of cells were removed after 20 brushes (average of 97% of the total TCC and 96% of the total ICC removed was achieved at this stage). After 30 brushes, on average, 100% of biofilm TCC and ICC had been removed from the coupons (additional brushing did not remove any further cells; Fig. 2C, D and S3†).
A greater number of brush strokes was needed to remove cells from mature biofilms developed on coupons (30 brushes) compared to BMD sections (15 brushes). This is likely due to the difference in the brush type (and strokes) required for the two sample types, which differ in surface type, shape and area being sampled (“top” coupon surface with area of 400 mm2; internal BMD pipe surface with area: 915.78 mm2). It could also be a function of the coupon biofilms being grown under different hydraulics (flow rate, shear stress) in the unpressurised bioreactor (0.4 L s−1, 0.12 Pa) compared to the pressurised BMD (1 L min−1, 0.16 Pa). Hydraulics have been repeatedly documented to influence biofilm volume, EPS composition, cohesive strength, morphology, growth rates and community composition.39–45 Biofilm–hydraulic trends do not converge in the literature, with some studies reporting increased density and adhesion/cohesion with increased shear stress or velocity41,45 and others observing the opposite, that adhesion/cohesion is reduced under greater ranges of velocity.42,44 The range of impacts of hydraulics on biofilm characteristics is likely due to different operating conditions and sampling procedures. This emphasises the need to establish robust, repeatable methods for studying and sampling biofilms from full-scale (and operational) systems in order to generate relevant data to enhance our understanding of the complex relationship between shear stress and drinking water biofilm formation.
The impact of brushing longevity on cell viability was evaluated via analysis of the proportion of ICC (as a percentage of TCC) for the mature biofilms sampled from BMDs and coupons. Biofilm suspensions generated from both sampling surfaces (BMD and coupons) fluctuated in their ICC proportion as the number of brush strokes increased and between replicates. No consistent trend between brushing longevity and ICC percentage was observed, which is a reflection of the biofilm heterogeneity. On average, for BMD biofilms, the ICC proportion was greatest in the initial suspension (average ICC% of 73% after 5 brushes), dropping and then increasing slightly (averages of 23% after 10 brushes and 32% after 15 brushes; Fig. S4†). It is possible that during the initial brushing stages intact cells were preferentially removed from the BMD if they were in less well-adhered or top layers of biofilm, which may have been removed first. In contrast, for the coupon biofilms the ICC proportion was fairly stable for the first three rounds of removal (average ICC% of 50% after 5 brushes, 56% after 10 brushes, 53% after 20 brushes) with a greater proportion of ICC in the suspension removed with 30 brushes (average ICC% of 82%, Fig. S5†). This could indicate that intact cells from the coupon biofilms were more prevalent in the deeper or more strongly-adhered areas of biofilms. Depth related viability profiles have not been clearly established for drinking water biofilms but studies of dental biofilms have reported similar contrasting trends, with non-viable cells often dominating either the very outer layer of the biofilm46 or the deeper layer of the biofilm.46–48 These trends could be governed by environmental parameters such as nutrient or oxygen concentration47 but the drivers governing viability depth profiles have yet to be established. It should be noted though that despite the average trends, both the coupon and BMD biofilms showed biological variation in the ICC proportions and as such any variation in ICC% with brushing longevity may purely be a reflection of biofilm heterogeneity (Table S3†). Nevertheless, the increase in the proportion of intact cells between 10 and 15 brushes for BMD biofilms, and 20 and 30 brushes for HDPE coupon biofilms, suggests that brushing was not having a kill effect. Therefore, the optimised brushing longevities should result in repeatable, high yield biofilm suspensions and preserve the integrity of intact cells to ensure that a representative biofilm sample is analysed.
3.2 Biofilm removal: brushing vs. sonication comparison
When comparing the efficiency of brushing (using the optimised protocol) and sonication with glass beads, as per Proctor, Gächter,20 for removing biofilms from a pipe section (Fig. 4), it was found that brushing resulted in a significantly greater cell yield (brush first vs. sonicate first TCC W = 25, p ≦ 0.01; ICC W = 25, p ≦ 0.01). On average, brushing removed 90% of the entire TCCs detected in a sample when used as the primary removal method (BMD pipes 1–5, Fig. 4A); subsequent sonication removed very few cells (an order of magnitude less). This demonstrates that brushing would remove the majority of the cells. In contrast, when sonication with glass beads was the primary removal method (BMD pipes 6–10), an average of 51% of all the TCCs removed were detached in the first removal phase with subsequent brushing removing a similar number of cells (on average 49% of TCCs). The average total number of TCCs removed (i.e. the sum of cells obtained from both removal phases per pipe) was similar for all pipe samples (pipes 1–5 average of 720 cells, pipes 6–10 average of 610 cells; Fig. 3A). This demonstrated no impact of the order of removal methods on overall cell yield from a sample.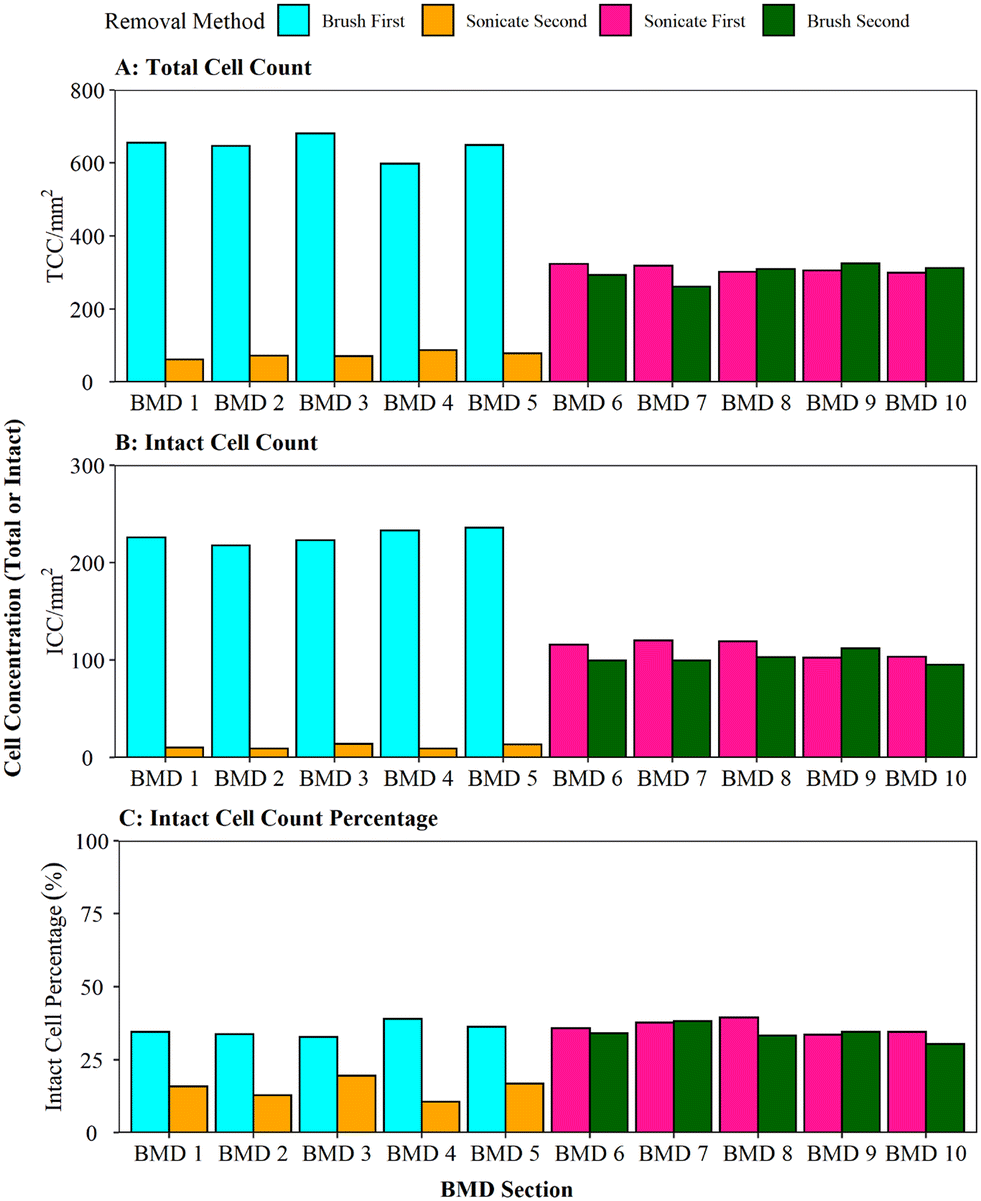 | ||
| Fig. 3 Comparison of brushing and sonication techniques for removing biofilm from a pipe section (biofilm monitoring device). (A) Total cell counts (TCC) and (B) intact cell counts (ICC) obtained from each BMD section are presented, along with (C) ICC as a proportion of the TCC. Brushing refers to the protocol optimised in this study. Sonication is with glass beads and a water bath. | ||
The same trends were seen with ICC (Fig. 3B): brushing as the primary removal method generated a significantly greater yield of cells than using sonication with glass beads as the primary removal method (95% vs. 52% of all cells detected, respectively). Brushing was therefore found to be the more efficient cell removal method. The total number of ICCs removed was conserved across all the pipes, irrespective of removal method order (pipes 1–5 average of 238 cells, pipes 6–10 average of 214 cells, Fig. 3B). When comparing ICC proportions (Fig. 3C), brushing first, sonicating first and brushing second all had similar ICC proportions (30–39%), but the cells removed in the sonication second treatment were less likely to be intact (average 15% ICC proportion; brush second vs. sonicate second TCC W = 25, p ≦ 0.01; ICC W = 25, p = 0.012). This shows that sonicating alone did not necessarily impact the ICC%, however the use of sonication after brushing may have had a kill effect because most of the biofilm (cells and EPS) had already been removed by brushing, hence any remaining cells may have been more exposed to the sonication impacts.
Whilst brushing has been evidenced to be the more efficient primary biofilm removal method, secondary removal via sonication did remove a few more cells but this data indicates that application of a dual removal approach should be undertaken with care if viability of the sample is being assessed. Peng, Shao49 combined swabbing with sonication for successful removal of biofilms from pipe sections for microbiome analysis, although it is unclear what proportion of biomass was removed with each method or the impact that the sonication had on cell integrity/viability.
Ascertaining the most efficient DWDS biofilm removal method with respect to yield and viability is of importance for any downstream quantification and characterisation to ensure that analytical techniques are being applied to a representative sample. The data presented herein demonstrates for the first time that brushing is more efficient than sonication with a water bath for biofilm removal as brushing yields greater cell counts and does not affect viability. Previous research has compared scraping, swabbing and stomaching for biofilm removal from polycarbonate coupons within a bench-top drinking water annual reactor found that stomaching consistently yielded a higher number of culturable and total bacterial cells, and was more repeatable between users (unlike scraping which was subject to variation between individual researchers).24 However, during stomaching the whole coupon is placed in a sterile bag with deionised water, consequently cells mobilised in the stomacher could include cells from the surfaces of the coupons that had not been exposed to the bulk water.24 Similarly, not all sample types (e.g. HDPE coupons or operational pipes) are suitable for glass-bead sonication, and instead the only practical options for biofilm removal are swabbing,50,51 or brushing.26,52 For downstream flow cytometry, brushing conveys additional benefits over swabbing or scraping as it limits debris, which has been previously observed to impact flow cytometry detection limits.53 As well as ensuring representative samples are analysed, the insights provided here on biofilm removal methods will help with increasing the yield of biomass recovered, which remains one of the greatest challenges in characterising drinking water biofilms. This is especially true with respect to DNA yields for microbiome analysis via metagenetic or metagenomics sequencing, as discussed in Peng, Shao,49 where sonication methods for improved DNA yield from drinking water biofilms are optimised. Results from this study suggest that brushing provides an even more efficient removal method than sonication as assessed via cell enumeration and viability, though there is no study directly comparing the impact of these two biofilm removal techniques on subsequent microbiome characterisation of drinking water derived samples.
3.3 Biofilm homogenisation: optimisation and method comparison
Where biofilms were removed using brushing, downstream vortexing for 0.5 to 8 minutes had no impact on the number of TCC or ICC recorded when compared with controls (Fig. 4A and B; brushing removal with vortexing vs. brushing removal control TCC W = 2.292, p = 0.130; ICC W = 2.048, p = 0.152). There were no significant differences in TCC or ICC singlet–doublet proportions (Table S2† vortex vs. control singlet–doublet percentage TCC W = 20, p = 0.151; ICC W = 17, p = 0.421, across all time points). Control samples (brushing removal, no vortexing) had singlet–doublet proportions TCC ≥ 97%, and ICC ≥ 99% (with one ICC control sample analysed after 8 minutes recording 88% classed as an outlier; see Table S2†). Samples that were vortexed from 0.5 to 8 minutes had average singlet–doublet proportions TCC ≥ 98% (with one TCC vortex sample analysed after 8 minutes recording 88% classes as an outlier; see Table S2†) and ICC average singlet–doublet proportions ≥98%. Therefore, when using brushing as the biofilm removal method samples were already well homogenised and there was very limited benefit of additional sample homogenisation using vortexing.
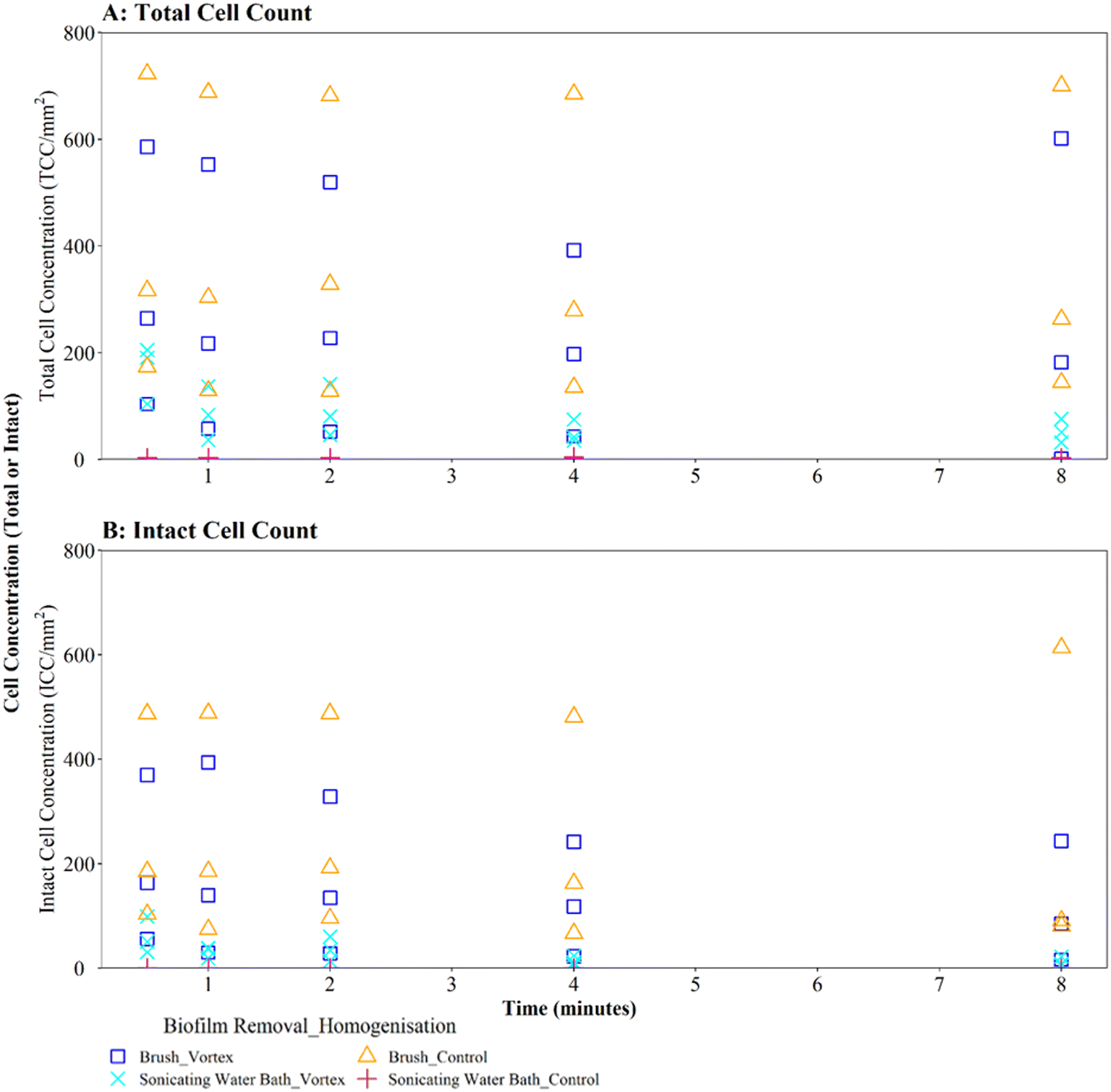 | ||
| Fig. 4 Cell counts obtained when using different vortexing longevities to homogenise drinking water biofilms removed from sample surfaces by brushing or sonication via a water bath (as indicated in key). Biofilms were 3-months old. Control samples were not vortexed. (A) Total cell counts (TCC) and (B) intact cell counts (ICC) obtained from each BMD section (n = 3) are presented in each panel. | ||
In contrast, where biofilms were removed using the sonicating water bath, vortexing for 0.5 to 8 minutes resulted in a significantly greater number of TCC or ICC compared with controls (Fig. 4A and B; sonicating removal with vortexing vs. sonicating removal control TCC, W = 225, p ≦ 0.01; ICC, W = 255 p ≦ 0.01). Control samples (no vortexing following sonication removal) had a singlet–doublet proportion of ≥87% compared to ≥98% for samples vortexed for 0.5 minutes, indicating a benefit of vortexing after biofilm removal using a sonicating water bath. However, the singlet–doublet percentage remained at an average of ≥99% when increasing the vortexing time up to 8 minutes, showing no additional benefit of vortexing biofilm samples for longer than 0.5 minutes (Table S2†).
Homogenisation via sonicating needle, following biofilm removal via brushing, had no significant impact on TCC or ICC when compared with controls (Fig. 5A and B; brushing removal with sonicating needle vs. brushing removal control TCC, W = 143, p = 0.217; ICC, W = 90, p = 0.367). The control samples (no sonication after brushing) had an average TCC and ICC singlet–doublet percentage of 99% at each time point, whereas the average singlet–doublet percentage of sonicated samples was ≥95% for TCC and ≥98% for ICC at each time point (Table S2†). However, the difference between sonicated and control singlet–doublet percentages was not significant (Table S2,† TCC, W = 10, p = 0.691; ICC, W = 11, p = 0.841). The control samples which had been removed using brushing already met the ≥98% singlet–doublet threshold for homogenised samples, so sonication provided no additional benefit. The control samples (no sonication after brushing) and sonicating samples had similar TCC and ICC singlet–doublet percentages from 0.5–2 minutes (≥98% for all samples), however after 4 minutes the singlet–doublet percentages for the sonicating samples started to drop indicating no homogenisation benefit from sonicating. Conversely, following biofilm removal using a sonicating water bath and glass beads, biofilm samples which were homogenised via sonication with a needle had significantly different TCC and ICC compared to controls which did not undergo the homogenisation sonication across all time points (Fig. 5A and B; TCC, W = 225, p ≦ 0.01; ICC, W = 225, p ≦ 0.01). Using a sonicating needle was found to help homogenise biofilm samples that were removed using sonication (water bath and glass beads) with sonicated samples having a higher singlet–doublet percentage (TCC ≥ 99%, ICC ≥ 97%) than controls (TCC ≥ 97%, ICC ≥ 91%), but the difference was not significant (TCC singlet–doublet percentage W = 14, p = 0.843; ICC singlet–doublet percentage W = 18, p = 0.301). Sonication with a needle provided some benefit up to 1 minute sonication, with ICC cell counts increasing for one of the triplicates (ICC increased from 75 to 78 mm2 triplicate 3). However, intact cell counts after 8 minutes of sonication were on average 65% lower than those after 2 minutes, indicating a kill effect when samples are sonicated for longer than 2 minutes.
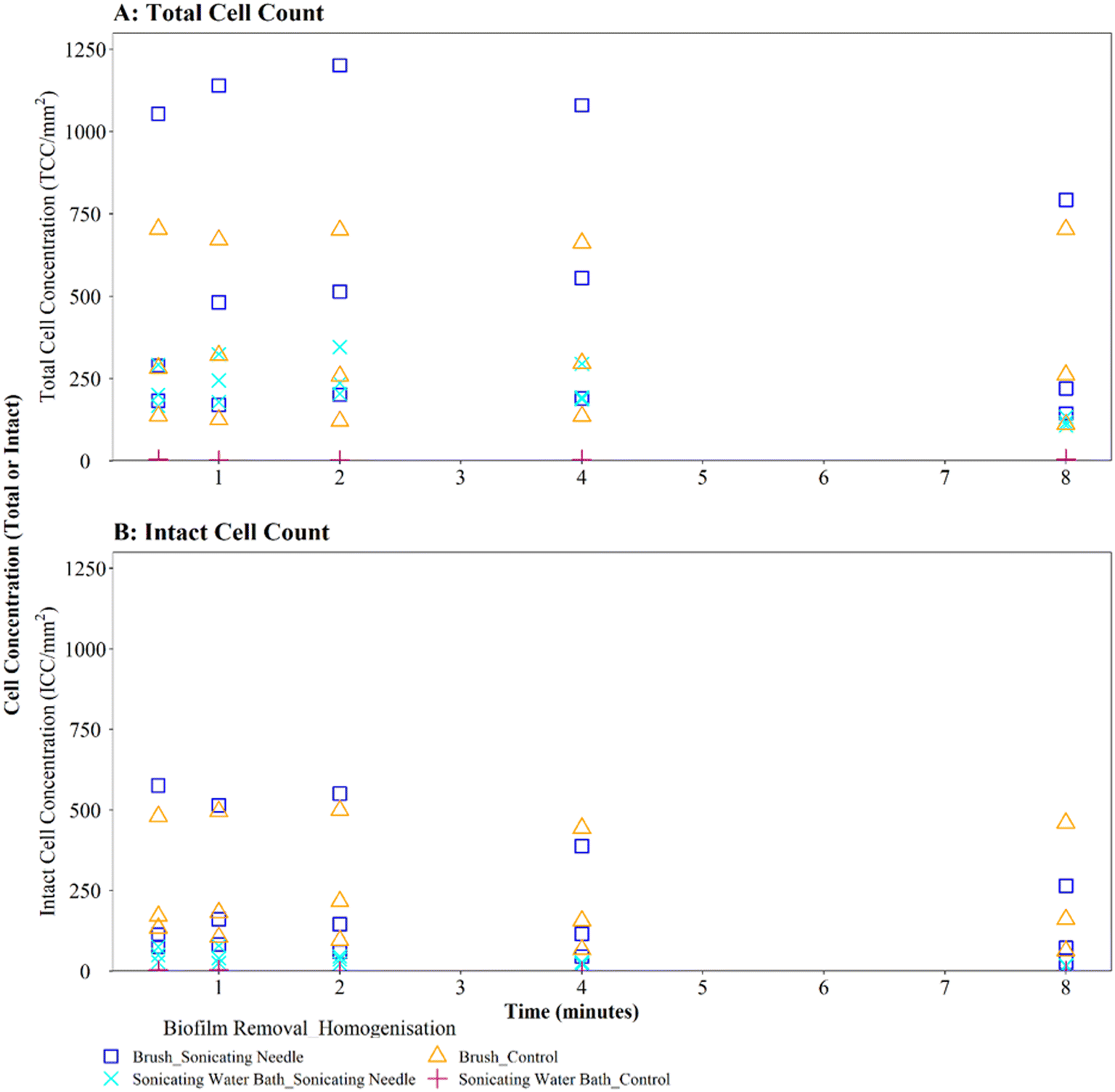 | ||
| Fig. 5 Cell counts obtained when using different sonication (needle) longevities to homogenise drinking water biofilms. 3-Month old biofilms were removed from a BMD by brushing or water bath sonication with glass beads as indicated. Control samples did not undergo a homogenisation step. (A) Total cell counts (TCC) and (B) intact cell counts (ICC) obtained from each pipe section (n = 3) are presented. | ||
Irrespective of vortexing, cell counts were lower (TCC and ICC <205 cells per mm2 across all time points) when biofilms were removed via sonication with glass beads, compared to those obtained when using brushing (TCC and ICC <723 cells per mm2 across all time points) (Fig. 4A and B). This further demonstrates that brushing was a more efficient biofilm removal method as well as conveying a benefit for homogenisation.
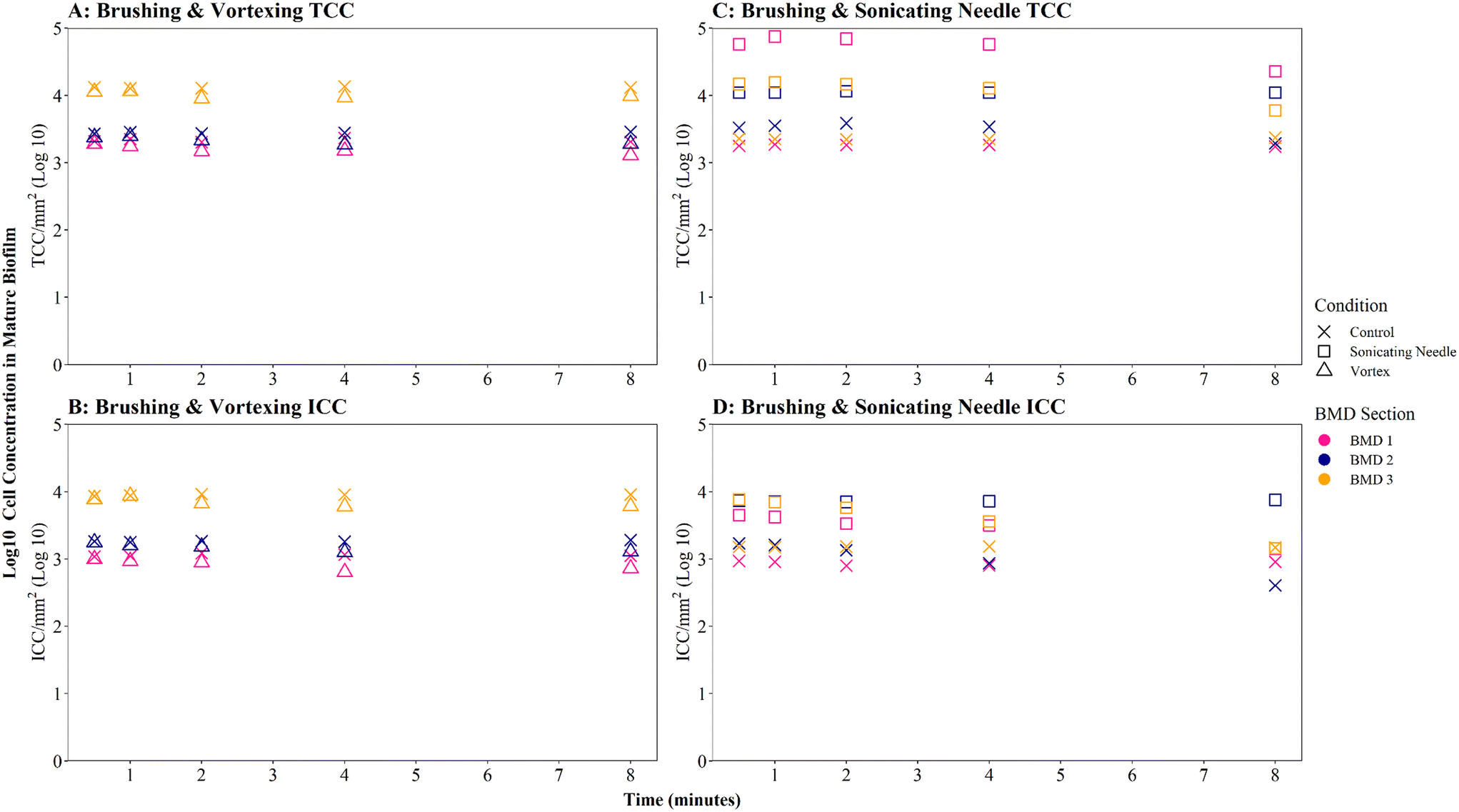 | ||
| Fig. 6 Cell counts obtained from mature biofilm samples following homogenisation via vortexing (A and B) or sonication (needle) (C and D). Control samples were not homogenised. Total cell counts (TCC) and intact cell counts (ICC) obtained from each BMD section (1–3) are presented as log transformed data. | ||
Brushed mature biofilm samples which were homogenised with a sonicating needle for 0.5 to 8 minutes displayed a statistically significant change with respect to TCC (Fig. 6C, W = 225, p ≦ 0.01) and ICC (Fig. 6D, W = 211, p ≦ 0.01) when compared with controls, across all time points, with sonicated samples having more cells. The longevity of needle sonication did have an impact on TCC, which initially increased from an average (mean) of 27910 cells per mm2 after 0.5 minutes to 34009 cells per mm2 after 1 minute but then remained mostly stable (Fig. 6C). However, longevity of needle sonication had a kill-effect on the samples as the mean ICC decreased by almost 50% from 6532 cells per mm2 at 0.5 minutes, to 3495 cells per mm2 after 8 minutes, suggesting that cells were lysed by sonication (Fig. 6D). Singlet–doublet analysis found that sonicated samples were slightly more homogenised than controls (no needle sonication) but that all samples had singlet–doublet percentages above the 96% (average singlet–doublet percentage sonicated TCC ≥ 99%, ICC ≥ 98% and controls TCC ≥ 98%, ICC ≥ 96%, Table S4†). The results conclude that brushing as a biofilm removal method was able to homogenise samples (singlet–doublet ≥98%), without the need for any additional downstream homogenisation such as sonication. Furthermore, sonicating drinking water biofilm samples for more than 0.5 minutes would results in cell lysis, indicating that if optimised brushing is used as the biofilm removal method there is no requirement for an additional homogenisation phase.
3.4 Verification of flow cytometry cell quantification for biofilms from a full-scale DWDS
Temporal dynamics of drinking water biofilm cell concentrations are presented in Fig. 7, for three chlorine regimes, quantified using flow cytometry (Fig. 7A) and qPCR methods (Fig. 7B and C). qPCR is a commonly used molecular method for quantifying planktonic and biofilm cells from drinking water treatment,55 wastewater treatment,56 premises plumbing,57 irrigation water58 and drinking water distribution systems.29 At day 0, there were typically no cells detected at the pipe wall and no differences between chlorine regimes with either quantification method (flow cytometry, χ2 ≥ 2.0, p ≥ 0.174; qPCR χ2 ≥ 4, p ≥ 0.117).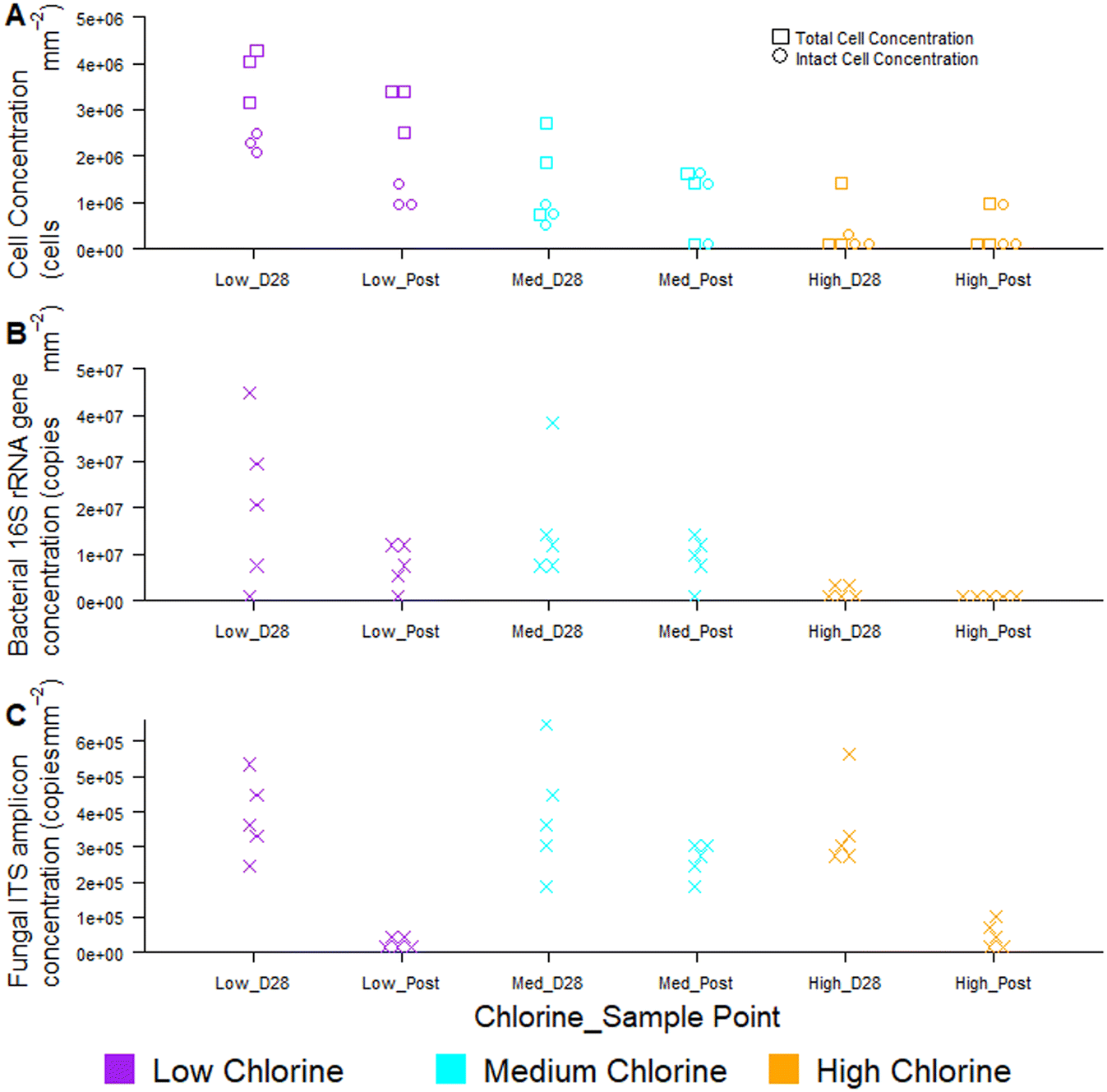 | ||
| Fig. 7 Impact of chlorine on drinking water biofilm cell quantities as ascertained by (A) flow cytometry, including viability assessment and molecular analysis of bacterial (B) and fungal genes (C) via qPCR. Raw data is shown in all plots, (A) n = 3, (B and C) n = 5. Average residual chlorine concentrations for each chlorine condition were: low = 0.05 mg L−1 (±0.06), “Med” = medium, 0.45 mg L−1 (±0.05), high = 0.80 mg L−1 (±0.16). | ||
Biofilm cell concentrations increased during the growth phase, as detected using both flow cytometry and qPCR (Fig. 7). Day 28 biofilm TCC (χ2 = 6.5, p = 0.039) and ICC (χ2 = 7.2, p = 0.027) decreased significantly with increasing chlorine concentrations at day 28 (Fig. 7A), demonstrating the same trends as observed for bacterial gene concentrations (χ2 = 8, p = 0.019, Fig. 7B). Similarly, biofilm TCC, ICC and bacterial gene copies were all a magnitude less in the high chlorine biofilms, compared to the low chlorine biofilms (Fig. 7A and B), suggesting a greater rate of biofilm growth under lower chlorine residual conditions. Cell concentrations were higher than reported with flow cytometry analysis of simulated DWDS previously59 and more similar to shower hose biofilm cell counts.20 This could be due to differences in methods, particularly biofilm removal and homogenisation, which was optimised and specifically assessed herein (homogenisation of 98% for all data in Fig. 7A, based on singlet–doublet analysis). Absolute concentrations understandably differed between analytical approaches due to the fundamental differences in how the methods quantify cells.
Fungi were present in the biofilms at lower abundances than bacteria (1–2 orders of magnitude less; Fig. 7C) and abundances did not differ between chlorine regimes (as discussed in Fish et al. 202022), as demonstrated by qPCR. Whilst flow cytometry did not facilitate a distinction between taxa, this method provided rapid insight to viability of cells in the biofilm. The proportion of ICC (as a percentage of TCC) was, on average, more similar between the low and medium chlorine regimes (55% and 61%, respectively) than the high chlorine regimes (average ICC% of 20%), though these trends were not significant (χ2 > 1, p > 0.15). Viability information was not available from the qPCR methods, as qPCR does not allow viable cells to be distinguished from dead cells.60,61 The use of qPCR along with propidium monoazide (PMA) (a photoreactive DNA-binding dye) can penetrate the membrane of compromised cells and block PCR amplification.60 Although the PMA approach has been applied to planktonic organisms, it has not yet been successfully applied to biofilms and the method has not yet been optimised with studies reporting different PMA efficiencies62,63 and a potential kill-effect as PMA is increasingly toxic at higher concentrations.62
Regardless of chlorine regime, biofilm cell counts reduced during the flushing intervention (post-flush biofilms had fewer TCC, ICC, bacterial and fungal gene copies than day 28 biofilms; Fig. 7) but did not return the pipes to day 0 conditions (day 0 vs. post-flush, qPCR: W = 0, p ≤ 0.01, flow cytometry W = 0, p ≤ 0.07). No clear or consistent changes in LNA or HNA ratios were detected, suggesting that the flushing intervention removed cells from each category in a similar way. Flow cytometry consistently showed the same trends in biofilm cell concentrations as qPCR, whilst establishing these trends more quickly and from a smaller sample area, thus verifying the use of the optimised biofilm removal and homogenisation method to prepare samples for downstream flow cytometry analysis to meaningfully characterise biofilm microbial loads. Using the drinking water biofilm sampling devices presented herein also ensures that the biofilm samples presented for analysis are representative of operational DWDS conditions. Combining flow cytometry with these sampling approaches provides an avenue for rapid cell concentration and viability analysis that goes beyond the bulk-water as is often currently studied. Although flow cytometry did not distinguish between taxa, emerging analytical approaches such as “Phenoflow” have been applied to demonstrate differences in microbiome in bulk-water64,65 and medical contexts.66 The “Phenoflow” approach demonstrates the potential to obtain more information from flow cytometry fingerprints of biofilm samples, although the method has not yet been applied to drinking water biofilms from full-scale or operational systems.
The data presented herein is based on analysis of drinking water biofilms from plastic pipes (HDPE being the most common material for new pipes in the UK and Europe), application to other materials would need to be further explored. However, a study by Waller, Packman53 compared TCCs, HPCs and biofilm bio-volume (as assessed by confocal laser scanning microscopy) to quantify biofilms grown on different materials within a pipe loop sampling device (and removed via sonication). Variability was reported for all measurements across the materials but biofilm TCCs assessed using flow cytometry showed the least variability (0.04 to 42% standard errors of measured counts for all three coupon materials).53 Ultimately, a multiple method approach, combining flow cytometry, and molecular based analyses (alongside in situ microscopy, e.g. fluorescent in situ hybridization, confocal laser scanning microscopy) would be recommended to characterise biofilms. Integration of these techniques would lead to a more comprehensive assessment of cell quantification (rapid with flow cytometry), cell viability, community composition (including screening for specific taxa or functional genes), and spatial distribution or biofilm architecture (including EPS). This holistic approach is integral to understanding biofilm dynamics within DWDS. However, this study has also demonstrated the critical importance of upstream biofilm removal and homogenisation methods in generating representative samples, with high yields to ensure downstream analysis is robust and repeatable.
4 Conclusions
The study presented here has developed a robust, optimised method for the preparation of drinking water biofilm samples (from different drinking water experimental systems) for flow cytometric enumeration of cells, without affecting cell integrity. Protocols for the removal of drinking water biofilms using brushing were optimised to maximise cell yield with no kill effect. The optimal brushing removal strategy differed slightly with respect to total number of brush strokes between different sampling surfaces: 30 brushes for coupons and 15 for pipe sections of the BMD. Compared to biofilm removal using sonication with glass beads, brushing can increase cell yield from pipe surfaces by up to nine times without affecting cell viability proportions, establishing brushing as the more efficient removal method. Using brushing and sonication with glass beads in sequence (where this is feasible), could yield a slightly higher cell yield but the secondary sonication impacts intact cell counts, hence this approach is not suitable for any samples that are under-going downstream viability analysis.Overall, brushing was the optimum method for removing the greatest number of biofilm cells from a drinking water pipe coupon or pipe section, with reduced (or no) cell lysis and also homogenisation of sample without need for further sample interrogation. This was confirmed for biofilms of different ages though in the case of more mature biofilms, the use of a sonicating needle for no more than 0.5 minutes may offer some benefit for sample homogenisation. When applied to biofilms from a full-scale pipe system, the optimised biofilm removal, sampling preparation and flow cytometry methods consistently showed the same trends in biofilm cell concentrations as obtained via molecular analysis (qPCR), but more quickly and from a smaller sample area.
The successful application of the removal and preparation method presented, to drinking water biofilms of different ages from different sampled surfaces (pipes and coupons), confirms this approach as a repeatable and robust way to assess biofilms within full-scale DWDS. Ensuring appropriate and optimised biofilm removal and sample preparation is critical for any subsequent characterisation or analysis of the samples to be representative and informative. In combination with advances in flow cytometry, application of the optimised sampling and preparation approach to biofilms from full-scale and operational DWDS has the potential to provide novel insights into biofilm fouling rates and assessment of operational interventions for managing microbial load.
Author contributions
FCP and KEF conceived, designed and performed the removal and homogenisation experiments, FCP analysed this data set, with input from KEF. KEF secured the funding for the flow cytometry and qPCR comparisons, designed and conducted these experiments, analysing and interpreting this dataset. FCP and KEF interpreted the results and wrote the paper.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
KEF and FCP were supported by the National Biofilm Innovation Centre (NBIC) grant (01POC18034), and the UKRI research grants “To Chlorinate or Not to Chlorinate?” (EP/W037270/1 & EP/W037475/1) and “TWENTY 65: Tailored Water Solutions for Positive Impact” (EP/N010124/1). Molecular analyses were performed by KEF at the Natural Environment Research Council (NERC) Environmental Omics Facility (NEOF) at the University of Sheffield (http://neof.org.uk/). The construction of the unpressurised bio-reactor was supported by NBIC (02POC19040). For the purpose of open access, the author has applied a creative commons attribution (CC BY) license to any author accepted manuscript versions arising. The authors of this paper would like to thank UK NEQAS, in particular Liam Whitby and Charlotte Wynn, for discussion around flow cytometry analysis and assistance with singlet–doublet gating.References
- E. I. Prest, F. Hammes, M. C. van Loosdrecht and J. S. Vrouwenvelder, Biological Stability of Drinking Water: Controlling Factors, Methods, and Challenges, Front. Microbiol., 2016, 7, 45 CrossRef PubMed.
- I. Douterelo, M. Jackson, C. Solomon and J. Boxall, Microbial analysis of in situ biofilm formation in drinking water distribution systems: implications for monitoring and control of drinking water quality, Appl. Microbiol. Biotechnol., 2016, 100(7), 3301–3311 CrossRef CAS PubMed.
- G. Liu, E. J. Van der Mark, J. Q. Verberk and J. C. Van Dijk, Flow cytometry total cell counts: a field study assessing microbiological water quality and growth in unchlorinated drinking water distribution systems, BioMed Res. Int., 2013, 2013, 595872 CAS.
- M. Berney, M. Vital, I. Hulshoff, H. U. Weilenmann, T. Egli and F. Hammes, Rapid, cultivation-independent assessment of microbial viability in drinking water, Water Res., 2008, 42(14), 4010–4018 CrossRef CAS PubMed.
- F. Hammes, M. Berney, Y. Wang, M. Vital, O. Koster and T. Egli, Flow-cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes, Water Res., 2008, 42(1–2), 269–277 CrossRef CAS PubMed.
- M. Gabrielli, A. Turolla and M. Antonelli, Bacterial dynamics in drinking water distribution systems and flow cytometry monitoring scheme optimization, J. Environ. Manage., 2021, 286, 112151 CrossRef CAS PubMed.
- L. C. Kennedy, S. E. Miller, R. S. Kantor and K. L. Nelson, Effect of disinfectant residual, pH, and temperature on microbial abundance in disinfected drinking water distribution systems, Environ. Sci.: Water Res. Technol., 2021, 7(1), 78–92 RSC.
- F. Hammes and T. Egli, Cytometric methods for measuring bacteria in water: advantages, pitfalls and applications, Anal. Bioanal. Chem., 2010, 397(3), 1083–1095 CrossRef CAS PubMed.
- Y. Wang, F. Hammes, K. De Roy, W. Verstraete and N. Boon, Past, present and future applications of flow cytometry in aquatic microbiology, Trends Biotechnol., 2010, 28(8), 416–424 CrossRef CAS PubMed.
- R. Cheswick, E. Cartmell, S. Lee, A. Upton, P. Weir and G. Moore, et al., Comparing flow cytometry with culture-based methods for microbial monitoring and as a diagnostic tool for assessing drinking water treatment processes, Environ. Int., 2019, 130, 104893 CrossRef PubMed.
- H. C. Flemming, S. L. Percival and J. T. Walker, Contamination potential of biofilms in water distribution systems, Water Sci. Technol.: Water Supply, 2002, 2(1), 271–280 CAS.
- K. E. Fish, A. M. Osborn and J. Boxall, Characterising and understanding the impact of microbial biofilms and the extracellular polymeric substance (EPS) matrix in drinking water distribution systems, Environ. Sci.: Water Res. Technol., 2016, 2(4), 614–630 RSC.
- S. Husband and J. Boxall, Predictive water quality modelling and resilience flow conditioning to manage discolouration risk in operational trunk mains, J. Water Supply: Res. Technol.--AQUA, 2015, 64(5), 529–542 CrossRef.
- J. El-Chakhtoura, P. E. Saikaly, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, Impact of Distribution and Network Flushing on the Drinking Water Microbiome, Front. Microbiol., 2018, 9, 2205 CrossRef PubMed.
- G. Quarini, E. Ainslie, M. Herbert, T. Deans, D. Ash and D. Rhys, et al., Investigation and development of an innovative pigging technique for the water-supply industry, Proc. Inst. Mech. Eng., Part E, 2010, 224(2), 79–89 CrossRef.
- F. C. Pick, K. E. Fish, S. Husband and J. B. Boxall, Non-invasive Biofouling Monitoring to Assess Drinking Water Distribution System Performance, Front. Microbiol., 2021, 12, 730344 CrossRef PubMed.
- P. Deines, R. Sekar, P. S. Husband, J. B. Boxall, A. M. Osborn and C. A. Biggs, A new coupon design for simultaneous analysis of in situ microbial biofilm formation and community structure in drinking water distribution systems, Appl. Microbiol. Biotechnol., 2010, 87(2), 749–756 CrossRef CAS PubMed.
- T. Juhna, D. Birzniece, S. Larsson, D. Zulenkovs, A. Sharipo and N. F. Azevedo, et al., Detection of Escherichia coli in biofilms from pipe samples and coupons in drinking water distribution networks, Appl. Environ. Microbiol., 2007, 73(22), 7456–7464 CrossRef CAS PubMed.
- K. Buckingham-Meyer, L. A. Miller, A. E. Parker, D. K. Walker, P. Sturman and I. Novak, et al., Harvesting and Disaggregation: An Overlooked Step in Biofilm Methods Research, J. Visualized Exp., 2022, 182, e62390 Search PubMed.
- C. R. Proctor, M. Gächter, S. Kötzsch, F. Rölli, R. Sigrist and J.-C. Walser, et al., Biofilms in shower hoses – choice of pipe material influences bacterial growth and communities, Environ. Sci.: Water Res. Technol., 2016, 2(4), 670–682 RSC.
- C. R. Proctor, M. Reimann, B. Vriens and F. Hammes, Biofilms in shower hoses, Water Res., 2018, 131, 274–286 CrossRef CAS PubMed.
- K. E. Fish, N. Reeves-McLaren, S. Husband and J. Boxall, Unchartered waters: the unintended impacts of residual chlorine on water quality and biofilms, npj Biofilms Microbiomes, 2020, 6(1), 34 CrossRef CAS PubMed.
- J. Liu, J. Li, L. Feng, H. Cao and Z. Cui, An improved method for extracting bacteria from soil for high molecular weight DNA recovery and BAC library construction, J. Microbiol., 2010, 48(6), 728–733 CrossRef CAS PubMed.
- G. A. Gagnon and R. M. Slawson, An efficient biofilm removal method for bacterial cells exposed to drinking water, J. Microbiol. Methods, 1999, 34(3), 203–214 CrossRef.
- P. S. Husband, J. B. Boxall and A. J. Saul, Laboratory studies investigating the processes leading to discolouration in water distribution networks, Water Res., 2008, 42(16), 4309–4318 CrossRef CAS PubMed.
- K. E. Fish, R. Collins, N. H. Green, R. L. Sharpe, I. Douterelo and A. M. Osborn, et al., Characterisation of the physical composition and microbial community structure of biofilms within a model full-scale drinking water distribution system, PLoS One, 2015, 10(2), e0115824 CrossRef PubMed.
- A. Cossarizza, H. D. Chang, A. Radbruch, M. Akdis, I. Andra and F. Annunziato, et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies, Eur. J. Immunol., 2017, 47(10), 1584–1797 CrossRef CAS PubMed.
- E. Gatza, F. Hammes and E. Prest, Assessing water quality with the BD Accuri™ C6 flow cytometer. White paper BD Biosciences, 2013.
- K. E. Fish and J. B. Boxall, Biofilm Microbiome (Re)Growth Dynamics in Drinking Water Distribution Systems Are Impacted by Chlorine Concentration, Front. Microbiol., 2018, 9, 2519 CrossRef PubMed.
- J. Zhou, M. A. Bruns and J. M. Tiedje, DNA Recovery from Soils of Diverse Composition, Appl. Environ. Microbiol., 1996, 62(2), 316–322 CrossRef CAS PubMed.
- D. J. Lane, 16S/23S rRNA sequencing, Nucleic acid techniques in bacterial systematics, 1991 Search PubMed.
- G. Muyzer, E. C. De Waal and A. Uitterlinden, Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA, Appl. Environ. Microbiol., 1993, 59(3), 695–700 CrossRef CAS PubMed.
- R. Vilgalys and M. Hester, Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species, J. Bacteriol., 1990, 172(8), 4238–4246 CrossRef CAS PubMed.
- M. Gardes and T. D. Bruns, ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts, Mol. Ecol., 1993, 2(2), 113–118 CrossRef CAS PubMed.
- F. C. Pick, K. E. Fish and J. B. Boxall, Assimilable organic carbon cycling within drinking water distribution systems, Water Res., 2021, 198, 117147 CrossRef CAS PubMed.
- G. Del Olmo, A. Ahmad, H. Jensen, E. Karunakaran, E. Rosales and C. Calero Preciado, et al., Influence of phosphate dosing on biofilms development on lead in chlorinated drinking water bioreactors, npj Biofilms Microbiomes, 2020, 6(1), 43 CrossRef CAS PubMed.
- M. W. Cowle, G. Webster, A. O. Babatunde, B. N. Bockelmann-Evans and A. J. Weightman, Impact of flow hydrodynamics and pipe material properties on biofilm development within drinking water systems, Environ. Technol., 2020, 41(28), 3732–3744 CrossRef CAS PubMed.
- S. Aggarwal, C. K. Gomez-Smith, Y. Jeon, T. M. LaPara, M. B. Waak and R. M. Hozalski, Effects of Chloramine and Coupon Material on Biofilm Abundance and Community Composition in Bench-Scale Simulated Water Distribution Systems and Comparison with Full-Scale Water Mains, Environ. Sci. Technol., 2018, 52(22), 13077–13088 CrossRef CAS PubMed.
- K. Fish, A. M. Osborn and J. B. Boxall, Biofilm structures (EPS and bacterial communities) in drinking water distribution systems are conditioned by hydraulics and influence discolouration, Sci. Total Environ., 2017, 593, 571–580 CrossRef PubMed.
- K. E. Fish, R. L. Sharpe, C. A. Biggs and J. B. Boxall, Impacts of temperature and hydraulic regime on discolouration and biofilm fouling in drinking water distribution systems, PLOS Water, 2022, 1(8), e0000033 CrossRef.
- H. Beyenal and Z. Lewandowski, Internal and external mass transfer in biofilms grown at various flow velocities, Biotechnol. Prog., 2002, 18(1), 55–61 CrossRef CAS PubMed.
- Y. Abe, S. Skali-Lami, J. C. Block and G. Francius, Cohesiveness and hydrodynamic properties of young drinking water biofilms, Water Res., 2012, 46(4), 1155–1166 CrossRef CAS PubMed.
- E. Tsagkari, S. Connelly, Z. Liu, A. McBride and W. T. Sloan, The role of shear dynamics in biofilm formation, npj Biofilms Microbiomes, 2022, 8(1), 33 CrossRef PubMed.
- S.-T. Khu, C. Xin, T. Wang, Y. Zhang and X. Zuo, Effects of hydraulic conditions on biofilm detached in drinking water distribution system, J. Water Process. Eng., 2023, 53, 103882 CrossRef.
- Y. Pechaud, M. Peyre Lavigne, Y. Bessiere, J. C. Ochoa, I. Queinnec and E. Paul, Influence of shear stress, organic loading rate and hydraulic retention time on the biofilm structure and on the competition between different biological aggregate morphotypes. Journal of Environmental, Chem. Eng., 2022, 10(3), 107597 CAS.
- C. K. Hope and M. Wilson, Biofilm structure and cell vitality in a laboratory model of subgingival plaque, J. Microbiol. Methods, 2006, 66(3), 390–398 CrossRef CAS PubMed.
- F. Dalwai, D. A. Spratt and J. Pratten, Modeling shifts in microbial populations associated with health or disease, Appl. Environ. Microbiol., 2006, 72(5), 3678–3684 CrossRef CAS PubMed.
- H. Beyenal, C. Yakymyshyn, J. Hyungnak, C. C. Davis and Z. Lewandowski, An optical microsensor to measure fluorescent light intensity in biofilms, J. Microbiol. Methods, 2004, 58(3), 367–374 CrossRef CAS PubMed.
- H. X. Peng, Y. Shao, Y. F. Zhang, R. W. Wang, D. Z. Zhu, H. Y. Chen and J. Q. Liu, Optimization of ultrasonic parameters for effective detachment of biofilm cells in an actual drinking water distribution system, J. Zhejiang Univ., Sci., A, 2020, 21(3), 167–178 CrossRef CAS.
- G. Liu, Y. Zhang, X. Liu, F. Hammes, W. T. Liu, G. Medema, P. Wessels and W. Van Der Meer, 360-Degree Distribution of Biofilm Quantity and Community in an Operational Unchlorinated Drinking Water Distribution Pipe, Environ. Sci. Technol., 2020, 54(9), 5619–5628 CrossRef CAS PubMed.
- D. Cheng, M. Leifels, C. Miccolis, S. Wuertz, J. R. Thompson, U. Szewzyk and A. J. Whittle, Factors Shaping Young and Mature Bacterial Biofilm Communities in Two Drinking Water Distribution Networks, bioRxiv, 2021, preprint, DOI:10.1101/2021.03.10.434709.
- C. Calero Preciado, J. Boxall, V. Soria-Carrasco, S. Martinez and I. Douterelo, Implications of Climate Change: How Does Increased Water Temperature Influence Biofilm and Water Quality of Chlorinated Drinking Water Distribution Systems?, Front. Microbiol., 2021, 12, 658927 CrossRef PubMed.
- S. A. Waller, A. I. Packman and M. Hausner, Comparison of biofilm cell quantification methods for drinking water distribution systems, J. Microbiol. Methods, 2018, 144, 8–21 CrossRef CAS PubMed.
- H. C. Flemming and J. Wingender, The biofilm matrix, Nat. Rev. Microbiol., 2010, 8(9), 623–633 CrossRef CAS PubMed.
- W. Lin, Z. Yu, H. Zhang and I. P. Thompson, Diversity and dynamics of microbial communities at each step of treatment plant for potable water generation, Water Res., 2014, 52, 218–230 CrossRef CAS PubMed.
- S. Perveen, C. Pablos, K. Reynolds, S. Stanley and J. Marugan, Growth and prevalence of antibiotic-resistant bacteria in microplastic biofilm from wastewater treatment plant effluents, Sci. Total Environ., 2023, 856(Pt 2), 159024 CrossRef CAS PubMed.
- H. Y. Buse, P. Ji, V. Gomez-Alvarez, A. Pruden, M. A. Edwards and N. J. Ashbolt, Effect of temperature and colonization of Legionella pneumophila and Vermamoeba vermiformis on bacterial community composition of copper drinking water biofilms, Microb. Biotechnol., 2017, 10(4), 773–788 CrossRef CAS PubMed.
- M. N. F. Shaheen, E. M. Elmahdy and M. Chawla-Sarkar, Quantitative PCR-based identification of enteric viruses contaminating fresh produce and surface water used for irrigation in Egypt, Environ. Sci. Pollut. Res., 2019, 26(21), 21619–21628 CrossRef CAS PubMed.
- J. Zhang, W. Li, J. Chen, F. Wang, W. Qi and Y. Li, Impact of disinfectant on bacterial antibiotic resistance transfer between biofilm and tap water in a simulated distribution network, Environ. Pollut., 2019, 246, 131–140 CrossRef CAS PubMed.
- S. Bonetta, C. Pignata, S. Bonetta, L. Meucci, D. Giacosa and E. Marino, Viability of Legionella pneumophila in Water Samples: A Comparison of Propidium Monoazide (PMA) Treatment on Membrane Filters and in Liquid, Int. J. Environ. Res. Public Health, 2017, 14(5), 467 CrossRef PubMed.
- S. Ditommaso, E. Ricciardi, M. Giacomuzzi, S. R. Arauco Rivera, A. Ceccarelli and C. M. Zotti, Overestimation of the Legionella spp. load in environmental samples by quantitative real-time PCR: pretreatment with propidium monoazide as a tool for the assessment of an association between Legionella concentration and sanitary risk, Diagn. Microbiol. Infect. Dis., 2014, 80(4), 260–266 CrossRef CAS PubMed.
- M. J. Taylor, R. H. Bentham and K. E. Ross, Limitations of Using Propidium Monoazide with qPCR to Discriminate between Live and Dead Legionella in Biofilm Samples, Microbiol. Insights, 2014, 7, 15–24 Search PubMed.
- M. Scaturro, S. Fontana, I. Dell'eva, F. Helfer, M. Marchio, M. V. Stefanetti, M. Cavallaro, M. Miglietta, M. T. Montagna, O. De Giglio and T. Cuna, A multicenter study of viable PCR using propidium monoazide to detect Legionella in water samples, Diagn. Microbiol. Infect. Dis., 2016, 85(3), 283–288 CrossRef CAS PubMed.
- K. Lautenschlager, C. Hwang, W. T. Liu, N. Boon, O. Koster and H. Vrouwenvelder, et al., A microbiology-based multi-parametric approach towards assessing biological stability in drinking water distribution networks, Water Res., 2013, 47(9), 3015–3025 CrossRef CAS PubMed.
- J. Heyse, F. Schattenberg, P. Rubbens, S. Müller, W. Waegeman, N. Boon and R. Props, Predicting the Presence and Abundance of Bacterial Taxa in Environmental Communities through Flow Cytometric Fingerprinting, mSystems, 2021, 6(5), e00551-21 CrossRef PubMed.
- P. Rubbens, R. Props, F. M. Kerckhof, N. Boon and W. Waegeman, Cytometric fingerprints of gut microbiota predict Crohn's disease state, ISME J., 2021, 15(1), 354–358 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00553d |
| This journal is © The Royal Society of Chemistry 2024 |