
Vinylidene rearrangements of internal borylalkynes via 1,2-boryl migration†
Takahiro
Iwamoto
 *ab,
Takuya
Mitsubo
a,
Kosuke
Sakajiri
a and
Youichi
Ishii
*a
*ab,
Takuya
Mitsubo
a,
Kosuke
Sakajiri
a and
Youichi
Ishii
*a
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo 112-8551, Japan. E-mail: iwamoto@kc.chuo-u.ac.jp; yo-ishii@kc.chuo-u.ac.jp
bFaculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, Goshokaido-cho, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: tiwamoto@kit.ac.jp
First published on 28th May 2024
Abstract
Vinylidene rearrangement of alkynes is a well-established and powerful method for alkyne transformations, while use of borylalkynes has remained largely unexplored. This paper describes vinylidene rearrangements of internal borylalkynes using a cationic ruthenium complex. This rearrangement is applicable to alkynes with both tri-(B(pin), B(dan)) and tetracoordinate (B(mida)) boryl groups, and the reaction rate is dramatically affected by the Lewis acidity of the boryl group. Mechanistic study revealed that the rearrangement proceeds via 1,2-boryl migration regardless of the coordination number of the boron center. The migration mode was elucidated by theoretical calculations to indicate that the migration of the tricoordinate boryl groups is an electrophilic process in contrast to the previous vinylidene rearrangements of internal alkynes with two carbon substituents.
Introduction
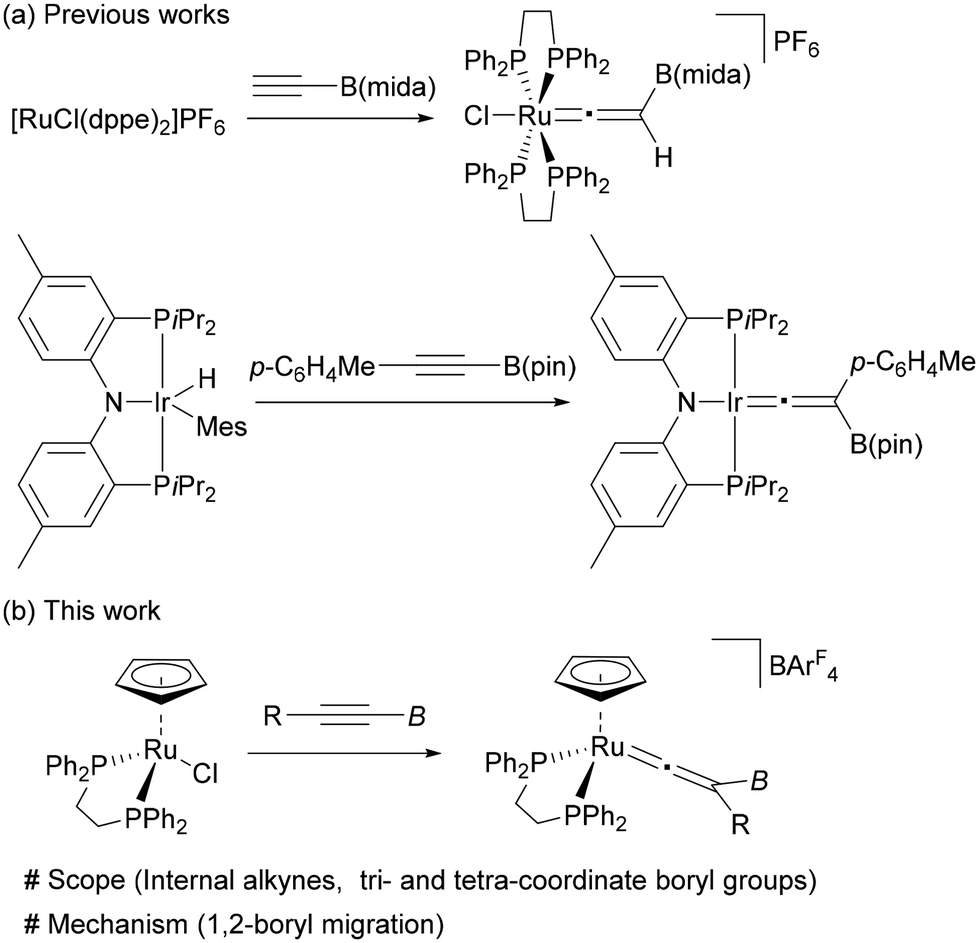
Since organoboron compounds are versatile synthetic intermediates in organic chemistry,1,2 substantial efforts have been devoted to the development of synthetic methods to expand their availability and diversity. Recently, significant advances have been made in the chemistry of boryl migrations, and various types of rearrangement reactions have been achieved by nucleophilic,3–13 electrophilic,14–17 and radical18–24 boryl migrations.25 These transformations provide useful synthetic tools to access organoboron compounds, which are complementary to the conventional approaches.Transition-metal mediated vinylidene rearrangement from an η2-alkyne ligand to the corresponding vinylidene species represents a powerful method for alkyne transformations.26–31 The most common is vinylidene rearrangement of terminal alkynes because of the high migration ability of a hydrogen atom.32 Over the last few decades, the substrate scope has been expanded to internal alkynes to demonstrate that a variety of carbon and heteroatom groups can participate in this rearrangement as a migrating group (Fig. 1a).33–48 However, vinylidene rearrangement of borylalkynes has been largely unexplored; the successful vinylidene rearrangements are limited to just a few examples (Fig. 1b).49,50 Hill and coworkers reported the vinylidene rearrangement of HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CB(mida) (mida = N-methyliminodiacetato) (Fig. 1b, top).49b Subsequently, the first instance of vinylidene rearrangement of internal borylalkyne, p-C6H4MeCCB(pin) (pin = pinacolato), was reported by Ozerov and coworkers.49c Braunschweig also described an equilibrium between rhodium-borylvinylidene and -(hydrido)(borylalkynyl) complexes, the latter of which was derived from a terminal borylalkyne.49a However, although Hill proposed that the boryl group would serve as a 1,2-migration group, no mechanistic information on the 1,2-migration process of η2-borylalkyne is provided in these reports.
CB(mida) (mida = N-methyliminodiacetato) (Fig. 1b, top).49b Subsequently, the first instance of vinylidene rearrangement of internal borylalkyne, p-C6H4MeCCB(pin) (pin = pinacolato), was reported by Ozerov and coworkers.49c Braunschweig also described an equilibrium between rhodium-borylvinylidene and -(hydrido)(borylalkynyl) complexes, the latter of which was derived from a terminal borylalkyne.49a However, although Hill proposed that the boryl group would serve as a 1,2-migration group, no mechanistic information on the 1,2-migration process of η2-borylalkyne is provided in these reports.
 | ||
| Fig. 1 Vinylidene rearrangements of borylalkynes. | ||
In this paper, we have investigated vinylidene rearrangements of internal borylalkynes by using [Ru(dppe)Cp]+, in which both tetra- and tricoordinate boryl groups are applicable (Fig. 1b, bottom). Isotopic labelling experiments provided the first experimental evidence showing that the boryl groups serve as a migrating group. Theoretical studies on a migration mode of the boryl group (i.e., either nucleophilic or electrophilic processes) is also reported. These results not only expand the scope of the vinylidene rearrangements but also add a new entry to the boryl group migration chemistry.
Results and discussion
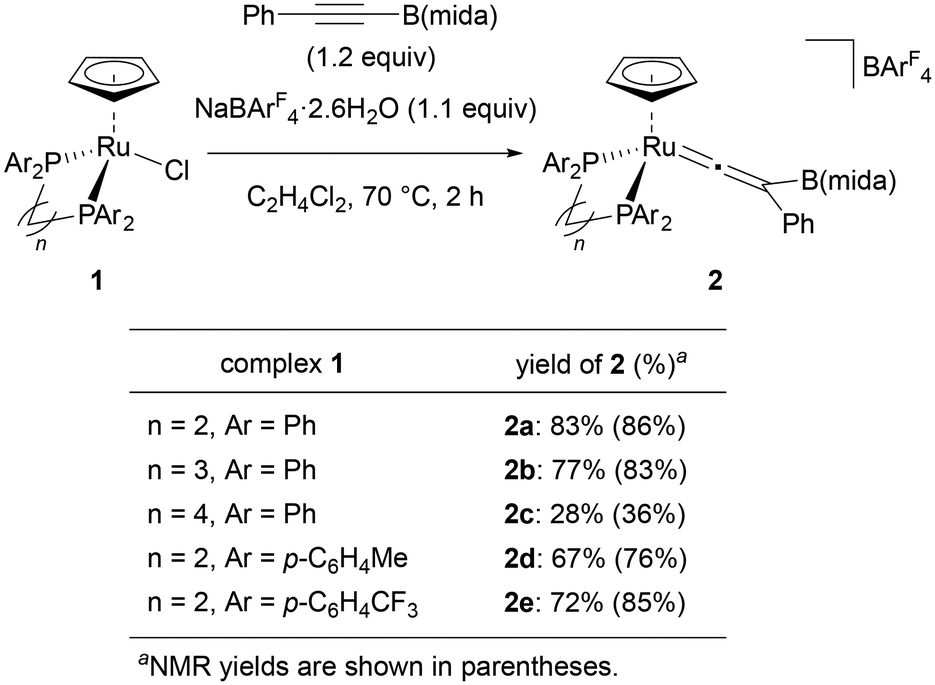
We selected PhCCB(mida) as a standard substrate and started our investigation with a ligand screening in a reaction of this internal boryl alkyne with [Ru(L)Cp]BArF4 (L = bidentate phosphine ligand) generated in situ (Fig. 2). When ruthenium complex [RuCl(dppe)Cp] (1) was treated with PhCCB(mida) in the presence of NaBArF4·2.6H2O (ArF = 3,5-bis(trifluoromethyl)phenyl) at 70 °C, vinylidene complex 2a was obtained in 87% yield after 1 h. Sterically more demanding ligands obviously diminished the yields (2aversus2b, 2c) indicating that the present rearrangement is sensitive to the steric effect. The coordination of a bulky internal boryl alkyne prior to the rearrangement may be hampered by dppb with a large bite angle. Electronically differentiated ligands provided the corresponding vinylidene complex, yet the yields were slightly low (2d and 2e).
 | ||
| Fig. 2 Ligand screening. | ||
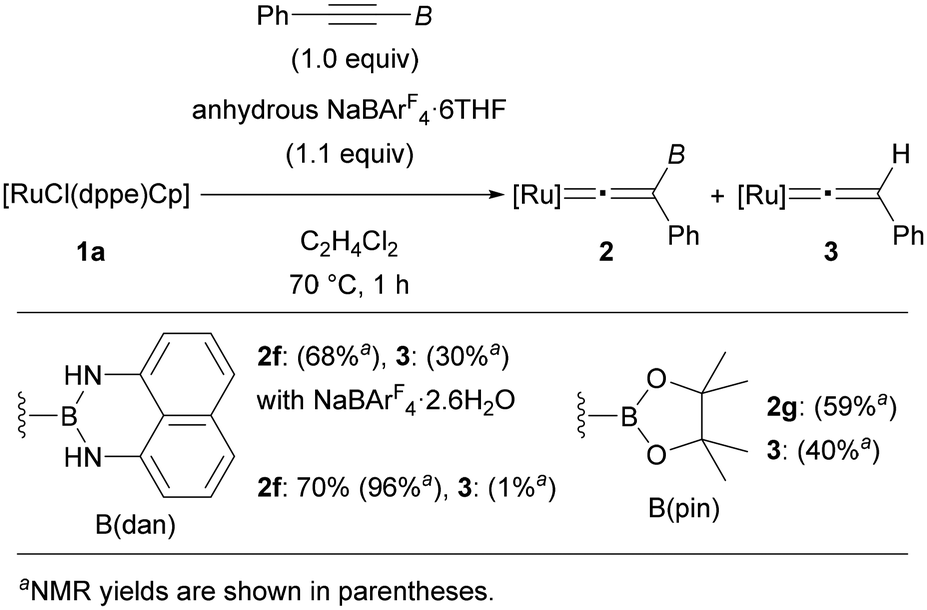
By using [RuCl(dppe)Cp] (1a) as the optimal precursor, we next conducted reactions of tricoordinate borylalkynes (Fig. 3). Treating PhCCB(dan) (dan = naphthalene-1,8-diaminato) under the same reaction conditions resulted in the complete consumption of 1a. However, a mixture of complex 2f and protodeboronated complex 3 was obtained in 68 and 30% yields, respectively. Through careful screening of the reagents and the reaction conditions, we found that use of anhydrous NaBArF4·6THF was effective to produce complex 2f in 70% isolated yield (96% NMR yield) along with formation of a small amount of 3. The susceptibility to the protodeboronation was more serious in the case of internal alkynes with B(pin). Treatment of PhCCB(pin) with [RuCl(dppe)Cp] in the presence of anhydrous NaBArF4·6THF provided a mixture of 2g and 3 in 59 and 40% NMR yield, respectively, and the high susceptibility toward H2O hampered isolation of complex 2g. Consequently, characterization of 2g was performed solely by NMR analyses, while several NMR signals characteristic of vinylidene complexes provided reliable information on the formation of vinylidene complex 2g (see the ESI†).
 | ||
| Fig. 3 Vinylidene rearrangements of tricoordinate borylalkynes ([Ru] = [Ru(dppe)Cp]+). | ||
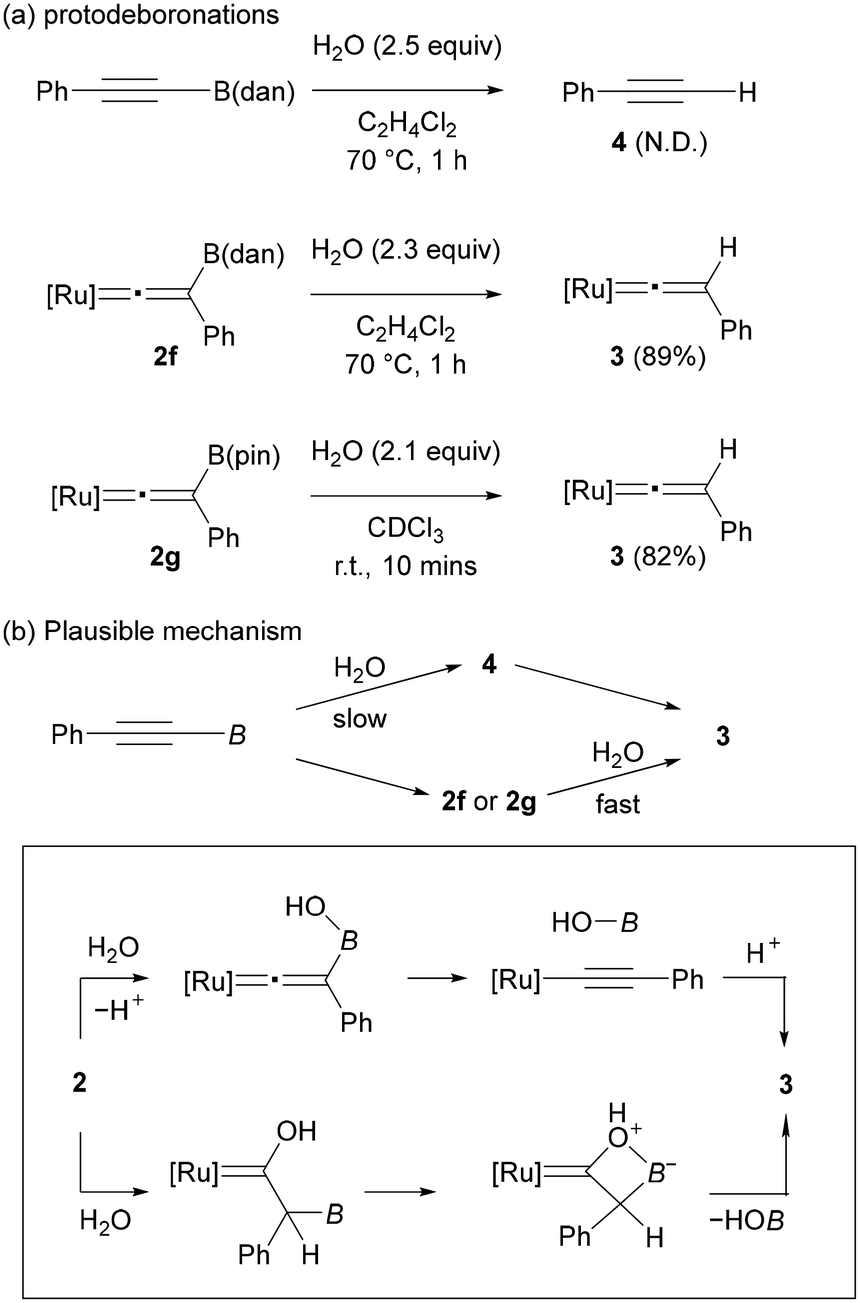
To gain insight into the formation of 3, we conducted brief studies on the protodeboronation. When a solution of PhCCB(dan) in C2H4Cl2 was treated with H2O (2.5 equiv.) at 70 °C for 1 h, PhCCH (4) was not formed, and the starting alkyne was fully recovered (Fig. 4a, up). In contrast, complex 2f underwent protodeboronation under similar conditions to give complex 3 in 89% yield. Protodeboronation of complex 2g proceeded much rapidly even at room temperature to afford 3 in 82% (Fig. 4a, bottom). Note that vinylidene complex 2a is stable under these reaction conditions. These results implied that the formation of complex 3 observed in the vinylidene rearrangements (Fig. 3) is derived from the protodeboronation of vinylidene complex 2, but not from the vinylidene rearrangement of terminal alkyne 4 (Fig. 4b). The remarkable susceptibility of vinylidene complex 2f and 2g to the protodeboronation likely results from their unique structure associated with the cationic vinylidene and the β-boryl functionalities.
 | ||
| Fig. 4 (a) Control experiments and (b) a proposed reaction mechanism for the protodeboronation ([Ru] = [Ru(dppe)Cp]+). | ||
Common organoboronate compounds are moderately stable toward hydrolysis. The high reactivity of 2f and 2g toward protodeboronation may be accounted for by two possible mechanisms: (1) addition of water at the boron atom followed by deprotonation and B–C bond cleavage, or (2) electrophilic addition of H2O to the vinylidene α-carbon followed by B−OH elimination like bora-Wittig-type reactions (Fig. 4b, bottom).51 Although a common protodeboronation mechanism (1) cannot be excluded, we assume that the latter mechanism (2) may be operative considering that the electron-deficient nature of the α-carbon in the cationic vinylidene complex 2 should be strongly enhanced by the Lewis acidic boryl group at the β-carbon.52 In any event, both mechanisms are in accordance with the actual reactivity order that the more Lewis acidic boryl group accelerates the protodeboronation.
To further assess the vinylidene rearrangements, we performed time course studies monitoring the formation of the vinylidene complexes by using 1H NMR analyses (Fig. 5). The vinylidene formation from PhCCB(pin) and 1a was found to proceed rapidly even at room temperature, and the vinylidene complex was formed almost quantitatively after 3 h. PhCCB(dan) also showed high activity, while the reaction rate was slightly lower compared with PhCCB(pin). In contrast, the reaction with PhCCB(mida) was sluggish at room temperature. Thus, the reaction rates of the vinylidene rearrangements are dramatically affected by the boryl groups and likely correlated with the Lewis acidity of the boryl groups (B(pin) > B(dan) > B(mida)).
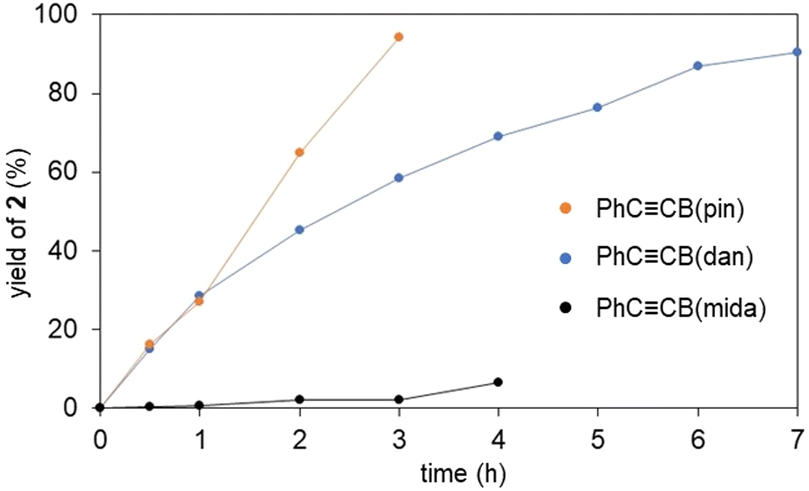 | ||
| Fig. 5 Time course studies on the formation of vinylidene complexes. Reactions were performed at room temperature in CDCl3 using an NMR tube. Yields of complexes 2 were determined by 1H NMR spectroscopy. | ||
To clarify the migrating group, 13C-labeling experiments were performed using 25% 13C-enriched alkyne, PhC13CB(mida) (Fig. 6). After a reaction under the optimal conditions, vinylidene complex 2–13Cα was obtained as the major product (2–13Cα![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2–13Cβ = 22.6:1), and the migration ratio of B(mida) and Ph was calculated to be 99:<1 on the basis of the product ratio 2–13Cα:2–13Cβ determined by 13C{1H} NMR. In the case of an alkyne bearing B(dan) (19.5% enriched), the isomer ratio was determined after protodeboronation of the vinylidene complex because of a partial overlap of 13C NMR signals of the boryl vinylidene complex. Again, B(dan) group was found to serve as the migrating group with >99% selectivity. These results represent the first observations of 1,2-boryl migration in vinylidene rearrangement.
:2–13Cβ = 22.6:1), and the migration ratio of B(mida) and Ph was calculated to be 99:<1 on the basis of the product ratio 2–13Cα:2–13Cβ determined by 13C{1H} NMR. In the case of an alkyne bearing B(dan) (19.5% enriched), the isomer ratio was determined after protodeboronation of the vinylidene complex because of a partial overlap of 13C NMR signals of the boryl vinylidene complex. Again, B(dan) group was found to serve as the migrating group with >99% selectivity. These results represent the first observations of 1,2-boryl migration in vinylidene rearrangement.
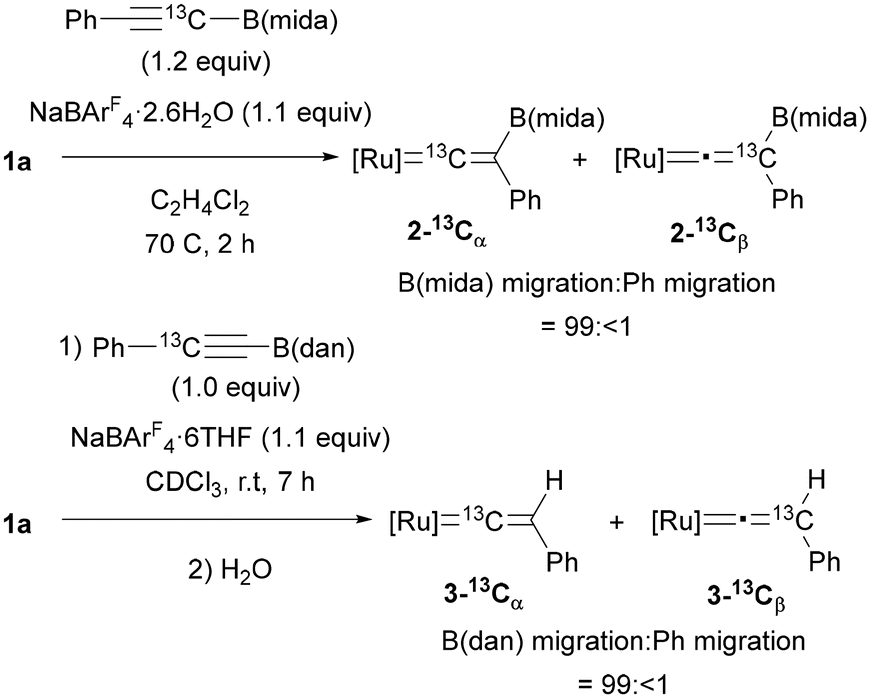 | ||
| Fig. 6 Isotopic labeling experiments ([Ru] = Ru(dppe)Cp+). | ||
Our previous mechanistic studies on the vinylidene rearrangements of internal alkynes with two carbon substituents revealed that the rearrangement proceeds via nucleophilic migration pathway to highlight the importance of nucleophilicity of a migration group.34,36 However, the observation of the rapid rearrangements of less nucleophilic B(pin) and B(dan) over B(mida) is contradiction to this mechanistic scenario. Thus, we performed validation of the migration mode. In the previous nucleophilic vinylidene rearrangements, the rate of the rearrangement was revealed to be remarkably dependent on the electron-donating ability of a non-migrating group (Fig. 7).36 Thus we compared the reactivities of alkynes with an electronically different non-migrating group. In reactions of p-RC6H4CCB(mida), the rearrangement of (p-C6H4OMe)CCB(mida) with an electron donating aryl group was faster than that of PhCCB(mida), while electron withdrawing p-C6H4CF3 group retarded the reaction significantly (Fig. 7a). The observed electronic effects were entirely identical with our previous observations in nucleophilic vinylidene rearrangements.36 On the other hand, in reactions of p-RC6H4CCB(dan), no obvious migration preference depending on the electronic character of the non-migrating group was observed (Fig. 7b). This different structure-reactivity relationship is indicative of the distinct migration mode with the tricoordinate borylalkynes.53
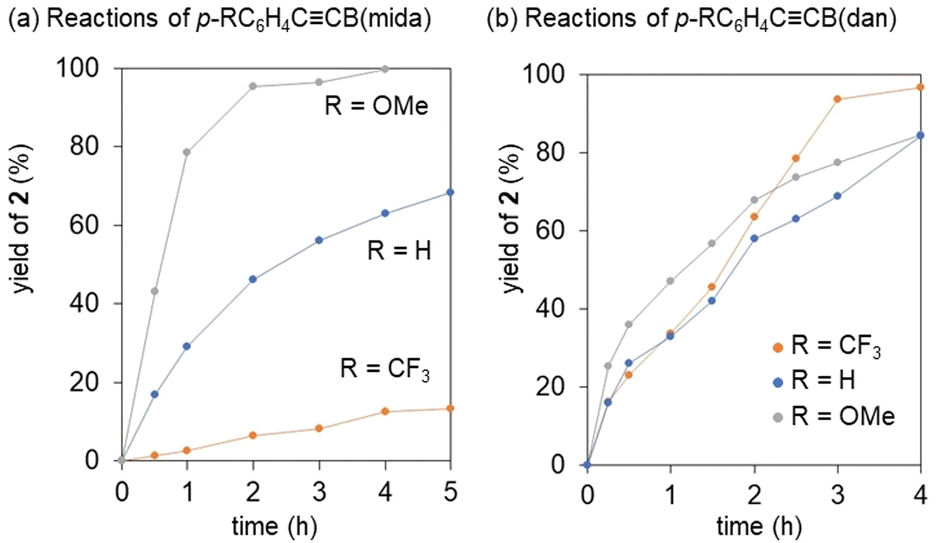 | ||
| Fig. 7 Vinylidene rearrangements of borylalkynes with an electronically differentiated aryl group. Reactions were performed at 50 °C for B(mida) group and at room temperature for B(dan) group. Yields of 2 were determined by 1H NMR spectroscopy. | ||
To further obtain an insight into the migration mode, the boryl migrations of B(dan) and B(mida) group were evaluated by DFT calculations at B3LYP/6-311G(d) + SDD (Fig. 8). Both process from η2-alkyne to the corresponding vinylidene species is endothermic with a ΔG value of 10.1 kcal mol−1. An activation barriers of B(dan) migration is significantly lower than that of B(mida) migration. These results are consistent with the experimental observation that the vinylidene rearrangement of B(dan) proceeds smoothly at room temperature, while that of B(mida) needs a higher reaction temperature. At the transition state (TS) for the B(dan) migration, the C1–B bond length (1.703 Å) is obviously shorter than C2–B bond length (1.929 Å), which corresponds to the nearly generating C–B bond. A linear alignment of C1, C2, and C3 atoms (177°) and a nearly orthogonal conformation of the non-migrating phenyl group toward the C1–B bond (100°) indicates that π-orbitals of the non-migrating phenyl group can effectively interact with the C1–C2 π bond, which should be directly involved in the boryl migration. Of interest, this TS structure is similar with that in 1,3-boryl migration of a (boryl)(alkynyl)osmium complex.50a In the TS of B(mida) migration, C1–B bond (1.911 Å) is shorter than C2–B bond (2.067 Å), while the difference in these bond lengths is smaller than that in the B(dan) migration. Furthermore, in contrast to the B(dan) migration, the angle of C1–C2–C3 is slightly bent (167°). Note that, in the B(mida) migration, a dative B–N bond remains during the whole calculated processes from A to B (N–B = 1.711–1.717 Å).
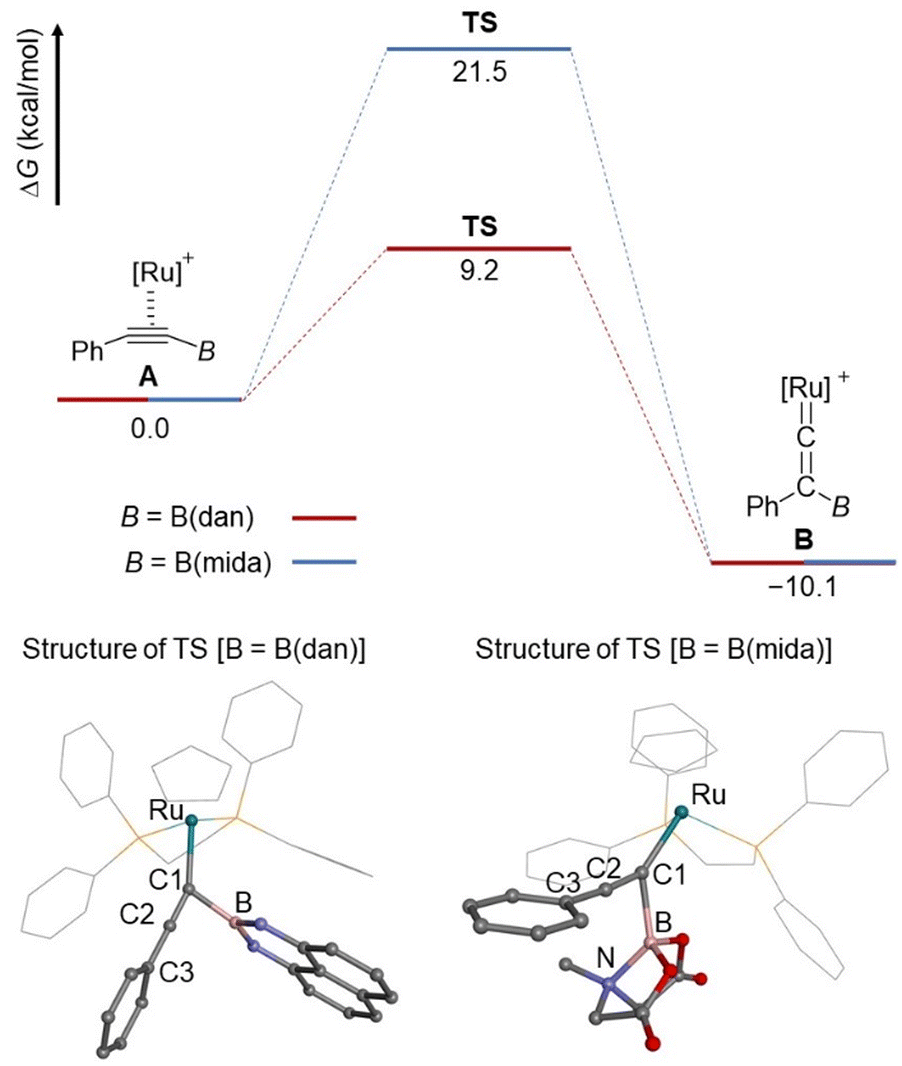 | ||
| Fig. 8 DFT calculations at the B3LYP/SDD (Ru) and 6-311G(d) (other atoms) level of theory ([Ru] = Ru(dppe)Cp). In the structures of TS, dppe and Cp ligands are shown by wireframe representations for clarity. Color code: grey, C; pink, B; green, Ru; purple, N; red, O; yellow, P. | ||
NBO analyzes were performed at HF/6-311G(d)+SDD for A, TS, and B. In the TS of B(dan) migration, a definite interaction is estimated between the C1–C2 π bond as a donor and the p orbital of the boron center as an acceptor (Fig. 9a). The donor acceptor interaction supports the electrophilic migration mode of B(dan) group. On the other hand, B(mida) migration hardly includes the same type of interaction, because the corresponding p orbital of the boron center participates in the dative bond formation with the nitrogen atom. Fig. 9b shows NBO charges of A, TS, and B. In B(dan) migration, a positive charge of the boron atom increases from intermediate A (1.020) to TS (1.169). Although C2 has a positive charge in A and TS, the value decreases from A to TS by 0.021. These changes of the charge distributions are completely different with our previous results for the nucleophilic vinylidene rearrangements,36 yet consistent with the other theoretical calculations of electrophilic vinylidene rearrangements.54 Similar electron flows are calculated in the case of the B(mida) migration. Although we cannot conclude either electrophilic or nucleophilic migration mode of B(mida) group based on the present experimental and theoretical studies,55 these calculations suggest the B(dan) migration proceeds via the electrophilic mode. We believe that the migration of a tricoordinate boryl group, B(pin), also follows the mechanistic scenario similar to that of B(dan). This conclusion is consistent with the high migration aptitude of Lewis acidic boryl groups.
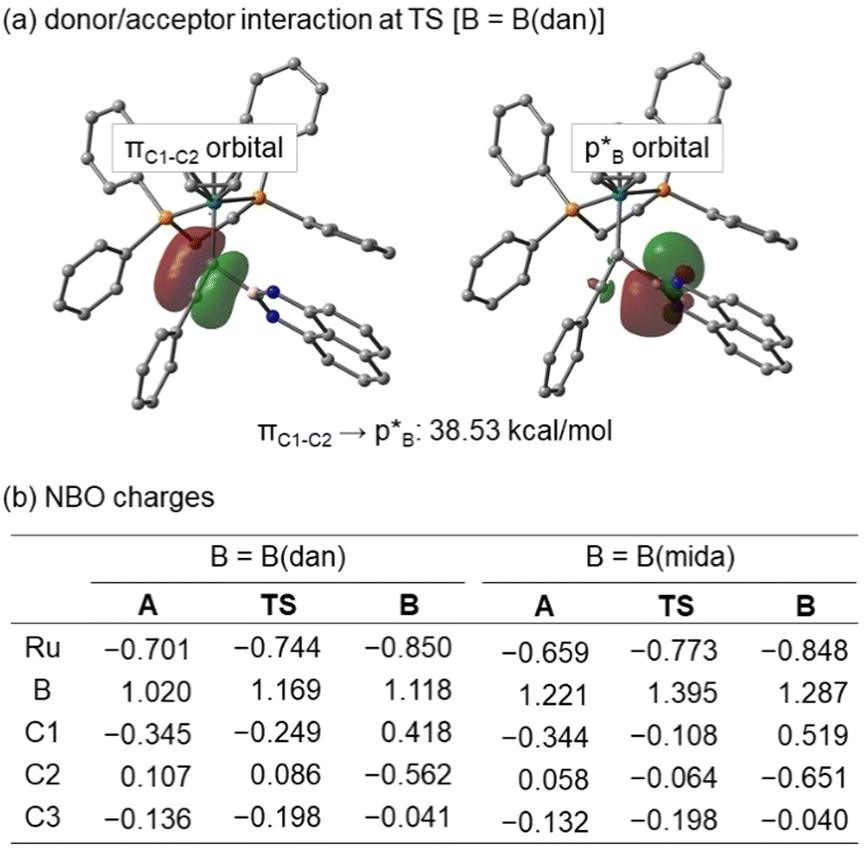 | ||
| Fig. 9 (a) NBO charges and (b) donor/acceptor interactions in TS of B(dan). | ||
We finally examined substrate scope by using RCCB(mida) (Fig. 10). Reactions of p-RC6H4CCB(mida) (R = Me and F) proceeded smoothly to provide the corresponding vinylidene complexes in 70 and 71% yield, respectively. Boryl alkynes with an aryl group substituted at o- and m-positions also provided vinylidene complexes in good yields, while a sterically hindered substrate with an o-methyl group diminished the yield. As indicated by the ligand screening, the present rearrangement is rather sensitive toward the steric hindrance. Reactions of alkynes bearing biphenyl and naphthyl proceeded in moderate yields. Electron-rich heteroaromatic groups were applicable to afford the corresponding vinylidene complexes in moderate yields. Unfortunately, a reaction with alkylalkyne was found to be sluggish.
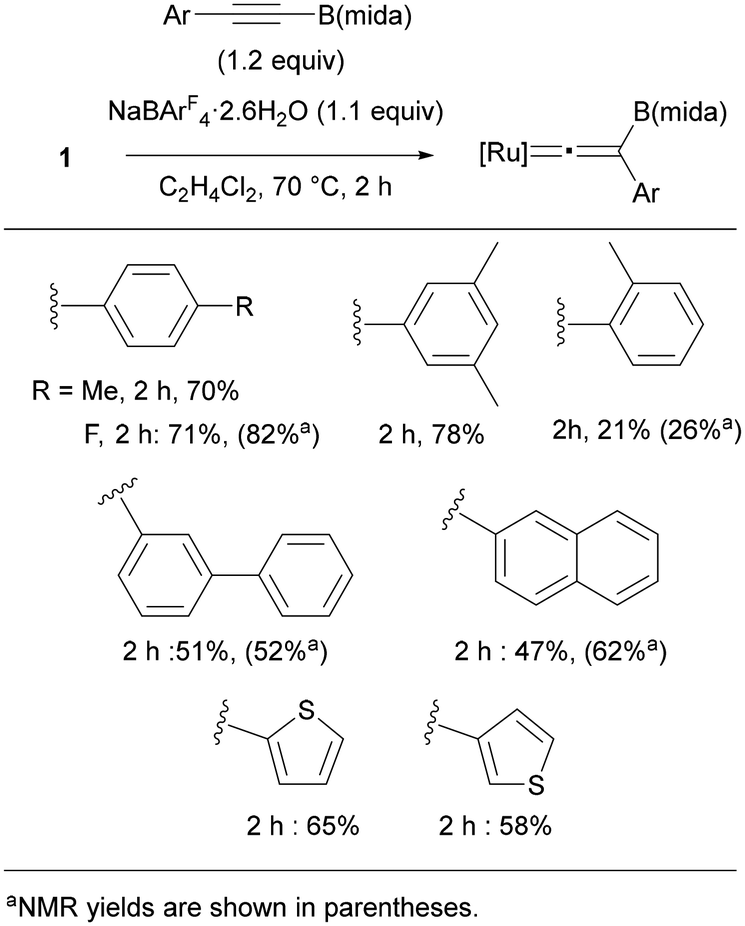 | ||
| Fig. 10 Substrate scope ([Ru] = [Ru(dppe)Cp]+). | ||
Conclusions
In conclusion, we have developed the vinylidene rearrangements of internal borylalkynes with tri- and tetra-coordinate boryl groups. Mechanistic studies revealed the selective migration of the boryl group regardless of the coordination number of the boron center, and that the migration rate was dramatically enhanced by the Lewis acidic tricoordinate boryl group. Theoretical calculation implied that the migration mode is an electrophilic process at least in the case of B(dan) group, which is completely different from the previous rearrangements of diarylalkynes. Therefore, vinylidene rearrangements can proceed through either nucleophilic or electrophilic migration modes depending on the migration groups. This allows for the utilization of various groups, including nucleophilic group 16 elements (S and Se) and electrophilic group 13 element (B) as well as electronically neutral groups (e.g., group 14 elements like C, Si and Sn). Note that the resulting vinylidene complexes with the tricoordinate boryl group are remarkably susceptible toward hydrolysis. We assumed that the observed rapid hydrolysis results from the high electrophilicity of the α-carbon of the vinylidene unit. This reactivity would be beneficial for their application to catalytic processes.Experimental
General
All manipulations were carried out under an argon atmosphere by using standard Schlenk techniques unless otherwise noted. 1,2-Dichloroethane was distilled over P4O10, degassed, and stored under an argon atmosphere. [RuCl(dppe)Cp], analogous ruthenium complexes,34 NaBArF4·2.6H2O,56 NaBArF4·6THF,56 and alkynes57–60 are synthesized according to the literature procedures. 1H (495 MHz), 13C{1H} (125 MHz), and 31P{1H} (200 MHz) NMR spectra were recorded on a JEOL ECA-500 spectrometer, except for those of complex 2g, which were recorded by Varian-400 spectrometer [1H (400 MHz) and 13C{1H}NMR (100 MHz)]. Chemical shifts are reported in δ and referenced to residual 1H and 13C signals of deuterated solvents as internal standards. Note that several signals assignable to the Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC could not be found, probably because it is overlapped with other signals and the quadrupolar boron is likely to further complicate its identification (vide infra). High-resolution mass spectra were measured on a JEOL JMS-T100LC AccuTOF spectrometer. Elemental analyses were performed on a PerkinElmer 2400 series II HCN analyzer. Procedures and data for other complexes mentioned in the text are given in the ESI.†
CC could not be found, probably because it is overlapped with other signals and the quadrupolar boron is likely to further complicate its identification (vide infra). High-resolution mass spectra were measured on a JEOL JMS-T100LC AccuTOF spectrometer. Elemental analyses were performed on a PerkinElmer 2400 series II HCN analyzer. Procedures and data for other complexes mentioned in the text are given in the ESI.†
Procedure for vinylidene rearrangements of alkynes with B(mida)
A mixture of [RuCl(L)Cp] (1.0 equiv.), PhCCB(mida) (1.2 equiv.), and NaBArF4·2.6H2O (1.1 equiv.) in 1,2-dichloroethane (2 mL) was heated at 70 °C for 2 h. The resulting orange suspension was filtered through a short pad of Celite and rinsed with 1,2-dichloroethane (ca. 1 mL). The filtrate was dried in vacuo, and the residue was purified by column chromatography on silica gel (dichloromethane). The eluate was further purified by recrystallization from dichloromethane/hexane to afford the vinylidene complex as yellow crystals. The NMR yields were determined by using tetrachloroethane as an internal standard.
Synthesis of [Ru(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CC(Ph){B(mida)})(dppe)Cp][BArF4] (2a)
CC(Ph){B(mida)})(dppe)Cp][BArF4] (2a)
Complex 2a was obtained as yellow crystals (71.6 mg, 0.0425 mmol, 83% yield) by the reaction of [CpRuCl(dppe)] (30.7 mg, 0.0512 mmol), PhCCB(mida) (15.6 mg, 0.0607 mmol), and NaBArF4·2.6H2O (51.1 mg, 0.0548 mmol).
1H NMR (acetone-d6): δ 7.79 (s, 8H, o-H of BArF4), 7.75 (m, 4H, o-H of Ph in dppe), 7.67 (s, 4H, p-H of BArF4), 7.45 (m, 4H, p-H of Ph in dppe × 2), 7.35 (m, 8H, m-H of Ph in dppe × 2), 7.24 (m, 4H, o-H of Ph in dppe), 7.01 (m, 3H, m- and p-H of RuCCPh), 6.88 (m, 2H, o-H of RuCCPh), 5.67 (s, 5H, Cp), 3.99 (d, 2H, 2JHH = 17.3 Hz, CH2 of mida), 3.39 (m, 2H, CH2 of dppe), 3.24 (d, 2H, 2JHH = 16.8 Hz, CH2 of mida), 3.03 (m, 2H, CH2 of dppe), 2.72 (s, 3H, CH3 of mida). 31P{1H} NMR (acetone-d6): δ 79.5 (s, dppe). 13C{1H} NMR (acetone-d6): δ 339.2 (t, 2JCP = 15.4 Hz, RuCC), 167.7 (s, CO of mida), 162.6 (q, 1JCB = 50.0 Hz, ipso-C of BArF4), 138.0 (m, ipso-C of Ph in dppe), 135.5 (br, o-C of BArF4), 134.9 (m, ipso-C of Ph in dppe), 133.9 (virtual t, o-C of Ph in dppe), 132.0 (m, o- and p-C of Ph in dppe), 131.5 (s, p-C of Ph in dppe), 130.8 (s, o-C of RuCCC6H5), 130.0 (brq, 2JCF = 34.4 Hz, m-C of BArF4), 129.7 (m, m-C of Ph in dppe × 2 and m-C of RuCCC6H5), 128.8 (s, ipso-C of RuCCC6H5), 127.3 (s, p-C of RuCCC6H5), 125.4 (q, 1JCF = 272.9 Hz, CF3 of BArF4), 118.4 (m, p-C of BArF4), 92.0 (s, Cp), 62.8 (s, CH2 of mida), 47.2 (s, CH3 of mida), 28.5 (m, PCH2). The signal assignable to the RuCC could not be found, probably because it is overlapped with other signals. Elemental analysis calcd for C76H53O4B2F24P2NRu·0.5CH2Cl2: C, 53.19; H, 3.09; N, 0.81. Found: C, 53.13; H, 3.06; N, 0.80.
Procedures for vinylidene rearrangements of tricoordinate borylalkynes
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CB(dan) with NaBArF4·2.6H2O.
A mixture of [RuCl(dppe)Cp] (30.1 mg, 0.0502 mmol), PhCCB(dan) (13.7 mg, 0.0511 mmol), and NaBArF4·2.6H2O (50.9 mg, 0.0546 mmol) in 1,2-dichloroethane (2 mL) was heated at 70 °C for 1 h. The resulting brown suspension was filtered through a short pad of Celite and rinsed with 1,2-dichloroethane (ca. 1 mL). The NMR yields were determined by using tetrachloroethane as an internal standard.
CB(dan) with anhydrous NaBArF4·6THF.
A reaction of PhCCB(dan) with anhydrous NaBArF4·6THF was performed according to the same procedure by using [RuCl(dppe)Cp] (29.9 mg, 0.0498 mmol), PhCCB(dan) (13.8 mg, 0.0515 mmol), and NaBArF4·6THF (81.1 mg, 0.0615 mmol). The NMR yield was determined by 1H NMR spectrum of the crude product (96% NMR yield). The combined filtrate was purified by recrystallization from dichloroethane/hexane to afford a vinylidene complex 2f as yellow crystals in 70% yield.
CB(dan) with NaBArF4·2.6H2O.
A mixture of [RuCl(dppe)Cp] (30.1 mg, 0.0502 mmol), PhCCB(dan) (13.7 mg, 0.0511 mmol), and NaBArF4·2.6H2O (50.9 mg, 0.0546 mmol) in 1,2-dichloroethane (2 mL) was heated at 70 °C for 1 h. The resulting brown suspension was filtered through a short pad of Celite and rinsed with 1,2-dichloroethane (ca. 1 mL). The NMR yields were determined by using tetrachloroethane as an internal standard.
CB(dan) with anhydrous NaBArF4·6THF.
A reaction of PhCCB(dan) with anhydrous NaBArF4·6THF was performed according to the same procedure by using [RuCl(dppe)Cp] (29.9 mg, 0.0498 mmol), PhCCB(dan) (13.8 mg, 0.0515 mmol), and NaBArF4·6THF (81.1 mg, 0.0615 mmol). The NMR yield was determined by 1H NMR spectrum of the crude product (96% NMR yield). The combined filtrate was purified by recrystallization from dichloroethane/hexane to afford a vinylidene complex 2f as yellow crystals in 70% yield.
1H NMR (CDCl3, see ESI† for the atom numbering): δ 7.74 (s, 8H, o-H of BArF4), 7.52 (s, 4H, p-H of BArF4), 7.38 (m, 12H, p- ×2, m- and o-H of Ph in dppe), 7.02 (m, 15H, o- and m-H of Ph in dppe and H3, H4, H6, H7), 6.62 (d, 2H, 3JHH = 6.9 Hz, H2), 5.90 (br, 2H, H5), 5.43 (s, 5H, Cp), 4.73 (br, 2H, NH of dan), 3.05 (m, 2H, CH2 of dppe), 2.87 (m, 2H, CH2 of dppe). 31P{1H} NMR (CDCl3): δ 78.0 (s, dppe). 13C{1H} NMR (CDCl3): δ 335.6 (t, 2JCP = 14.8 Hz, RuCC), 161.8 (q, 1JCB = 50.0 Hz, ipso-C of BArF4), 140.0 (s, C10), 136.2 (br, C8), 135.0 (m, o-C of BArF4 and ipso-C of Ph in dppe), 133.1 (m, ipso-C of Ph in dppe), 132.0 (m, p- × 2 and o-C of Ph in dppe), 131.4 (m, o-C of Ph in dppe), 129.0 (m, m-C × 2 of Ph in dppe, and C2, C3, and m-C of BArF4), 127.3 (s, C6), 127.0 (s, C4), 125.8 (s, C1), 124.7 (q, 1JCF = 273.6 Hz, CF3 of BArF4), 119.6 (s, C9), 118.4 (s, C7), 117.6 (s, p-C of BArF4), 117.1 (br, RuCC), 106.5 (s, C5), 91.3 (s, Cp), 27.8 (m, PCH2). High resolution mass measurement and elemental analysis have failed due to the high susceptibility toward hydrolysis.
CB(pin) with anhydrous NaBArF4·6THF.
A mixture of [RuCl(dppe)Cp] (29.9 mg, 0.0498 mmol), PhCCB(pin) (11.6 mg, 0.0509), and NaBArF4·6THF (81.8 mg, 0.0620 mmol) in 1,2-dichloroethane (2 mL) was heated at 70 °C for 1 h. The resulting dark orange suspension was filtered through a short pad of Celite and rinsed with 1,2-dichloroethane (ca. 1 mL). The combined filtrate was dried in vacuo. The NMR yield was determined by using 1,3,5-trimethoxybenzene as an internal standard. Isolation of the corresponding vinylidene complex cannot be performed due to the instability toward hydrolysis. However, the efficient formation of the vinylidene complex 2g in a reaction using NMR tube provided reliable information on the formation of the vinylidene complex (for the procedure of the reaction using NMR tube, see the section of time course studies shown below).
1H NMR (CDCl3): δ 7.74 (s, 8H, o-H of BArF4), 7.53 (s, 4H, p-H of BArF4), 7.48–7.28 (m, 12H, p- ×2, m- and o-H of Ph in dppe), 7.24 (m, 4H, m-H of Ph in dppe), 7.10 (m, 7H, o-H of Ph in dppe and m- and p-H of RuCCPh), 6.69 (m, 2H, o-H of RuCCPh), 5.22 (s, 5H, Cp), 2.98 (m, 4H, CH2 of dppe × 2), 0.93 (s, 12H, CH3 of pin). 31P{1H} NMR (CDCl3): δ 78.3 (s, dppe). 13C{1H} NMR (CDCl3): δ 347.4 (t, 2JCP = 15.4 Hz, RuCC), 161.8 (q, 1JCB = 50.0 Hz, ipso-C of BArF4), 136.1 (m, ipso-C of Ph in dppe), 134.9 (br s, o-C of BArF4), 132.8 (m, o-C of Ph in dppe), 132.7 (m, ipso-C of Ph in dppe), 131.8 (s, p-C of Ph in dppe), 131.6 (s, p-C of Ph in dppe), 131.0 (m, o-C of Ph in dppe), 129.2 (m, m-C of Ph in dppe), 129.0 (brq, 2JCF = 29.7 Hz, m-C of BArF4), 128.9 (m, m-C of Ph in dppe and m- and o-C of RuCCC6H5), 126.7 (s, p-C of RuCCC6H5), 124.6 (q, 1JCF = 271.0 Hz, CF3 of BArF4), 117.5 (s, p-C of BArF4), 91.5 (s, Cp), 84.0 (s, CO of pin), 28.9 (m, PCH2), 24.4 (s, CH3 of pin). The signal assignable to the RuCC and ipso-C of RuCCC6H5 could not be found, probably because it is overlapped with other signals. High resolution mass measurement and elemental analysis have failed due to the high susceptibility toward hydrolysis.
Procedures for the protodeboronation
A 20 ml Schlenk was charged with PhCCB(dan) (13.9 mg, 0.0518 mmol) and H2O (2.3 mmol, 0.1276 mg) in 1,2-dichloroethane (2 ml) under an argon atmosphere. The mixture was heated 70 °C for 1 h. The brown solution was dried in vacuo and analyzed by 1H NMR. The NMR yield was determined by using 1,3,5-trimethoxybenzene as an internal standard.
A J Young NMR tube was charged with [RuCl(dppe)Cp] (30.0 mg, 0.0500 mmol), PhCCB(dan) (13.6 mg, 0.0507 mmol), and NaBArF4·6THF (81.7 mg, 0.0619 mmol) in CDCl3 (0.5 mL). After 7 h at room temperature, vinylidene complex 2f was formed in 94% NMR yield. After solvent was removed under reduced pressure, H2O (1.8 mg, 0.10 mmol) and 1,2-dichloroethane (0.5 ml) were added. The mixture was heated 70 °C for 1 h. The black solution was dried in vacuo and analyzed by 1H NMR spectrum. The NMR yield was determined by using 1,3,5-trimethoxybenzene as an internal standard. The same procedure was used for the protodeboronation of vinylidene complex 2g.
Syntheses of the byproduct 3 formed by protodeboronation
To characterize byproducts in the vinylidene rearrangements, we performed synthesis of the protodeboronated complex 3 as follows; A mixture of [RuCl(dppe)Cp] (60.3 mg, 0.1005 mmol), PhCCH (10.4 mg, 0.1018 mmol), and NaBArF4·2.6H2O (102.1 mg, 0.1094 mmol) in 1,2-dichloroethane (2 mL) was heated at 70 °C for 1 h. The resulting brown suspension was filtered through a short pad of Celite and rinsed with 1,2-dichloroethane (ca. 1 mL). The filtrate was dried in vacuo, and the residue was purified by column chromatography on silica gel (dichloromethane/hexane = 1:1). Complex 3 was obtained as a red oil (124.2 mg, 0.08812 mmol).
1H NMR (CDCl3): δ 7.76 (s, 8H, o-H of BArF4), 7.53 (s, 4H, p-H of BArF4), 7.40 (m, 16H, p- ×2, m- ×2 and o-H of Ph in dppe), 7.15 (m, 4H, o-H of Ph in dppe), 6.92 (m, 1H, p-H of RuCCPh), 6.86 (m, 2H, m-H of RuCCPh), 6.23 (m, 2H, o-H of RuCCPh), 5.52 (s, 5H, Cp), 4.68 (s, 1H, RuCCH), 3.00 (m, 2H, CH2 of dppe), 2.71 (m, 2H, CH2 of dppe). 31P{1H} NMR (CDCl3): δ 77.3 (s, dppe). 13C{1H} NMR (CDCl3): δ 354.1 (t, 2JCP = 16.1 Hz, RuCC), 161.9 (q, 1JCB = 50.0 Hz, ipso-C of BArF4), 135.0 (br, o-C of BArF4), 134.7 (m, ipso-C of Ph in dppe), 133.6 (m, ipso-C of Ph in dppe), 132.1 (m, p- ×2 and o-C of Ph in dppe), 131.4 (virtual t, o-C of Ph in dppe), 129.4 (m, m-C of Ph in dppe × 2), 129.0 (brq, 2JCF = 34.0 Hz, m-C of BArF4), 128.8 (s, m-C of RuCCC6H5), 126.9 (s, p-C of RuCCC6H5), 125.8 (s, o-C of RuCCC6H5), 125.1 (s, ipso-C of RuCCC6H5), 124.7 (q, 1JCF = 273.5 Hz, CF3 of BArF4), 117.9 (s, RuCC), 117.6 (m, p-C of BArF4), 92.0 (s, Cp), 27.2 (m, PCH2). HRMS m/z: [M + Na]+ calcd for RuP2C39H35+: 667.12575; found 667.12595.
Procedure for time course studies
A J Young NMR tube was charged with [RuCl(dppe)Cp] (1.0 equiv.), PhCCB (1.0 equiv.), and NaBArF4·6THF (1.1 equiv.) in CDCl3 (0.5 mL). The reaction was monitored by means of 1H NMR analysis, and the NMR yields were determined by using 1,3,5-trimethoxybenzene as an internal standard.
Author contributions
T. M. and K. S. performed all experiments under supervision by T. I. and Y. I. T. I. carried out theoretical studies. The manuscript was written by T. I. and Y. I. and approved by all authors prior to publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research C (21K05101 and 18K05154) from the JSPS. This work was also supported in part by Grant-in-Aid for Scientific Research C (20K05493) from the JSPS. Computation time was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University.References
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, Sandford, 2011 Search PubMed.
- V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- T. Hata, H. Kitagawa, H. Masai, T. Kurahashi, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 790–792 CrossRef CAS PubMed.
- Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770–13773 CrossRef CAS PubMed.
- J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774–13777 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943–2946 CrossRef CAS PubMed.
- R. K. Shiroodi, O. Koleda and V. Gevorgyan, J. Am. Chem. Soc., 2014, 136, 13146–13149 CrossRef CAS PubMed.
- A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed.
- L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef CAS PubMed.
- C. Frank Lee, D. B. Diaz, A. Holownia, S. J. Kaldas, S. K. Liew, G. E. Garrett, T. Dudding and A. K. Yudin, Nat. Chem., 2018, 10, 1062–1070 CrossRef PubMed.
- V. Fasano, J. Cid, R. J. Procter, E. Ross and M. J. Ingleson, Angew. Chem., Int. Ed., 2018, 57, 13293–13297 CrossRef CAS PubMed.
- D. Yukimori, Y. Nagashima, C. Wang, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2019, 141, 9819–9822 CrossRef CAS PubMed.
- Q. Wang, M. Biosca, F. Himo and K. J. Szabó, Angew. Chem., Int. Ed., 2021, 60, 26327–26331 CrossRef CAS PubMed.
- H. Kisu, H. Sakaino, F. Ito, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2016, 138, 3548–3552 CrossRef CAS PubMed.
- Z. Li, L. Zhang, M. Nishiura and Z. Hou, ACS Catal., 2019, 9, 4388–4393 CrossRef CAS.
- F. P. Wu and X. F. Wu, Angew. Chem., Int. Ed., 2021, 60, 695–700 CrossRef CAS PubMed.
- P. Dominguez-Molano, R. Weeks, R. J. Maza, J. J. Carbό and E. Fernández, Angew. Chem., Int. Ed., 2023, 62, e202304791 CrossRef CAS PubMed.
- R. A. Batey and D. V. Smil, Angew. Chem., Int. Ed., 1999, 38, 1798–1800 CrossRef CAS PubMed.
- D. Kaiser, A. Noble, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 14104–14109 CrossRef CAS PubMed.
- D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2020, 142, 9119–9123 CrossRef CAS PubMed.
- (a) K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CrossRef CAS; (b) K. Yang, Z. Kuang and Q. Song, Chem, 2020, 6, 330–331 CrossRef CAS.
- K. Jana and A. Studer, Org. Lett., 2022, 24, 1100–1104 CrossRef CAS PubMed.
- X. Tao, S. Ni, L. Kong, Y. Wang and Y. Pan, Chem. Sci., 2022, 13, 1946–1950 RSC.
- H. Wang, J. Wu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202202061 CrossRef CAS PubMed.
- X.-M. Jiang, X.-R. Liu, A. Chen, X.-Z. Zou, J.-F. Ge and D.-W. Gao, Eur. J. Org. Chem., 2022, e202101463 CrossRef CAS.
- C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS.
- F. E. McDonald, Chem. – Eur. J., 1999, 5, 3103–3106 CrossRef CAS.
- B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067–2096 CrossRef CAS PubMed.
- C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176–2203 CrossRef CAS PubMed.
- B. M. Trost and A. McClory, Chem. - Asian J., 2008, 3, 164–194 CrossRef CAS PubMed.
- J. A. Varela, C. González-Rodríguez and C. Saá, Top. Organomet. Chem., 2014, 48, 237–287 CrossRef.
- For a review, see: S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 CrossRef CAS PubMed.
- Y. Ikeda, T. Yamaguchi, K. Kanao, K. Kimura, S. Kamimura, Y. Mutoh, Y. Tanabe and Y. Ishii, J. Am. Chem. Soc., 2008, 130, 16856–16857 CrossRef CAS PubMed.
- Y. Mutoh, Y. Ikeda, Y. Kimura and Y. Ishii, Chem. Lett., 2009, 38, 534–535 CrossRef CAS.
- Y. Mutoh, Y. Kimura, Y. Ikeda, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2012, 31, 5150–5158 CrossRef CAS.
- M. Otsuka, N. Tsuchida, Y. Ikeda, Y. Kimura, Y. Mutoh, Y. Ishii and K. Takano, J. Am. Chem. Soc., 2012, 134, 17746–17756 CrossRef CAS PubMed.
- Y. Ikeda, Y. Mutoh, K. Imai, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2013, 32, 4353–4358 CrossRef CAS.
- M. Otsuka, N. Tsuchida, Y. Ikeda, N. Lambert, R. Nakamura, Y. Mutoh, Y. Ishii and K. Takano, Organometallics, 2015, 34, 3934–3943 CrossRef CAS.
- Y. Ikeda, S. Kodama, N. Tsuchida and Y. Ishii, Dalton Trans., 2015, 44, 17448–17452 RSC.
- R. Sun, S. Zhang, X. Chu and B. Zhu, Organometallics, 2017, 36, 1133–1141 CrossRef CAS.
- X. Chu, S. Zhang, Z. Wang, T. Li and B. Zhu, RSC Adv., 2018, 8, 7164–7172 RSC.
- K. Yoshida and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 338–342 CrossRef CAS PubMed.
- M. Korb, S. Moggach and P. J. Low, Chem. Commun., 2021, 57, 4251–4254 RSC.
- T. Watanabe, Y. Mutoh and S. Saito, J. Am. Chem. Soc., 2017, 139, 7749–7752 CrossRef CAS PubMed.
- T. Watanabe, H. Abe, Y. Mutoh and S. Saito, Chem. – Eur. J., 2018, 24, 11545–11549 CrossRef CAS PubMed.
- T. Watanabe, Y. Mutoh and S. Saito, Org. Biomol. Chem., 2020, 18, 81–85 RSC.
- For the pioneering works on vinylidene rearrangement of Si/Sn-substituted alkynes, see: (a) H. Werner, M. Baum, D. Schneider and B. Windmüller, Organometallics, 1994, 13, 1089–1097 CrossRef CAS; (b) K. Venkatesan, O. Blacque, T. Fox, M. Alfonso, H. W. Schmalle, S. Kheradmandan and H. Berke, Organometallics, 2005, 24, 920–932 CrossRef CAS.
- For vinylidene rearrangement of S/Se/P/I-substituted alkynes, see: (a) D. C. Miller and R. J. Angelici, Organometallics, 1991, 10, 79–89 CrossRef CAS; (b) A. F. Hill, A. G. Hulkes, A. J. P. White and D. J. Williams, Organometallics, 2000, 19, 371–373 CrossRef CAS; (c) H. Kim and C. Lee, J. Am. Chem. Soc., 2005, 127, 10180–10181 CrossRef CAS PubMed; (d) W. Lim and Y. H. Rhee, Eur. J. Org. Chem., 2013, 460–464 CrossRef CAS; (e) T. Kuwabara, Y. Aoki, K. Sakajiri, K. Deguchi, S. Takamori, A. Hamano, K. Takano, H. Houjou and Y. Ishii, Organometallics, 2020, 39, 711–718 CrossRef CAS; (f) C. Löwe, H.-U. Hund and H. Berke, J. Organomet. Chem., 1989, 371, 311–319 CrossRef; (g) T. Miura and N. Iwasawa, J. Am. Chem. Soc., 2002, 124, 518–519 CrossRef CAS PubMed.
- For vinylidene rearrangement of B-substituted alkynes, see: (a) H. Braunschweig, C. K. L. Brown, R. D. Dewhurst, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher and B. Pfaffinger, Chem. – Eur. J., 2014, 20, 1427–1433 CrossRef CAS PubMed; (b) A. F. Hill, C. D. Stewart and J. S. Ward, Dalton Trans., 2015, 44, 5713–5726 RSC; (c) C.-I. Lee, J. C. DeMott, C. J. Pell, A. Christopher, J. Zhou, N. Bhuvanesha and O. V. Ozerov, Chem. Sci., 2015, 6, 6572–6582 RSC.
- Boryl vinylidene complexes of osmium were reported to be formed through 1,3-boryl migration of a (boryl)(alkynyl)osmium complex; (a) M. L. Buil, M. A. Esteruelas, K. Garcés and E. Oñate, J. Am. Chem. Soc., 2011, 133, 2250–2263 CrossRef CAS PubMed; (b) M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, Organometallics, 2012, 31, 2965–2970 CrossRef CAS.
- A. B. Cuenca and E. Fernndez, Chem. Soc. Rev., 2021, 50, 72–86 RSC.
- We thank the reviewers for this discussion on the mechanism of the protodeboronation.
- Formation of η2-alkyne complexes are considered to be more facile than vinylidene formation from the η2-alkyne complexes with the [CpRuCl(phosphine)2] + NaBArF4 system; for examples, see ref. 34 and 35.
- (a) V. K. Singh, E. Bustelo, I. de los Rios, I. Macias-Arce, M. C. Puerta, P. Valerga, M. A. Ortuno, G. Ujaque and A. Lledos, Organometallics, 2011, 30, 4014–4031 CrossRef CAS; (b) J. Silvestre and R. Hoffmann, Helv. Chim. Acta, 1985, 68, 1461–1506 CrossRef CAS.
- Although we must await further theoretical study, migration of B(mida) group may require dissociation of the B–N dative bond. We thank a reviewer for this mechanistic discussion.
- (a) N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed; (b) A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC.
- L. Britton, M. Skrodzki, G. S. Nichol, A. P. Dominey, P. Pawluc, J. H. Docherty and S. P. Thomas, ACS Catal., 2021, 11, 6857–6864 CrossRef CAS.
- T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Adv. Synth. Catal., 2015, 357, 77–82 CrossRef CAS.
- H. E. Ho, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4670–4673 CrossRef CAS PubMed.
- W.-X. Fan, J.-L. Li, W.-X. Lv, L. Yang, Q. Li and H. Wang, Chem. Commun., 2020, 56, 82–85 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01042f |
| This journal is © The Royal Society of Chemistry 2024 |