DOI:
10.1039/D3CS00762F
(Review Article)
Chem. Soc. Rev., 2024,
53, 3457-3484
Recent advances in Rh(I)-catalyzed enantioselective C–H functionalization
Received
12th September 2023
First published on 27th February 2024
Abstract
Chiral carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds are pervasive and very essential in natural products, bioactive molecules, and functional materials, and their catalytic construction has emerged as one of the hottest research fields in synthetic organic chemistry. The last decade has witnessed vigorous progress in Rh(I)-catalyzed asymmetric C–H functionalization as a complement to Rh(II) and Rh(III) catalysis. This review aims to provide the most comprehensive and up-to-date summary covering the recent advances in Rh(I)-catalyzed C–H activation for asymmetric functionalization. In addition to the development of diverse reactions, chiral ligand design and mechanistic investigation (inner-sphere mechanism, outer-sphere mechanism, and 1,4-Rh migration) will also be highlighted.

Yue Zhang
| Yue Zhang received her BS degree in Chemical Biology in 2015 from Tianjin Normal University (China). She completed her PhD in 2020 under the supervision of Professor Jun-An Ma in Tianjin University (China). Currently, she is an assistant researcher at Institute of Botany, Jiangsu Province and Chinese Academy of Sciences. From 2022 to 2023, she worked as visiting scholar at Nanjing Forestry University. Her research interests focus on chiral synthesis, isolation and extraction of natural products from plants and analysis of their biosynthetic pathways. |

Jing-Jing Zhang
| Jing-Jing Zhang received her Bachelor's degree in Chemistry and Master's degree in Organic Chemistry from Nanjing University (China) in 2006 and 2009, respectively. Thereafter, she got her PhD degree in Chemical Biology from the University of Hong Kong in 2014 (China). From 2014 to 2016, she worked at Technische Universitat Braunschweig (Germany) and Universitat Heidelberg (Germany) and was sponsored by Alexander von Humboldt Research Fellowship. From 2016 to 2022, she worked as an associate researcher at China Pharmaceutical University. She currently works at Nanjing Forestry University. Her research interests include the design and synthesis of novel compounds with multiple biological activities and the study of the underlying mechanism. |

Lujun Lou
| Lujun Lou received her BS degree in Chemical Engineering and Technology in 2023 from Taizhou University. She is now in her first year of postgraduate study under the supervision of Prof. Zhen Chen and Jing-Jing Zhang. Her research interest is the development of catalysis and organofluorine chemistry. |

Ruofan Lin
| Ruofan Lin received her BS degree in Biology and Medicine in 2021 from Nanjing Forestry University. She is now in her second year of postgraduate study under the supervision of Prof. Zhen Chen. Her research interest is the development of CNS PET ligands and organofluorine chemistry. |

Nicolai Cramer
| Nicolai Cramer earned his PhD in 2005 at the University of Stuttgart under the guidance of Sabine Laschat. After a postdoctoral stint with Barry Trost at Stanford, he completed his habilitation at ETH Zurich from 2007 to 2010 with Erick Carreira. Subsequently, he moved to EPFL as assistant professor, was tenured in 2013 and promoted to full professor in 2015. His research encompasses sustainable enantioselective metal-catalyzed transformations and their implementation in synthesis. A focus is placed on asymmetric C−H functionalizations enabled by novel ligand designs. |

Shou-Guo Wang
| Shou-Guo Wang obtained his PhD from the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences in 2015 under the supervision of Prof. Shu-Li You before joining the lab Prof. Nicolai Cramer at École Polytechnique Fédérale de Lausanne (EPFL) as postdoctoral researcher. In 2020, he joined the Shenzhen Institutes of Advanced Technology (SIAT), Chinese Academy of Sciences, as a principal investigator. His main research interest encompasses the discovery of new catalytic systems in organic chemistry and beyond to enable their implementation in the synthesis of biologically active molecules. |

Zhen Chen
| Zhen Chen received his BS degree in Applied Chemistry in 2013 from Tianjin University (China). He completed his PhD in 2019 under the supervision of Prof. Jun-An Ma at Tianjin University (China). From 2017 to 2020, he studied radiochemistry as an exchange PhD student and postdoctoral fellow with Prof. Steven H. Liang at Harvard Medical School/Massachusetts General Hospital (HMS/MGH, USA). From 2020 to 2021, he worked as a postdoctoral research fellow in the group of Prof. Nicolai Cramer at EPFL (Switzerland). Currently, he is a full professor at Nanjing Forestry University. His research interests focus on the development of novel catalytic synthetic methodologies and PET biomarkers. |
1. Introduction
Chiral carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds are pervasive and very essential in natural products, bioactive molecules, and functional materials, and their catalytic construction has emerged as one of the hottest research fields in synthetic organic chemistry.1–3 Among various methodologies, direct asymmetric functionalization of C–H bonds, which are ubiquitous in organic molecules, via transition metal catalysis represents one of the most powerful approaches in this field benefiting from its atom- and step-economy.4–8 Additionally, such an approach offers a new synthetic opportunity as well as an unconventional and complementary retro-synthetic disconnection compared with traditional methodologies requiring pre-activation or pre-functionalization of both reaction partners. Although C–H bonds are more difficult to be implemented than their pre-functionalized counterparts, they are, virtually, not completely inert, and could be cleaved by an appropriate catalyst to serve as latent surrogates of various functional groups.
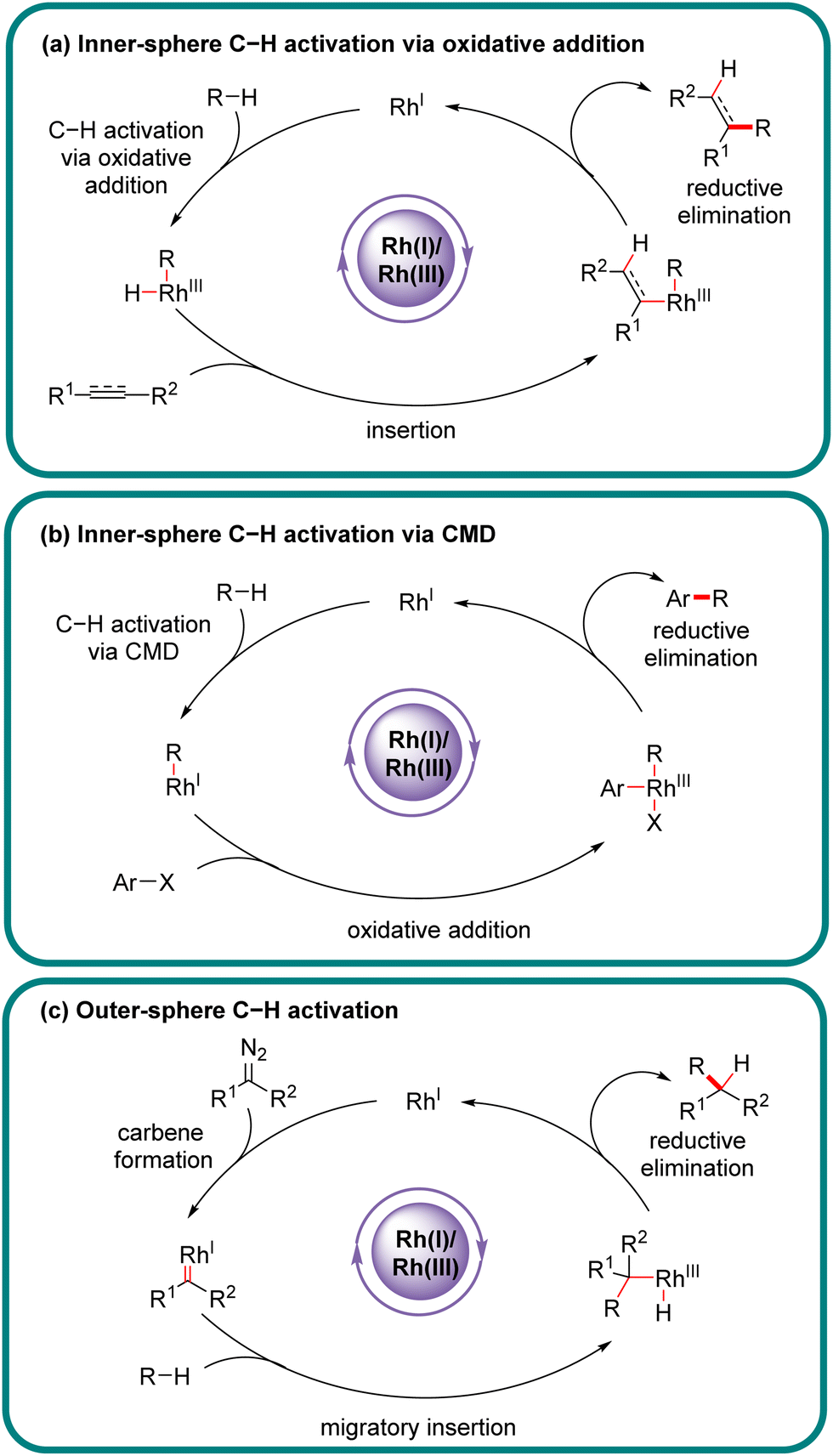
Actually, the past few decades have witnessed the successful implication of diverse transition metals in asymmetric C–H bond functionalization, including first, second, and third-row transition metals, as well as lanthanides.9–31 Compared to other transition metals, one characteristic feature of rhodium catalysts resides in the availability of multifarious oxidation states (0, 1, 2, and 3), offering an opportunity to effect various catalytic reactions. In particular, rhodium catalysts have been extensively investigated in asymmetric C–H functionalization with diverse mechanisms.32–45 From the viewpoint of substrate activation modes, Rh-catalyzed C–H functionalization could be classified into two categories: inner-sphere mechanism and outer-sphere mechanism, which differ in the mechanism of the C–H activation step (Scheme 1).46,47 Generally, Rh(III)-catalyzed asymmetric C–H functionalization adopts the inner-sphere mechanism by taking advantage of a chiral cyclopentadienyl (Cp) ligand,35,38,39,48–51 whereas Rh(II) usually coordinates with a chiral carboxylic acid to achieve the effect of asymmetric C–H functionalization via the outer-sphere mechanism,14,52–54 both of which have been well reviewed elsewhere.
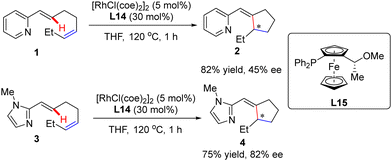 |
| | Scheme 1 Rh(I)-Catalyzed enantioselective intramolecular C(sp2)–H alkylation of alkenes. | |
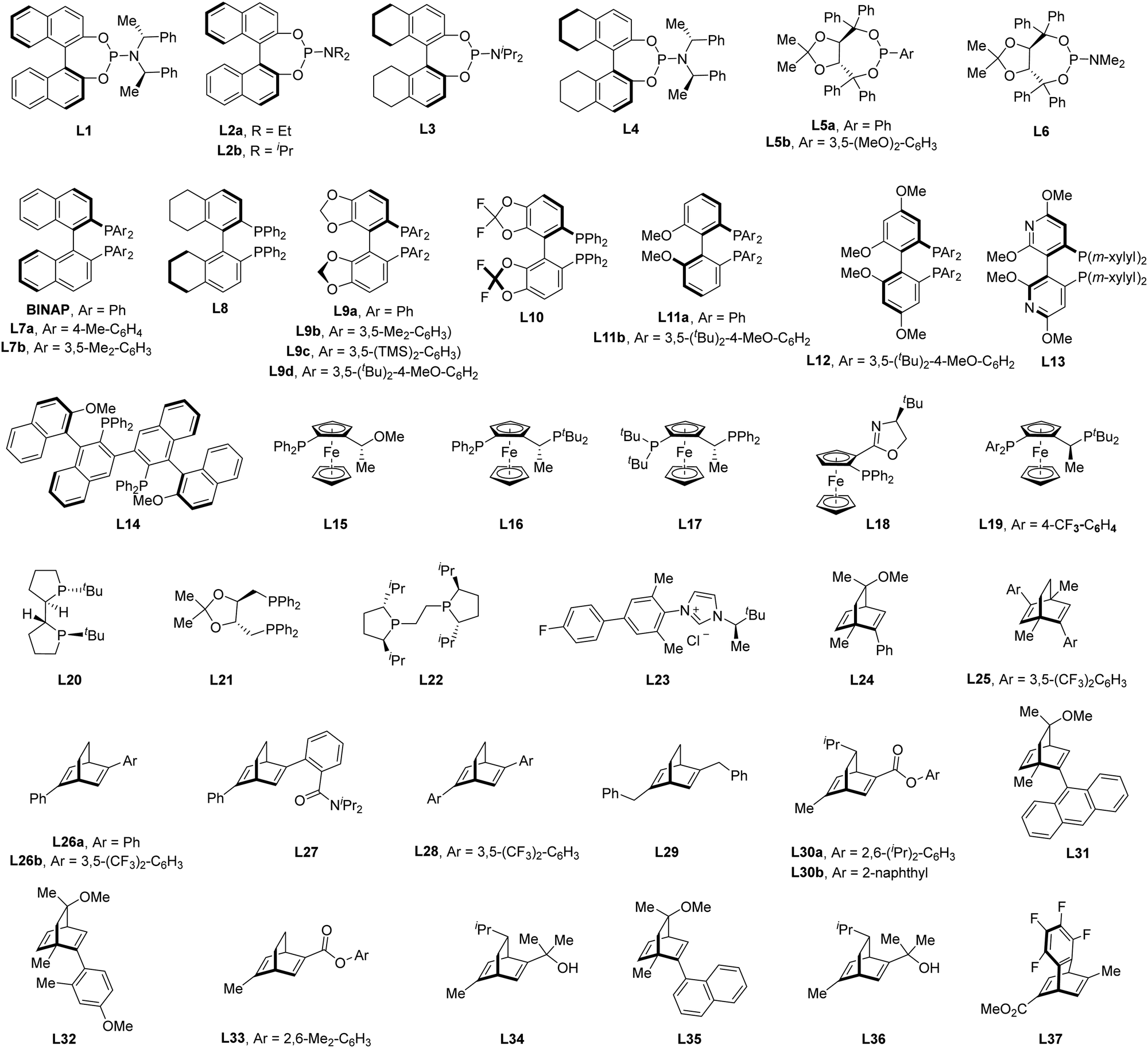
Of note, the last decade has witnessed vigorous progress in Rh(I)-catalyzed asymmetric C–H functionalization as a complement to Rh(II) and Rh(III) catalysis. In the asymmetric Rh(I) catalysis, both inner-sphere and outer-sphere mechanisms could be involved, despite that the inner-sphere dominates (Fig. 1). Additionally, a special 1,4-Rh migration pathway may also be involved in the asymmetric Rh(I) catalysis. Notably, various types of chiral ligands could be implemented including phosphoramidite, phosphonate, carbodicarbene, bisoxazolinephosphane, BINAP, Segphos, Tangphos, Josiphos, diene, and so on (Fig. 2). As a consequence, a diversity of asymmetric C–H transformations, such as alkylation, alkenylation, allylation, arylation, and silylation, have been developed. Despite these advances, a review comprehensively summarizing Rh(I)-catalyzed asymmetric C–H functionalization is elusive. Herein, we aim to provide the most comprehensive and up-to-date summary covering the recent advances in Rh(I)-catalyzed asymmetric functionalization of both C(sp2)–H and C(sp3)–H bonds. Rh(I)-catalyzed hydroacylations and Friedel–Crafts reactions have been the subject of multiple reviews and are therefore not covered here.55,56 We believe that this review will gain much attention from researchers with C–H functionalization and asymmetric transition metal catalysis backgrounds, stimulate more future efforts in this area, and facilitate the synthetic application of these novel reactions. We have structured the literature according to substrate activation modes and reaction types.
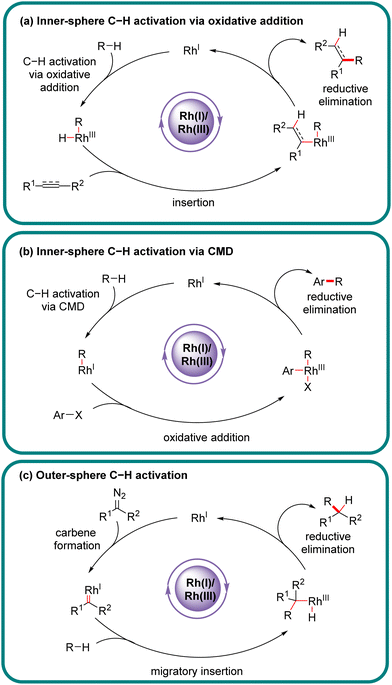 |
| | Fig. 1 Catalytic cycles for Rh(I)-catalyzed enantioselective C–H functionalization. | |
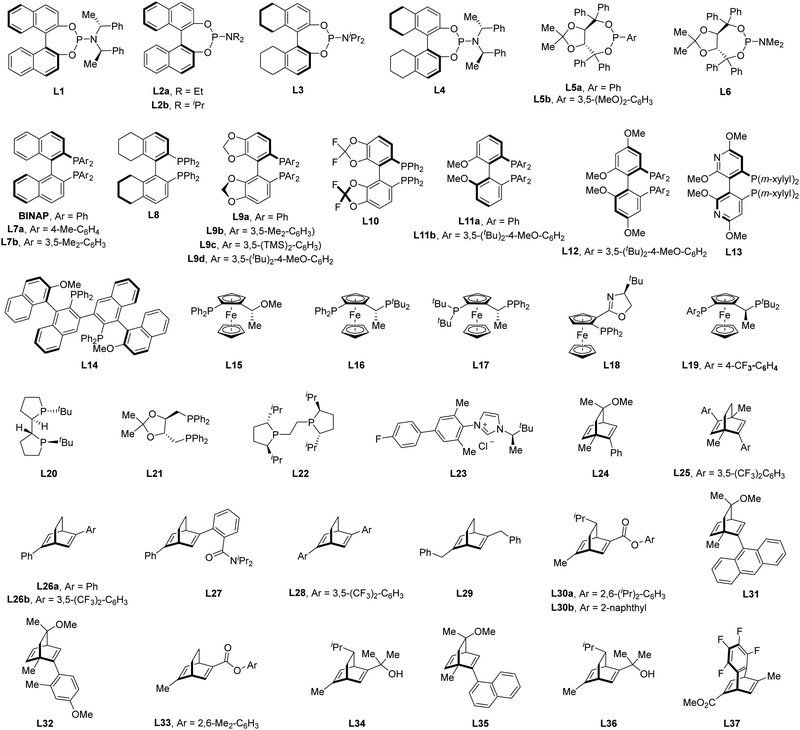 |
| | Fig. 2 The generally used chiral ligands in Rh(I)-catalyzed asymmetric C–H functionalization. | |
2. Inner-sphere C–H functionalization
In the inner-sphere mechanism, the metal directly interacts with a specific C–H bond to effect its activation and cleavage, leading to a discrete organo-rhodium species to proceed with follow-up functionalization. For Rh(I)-catalyzed asymmetric C–H functionalization, the inner-sphere mechanism represents the most prevalent C–H activation pathway. Owing to the fundamental differences in substrate activation modes, the selectivity of multiple C–H bonds differs. Typically, the inner-sphere activation mode is highly sensitive to the steric factor surrounding C–H bonds.
2.1 Functionalization of C(sp2)–H bonds
Compared with C(sp3)–H bonds, the sp2-hybridized counterparts are generally more sterically accessible and acidic, leading to more vigorous progress in C(sp2)–H functionalization. Additionally, rhodium usually possesses a good coordination affinity to unsaturated double and triple C–C bonds. Therefore, the use of alkenes, allenes, or alkynes as electrophiles can provide a good solution for the Rh(I)-catalyzed asymmetric functionalization of C(sp2)–H bonds.
2.1.1 Asymmetric addition of C(sp2)–H bonds to unsaturated bonds.
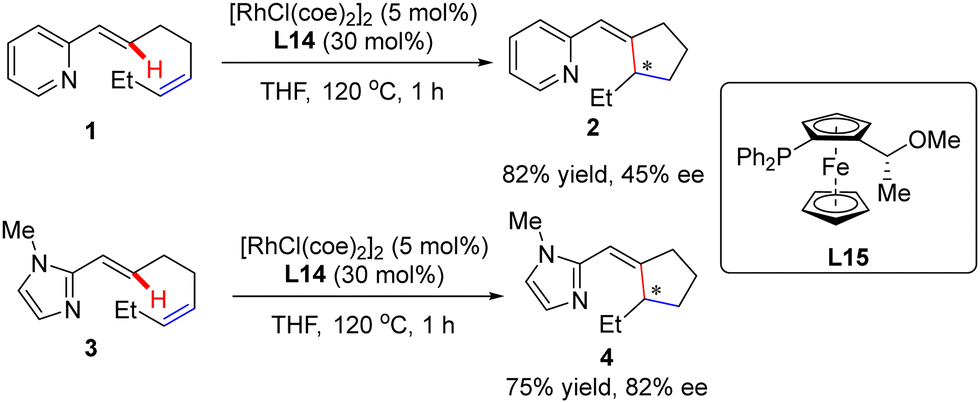
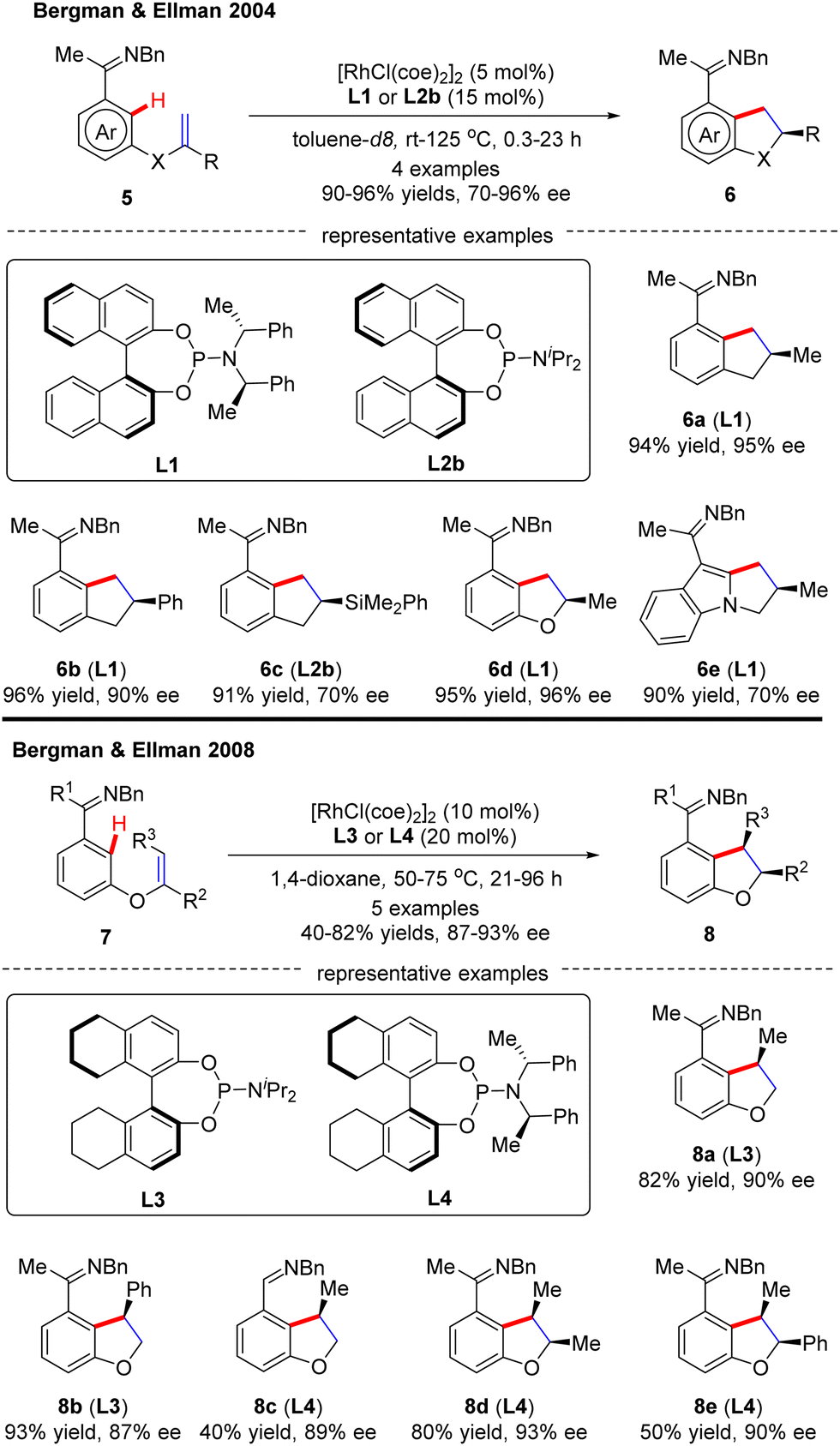
In this part, Murai and co-workers pioneered a Rh(I)-catalyzed asymmetric intramolecular alkene C(sp2)–H activation/olefin coupling reaction of 1,5-dienes with a 2-pyridyl (1) or 2-imidazolyl (3) directing group in 1997 (Scheme 1).57 By the use of monodentate planar chiral ferrocene phosphine ligand L15, the desired cyclopentane derivatives 2 and 4 were attained in 45% and 82% ee, respectively. Following this pioneering work, an elegant example was communicated at the beginning of this century, when Bergman and Ellman effected an imine-directed intramolecular addition of aromatic C–H bonds to 1,1-disubstituted alkenes (Scheme 2).58 Using [RhCl(coe)2]2 as the catalyst and (S)-binaphthol (BINOL)-derived monodentate phosphoramidite L1 or L2b as the chiral ligand, several five-membered carbo- and heterocycles 6 were accessed with impressive yields and enantioselectivities (up to 96% yield and 96% ee). Subsequently, the same group further expanded the substrate scope to 1,2-disubstituted as well as 1,1,2-trisubstituted alkenes by taking advantage of (S)-octahydrobinaphthol-based phosphoramidite L3 as a chiral ligand, thus giving rise to 2,3-dihydrobenzofuran derivatives 8 in moderate to excellent yields (40–82%) and excellent enantioselectivities (87–93% ee).59 For the cyclization of aldimine or 1,1,2-trisubstituted alkene substrates, ligand L4 provided better enantiocontrol. Notably, only syn-selective products were achieved, owing to that the E-alkenes, which would lead to anti-selective products, were unreactive. More importantly, the application of this approach was validated by the total synthesis of a PKC inhibitor.60
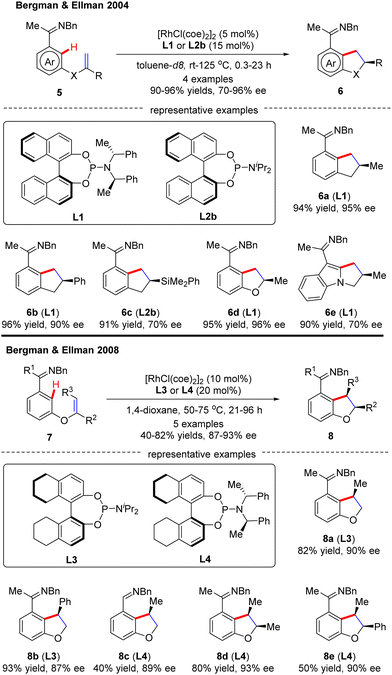 |
| | Scheme 2 Rh(I)-Catalyzed enantioselective imine-directed intramolecular C(sp2)–H alkylation. | |
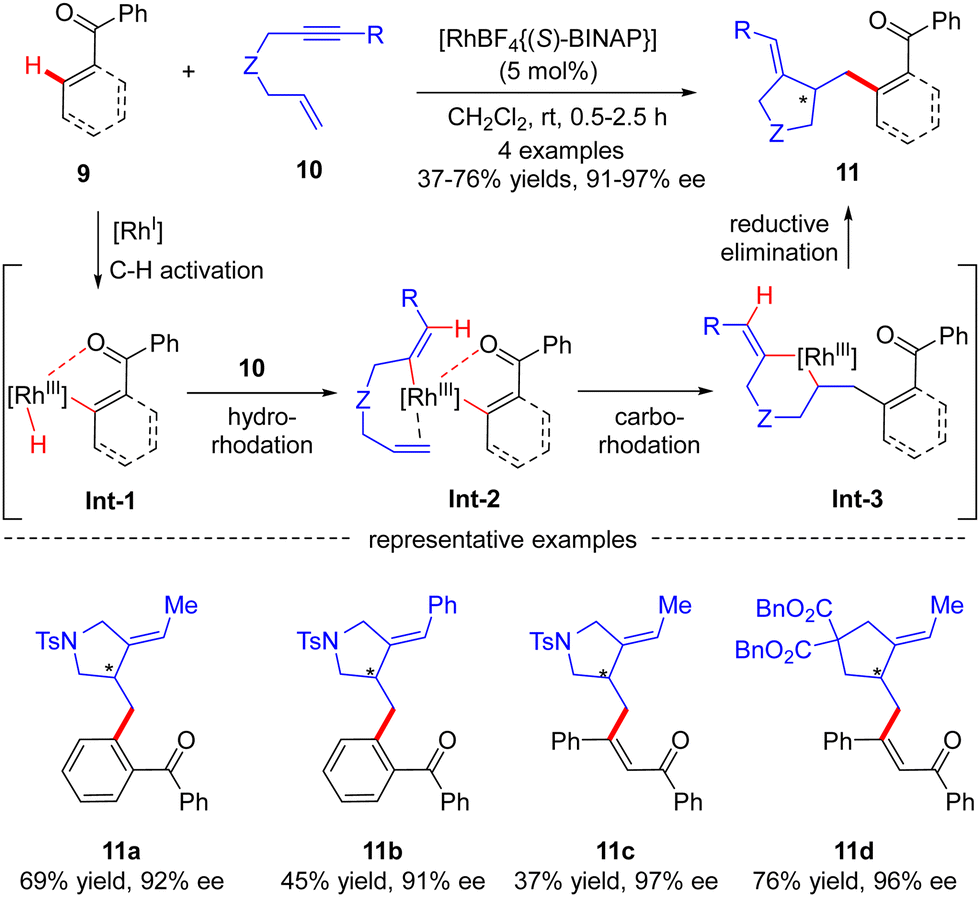
Aside from several reports of imine-directed C(sp2)–H functionalization, carbonyl could also serve as an effective directing group to enable similar transformations. For example, in 2007, Shibata and co-workers described a carbonyl-directed cascade C(sp2)–H activation and cyclization with enynes via asymmetric Rh(I) catalysis.61 With the chiral Rh-(S)-BINAP complex as a catalyst, both alkyl and aryl substituted N-tethered enynes readily reacted with the C(sp2)–H bond of benzophenone to furnish products 11a and 11b with 92% and 91% ee, respectively (Scheme 3). Apart from benzophenone, vinylic C–H functionalization of chalcone was also effected, thus affording the corresponding dienones 11c and 11d in excellent enantiocontrol. For the mechanism, the reaction was speculated to commence with carbonyl-directed C–H oxidative addition onto Rh(I) to provide aryl- or alkenyl-rhodium complex Int-1. Hydro-rhodation of Int-1 to the alkyne moiety of enyne 10 afforded intermediate Int-2, which then underwent intramolecular carbo-rhodation to the alkene moiety to afford metallacyclohexane Int-3. Finally, reductive elimination of Int-3 led to the release of the desired product 11.
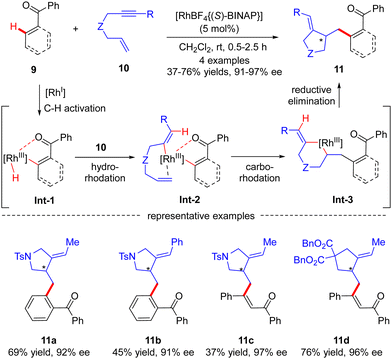 |
| | Scheme 3 Rh(I)-Catalyzed cyclization of enynes initiated by carbonyl-directed activation of aromatic and vinylic C–H bonds. | |
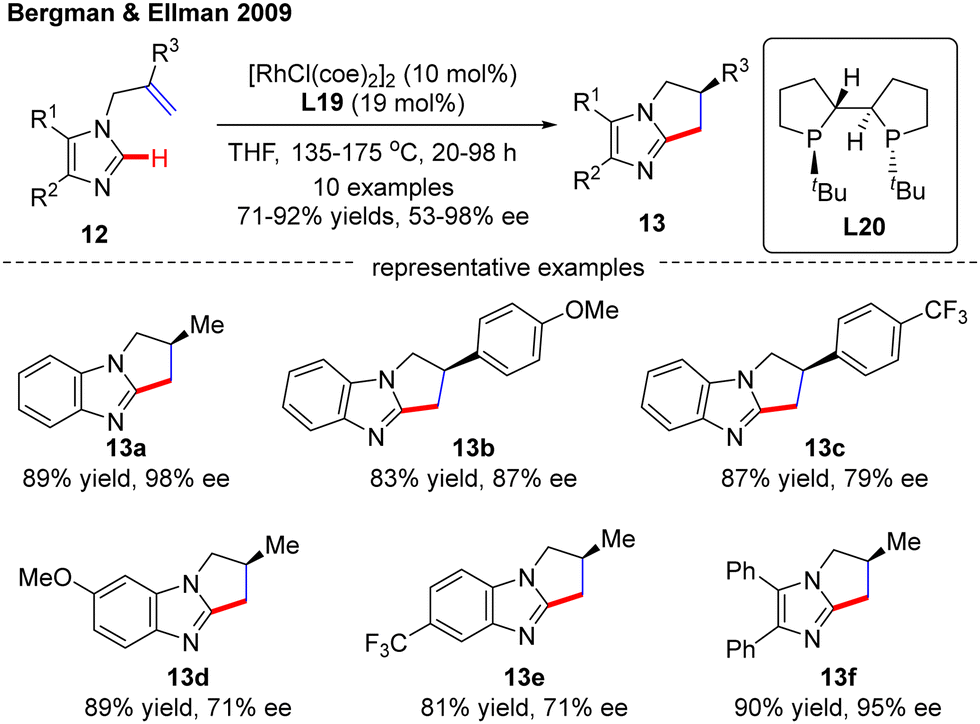
In 2009, Bergman and Ellman documented a Rh(I)-catalyzed intramolecular asymmetric C(sp2)–H alkylation of imidazoles (Scheme 4).62 While most bidentate ligands failed to deliver the desired product according to their previous work,63 Tangphos (L20) represented an exception. At elevated temperatures (135–175 °C), the intramolecular cyclization of allyl-substituted imidazoles 12 readily proceeded to forge various dihydro-pyrrolo-imidazoles 13 in moderate to excellent yields (71–92%) and enantioselectivities (53–98% ee).
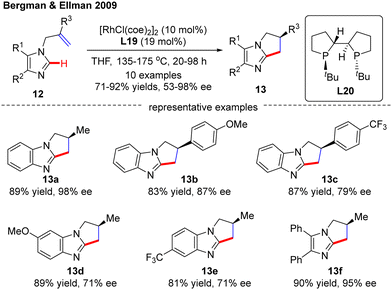 |
| | Scheme 4 Rh(I)-Catalyzed enantioselective intramolecular C(sp2)–H alkylation of imidazoles. | |
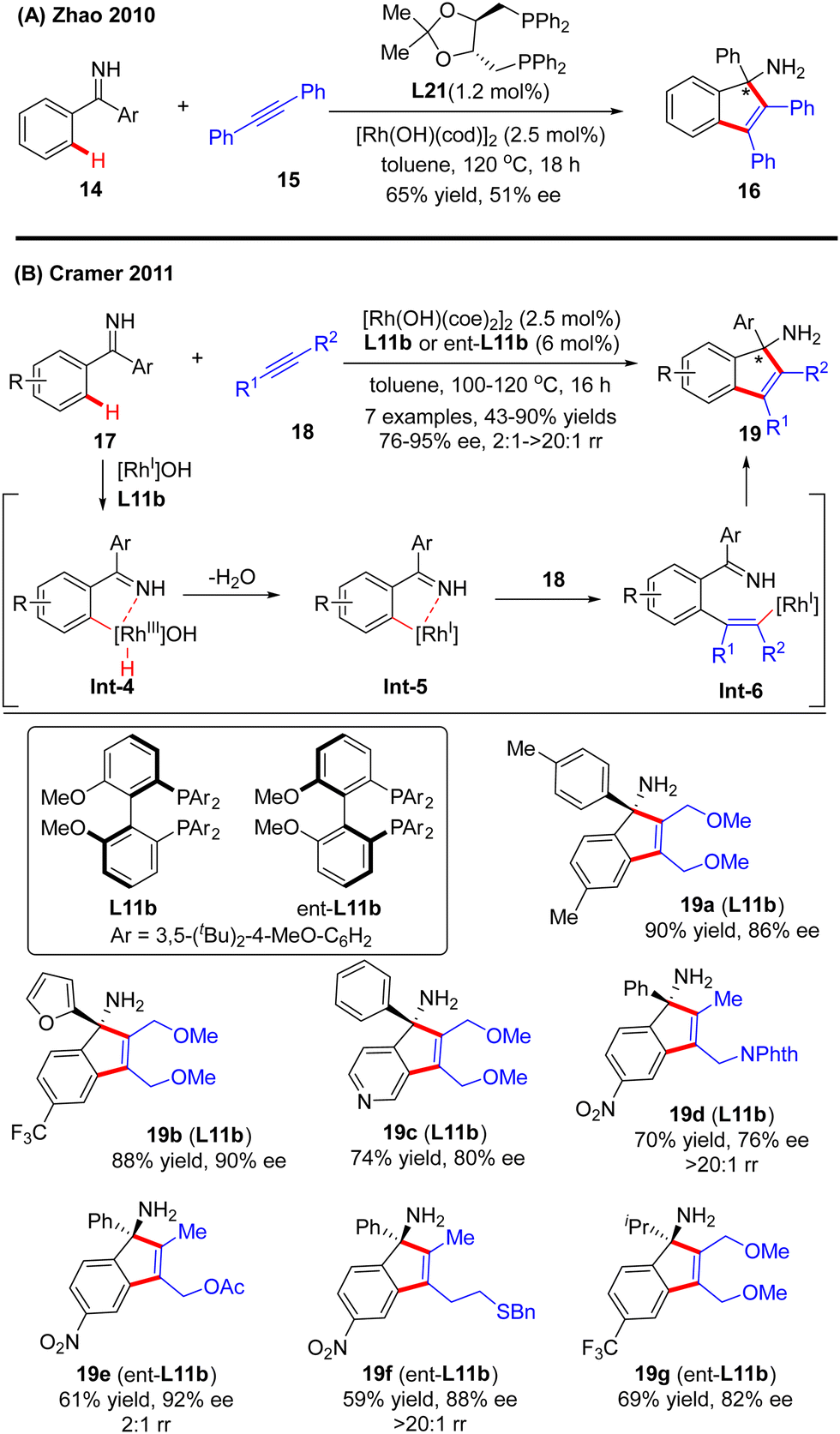
In 2010, Zhao and co-workers reported a single example of a Rh(I)-catalyzed [3+2] annulation reaction of benzophenone imine 14 with diphenylacetylene 15.64 In this reaction, the imine group in benzophenone imine 14 not only served as a directing group to enable C(sp2)–H activation at the ortho-position, but also could be transformed into an amino group by an addition reaction across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. To their disappointment, with bidentate phosphine (R,R)-DIOP L21 as the chiral ligand, only moderate enantioselectivity (51% ee) was achieved for the production of the tertiary 1H-inden-1-amine 16. Subsequently, the Cramer group disclosed an elegant approach for the [3+2] annulation reaction of aromatic ketimines 17 with internal dialkyl or arylalkyl alkynes 18 by taking advantage of the biphenyl diphosphine ligand DTBM-MeOBiphep (L11b) (Scheme 5).65 This approach allowed for the synthesis of a broad set of tertiary 1H-inden-1-amine derivatives 19 in 43–90% yields and good to excellent enantioselectivities (76–95% ee). The reaction was envisioned to be initiated by imine-directed ortho C(sp2)–H activation to form the cyclometalated rhodium complex Int-4. Notably, the C(sp2)–H activation step revealed a good positional preference for the more electron-poor aromatic substituent of the ketimine substrates. Subsequent removal of hydrogen from the metal center in Int-4 provided intermediate Int-5, which was essential to avoid undesirable hydroarylation reactivity. Migratory insertion of Int-5 into internal alkyne 18 then occurred to furnish an alkenyl-rhodium species Int-6, wherein the regioselectivity control could be achieved by using a potentially coordinating functional group in unsymmetrically substituted alkynes, such as ether (19a–c, 19g), ester (19e), phthalimide (19d), and sulfur-containing group (15f). Finally, enantioselective addition of Int-6 across the ketimine group set the desired product 19.
N bond. To their disappointment, with bidentate phosphine (R,R)-DIOP L21 as the chiral ligand, only moderate enantioselectivity (51% ee) was achieved for the production of the tertiary 1H-inden-1-amine 16. Subsequently, the Cramer group disclosed an elegant approach for the [3+2] annulation reaction of aromatic ketimines 17 with internal dialkyl or arylalkyl alkynes 18 by taking advantage of the biphenyl diphosphine ligand DTBM-MeOBiphep (L11b) (Scheme 5).65 This approach allowed for the synthesis of a broad set of tertiary 1H-inden-1-amine derivatives 19 in 43–90% yields and good to excellent enantioselectivities (76–95% ee). The reaction was envisioned to be initiated by imine-directed ortho C(sp2)–H activation to form the cyclometalated rhodium complex Int-4. Notably, the C(sp2)–H activation step revealed a good positional preference for the more electron-poor aromatic substituent of the ketimine substrates. Subsequent removal of hydrogen from the metal center in Int-4 provided intermediate Int-5, which was essential to avoid undesirable hydroarylation reactivity. Migratory insertion of Int-5 into internal alkyne 18 then occurred to furnish an alkenyl-rhodium species Int-6, wherein the regioselectivity control could be achieved by using a potentially coordinating functional group in unsymmetrically substituted alkynes, such as ether (19a–c, 19g), ester (19e), phthalimide (19d), and sulfur-containing group (15f). Finally, enantioselective addition of Int-6 across the ketimine group set the desired product 19.
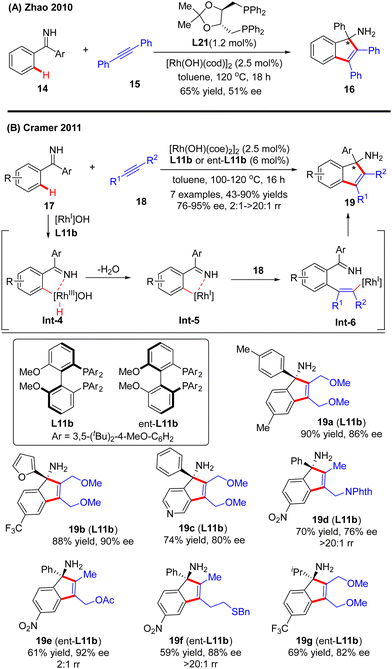 |
| | Scheme 5 Rh (I)-Catalyzed [3+2] annulations of aromatic ketimines with internal alkynes. | |
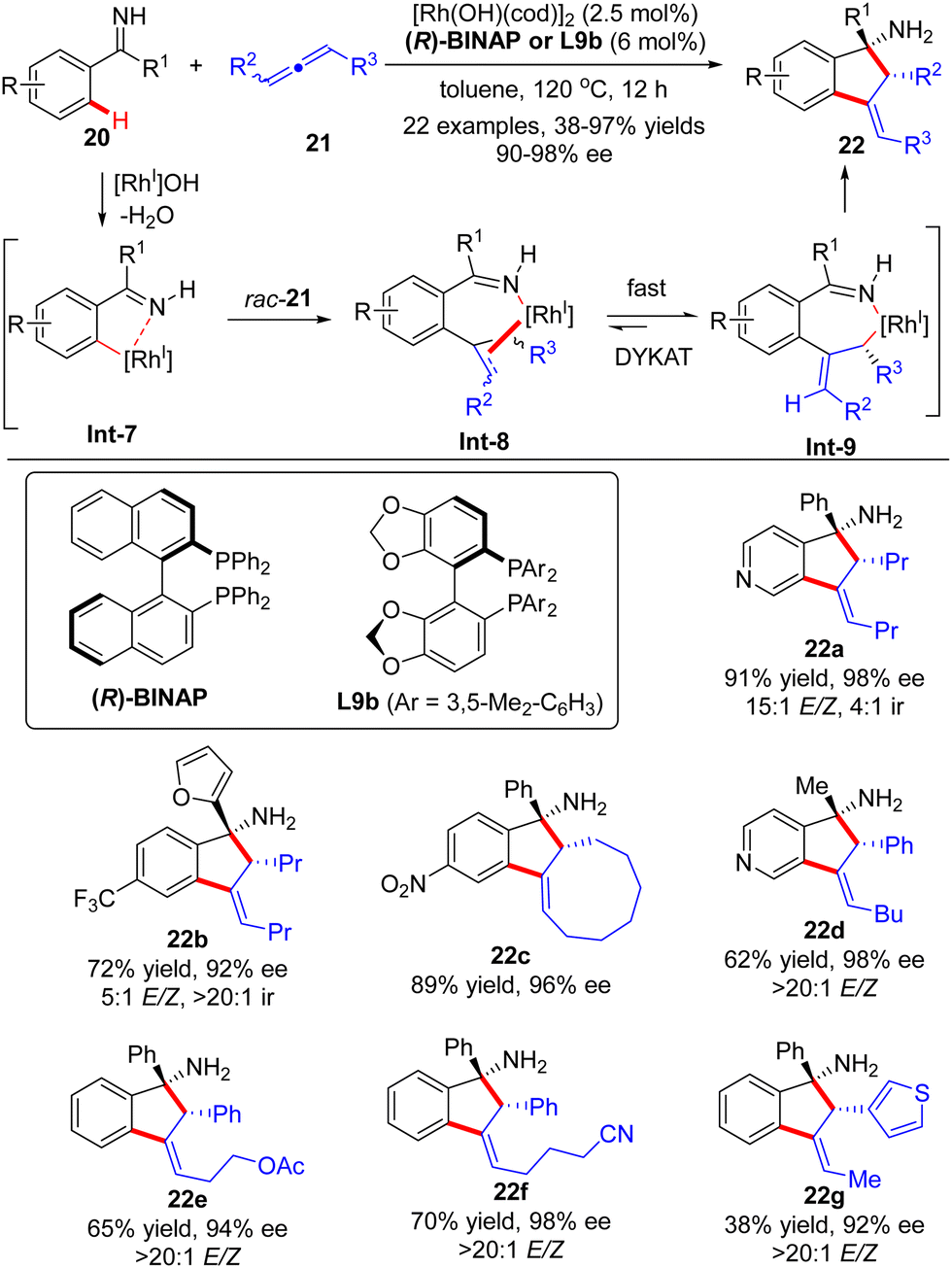
Soon after, the Cramer group further devised a Rh(I)-catalyzed dynamic kinetic asymmetric transformation (DYKAT) of racemic allenes 21 with ketimines 20 (Scheme 6).66,67 Similar to the above-described process with alkynes, this [3+2] annulation version of allenes with ketimines exhibited excellent regio-selectivity ranging from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (22a) to >20:1 (22b). The regio-selectivity resulted from the different positions of C–H activation of ketimines, which favored the electron-poor aryl and heteroaryl substituents. The coordination and migratory insertion of the initially generated cyclometalated species Int-7 into a racemic allene 21 gave rise to eight stereoisomers Int-8, which rapidly equilibrated and isomerized to reach the appropriate (E)-allyl isomer Int-9 by either a β-H elimination/re-addition sequence or a σ–π–σ mechanism.68 Follow-up intramolecular 1,2-addition across the imine moiety in Int-9 prevailed to arrive at the desired indanylamine 22 with excellent diastereomeric (syn-diasteroselectivity) and enantiomeric control (90–98% ee). For unsymmetrically 1,3-disubstituted allenes, the selectivity in the equilibration process of Int-8 was intensively governed by electronic factors with a preference for the formation of a benzylic allyl rhodium intermediate over the alternative alkyl allyl intermediate. Notably, the ketimine part did not exert a significant effect on the enantioselectivity, and both aryl aryl and aryl alkyl ketimines were well suited for this reaction.
:1 (22a) to >20:1 (22b). The regio-selectivity resulted from the different positions of C–H activation of ketimines, which favored the electron-poor aryl and heteroaryl substituents. The coordination and migratory insertion of the initially generated cyclometalated species Int-7 into a racemic allene 21 gave rise to eight stereoisomers Int-8, which rapidly equilibrated and isomerized to reach the appropriate (E)-allyl isomer Int-9 by either a β-H elimination/re-addition sequence or a σ–π–σ mechanism.68 Follow-up intramolecular 1,2-addition across the imine moiety in Int-9 prevailed to arrive at the desired indanylamine 22 with excellent diastereomeric (syn-diasteroselectivity) and enantiomeric control (90–98% ee). For unsymmetrically 1,3-disubstituted allenes, the selectivity in the equilibration process of Int-8 was intensively governed by electronic factors with a preference for the formation of a benzylic allyl rhodium intermediate over the alternative alkyl allyl intermediate. Notably, the ketimine part did not exert a significant effect on the enantioselectivity, and both aryl aryl and aryl alkyl ketimines were well suited for this reaction.
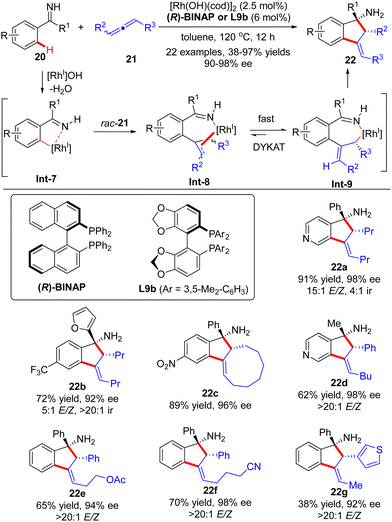 |
| | Scheme 6 Rhodium(I)-Catalyzed [3+2] annulations of aromatic ketimines with allenes. | |
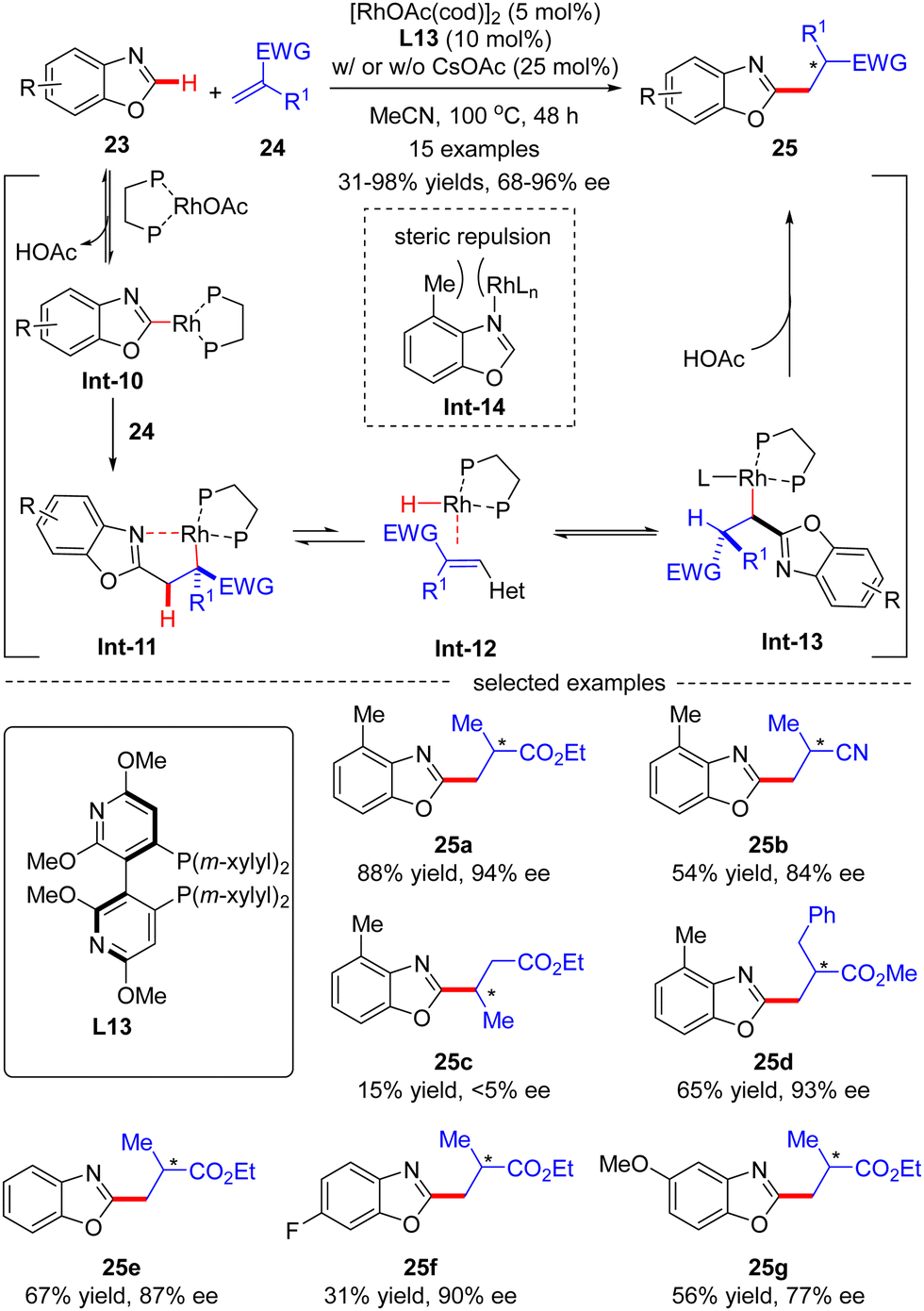
In 2015, Rovis and co-workers disclosed an asymmetric intermolecular C(sp2)–H alkylation of benzoxazole 23 with acrylates 24via Rh(I)-catalysis (Scheme 7).69 Mechanistic studies indicated that this reaction was initiated by reversible C–H activation of benzoxazole 23 with the aid of the rhodium acetate catalyst, thus providing Rh-benzoxazole complex Int-10. Migratory insertion of Int-10 across acrylate 24 furnished Rh-enolate Int-11, which isomerized into heterobenzylic rhodium species Int-13via a β-hydride elimination/hydrorhodation sequence. Protonation of Int-13 then occurred to set the alkylated benzoxazole 25, which was assumed to be the turnover limiting step. It was also demonstrated that 4-methylbenzoxazole, characteristic of a [1,3]-strain, disfavored strong coordination to rhodium in Int-14, thus exhibiting higher reactivity compared with its 6-substituted or unsubstituted counterparts (25avs.25evs.25f). In this scenario, the authors took advantage of a bulky Segphos derivative CTH-(R)-Xylyl-P-Phos (L13) as chiral ligand to surmount the possible deleterious coordination of the heteroarene and harness the reactivity. As a consequence, a diversity of 2-alkylated benzoxazoles 25 were attained in moderate to excellent yields (31–98%) and good to excellent enantioselectivities (68–96% ee). This reaction was tolerant of α-methyl substituted acrylate derivatives, whereas using β-substituted acrylate derivative crotonate as an alkylation reagent led to the racemic product 25c in extremely poor results (15% yield and <5% ee).
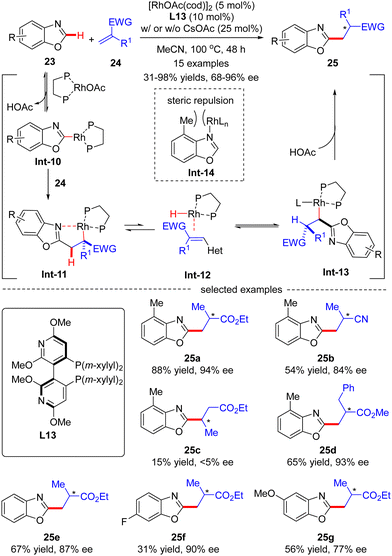 |
| | Scheme 7 Rh(I)-Catalyzed asymmetric intermolecular C(sp2)–H alkylation of benzoxazoles with acrylates. | |
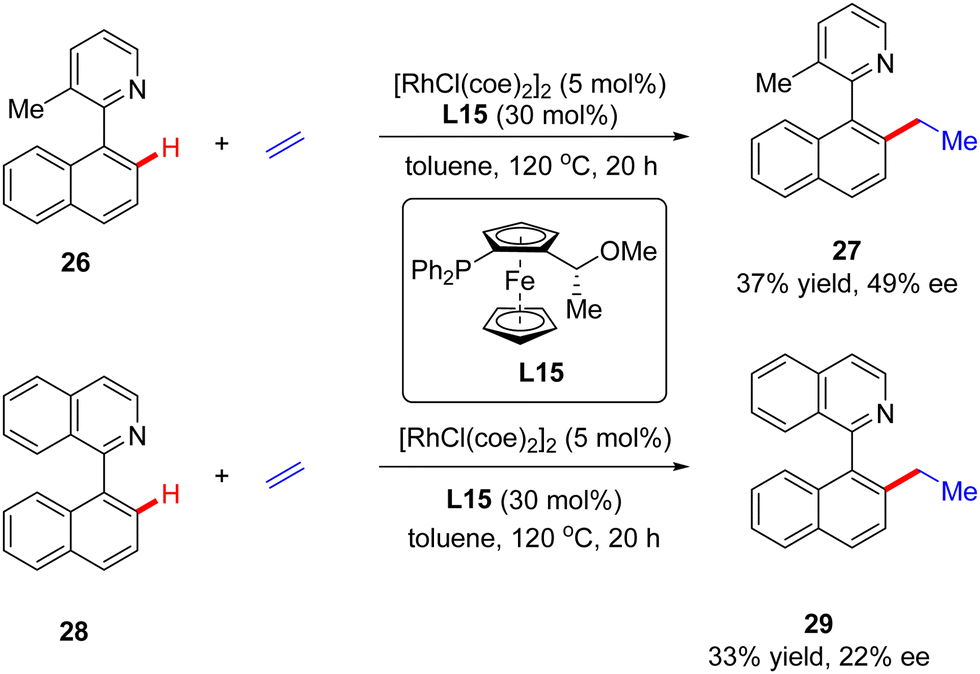
In addition to a number of reports on constructing central chirality, Rh(I)-catalyzed addition of C(sp2)–H to unsaturated bonds could also be applied in effecting axial chirality. In 2000, Murai and co-workers, for the first time, disclosed the construction of axially chiral biaryls 27 and 29via Rh(I)-catalyzed C(sp2)–H alkylation of naphthylpyridine derivatives 26 & 28 with ethylene (Scheme 8).70 Unfortunately, only moderate yields and enantioselectivities could be achieved (up to 37% yield and 49% ee) with [RhCl(coe)2]2 as the catalyst and chiral ferrocenyl phosphine L15 as the ligand.
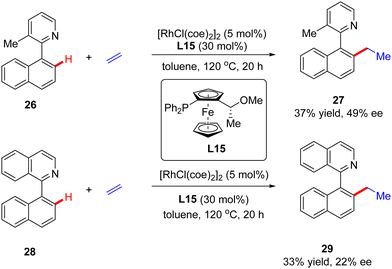 |
| | Scheme 8 Rh(I)-Catalyzed atroposelective C(sp2)–H alkylation of naphthylpyridine derivatives with ethylene. | |
2.1.2 Asymmetric arylation of C(sp2)–H bonds.
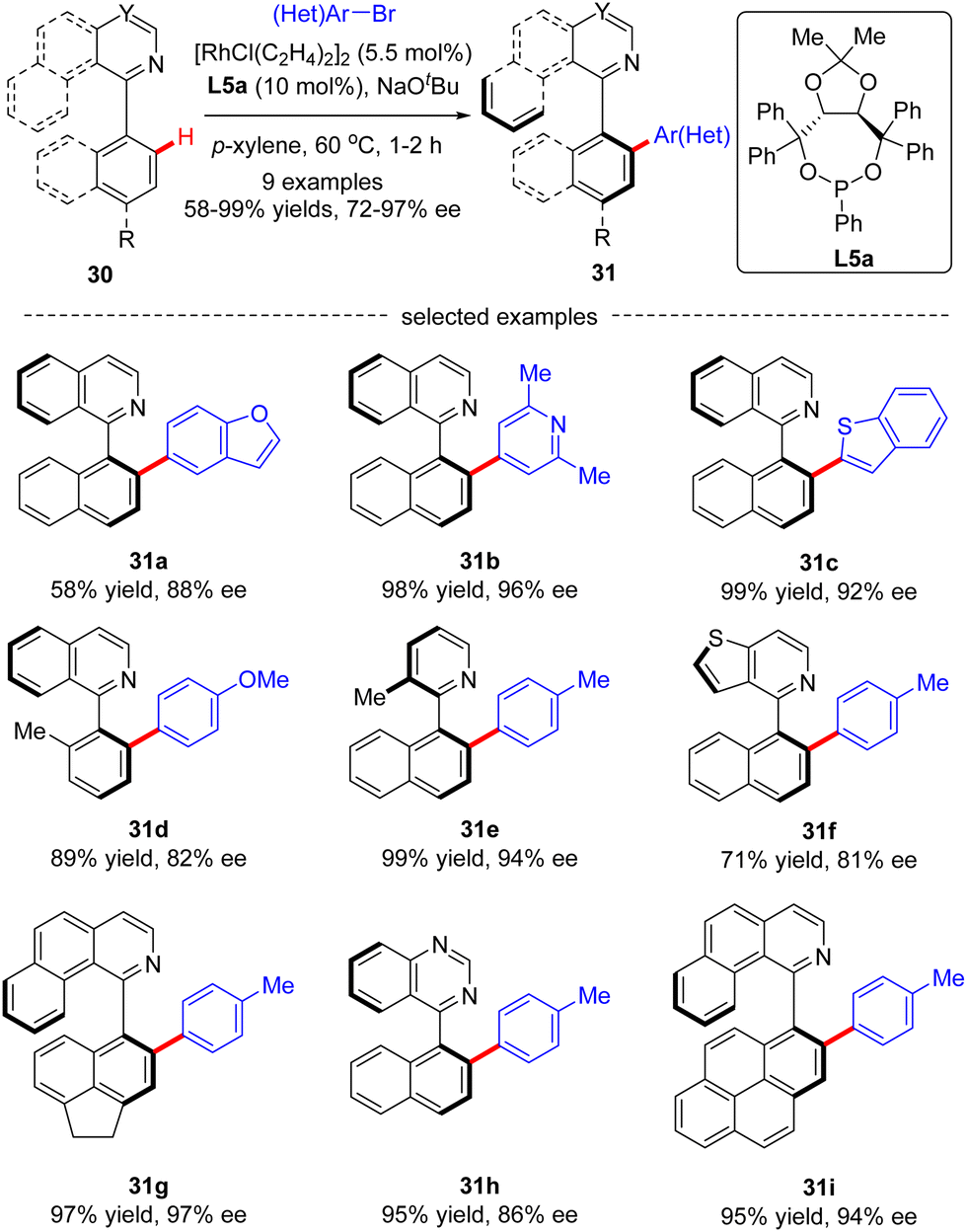
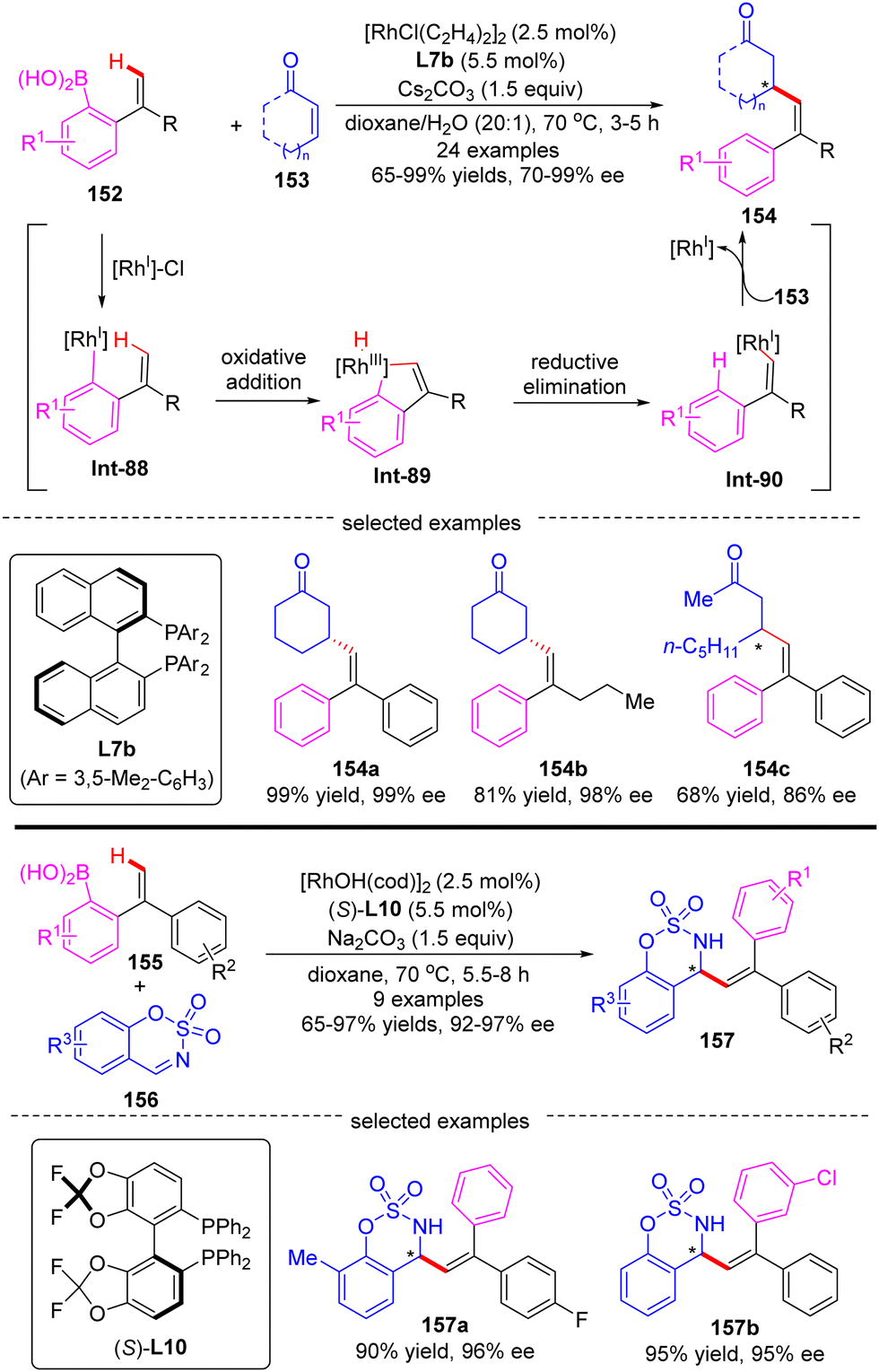
In 2019, You and co-workers reported a Rh(I)-catalyzed atroposelective C(sp2)–H arylation of heterobiaryls 30 with aryl and heteroaryl bromides (Scheme 9).71 By taking advantage of TADDOL-derived monodentate phosphonite L10 as the chiral ligand and N-heterocycles as directing groups, a diverse set of axially chiral heterobiaryls 31 were released in moderate to excellent yields (58–99%) with high to excellent enantioselectivities (72–99% ee). Remarkably, this approach lays a foundation to synthesize various axially chiral heterobiaryl catalysts.
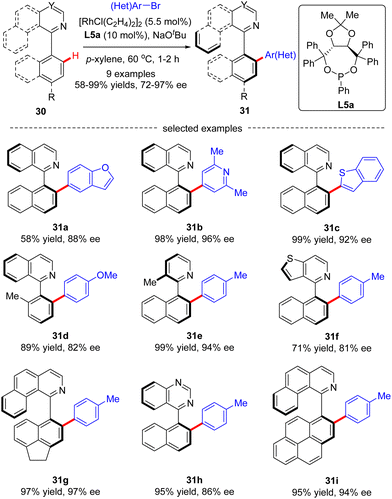 |
| | Scheme 9 Rh(I)-Catalyzed atroposelective C(sp2)–H arylation of heterobiaryls with aryl and heteroaryl bromides. | |
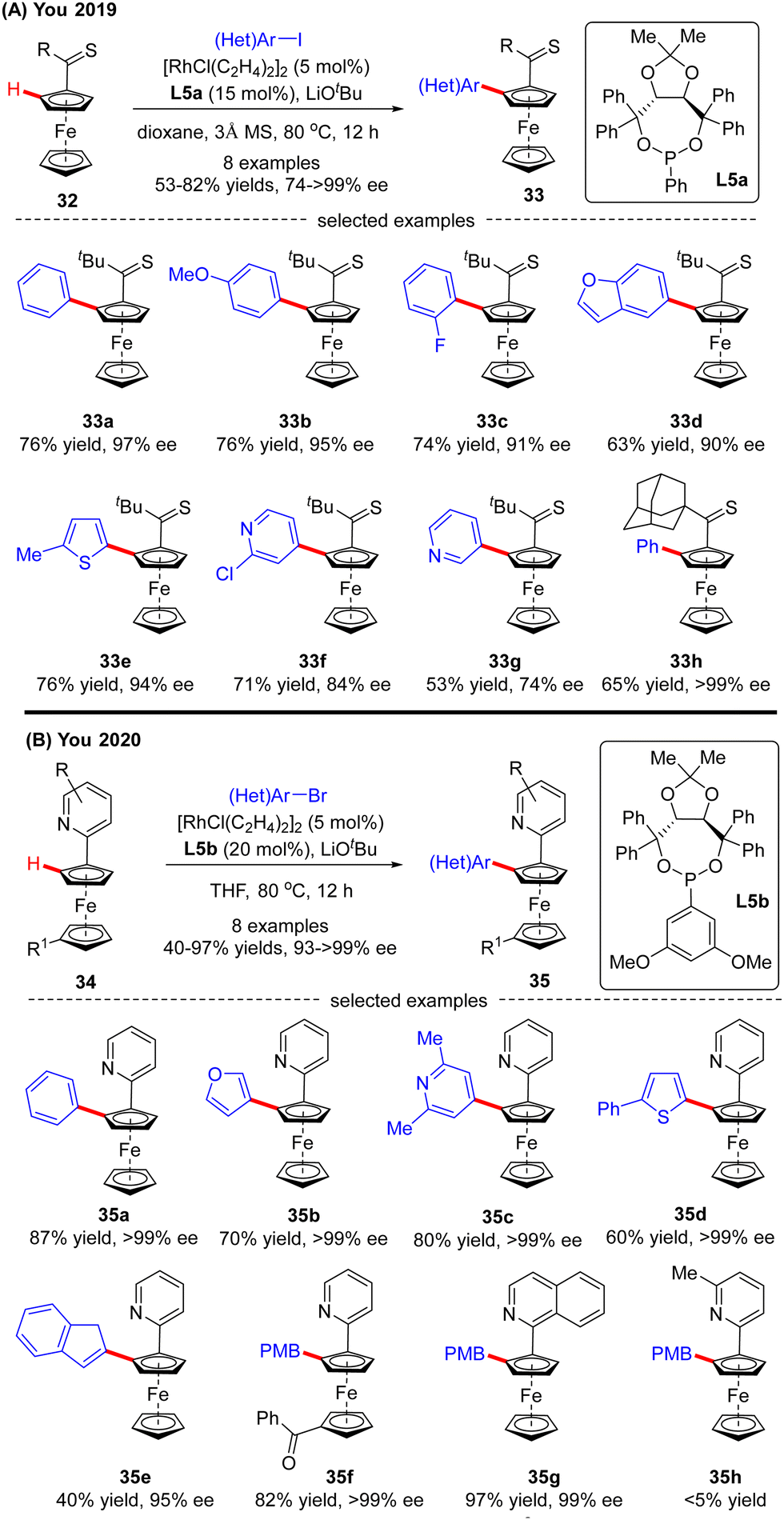
Recently, the implementation of planar chirality in Rh(I)-catalyzed C(sp2)–H functionalization was also demonstrated by You and co-workers (Scheme 10A).72 With thioketone as the directing group and L5a as the chiral ligand, a range of aryl and heteroaryl iodides were readily incorporated into thiocarbonylferrocene 32, thus providing direct access to various planar chiral ferrocenes 33 in moderate to good yields (53–82%) and excellent enantioselectivities (up to >99% ee). Subsequently, the same group further extended the ferrocene scope to pyridylferrocenes 34 by use of the Rh(I)/phosphonite catalytic system, wherein pyridine served as the directing group (Scheme 10B).73 As a consequence, a variety of (hetero)aryl bromides were readily coupled with pyridylferrocenes 34 to afford planar chiral ferrocene-based pyridine derivatives 35 in excellent yields (up to 97%) and enantioselectivities (93–>99% ee). Additionally, aryl iodide and aryl chloride were competent coupling partners, leading to the formation of 35a in comparable results (95% and >99% ee, respectively). Notably, aside from (hetero)aryl halides, alkenyl bromide also proved compatible with this reaction, giving product 35e in 40% yield and 95% ee. Meanwhile, substitution on the pyridine ring or ferrocene part was also tolerated (35f, 35g). However, 5-substituted pyridine was not a suitable substrate, which was probably attributed to its weak coordination with the rhodium catalyst (35h).
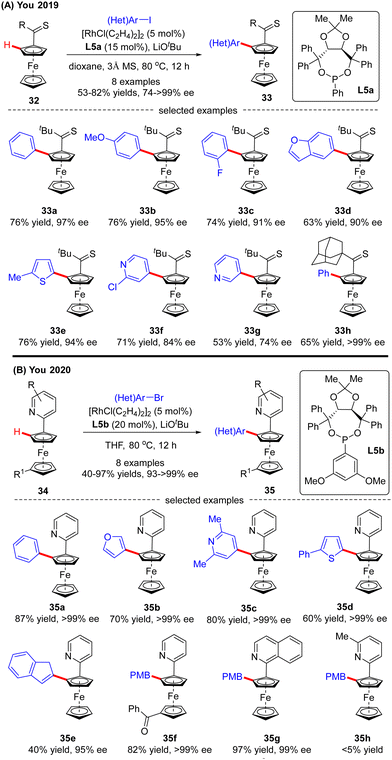 |
| | Scheme 10 Rh(I)-Catalyzed enantioselective C(sp2)–H functionalization of ferrocenes (PMB = p-methoxybenzyl). | |
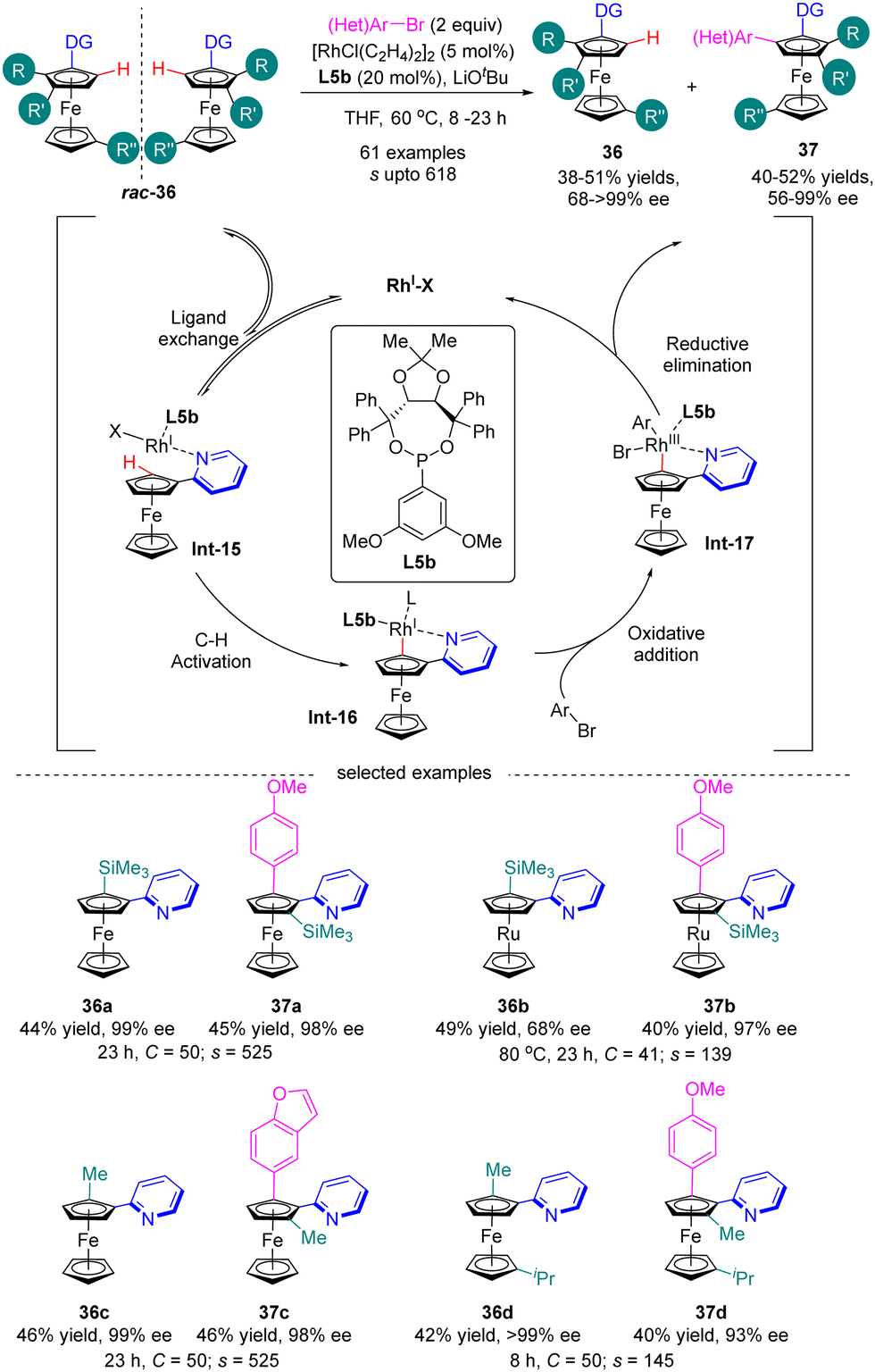
Most recently in 2023, You and co-workers further achieved the Rh(I)-catalyzed enantioselective C(sp2)–H arylation of metallocenes for the synthesis of both planar chiral 1,2-disubstituted and 1,3-disubstituted metallocenes via kinetic resolution (Scheme 11).74 In the presence of Rh(I) catalyst and 3,5-(MeO)2-C6H3-substituted TADDOL-derived monodentate phosphonite L5b as the chiral ligand, a broad range of aryl and heteroaryl bromides were readily incorporated into various directing group (e.g. 3- or 4-methyl, 4-phenyl and 4-methoxy pyridine) functionalized 1,2-disubstituted or 1,3-disubstituted ferrocenes and ferrocenes rac-36 containing diverse substituents, including alkyl groups, aryl groups, and even heteroatoms, thus giving rise to arylated products 37 with satisfactory selectivity factor values (s up to 618) and excellent enantiomeric control (up to 99% ee). For instance, ferrocene 36a and ruthenocene 36b with ortho-trimethylsilyl substituent acted as applicable substrates with outstanding selectivity (s = 525 for 37a and s = 139 for 37b). Additionally, the calculated conversion (C) matched well with the isolated yields. The underlying mechanism of this reaction has been studied using 2-pyridinylferrocenes and aryl bromides as representative reagents by both experimental and computational studies (Scheme 11).75 The reaction was proposed to be initiated by the coordination of rhodium with 2-pyridinylferrocenes to afford intermediate Int-15. Subsequently, C–H activation of Int-15 occurred to generate intermediate Int-16, which then proceeded to oxidative addiction with aryl bromide to provide Int-17. Finally, reductive elimination of Int-17 readily proceeded to arrive at the expected product and release the rhodium catalyst.
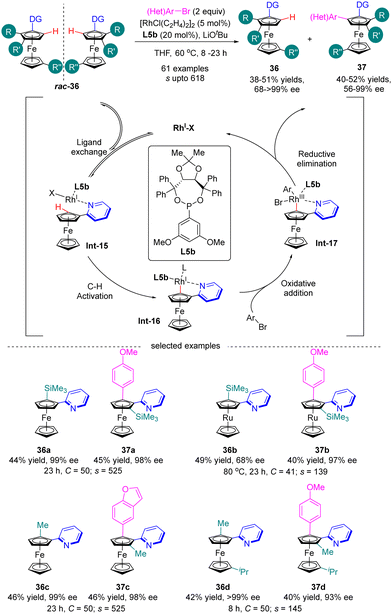 |
| | Scheme 11 Rh(I)-Catalyzed enantioselective C(sp2)–H arylation of metallocenes via kinetic resolution. | |
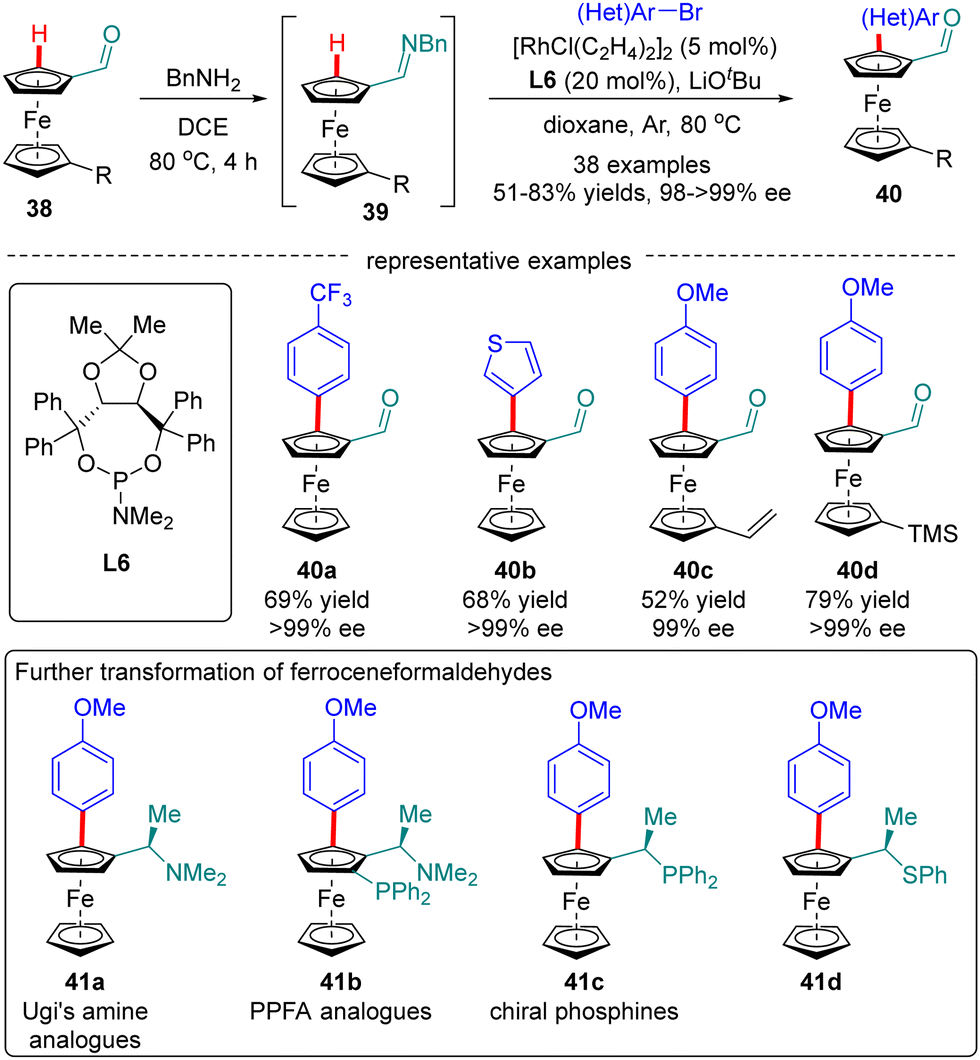
Ferroceneformaldehydes constitute a vital type of platform molecules, which can be smoothly converted into diverse functionalities to provide facile access to chiral ligands and catalysts. However, it had been a long-standing challenge to synthesize enantiopure planar chiral ferroceneformaldehydes via a straightforward and highly efficient approach. To surmount this obstacle, You and co-workers recently disclosed a Rh(I)-catalyzed enantioselective C(sp2)–H arylation of ferroceneformaldehydes 38 with the assistance of a chiral phosphoramidite ligand L6 (Scheme 12).76 It was demonstrated that the formation of imine intermediates 39 was critical for the success of this transformation. A broad set of readily available aryl halides such as aryl iodides, aryl bromides, and even aryl chlorides are found to be competent substrates for this reaction, providing access to chiral ferroceneformaldehydes 40 in uniformly excellent enantioselectivity (98–>99% ee). Among various functionalities derived from ferroceneformaldehydes by an elegant decoration of the aldehyde group, enantiopure Ugi's amine 41a and PPFA analogues 41b were particularly noteworthy, as the latter proved as a highly efficient ligand in Pd-catalyzed asymmetric allylic alkylation reactions.
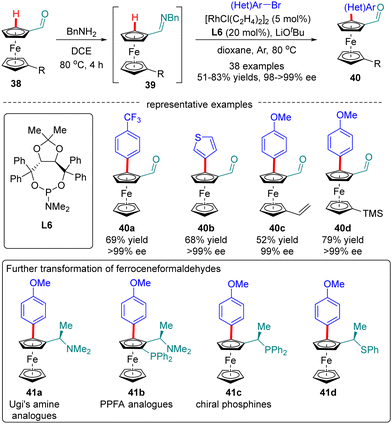 |
| | Scheme 12 Rh(I)-Catalyzed enantioselective C(sp2)–H arylation of ferroceneformaldehydes. | |
2.1.3 Asymmetric silylation of C(sp2)–H bonds.
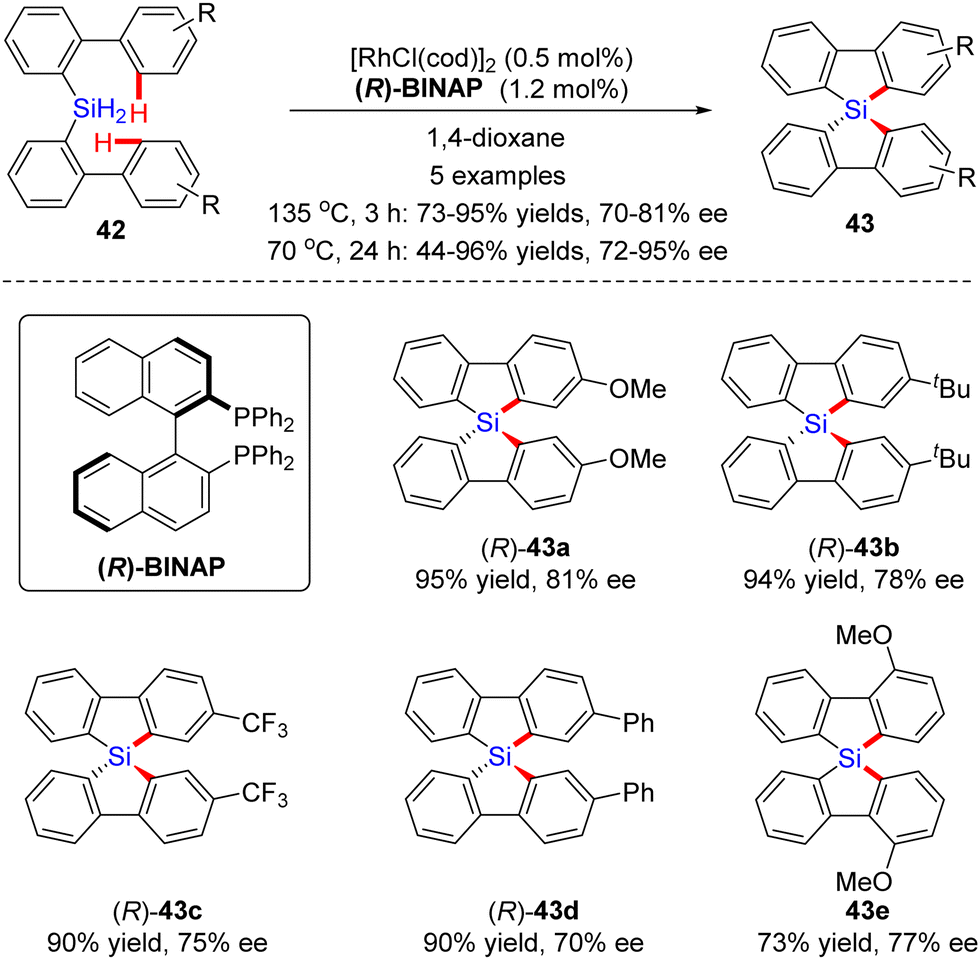
In 2013, Takai and collaborators presented a Rh(I)-catalyzed dual dehydrogenative cyclization of bis(biphenyl)silanes 42via C–H silylation (Scheme 13).77,78 By the use of [RhCl(cod)]2 as a catalyst and (R)-BINAP as a ligand, several chiral spirosilabifluorenes 43 were constructed in favorable yields (73–85%) and enantiomeric control (70–81% ee). Subsequently, the same group found that the enantioselectivities of the products could be further enhanced to 72–95% ee without sacrificing the yields by lowering the temperature from 135 °C to 70 °C and extending the reaction time to 24 h.79
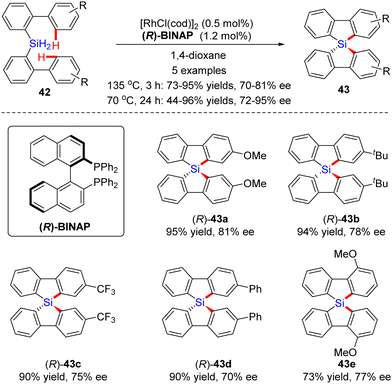 |
| | Scheme 13 Rh(I)-Catalyzed dual dehydrogenative cyclization of bis(biphenyl)silanes. | |
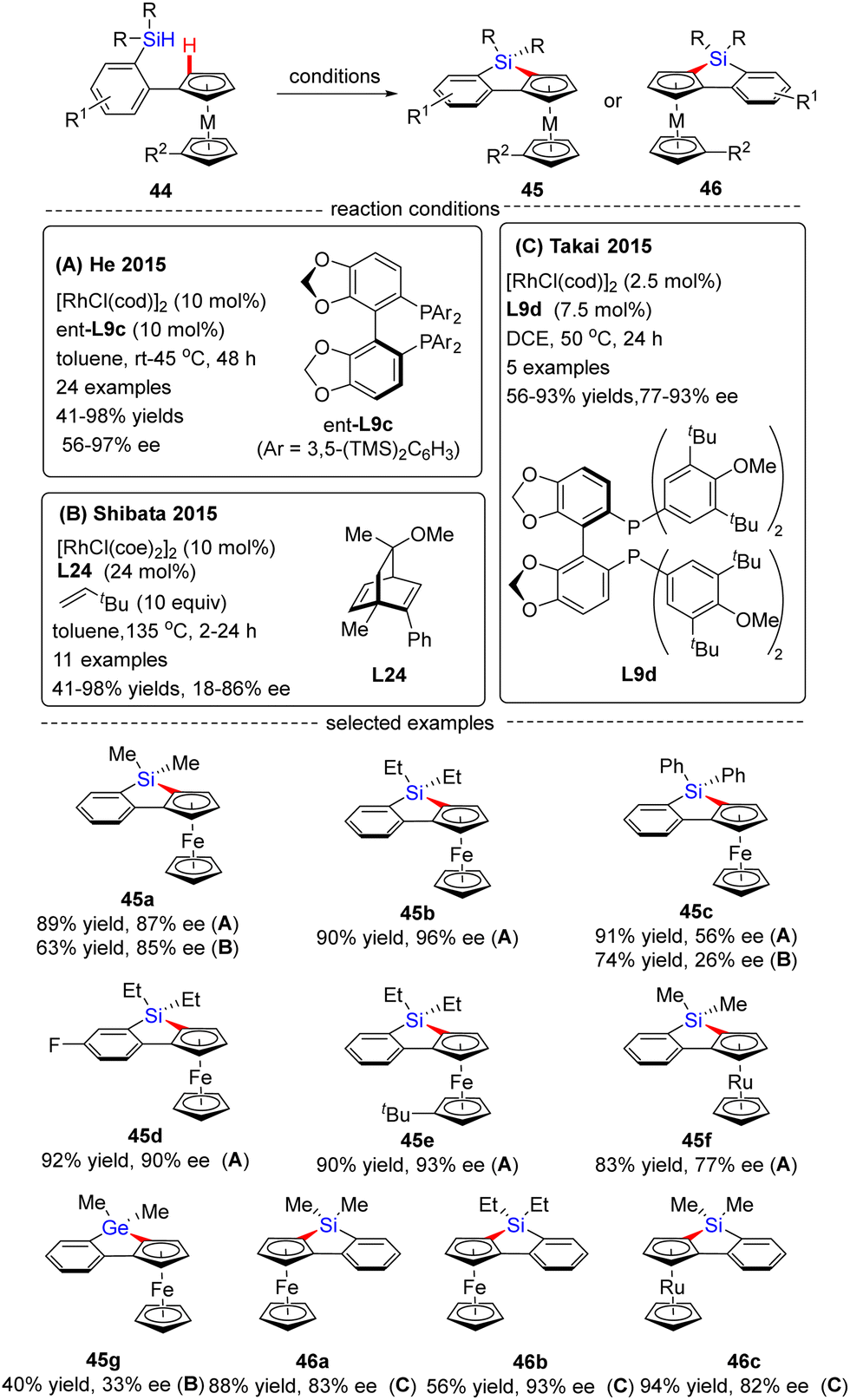
In 2015, He and co-workers opened the door to the untamed Rh(I)-catalyzed asymmetric C–H bond silylation under much milder reaction conditions (Scheme 14A).80 With the aid of chiral ligand (S)-TMS-Segphos (ent-L9c), a plethora of Fe and Ru metallocenes 44 proceeded intramolecular dehydrogenative cyclization at temperatures between room temperature and 45 °C, thus providing direct access to planar-chiral ferrocene and ruthenocene siloles 45 in good yields (up to 98%) with high to excellent enantioselectivities (up to 97% ee). In general, the enantioselectivity was largely independent of the substituents on the aryl or metallocene part. In contrast, the substituents on the silicon atom exerted a profound effect on the enantioselectivity, and substrates with two ethyl or propyl groups on the silicon atom gave rise to better results (45b) compared with that with two methyl groups (45a). Besides, only moderate enantiocontrol (56% ee) was achieved for diphenyl silane 45c. Meanwhile, the Takai group realized the same transformation by using (R)-DTBM-Segphos L9d as a chiral ligand, which provided several planar-chiral ferrocene and ruthenocene siloles 46 in comparable enantioselectivities (77–93% ee) but with opposite absolute configuration (Scheme 14C).81,82 Remarkably, apart from Segphos-derived ligand, a chiral diene ligand L24 was also competent to effect this transformation, but with much lower yields (39–75%) and enantioselectivities (18–86% ee), as described by Shibata and co-workers (Scheme 14B).83 Additionally, this catalytic system was also applied for the intramolecular dehydrogenative coupling of the C(sp2)–H bond with the Ge–H bond in (dimethylhydrogermyl)phenylferrocene 45g. Unfortunately, the corresponding benzogermoloferrocene 45g was afforded with only 40% yield and 33% ee. It should be noted that an excess of 3,3-dimethylbut-1-ene as addictive was crucial for the maintenance of enantiocontrol.
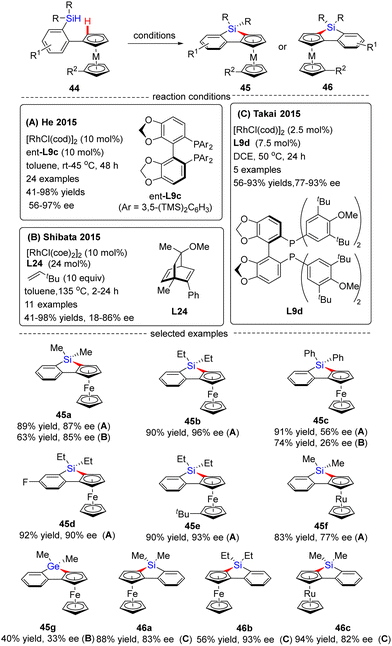 |
| | Scheme 14 Rh(I)-Catalyzed enantioselective intramolecular C–H silylation for the synthesis of planar-chiral metallocene siloles and germolane. | |
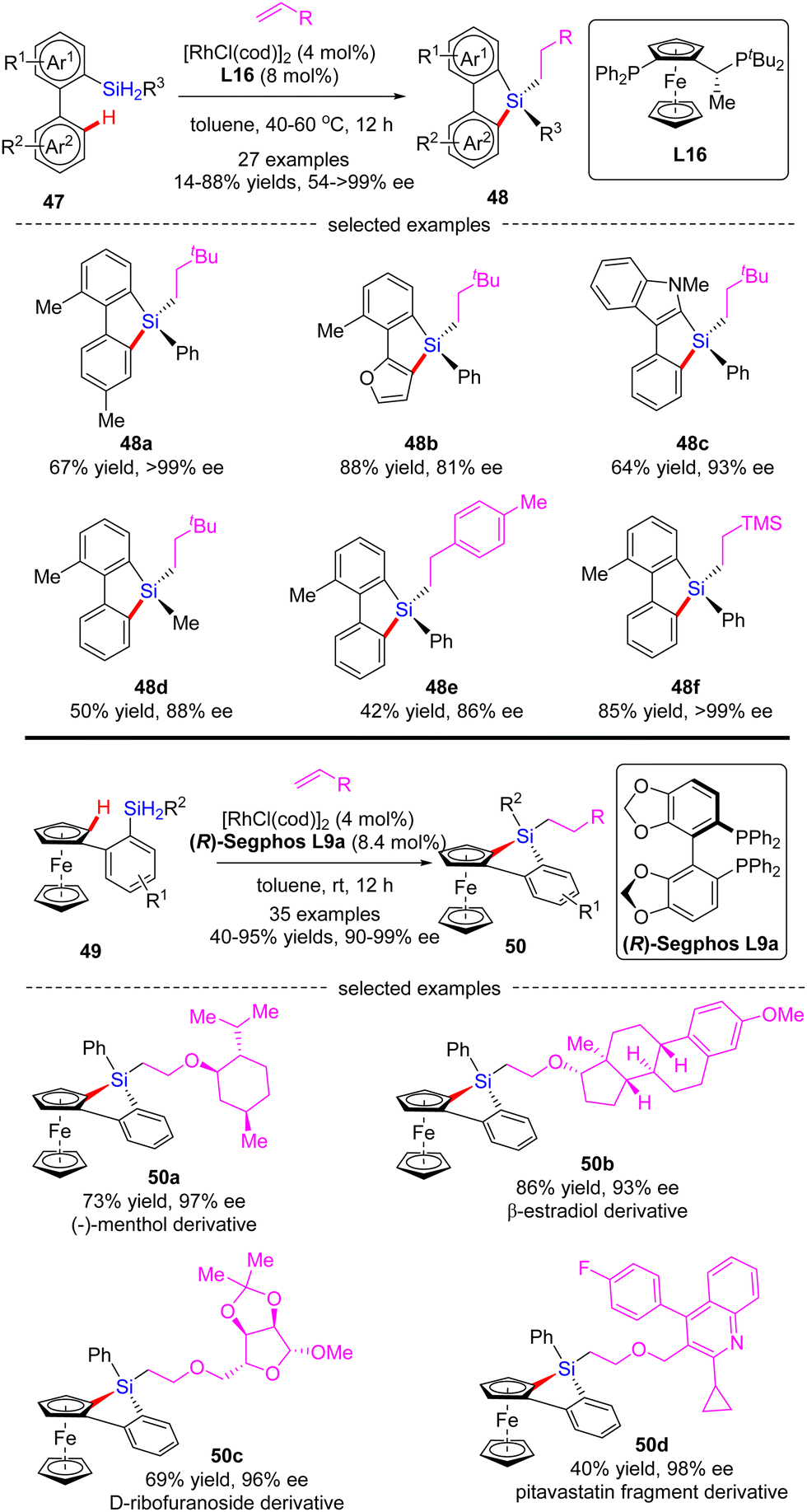
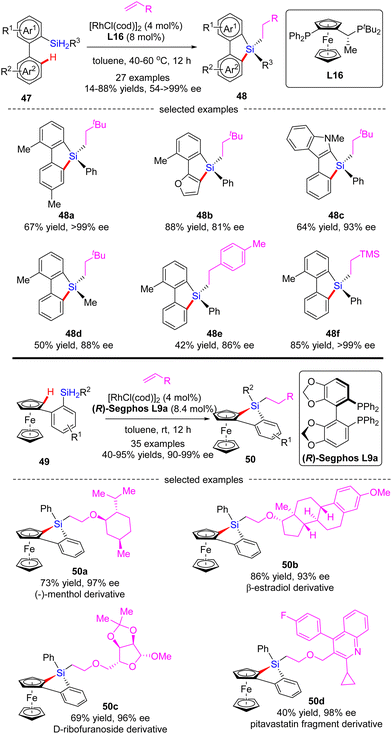
In 2020, He and co-workers developed a Rh(I)-catalyzed cascade enantioselective C(sp2)–H silylation/alkene hydrosilylation of dihydrosilanes (Scheme 15).84 By the use of Josiphos-derived chiral ligand L16, a diverse set of bi-(hetero)aryl dihydrosilanes 47 with different substituents at different positions were smoothly converted to silicon-stereogenic tetra-substituted silanes 48 in 55–88% yields and good to excellent enantiocontrol (81–99% ee). For the scope of alkene partners, alkyl, aryl and heteroatom-substituted alkenes all proved as competent substrates. Alternatively, with (R)-Segphos as a chiral ligand, various ferrocene dihydrosilanes 49 were compatible with this transformation, which enabled streamlined construction of tetrasubstituted silicon-stereogenic and planar chiral benzosiloloferrocenes 50 as single diastereomers (dr > 20:1) with excellent enatioselectivities (90–99% ee). It is worth mentioning that several core scaffolds from pharmaceuticals, bioactive molecules, and materials, such as (−)-menthol, β-estradiol, D-ribofuranoside, and pitavastatin fragment, were well accommodated in this reaction to furnish the corresponding benzosiloloferrocenes 50a–d in 40–86% yields and 93–98% ee.
 |
| | Scheme 15 Rh(I)-Catalyzed tandem enantioselective C–H silylation/alkene hydrosilylation of dihydrosilanes. | |
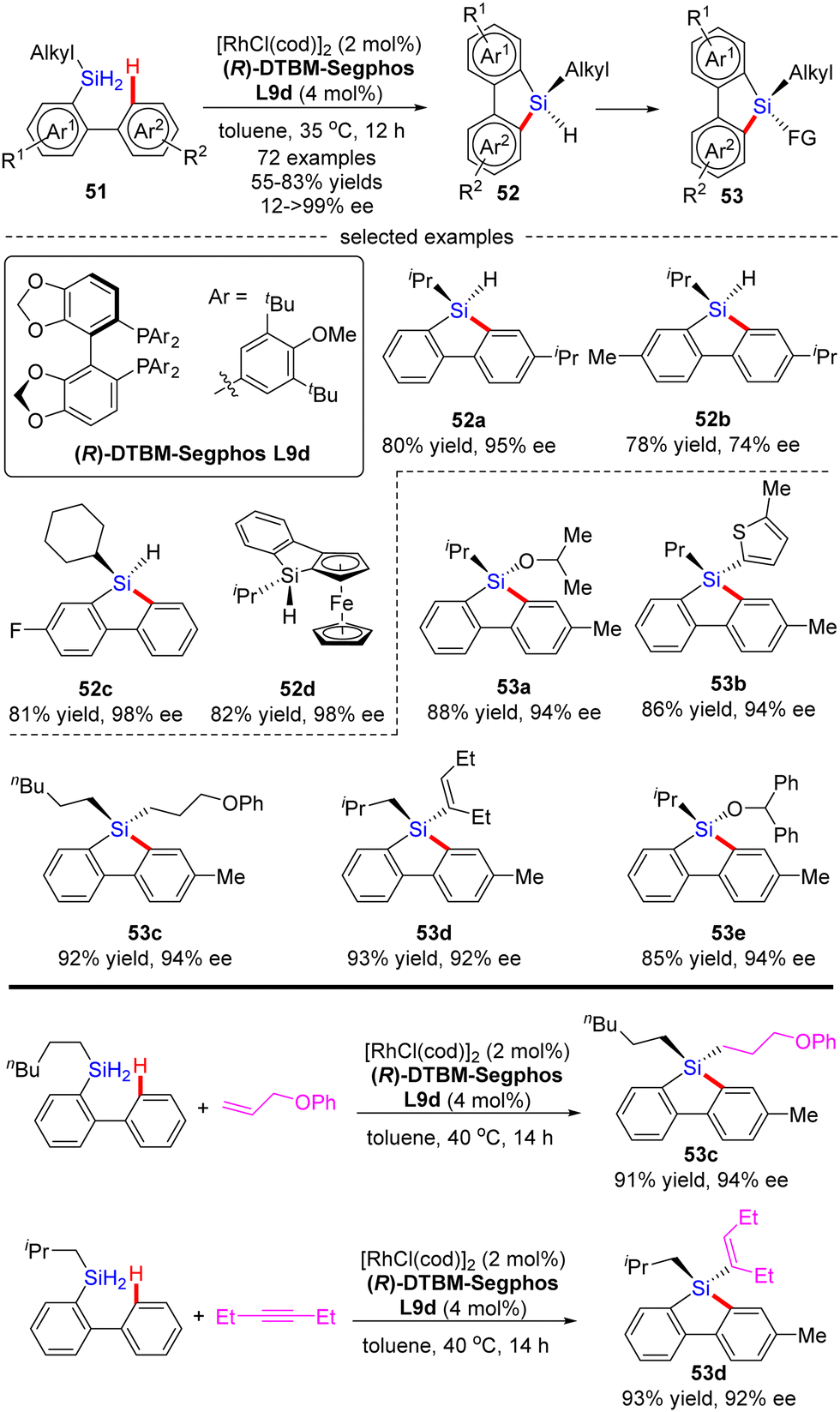
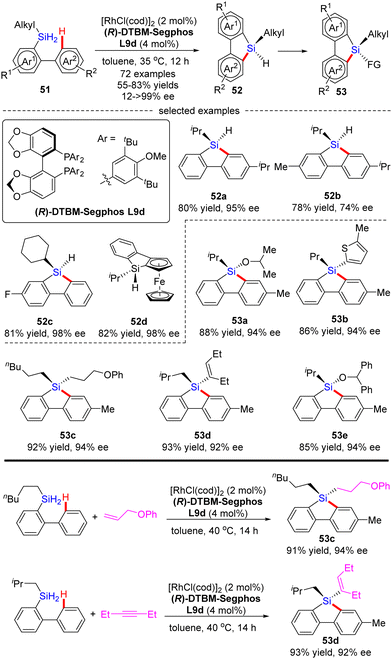
Most recently, He and co-workers described an elegant synthesis of chiral monohydrosilanes by Rh(I)-catalyzed intramolecular C–H silylation of dihydrosilanes (Scheme 16).85 Under mild conditions, a diverse set of chiral monohydrosilanes 52 were obtained in 55–83% yields with favorable enantioselectivities (up to >99% ee). Substrates with substituents on both aryl rings were also compatible with this transformation, resulting in decreased yields and enantioselectivities (52b). Furthermore, a wide array of silicon-stereogenic and planar chiral ferrocene monohydrosilanes were successfully accessed by this approach with consistently excellent enantioselectivities (93–99% ee, i.e.52d). Unfortunately, only aryl alkyl dihydrosilanes proved as competent substrates, whereas the use of diaryl dihydrosilanes was not successful in delivering the corresponding products, due to their decomposition issue in the presence of highly reactive rhodium catalysts. It is noteworthy that the resulting chiral monohydrosilanes 52 served as critical building blocks to effect the facile synthesis of different stereogenic tetraorganosilanes 53. While alcoholysis with isopropanol and C–H functionalization with 2-methylthiophene furnished products 53a and 53b, respectively, hydrosilylation with π bond compounds such as alkenes, alkynes, and ketones, delivered chiral tetraorganosilanes 53c–evia the construction of Si–C(sp2), Si–C(sp3), and Si–O bonds. In all cases, high yields were achieved with intact enantiocontrol. Remarkably, tandem C(sp2)–H silylation/hydrosilylation of dihydrosilanes with alkenes and alkynes also proved successful, thus providing 53c and 53d in excellent yields and enantioselectivities.
 |
| | Scheme 16 Rh(I)-Catalyzed intramolecular C–H silylation of aryl alkyl dihydrosilanes. | |
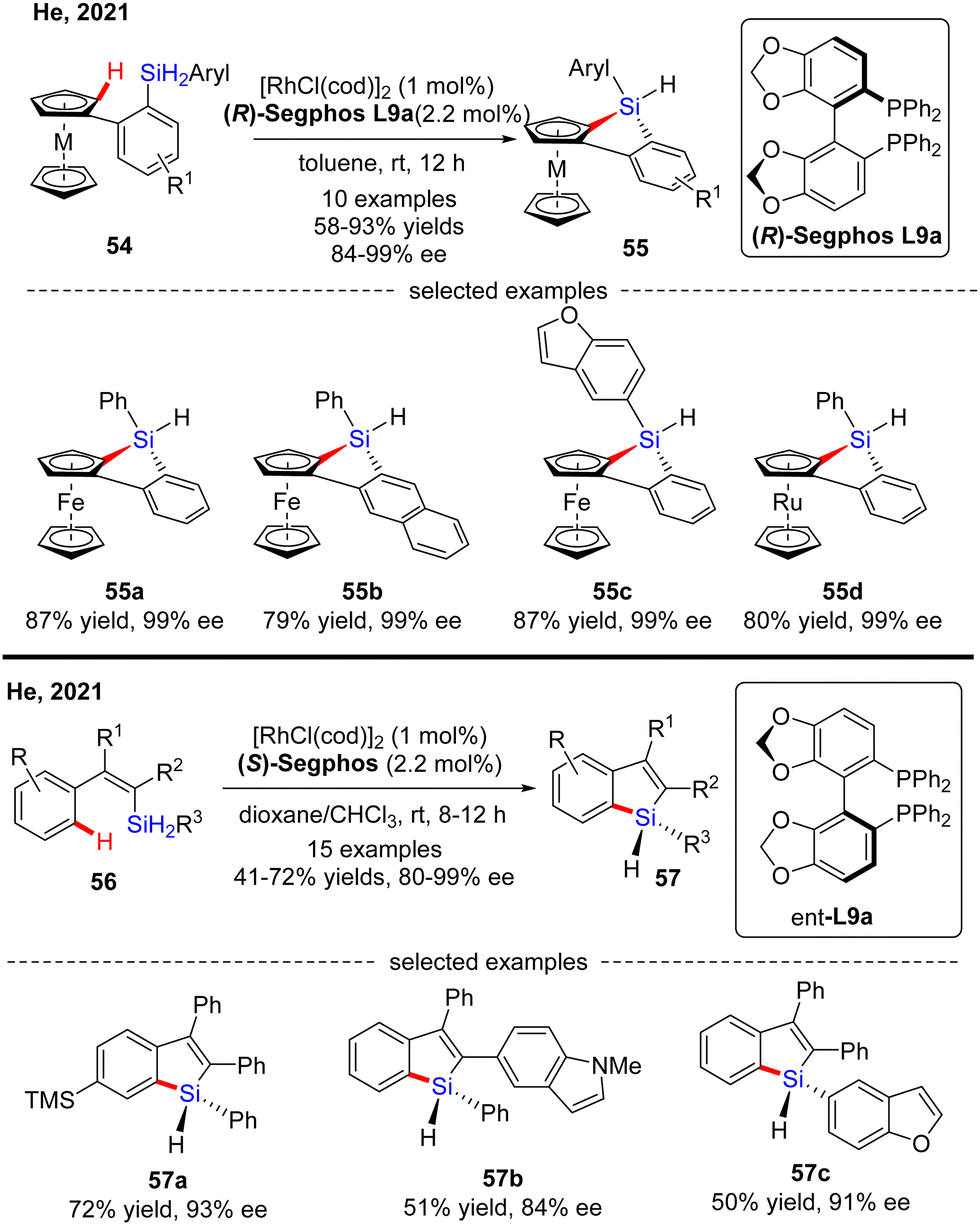
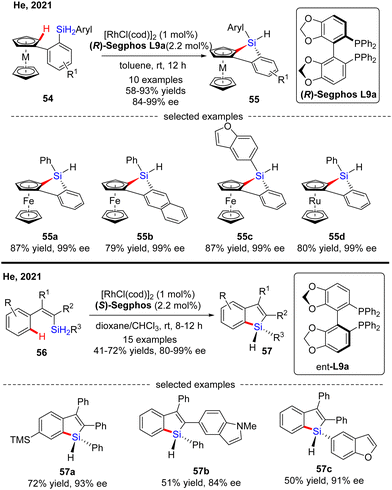
Subsequently, He's group further expanded the substrate scope of the Rh(I)-catalyzed intramolecular C–H silylation reaction to diaryl dihydrosilanes (Scheme 17).86 The key to the success of this transformation relied on decreased rhodium catalyst loading (1 mol%) to address the decomposition issue of the monohydrosilane products. To the authors’ delight, a variety of silicon-stereogenic and planar chiral trisubstituted 1H-benzosiloloferrocenes 55 were achieved as single diastereomers with excellent enantioselectivities (84–99% ee). Furthermore, a ruthenocene-containing substrate was also compatible with this transformation, thus affording 55d in 80% yield and 99% ee. Meanwhile, with this catalytic system, He and co-workers effected asymmetric intramolecular dehydrogenative C–H silylation of alkenyl dihydrosilanes (Scheme 17).86 With 1 mol% of rhodium catalyst, various alkenyl dihydrosilanes containing a diverse set of substituents at different positions were readily transformed into trisubstituted silicon-stereogenic 1H-benzosiloles 57 in moderate to decent yields (41–72%) and good to excellent enantioselectivities (80–99% ee).
 |
| | Scheme 17 Rh(I)-catalyzed intramolecular C–H silylation of diaryl and alkenyl dihydrosilanes. | |
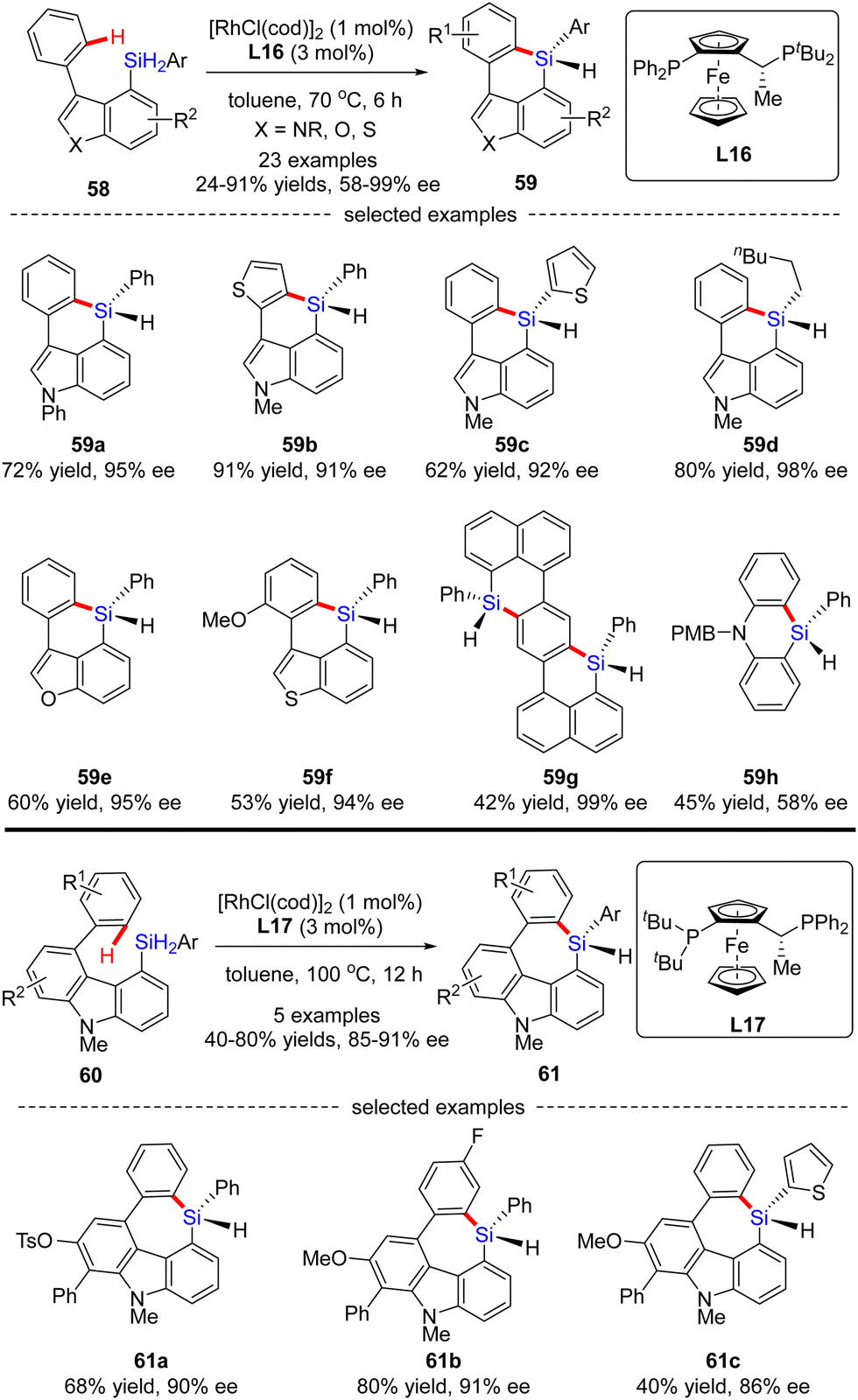
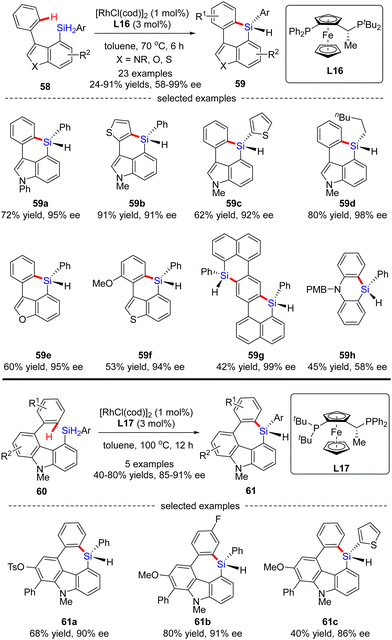
Aside from a number of reports on the Rh(I)-catalyzed enantioselective construction of five-membered silicon-stereogenic silanes, most recently, He and co-workers disclosed a novel synthesis of six and seven-membered counterparts via asymmetric Rh(I) catalysis (Scheme 18).87 Under the catalysis of [RhCl(cod)]2 and Josiphos ligand L16, various silicon-bridged biaryls 58 consisting of benzene and indole moieties smoothly underwent dehydrogenative C–H silylation to provide a diverse set of six-membered silicon-stereogenic triorgano-substituted silanes 59 in 24–91% yields with excellent enantiocontrol (87–98% ee). It was found that high temperature was crucial to achieve high reactivity in this transformation. In addition to the indole scaffold, benzofurane, benzothiophene, and naphthalene counterparts were also well incorporated to afford silicon-stereogenic monohydrosilanes 59e–g in 94–99% ee. Furthermore, chiral silicon-bridged diphenylamine 59h could also be accessed by this approach, despite a much lower enantioselectivity (58% ee). It is noteworthy that several seven-membered carbazole-based silicon-stereogenic triorgano-substituted silanes 61 were readily obtained in moderate to good yields (40–80%) and decent enantioselectivities (85–91% ee) by simply replacing the chiral ligand L16 with L17.
 |
| | Scheme 18 Rh(I)-Catalyzed synthesis of six- and seven-membered silicon-stereogenic triorgano-substituted silanes. | |
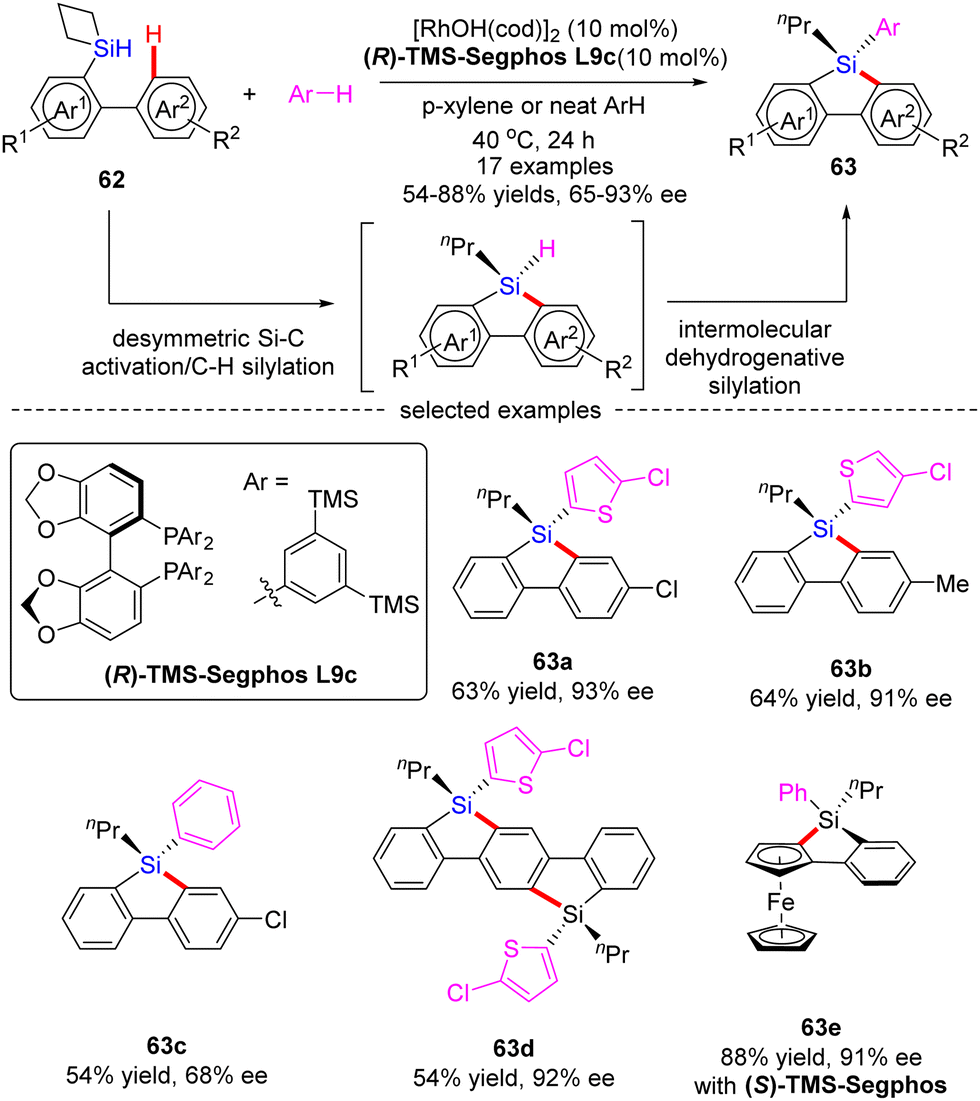
Subsequently, He and co-workers disclosed a cascade desymmetric Si–C activation/C–H silylation and intermolecular dehydrogenative silylation reaction of silacyclobutanes (SCB) 62 with arenes via asymmetric Rh(I) catalysis (Scheme 19).88 As a result, a variety of quaternarily silicon-stereogenic dibenzosiloles 63 were attained in moderate to high yields (54–88%) and good enantioselectivities (65–93% ee). Aside from thiophene derivatives, benzene also proved as capable substrate to furnish the corresponding silole 63c in comparable yield, but diminished enantiocontrol (68% ee). Notably, by taking advantage of this catalytic system, bis-silole 63d and ferrocene silole 63e were achieved in 92% ee and 91% ee, respectively.
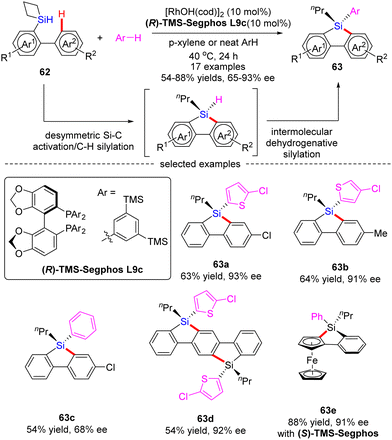 |
| | Scheme 19 Rh(I)-Catalyzed asymmetric desymmetrization/C–H silylation/dehydrogenative silylation of silacyclobutanes. | |
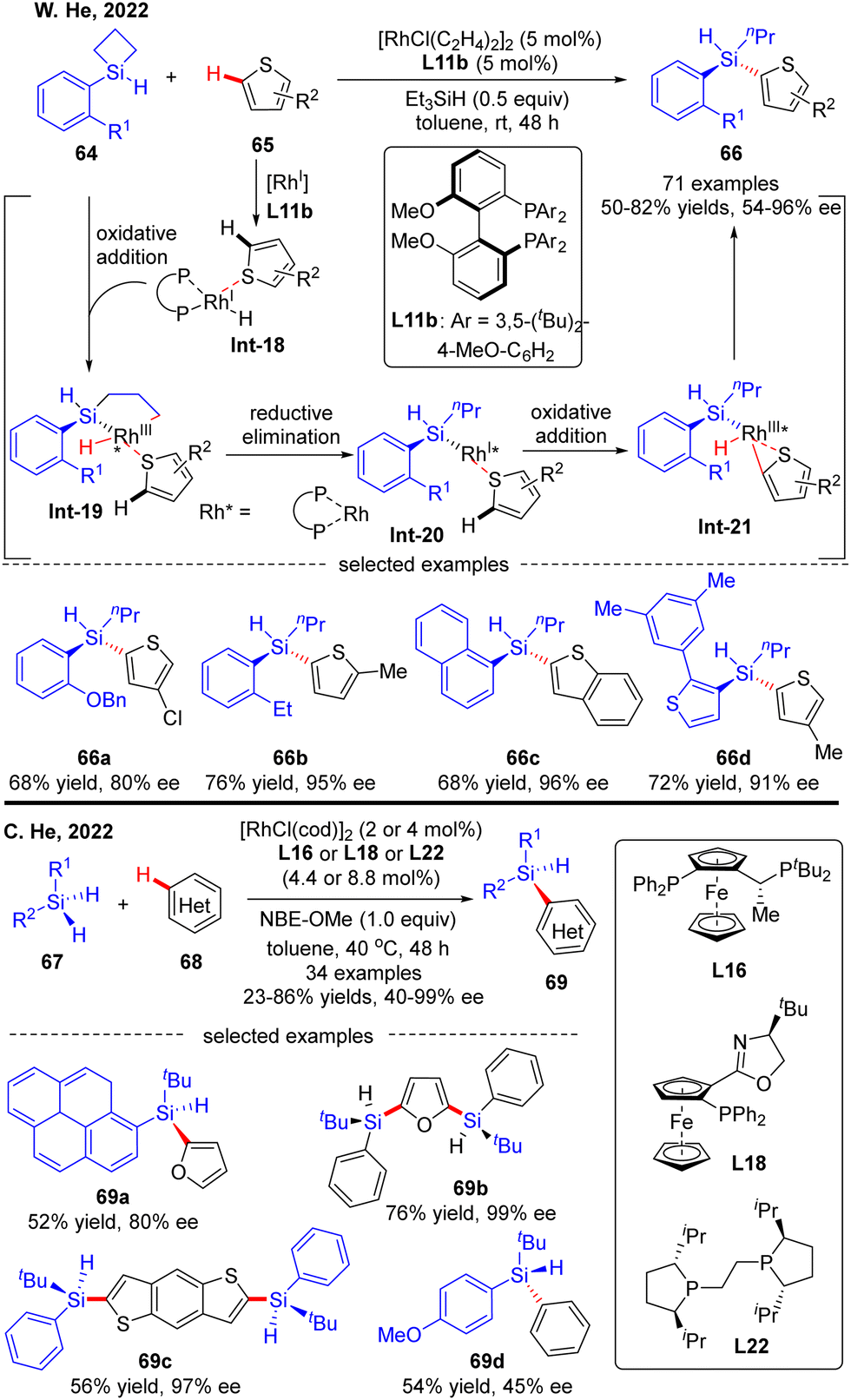
Most recently, the Rh(I)-Catalyzed intermolecular asymmetric C–H silylation was independently successfully realized by W. He and C. He (Scheme 20).89 As described by W. He and co-workers, with biphenylphosphine L11b as a chiral ligand, various silacyclobutanes 64 were smoothly coupled with thiophene derivatives 65, forging chiral acyclic monohydrosilanes 66 in moderate to excellent yields (50–82%) and stereo-control (54–96% ee). The reaction was proposed to commence with the generation of intermediate Int-18via thiophene coordination to the pre-formed [Rh]–H catalyst. Subsequent oxidative addition of silacyclobutane 64 onto Rh(I) in Int-18 proceeded to afford a five-membered rhodacycle Int-19. Reductive elimination of rhodacycle Int-19 occurred to form Int-20, which then underwent C–H activation to furnish oxidative addition intermediate Int-21. Finally, the desired acyclic stereogenic monohydrosilane 66 was obtained via a second reductive elimination of Int-21. Almost at the same time, C. He and co-workers also communicated a Rh(I)-catalyzed intermolecular asymmetric C–H silylation of heteroarenes via a direct dehydrogenative C–H/Si–H cross-coupling (Scheme 20).90 As a result, a diversity of acyclic heteroarylated silicon-stereogenic monohydrosilanes 69 including bis-Si-stereogenic silanes (69b, 69c) were generated in decent yields and enantioselectivities. Notably, apart from furan and thiophene derivatives, simple arenes were also compatible with this reaction, despite moderate stereo-control (69d). It was worth mentioning that the use of norbornene derivative NBE-OMe as a hydrogen acceptor was critical to maintaining the reactivity.
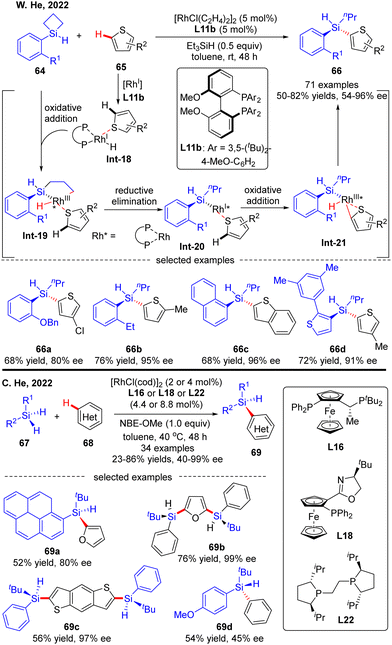 |
| | Scheme 20 Rh(I)-Catalyzed intermolecular asymmetric C–H silylation. | |
2.2 Functionalization of C(sp3)–H bonds
2.2.1 Functionalization of activated C(sp3)–H bonds.
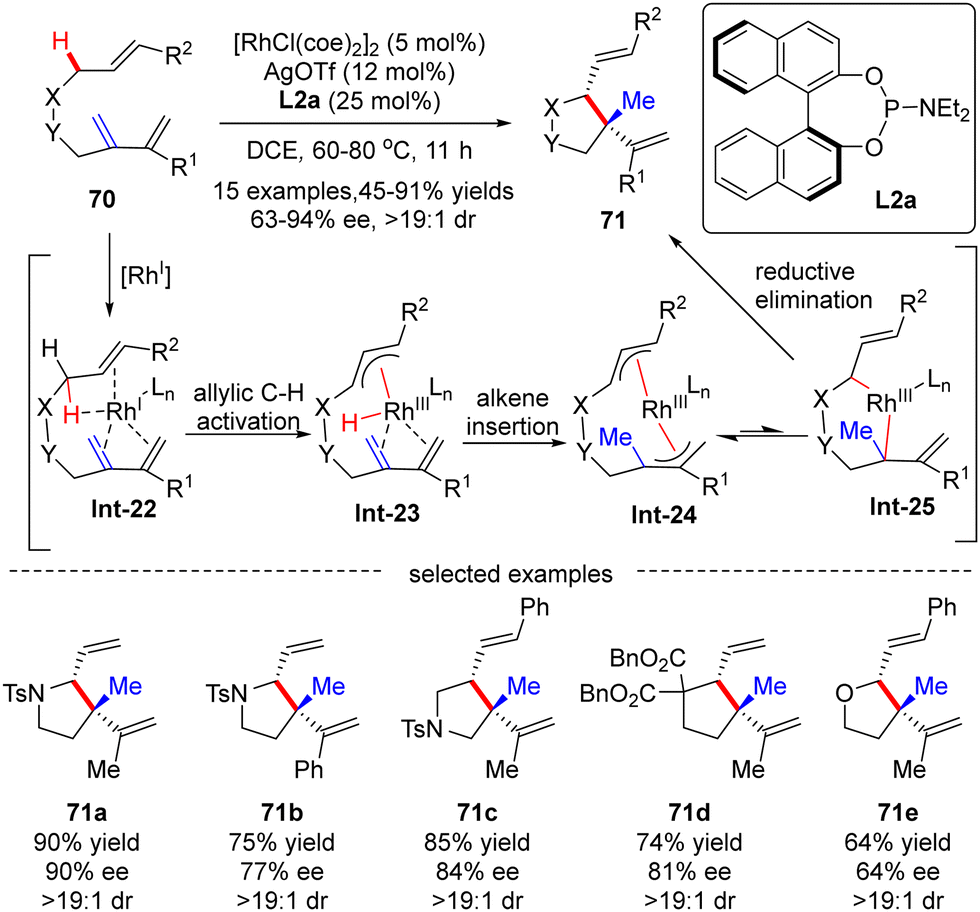
In 2012, Yu's group reported a Rh(I)-catalyzed asymmetric functionalization of activated allylic C(sp3)–H bonds for intramolecular addition to conjugated 1,3-dienes.91 Various nitro-tethered ene-2-dienes 70 containing either alkyl or aryl substituents were readily converted to pyrrolidine derivatives 71 in 45–91% yields with moderate to excellent enantioselectivities (63–94% ee), using [RhCl(coe)2]2 as a catalyst, AgOTf as a co-catalyst and (S)-binaphthol (BINOL)-derived monodentate phosphoramidite L2a as a chiral ligand. Moreover, carbon- and oxygen-tethered ene-2-dienes were also suited to this transformation, providing cyclopentane and tetrahydrofuran derivatives 70d and 70e in 81% and 64% ee, respectively. Notably, two adjacent stereo-centers with one quaternary carbon center were constructed in excellent diastereoselectivities (dr > 19:1). The underlying mechanism was next thoroughly investigated based on the DFT (density functional theory) calculation (Scheme 21).92 As depicted in Scheme 21, the reaction was initiated by the coordination of the rhodium catalyst with the substrate, leading to the formation of Rh-complex Int-22. Follow-up allylic C–H activation provided rhodium-hydride species Int-23, which then underwent alkene insertion to furnish bis-allylic Rh-complex Int-24. Finally, di-π-allyl-assisted reductive elimination of Int-25 occurred to release the desired product.
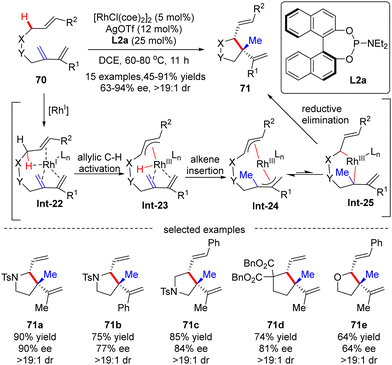 |
| | Scheme 21 Rh(I)-Catalyzed allylic C–H activation and addition to conjugated dienes. | |
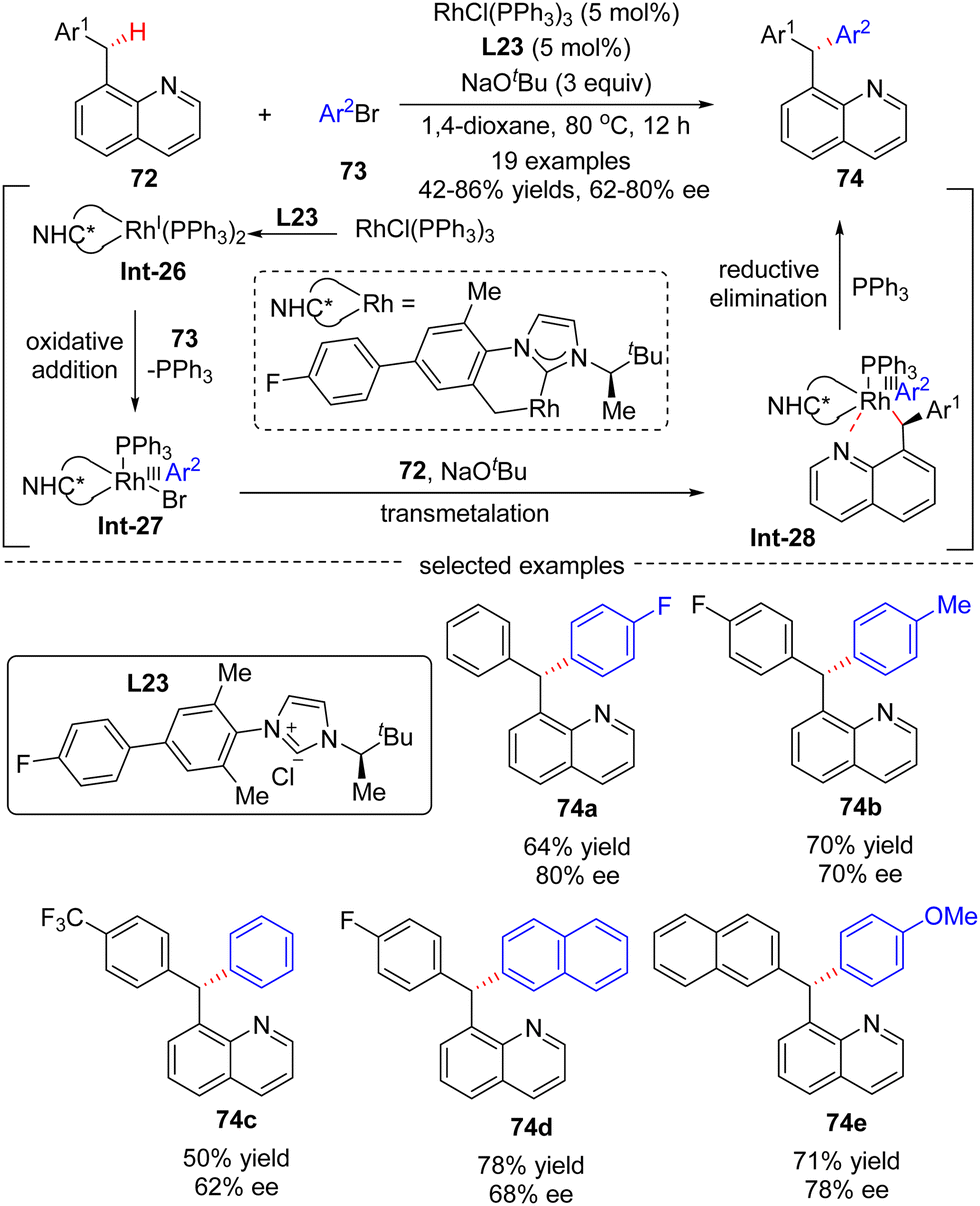
In 2016, Glorius's group reported a Rh(I)-catalyzed asymmetric approach for the site- and enantio-selective intermolecular arylation of benzylic C(sp3)–H bonds (Scheme 22).93 Diverse triarylmethanes 74 were achieved in 42–86% yields and with moderate to good enantioselectivities (62–80% ee) by reacting 8-benzyl quinolones 72 with aryl bromides 73 under the catalysis of RhCl(PPh3)2 and NHC (N-heterocyclic carbene) pre-ligand L23. The underlying plausible reaction mechanism was proposed as depicted in Scheme 22. First, reactive intermediate Rh(I)-complex Int-26 was generated by the coordination of in situ generated free NHC to rhodium, followed by intramolecular activation of a benzylic C–H bond on the carbene ligand. Then, Rh(III) species Int-27 was formed by oxidative addition of aryl bromides into Int-26, with concomitant elimination of associated ligand PPh3. Afterwards, Rh-complex Int-28 was produced by a trans-metalation reaction of Int-27 with deprotonated substrate 72, which was generated by an excess amount of NaOtBu. Finally, the desired product 74 was achieved via reductive elimination of Int-28.
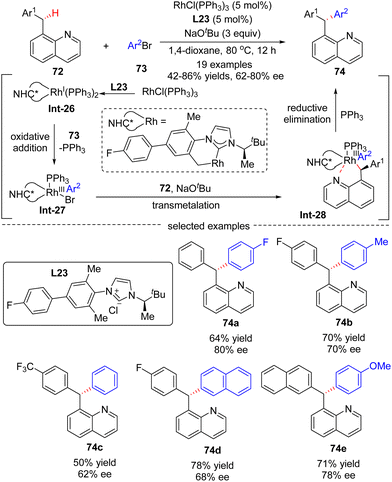 |
| | Scheme 22 Rh(I)-Catalyzed intermolecular C–H arylation of 8-benzyl quinolones and aryl bromides. | |
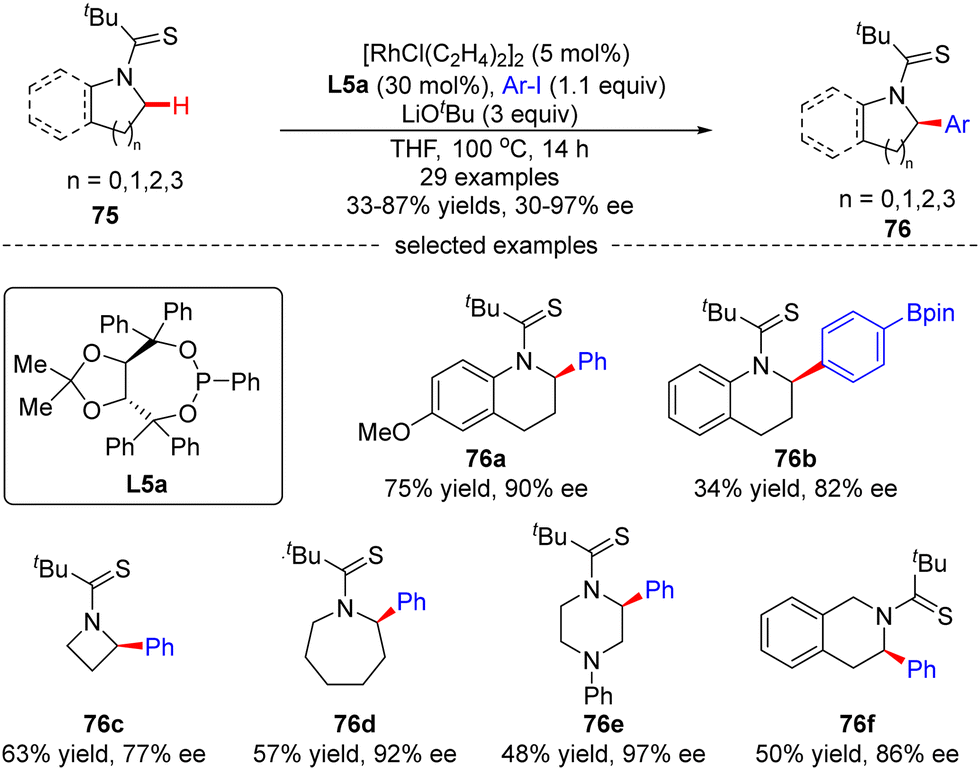
Glorius's group later explored another Rh(I)-catalyzed approach for the enantioselective intermolecular C(sp3)–H arylation of tetrahydroquinolines and other saturated N-heterocycles with aryl iodides, without observation of the arylation of the adjacent C(sp2)–H bonds on tetrahydroquinolines (Scheme 23).94 In this reaction, the combination of [RhCl(C2H4)2]2 and chiral phosphonite ligand L5a acted as a powerful catalytic system producing a diversity of α-arylated N-heterocycles with high regio- and enantio-selectivity, while using tert-butyl thioamide as a directing group. Both electron-donating and electron-withdrawing substituents on the phenyl ring were well tolerated when the tetrahydroquinoline scope was concerned. Substituents such as cyano, thioanisoles, trifluoromethyl, and arylboronic pinacol esters were also compatible in the case of aryl iodides. Remarkably, the scope of saturated aza-heterocycles could be extended to piperidine, pyrrolidine, piperazine, azepane and azetidine, and the corresponding α-N-arylated products 76c–f were obtained in good regio- and enantio-selectivities (up to 97% ee).
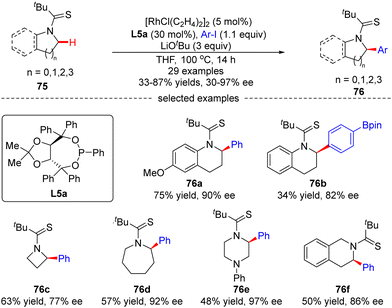 |
| | Scheme 23 Rh(I)-Catalyzed intermolecular C–H arylation of tetrahydroquinolines and saturated aza-heterocycles. | |
2.2.2 Functionalization of unactivated C(sp3)–H bonds.
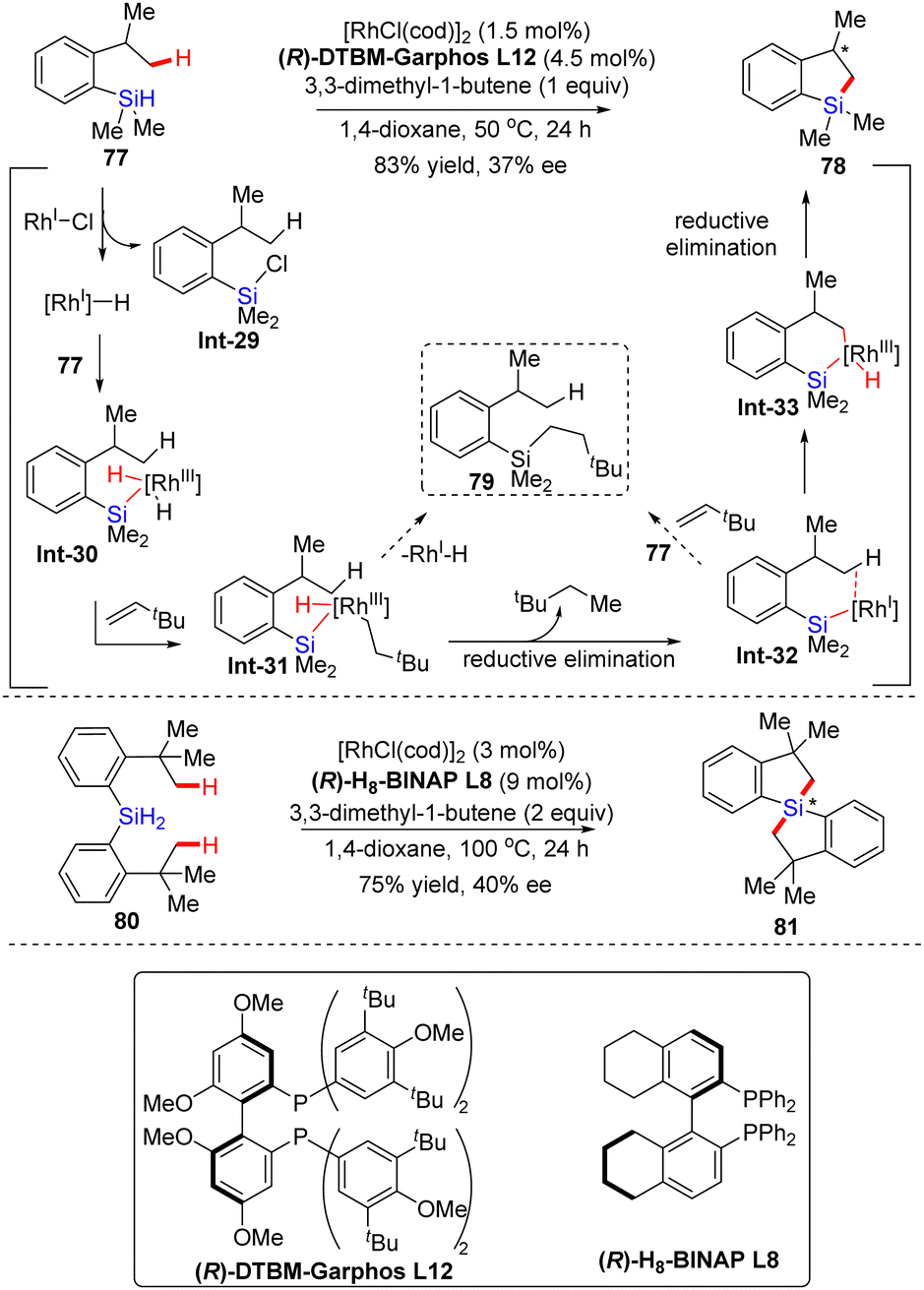
In 2015, Takai's group shared their pioneering work on the enantioselective rhodium-catalyzed dehydrogenative silylation of unactivated C(sp3)–H bonds (Scheme 24).95 In the presence of [RhCl(cod)]2, C2-symmetric diphosphine ligand (R)-DTBM-Garphos L12 and hydrogen acceptor 3,3-dimethyl-1-butene, 2-isopropylphenylsilane 77 was converted to 2,3-dihydrobenzo[b]silole 78 in 83% yield without any formation of hydrosilylated product 79, in spite of the low enantiocontrol (37% ee) (Scheme 24). While ligand (R)-DTBM-Garphos was replaced with (R)-H8-BINAP L8, 1,1′-spirosilabiindane 81 with a new tetrasubstituted silicon-stereogenic center was achieved in comparable yield (75%) and enantioselectivity (40% ee) via dual asymmetric alkylation of dihydrosilane 80 at elevated temperature with increased catalyst and hydrogen acceptor loading. The mechanism of this unique reaction has also been proposed as given in Scheme 24. First, hydrosilane 77 was oxidatively added to the Rh(I)–Cl pre-catalyst, furnishing a Rh(I)–H species involving the reductive elimination of chlorosilane Int-29. The Rh(I)–H species was then added to the Si–H bond of 77, and the resulting intermediate Int-30 further reacted with 3,3-dimethyl-1-butene to produce intermediate Int-31. Instead of the hydrosilylated product 79, Rh-silyl species Int-32 was preferably formed by reductive elimination of H and 3,3-dimethylbutane groups on the rhodium center of Int-31, which might be attributed to the tendency of 3,3-dimethylbutyl and sterically bulky silyl groups to keep distance from each other. The hydrosilylated product 79 might be generated by the reaction of Int-32 with 3,3-dimethyl-1-butene via silylrhodation followed by the Sigma-bond metathesis with 77. However, the electron-rich Rh center in Int-32 favored intramolecular oxidative addition over the silylrhodation to provide Int-33, which then underwent reductive elimination to generate the desired product 78.
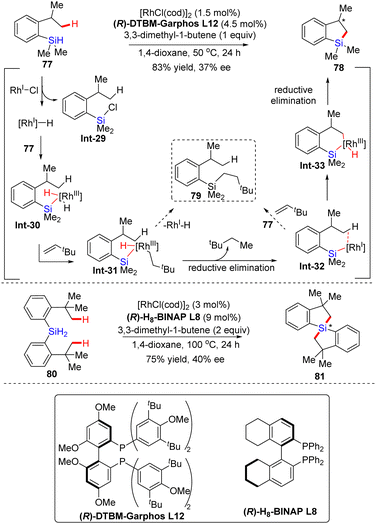 |
| | Scheme 24 Rh(I)-Catalyzed intramolecular C–H silylation of 2-isopropylphenylsilane and dihydrosilane. | |
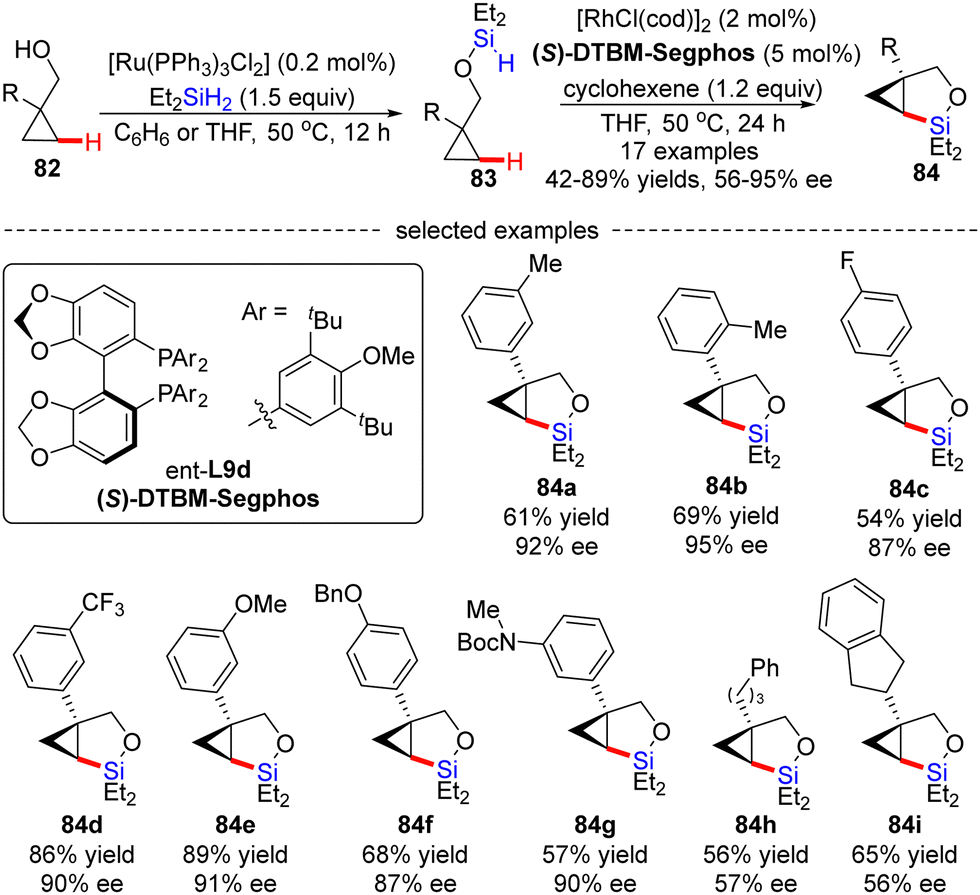
Soon after, Hartwig's group described an unprecedented enantioselective intramolecular silylation of cyclopropyl C–H bonds in the presence of a Rh catalyst.96 As displayed in Scheme 25, silyl ether 83 was in situ generated by Ru-catalyzed dehydrogenative coupling of cyclopropylmethanols 82 with diethylsilane and was used in the follow-up reaction without further purification. In the presence of [RhCl(cod)]2/(S)-DTBM-Segphos and a hydrogen acceptor cyclohexene, the intramolecular silylation of silyl ethers 83 readily occurred to generate various bicyclic silylcyclopropanes 84 in decent to high yields (42–89%) with moderate to good enantiomeric excesses (56–95% ee). Generally, aryl-substituted cyclopropylmethanols provided higher enantioselectivities compared with alkyl-substituted substrates (84a–gvs.84h–i), which was contributed by an additional π–π interaction between the aryl moieties of the substrate and the Rh(I) catalyst. Mechanistic studies suggested that the C–H cleavage might be the turnover-limiting step and determined the configuration of the product.
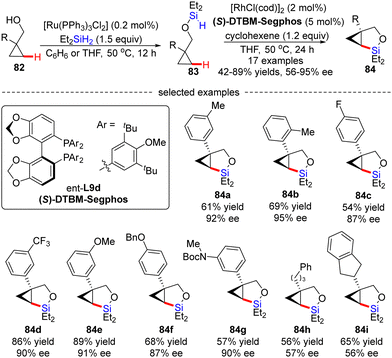 |
| | Scheme 25 Rh(I)-Catalyzed intramolecular C–H silylation of hydrosilyl ethers. | |
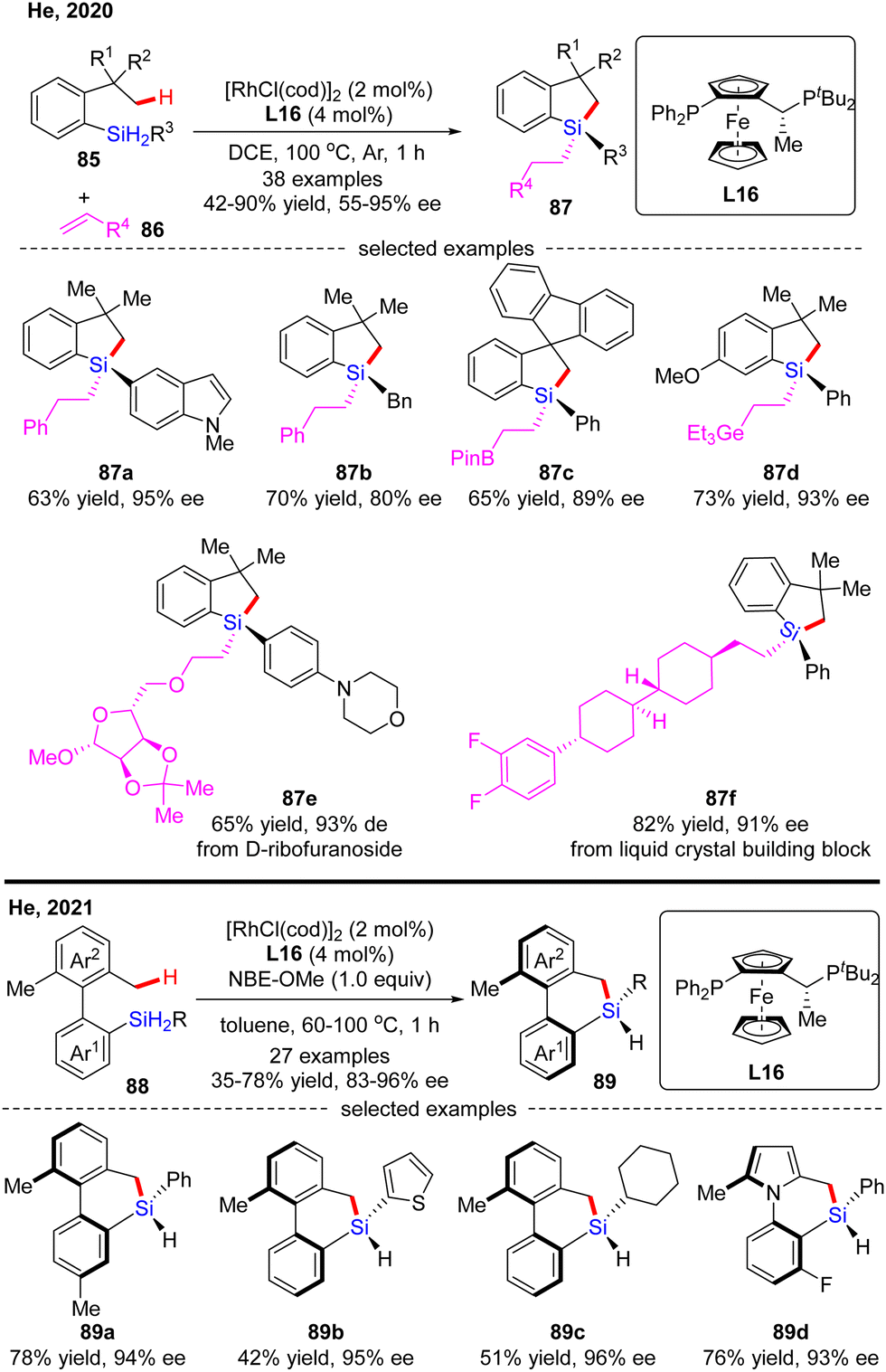
In 2020, He and co-workers developed a Rh(I)-catalyzed enantioselective silylation of unactivated C(sp3)–H bonds, thus providing an elegant access to silicon-stereogenic dihydrobenzosiloles (Scheme 26).97 This reaction comprises a two-step sequence, namely a highly asymmetric silylation of C(sp3)–H bonds in dihydrosilanes and a follow-up stereospecific alkene hydrosilylation. With (R,Sp)-Josiphos L16 as a chiral ligand, a diverse set of dihydrosilanes 85 and alkenes 86 were well compatible with this reaction to afford various silicon-stereogenic tetrasubstituted silanes 87 in good to excellent yields (42–90%) and enantioselectivities (55–95% ee). Shortly after, He and co-workers further disclosed an elegant approach to axially chiral 6-member-bridged biaryl derived dihydrodibenzosilines with a silicon-stereogenic center via Rh(I)/L16 catalysis.98 A diverse set of silicon-centrally and axially dihydrodibenzosilines 89 were conveniently generated in 35–78% yields and 83–96% ee. It was found that the presence of a hydrogen acceptor such as a norbornene derivative (NBE-OMe) was crucial to enhance the reactivity.
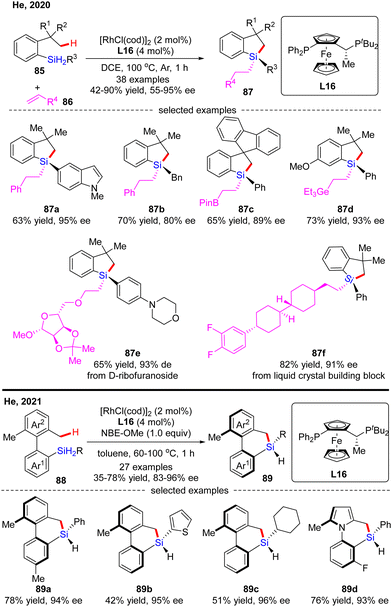 |
| | Scheme 26 Rh(I)-Catalyzed asymmetric silylation of unactivated C(sp3)–H bonds. | |
3. Outer-sphere C–H functionalization
The outer-sphere mechanism does not involve the direct interaction of the metal with the C–H bond, but instead commences with the formation of a rhodium-carbenoid, -nitrenoid, or -oxo species, followed by the reaction of this species with the C–H bond via either a direct insertion or a hydrogen atom transfer (HAT)/radical rebound pathway.
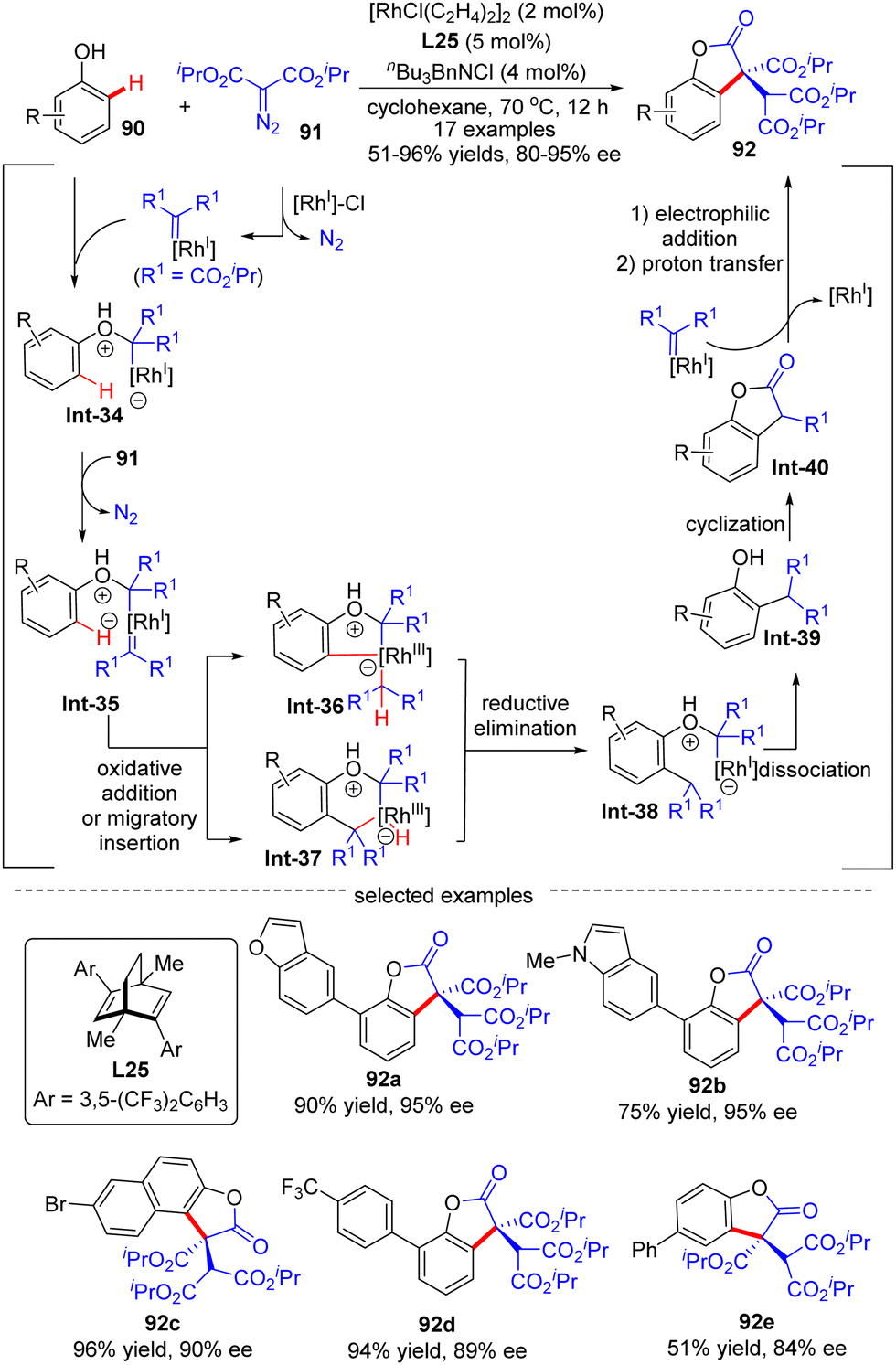
In 2020, Wang's group demonstrated an example of carbene ortho-selective C(sp2)–H insertion of phenols with diisopropyl diazomalonate via Rh(I)/diene catalysis (Scheme 27).99 With a transient oxonium ylide as the directing group, the final tris-carboxylate-substituted 2-benzofuranones 92 bearing a quaternary all-carbon center were produced in good yields (51–96%) and enantioselectivities (80–95% ee). The substrate scope investigation showed that various substituents at the ortho-, meta- and para-position were all well tolerated, indicating the good functional-group tolerance of this reaction. The underlying mechanism was proposed as shown in Scheme 27. Generally, this reaction was initiated by Rh(I)-triggered N2 extrusion from 91 to form a Rh-carbene intermediate, which then reacted with phenol to give an oxonium ylide Int-34. Int-34 promoted the extrusion of N2 from a second 91, giving access to another Rh-carbene species Int-35. Subsequently, Int-35 underwent ortho-C(sp2)–H oxidative addition/migratory insertion sequence to produce Int-36 or Int-37, which then experienced a reductive elimination to provide Int-38. Afterwards, Int-40 was formed by dissociation of the oxonium ylide Int-38 and follow-up cyclization of Int-39. Finally, Int-40 proceeded with electrophilic addition to another Rh-carbene, followed by proton transfer to release the desired product 92.
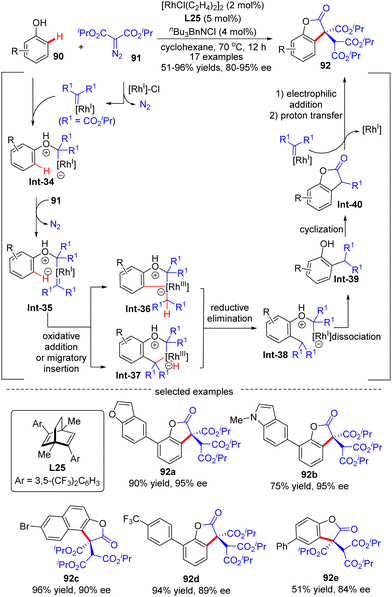 |
| | Scheme 27 Rh(I)-Catalyzed enantioselective C(sp2)–H functionalization of phenols with diisopropyl diazomalonate. | |
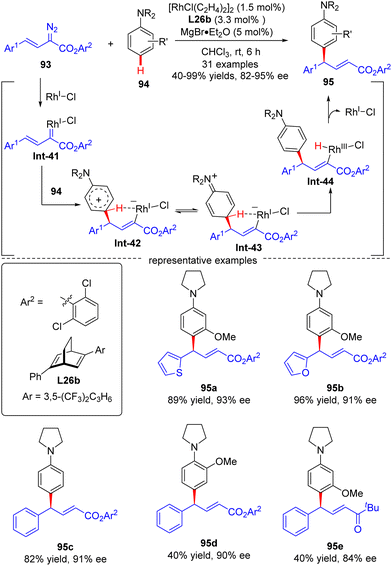 |
| | Scheme 28 Rh(I)-Catalyzed enantioselective arylvinylcarbene insertion of the C–H Bond of aniline derivatives. | |
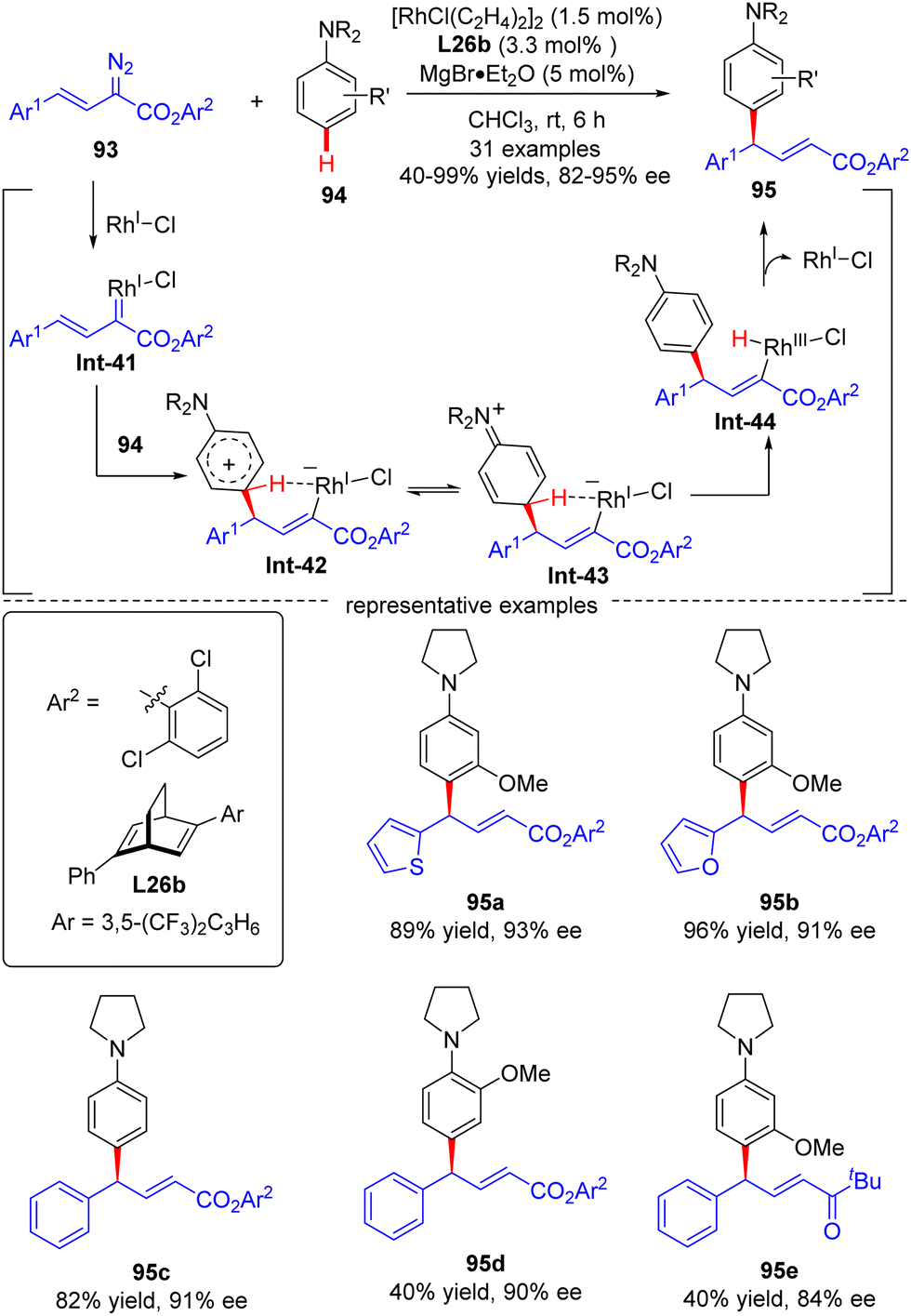
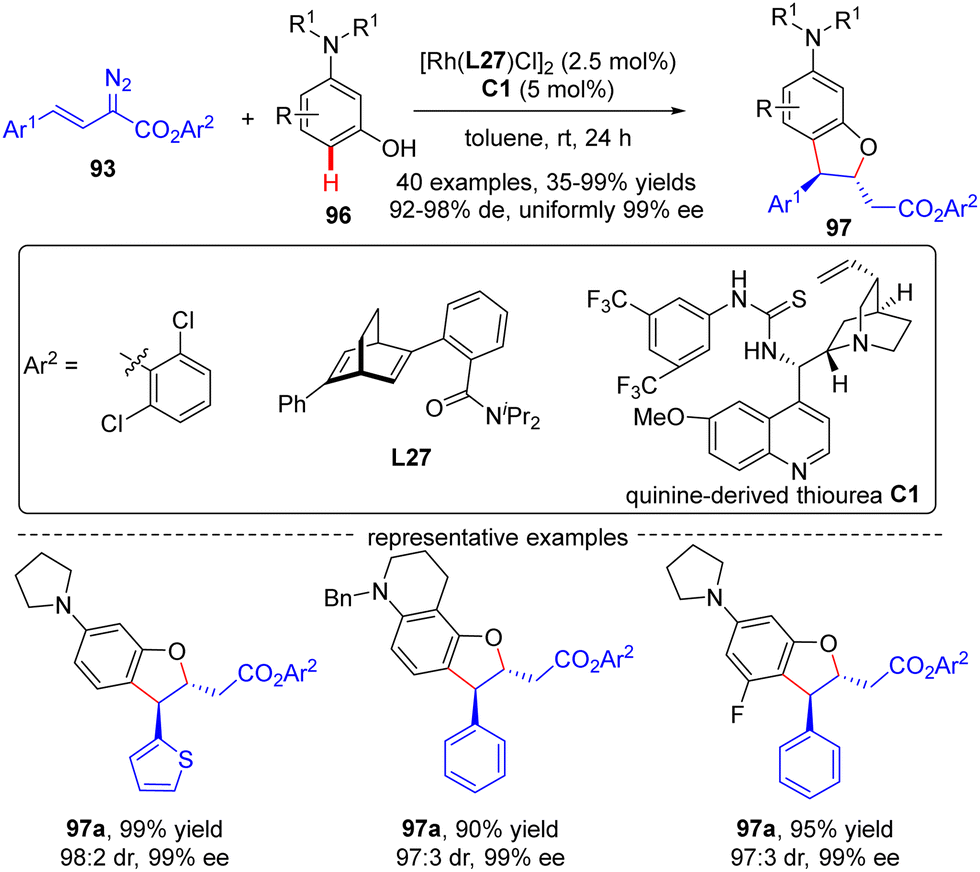
In 2021, Xu and co-workers developed an elegant Rh(I)/diene-catalyzed regiospecific and enantioselective arylvinylcarbene insertion of the C(sp2)–H bond of aniline derivatives (Scheme 28).100 The reaction was characterized by multiple merits, such as unusual regio-selectivity at the vinyl terminus site, excellent E-selectivity and enantioselectivities (82–95% ee), simple and mild conditions, and broad functional group tolerance, offering access to a diversity of chiral γ,γ-gem-diarylsubstituted α,β-unsaturated esters 95. Synthesis applications of this protocol were featured by several versatile product transformations. Apart from arylvinyldiazoacetates, a more challenging arylvinyldiazoketone substrate was also found to be compatible with this transformation, giving rise to the expected C–H functionalization product 95e in moderate yield (40%) and good enantioselectivity (84% ee). Combined experimental and computational studies were carried out to elucidate the reaction mechanism. Initially, arylvinyldiazoacetate reacted with the active monorhodium catalyst Rh-Cl to afford the Rh(I)-arylvinylcarbenoid intermediate Int-41. The ensuring addition of the electron-rich phenyl ring of aniline onto Int-41 site-selectively occurred at the vinyl terminus site to furnish zwitterionic species Int-42, which then proceeded a 1,5-proton transfer event to provide Rh(III) species Int-44. Finally, reductive elimination of Int-44 happened, leading to the desired product 95 and the regeneration of the monorhodium catalyst. Of note, an inverse deuterium kinetic isotope effect was observed in this reaction, supporting the C–C bond-formation step as a rate-determining step. Encouraged by this elegant work, Xu and co-workers further realized a one-pot rhodium-catalyzed C–H functionalization/organocatalyzed oxa-Michael addition cascade reaction of 3-hydroxylaniline derivatives 96 with arylvinyldiazoacetate 93 (Scheme 29).101 Under mild conditions, a diverse set of 2,3-disubstituted dihydrobenzofurans 97 were attained in good yields (up to 99%) and excellent stereoselectivity (92–98% de, uniformly 99% ee). It was noteworthy that the two contiguous stereogenic centers of product 97 were independently controlled by two chiral catalysts via a one-pot two-step operation. As a consequence, the full complement of stereoisomers could be accessed at will by appropriate permutations of the two chiral catalysts.
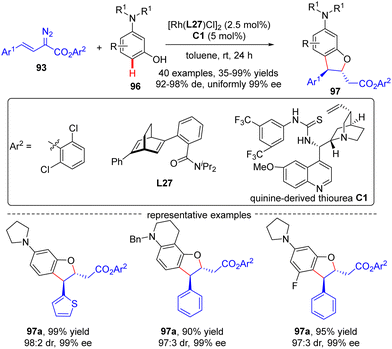 |
| | Scheme 29 Rh(I)-Catalyzed enantioselective C–H functionalization/oxa-Michael addition cascade. | |
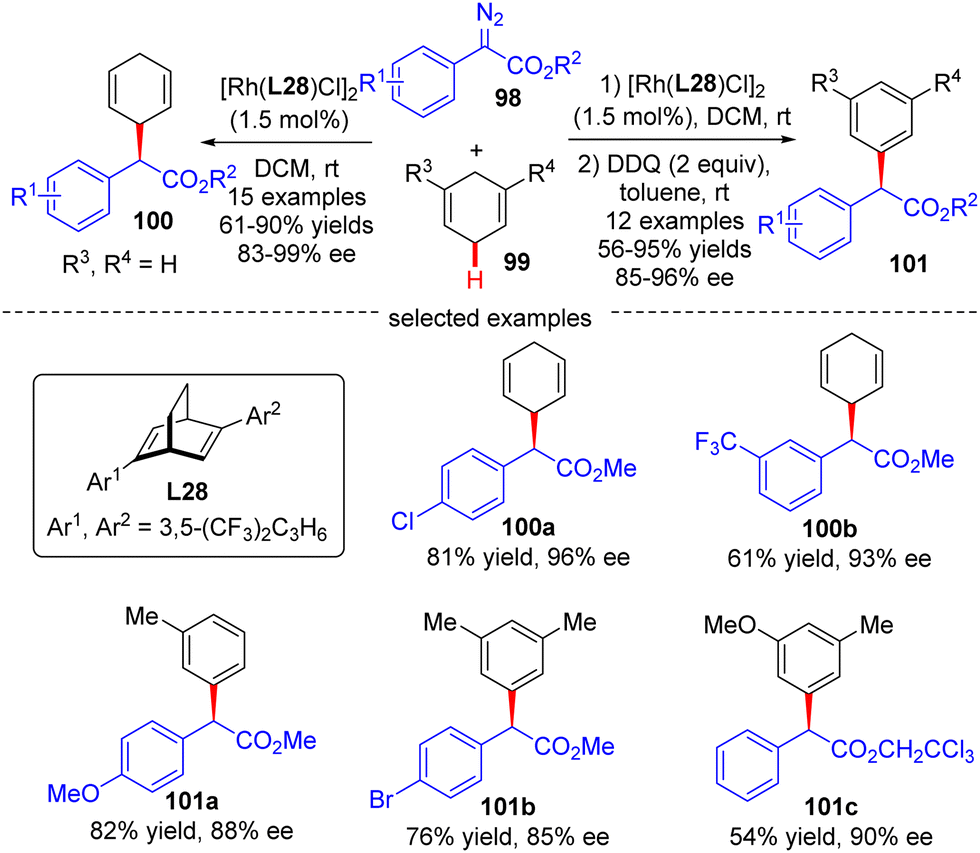
In 2021, Xu's group developed an asymmetric Rh(I)/diene-catalyzed C(sp3)–H functionalization of 1,4-cyclohexadiene 99 with α-aryl-diazoacetate 98, which provided a direct access to various chiral 2-arylacetates 100 in good yields (up to 90%) and enantiomeric excess (up to 99%) (Scheme 30).102 The application of this reaction was further highlighted by the follow-up oxidation of the products with DDQ with the generation of diverse enantioenriched α,α-diarylacetates 101. Notably, the steric factor had a great impact on the reactivity of different C(sp2)–H bonds, and only the less sterically hindered C(sp2)–H bonds were functionalized.
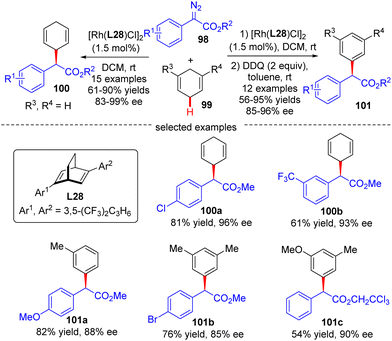 |
| | Scheme 30 Rh(I)-Catalyzed enantioselective C(sp3)–H functionalization of 1,4-cyclohexadiene with α-aryl-diazoacetates. | |
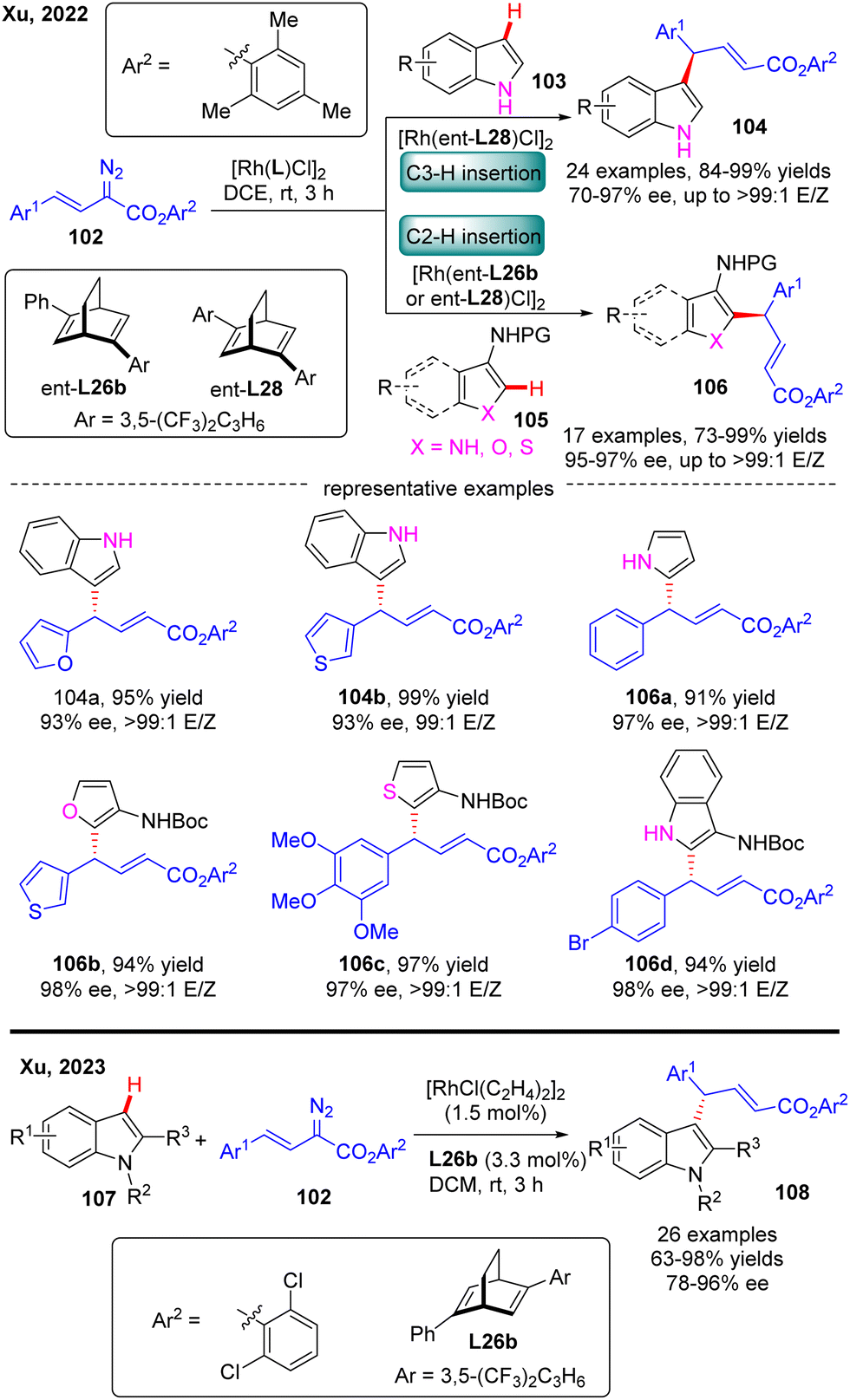
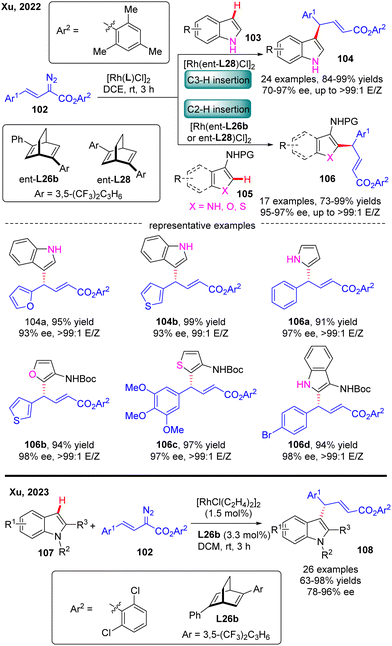
In 2022, Xu and co-workers communicated an elegant Rh(I)-diene catalyzed highly enantioselective C(sp2)–H allylation of simple unprotected indoles, pyrroles, and their common analogues such as furans, thiophenes, and benzofurans with arylvinyldiazoesters, offering a facile and reliable access to a diversity of diarylmethane-containing α,β-unsaturated esters 104 & 106 in favorable yields (73–99%), and excellent enantioselectivities (70–97% ee) (Scheme 31).103 For unprotected indole substrates 103 without a substituent at the C3 position, the carbene insertion site selectively occurred at the C3 position of indoles. In contrast, when unprotected pyrroles were used or the C3 position of indoles, furans, thiophenes and benzofurans (105) were occupied by a protected amino group, only C2 carbene insertion proceeded. Notably, the bulky 2,4,6-trimethylphenyl substituent on arylvinyldiazoesters was found to be critical to guarantee the excellent enantioselectivity of this reaction and this reaction was also characterized by an uncommon site-selectivity at the vinyl terminus of arylvinylcarbene. Mechanistic experiments and DFT calculations indicated that the C–C addition barrier was lowered by the more electron-rich indole substrate, thus changing the rate-determining step to the reductive elimination step. Soon after, Xu and co-workers further extended the substrate scope of Rh(I)-diene catalysed arylvinylcarbene insertion process to N-protected indoles 107, giving rise to an interesting class of chiral indole scaffolds 108 containing a gem-diaryl carbon stereocenter in good yields (up to 99%) and excellent enantioselectivities (up to 96% ee).104
 |
| | Scheme 31 Rh(I)-Catalyzed enantioselective C(sp2)–H functionalization of simple unprotected indoles, pyrroles and heteroanalogues and protected indoles. | |
4. C–H functionalization via 1,4-Rh migration
Aside from the above-described Rh(I)-catalyzed direct C–H activation via either an inner- or outer-sphere mechanism, 1,4-Rh migration represents an alternative approach to promote C–H activation to form unique carbon–Rh bonds, which could be further transformed into a diversity of valuable scaffolds. More importantly, 1,4-Rh migration could facilely introduce a rhodium moiety into a specific position of an organic molecular skeleton, which would be otherwise difficult to be obtained via classic approaches. Currently, the most prevalent 1,4-Rh migration for asymmetric C–H functionalization is from alkenyl to aryl, despite that several other modes such as alkyl-to-aryl, aryl-to-alkenyl, allyl-to-allyl, and alkenyl-to-allyl 1,4-Rh migration may also exist.
4.1. C(sp2)–H functionalization via 1,4-Rh migration
4.1.1 C(sp2)–H functionalization via alkenyl-to-aryl 1,4-Rh migration.
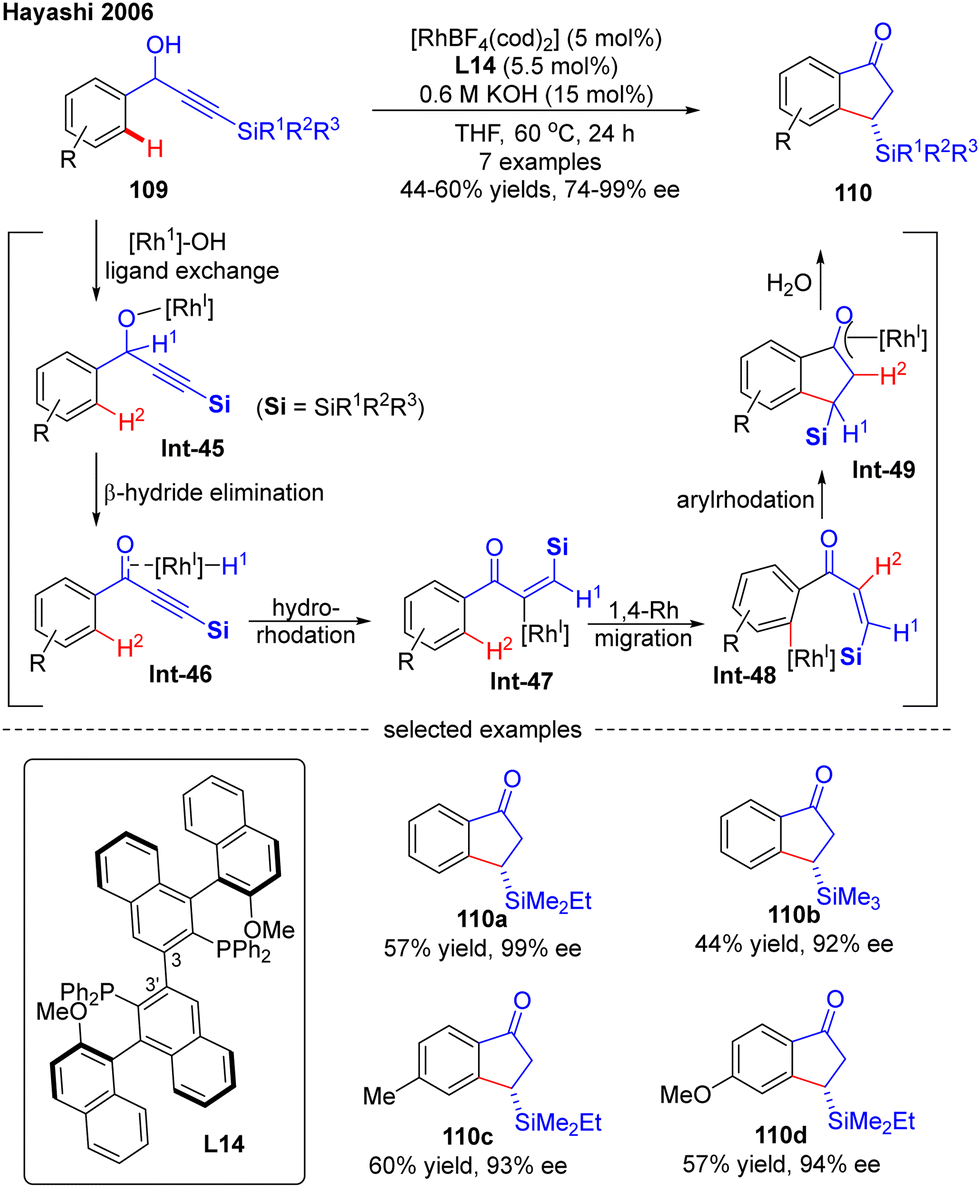
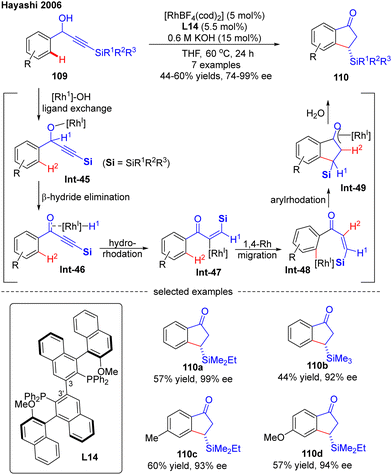
In 2006, Hayashi's research group developed an effective Rh(I)-catalyzed asymmetric synthesis of 3-substituted 1-indanones via an alkenyl-to-aryl 1,4-Rh migration (Scheme 32).105 By employing a newly designed axially chiral bisphosphine ligand L14, wherein the absolute configuration of the 3-3′ axis was fixed to (R) after complexation to transition metal rhodium, they successfully prepared several indanones 110 in 44–60% yields with 92–99% ee via an asymmetric isomerization of racemic α-arylpropargyl alcohols 109. The underlying mechanism was studied, suggesting that this reaction was initiated by the ligand exchange of 109 by Rh(I) catalyst to generate alkoxylrhodium Int-45. Subsequent β-H elimination of Int-45 followed by conjugate hydrorhodation resulted in alkenylrhodium species Int-47, which further underwent 1,4-rhodium migration to provide Int-48. Afterwards, Int-49 with a new emerging stereocenter was produced by intramolecular 1,4-addition of Int-48. The Rh/(R,R)-L13 complex played an important role in this step, as it could recognize the enantiotopic face of the C–C double bond in Int-48 to create a new stereocenter with high enantioselectivity. The desired product 110 was finally obtained by the hydrolysis of Int-49.
 |
| | Scheme 32 Rh(I)-Catalyzed enantioselective isomerization of racemic α-arylpropargyl alcohols. | |
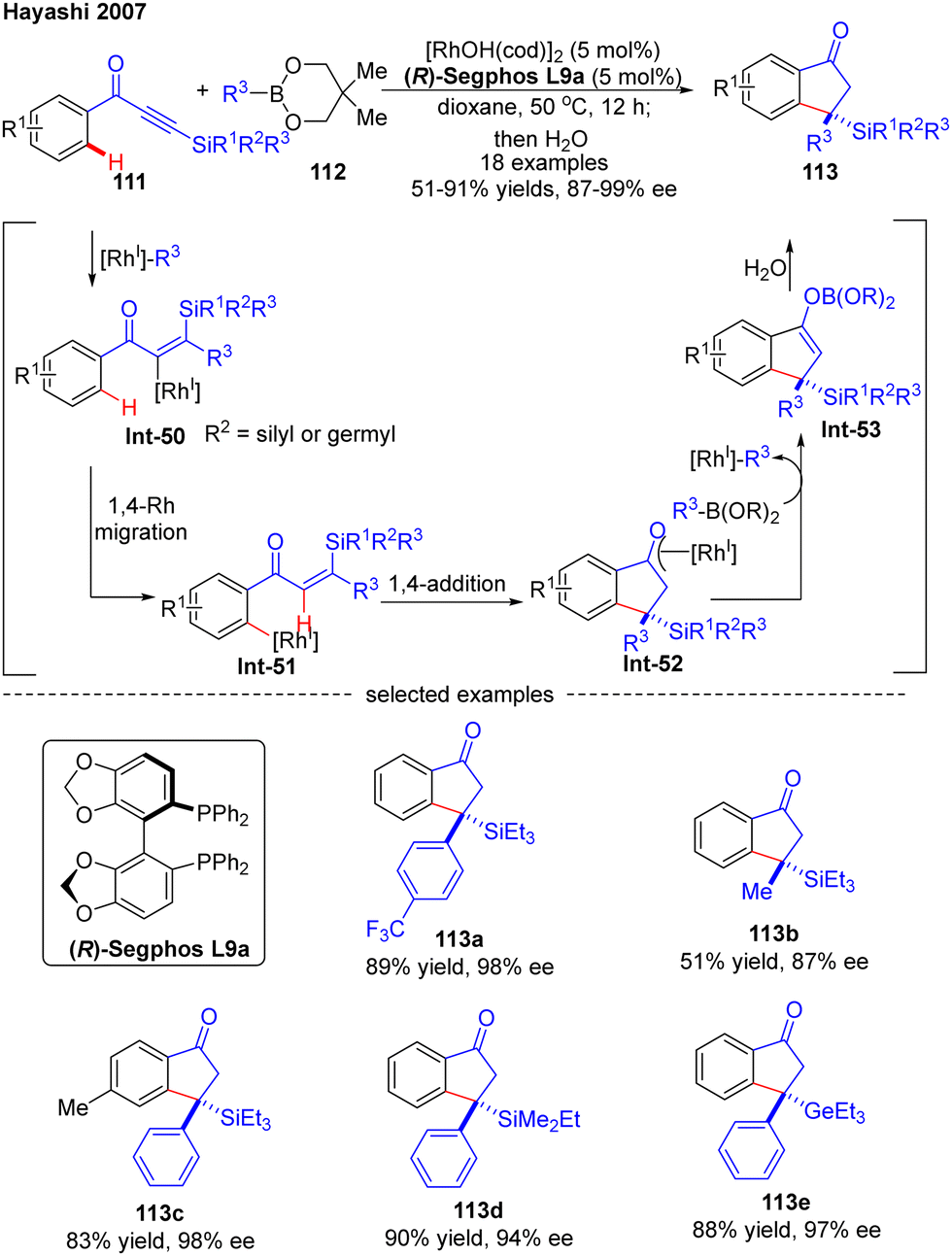
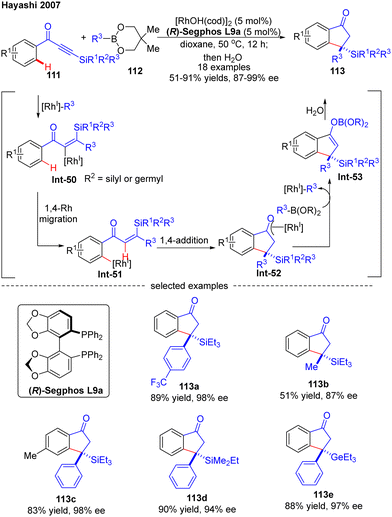
In 2007, Hayashi and co-workers further reported a [RhOH(cod)]2/(R)-Segphos catalytic system for high enantioselective addition of boronates 112 to aryl silylethynyl ketones 111 (Scheme 33).106 With this system, a variety of 3,3-disubstituted 1-indanones 113 were obtained in impressive yields (51–91%) and excellent enantiomeric excess (87–99%). Aside from the silyl substituents on the ethynyl ketones, a germyl substituted substrate was also compatible with this reaction to give the corresponding product 113e in 88% yield and 97% ee. The underlying mechanism of the reaction was also proposed (Scheme 33). First, insertion of the alkyne group in 111 into the [Rh]–carbon bond generated the alkenyl rhodium species Int-50, which underwent a 1,4-Rh migration process to produce the aryl rhodium intermediate Int-51. Subsequent 1,4-intramolecular addition in Int-51 occurred to deliver the oxa-π-allyl rhodium species Int-52, wherein the stereochemistry was determined. Follow-up transmetalation of Int-52 with boronates 112 released the boron enolate Int-53 along with the [Rh]–R3 species for the next cycle. Finally, the desired product 113 was achieved by hydrolysis of Int-53.
 |
| | Scheme 33 Rh(I)-Catalyzed enantioselective addition of aryl boronates to aryl alkynyl ketones. | |
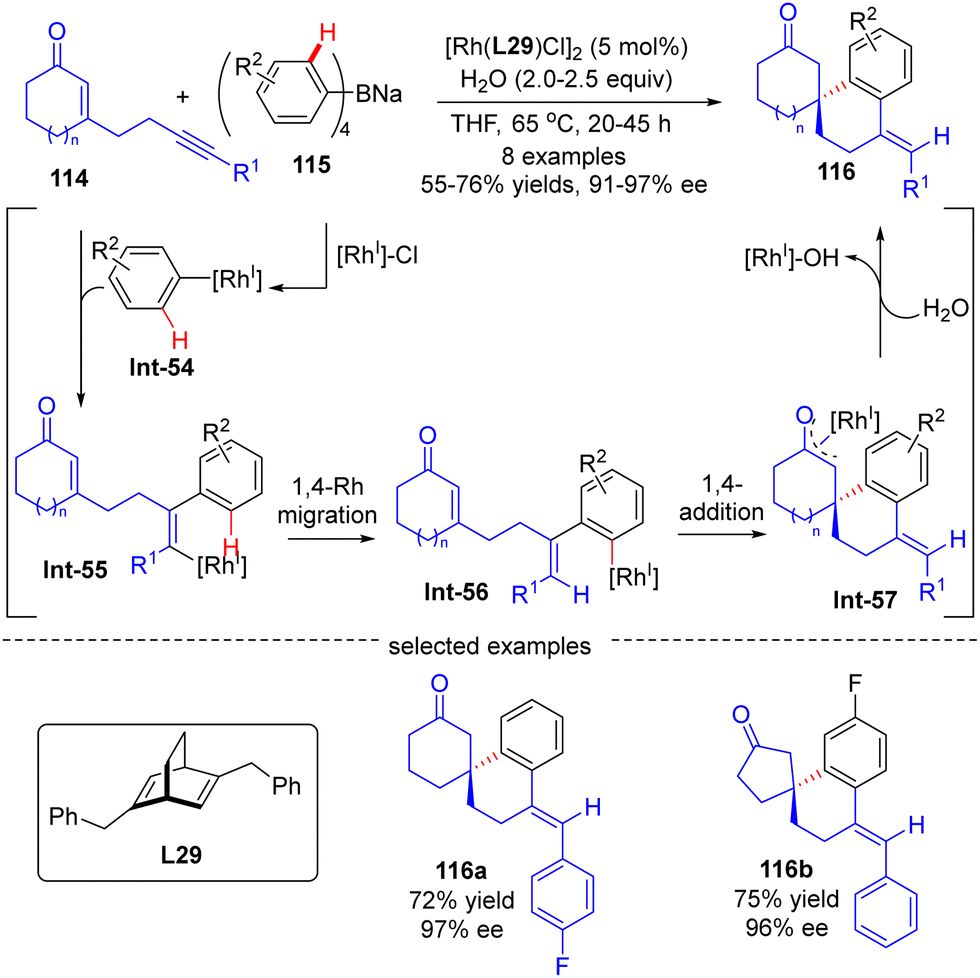
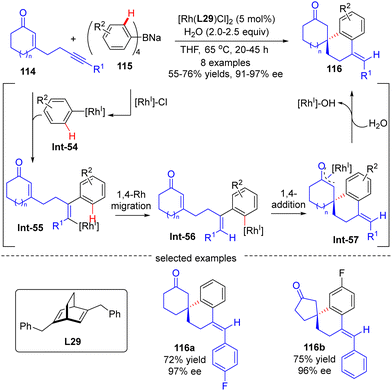
The asymmetric synthesis of enantiopure spirocabocycles, a class of prevalent scaffolds in natural products, biologically active molecules and chiral ligands, has gained great attention in organic chemistry. In 2010, Hayashi's group communicated a facile access to chiral spirocarbocycles via a Rh(I)/diene catalyzed enantioselective addition of sodium tetraarylborates 115 to alkyne-tethered 2-cycloalken-1-ones 114 (Scheme 34).107 With sodium tetraarylborates 115 as surrogates of 1,2-dimetalloarenes and the chiral diene L28 as a ligand, a variety of spirocycles 116 bearing a quaternary carbon stereocenter were prepared in good yields (55–76%) and a decent enantiomeric excess (91–97%). This reaction was proposed to be initiated by the generation of an aryl-Rh species Int-54via the transmetalation of 115 with [Rh]–Cl. Int-54 underwent insertion of the alkyne group in 114 to generate alkenyl-Rh intermediate Int-55, which was then converted to aryl-Rh species Int-56via a 1,4-Rh migration process. Successive intramolecular 1,4-addition onto the tethered enone moiety in Int-56 resulted in oxa-π-allyl-Rh intermediate Int-57. The final hydrolysis of Int-57 released the desired product 116 along with concurrent regeneration of the Rh(I) catalyst.
 |
| | Scheme 34 Rh(I)-Catalyzed enantioselective addition of sodium tetraarylborates to alkyne-tethered 2-cycloalken-1-ones. | |
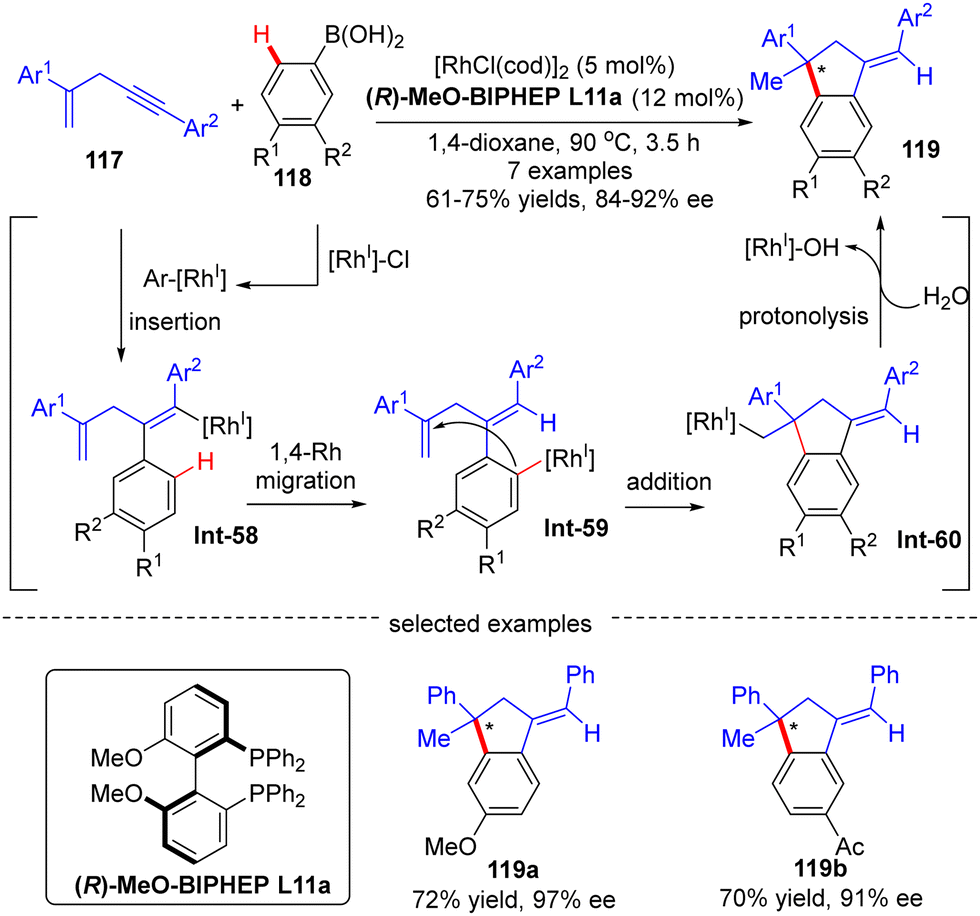
Afterwards, Matsuda and co-workers reported the asymmetric synthesis of indanes 119 possessing a quaternary all-carbon center from 1,4-enynes 117 with arylboronic acids 118via a Rh(I)-catalyzed arylation/annulation process (Scheme 35).108 The desired indanes 119 were readily obtained with yields of 61–75% and enantiomeric excess of 84–92% by taking advantage of the [Rh(cod)OH]2/(R)-MeO-BIPHEP catalytic system. As far as this reaction mechanism was concerned, four key steps including intermolecular alkyne insertion, 1,4-Rh migration, intramolecular addition onto the tethered alkene moiety, and protonolysis were proposed to be involved.
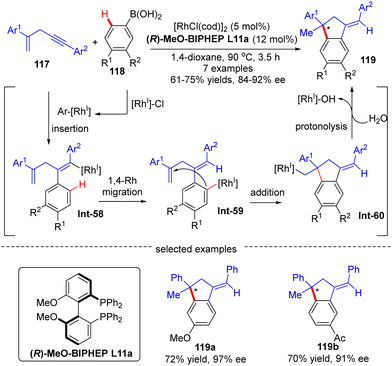 |
| | Scheme 35 Rh(I)-Catalyzed asymmetric indane synthesis from 1,4-enynes and arylboronic acids. | |
In 2012, Hayashi's group reported a tandem 1,4-Rh migration/1,4-addition process of 2-arylethenylboronic acid 120 to enones 121 by use of chiral ligand L30a, a new chiral diene derivative from (R)-phellandrene (Scheme 36).109 With regard to the variability of the substrates, both cyclic and acyclic enones 121 were tolerated to produce the rearranged products 122 with high yields and excellent regio- and enantio-selectivities. The catalytic cycle of this reaction involved a transmetalation to form the 2-phenylethenyl-Rh(I) intermediate Int-61, 1,4-Rh migration to give Int-63, and enantioselective 1,4-addition onto enones 121 to arrive at the desired products 122. Density functional theory (DFT) studies indicated that the 1,4-Rh migration proceeded via an oxidative addition onto the aryl C–H bond in Int-61, leading to a distorted square-pyramidal Rh(III)–H species Int-62, and followed by reductive elimination of Int-62 to afford Int-63.
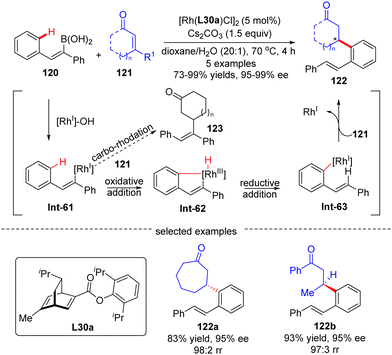 |
| | Scheme 36 Rh(I)-Catalyzed 1,4-migration/1,4-addition of 2-arylethenylboronic acid to enones. | |
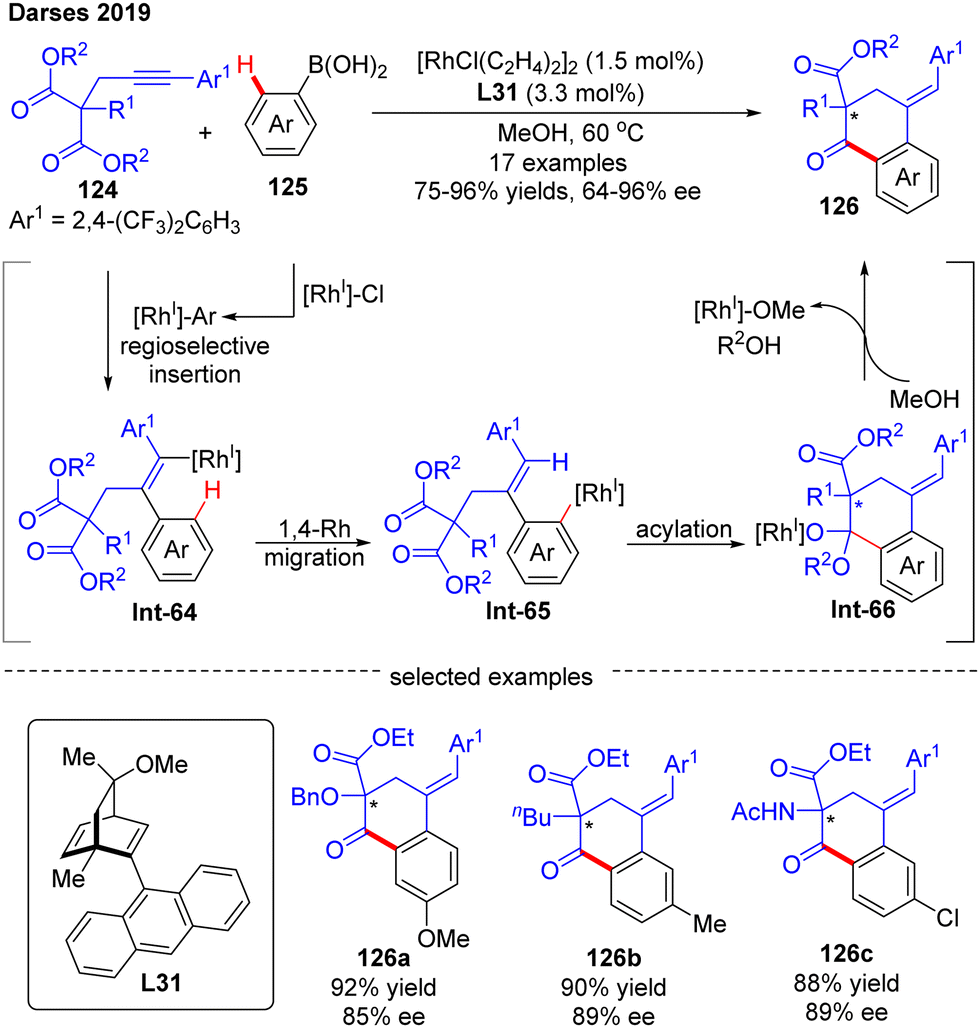
In 2019, by using the chiral diene derivative L31 with an anthracen-9-yl substituent as a ligand, Darses’ group successfully developed an asymmetric arylative cyclization of propargylic malonates 124 with arylboronic acids 125, giving access to chiral 1-tetralones 126 with a quaternary carbon stereocenter via the desymmetrization of the malonate moiety (Scheme 37).110 This reaction involved a regioselective alkyne insertion, an alkenyl-to-aryl 1,4-Rh migration and an acylation process. The 2,4-bis(trifluoromethyl)phenyl substituent on propargyl malonates 124 played an important role in controlling the regioselectivity of alkyne insertion (>99:1). Alkyl-, alkoxyl, and amino-substituted alkynylmalonates were all competent substrates to generate the desired 1-tetralones 126 in favorable yields (up to 96%) and enantioselectivities (up to 96% ee). Of note, one enantioselective variant of 1-tetralone was also prepared from bis(2,2,2-trifluorethyl)-malonate by Lam's group under the catalysis of [RhCl(C2H4)2]2 and (R)-MeO-BIHEP.111
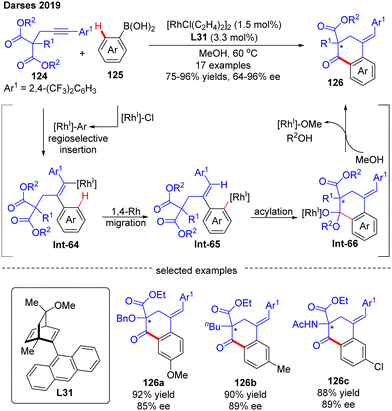 |
| | Scheme 37 Rh(I)-Catalyzed arylative cyclization of propargylic malonates with arylboronic acids. | |
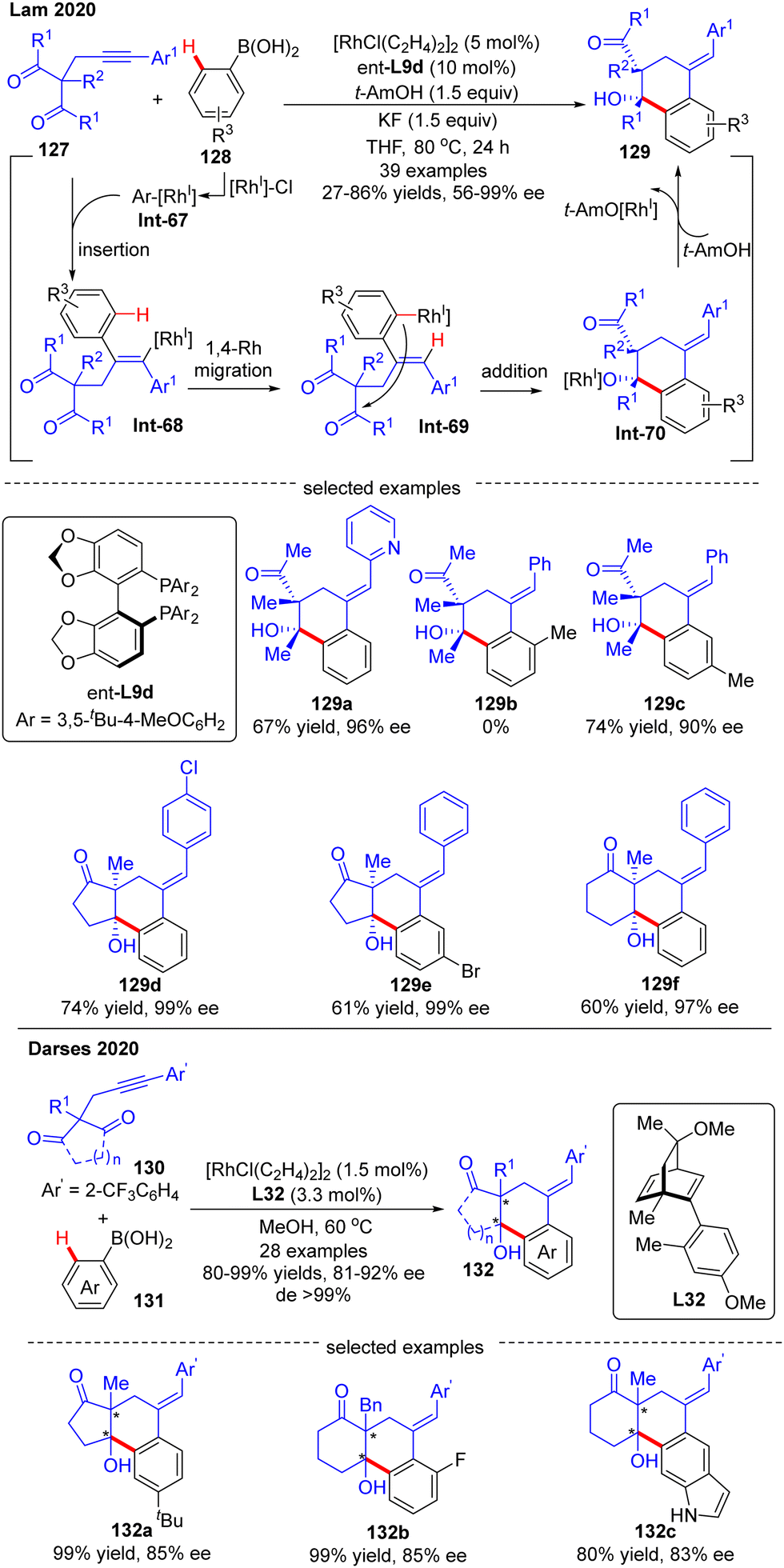
In 2020, Lam's group developed a Rh(I)-catalyzed asymmetric arylation/annulation reaction of 2-propargyl-1,3-diketones 127 with arylboronic acids 128 involving an alkenyl-to-aryl 1,4-Rh migration as the key step, thus giving access to densely functionalized polycarbocycles 129 containing two contiguous quaternary stereogenic centers (Scheme 38).112 By the use of the [RhCl(C2H4)2]2 catalyst combined with (S)-DTBM-Segphos ligand ent-L9d, the desired products 129 were achieved in excellent enantioselectivities (up to 99% ee) and diastereoselectivities (in most cases dr > 19:1). A broad range of functional groups on both reaction partners were well tolerated regardless of the electronic effect, whereas the steric factor played a critical role in the reactivity as reflected by the failure of 2-methylphenylboronic acid as a substrate (129b). It was also found that 3-substituted arylboronic acids favored affording the less sterically hindered products (129c). When cyclic 1,3-diketones were used as a substrate, favorable results were also achieved to give 129d–f in 60–74% yields and 97–99% ee. It was hypothesized that this reaction might experience a transmetalation/migratory insertion/1,4-Rh migration/1,2-addition sequence. The similar transformation between 1,3-diketones and aryl boronic acids was also realized by Darses and co-workers by use of chiral diene L32 as a ligand, providing straightforward access to 1-tetralols 132 containing two contiguous quaternary stereogenic centers in good enantioselectivities (81–92% ee) and diastereoselectivities (>99% de) (Scheme 38).113 Diverse 2-propargyl-1,3-diketones including cyclic and acyclic substrates with various functional groups were all well tolerated in this reaction. It is worth mentioning that the 2-trifluoromethyl-phenyl substituent (Ar′) on the alkyne moiety of 130 was essential to achieve high regioselectivity for alkyne insertion.
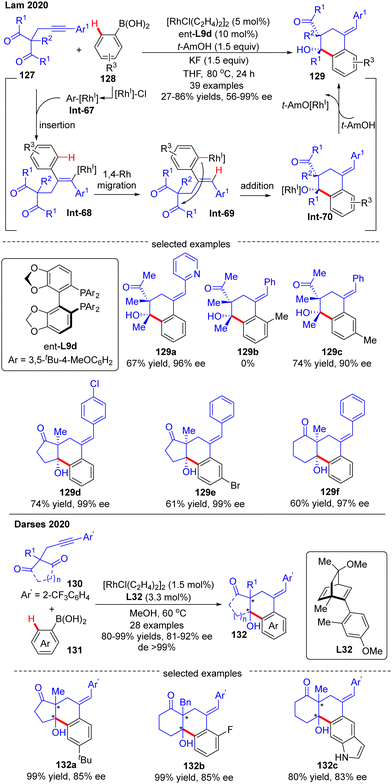 |
| | Scheme 38 Rh(I)-Catalyzed arylative cyclization of 2-propargyl-1,3-diketones with arylboronic acids. | |
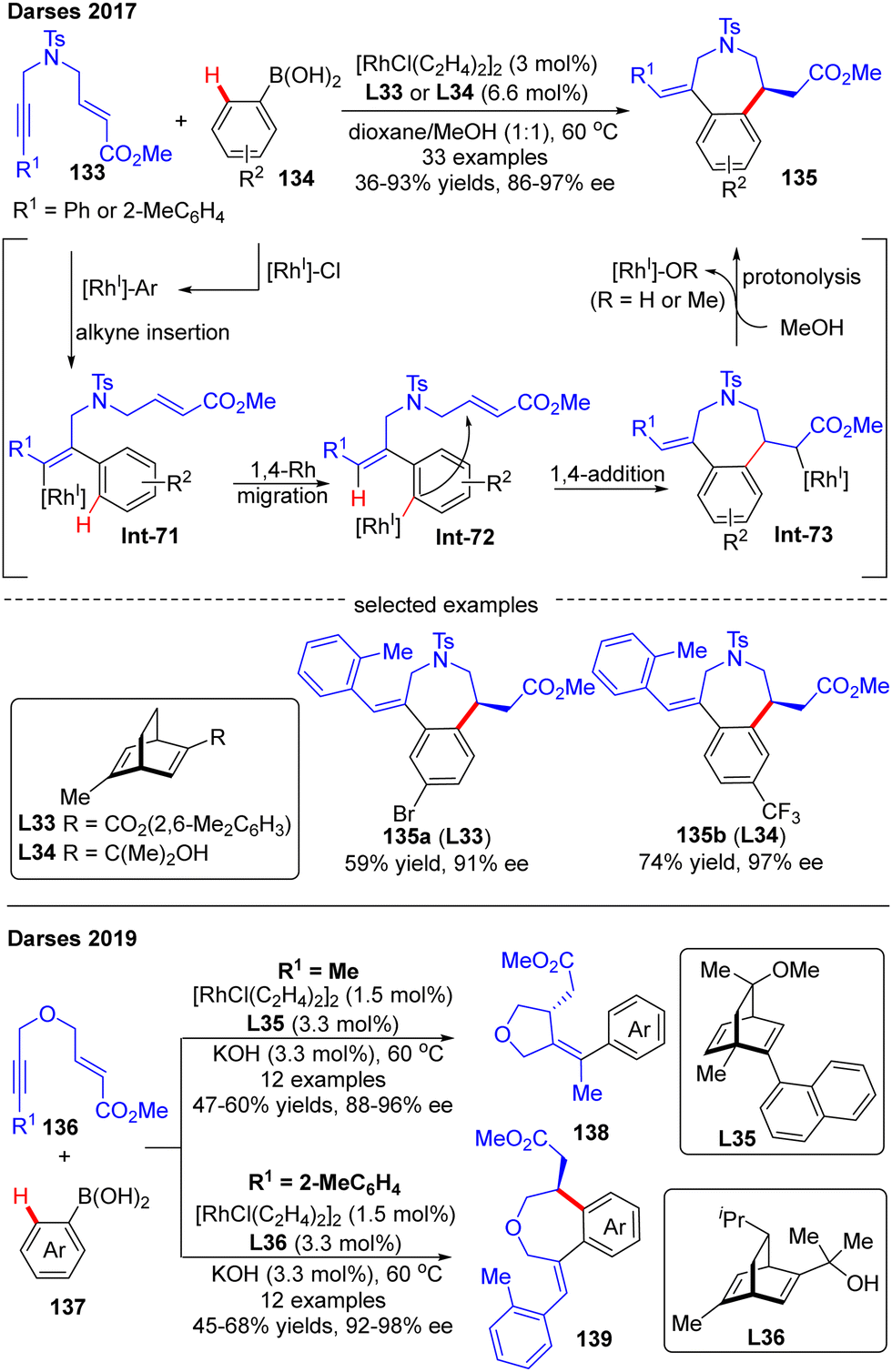
Darses's research group also investigated the cascade reactions of eneyne derivatives with arylboronic acids by taking advantage of asymmetric catalysis with Rh(I)/chiral diene complexes. In 2017, they disclosed the preparation of various 7-membered chiral azepane 135 in moderate to high yields (36–93%) and good enantiomeric excess (86–97%) by using the N-tethered alkyne enoates 133 and aryl boronic acids 134 as substrates and chiral diene L32 or L33 as a ligand (Scheme 39).114 This reaction displayed good functional group tolerance. Shortly after, Darses and co-workers further developed an efficient access to chiral tetrahydrofurans 138 and tetrahydrobenzo[d]oxepines 139 in decent stereo-control from simple O-tethered alkyne enoates 136.115 When the R1 substituent in 136 was methyl, the chiral 5-membered tetrahydrofurans 138 were obtained, whereas the enantioenriched 7-membered tetrahydrobenzo[d]oxepines 139 was produced when the R1 substituent was aryl. The differed product structures were ascribed to a switch of the regioselectivity of alkyne insertion by the alkyne substituent, suggesting that the regioselectivity of the alkyne insertion may be impacted by the steric and electronic properties of R1 substituents.116
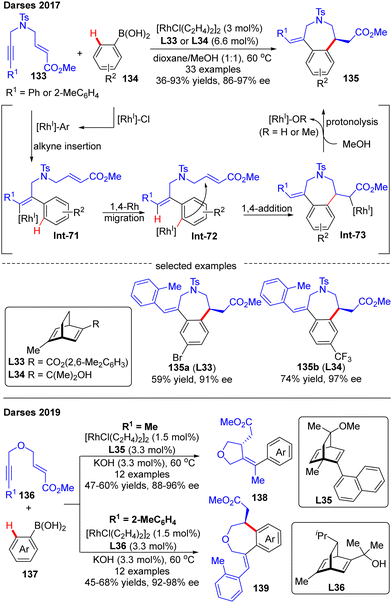 |
| | Scheme 39 Rh(I)-Catalyzed arylative cyclization of N- or O-tethered alkyne enoates with arylboronic acids. | |
4.1.2 C(sp2)–H functionalization via alkyl-to-aryl 1,4-Rh migration.
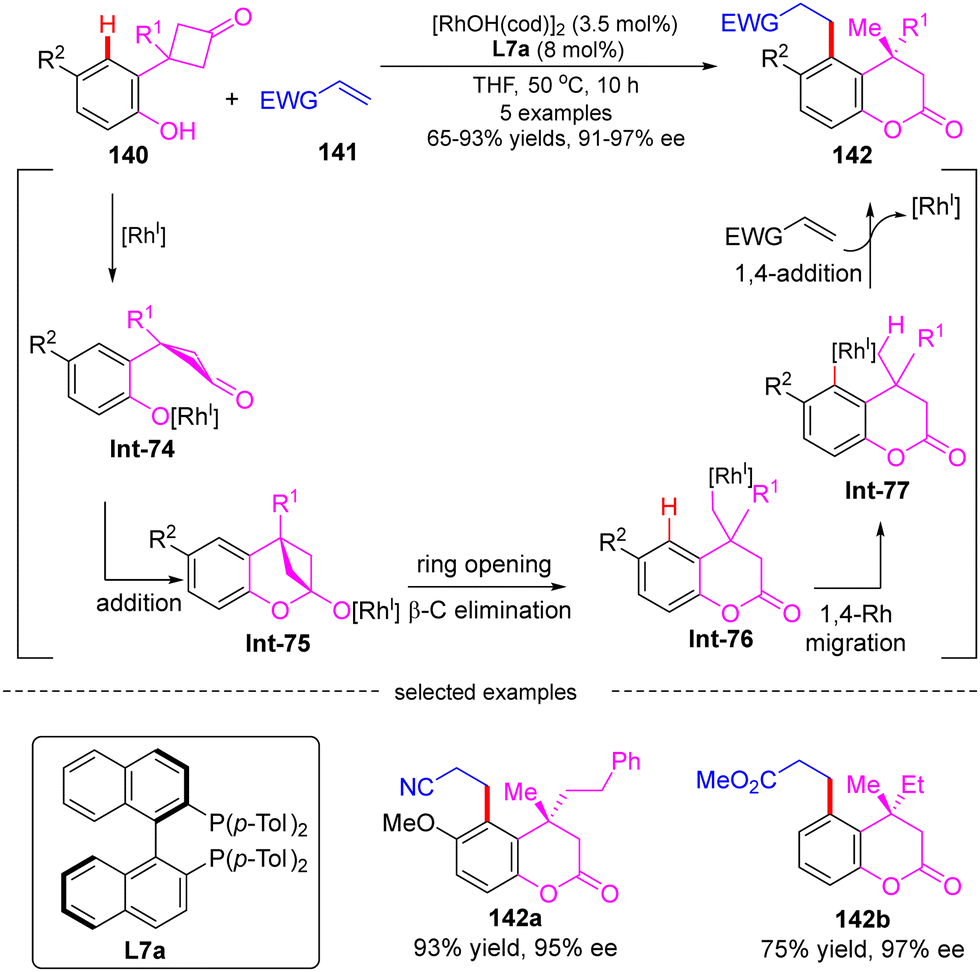
In 2007, Murakami's group developed a Rh(I)-catalyzed cascade reaction of 3-(2-hydroxyphenyl)-cyclobutanones 140 with electron-deficient alkenes 141via an alkyl-to-aryl 1,4-Rh migration (Scheme 40).117 Using [RhOH(cod)]2 as a catalyst and (R)-Tol-BINAP L7a as a chiral ligand, several 4,4-disubstituted 3,4-dihydrocoumarins 142 were obtained in high yields (65–93%) and enantioselectivities (91–97% ee). It was proposed that this cascade reaction might involve a carbonyl addition, a ring opening via β-C elimination, a 1,4-Rh migration, and an intermolecular 1,4-addition. Specifically, rhodium aryloxide Int-74 was first generated from 140 with [RhOH(cod)]2. Intramolecular addition onto the carbonyl group in Int-74 happened to produce the rhodium cyclobutanolate Int-75, which readily proceeded β-C elimination, forging ring-opening alkyl rhodium species Int-76. This step was believed as the stereochemistry-determining step of this reaction. Afterwards, the arylrhodium species Int-77 was then generated from Int-76via a 1,4-rhodium migration process. A final intermolecular 1,4-addition of Int-77 onto electron-deficient alkenes 141 arrived at the enantioenriched 3,4-dihydrocoumarins 142.
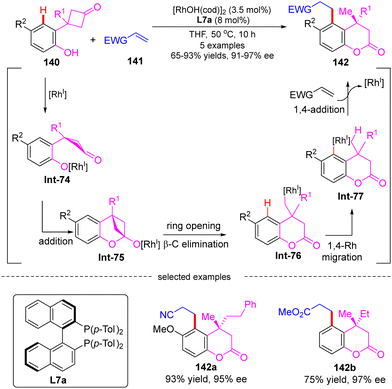 |
| | Scheme 40 Rh(I)-Catalyzed cascade reaction of 3-(2-hydroxyphenyl)cyclobutanones with electron-deficient alkenes. | |
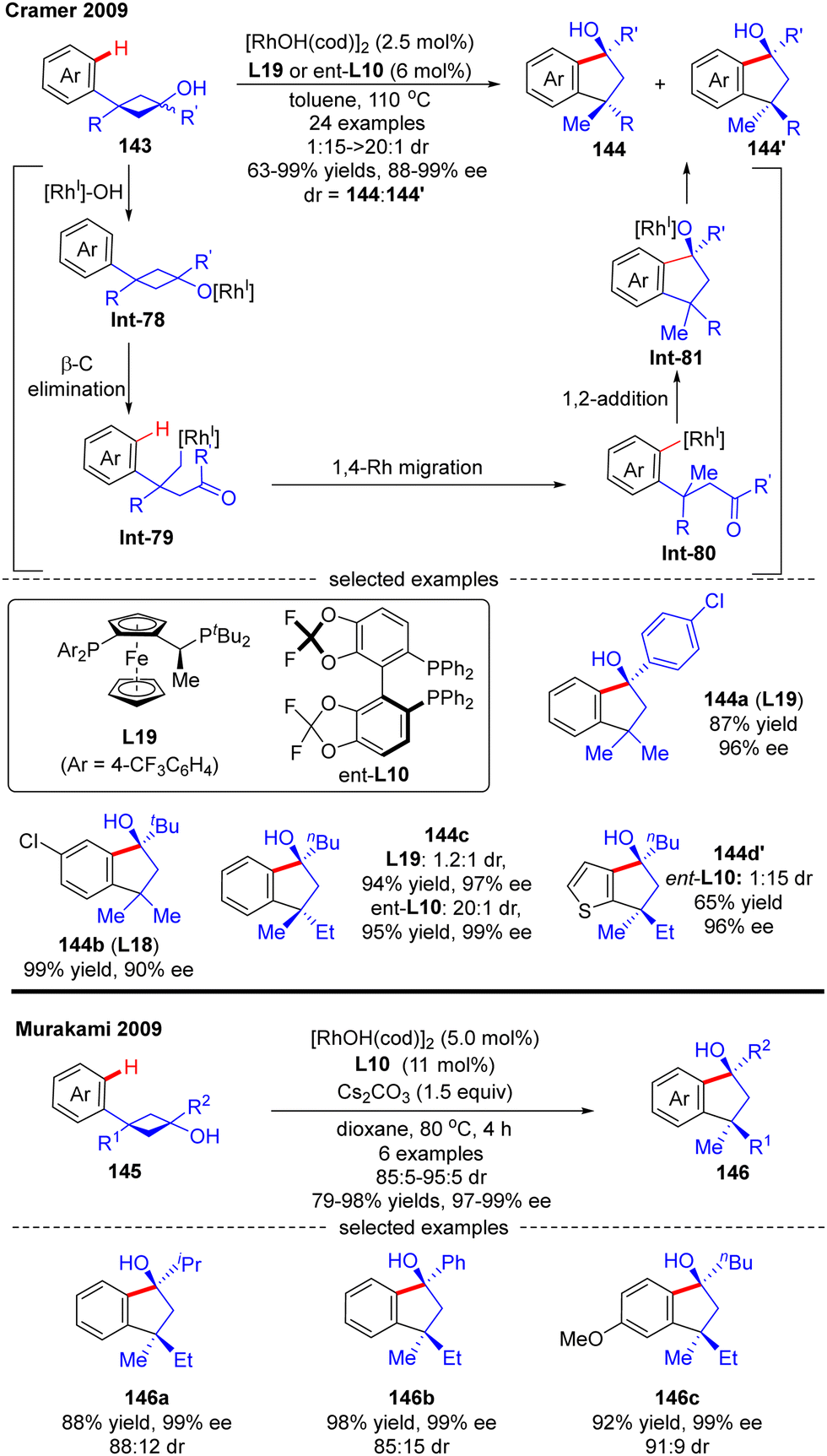
In 2009, Cramer and co-workers successfully prepared enantioenriched 1-indanols 144 from cyclobutanols 143 under the catalysis of [RhOH(cod)]2 (Scheme 41).118 When 3-aryl-3′-methyl-cyclobutanols were used as substrates, Josiphos-type ligand L19 with an electron-withdrawing trifluoromethyl group demonstrated as the optimal, providing products 144a and 144b as a single stereoisomer with decent yields and stereo-control. Interestingly, when the 3′-position of cyclobutanols was not methyl, diastereo-isomers existed and the chiral ligand had a great influence on the diastereomeric ratios, as reflected by the increased diastereoselectivity by taking advantage of the difluorophos ligand ent-L10 (144c, 20:1 vs. 1.2:1 with L19 and ent-L10, respectively). As illustrated in Scheme 41, the reaction was proposed to be initiated by the formation of the rhodium cyclobutanolate Int-78via deprotonation of 143. Fragmentation of the cyclobutane ring in Int-78 occurred to form the alkyl rhodium species Int-79via β-C elimination. Following a 1,4-Rh migration process of Int-79, the aryl rhodium species Int-80 bearing one quaternary carbon was produced, which was further turned into Int-81 containing two quaternary carbons through intramolecular 1,2-addition. The desired product 1-indanols 144 was finally released by protonation of Int-81. A similar approach to stereoselective restructuring of 3-arylcyclobutanols 145 into 1-indanols 146 was also reported by Murakami's group,119 and the underlying mechanism was systematically studied by Dang's group with DFT calculations in 2014.120 The results showed that the stereoselectivity of this reaction was contributed by three main factors including the relative stability of β-C elimination product, the kinetic feasibilities of 1,4-Rh migration, and the relative free energies of CO insertion step.
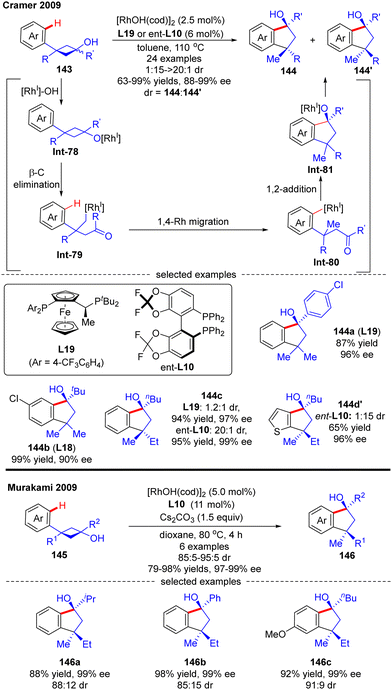 |
| | Scheme 41 Rh(I)-Catalyzed asymmetric synthesis of 1-indanols from 3-arylcyclobutanols. | |
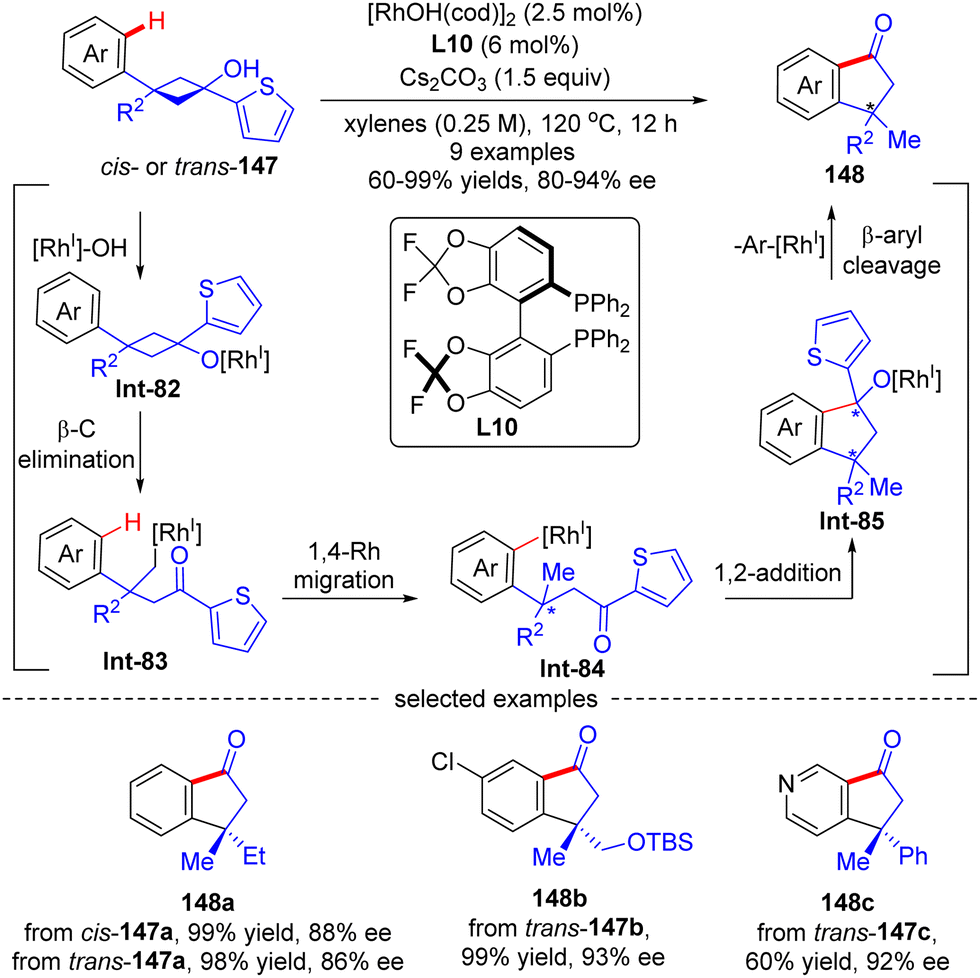
As an extension of the above-described transformation, in 2010, Cramer's group further achieved the asymmetric synthesis of indanones 148 bearing a quaternary stereogenic center from 1-arylcyclobutanols 147 (Scheme 42).121 Similar to the previous transformations, rhodium alkoxide Int-85 was also generated in this reaction through a four-step sequence comprising Rh-catalyzed deprotonation of 1-arylcyclobutanols 147, cyclobutane fragmentation via β-C elimination, alkyl-to-aryl 1,4-Rh migration, and intramolecular 1,2-addition. Next, by contrast to the direct protonolysis in previous transformations, herein Int-85 underwent a further β-aryl cleavage, leading to indanones 148 with a quaternary carbon center. The aryl substituent on prototype cyclobutanols 147 had a great impact on the reaction pathway of Int-85, and the 2-thienyl substituent performed the best to selectively furnish indanones 148via β-aryl cleavage instead of indanols via direct protonation. This reaction tolerated a broad range of functional groups, giving the corresponding indanones 148 in 60–99% yields and 80–94% ee. Notably, the geometry of substrates 147 revealed minimal effect on the stereo-control of the desired products (148a).
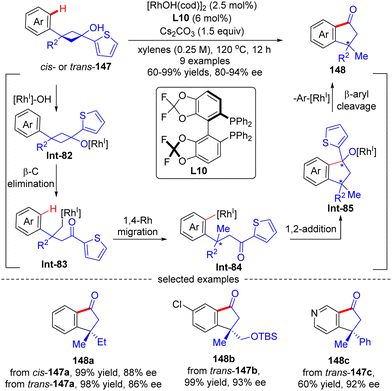 |
| | Scheme 42 Rh(I)-Catalyzed asymmetric synthesis of indanones from 1-(2-thienyl)cyclobutanols. | |
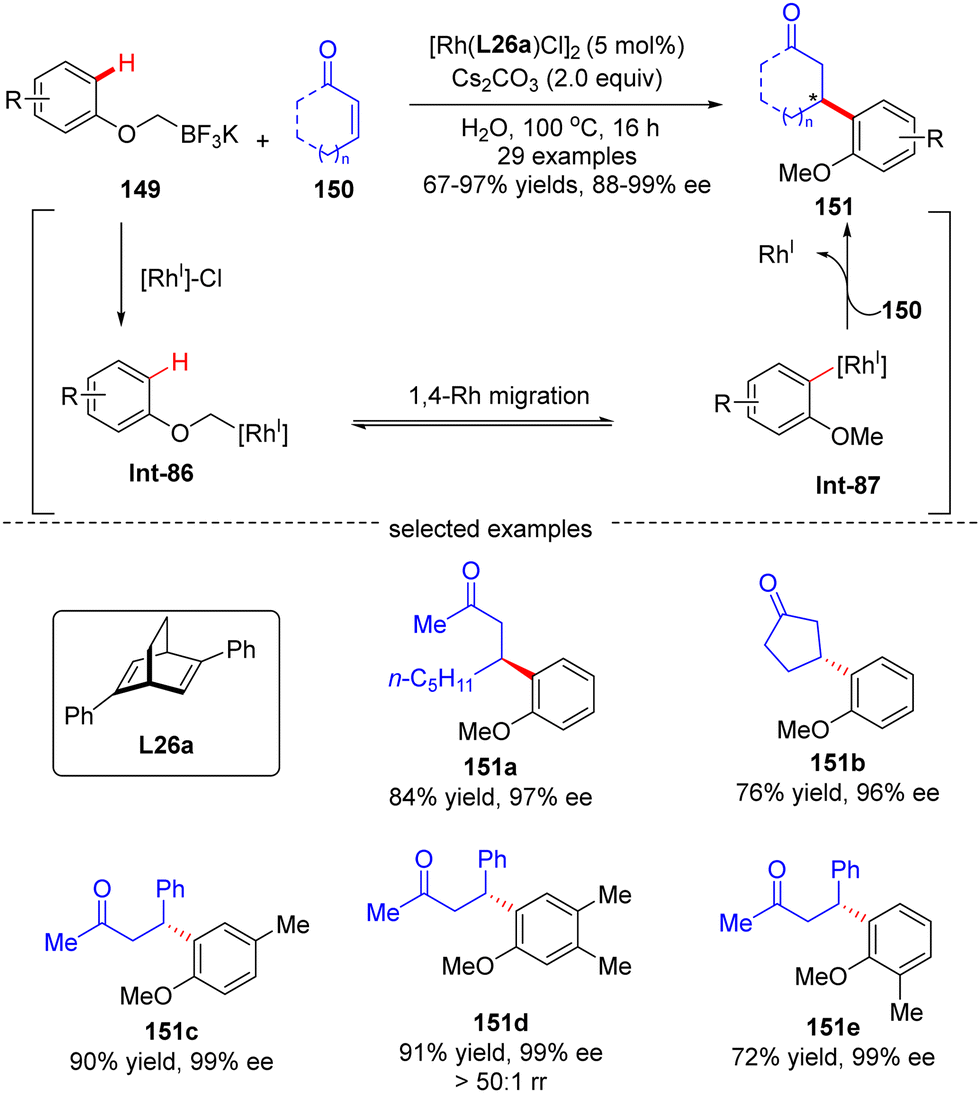
In 2016, Hayashi's group further disclosed a sequential 1,4-Rh migration/arylation reaction of enones 150 with potassium aryloxymethyltrifluoroborates 149 to yield β-(2-methoxylaryl)-substituted ketones 151 in high yields (67–97%) and excellent enantioselectivities (88–99% ee) via asymmetric Rh(I)/diene catalysis (Scheme 43).122 Both cyclic and linear enones displayed good compatibility. The mechanistic investigation suggested that this reaction involved an alkyl-to-aryl 1,4-Rh migration of aryloxymethyl-Rh intermediate Int-86, which forged 2-methoxyaryl-Rh species Int-87. This 1,4-Rh migration between C(sp3)–H and C(sp2)–H was reversible as revealed by the deuterium-labeling studies, and C–H bond cleavage was not the turnover-limiting step. Notably, the regioselectivity for 1,4-Rh migration was impacted by the steric factor, as the less hindered isomer (151d) was preferably formed.
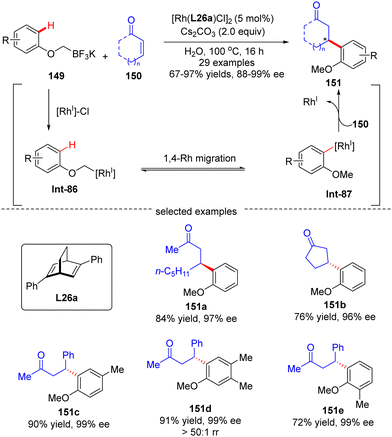 |
| | Scheme 43 Rh(I)-Catalyzed conjugate arylation of enones with potassium aryloxymethyltrifluoroborates. | |
4.1.3 C(sp2)–H functionalization via aryl-to-alkenyl 1,4-Rh migration.
Aside from the intensively studied alkenyl-to-aryl and alkyl-to-aryl 1,4-Rh migration process, a novel aryl-to-alkenyl 1,4-Rh migration process was also developed by Lin and co-workers to effect enantioselective C(sp2)–H functionalization. In 2019, they reported a Rh(I)/BINAP-catalyzed asymmetric alkenylation of enones 153 with 2-ethenyl-arylboronic acids 152, providing facile access to a wide range of β-alkenylated ketones 154 in high yields (65–99%) and excellent enantiomeric excess (70–99% ee) (Scheme 44).123 In addition to enones, cyclic aldimines 156 were also compatible with this strategy to prepare a set of chiral allylic sulfonamides 157 by employing (S)-difluorphos L10 as a chiral ligand. The key to the success of this reaction relied on a highly efficient aryl-to-alkenyl 1,4-Rh migration, which offered a new mode to prepare stereodefined alkenyl-rhodium species Int-90. DFT calculations revealed that the aryl-to-alkenyl 1,4-Rh migration was a kinetically favored process, comprising a C–H oxidative addition to form distorted Rh–H species Int-89 and follow-up reductive elimination to generate a stereodefined (E)-alkenylrhodium intermediate Int-90.
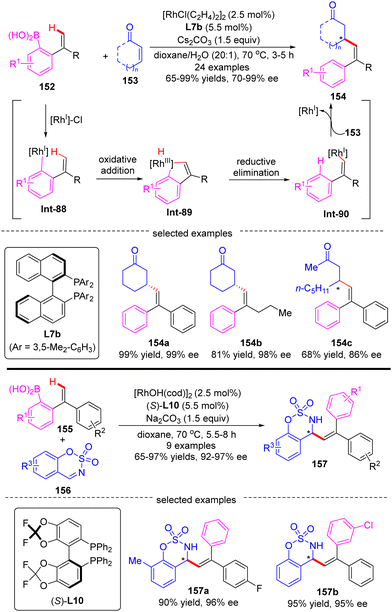 |
| | Scheme 44 Rh(I)-Catalyzed alkenylation of enones and cyclic imines with 2-ethenyl-arylboronic acids. | |
4.2 C(sp3)–H functionalization via 1,4-Rh migration
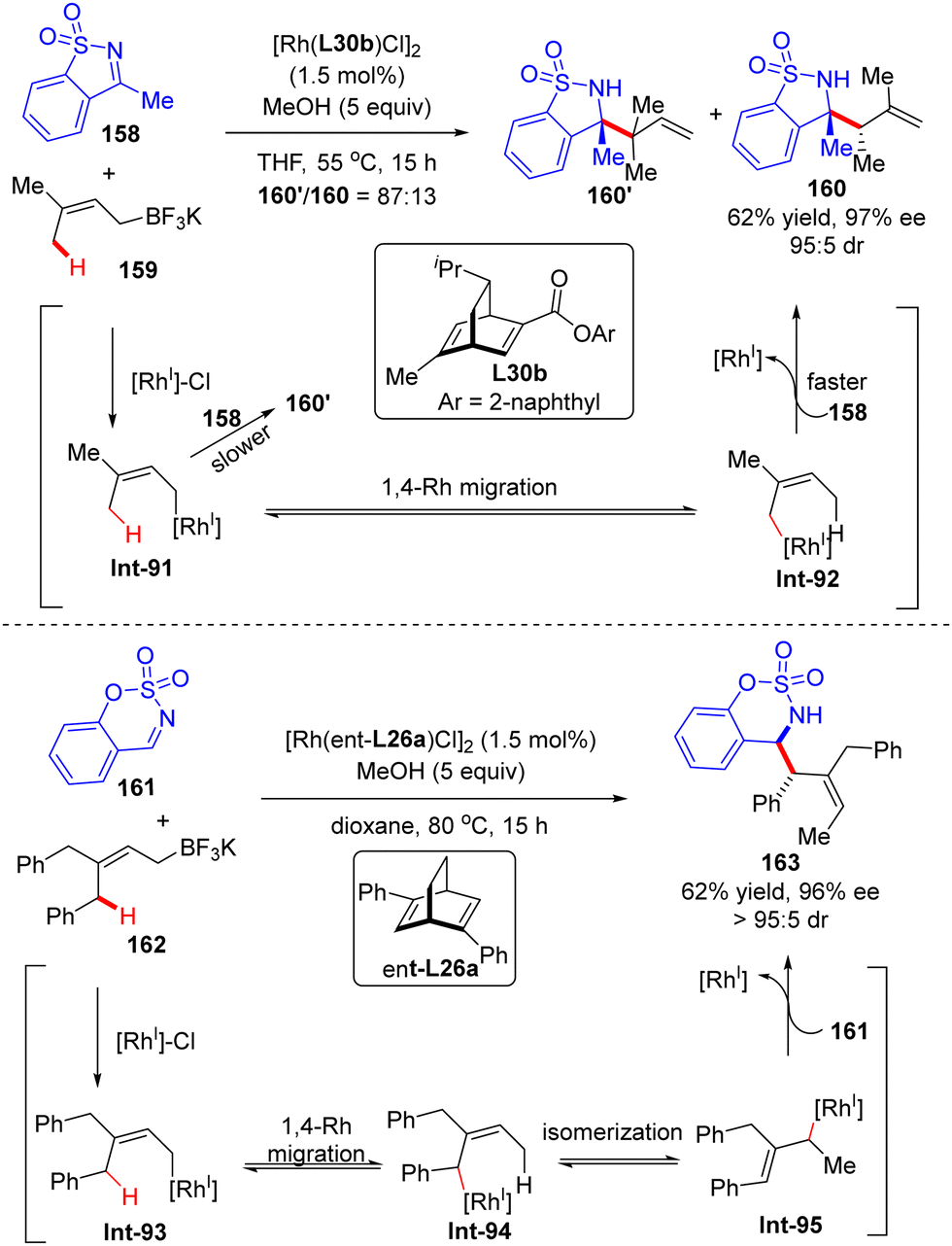
In 2014, Lam's group described a facile synthesis of chiral homoallylic sulfonamide 160 from 5-membered cyclic imine 158 and potassium prenyltrifluoroborate 159via a Rh(I)/diene asymmetric catalysis (Scheme 45).124 Allyl-Rh species Int-91 was first generated through transmetalation of potassium prenyltrifluoroborate 159 with [Rh]–Cl. With the less reactive saccharin-derived imine 158, it is disfavored to forge the reverse prenylation product 160′ with sterical congestion via direct addition of Int-91 to the imine substrate as per the previous report.125 Instead, an allyl-to-allyl 1,4-Rh migration occurred, furnishing another alkyl-Rh species Int-92. Finally, nucleophilic addition of Int-92 onto cyclic imine 158 proceeded to arrive at the desired homoallylic sulfonamide 160 as the major isomer in decent yield and excellent enantio-control. Notwithstanding, the minor isomer 160′ was also observed via the direct addition of Int-91 onto cyclic imine 158. It is worth mentioning that the 1,4-Rh migration was a reversible process and the steric hindrance of allyltrifluoroborates may promote the following isomerization of allyl-Rh species Int-94 to afford Int-95. Indeed, with the more sterically hindered allyltrifluoroborates 162 and chiral ligand ent-L26a, homoallylic sulfonamide 163 was produced as a single isomer via nucleophilic addition of Int-93 onto more reactive aldimine 161.
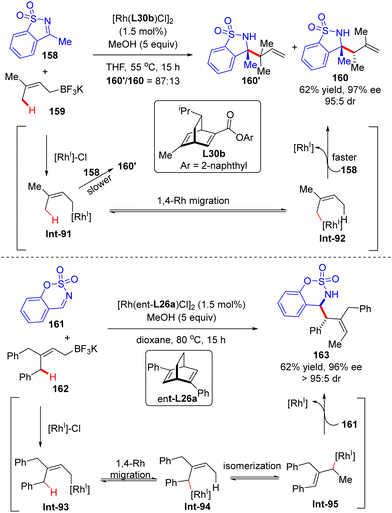 |
| | Scheme 45 Rh(I)-Catalyzed allylation of cyclic imines with potassium prenyltrifluoroborates. | |
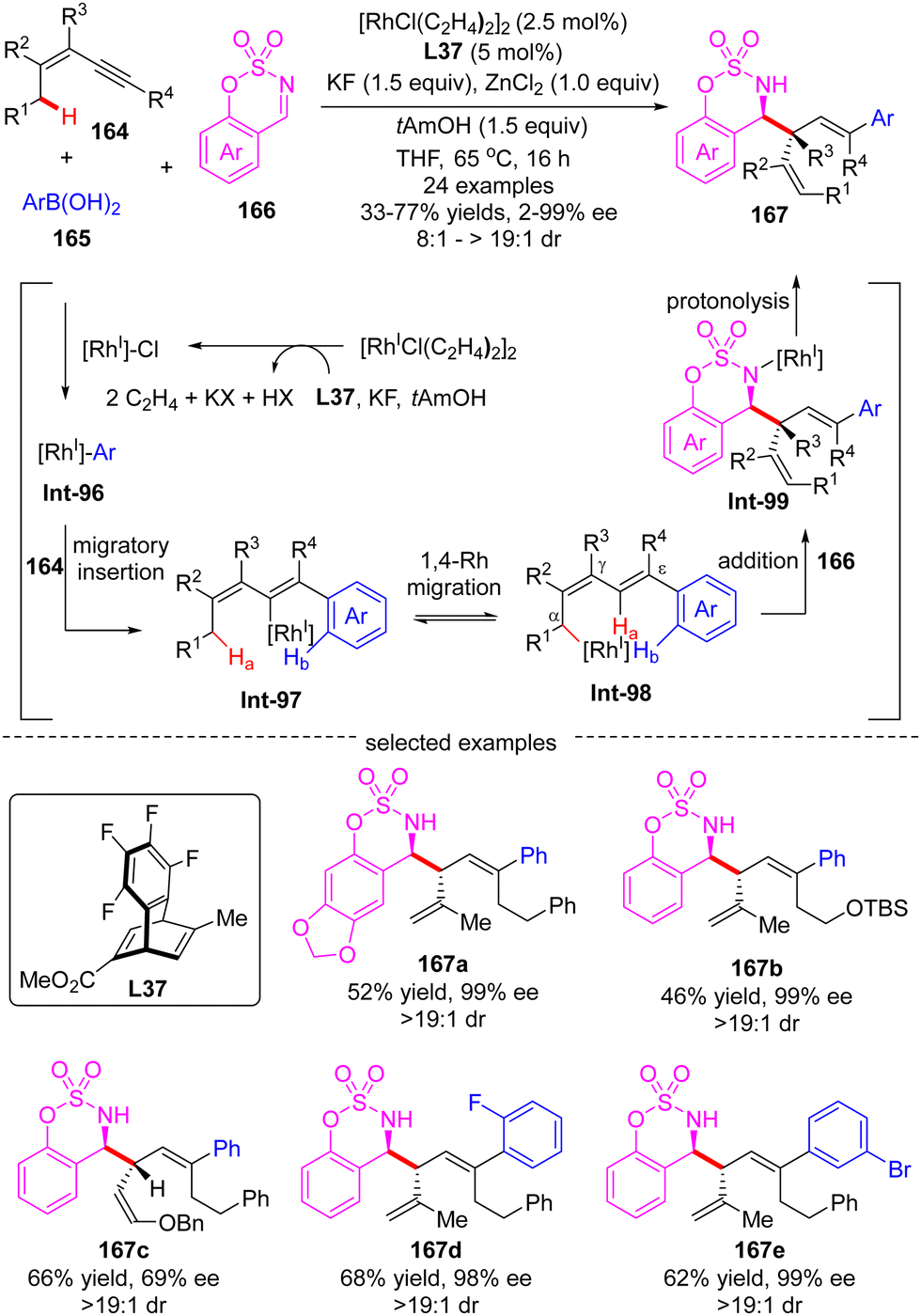
In 2017, Lam's group further reported an enantioselective Rh(I)-catalyzed three-component coupling of 1,3-enynes 164, arylboronic acids 165 and cyclic aldimines 166 (Scheme 46).126 The efficient catalytic system of [RhCl(C2H4)2]2/chiral tetrafluorobenzobarrelene L37 enabled a straightforward access to a diverse of branched homoallylic sulfamates 167 bearing tertiary or quaternary carbon stereogenic centers in decent yields (33–77%), high enantioselectivities (up to 99% ee) and excellent diastereoselectivities (in most cases >19:1). The underlying mechanism was proposed as exhibited in Scheme 46. First, transmetalation of arylboronic acid 165 with in situ generated [Rh]–Cl produced the arylrhodium species Int-96. Then, the migratory insertion of Int-96 across 1,3-enynes 164 occurred, furnishing alkenylrhodium species Int-97. Afterwards, Int-97 underwent a reverse alkenyl-to-allyl 1,4-Rh migration to produce Int-98, which was considered as the key step of this reaction. Subsequent intermolecular nucleophilic addition of Int-98 to cyclic imine 166 proceeded to form Int-99. The final protonolysis of Int-99 released the homoallylated product 167 with the regeneration of the rhodium complex.
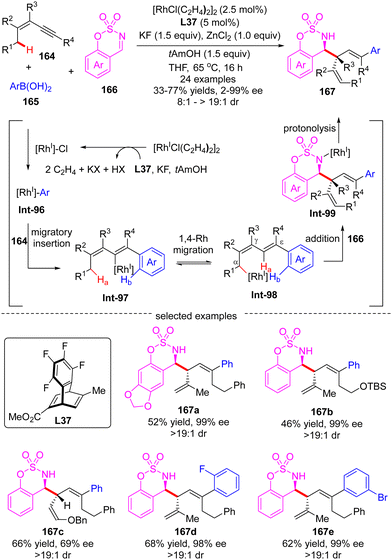 |
| | Scheme 46 Rh(I)-Catalyzed three-component allylation of cyclic imines with arylboronic acids and 1,3-enynes. | |
5. Conclusion and outlook
Among the various rhodium complexes, Rh(I) has mainly been utilized for the direct functionalization of C–H bonds via oxidation addition in both directed and undirected patterns. Although some related reactions could also be effected by other transition metals, Rh(I) catalysis provides the advantage of fine-tuning due to the commercial availability of a diversity of chiral ligands. As illustrated in this review, organic chemists have successfully used Rh(I) catalysis to enable a plethora of asymmetric C–H functionalization reactions including arylation, alkylation, and silylation, which serve as a versatile and efficient synthetic complement to Rh(II) and Rh(III) catalysis. Despite these impressive progresses, several major limitations still remain in the realm of Rh(I)-catalyzed C–H activation for asymmetric functionalization, such as the requisite of directing group for C–H functionalization, the confine of unactivated C(sp3)–H functionalization into an intramolecular fashion, the lack of applications in forging carbon–heteroatom bonds (e.g., C–O, C–N, C–S, C–X, and C–P) aside from C–Si bonds (partially owing to the competing coordination of heteroatoms with the chiral ligands), and the elusive 1,4-Rh migration to general C(sp3)–H bonds. Therefore, it is still of immense demand to devote more efforts to developing transient directing group strategy or non-directed Rh(I)-catalyzed asymmetric C–H functionalization, uncovering intermolecular asymmetric silylation and unactivated C(sp3)–H functionalization reactions, and designing novel chiral ligands to circumvent the catalyst poison issue by heteroatoms. More importantly, a deeper understanding of the mechanism in Rh(I)-catalyzed asymmetric C–H functionalization reactions is highly desirable to facilitate the rational design of new asymmetric reactions. Additionally, the synthetic implementation in the late-stage functionalization of natural products or other complex molecules also needs to be validated. As such, one can forecast that more novel Rh(I)-catalyzed asymmetric C–H functionalization reactions will be well-designed and applicable in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Basic Research Program of Jiangsu Province (BK20220408), the National Natural Science Foundation of China (22001260), Guangdong Basic and Applied Basic Research Foundation (2021A1515010016), and Shenzhen Science and Technology Innovation Committee (JCYJ20210324101810028, JCYJ20220818101601004, JCYJ20220818095801004).
References
-
E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1989 Search PubMed.
-
T. Hudlicky and J. W. Reed, The Way of Synthesis: Evolution of Design and Methods for Natural Products, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
-
E. J. Corey and L. Kürti, Enantioselective Chemical Synthesis: Methods, Logic, and Practice, ed. E. J. Corey and L. Kürti, Direct Book Publishing, Dallas, TX, 2010 Search PubMed.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- Y. Qiu and S. Gao, Nat. Prod. Rep., 2016, 33, 562–581 RSC.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC.
- D.-Y. Zhu, P. Chen and J.-B. Xia, ChemCatChem, 2016, 8, 68–73 CrossRef CAS.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed.
- Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC.
- J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC.
- R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720–743 CrossRef CAS.
- Ł. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef.
- T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS.
- C.-X. Liu, Q. Gu and S.-L. You, Trends Chem., 2020, 2, 737–749 CrossRef CAS.
- J. Thongpaen, R. Manguin and O. Baslé, Angew. Chem., Int. Ed., 2020, 59, 10242–10251 CrossRef CAS PubMed.
- Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. Madron du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed.
- K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 11, 12616–12632 RSC.
- X.-L. Liu, L.-B. Jiang, M.-P. Luo, Z. Ren and S.-G. Wang, Org. Chem. Front., 2022, 9, 265–280 RSC.
- C. Pan, S.-Y. Yin, Q. Gu and S.-L. You, Org. Biomol. Chem., 2021, 19, 7264–7275 RSC.
- J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2013, 19, 14010–14017 CrossRef CAS PubMed.
- T. Yoshino, S. Satake and S. Matsunaga, Chem. – Eur. J., 2020, 26, 7346–7357 CrossRef CAS PubMed.
- M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS.
- G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed.
- B.-F. S. Ya-Lan Feng, Chin. J. Org. Chem., 2021, 41, 3753–3770 CrossRef.
- Q. Zhang and B.-F. Shi, Acc. Chem. Res., 2021, 54, 2750–2763 CrossRef CAS PubMed.
- S. Motevalli, Y. Sokeirik and A. Ghanem, Eur. J. Org. Chem., 2016, 1459–1475 CrossRef CAS.
- W.-W. Chen and M.-H. Xu, Org. Biomol. Chem., 2017, 15, 1029–1050 RSC.
- X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799–2808 CrossRef CAS PubMed.
- E. A. Trifonova, N. M. Ankudinov, A. A. Mikhaylov, D. A. Chusov, Y. V. Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2018, 57, 7714–7718 CrossRef CAS PubMed.
-
Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed.
- S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS PubMed.
- B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed.
- J. Mas-Roselló, A. G. Herraiz, B. Audic, A. Laverny and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 13198–13224 CrossRef PubMed.
-
Y. Cao and K. G. M. Kou, Rhodium-Catalyzed Heterocycle Synthesis by C–H Functionalization, Handbook of C–H Functionalization, 2022, pp. 1–39 Search PubMed.
-
N. Chatani, Rhodium-Catalyzed C(sp3)–H Functionalization, Handbook of C–H Functionalization, 2022, pp. 1–34 Search PubMed.
-
V. Hanchate, M. S. Sherikar, A. Kumar and K. R. Prabhu, Rh-Catalyzed C–H Functionalization with Alkene, Handbook of C–H Functionalization, 2022, pp. 1–44 Search PubMed.
-
S. Mahato, W. Sarkar, K. Naskar and I. Deb, Rhodium-Catalyzed C–H Functionalization to Construct Annulated Molecules, Handbook of C–H Functionalization, 2022, pp. 1–39 Search PubMed.
-
A. Singh and C. M. R. Volla, Rh-Catalyzed C–H Functionalization with Allenes, Handbook of C–H Functionalization, 2022, pp. 1–29 Search PubMed.
- C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS PubMed.
- R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2001, 2437–2450 RSC.
- A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS.
-
C. G. Newton and N. Cramer, Chiral Cp Ligands for Rhodium(III)-Catalyzed Asymmetric Carbon–Hydrogen Bond Functionalization, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 629–644 Search PubMed.
-
F. Romanov-Michailidis, E. J. T. Phipps and T. Rovis, Sterically and Electronically Tuned Cp Ligands for Rhodium(III)-Catalyzed Carbon–Hydrogen Bond Functionalization, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 593–628 Search PubMed.
-
T. Satoh and M. Miura, Rhodium(III)-Catalyzed Annulative Carbon–Hydrogen Bond Functionalization, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 487–519 Search PubMed.
-
F. Xie and X. Li, Rhodium(III)-Catalyzed Non-annulative Carbon–Hydrogen Bond Functionalization, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 521–592 Search PubMed.
-
T. G. Driver, Rhodium(II)-Catalyzed Nitrogen-Atom Transfer for Oxidation of Aliphatic C–H Bonds, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 373–432 Search PubMed.
-
T. Miura and M. Murakami, Reactions of α-Imino Rhodium(II) Carbene Complexes Generated from N-Sulfonyl-1,2,3-Triazoles, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 449–470 Search PubMed.
-
S. M. Wilkerson-Hill and H. M. L. Davies, Rhodium(II) Tetracarboxylate-Catalyzed Enantioselective C–H Functionalization Reactions, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 341–372 Search PubMed.
-
Z. Chen and V. M. Dong, Rhodium(I)-Catalyzed Hydroformylation and Hydroamination, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 49–62 Search PubMed.
-
M. Fernández and M. C. Willis, Rhodium(I)-Catalyzed Hydroacylation, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH, Weinheim, Germany, 2019, pp. 63–84 Search PubMed.
- N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 425–426 CrossRef CAS.
- R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192–7193 CrossRef CAS PubMed.
- H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772–6779 CrossRef CAS PubMed.
- R. M. Wilson, R. K. Thalji, R. G. Bergman and J. A. Ellman, Org. Lett., 2006, 8, 1745–1747 CrossRef CAS PubMed.
- K. Tsuchikama, Y. Kuwata, Y.-K. Tahara, Y. Yoshinami and T. Shibata, Org. Lett., 2007, 9, 3097–3099 CrossRef CAS PubMed.
- A. S. Tsai, R. M. Wilson, H. Harada, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3910–3912, 10.1039/B902878A.
- S. H. Wiedemann, J. C. Lewis, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2006, 128, 2452–2462 CrossRef CAS PubMed.
- Z.-M. Sun, S.-P. Chen and P. Zhao, Chem. – Eur. J., 2010, 16, 2619–2627 CrossRef CAS PubMed.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098–11102 CrossRef CAS PubMed.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181–8184 CrossRef CAS PubMed.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630–10634 CrossRef CAS PubMed.
- M. Tsutsui, M. Hancock, J. Ariyoshi and M. N. Levy, Angew. Chem., Int. Ed. Engl., 1969, 8, 410–420 CrossRef CAS.
- C. M. Filloux and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508–517 CrossRef CAS PubMed.
- F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647–2651 CrossRef CAS.
- Q. Wang, Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2019, 141, 9504–9510 CrossRef CAS PubMed.
- Z.-J. Cai, C.-X. Liu, Q. Wang, Q. Gu and S.-L. You, Nat. Commun., 2019, 10, 4168 CrossRef PubMed.
- C.-X. Liu, Z.-J. Cai, Q. Wang, Z.-J. Wu, Q. Gu and S.-L. You, CCS Chem., 2020, 2, 642–651 CrossRef CAS.
- C.-X. Liu, F. Zhao, Z. Feng, Q. Wang, Q. Gu and S.-L. You, Nat. Syn., 2023, 2, 49–57 CrossRef.
- C.-X. Liu, P.-P. Xie, F. Zhao, Q. Wang, Z. Feng, H. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2023, 145, 4765–4773 CrossRef CAS PubMed.
- C.-X. Liu, F. Zhao, Q. Gu and S.-L. You, ACS Cent. Sci., 2023, 9, 2036–2043 CrossRef CAS PubMed.
- Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520–1522 CrossRef CAS PubMed.
- Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2016, 55, 1948 CrossRef CAS PubMed.
- M. Murai, Y. Takeuchi, K. Yamauchi, Y. Kuninobu and K. Takai, Chem. – Eur. J., 2016, 22, 6048–6058 CrossRef CAS PubMed.
- Q.-W. Zhang, K. An, L.-C. Liu, Y. Yue and W. He, Angew. Chem., Int. Ed., 2015, 54, 6918–6921 CrossRef CAS PubMed.
- M. Murai, K. Matsumoto, Y. Takeuchi and K. Takai, Org. Lett., 2015, 17, 3102–3105 CrossRef CAS PubMed.
- M. Murai, K. Matsumoto, Y. Takeuchi and K. Takai, Org. Lett., 2016, 18, 3041 CrossRef CAS PubMed.
- T. Shibata, T. Shizuno and T. Sasaki, Chem. Commun., 2015, 51, 7802–7804 RSC.
- D. Mu, W. Yuan, S. Chen, N. Wang, B. Yang, L. You, B. Zu, P. Yu and C. He, J. Am. Chem. Soc., 2020, 142, 13459–13468 CrossRef CAS PubMed.
- W. Ma, L.-C. Liu, K. An, T. He and W. He, Angew. Chem., Int. Ed., 2021, 60, 4245–4251 CrossRef CAS PubMed.
- W. Yuan, L. You, W. Lin, J. Ke, Y. Li and C. He, Org. Lett., 2021, 23, 1367–1372 CrossRef CAS PubMed.
- S. Chen, D. Mu, P.-L. Mai, J. Ke, Y. Li and C. He, Nat. Commun., 2021, 12, 1249 CrossRef CAS PubMed.
- Q.-W. Zhang, K. An, L.-C. Liu, Q. Zhang, H. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125–1129 CrossRef CAS PubMed.
- K. An, W. Ma, L.-C. Liu, T. He, G. Guan, Q.-W. Zhang and W. He, Nat. Commun., 2022, 13, 847 CrossRef CAS PubMed.
- S. Chen, J. Zhu, J. Ke, Y. Li and C. He, Angew. Chem., Int. Ed., 2022, 61, e202117820 CrossRef CAS PubMed.
- Q. Li and Z.-X. Yu, Angew. Chem., Int. Ed., 2011, 50, 2144–2147 CrossRef CAS PubMed.
- Q. Li and Z.-X. Yu, Organometallics, 2012, 31, 5185–5195 CrossRef CAS.
- J. H. Kim, S. Greßies, M. Boultadakis-Arapinis, C. Daniliuc and F. Glorius, ACS Catal., 2016, 6, 7652–7656 CrossRef CAS.
- S. Gressies, F. J. R. Klauck, J. H. Kim, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 9950–9954 CrossRef CAS PubMed.
- M. Murai, H. Takeshima, H. Morita, Y. Kuninobu and K. Takai, J. Org. Chem., 2015, 80, 5407–5414 CrossRef CAS PubMed.
- T. Lee and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 8723–8727 CrossRef CAS PubMed.
- B. Yang, W. Yang, Y. Guo, L. You and C. He, Angew. Chem., Int. Ed., 2020, 59, 22217–22222 CrossRef CAS PubMed.
- Y. Guo, M.-M. Liu, X. Zhu, L. Zhu and C. He, Angew. Chem., Int. Ed., 2021, 60, 13887–13891 CrossRef CAS PubMed.
- R.-T. Guo, Y.-L. Zhang, J.-J. Tian, K.-Y. Zhu and X.-C. Wang, Org. Lett., 2020, 22, 908–913 CrossRef CAS PubMed.
- D.-X. Zhu, H. Xia, J.-G. Liu, L. W. Chung and M.-H. Xu, J. Am. Chem. Soc., 2021, 143, 2608–2619 CrossRef CAS PubMed.
- D.-X. Zhu, J.-G. Liu and M.-H. Xu, J. Am. Chem. Soc., 2021, 143, 8583–8589 CrossRef CAS PubMed.
- B. Liu and M.-H. Xu, Chin. J. Chem., 2021, 39, 1911–1915 CrossRef CAS.
- T.-Y. Wang, X.-X. Chen, D.-X. Zhu, L. W. Chung and M.-H. Xu, Angew. Chem., Int. Ed., 2022, 61, e202207008 CrossRef CAS PubMed.
- D.-X. Zhu and M.-H. Xu, J. Org. Chem., 2023, 88, 7844–7848 CrossRef CAS PubMed.
- R. Shintani, K. Yashio, T. Nakamura, K. Okamoto, T. Shimada and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 2772–2773 CrossRef CAS PubMed.
- R. Shintani, K. Takatsu and T. Hayashi, Angew. Chem., Int. Ed., 2007, 46, 3735–3737 CrossRef CAS PubMed.
- R. Shintani, S. Isobe, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3795–3798 CrossRef CAS PubMed.
- T. Matsuda and S. Watanuki, Org. Biomol. Chem., 2015, 13, 702–705 RSC.
- K. Sasaki, T. Nishimura, R. Shintani, E. A. B. Kantchev and T. Hayashi, Chem. Sci., 2012, 3, 1278–1283 RSC.
- A. Selmani and S. Darses, Org. Lett., 2019, 21, 8122–8126 CrossRef CAS PubMed.
- L. O'Brien, S. N. Karad, W. Lewis and H. W. Lam, Chem. Commun., 2019, 55, 11366–11369 RSC.
- A. Groves, J. Sun, H. R. I. Parke, M. Callingham, S. P. Argent, L. J. Taylor and H. W. Lam, Chem. Sci., 2020, 11, 2759–2764 RSC.
- A. Selmani and S. Darses, Org. Lett., 2020, 22, 2681–2686 CrossRef CAS PubMed.
- A. Claraz, F. Serpier and S. Darses, ACS Catal., 2017, 7, 3410–3413 CrossRef CAS.
- A. Selmani, F. Serpier and S. Darses, J. Org. Chem., 2019, 84, 4566–4574 CrossRef CAS PubMed.
- T. Johnson, K.-L. Choo and M. Lautens, Chem. – Eur. J., 2014, 20, 14194–14197 CrossRef CAS PubMed.
- T. Matsuda, M. Shigeno and M. Murakami, J. Am. Chem. Soc., 2007, 129, 12086–12087 CrossRef CAS PubMed.
- T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320–6323 CrossRef CAS PubMed.
- M. Shigeno, T. Yamamoto and M. Murakami, Chem. – Eur. J., 2009, 15, 12929–12931 CrossRef CAS PubMed.
- H. Yu, C. Wang, Y. Yang and Z.-M. Dang, Chem. – Eur. J., 2014, 20, 3839–3848 CrossRef CAS PubMed.
- T. Seiser, G. Cathomen and N. Cramer, Synlett, 2010, 1699–1703 CAS.
- J. Ming and T. Hayashi, Org. Lett., 2016, 18, 6452–6455 CrossRef CAS PubMed.
- S.-S. Zhang, T.-J. Hu, M.-Y. Li, Y.-K. Song, X.-D. Yang, C.-G. Feng and G.-Q. Lin, Angew. Chem., Int. Ed., 2019, 58, 3387–3391 CrossRef CAS PubMed.
- H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605–11610 CrossRef CAS PubMed.
- Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309–8313 CrossRef CAS PubMed.
- M. Callingham, B. M. Partridge, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 16352–16356 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Jing-Jing
Zhang†
ab,
Jing-Jing
Zhang†