
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic micelles as nanocarriers for enhancing intra-cartilage drug penetration and retention†
Chenxian
Zhu
 a,
Zhongxing
Zhang
*a,
Yuting
Wen
ab,
Xia
Song
a,
Jingling
Zhu
ac,
Yifei
Yao
*d and
Jun
Li
*abc
a,
Zhongxing
Zhang
*a,
Yuting
Wen
ab,
Xia
Song
a,
Jingling
Zhu
ac,
Yifei
Yao
*d and
Jun
Li
*abc
aDepartment of Biomedical Engineering, National University of Singapore, 15 Kent Ridge Crescent, Singapore 119276, Singapore. E-mail: biezhozh@nus.edu.sg; jun-li@nus.edu.sg
bNational University of Singapore (Chongqing) Research Institute, 2 Huizhu Road, Yubei District, Chongqing 401120, China
cNUS Environmental Research Institute (NERI), National University of Singapore, 5A Engineering Drive 1, Singapore 117411, Singapore
dSchool of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai 200030, China. E-mail: yifeiyao@sjtu.edu.cn
First published on 14th December 2022
Abstract
There is a tremendous unmet medical need for osteoarthritis (OA) treatment around the world, and pharmacological management is the most common option but presents a limited and short efficacy. Insufficient drug delivery to articular cartilage is the key cause. It is widely accepted that the complex structure of articular cartilage and the rapid clearance of joint liquids largely hinder drug penetration and retention in the cartilage. To address these obstacles, we designed and prepared a positively charged micellar system that can effectively deliver a model drug to the deep zone of the cartilage and prolong the drug retention time. In this work, a triblock copolymer composed of cationic poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) and poly(ε-caprolactone) (PCL), denoted as PDMAEMA–PCL–PDMAEMA, was synthesized. A triblock copolymer composed of brush poly[poly(ethylene glycol) methacrylate] (pPEGMA) and PCL, denoted as pPEGMA–PCL–pPEGMA, was prepared for comparison. The two types of triblock copolymers were self-assembled in an aqueous environment to form cationic and neutral micelles, respectively. A hydrophobic fluorescent dye as a model drug was loaded into micelle cores, and the dye-loaded micelles were evaluated for intra-cartilage penetration and retention using porcine knee cartilage explants. The PDMAEMA–PCL–PDMAEMA cationic micelles were found to significantly enhance the intra-cartilage penetration and retention capability due to the electrostatic interaction between the micelles and the negatively charged cartilage extracellular matrix. The confocal microscopy study showed that the cationic micelles could penetrate the full-thickness porcine cartilage explants (around 1.5 mm) within 24 hours. Up to 87% of the cationic micelles were taken up by porcine cartilage explants, and 71% of the absorbed micelles were retained in the tissue for at least 4 days. Although the pPEGMA–PCL–pPEGMA neutral micelles were able to penetrate the full-thickness cartilage, this type of micelle showed lower uptake (44%) and retention (44%) rates. This observation implied that the surface charge of micelles could play an important role in efficient intra-cartilage drug delivery. This study verified the feasibility and effectiveness of the PDMAEMA–PCL–PDMAEM cationic micelles in intra-cartilage drug delivery, showing that cationic micelles could be promising carriers for OA treatment.
1. Introduction
Osteoarthritis (OA) is a common joint disease, and 303 million people globally suffered from OA in 2017.1 The key changed features associated with OA are irreversible cartilage damage or loss.2 OA is accompanied by severe pain, impairment, reduced life quality, and even disability.2 Despite such massive medical needs, unfortunately, there is no FDA-approved disease modifying OA drugs (DMOADs) available on the market,3 and the main therapeutic principles are symptomatic pain relief and anti-inflammatory treatment.The drug target site of OA therapy is mainly articular cartilage, which is an avascular, alymphatic, and aneural tissue.4 Thus, systemic administration is less effective for OA treatment. Intra-articular (IA) injection as the local administration is increasing as a promising alternative, which increases drug bioavailability and reduces systemic side effects.5 Chondrocytes, as the only cell type in cartilage that secretes pro-inflammatory cytokines in the OA progression, mainly locate within the middle and deep zones of cartilage.4 Thus, it is necessary to deliver drugs to the middle and deep zones of cartilage. Although some IA therapeutics have been approved for the clinical treatment of OA,6 such as corticosteroids, they show limited efficacy and offer only short-term relief of pain and inflammation, which is rooted in inadequate drug delivery.5 Poor drug intra-cartilage penetration and retention lead to insufficient therapeutic concentrations and duration. There are two main reasons for the obstacles. First, the highly complex structure of cartilage should be responsible for the low drug penetration. The densely organized extracellular matrix (ECM) structure, with a pore size of about 60–200 nm, sterically hinders the penetration of large-sized solutes.4 Besides, the cartilage contains highly negatively charged glycosaminoglycan (GAG) chains, which impair or prevent the penetration of negatively charged solutes. Second, rapid clearance in the joint is another huge challenge. Both large and small solutes in the joint space are quickly cleared by the lymphatics and small blood vessels, respectively.5 Previous reports indicated that the IA half-life for the commonly used drugs is 1–5 hours.7 Furthermore, some drugs may transport with trans-synovial flow into synovial lymph vessels, finally into the systemic circulation via superior vena cava, which may cause off-target side effects.8
The positively charged nanocarrier is a promising strategy to overcome the obstacles of intra-cartilage drug delivery and has attracted enormous attention in recent years. Based on the fact that cartilage contains abundant negatively charged GAGs and its density increases with cartilage depth, cationic carriers with an appropriate size and charge can effectively penetrate the full-thickness cartilage and remained inside due to reversible electrostatic interactions between the cationic carriers and the negatively charged ECM.4 Several cationic nanocarriers have been explored for OA treatment, for example, avidin-based carriers,9,10 chitosan-based carriers,11 PEGylated PAMAM dendrimers,12 cationic peptide carriers,13 and positively charged liposomes.14 Although they exhibit good intra-cartilage penetration and retention capabilities, in most systems, therapeutic agents were conjugated to cationic materials. Such drug modifications may cause altered drug efficacy and drug release problems.14 Among the drug carriers, polymeric micelles have emerged as attractive nanocarriers for drug delivery due to their advantages, such as nanosize, easy cell internalization, simple functionalization, increased bioavailability of hydrophobic drugs, physical encapsulation without drug modification, and flexibility.15–20 To our knowledge, there are few studies that investigate the feasibility and effectiveness of cationic micelles in intra-cartilage drug delivery.
Herein, we propose to apply cationic micelles formed with a triblock copolymer composed of poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) and poly(ε-caprolactone) (PCL), i.e., the PDMAEMA–PCL–PDMAEMA triblock copolymer, for intra-cartilage drug delivery. Among cationic polymers, PDMAEMA presents relatively low toxicity thus gaining numerous attention for drug delivery and gene delivery,21–25 and its toxicity could be further reduced by combining with biodegradable PCL.26,27 The triblock copolymers can self-assemble into micelles with a hydrophobic core and a hydrophilic shell in an aqueous environment, while hydrophobic molecules can be loaded into the hydrophobic core because of the hydrophobic interactions; thus, the micelles provide physical encapsulation for hydrophobic drugs. This cationic micelle delivery system is expected to deeply penetrate cartilage under the guidance of the inwardly pointing electric field,4 and then retain in the tissue due to electrostatic binding between the micelles and the negatively charged ECM, which can help in the effective retention of the encapsulated drug (Fig. 1).
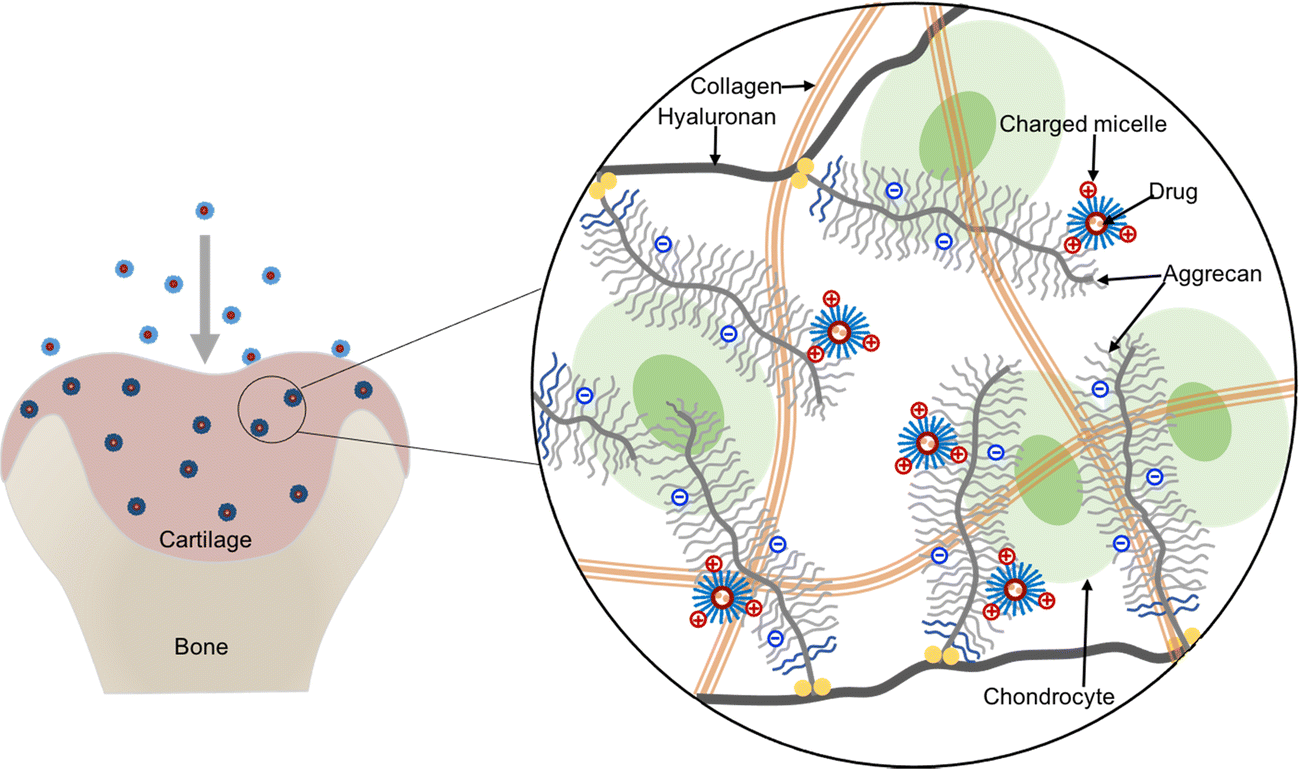 | ||
| Fig. 1 Illustration of electrostatic interactions between the cationic micelles and the negatively charged cartilage aggrecan. Micelles with an appropriate positive charge can penetrate through the full-thickness cartilage under the charge guidance and retained inside the tissue. | ||
In this study, we aimed at developing a suitable polymeric micellar system for delivering a model drug into the deep zone of cartilage and retaining the delivered model drug within the cartilage over long periods, for potentially sustained and efficient treatment of OA. We synthesized PDMAEMA–PCL–PDMAEMA triblock copolymers and characterized their formation of cationic micelles. A hydrophobic fluorescent dye as the model drug was loaded into the cationic micelles, which were evaluated for intra-cartilage penetration and retention using porcine cartilage explants. Besides, a neutral micellar system formed with the triblock copolymer composed of brush poly[poly(ethylene glycol) methacrylate] (pPEGMA) and PCL, denoted as pPEGMA–PCL–pPEGMA, was prepared for comparison. The results showed that the cationic micelles as nanocarriers could help overcome the intra-cartilage barriers to promote the delivery of drugs deep into the cartilage.
2. Materials and methods
2.1 Materials
Poly(ε-caprolactone) diol (PCL-diol, 2000 Da), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%), poly(ethylene glycol) methacrylate (PEGMA, 360 Da), 2-bromoisobutyryl bromide (BIBB, 98%), branched polyethylenimine (bPEI, 25 kDa), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 99%), copper(I) bromide (CuBr, 99%), triethylamine (TEA, >99%), 1,4-dioxane (>99%), and difluoro(2-(1-(3,5-dimethyl-2H-pyrrol-2-ylidene-N)ethyl)-3,5-dimethyl-1H-pyrrolato-N)boron (BODIPY) were purchased from Sigma-Aldrich. Cell counting kit-8 (CCK-8) was obtained from Dojindo Laboratories. Hoechst was purchased from Life Technologies.2.2 Polymer synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DMAEMA]:[CuBr]:[HMTETA] = 1:20:2:4; 1:40:2:4; and 1:60:2:4. The reaction with the first feed ratio is described below as an example. First, 1.6 mmol of Br-PCL-Br macroinitiators were introduced into the 100 mL Schlenk tube containing 20 mL of 1,4-dioxane. After the macroinitiator dissolved completely with stirring, DMAEMA monomers (32 mmol, 5.40 mL) and HMTETA (6.4 mmol, 1.74 mL) were added into the Schlenk tube, and then the reaction mixture was degassed by bubbling nitrogen through the reaction mixture for 30 min. After degassing, CuBr was mixed with 2 mL of 1,4-dioxane and was then transferred into the Schlenk tube under a nitrogen atmosphere, followed by further degassing for 10 min. Then, the Schlenk tube was placed in the oil bath to allow the polymerization reaction to proceed at 45 °C with continuous stirring for 16 h. The reaction was terminated by diluting with 45 mL of tetrahydrofuran (THF) and continued stirring at room temperature for 1 h. After this, the catalyst complex was removed by passing the dilute reaction mixture through a short aluminum oxide column. The colorless solution was collected and further concentrated by rotary evaporation, and then precipitated in hexanes to remove the unreacted DMAEMA monomers three times. The final product was collected by redissolving using a small amount of THF and then dried at 45 °C under vacuum. 1H NMR (600 MHz, CDCl3): δ 4.20–4.00 (m, –OCH2CH2CH2CH2CH2CO– and –OCH2CH2N(CH3)2), 2.58 (b, –OCH2CH2N(CH3)2), 2.30 (m, –OCH2CH2CH2CH2CH2CO– and –N(CH3)2), 1.95–1.76 (b, –CH2C(CH3)(COO–)–), 1.73–1.56 (m, –OCH2CH2CH2CH2CH2CO–), 1.38 (m, –OCH2CH2CH2CH2CH2CO–), 1.10–0.70 (b, –CH2C(CH3)(COO–)–).
:[PEGMA]:[CuBr]:[HMTETA] = 1:20:2:4. The synthesis procedures were similar to those of PDMAEMA–PCL–PDMAEMA triblock copolymers, but the reaction time was longer (24 h). After precipitation with hexanes, the precipitate was redissolved in acetone and transferred to a dialysis bag (MWCO 1 kDa) against deionized (DI) water for 3 days. 1H NMR (600 MHz, CDCl3): δ 4.31–3.93 (b, –OCH2CH2CH2CH2CH2CO– and –COOCH2CH2O–), 3.85–3.45 (m, –OCH2CH2O–), 2.30 (t, –OCH2CH2CH2CH2CH2CO–), 2.10–1.71 (b, –CH2C(CH3)(COO–)–), 1.73–1.56 (m, –OCH2CH2CH2CH2CH2CO–), 1.38 (m, –OCH2CH2CH2CH2CH2CO–), 1.09–0.70 (b, –CH2C(CH3)(COO–)–).
:[DMAEMA]:[CuBr]:[HMTETA] = 1:20:2:4; 1:40:2:4; and 1:60:2:4. The reaction with the first feed ratio is described below as an example. First, 1.6 mmol of Br-PCL-Br macroinitiators were introduced into the 100 mL Schlenk tube containing 20 mL of 1,4-dioxane. After the macroinitiator dissolved completely with stirring, DMAEMA monomers (32 mmol, 5.40 mL) and HMTETA (6.4 mmol, 1.74 mL) were added into the Schlenk tube, and then the reaction mixture was degassed by bubbling nitrogen through the reaction mixture for 30 min. After degassing, CuBr was mixed with 2 mL of 1,4-dioxane and was then transferred into the Schlenk tube under a nitrogen atmosphere, followed by further degassing for 10 min. Then, the Schlenk tube was placed in the oil bath to allow the polymerization reaction to proceed at 45 °C with continuous stirring for 16 h. The reaction was terminated by diluting with 45 mL of tetrahydrofuran (THF) and continued stirring at room temperature for 1 h. After this, the catalyst complex was removed by passing the dilute reaction mixture through a short aluminum oxide column. The colorless solution was collected and further concentrated by rotary evaporation, and then precipitated in hexanes to remove the unreacted DMAEMA monomers three times. The final product was collected by redissolving using a small amount of THF and then dried at 45 °C under vacuum. 1H NMR (600 MHz, CDCl3): δ 4.20–4.00 (m, –OCH2CH2CH2CH2CH2CO– and –OCH2CH2N(CH3)2), 2.58 (b, –OCH2CH2N(CH3)2), 2.30 (m, –OCH2CH2CH2CH2CH2CO– and –N(CH3)2), 1.95–1.76 (b, –CH2C(CH3)(COO–)–), 1.73–1.56 (m, –OCH2CH2CH2CH2CH2CO–), 1.38 (m, –OCH2CH2CH2CH2CH2CO–), 1.10–0.70 (b, –CH2C(CH3)(COO–)–).
:[PEGMA]:[CuBr]:[HMTETA] = 1:20:2:4. The synthesis procedures were similar to those of PDMAEMA–PCL–PDMAEMA triblock copolymers, but the reaction time was longer (24 h). After precipitation with hexanes, the precipitate was redissolved in acetone and transferred to a dialysis bag (MWCO 1 kDa) against deionized (DI) water for 3 days. 1H NMR (600 MHz, CDCl3): δ 4.31–3.93 (b, –OCH2CH2CH2CH2CH2CO– and –COOCH2CH2O–), 3.85–3.45 (m, –OCH2CH2O–), 2.30 (t, –OCH2CH2CH2CH2CH2CO–), 2.10–1.71 (b, –CH2C(CH3)(COO–)–), 1.73–1.56 (m, –OCH2CH2CH2CH2CH2CO–), 1.38 (m, –OCH2CH2CH2CH2CH2CO–), 1.09–0.70 (b, –CH2C(CH3)(COO–)–).
2.3 Molecular characterization
The chemical structures of PDMAEMA–PCL–PDMAEMA and pPEGMA–PCL–pPEGMA triblock copolymers were confirmed using the proton nuclear magnetic resonance (1H NMR) spectra and Fourier transform infrared spectroscopy (FTIR) spectra. The 1H NMR spectra were recorded using a Varian 600 MHz NMR spectrometer at room temperature. The chemical shift was referenced to a residual solvent peak (δ = 7.26 ppm for CHCl3). The FTIR spectra were recorded using a SHIMADZU IRPrestige21 spectrometer in the range of 4000–400 cm−1, at a resolution of 4 cm−1.2.4 Preparation of empty micelles and dye loaded micelles
Micelles were prepared by a simple nanoprecipitation method. Briefly, 10 mg of the triblock copolymer (PDMAEMA–PCL–PDMAEMA or pPEGMA–PCL–pPEGMA copolymers) was dissolved in 1 mL of acetone. The mixture was added dropwise into 10 mL of phosphate buffered saline (PBS) solution (1× or 0.1× PBS, pH 7.4) under intense stirring. The dispersion was maintained under stirring in an open vial for 20 h at room temperature to allow acetone evaporation. After stirring, a certain volume of DI water was added to the vial to resume the volume of PBS. Then the resulting dispersion was filtered through a 0.22 μm filter to yield the final micelle solution with a copolymer concentration of 1 mg mL−1.A fluorescent dye, BODIPY (Mn = 262.1 g mol−1), was selected as the model drug due to its neutral charge for further evaluation. The BODIPY-loaded micelles were produced by the nanoprecipitation method. A known amount (10 mg) of the triblock copolymer (PDMAEMA–PCL–PDMAEMA or pPEGMA–PCL–pPEGMA) and the BODIPY dye (0.03 mg and 0.65 mg, respectively) were co-dissolved in 1 mL of acetone, and the mixture was slowly dropped into 10 mL of 1× PBS. The following procedures were the same as those for the empty micelle preparation. The final concentration of the copolymer was 1 mg mL−1.
2.5 Characterization of micelles
The sizes and the zeta potentials of the self-assembled micelles were determined by dynamic light scattering (DLS, Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). The size measurement of micelles in 1× PBS (pH 7.4) was performed at 25 °C in triplicate and the Z-average hydrodynamic diameters of the particles were obtained. The zeta potential measurement of micelles was performed in 0.1× PBS (pH 7.4) at 25 °C to avoid testing cuvette damage caused by high salt concentration. The zeta potential measurements were carried out in triplicate. The zeta potential of micelles was also measured in 0.1× PBS with different pH values (5, 7, and 9) at 25 °C. Furthermore, the particle sizes and size distributions of the empty and dye-loaded micelles were observed by transmission electron microscopy (TEM). TEM samples were prepared by depositing micelle solution (in 1× PBS) on a 200-mesh carbon-coated copper grid. After samples were dried overnight, the dried samples were stained with 2 wt% of phosphotungstic acid.The critical micelle concentration (CMC) was measured using pyrene as a fluorescent probe.29 The concentration of triblock copolymers varied from 1 × 10−5 to 0.5 mg mL−1 and the concentration of pyrene was set at 0.6 μM.26 The fluorescence spectra were recorded using a Cary Eclipse Fluorescence Spectrometer with an emission wavelength of 390 nm. The excitation fluorescence at 337 and that at 344 nm were recorded at 25 °C. The CMC was estimated by plotting the intensity ratio I337/I344versus the logarithmic concentration.
The concentration of the loaded BODIPY dye in micelles was determined using a microplate reader (Infinite M200 PRO, TECAN), with an excitation wavelength at 493 nm and emission at 530 nm. For each quantification, a standard curve was established for the emission intensity of BODIPY at 530 nm versus the freshly prepared BODIPY concentration in a mixture of DI water and acetone (20/80, v/v). In penetration experiments, 200 μL of collected baths were mixed with 800 μL of acetone followed by sonication and centrifugation. The fluorescence signal in the supernatant was determined using a microplate reader, and then the dye concentration was calculated from the established standard curve.
2.6 Cytotoxicity
The cytotoxicity of empty micelles was estimated using a CCK-8 viability assay against L929 mouse fibroblast cells (purchased from ATCC). L929 cells were seeded onto 96-well plates at a density of 1 × 104 cells per well in 100 μL of the culture medium (DMEM supplemented with 10% v/v fetal bovine serum and 1% v/v penicillin/streptomycin) for 24 h at 37 °C in a humidified incubator with 5% CO2. Then, the cells were treated with 100 μL of the medium containing different concentrations of empty micelles for 24 h at 37 °C. bPEI (25 kDa) was used as a control group. After incubation, the cell morphology was observed by phase contrast microscopy in a bright field at 10× magnification. Finally, the number of viable cells was evaluated using the CCK-8 cell viability assay. The CCK-8 solution (10 μL) was added to wells and incubated for 1 h at 37 °C. The absorbance at 450 nm was determined using a microplate reader.2.7 Porcine cartilage explant harvest
Cartilage was obtained from fresh porcine femur purchased from a local butcher. The cartilage explants were harvested from the trochlear groove of the porcine femur with a 9 mm diameter biopsy punch. The cylindrical cartilage plugs were further cut to obtain 1.5 mm-thick cartilage disks containing the intact surface. After harvesting, the disks were washed and equilibrated three times with sterile PBS solution, one hour for each equilibration.2.8 Transport studies of micelle in cartilage explants
The BODIPY-loaded PDMAEMA–PCL–PDMAEMA micelle and BODIPY-loaded pPEGMA–PCL–pPEGMA micelle solutions were prepared as the aforementioned nanoprecipitation method. Since the two types of micelles have distinctive loading capacities for the dye, the initial concentrations of loaded dyes in the two different micellar solutions were adjusted to a similar level. For comparison, a free BODIPY group was designed, the dye was dissolved in 1× PBS supplemented with 20% v/v dimethyl sulfoxide (DMSO), and then filtered through a 0.45 μm filter to remove the undissolved dye (herein, DMSO was to enhance the solubility of this hydrophobic dye in an aqueous solution).Porcine cartilage explants (size: ∅ 9 × 1.5 mm) were equilibrated in 300 μL of BODIPY-loaded cationic and neutral micelles for 24 h in a 48-well plate at 37 °C with gentle shaking to avoid forming stagnant layers. The empty wells in the plate were filled with DI water and the plate was wrapped with parafilm to minimize evaporation. After absorption, the disks were carefully removed from the well, the surfaces of the disks were quickly rinsed with 1× PBS and wiped with Kimwipe to remove unbound molecules, and the absorption bath was collected for fluorescent quantitation. Following 24 h absorption, the explant disks were then desorbed in 300 μL of sterile 1× PBS in a new 48-well plate for 4 days at 37 °C with gentle shaking. The desorption bath was collected and replaced with fresh 1× PBS every day. Next, 200 μL of the collected absorption/desorption baths were mixed with 800 μL of acetone followed by sonication and centrifugation. The fluorescence signal in the supernatant was determined using a microplate reader, and then the dye concentration was calculated from the established standard curve. The solute content absorbed into the cartilage disk was determined by the fluorescence signal difference of the absorption bath before and after incubation. The solute amount retained inside the cartilage disk was calculated by subtracting the daily desorption amount from the total absorbed amount. The uptake rate was defined as the ratio of the amount of solute inside the tissue after 24 h absorption to that of the solute in the initial bath. The retention rate is defined as the ratio of the amount of solute retained inside the tissue after desorption to the total amount of absorbed solute.
2.9 Confocal imaging to estimate the penetration depth
A special transport setup was designed to evaluate the one-way transport of solute in cartilage, which only allowed the solute to transport from the cartilage surface to the deep zone. The porcine cartilage explant (size: ∅ 9 × 1.5 mm) was fixed on a 0.5 mL tube using a parafilm. Then, 300 μL of the BODIPY-loaded micelle solutions were injected into the tube using a syringe, and the tube was inverted in a covered 5 mL tube containing PBS to prevent evaporation and placed on a 37 °C incubator with gentle shaking. After specific transport times, the disk was detached from the tubes, rinsed with 1× PBS, and wiped with Kimwipe to remove unbound molecules. Then, a section with a thickness of around 100 μm was longitudinally cut from the center of the cartilage disks and immediately imaged using a confocal microscope (Zeiss LSM710) at 5× magnification. Z-Stack multilayer images were acquired to visualize the distribution of the BODIPY-loaded micelles in the cartilage tissue. The maximum intensity projections of z-stack images were performed on 80 μm sections. To visualize the location of the nucleus, sections were mounted on glass slides and stained with Hoechst for 15 min, then quickly washed by 1× PBS and wiped with Kimwipe, and imaged using a confocal microscope at 100× magnification. BODIPY was excited using a 493 nm laser light and Hoechst was excited using a 405 nm laser light.2.10 Statistics
Quantitative data are presented as mean ± standard deviation. Statistical analysis was carried out by one-way analysis of variance (ANOVA) followed by the Tukey test. Differences were considered statistically significant when the p value is ≤ 0.05. *p < 0.05, **p <0.01, ***p < 0.001.3. Results and discussion
3.1 Copolymer synthesis and characterization
Amphiphilic triblock PDMAEMA–PCL–PDMAEMA copolymers were synthesized by a two-step process (Fig. 2(A)). First, the commercially available PCL-diol was converted to the Br-PCL-Br macroinitiator through the reaction of the terminal hydroxyl end groups of PCL-diol with BIBB. The reaction was confirmed by 1H NMR spectroscopy (Fig. 3). The NMR signals of the bromoisobutyryl group appeared at 4.17 ppm, while the peak at 3.64 ppm of the hydroxy methylene groups of PCL disappeared (Fig. 3(A) and (B)). In addition, a new peak at 1.92 ppm signed to the methyl protons of the BIBB was observed. The integral ratio between the peaks at 1.92 ppm and 1.38 ppm (the methylene protons of the main chain of PCL) was close to 6:17, indicating the successful synthesis of Br-PCL-Br. The molecular weight of Br-PCL-Br based on the NMR results was about 2200 Da.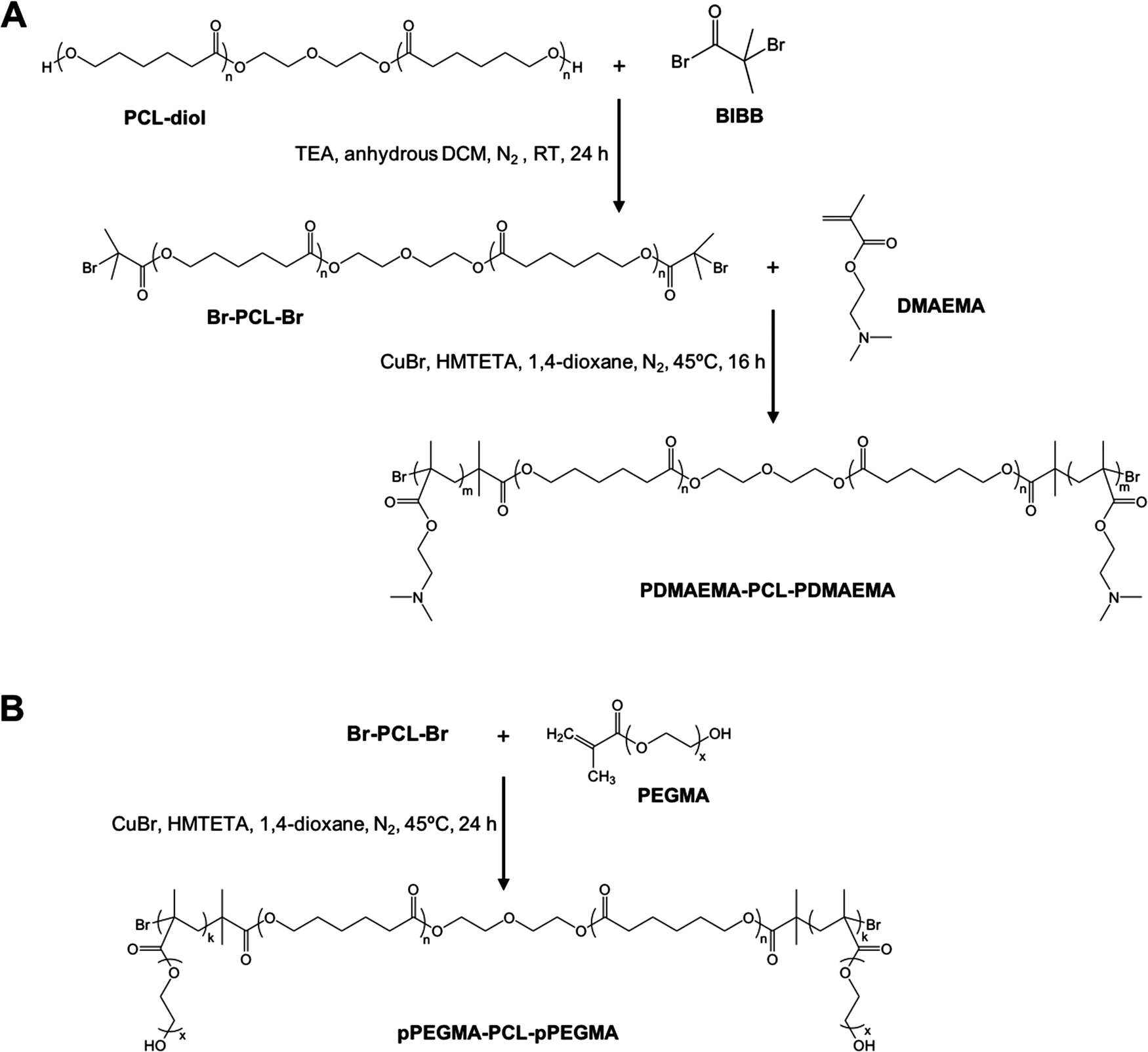 | ||
| Fig. 2 Synthesis of (A) PDMAEMA–PCL–PDMAEMA and (B) pPEGMA–PCL–pPEGMA triblock copolymers. | ||
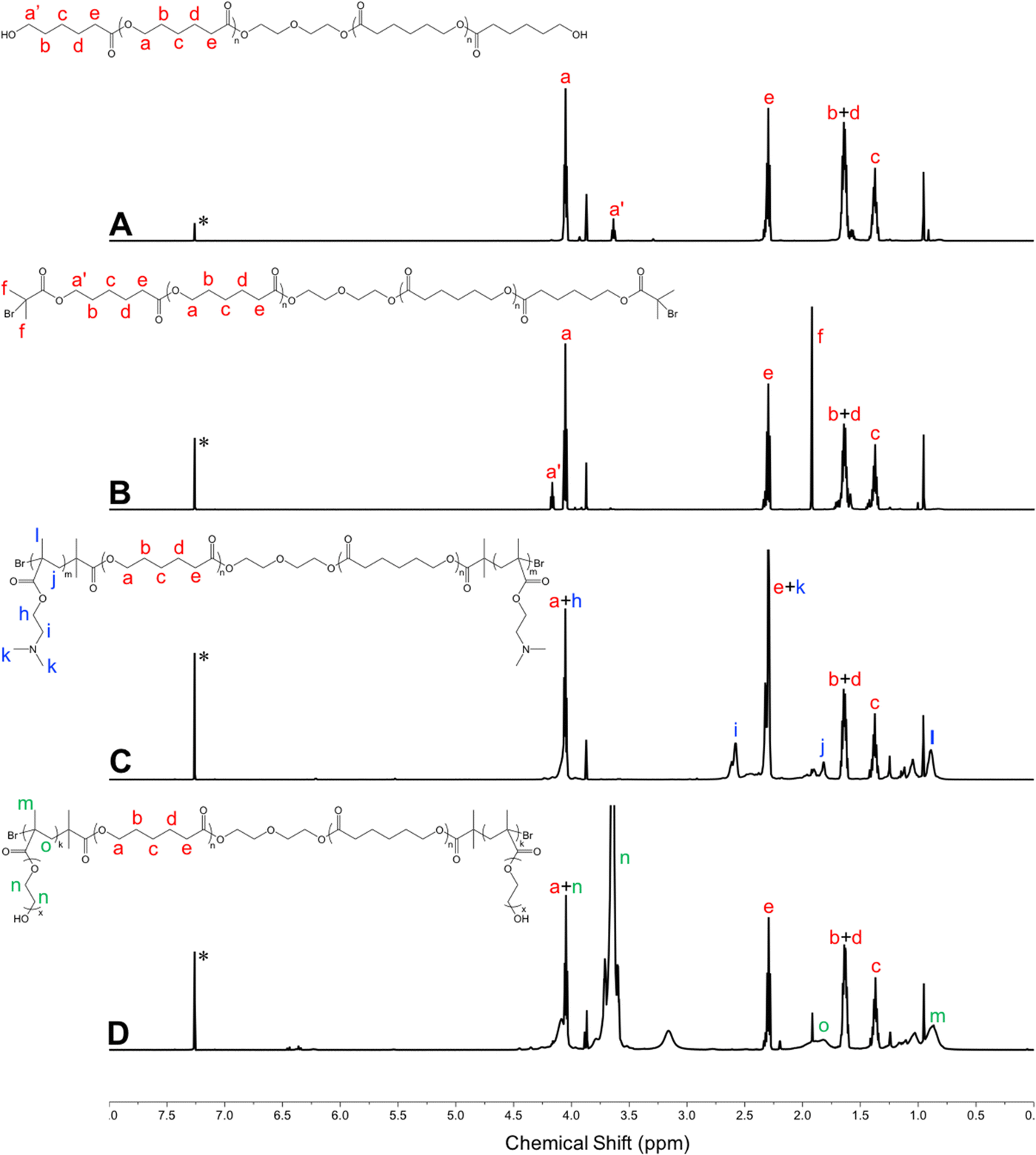 | ||
| Fig. 3 1H NMR spectra of polymers in CDCl3 (600 MHz, 25 °C). (A) PCL-diol; (B) Br-PCL-Br; (C) PDMAEMA–PCL–PDMAEMA; and (D) pPEGMA–PCL–pPEGMA. *Denotes the signal of CHCl3. | ||
The PDMAEMA–PCL–PDMAEMA triblock copolymers were synthesized via the ATRP of DMAEMA using the Br-PCL-Br macroinitiator in 1,4-dioxane at 45 °C under a nitrogen atmosphere. A series of triblock copolymers with different PDMAEMA block lengths were produced by changing the molar feed ratio of monomer to macroinitiator (Table 1). The 1H NMR spectrum of the D9CL17D9 triblock copolymer is shown in Fig. 3(C), while those of D16CL17D16 and D24CL17D24 triblock copolymers are shown in Fig. S1 (ESI†). The connection of PDMAEMA was confirmed by a clear signal of 2.58 ppm which was assigned to the methylene protons next to the amine group of DMAEMA. Besides, the other characteristic peaks of the PDMAEMA segment were observed at δ 1.10–0.70, 1.95–1.76, 2.30 and 4.20–4.00 ppm, respectively, which is consistent with previously reported results.30–33 The peaks attributed to the methylene groups in the PCL segment (δ 1.38, 1.73–1.56, 2.30, and 4.05 ppm) could also be found in the spectra. The molecular weights of the triblock copolymers were determined by comparing the integral of the methylene protons of PDMAEMA at 2.58 ppm to the integral of the methylene protons in the main chain of PCL at 1.38 ppm. The produced three triblock copolymers had molecular weights varying from 5068 Da for D9CL17D9, 7269 Da for D16CL17D16, to 9784 Da for D24CL17D24.
| Copolymera | Initial monomer/PCLb | M n (Da) | Micelle characteristics | ||
|---|---|---|---|---|---|
| Sized (nm) | PDId | Zeta potentiale (mV) | |||
| a PDMAEMA–PCL–PDMAEMA triblock copolymers are denoted as DmCLnDm, where m is the number of repeat unit of DMAEMA, and n is the number of repeat unit of CL. The pPEGMA–PCL–pPEGMA triblock copolymer is denoted as EkCLnEk, where k is the number of repeat unit of PEGMA, and n is the number of repeat unit of CL. b Molar feed ratio of the DMAEMA monomer or the PEGMA monomer to the Br-PCL-Br macroinitiator. c Calculated from 1H NMR results. d Determined by DLS. Micelles were prepared by the nanoprecipitation method in 1× PBS at a concentration of 1 mg mL−1 (mean ± SD, n = 3). The polydispersity index is denoted as PDI. e Measured by DLS. Micelles were prepared by the nanoprecipitation method in 0.1× PBS at a concentration of 1 mg mL−1 (mean ± SD, n = 3). | |||||
| D9CL17D9 | 20/1 | 5068 | 25.9 ± 2.5 | 0.31 ± 0.07 | 17.5 ± 0.1 |
| D16CL17D16 | 40/1 | 7269 | 22.6 ± 1.1 | 0.30 ± 0.04 | 15.0 ± 1.3 |
| D24CL17D24 | 60/1 | 9784 | 21.7 ± 0.6 | 0.24 ± 0.02 | 13.8 ± 1.3 |
| E9CL17E9 | 20/1 | 8718 | 34.4 ± 4.7 | 0.32 ± 0.07 | −1.6 ± 0.4 |
Similarly, the pPEGMA–PCL–pPEGMA triblock copolymer composed of brush PEG and linear PCL was prepared using the PEGMA monomer (360 Da) and the Br-PCL-Br macroinitiator (Fig. 2(B)). The polymerization degree and structure were confirmed by 1H NMR spectra, as shown in Fig. 3(D). The presences of the methyl and methylene proton signals in PEG segments were observed at around δ 1.09–0.70, 2.10–1.71, 3.85–3.45, and 4.31–3.93 ppm. The molecular weight was calculated by comparing the integral of the methylene protons of PEG at δ 3.85–3.45 ppm to that of the methylene protons in the main chain of PCL at 1.38 ppm. Thus, the composition of the triblock copolymer is E9CL17E9, and the molecular weight is 8718 Da.
FTIR spectroscopy was performed to further confirm the chemical structures of copolymers, and the results are shown in Fig. S2 (ESI†). The characteristic peaks of PDMAEMA appeared at 2769 and 2821 cm−1 were assigned to the C–H stretching of the –N(CH3)2 group.34,35 Also, the signal at 1147 cm−1 was attributed to the C–N stretching of the –N(CH3)2 group.34 A reported peak at around 1639 cm−1 was ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group of the DMAEMA monomer disappeared in the spectrum.36 Taken together, the FTIR spectrum indicated the successful synthesis of the PDMAEMA–PCL–PDMAEMA triblock copolymer. Moreover, the FTIR spectrum also shows the signals at 2944 and 2866 cm−1 which were assigned to the C–H stretching of the methylene (–CH2) group, and the signal at 1724 cm−1 was ascribed to the carboxylic ester (CO) groups. For the characterization of pPEGMA–PCL–pPEGMA, the signals that appeared at 1724 and 1100 cm−1 from the FTIR spectrum were assigned to the carboxylic ester (CO) and ether (C–O) groups.37 A broad peak at around 3500 cm−1 was attributed to the stretching of hydroxyl groups (O–H) from the PEG segment. Thereby, the FTIR spectrum supported the successful formation of the pPEGMA–PCL–pPEGMA copolymer.
C group of the DMAEMA monomer disappeared in the spectrum.36 Taken together, the FTIR spectrum indicated the successful synthesis of the PDMAEMA–PCL–PDMAEMA triblock copolymer. Moreover, the FTIR spectrum also shows the signals at 2944 and 2866 cm−1 which were assigned to the C–H stretching of the methylene (–CH2) group, and the signal at 1724 cm−1 was ascribed to the carboxylic ester (CO) groups. For the characterization of pPEGMA–PCL–pPEGMA, the signals that appeared at 1724 and 1100 cm−1 from the FTIR spectrum were assigned to the carboxylic ester (CO) and ether (C–O) groups.37 A broad peak at around 3500 cm−1 was attributed to the stretching of hydroxyl groups (O–H) from the PEG segment. Thereby, the FTIR spectrum supported the successful formation of the pPEGMA–PCL–pPEGMA copolymer.
3.2 Preparation and characterization of micelles
Micelles were prepared from the triblock copolymers (PDMAEMA–PCL–PDMAEMA and pPEGMA–PCL–pPEGMA) using nanoprecipitation, as shown in Fig. 4(A). These amphiphilic triblock copolymers can self-assemble into micelles with a hydrophilic shell and a hydrophobic core in the aqueous environment, while hydrophobic molecules could be encapsulated in the core due to hydrophobic interactions. The core imparted the loading capacity of the micelles as the hydrophobic drug carriers. Acetone was used to assist the triblock copolymers to disperse better in an aqueous solution. Vigorous stirring for 20 h facilitated not only the micelle formation of the copolymers but also the evaporation of acetone from the system. We detected the UV absorption value of acetone in the micelle solution after stirring, and the results showed that the micelle solution contained only a trace of acetone, at about 0.08% (v/v). Bubbling could further reduce the residue of acetone (data not shown).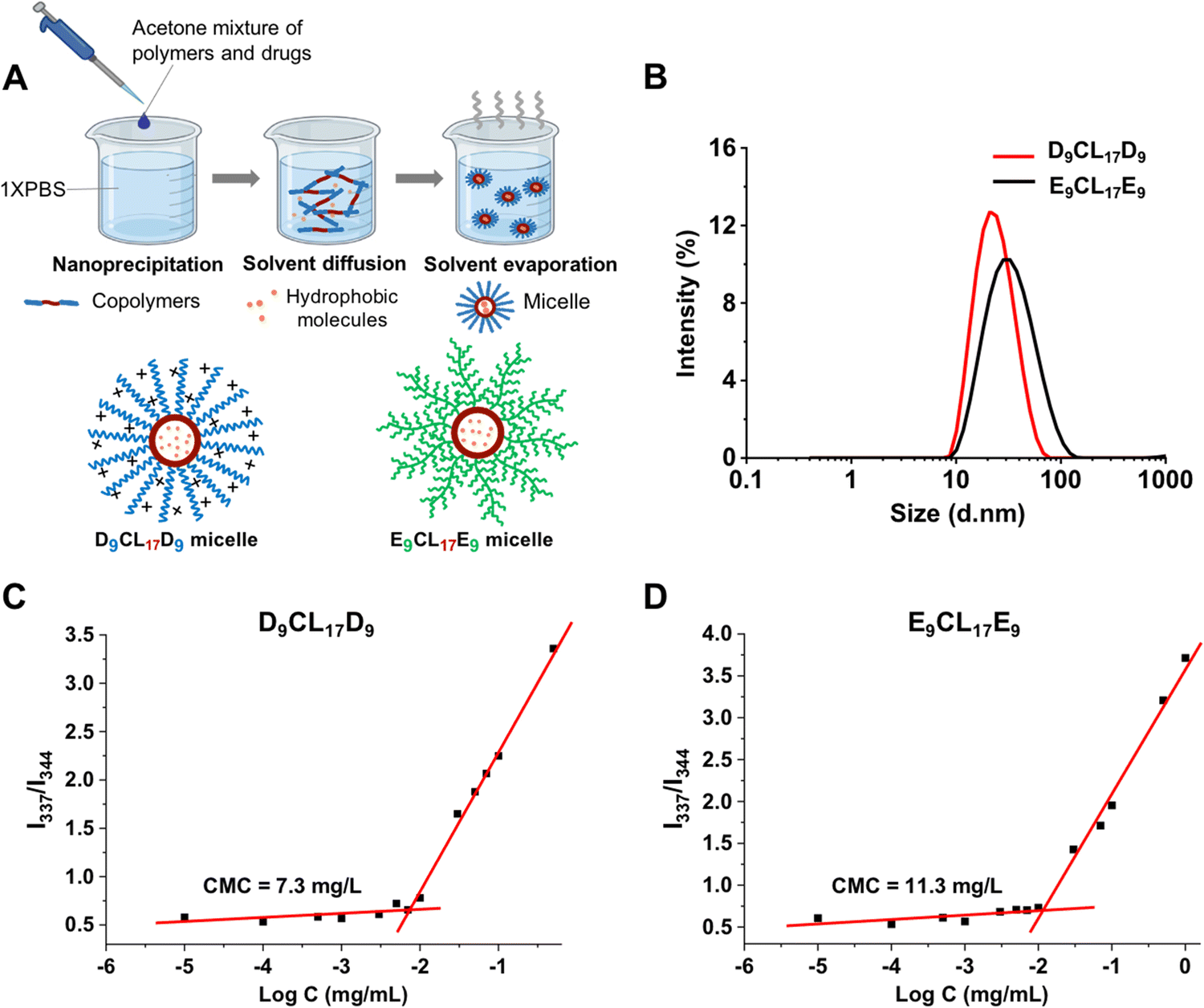 | ||
| Fig. 4 Preparation and characterization of micelles. (A) Micelles were prepared via nanoprecipitation; D9CL17D9 was taken as an example. (B) The size distribution of empty micelles in an aqueous solution. (C) CMC value of D9CL17D9 (emission wavelength = 390 nm). (D) CMC value of E9CL17E9 (emission wavelength = 390 nm). | ||
The sizes and zeta potentials of micelles were typically determined by DLS (Fig. 4(B) and Table 1). The three micelles prepared with the PDMAEMA–PCL–PDMAEMA triblock copolymers containing different lengths of PDMAEMA segments had a similarly small size of about 25 nm, which was smaller than the reported PDMAEMA–PCL–PDMAEMA micelles.26,30,31 The size of micelles is one of the advantages that determines its applications, and small micelles show a remarkable advantage in site-specific drug delivery. The E9CL17E9 micelle also indicated a small size of around 34 nm. Furthermore, the surface potential of micelles is another vital property, which significantly affects the micelle's behaviors. The surface potential regulates the stability of micelles in the physiological environment and largely affects the interactions between micelles and different components in vivo.38,39 The structure and composition of the incorporated polymers determine the micelle surface charge. For the micelles self-assembled from PDMAEMA–PCL–PDMAEMA, the cationic nature of the PDMAEMA block gave the micelles a positive charge. Out of expectation, the zeta potential of micelles slightly decreased with the increasing PDMAEMA block length, ranging from 17.5 to 13.8 mV (pH 7.4), which could be due to the decrease of the micelle size with the increasing PDMAEMA block length. This observation was similar to the result reported by Zhu et al.26 By comparison, the E9CL17E9 micelle exhibited a nearly neutral charge of −1.6 ± 0.4 mV. Besides, the zeta potential of micelles in the aqueous environment (0.1× PBS) under different pH conditions (pH = 5, 7, or 9) was measured. The zeta potential of the D9CL17D9 micelle decreased with the increase of pH, while the zeta potential of the E9CL17E9 micelle remains neutral (Table S1, ESI†). TEM images (Fig. S3, ESI†) show that D9CL17D9 and E9CL17E9 micelles were spherical with a diameter of around 10 nm, and dispersed evenly without aggregation. After dye loading, the particle sizes of the two micelles slightly increased to around 15 nm. The micelle size in TEM images was smaller than that measured by DLS, because the micelles shrank during drying for the TEM measurement.
CMC is a parameter to estimate the stability of micelles. Pyrene was employed as the fluorescent probe to measure the CMC value of micelles.29Fig. 4(C) and (D) show the intensity ratio I337/I344 of pyrene excitation spectra as a function of the logarithmic copolymer concentration, and the CMC value was determined as the cross-point of the curve. The CMC of the triblock copolymer with the shortest PDMAEMA segments (D9CL17D9) was 7.3 mg L−1, which was lower than that of E9CL17E9 (11.3 mg L−1). The low CMC is an important advantage of the polymeric micelle which indicates high thermodynamic and kinetic stability. The low CMC allows a series of dilution and still maintains stable a micellar structure to ensure the delivery of loaded drugs to the target sites.40
3.3 Cytotoxicity of micelles
Biocompatibility is a critical factor in determining the practical application of cationic polymeric carriers. In this study, the cytotoxicity of different copolymers was investigated in L929 mouse fibroblast cells by CCK-8 assay. The commonly used cationic polymer, bPEI (25 kDa), was employed as a control. The results indicated that bPEI had strong cytotoxicity leading to low cell viability and abnormal cell morphology even at low concentrations (Fig. 5 and Fig. S3, ESI†). Conversely, E9CL17E9 micelles showed good biocompatibility that maintained over 80% of cell viability and normal cell morphology at all tested concentrations (5–80 μg mL−1). Notably, the cytotoxicity of PDMAEMA–PCL–PDMAEMA was between bPEI and E9CL17E9, which decreased with the reduction of the PDMAEMA segment length, and this trend was consistent with a previous report.26 The D24CL17D24 copolymer retained over 80% of the cell viability at concentrations below 30 μg mL−1, and few cell debris was observed at a concentration of 40 μg mL−1, suggesting that the copolymer at this concentration showed noticeable cytotoxicity for L929. This was followed by D16CL17D16, which began to show obvious cytotoxicity at a concentration of 60 μg mL−1. It should be noted that the copolymer D9CL17D9 with the shortest PDMAEMA segments had relatively low cytotoxicity, maintained more than 80% of cell viability at a concentration of 60 μg mL−1, and 74% of cell viability and few cell debris were observed at a concentration of 80 μg mL−1. These PDMAEMA–PCL–PDMAEMA triblock copolymers showed lower cytotoxicity compared to bPEI (25 kDa) due to their low molecular weights and the combination of PCL. The biodegradability of the copolymer is likely to further reduce long-term toxicity in vivo.26 Among the cationic triblock copolymers, D9CL17D9 had advantages in higher biocompatibility and the surface charge of the formed micelles, thereby it was selected for subsequent studies.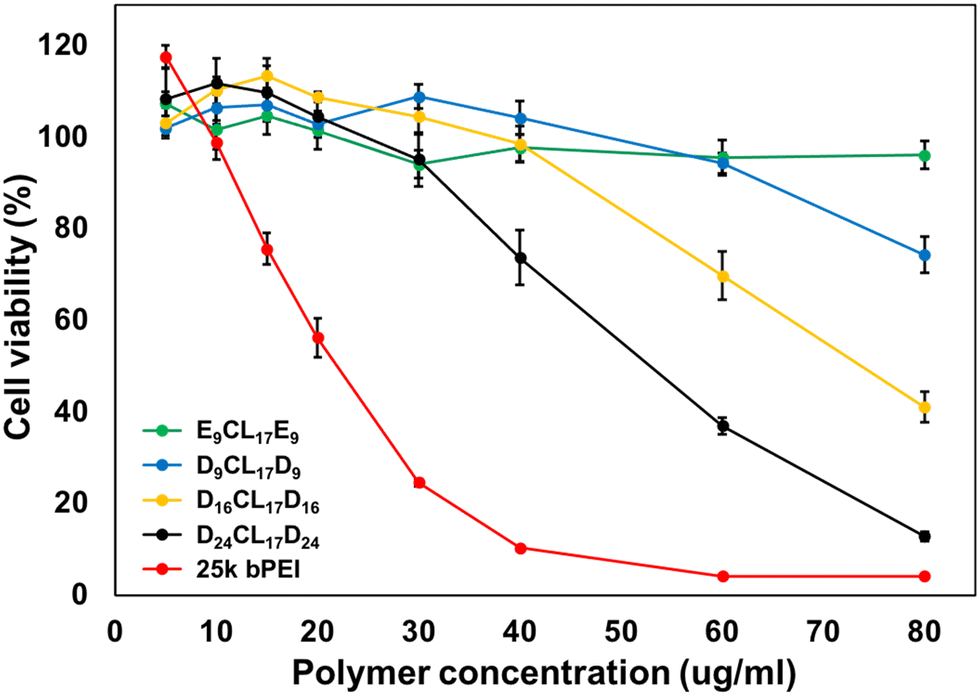 | ||
| Fig. 5 Cytotoxicity of PDMAEMA–PCL–PDMAEMA and pPEGMA–PCL–pPEGMA empty micelles in L929 cells. The metabolic activity of cells was evaluated by CCK assay after 24 h treatment (mean ± S. D., n = 6). | ||
3.4 Uptake rate of micelles in cartilage
To evaluate the penetration and retention capability of micelles in cartilage, a fluorescence dye BODIPY was employed as the model drug that encapsulated into the micelle core. Since D9CL17D9 and E9CL17E9 micelles had distinctive loading capacities for the BODIPY dye, the initial concentrations of loaded dyes in the two different micellar solutions were adjusted to a similar level (0.54 μg mL−1 for the D9CL17D9 micelle and 0.64 μg mL−1 for the E9CL17E9 micelle, respectively). The comparison of the transport properties between different groups was conducted by uptake and retention ratios rather than absolute values. The Z-average hydrodynamic diameters of the BODIPY-loaded D9CL17D9 and E9CL17E9 micelles were 31.2 ± 7.4 nm and 59.6 ± 9.3 nm, respectively. The zeta potentials were 18.7 ± 2.8 mV and −1.8 ± 0.5 mV, respectively.The transport properties of micelles in the porcine cartilage explants were assessed using the aforementioned method (Section 2.8), as shown in Fig. 6(A). The cartilage disks (∅ 9 × 1.5 mm) were exposed to three dye solutions for 24 h at 37 °C: BODIPY-loaded D9CL17D9 (1 mg mL−1) micelles in 1× PBS, BODIPY-loaded E9CL17E9 (1 mg mL−1) micelles in 1× PBS, and free BODIPY in the mixture of DMSO and PBS (20/80, v/v). In this experiment, the fluorescence signal differences in the bath before and after incubation were monitored using a microplate reader, and then, the amount of dye absorbed and retained in cartilage disks were calculated based on the established standard curves (Fig. S4, ESI†). It should be noted that a large amount of acetone (800 μL) was mixed into the collected bath (200 μL), which diluted the bath solution and ensure that the fluorescence intensity was within the linear range. At the same time, the addition of acetone promoted the micelle dissociation to release the dye to ensure that the dye was quantified in the same environment. The uptake rates of the three groups are shown in Fig. 6(B). Notably, the D9CL17D9 micelles showed the highest uptake rate as expected, with 87% of solutes in the bath absorbed into the cartilage explants after 24 h. Followed by the free dye group, the uptake rate was 55%. By comparison, the neutral E9CL17E9 micelle group showed the lowest uptake rate of around 44%. Therefore, the D9CL17D9 micelle that has the best penetration capability was about 1.6 fold and 2 fold of uptake rate as compared to the free dye and E9CL17E9 micelle groups, respectively.
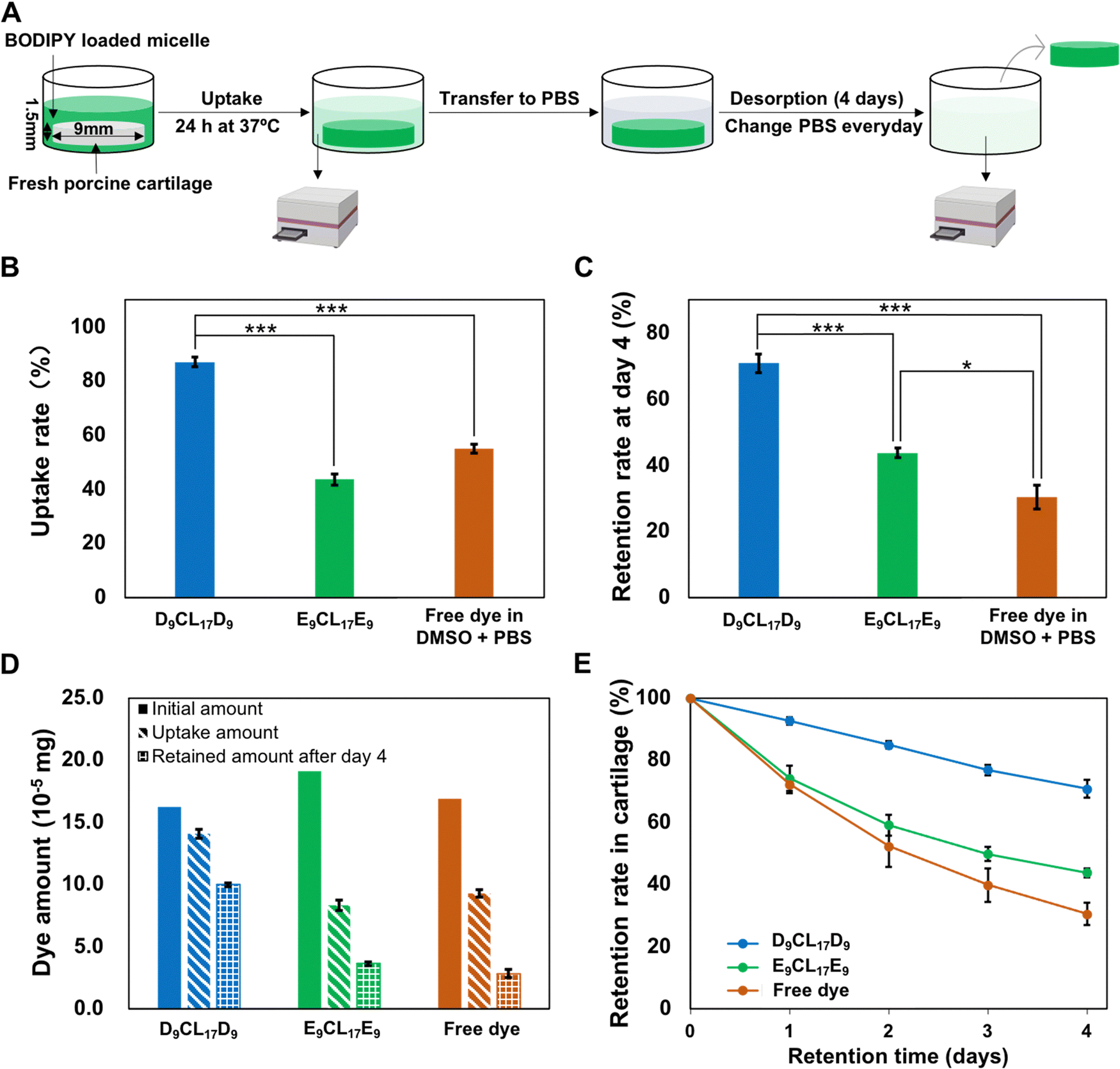 | ||
| Fig. 6 Penetration and retention of micelles in porcine cartilage explants. (A) Schematic of the absorption and desorption of dye-loaded micelles in porcine cartilage disks. (B) Fluorescent dye-loaded micelles uptake rates in porcine cartilage explants after 24 h incubation (mean ± SD, n = 3). (C) The retention rates of micelles inside cartilage following desorption in 1× PBS after 4 days. (D) The absolute amounts of the initial, absorbed, and retained dyes for each group (mean ± SD, n = 3). (E) The retention rates of micelles inside cartilage following desorption in 1× PBS over 4 days. *p < 0.05, **p <0.01, ***p < 0.001. | ||
Regarding the effect of the core–shell structure of micelles on the intra-cartilage transport of hydrophobic solutes, both the core and the shell played their respectively positive roles. First, the core provided an area for the incorporation of hydrophobic molecules via hydrophobic interactions, and this physical encapsulation was beneficial to protecting the intact structure and efficacy of drugs. Conversely, drugs were covalently combined with cationic materials by chemical bonds to promote drug penetration in cartilage in most existing systems.10–12 However, these drug modifications may cause reduced or altered drug efficacy14 and drug release problems.11 On the other hand, the micelle shell was formed by the hydrophilic segments, which had a very vital effect on the micelle's behaviors in vivo, in particular for the steric stabilization and cell interaction.41 The shell protected the hydrophobic part from the biological invasion, thus ensuring that the micelle could remain stable in the physiological environment.42 Therefore, the unique core–shell structure of micelles can improve their bioavailability and stability of poorly soluble drugs.41
Importantly, the hydrophilic shell of the micelle can contact and react with the environment, and the nature of the shell determines the transport performances of micelles within the cartilage. In this study, D9CL17D9 and E9CL17E9 had the same PCL core but showed distinct performances in cartilage transport, with the former having a remarkably higher uptake rate (87%) while the latter having a lower uptake rate (44%). This was largely related to the different nature of their shells. The nature of the PDMAEMA segments gave the D9CL17D9 micelles the positive charge, while the brush PEG segments of the E9CL17E9 micelle resulted in a neutrally charged shell. It is important to mention that the density of negatively charged glycosaminoglycans (GAGs) increases with the cartilage depth, leading to a decrease in the electrical potential from the surrounding bath to cartilage, thus creating a strong, inwardly pointing electric field.4 The driving force can attract cationic solutes into deeper zones of cartilage causing a sharp increase in the cationic carrier concentration within the cartilage.4 The phenomenon is known as Donnan partitioning.4 The result of Donnan partitioning as the total net charge inside the cartilage must be zero.4 Since the tissue contains an extremely high density of negative charge (a fixed charge density of −158 to −182 mM),43 such an equilibrium requires a large amount of cationic solutes, thus constantly attracting cationic solutes into the cartilage. Consequently, the effective penetration of cationic D9CL17D9 micelles into the cartilage was mainly driven by the inwardly pointing electric field.
Nevertheless, the penetration of E9CL17E9 micelles in the cartilage largely depended on the concentration gradient. The electrically neutral micelles were not sterically hindered by the cartilage ECM and could freely penetrate into the cartilage, which was also supported by the fluorescence images presented in Section 3.6. We suggested that the difference in the solute concentration between the bath and cartilage was the driving force for the inward penetration of the neutral micelles. When in equilibrium, the solute concentration in the surrounding bath and the tissue are closely equal, and the neutral micelles no longer diffuse into the cartilage.4 Based on the uptake rates, it could be found that this driving force from the concentration gradient was limited and significantly weaker than the charge guidance. To establish a control group for the free dye, 20% DMSO was added to PBS to increase the solubility of the hydrophobic dye. It should be noted that amphiphilic DMSO is the widely used penetration enhancer for solutes in drug delivery, which have been reported to enhance drug permeation by modifying the tissue structure.44 Hence, the addition of DMSO may promote the penetration of the small-molecule dye in the cartilage tissue to a certain extent. However, even with the assistance of the penetration enhancer, the small molecular dye, BODIPY (262 Da), showed only a 55% uptake rate in the cartilage significantly lower than the D9CL17D9 micelle group.
3.5 Intra-cartilage retention of micelles
Our results have shown that electrostatic interactions could enhance the penetration of the cationic micelles into the cartilage. However, the effective penetration into the cartilage may not guarantee effective retention because partitioning and binding are separate processes.4 To investigate whether electrostatic interactions can simultaneously guarantee the inward penetration and retention of micelles within the cartilage, the absorbed cartilage disks were desorbed in the 1× PBS bath for 4 days. The 1× PBS bath was replaced every day to avoid the concentration equilibrium. The desorption studies showed that the majority (93%) of the absorbed dye-loaded cationic D9CL17D9 micelles retained within the cartilage after 24 h, while the dye-loaded neutral E9CL17E9 micelles and free dye retained about 74% and 72%, respectively (Fig. 6(E)). After 4 days of desorption, 71% of the absorbed dye-loaded D9CL17D9 micelles continued to remain inside the cartilage disks. However, the retention rate of the free dye group was greatly reduced to 31%, and the retention rate of E9CL17E9 micelles also notably decreased to 44% (Fig. 6(C)). Hence, the D9CL17D9 micelles showed remarkably excellent retention capability in the cartilage for at least a 4 day period, which was 2.3 folds and 1.6 folds of the free dye group and E9CL17E9 micelle group, respectively. It should be noted that the dye-loaded E9CL17E9 micelle group also showed a significantly higher retention rate than the free dye group (*p < 0.05).The retention superiority of the D9CL17D9 micelles was mainly derived from its positively charged shell, the dimethylamino groups of PDMAEMA bind negatively charged chondroitin sulfate monomers via electrostatic interactions. Intra-cartilage binding relied on the precise chemical structure of the binding sites and solutes,4 and various binding strategies have been reported to enhance intra-cartilage drug delivery, such as electrostatic interactions, collagen type II binding peptide,45 aggrecan binding peptide,46 and chondrocyte binding peptide.47 Although the electrostatic binding is considered to be weak, the cartilage contains a large number of chondroitin sulfate monomers, which provide numerous sites for electrostatic binding, and the sum of the single electrostatic interaction is sufficient to guarantee an effective binding for a period.4,11 Conversely, free dyes diffused out due to their inability to bind tissue during desorption. Most of the neutral micelles desorbed from the tissue due to the absence of electrostatic binding, but showed higher retention capability than free dyes, suggesting the positive role of the micelle structure on retention. Micelles as the carriers help the encapsulated molecules to retain in the cartilage to a certain extent.
3.6 Distribution study using confocal microscopy
To study the distribution of micelles inside the cartilage, a special setup was used to achieve one-way diffusion of the solutes in the cartilage (Fig. 7(A)), and then the dye-loaded micelle distribution was visualized by confocal microscopy. Fig. 7(B) reveals that D9CL17D9 and E9CL17E9 micelles rapidly penetrated nearly half of the tissue thickness within 3 h with an obvious intensity gradient. Besides, strong fluorescent signals were found on the cartilage surface for both micelle groups, revealing that most micelles were entering and penetrating the surface zone. At the 12 h time point, both micelles nearly penetrated through the full thickness of cartilage, but slight gradients were still observed in the plotted intensity profiles. Finally, the fluorescence images of the two micelles showed the homogeneous fashion within the cartilage, suggesting that two micelles could penetrate the 1.5 mm-thick cartilage in 24 h. Thus, neither D9CL17D9 nor E9CL17E9 micelles were sterically hindered on the cartilage surface by dense cartilage ECM networks. Moreover, Fig. 7(C) shows that two micelles were mainly distributed in the cartilage ECM, and some were densely bound to the cell membrane to form bright spots.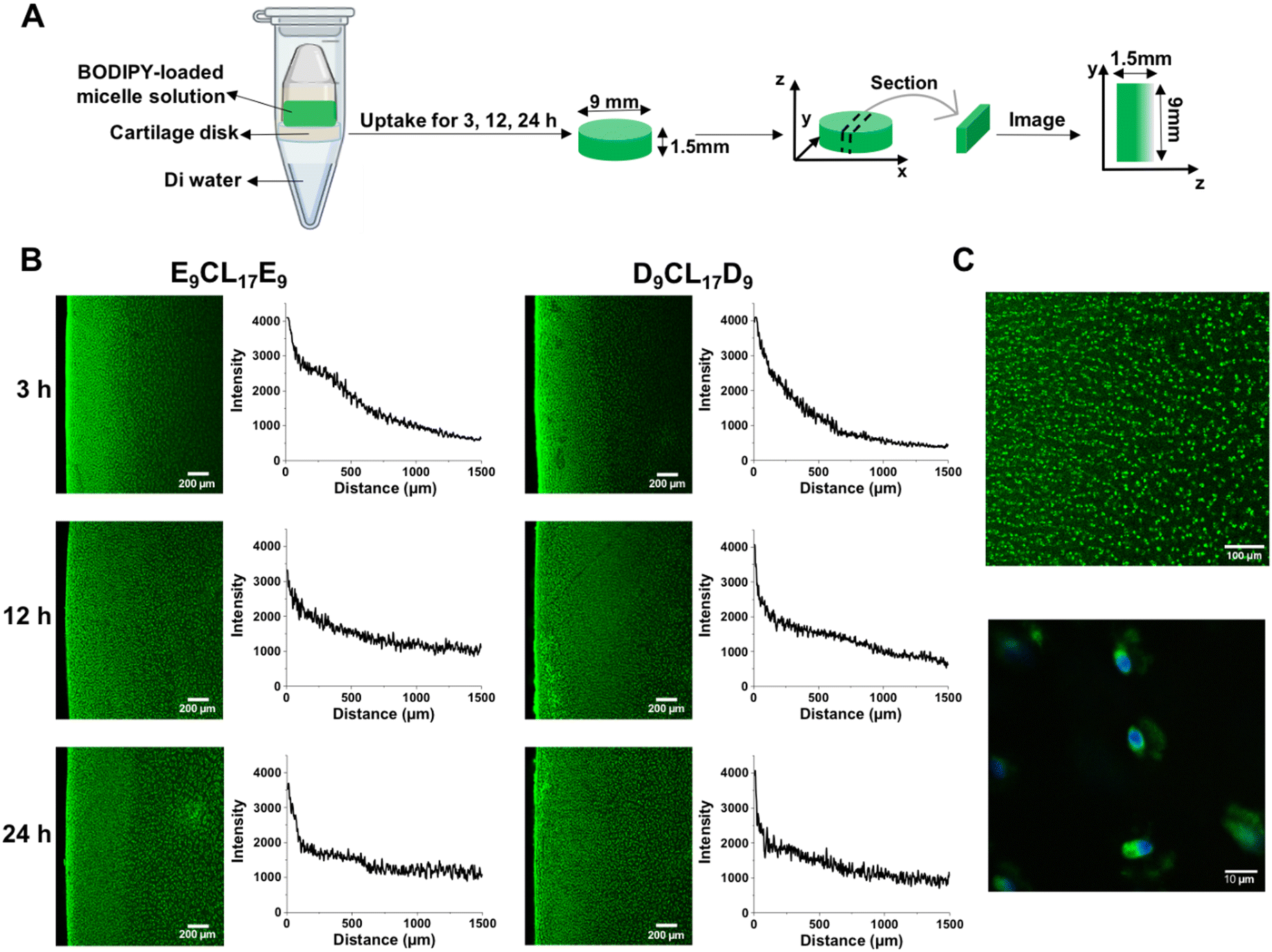 | ||
| Fig. 7 D9CL17D9 and E9CL17E9 micelles exhibited the full penetration of the porcine cartilage within 24 h. (A) Schematic of the one-way diffusion experiment. (B) Confocal images of the concentration profile inside porcine cartilage explants. BODIPY-loaded E9CL17E9 and D9CL17D9 micelles penetrated the cartilage for 3, 12 and 24 h. Diffuse from left (superficial zone) to right (deep zone). Scale bar = 200 μm. Average fluorescence intensities across the thickness were plotted as a function of the distance from the cartilage surface. (C) The distribution of BODIPY-loaded D9CL17D9 micelles in the porcine cartilage tissue was visualized via confocal microscopy at 5× (top) and 100× (bottom) magnifications. Green channel indicated BODIPY-loaded micelles that bind to the ECM and densely to the cellular membrane. The blue channel (Hoechst) demonstrated the nucleus. | ||
An appropriate charge is important for the penetration of cationic solutes in order to ensure the electrostatic interactions between cationic solutes and the ECM is weak and reversible. A strong charge can cause permanent immobilization of cationic solutes on the cartilage superficial zone thus hindering their penetration.13 However, if the charge of the solutes is too weak, it will lead to invalid retention.13 Our experimental results suggested that the D9CL17D9 micelles possessed the appropriate surface charge for penetration and retention in the cartilage explant.
Bajpayee et al. found that 15 nm quantum dots were unable to penetrate beyond the cartilage surface and 7 nm cationic avidin were allowed to enter cartilage. Thus, they claimed that the solutes with a hydrodynamic diameter of ≤10 nm were allowed to penetrate into the cartilage, while solutes with a diameter of more than 15 nm were trapped on the surface and cannot penetrate the cartilage.9 Interestingly, the Z-average hydrodynamic diameters of D9CL17D9 and E9CL17E9 micelles exceeded 15 nm; however, they were proved to penetrate the full-thickness cartilage by confocal images. The same phenomenon of the nanoparticle penetration has been reported in the skin. Šmejkalová et al. reported that 200 nm hyaluronan polymeric micelles were found to penetrate the skin,48 while rigid polystyrene nanoparticles with a particle size of around 30 nm were unable to penetrate the stratum corneum.49 This may be explained by the flexible and deformable structure of micelles.48 Micelles are flexible nanoparticles,50 and the incorporated chains may allow the particles to deform in shape to pass through the dense tissue. It was reported that the deformable liposome can squeeze between the cells in the stratum corneum and then intactly penetrate the skin, even with a large size.51,52 A positively charged liposome with a size of 91.2 nm was reported to penetrate deeply into the rabbit cartilage.14 Therefore, we speculated that the steric hindrance of the ECM had a significant impact on the penetration of rigid nanoparticles, but less on flexible nanoparticles such as micelles or liposomes, which can deform shape to pass through the dense network structure and remain intact in the tissue. The flexibility of micelles showed their superiority in overcoming the obstacles of the dense cartilage structure.
In this study, the cationic D9CL17D9 micelle has been shown to remarkably enhance the penetration and retention of the model drug in porcine cartilage explants, which is a potential drug carrier to overcome the obstacles of intra-cartilage drug delivery. Nevertheless, it is important to mention that there are some limitations of this study that need to be further investigated. Firstly, biocompatibility is one of the most important concerns for cationic materials. The proposed D9CL17D9 micelles had good biocompatibility in a certain range of concentrations (5–60 μg mL−1), but still showed toxicity at high concentrations (80 μg mL−1), which may be further reduced by modifications. Moreover, during the penetration testing period of 24 h, it is still possible that the cationic carriers are to be cleared by the rapid synovial fluid turnover. Therefore, faster penetration may be more effective to resist the rapid clearance. Finally, unspecific electrostatic interactions between cationic micelles and other negatively charged components of the joint may lead to off-target delivery. In response to these limitations, we further modify our micellar systems to achieve safe and effective intra-cartilage drug delivery.
4. Conclusions
We have successfully prepared and characterized neutral and cationic micelles via nanoprecipitation using amphiphilic pPEGMA–PCL–pPEGMA and PDMAEMA–PCL–PDMAEMA triblock copolymers, all of which showed a small size (around 20–30 nm). Among a series of cationic micelles, D9CL17D9 micelles with the shortest PDMAEMA segments had relatively good biocompatibility and was selected for further transport studies. Our current work verified that the positively charged D9CL17D9 micelles showed a largely increased penetration and retention rates in cartilage compared with neutral micelles and free model drugs, which can penetrate the full-thickness cartilage and remain in cartilage for at least 4 days. The cationic micellar system in our work showed advantages in intra-cartilage drug delivery, such as nanosize, easy fabrication, physical encapsulation, flexibility, and excellent penetration and retention, enabling it to be a promising drug carrier to overcome the obstacles of intra-cartilage drug delivery in the negatively charged tissue. Our future work will focus on further improvement of the biocompatibility and verification of the therapeutic efficacy of drug-loaded cationic micellar systems in vitro and in vivo.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Singapore Ministry of Education Academic Research Funds (grant no. A-0005508-01-00 and A-0009388-01-00). We acknowledge that Fig. 4(A), 6(A) and 7(A) were created with BioRender.com.References
- M. Kloppenburg and F. Berenbaum, Osteoarthr. Cartil., 2020, 28, 242–248 CrossRef PubMed.
- D. Umlauf, S. Frank, T. Pap and J. Bertrand, Cell. Mol. Life Sci., 2010, 67, 4197–4211 CrossRef PubMed.
- A. Latourte, M. Kloppenburg and P. Richette, Nat. Rev. Rheumatol., 2020, 16, 673–688 CrossRef PubMed.
- A. G. Bajpayee and A. J. Grodzinsky, Nat. Rev. Rheumatol., 2017, 13, 183–193 CrossRef PubMed.
- C. H. Evans, V. B. Kraus and L. A. Setton, Nat. Rev. Rheumatol., 2014, 10, 11–22 CrossRef PubMed.
- N. Gerwin, C. Hops and A. Lucke, Adv. Drug Delivery Rev., 2006, 58, 226–242 CrossRef CAS PubMed.
- L. M. Mancipe Castro, A. J. García and R. E. Guldberg, J. Biomed. Mater. Res., Part A, 2021, 109, 426–436 CrossRef CAS PubMed.
- M. L. Kang and G.-I. Im, Expert Opin. Drug Delivery, 2014, 11, 269–282 CrossRef.
- A. G. Bajpayee, C. R. Wong, M. G. Bawendi, E. H. Frank and A. J. Grodzinsky, Biomaterials, 2014, 35, 538–549 CrossRef.
- T. He, C. Zhang, A. Vedadghavami, S. Mehta, H. A. Clark, R. M. Porter and A. G. Bajpayee, J. Controlled Release, 2020, 318, 109–123 CrossRef.
- F. A. Formica, G. Barreto and M. Zenobi-Wong, J. Controlled Release, 2019, 295, 118–129 CrossRef PubMed.
- B. C. Geiger, S. Wang, R. F. Padera Jr, A. J. Grodzinsky and P. T. Hammond, Sci. Transl. Med., 2018, 10, eaat8800 CrossRef.
- A. Vedadghavami, E. K. Wagner, S. Mehta, T. He, C. Zhang and A. G. Bajpayee, Acta Biomater., 2019, 93, 258–269 CrossRef PubMed.
- F. Lin, Z. Wang, L. Xiang, L. Deng and W. Cui, Adv. Funct. Mater., 2021, 31, 2107678 CrossRef.
- Y. T. Wen and J. Li, Nat. Biomed. Eng., 2018, 2, 273–274 CrossRef PubMed.
- D. Hwang, J. D. Ramsey and A. V. Kabanov, Adv. Drug Delivery Rev., 2020, 156, 80–118 CrossRef.
- X. Song, J. L. Zhu, Y. T. Wen, F. Zhao, Z. X. Zhang and J. Li, J. Colloid Interface Sci., 2017, 490, 372–379 CrossRef.
- X. Song, Z. X. Zhang, J. L. Zhu, Y. T. Wen, F. Zhao, L. J. Lei, P. T. Nhan, B. C. Khoo and J. Li, Biomacromolecules, 2020, 21, 1516–1527 CrossRef.
- W. W. M. Soh, R. Y. P. Teoh, J. Zhu, Y. Xun, C. Y. Wee, J. Ding, E. S. Thian and J. Li, Biomacromolecules, 2022, 23, 3477–3492 CrossRef.
- M. Zhang, Z. Zhang, X. Song, J. Zhu, J. A. Sng, J. Li and Y. Wen, Biomacromolecules, 2022, 23, 4586–4596, DOI:10.1021/acs.biomac.2c00838.
- Y. T. Wen, Z. X. Zhang and J. Li, Adv. Funct. Mater., 2014, 24, 3874–3884 CrossRef.
- X. H. Liu, H. Yin, Z. X. Zhang, B. S. Diao and J. Li, Colloids Surf., B, 2015, 125, 230–237 CrossRef PubMed.
- Y. T. Wen, H. Z. Bai, J. L. Zhu, X. Song, G. P. Tang and J. Li, Sci. Adv., 2020, 6, eabc2148 CrossRef PubMed.
- Z. X. Zhang, Y. T. Wen, X. Song, J. L. Zhu and J. Li, ACS Appl. Bio Mater., 2021, 4, 5057–5070 CrossRef PubMed.
- X. Liu, H. Yin, X. Song, Z. Zhang and J. Li, Biomolecules, 2022, 12, 102 CrossRef PubMed.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef PubMed.
- V. Tzankova, C. Gorinova, M. Kondeva-Burdina, R. Simeonova, S. Philipov, S. Konstantinov, P. Petrov, D. Galabov and K. Yoncheva, Food Chem. Toxicol., 2016, 97, 1–10 CrossRef PubMed.
- X. J. Loh, Y.-L. Wu, W. T. J. Seow, M. N. I. Norimzan, Z.-X. Zhang, F.-J. Xu, E.-T. Kang, K.-G. Neoh and J. Li, Polymer, 2008, 49, 5084–5094 CrossRef.
- J. Li, X. P. Ni, X. Li, N. K. Tan, C. T. Lim, S. Ramakrishna and K. W. Leong, Langmuir, 2005, 21, 8681–8685 CrossRef PubMed.
- K. Yoncheva, K. Kamenova, T. Perperieva, V. Hadjimitova, P. Donchev, K. Kaloyanov, S. Konstantinov, M. Kondeva-Burdina, V. Tzankova and P. Petrov, Int. J. Pharm., 2015, 490, 298–307 CrossRef PubMed.
- I. L. Diaz and L. D. Perez, Colloid Polym. Sci., 2015, 293, 913–923 CrossRef.
- S. Motala-Timol and D. Jhurry, Eur. Polym. J., 2007, 43, 3042–3049 CrossRef.
- Y.-L. Lo, G.-J. Chen, T.-H. Feng, M.-H. Li and L.-F. Wang, RSC Adv., 2014, 4, 11089–11098 RSC.
- L. C. Bonkovoski, A. F. Martins, I. C. Bellettini, F. P. Garcia, C. V. Nakamura, A. F. Rubira and E. C. Muniz, Int. J. Pharm., 2014, 477, 197–207 CrossRef.
- J. J. Yin, F. Wahid, Q. Zhang, Y. C. Tao, C. Zhong and L. Q. Chu, Macromol. Mater. Eng., 2017, 302, 1700069 CrossRef.
- X. Fan, X. Ren, T.-S. Huang and Y. Sun, RSC Adv., 2016, 6, 42600–42610 RSC.
- H. K. Manjili, H. Malvandi, M. S. Mousavi, E. Attari and H. Danafar, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 926–936 CrossRef PubMed.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. Del Favero, L. Cantù and S. Nicoli, J. Controlled Release, 2021, 332, 312–336 CrossRef PubMed.
- X. Bu, N. Ji, L. Dai, X. Dong, M. Chen, L. Xiong and Q. Sun, Trends Food Sci. Technol., 2021, 114, 386–398 CrossRef CAS.
- W. Xu, P. Ling and T. Zhang, J. Drug Delivery, 2013, 2013, 340315 Search PubMed.
- K. Yoncheva, P. Calleja, M. Agüeros, P. Petrov, I. Miladinova, C. Tsvetanov and J. M. Irache, Int. J. Pharm., 2012, 436, 258–264 CrossRef CAS.
- Z. Ahmad, A. Shah, M. Siddiq and H.-B. Kraatz, RSC Adv., 2014, 4, 17028–17038 RSC.
- E. M. Shapiro, A. Borthakur, A. Gougoutas and R. Reddy, Magn. Reson. Med., 2002, 47, 284–291 CrossRef PubMed.
- A. Otterbach and A. Lamprecht, Pharmaceutics, 2021, 13, 320 CrossRef CAS.
- D. A. Rothenfluh, H. Bermudez, C. P. O’Neil and J. A. Hubbell, Nat. Mater., 2008, 7, 248–254 CrossRef CAS PubMed.
- C. S. Cheung, J. C. Lui and J. Baron, J. Orthop. Res., 2013, 31, 1053–1058 CrossRef CAS PubMed.
- Y. Pi, X. Zhang, J. Shi, J. Zhu, W. Chen, C. Zhang, W. Gao, C. Zhou and Y. Ao, Biomaterials, 2011, 32, 6324–6332 CrossRef CAS PubMed.
- D. Šmejkalová, T. Muthný, K. Nešporová, M. Hermannová, E. Achbergerová, G. Huerta-Angeles, M. Svoboda, M. Čepa, V. Machalová and D. Luptáková, Carbohydr. Polym., 2017, 156, 86–96 CrossRef.
- X. Wu, G. J. Price and R. H. Guy, Mol. Pharmaceutics, 2009, 6, 1441–1448 CrossRef PubMed.
- F. M. Menger, Acc. Chem. Res., 1979, 12, 111–117 CrossRef.
- A. Gillet, F. Lecomte, P. Hubert, E. Ducat, B. Evrard and G. Piel, Eur. J. Pharm. Biopharm., 2011, 79, 43–53 CrossRef PubMed.
- G. Cevc, A. Schätzlein and H. Richardsen, Biochim. Biophys. Acta, Biomembr., 2002, 1564, 21–30 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: 1H NMR spectra of polymers in CDCl3. (A) PCL-diol; (B) D16CL17D16; (C) D24CL17D24; Fig. S2: FTIR spectra of PCL-diol, E9CL17E9, and D9CL17D9; Fig. S3: TEM images of empty and dye-loaded D9CL17D9 and E9CL17E9 micelles. Scale bar = 50 nm; Fig. S4: cell morphology after treatment with different micelles and bPEI was observed by microscopy at bright field at 10 × magnification. Scale bar = 100 μm; Fig. S5: the standard curve of BODIPY with two concentration ranges in a mixture of 20% v/v DI water and 80% v/v acetone; Fig. S6: the photos of excised porcine cartilage disks. (A) After harvesting, the disks (∅ 9 × 1.5 mm) were washed and equilibrated with a sterile PBS solution. (B) For the absorption or desorption experiments, cartilage disks were equilibrated in 300 μL of absorption or desorption baths in the 48-well plate; Table S1: zeta potential of D9CL17D9 and E9CL17E9 micelles at different pH values. Micelles were prepared by the nanoprecipitation method in 0.1× PBS at a concentration of 1 mg mL−1. See DOI: https://doi.org/10.1039/d2tb02050e |
| This journal is © The Royal Society of Chemistry 2023 |