
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHolistic functional biomimetics: a key to make an efficient electrocatalyst for water oxidation†
Lizhou
Fan
a,
Yuxiang
Song
d,
Fan
Zhang
 b,
Brian J. J.
Timmer
a,
Alexander
Kravberg
a,
Biaobiao
Zhang
*ad and
Licheng
Sun
acd
b,
Brian J. J.
Timmer
a,
Alexander
Kravberg
a,
Biaobiao
Zhang
*ad and
Licheng
Sun
acd
aDepartment of Chemistry, KTH Royal Institute of Technology, 10044 Stockholm, Sweden. E-mail: zhangbiaobiao@westlake.edu.cn
bDivision of Surface and Corrosion Science, KTH Royal Institute of Technology, 10044 Stockholm, Sweden
cState Key Laboratory of Fine Chemicals, Institute of Artificial Photosynthesis, DUT-KTH Joint Education and Research Center on Molecular Devices, Dalian University of Technology (DUT), 116024 Dalian, People's Republic of China
dCenter of Artificial Photosynthesis for Solar Fuels and Department of Chemistry, School of Science and Research Center for Industries of the Future, Westlake University, Hangzhou 310024, Zhejiang, People's Republic of China
First published on 20th April 2023
Abstract
Water oxidation is the holy grail reaction of natural and artificial photosynthesis. How to design an efficient water-oxidation catalyst remains a long-term challenge for solar fuel production. The rate of water oxidation in photosystem II by the oxygen-evolving complex (OEC) Mn4CaO5 cluster is as high as 100–400 s−1. Mimicking the structures of the OEC is a straightforward strategy to design water-oxidation catalysts. However, the high efficiency of the OEC relies on not only its highly active site but also its holistic system for well-organized electron transfer and proton transport. Lacking such a holistic functional system makes δ-MnO2 a poor water-oxidation catalyst, although the local structure of δ-MnO2 is similar to that of the Mn4CaO5 cluster. Electrocatalysts simultaneously imitating the catalytically active sites, fast electron transfer, and promoted proton transport in a natural OEC have been rarely reported. The significance of the synergy of a holistic system is underrated in the design of water-oxidation catalysts. In this work, we fabricated holistic functional biomimetic composites of two-dimensional manganese oxide nanosheets and pyridyl-modified graphene (MnOx-NS/py-G) for electrocatalytic water oxidation. MnOx-NS/py-G simultaneously imitates the synergy of catalytically active sites, fast electron transfer, and promoted proton transport in a natural OEC, resulting in overall 600 times higher activity than that of typical δ-MnO2. This work demonstrates the significance of holistic functional biomimetic design and guides the development of highly active electrocatalysts for small molecule activation related to solar energy storage.
Introduction
The development of efficient water-oxidation catalysts is of significance to realize high-performance artificial photosynthesis for solar energy conversion and fuel production.1,2 In photosystem II (PSII), water is oxidized by the oxygen-evolving complex (OEC), a Mn4CaO5 cluster surrounded by amino acid residues, two Cl− ions, and many H2O molecules.3–5 The Mn4CaO5 cluster, which can be regarded as a nanosized Mn oxide in a protein environment, catalyzes water oxidation with a low overpotential of around 160 mV and a high rate of 100–400 s−1.6 Mimicking the structure of the OEC in PSII is a straightforward strategy to design water-oxidation catalysts.2,7 A successful example is the introduction of carboxylic groups into molecular catalysts, leading to the breakthrough of water oxidation performance.8 However, the structural similarity to the natural OEC structure is not always related to the catalytic performance. δ-MnO2 possess similar structural properties to the Mn4CaO5 cluster in PSII, e.g., the cabane-like local structure, and the mixing of Mn3+ and Mn4+ oxidation states, therefore, can be considered structural models for the OEC.9–11 But the performance of typical δ-MnO2 for water oxidation is poor, far less than that of natural enzymes.The high efficiency of the OEC relies on its well-organized holistic system, which includes not only highly active sites for the formation of reactive high-valent metal-oxo and O–O bonds but also effective electron transfer pathways via nearby tyrosine residues12 and proton transport channels via the Ca2+ ion,13 namely the synergy of the catalytically active site, electron transfer, and proton transport (Scheme 1). However, δ-MnO2 only imitates the core structure of the Mn4CaO5 cluster but cannot imitate the OEC's secondary environment.9 Although its local structural unit is close to that of the Mn4CaO5 cluster, the water-oxidation performance of δ-MnO2 is heavily limited due to the lacking exposure of catalytic sites and the components that facilitate electron transfer and proton transport.10 In addition to the structure of the active site, the inspiration that the design of water-oxidation catalysts can take from a natural OEC is the synergy of its holistic system.11 To date, the significance of the synergy of a holistic system is underrated in the design of water-oxidation catalysts. Electrocatalysts simultaneously imitating the catalytically active sites, fast electron transfer, and promoted proton transport in a natural OEC have been rarely reported.
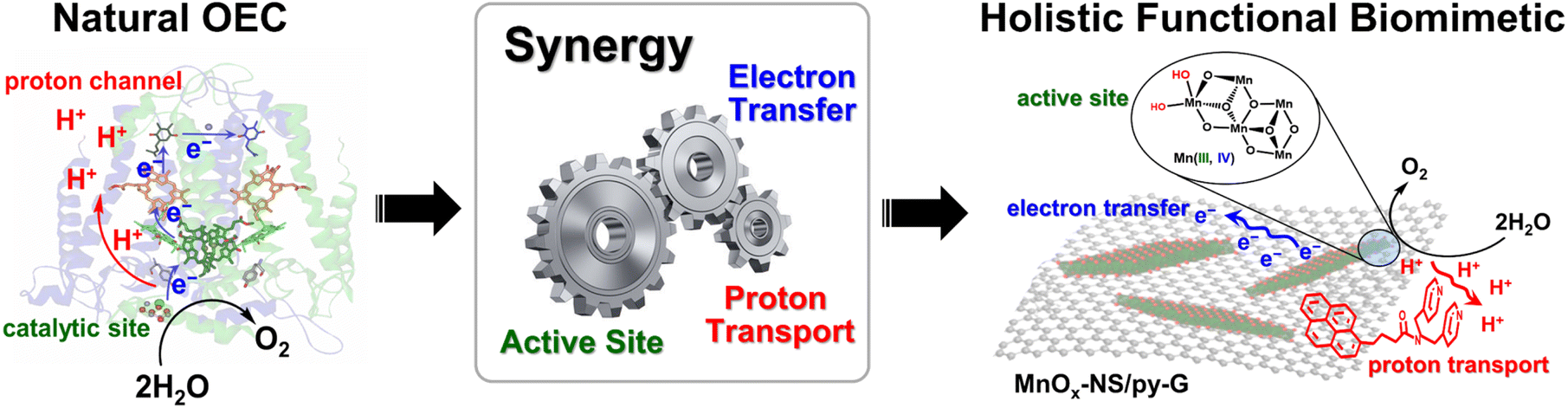 | ||
| Scheme 1 Concept of holistic functional biomimetics inspired by the natural OEC's synergy of the catalytically active site, electron transfer, and proton transport. | ||
In this work, we report holistic functional biomimetic inorganic–organic composites with clearly defined structure–function relationships to display the significance of the synergy of the catalytically active site, electron transfer, and proton transport in the design of water-oxidation electrocatalysts (Scheme 1). Coordinatively unsaturated edge, corner, and defect sites on two-dimensional (2D) manganese oxide nanosheets (MnOx-NS), which can be obtained from structural exfoliation of δ-MnO2, serve as biomimetic active sites. To achieve fast electron transfer between local active sites, material bulk, and the electrode interface, MnOx-NS was assembled with graphene to form a nanocomposite structure. Proton transport is essential for the formation of water-oxidation key intermediates, such as MnVII-oxo,14 MnIVO,15 and MnV-oxo.16 Inspired by Nakamura and co-workers’ study,17 we further modified the graphene surface with pyridyl groups with a pKa value of around 5 to accelerate proton transfer.
The fabricated holistic functional biomimetic composites of MnOx-NS and pyridyl-modified graphene (MnOx-NS/py-G) showed 600-fold higher catalytic activity than δ-MnO2, being ranked on top in highly active manganese-based water-oxidation catalysts. Further in-depth studies revealed the structure–activity relationship of the MnOx-NS/py-G composites. The activity improvement results from the synergy of sufficient exposure of catalytically active sites, fast electron transport, and efficient proton transport. Each of the factors contributes 30 times, 10 times, and 2 times to the overall activity increment, respectively. These results proved the effectiveness of the holistic functional biomimetic for electrocatalyst design.
Results and discussion
Preparation and characterization
General synthetic procedures of MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G are demonstrated in Scheme 2. Control samples δ-MnO2/G and δ-MnO2/py-G were also prepared by similar methods. The graphene used is commercial, and the pyridyl molecule for proton transport is from laboratory synthesis (Fig. S1 and S2†). A colloidal solution of 2D MnOx-NS was first obtained from the exfoliation of δ-MnO2 by an ion-exchange approach.18–20 A typical Tyndall effect is observed in the brown and transparent solution under laser beam illumination, demonstrating its good dispersion and colloidal properties (inset of Fig. 1a). Fig. 1a shows the powder X-ray diffraction (XRD) spectrum of δ-MnO2 and MnOx-NS. In contrast to the XRD pattern of δ-MnO2 with typical sharp peaks at 2θ = 12.4°, which is related to the layered structure with an interlayer spacing of 0.7 nm, the XRD spectrum of MnOx-NS presents no diffraction peak. The disappearance of the peak at 12.4° illustrates the delamination of the long-range ordered layered structure to individual irregularly oriented nanosheets after exfoliation.18,20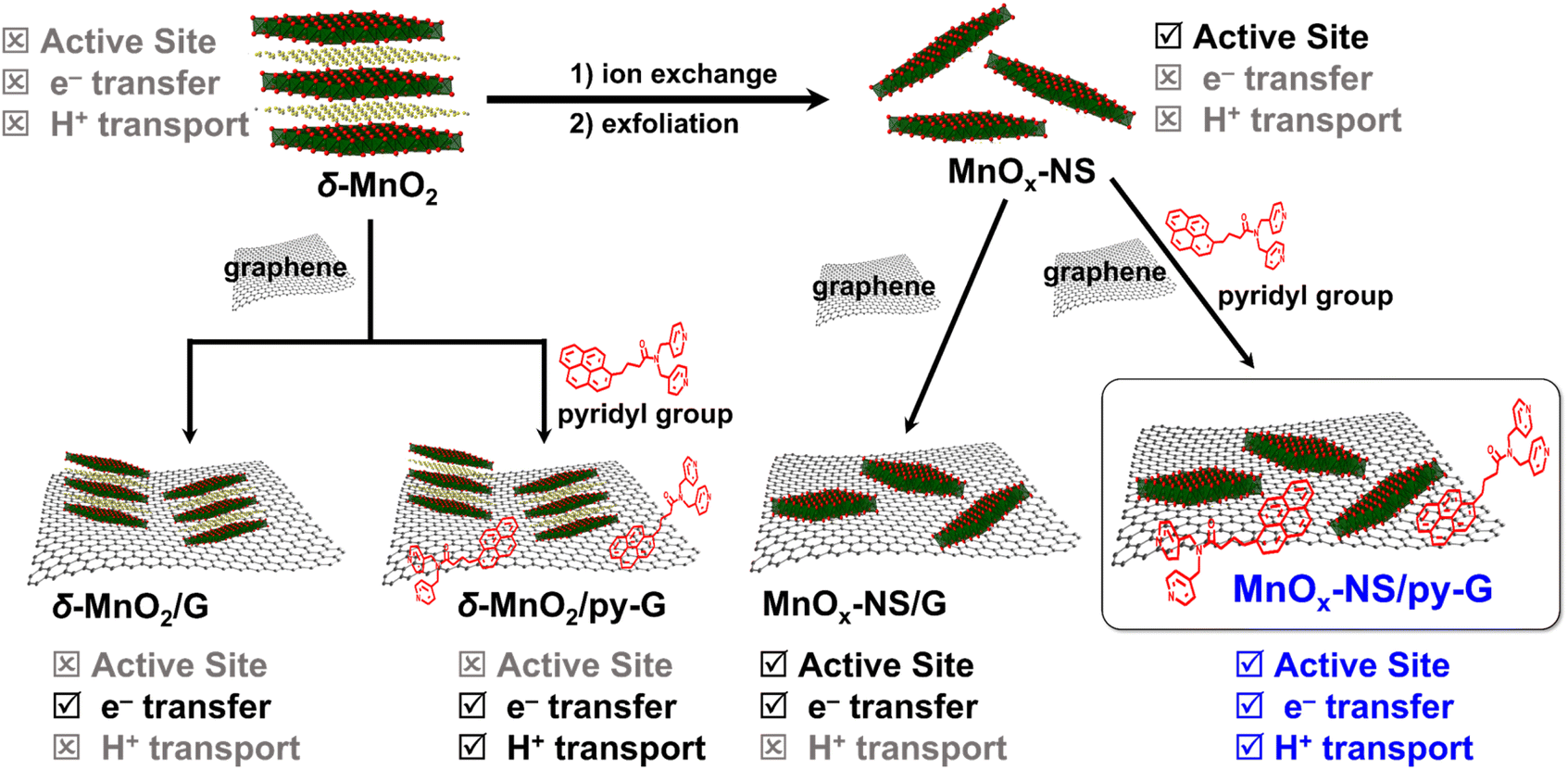 | ||
| Scheme 2 Schematic illustration of the synthetic procedure of MnOx-NS/G, MnOx-NS/py-G, δ-MnO2/G, and δ-MnO2/py-G. | ||
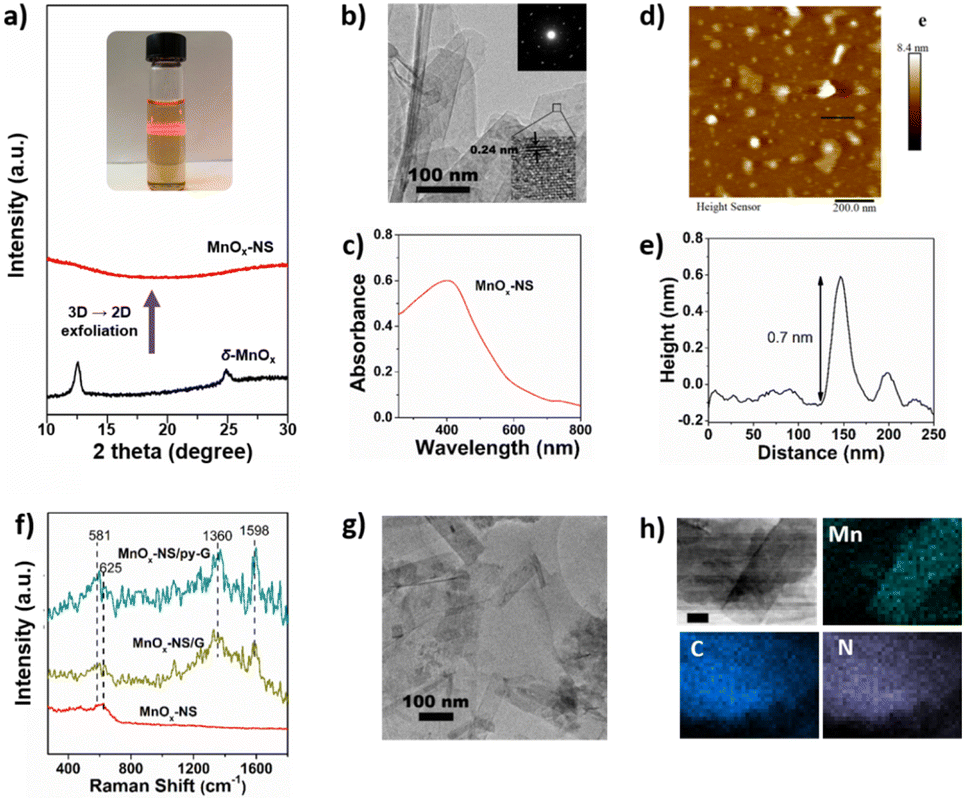 | ||
| Fig. 1 (a) Optical image of the colloidal solution of exfoliated MnOx-NS and XRD pattern of δ-MnO2 and MnOx-NS. (b) The TEM image of MnOx-NS. (c) The UV-vis absorption spectra of the colloidal suspension of MnOx-NS. (d) The AFM image and (e) the height profile of MnOx-NS. (f) The Raman spectrum of MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G. (g) The TEM image of MnOx-NS/py-G. (h) STEM-EDX elemental mapping of Mn, C, and N of MnOx-NS/py-G (scale bar is 20 nm). | ||
MnOx-NS was characterized in detail by transmission electron microscopy (TEM), ultraviolet-visible (UV-vis) spectroscopy, and atomic force microscopy (AFM). The TEM image of MnOx-NS displays a clear single-layer nanosheet morphology (Fig. 1b). The selected area electron diffraction (SAED) pattern of MnOx-NS exhibits hexagonally arranged diffraction spots (inset of Fig. 1b). All these results are in sharp contrast to δ-MnO2, which exhibits a multi-layered morphology and polycrystal SAED image with diffraction rings, showing the exfoliation of a multi-layered structure to monolayer nanosheets (Fig. S3†).20 Notably, the SAED results of δ-MnO2 and MnOx-NS present hexagonal reciprocal lattices, indicating that MnOx-NS retains a similar crystalline structure to the δ-MnO2 crystal. The HRTEM image of MnOx-NS presents a lattice fringe with an interlayer spacing of 0.24 nm, which can be assigned to the {100} plane of δ-MnO2.21,22 The UV-vis spectrum of MnOx-NS shows a broad absorption peak at around 400 nm, which can be attributed to the d–d transition of Mn ions in MnO6 octahedra (Fig. 1c).18,20 At last, the successful exfoliation of δ-MnO2 to 2D MnOx monolayer nanosheets was confirmed by AFM imaging of MnOx-NS (Fig. 1d and e). A thickness of 0.7 nm was determined for MnOx-NS, which is consistent with the thickness of the MnOx monolayer.18,20
An apparent change of the electronic structure in MnOx-NS compared to δ-MnO2 is the increased ratio of Mn3+/Mn4+ oxidation states, which was determined by X-ray photoelectron spectroscopy (XPS). δ-MnO2 shows Mn peaks at 643.0, 642.1 eV, and 640.7 eV, which can be assigned to Mn4+, Mn3+, and a minimal amount of Mn2+, respectively (Fig. S4a†).23–26 Based on the peak integration, its Mn3+/Mn4+ content ratio was calculated to be 0.44. Similar Mn 2p XPS peaks were presented for MnOx-NS. However, the ratio of Mn3+/Mn4+ oxidation states increased to 0.67, much larger than the 0.44 of δ-MnO2. Moreover, the ratio of Mn–OH (531.0 eV)/Mn–O–Mn (529.7 eV) is calculated to be 0.46 for MnOx-NS, much higher than 0.34 for δ-MnO2 (Fig. S4b†).23–25 In general, Mn–OH is a marker of active sites since the Mn ions with several coordinating –OH groups in the Mn4CaO5 cluster, usually labeled as Mn4, are considered to be the active sites for O–O bond formation.27 These XPS results demonstrate that abundant Mn3+ species and coordinatively unsaturated sites are exposed on MnOx-NS upon 3D → 2D structural exfoliation, serving as biomimetic active sites.
Then, single-layer MnOx-NS were assembled with graphene and pyridyl-modified graphene (py-G) to give the MnOx-NS/G and MnOx-NS/py-G samples. The Raman spectra of MnOx-NS/G and MnOx-NS/py-G are presented in Fig. 1f. Both the samples show additional scattering bands at 1360 and 1598 cm−1 compared with MnOx-NS, which are the typical Raman signals of graphene.28,29 The TEM images of MnOx-NS/G and MnOx-NS/py-G (Fig. 1g and S5†) and the energy dispersive spectroscopy (EDS) mapping (Fig. 1h) of MnOx-NS/py-G display randomly distributed MnOx-NS on the surface of graphene without apparent aggregation. A nitrogen fluorescence signal was presented in the EDS mapping of MnOx-NS/py-G, confirming the homogeneous modification by the pyridyl molecule. The presence of the pyridyl molecule in MnOx-NS/py-G can be further depicted by the electrochemical cyclic voltammetry (CV) study. A broad reduction wave was distinctly presented between −1.0 and −1.35 V (vs. Ag/AgCl) for MnOx-NS/py-G, which is the typical redox peak of the pyridine moiety (Fig. S6†).30,31 Overall, these above characterization studies thoroughly demonstrate the successful assembly of MnOx-NS/py-G composites.
Electrocatalytic water oxidation
Electrocatalytic oxygen evolution reaction (OER) performances of δ-MnO2, MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G were evaluated by loading on glass carbon (GC) electrodes under alkaline conditions (1 M KOH). δ-MnO2 shows negligible activity within a wide potential range, which is consistent with the generally low OER activity characteristics of δ-MnO2 (Fig. 2a).32–34 MnOx-NS displays significantly enhanced OER activity, which provides approximately 30 times higher current densities compared with δ-MnO2 (Fig. 2a). Notably, after assembling with graphene, the catalytic current density of MnOx-NS/G increases further by 10-fold compared to that of MnOx-NS (Fig. 2b). Then, with the effect of the pyridyl molecule modification, 2 times improvement in OER performance was additionally achieved for the final MnOx-NS/py-G catalyst compared to that of MnOx-NS/G. In contrast to the initial δ-MnO2, MnOx-NS/py-G eventually displays 600 times higher OER activity due to the synergy of active site exposure, nanocomposite structure, and molecular modification, being ranked as a highly active manganese-based water oxidation catalyst (Fig. S7†).35–41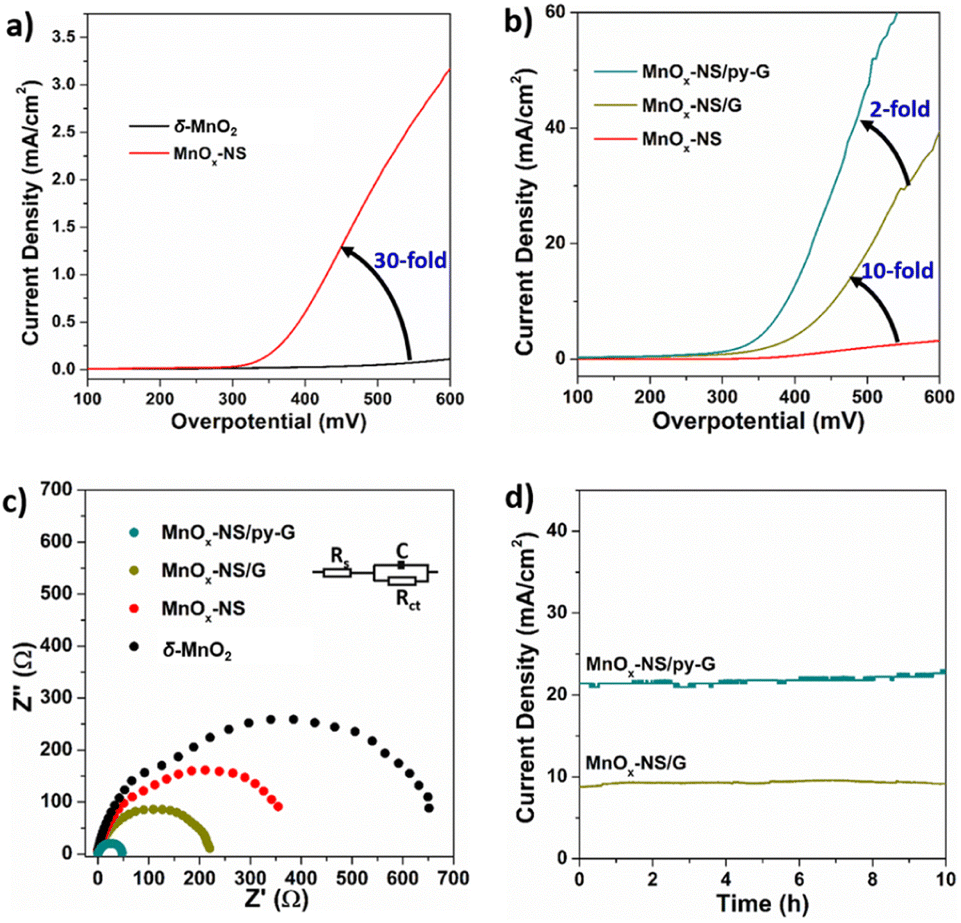 | ||
| Fig. 2 (a) Polarization curves of δ-MnO2 and MnOx-NS. (b) Polarization curves of MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G. (c) Nyquist diagrams of δ-MnO2, MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G. (d) Chronoamperometry curve of MnOx-NS/G and MnOx-NS/py-G at a 450 mV overpotential. | ||
The electrochemical impedance spectroscopy (EIS) measurements under water oxidation conditions can further reflect the improved catalytic rate. MnOx-NS/py-G exhibits a charge transfer resistance (Rct) of 41.09 Ω, which is 20 times lower compared with 584.8 Ω of δ-MnO2 (Fig. 2c). A low Tafel slope of 86 mV dec−1 was determined for MnOx-NS/py-on the MnOx-NS/py-G catalyst to assess the catalytic stability (Fig. S8†). A stable current density of 21 mA cm−2 was maintained during 10 h electrolysis at a 450 mV overpotential, indicating the excellent electrocatalytic durability of MnOx-NS/py-G (Fig. 2d). However, due to the bubble removal, mass transfer, and mechanical stability problems, long-term electrolytic assessment at a large current density (e.g. 200 mA cm−2) cannot be carried out using a GC working electrode in this study.42 For a powder catalyst, a flow electrochemical cell and anion exchange membrane electrolyzer are better and important evaluation methods to achieve the performance of the catalyst in practical application.42,43
TEM, EDS-mapping, and electrochemical cyclic voltammetry of the MnOx-NS/py-G catalyst after reaction shows comparable results with the as-synthesized catalyst, which confirm its good catalytic stability (Fig. S9 and S10†). In a separate experiment, the OER faradaic efficiency was determined to be 98% for MnOx NS/py-G under 400 mV overpotential electrolysis, verifying that a vast majority of charges was contributed to water oxidation (Fig. S11†).
Following our design strategy in Scheme 1, the OER activities of the δ-MnO2, MnOx-NS, MnOx-NS/G, and MnOx-NS/py-G catalysts have been successively advanced owing to the directed improvement in each sample, achieving a remarkable OER performance for the final MnOx-NS/py-G. To understand the activity enhancement from the starting inactive δ-MnO2 to the final highly active MnOx-NS/py-G, we then thoroughly examined the specific effects of structural exfoliation, graphene substrate assembly, and pyridyl molecule modification on electrocatalytic water oxidation.
Effects of the exposure of the active site
Compared to δ-MnO2, the activity of MnOx-NS increased by 30 times, indicating the significant effects of the structural exfoliation. Two possible ways may induce this dramatic improvement: (i) enlarged electrochemically active surface area (ECSA) caused by a morphology change; (ii) increased numbers of catalytic sites due to 3D → 2D structural changes. To identify the effects of structural exfoliation, we first estimated the ECSA of δ-MnO2 and MnOx-NS from electrochemical double-layer capacitance (Cdl, Fig. 3a and S12†). The linear slope of MnOx-NS was calculated to be 0.045 mF cm−2, which is only 18% higher than the 0.038 mF cm−2 of δ-MnO2. The similar ECSA of δ-MnO2 and MnOx-NS indicates that the layered structure of δ-MnO2 is already widely accessible for electrochemical charging, which has also been reported on double layer hydroxide materials.44 This slight increase in ECSA contributes negligibly to the 30 times increase in activity upon exfoliation of δ-MnO2 to MnOx-NS.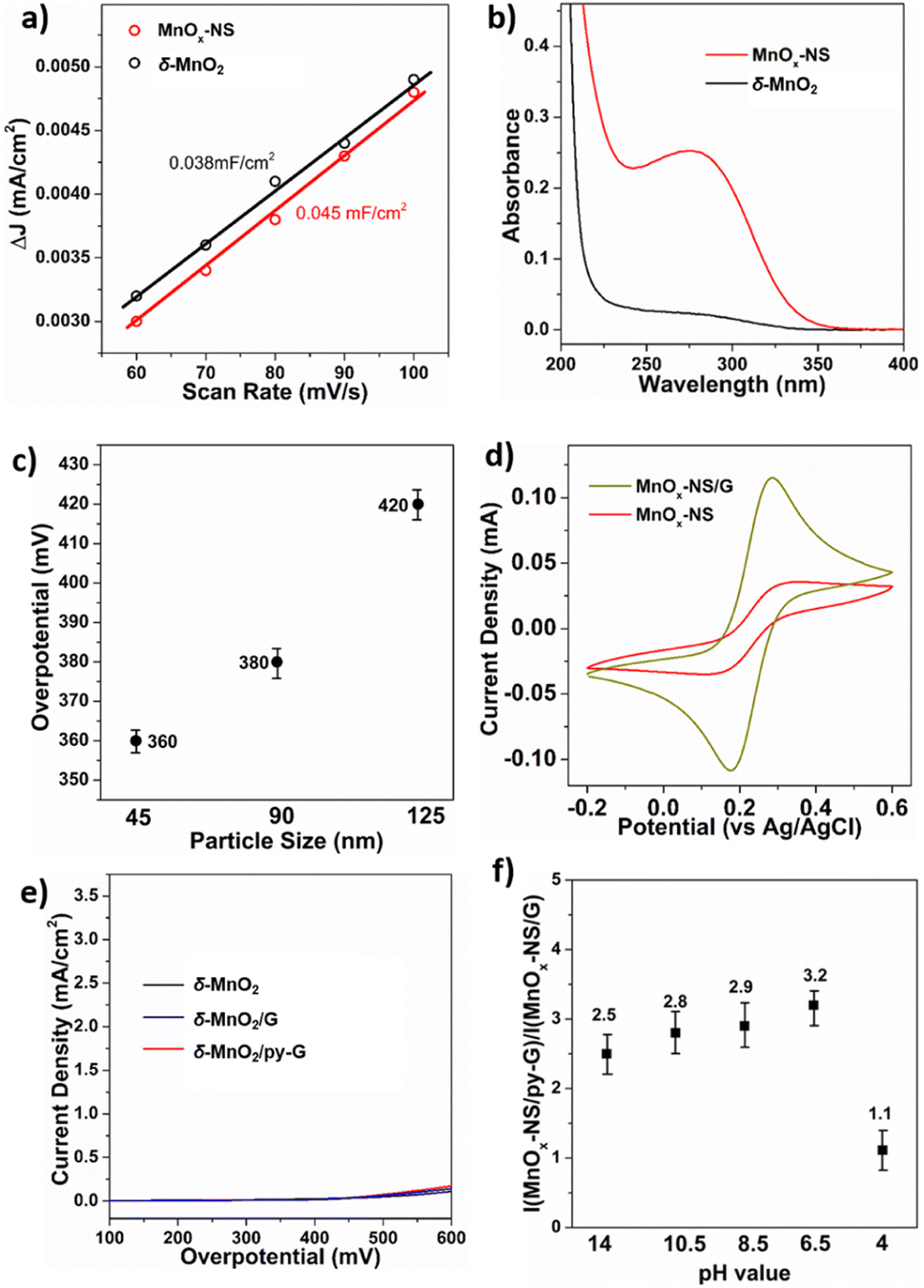 | ||
| Fig. 3 (a) The ECSA measurement: ΔJ (Ja − Jc) from the CV curves of δ-MnO2 and MnOx-NS plotted against scan rates. (b) UV-vis absorption spectra of a 20 mM sodium pyrophosphate solution after dipping δ-MnO2 and MnOx-NS. (c) Comparison of the required overpotential for 0.5 mA cm−2 current density on MnOx-NS with a particle size of 45, 90, and 125 nm. (d) CV curves of MnOx-NS and MnOx-NS/G in 0.1 M KCl electrolyte containing 10 mM Fe(CN)63−/4− ions. (e) LSV curves of δ-MnO2, δ-MnO2/G, and δ-MnO2/py-G. (f) Comparison of I(MnOx-NS/py-G)/I(MnOx-NS/G) under pH 14, pH 10.5, pH 8.5, pH 6, and pH 4 conditions. | ||
Since the change of ECSA does not account for activity improvement, the remaining possibility is the increment of catalytic sites. Mn3+ species have been commonly recognized as an indicator of active sites for Mn-based water oxidation catalysts.45,46 To investigate the promotion effect of exfoliation o active site exposure, we introduce a pyrophosphate (PP) molecule to the solution of δ-MnO2 and MnOx-NS, which can extract Mn3+ from the catalyst through coordination (Fig. 3b).46–48 The resulting Mn3+-PP species was then monitored by using the UV-vis absorption spectrum. MnOx-NS exhibits a much higher Mn3+-PP absorption peak at around 258 nm compared with δ-MnO2. Consistent with XPS, these results demonstrate the abundance of Mn3+ upon exfoliation. These Mn3+ species can be positively involved in water oxidation as highly reactive sites, improving the catalytic activity.
With further investigations, we could propose two aspects that effectively increase catalytically active sites by exfoliation. On the one hand, complete structural exfoliation can spontaneously promote active site exposure at the surface of the monolayer nanosheets. To verify this, Na+ was introduced into the MnOx-NS colloidal suspension to promote the self-reassembly of MnOx-NS, regenerating layered δ-MnO2.20 The XRD pattern of the re-assembled sample presents a typical diffraction peak of a layered structure with an interlayer space of 0.73 nm, indicating the reformation of the layered δ-MnO2 (Fig. S13a†). The electrocatalytic performance of this reformed δ-MnO2 dramatically decreased compared with that of MnOx-NS (Fig. S13b†). The remarkable activity difference between MnOx-NS and δ-MnO2 and our previous active “c-disordered” δ-MnOx demonstrates the importance of the exposure of the MnO2 nanosheet for efficient electrocatalytic water oxidation. However, we cannot exactly explain why δ-MnO2 has the same charge transfer properties as MnOx-NS, i.e., the same ECSA, but the ordered MnO2 nanosheet has deficient catalytic performance for water oxidation.
On the other hand, based on the TEM images, the average particle size of MnOx-NS was estimated to be around 90 nm (Fig. S14†), which is much smaller than the 240 nm of δ-MnO2. Without a well-ordered layered structure, the MnOx monolayer will collapse and break, leading to a decrease in particle size. The formed smaller nanosheets promoted the exposure of coordinatively unsaturated edge- and corner-sites, which are believed to be involved in water oxidation as highly reactive catalytic sites.44,49 To further verify this, MnOx-NS with different lateral sizes were separated by controlled centrifugation, and denoted as MnOx-45 and MnOx-125 (Fig. S15†). A close relationship is presented between the lateral size and OER activity: where MnOx-45 with a smaller particle size shows higher activity than MnOx-90; whereas MnOx-125 with a larger particle size displays lower activity compared with MnOx-90 (Fig. 3c and S16†). These results demonstrate OER activity enhancement with particle size decrement. By the exfoliation process, plenty of coordinated unsaturated edge sites were generated on MnOx-NS (Fig. S17†), which are much easier to be oxidized to high-valent key intermediate states for water oxidation.14,47,48,50–53
Effects of fast electron transfer
MnOx-NS/G shows a 10 times higher activity compared with MnOx-NS. LSV and long-term electrolysis were used to show that bare graphene displays minor water oxidation. This indicates that the graphene substrate promotes the catalytic system rather than introducing more active sites (Fig. S18†). Graphene serves as a loading substrate to promote the uniform distribution of MnOx-NS with less stacking, as displayed in the XRD and TEM images of MnOx-NS/G and MnOx-NS/py-G (Fig. 1g and S19†). More importantly, the graphene substrate with excellent charge transportation capacity can facilitate electron transfer during the OER process.54,55 To probe the enhancement in electron transfer by graphene, CV of K4Fe(CN)6 was performed using a MnOx-NS and MnOx-NS/G covered GC electrode as the working electrode. In contrast to the broad redox peak of Fe2+/Fe3+ for MnOx-NS/GC, a sharp Fe2+/Fe3+ peak was observed in the CV curve measured with MnOx-NS/G/GC (Fig. 3d). The promoted Fe2+/Fe3+ redox couple on MnOx-NS/G/GC illustrates that the combination of MnOx-NS with graphene effectively solves the slow charge transfer problems. The lowered Tafel slopes can also demonstrate the acceleration of electron transfer after graphene introduction (Fig. S8†). The Tafel slopes were calculated to be 89 mV dec−1 and 86 mV dec−1 (approaching 60 mV dec−1) for MnOx-NS/G and MnOx-NS/py-G, which are much smaller than the 134 mV dec−1 (∼120 mV dec−1) of pristine MnOx-NS, indicating that the rate-determining step has been changed from the first electron transfer to the chemical reaction that takes place after the one-electron transfer,48,56,57 demonstrating the improvement in catalytic kinetics.44,47,58The synergy of active site exposure and fast electron transfer can be displayed by the catalytic performance of δ-MnO2/G and δ-MnO2/py-G. No noticeable activity enhancement was observed when δ-MnO2 was assembled with graphene or py-graphene, indicating that the promoting effect of graphene requires optimal assembly between graphene and an exfoliated 2D nanosheet structure (Fig. 3e). These results demonstrate that the further 10-fold enhancement of the OER activity of MnOx-NS/G compared with that of MnOx-NS arises from the synergy of 3D → 2D structural exfoliation and fast electron transfer of graphene.
Effects of molecular pyridyl modification
It has been reported that introducing pyridine into electrolytes can prominently improve the catalytic performance of MnOx electrocatalysts17 and α-Fe2O3 photoanodes.59 In these systems, pyridine can work as a proton transfer relay, facilitating the proton-coupled electron transfer in the catalytic process. For MnOx-NS/py-G, we directly modified the graphene surface, i.e., the catalytic environment, with a pyridyl molecule to accelerate proton transfer during water oxidation. The MnOx-NS loading on graphene afforded a two-fold increase in activity. A pH dependence of OER activity was established to evaluate the proton transfer properties in the catalytic process (Fig. S20†). Fig. 3f shows the OER activities of MnOx-NS/G and MnOx-NS/py-G in electrolytes at different pH values (pH ranging from 4 to 14). In contrast to the significantly enhanced performance under pH ≥ 6.5 conditions, the pyridine modification shows minor enhancements in the OER at pH 4. The absence of increased activity at pH 4 can be attributed to the inhibited proton transfer relay effect of the pyridine group, which possesses a pKa value around 5 and is thus mainly present as its conjugate acid. The deuterium kinetic isotope effect (KIE) was evaluated to study the role of proton transfer in the rate determining step (RDS). The KIE values of MnOx-NS/py-G and MnOx-NS/G were determined to be 3.4 and 4.3, respectively, which exhibit primary deuterium kinetic isotope effects, indicating that the rate-determining step (RDS) during water oxidation involves proton transfer. The lower KIE value of MnOx-NS/py-G compared with that of MnOx-NS/G illustrates the facilitated proton transfer by the introduced pyridyl functionality (Fig. S21†). These pH-dependence and KIE studies demonstrate the proton transfer relay effect of pyridine modification.60,61The above analyses elucidate that the origin of the dramatically enhanced catalytic activity of x-NS/py-G is a combined result of 3D → 2D structural exfoliation, the graphene substrate, and pyridyl molecule modification. The 2D structure of MnOx-NS achieved by exfoliation significantly increased the number of catalytically active sites in the MnOx-NS/py-G catalyst. The nanocomposite structure between 2D MnOx-NS and the graphene substrate provided a fast electron transfer pathway. And the pyridyl group modification accelerated proton transport during water oxidation. The synergy of catalytically active site exposure, fast electron transport, and efficient proton transport accounts for the increased activity by 600 times (Scheme 3), respectively contributing 30 times, 10 times, and 2 times to the overall activity enhancement (Fig. 2a and b). The clear revelation of the structure–activity relationship of the MnOx-NS/py-G composites demonstrates the significance of the holistic functional biomimetic in electrocatalyst design, as the impact of each factor on the overall catalytic performance is multiplied.
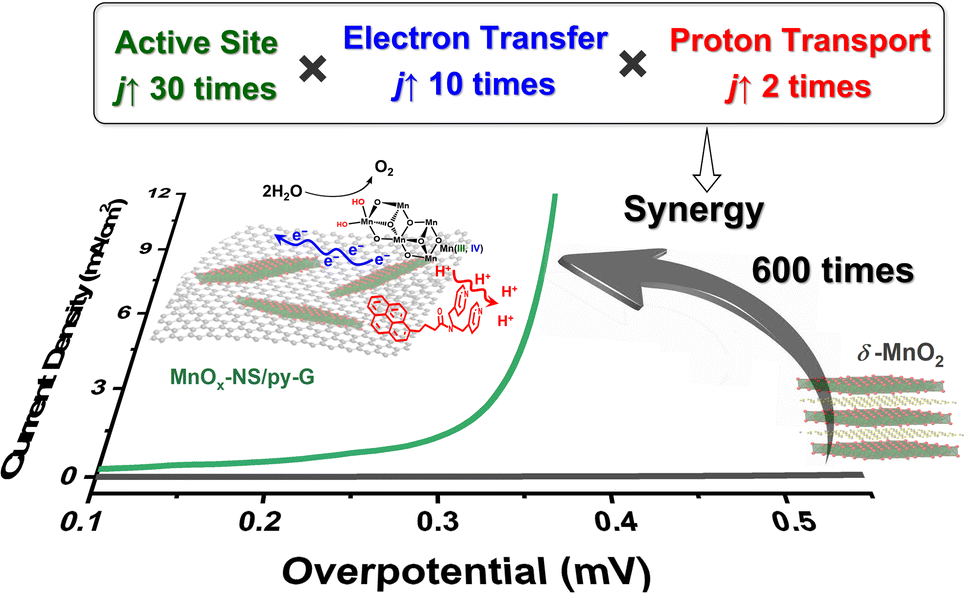 | ||
| Scheme 3 Schematic illustration of the origin of the dramatic activity enhancement owing to the synergy of catalytically active site exposure, fast electron transport, and efficient proton transport. | ||
Conclusions
A natural OEC catalyzes water oxidation with a low overpotential and high turnover frequency because it is an integrated system that realizes the synergy of water molecule activation, O–O bond formation, electron transfer, and proton transport. The design of water oxidation electrocatalysts need to follow nature's course, not only simulating the catalytic site structure but also creating an overall synergistic system. Starting from typical δ-MnO2, we developed MnOx-NS/py-G composites with a 600-fold activity improvement owing to the synergy of active site exposure, fast electron transport, and facilitated proton transport. Further studies indicate that the influence of each of the above factors on the overall catalytic performance is multiplied; therefore, outstanding activity can be achieved when the catalyst possesses efficient active sites, electron transfer, and proton transport simultaneously. In the development of water oxidation catalysts, in-depth analysis of factors leading to low catalytic activity can guide precise modification of the catalyst design for significantly improving the catalytic performance. This work demonstrates the success and significance of holistic functional biomimetics in electrocatalyst design, thereby providing a universal and de novo electrocatalyst design concept.Author contributions
L. S., B. Z. and L. F. conceived and designed the idea. B. Z. and L. F. designed the experiments. L. F. collected and analyzed the data. Y. S., B. J. J. T., and A. K. assisted with the experiments and characterization. F. Z. carried out the AFM measurements. L. F. and B. Z. discussed the results and wrote the manuscript. L. S. revised the manuscript. All the authors reviewed and contributed to this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Prof. Licheng Sun on his 60th birthday. We acknowledge the financial support of this work from the National Key R&D Program of China (2022YFC3401802), the Swedish Energy Agency, the Knut and Alice Wallenberg Foundation (KAW 2016.0072), the National Natural Science Foundation of China (22279105), the Kunpeng research fund from Zhejiang Province, the Research Center for Industries of The Future at Westlake University, and Zhejiang Laboratory for Energy and Carbon Neutralization. We sincerely thank Prof. Jinshan Pan at the KTH Royal Institute of Technology for assistance with the AFM measurements. We sincerely thank Prof. Xin Zhang at Westlake University for the discussions on the manuscript. L. Fan acknowledges financial support from the Swedish Research Council (Vetenskapsrådet) for an International Postdoc Fellowship (2021-00282).Notes and references
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- L. Hammarström and S. Styring, Energy Environ. Sci., 2011, 4, 2379–2388 RSC.
- S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250–3256 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- P. Garrido-Barros, C. Gimbert-Surinach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Holynska, A. N. Moghaddam, E. M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J. R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- S. Park, Y. H. Lee, S. Choi, H. Seo, M. Y. Lee, M. Balamurugan and K. T. Nam, Energy Environ. Sci., 2020, 13, 2310–2340 RSC.
- M. Wiechen, M. M. Najafpour, S. I. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203–2212 RSC.
- J. G. Metz, P. J. Nixon, M. Rogner, G. W. Brudvig and B. A. Diner, Biochemistry, 1989, 28, 6960–6969 CrossRef CAS PubMed.
- R. Hussein, M. Ibrahim, A. Bhowmick, P. S. Simon, R. Chatterjee, L. Lassalle, M. Doyle, I. Bogacz, I. S. Kim, M. H. Cheah, S. Gul, C. de Lichtenberg, P. Chernev, C. C. Pham, I. D. Young, S. Carbajo, F. D. Fuller, R. Alonso-Mori, A. Batyuk, K. D. Sutherlin, A. S. Brewster, R. Bolotovsky, D. Mendez, J. M. Holton, N. W. Moriarty, P. D. Adams, U. Bergmann, N. K. Sauter, H. Dobbek, J. Messinger, A. Zouni, J. Kern, V. K. Yachandra and J. Yano, Nat. Commun., 2021, 12, 6531 CrossRef CAS PubMed.
- B. Zhang, Q. Daniel, L. Fan, T. Liu, Q. Meng and L. Sun, iScience, 2018, 4, 144–152 CrossRef CAS PubMed.
- K. H. Cho, S. Park, H. Seo, S. Choi, M. Y. Lee, C. Ko and K. T. Nam, Angew. Chem., Int. Ed., 2021, 60, 4673–4681 CrossRef CAS PubMed.
- R. D. Britt, D. L. Suess and T. A. Stich, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5265–5266 CrossRef CAS PubMed.
- A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Nat. Commun., 2014, 5, 4256 CrossRef CAS PubMed.
- B. Ma, W. Hou, Y. Han, R. Sun and Z.-H. Liu, Solid State Sci., 2008, 10, 141–147 CrossRef CAS.
- Q. Feng, E.-H. Sun, K. Yanagisawa and N. Yamasaki, J. Ceram. Soc. Jpn., 1997, 105, 564–568 CrossRef CAS.
- X. Yang, Y. Makita, Z. Liu, K. Sakane and K. Ooi, Chem. Mater., 2004, 16, 5581–5588 CrossRef CAS.
- X. Liang, Z. Zhao, M. Zhu, F. Liu, L. Wang, H. Yin, G. Qiu, F. Cao, X. Liu and X. Feng, Environ. Sci.: Nano, 2017, 4, 1656–1669 RSC.
- G. Zhao, J. Li, X. Ren, J. Hu, W. Hu and X. Wang, RSC Adv., 2013, 3, 12909 RSC.
- H.-Q. Wang, J. Chen, S.-J. Hu, X.-H. Zhang, X.-P. Fan, J. Du, Y.-G. Huang and Q.-Y. Li, RSC Adv., 2015, 5, 72495–72499 RSC.
- H. Liu, Z. Hu, L. Tian, Y. Su, H. Ruan, L. Zhang and R. Hu, Ceram. Int., 2016, 42, 13519–13524 CrossRef CAS.
- H. Kim, A. Watthanaphanit and N. Saito, RSC Adv., 2016, 6, 2826–2834 RSC.
- H. Zhu, Q. Liu, J. Liu, R. Li, H. Zhang, S. Hu and Z. Li, Electrochim. Acta, 2015, 178, 758–766 CrossRef CAS.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- J. Zabel, R. R. Nair, A. Ott, T. Georgiou, A. K. Geim, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 617–621 CrossRef CAS PubMed.
- X. Y. Yan, X. L. Tong, Y. F. Zhang, X. D. Han, Y. Y. Wang, G. Q. Jin, Y. Qin and X. Y. Guo, Chem. Commun., 2012, 48, 1892–1894 RSC.
- A. J. Lucio and S. K. Shaw, J. Phys. Chem. C, 2015, 119, 12523–12530 CrossRef CAS.
- E. Lebegue, J. Agullo and D. Belanger, ChemSusChem, 2018, 11, 219–228 CrossRef CAS PubMed.
- Q. Kang, L. Vernisse, R. C. Remsing, A. C. Thenuwara, S. L. Shumlas, I. G. McKendry, M. L. Klein, E. Borguet, M. J. Zdilla and D. R. Strongin, J. Am. Chem. Soc., 2017, 139, 1863–1870 CrossRef CAS PubMed.
- A. C. Thenuwara, E. B. Cerkez, S. L. Shumlas, N. H. Attanayake, I. G. McKendry, L. Frazer, E. Borguet, Q. Kang, R. C. Remsing, M. L. Klein, M. J. Zdilla and D. R. Strongin, Angew. Chem., Int. Ed., 2016, 55, 10381–10385 CrossRef CAS PubMed.
- A. C. Thenuwara, S. L. Shumlas, N. H. Attanayake, Y. V. Aulin, I. G. McKendry, Q. Qiao, Y. Zhu, E. Borguet, M. J. Zdilla and D. R. Strongin, ACS Catal., 2016, 6, 7739–7743 CrossRef CAS.
- D. A. Vermaas, S. Bajracharya, B. B. Sales, M. Saakes, B. Hamelers and K. Nijmeijer, Energy Environ. Sci., 2013, 6, 643–651 RSC.
- K. L. Pickrahn, S. W. Park, Y. Gorlin, H. B. R. Lee, T. F. Jaramillo and S. F. Bent, Adv. Energy Mater., 2012, 2, 1269–1277 CrossRef CAS.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 RSC.
- C. Walter, P. W. Menezes, S. Loos, H. Dau and M. Driess, ChemSusChem, 2018, 11, 2554–2561 CrossRef CAS PubMed.
- Y. Meng, W. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- K. L. Pickrahn, Y. Gorlin, L. C. Seitz, A. Garg, D. Nordlund, T. F. Jaramillo and S. F. Bent, Phys. Chem. Chem. Phys., 2015, 17, 14003–14011 RSC.
- J. Knöppel, M. Möckl, D. E. López, K. Stojanovski, M. Bierling, T. Böhm, S. Thiele, M. Rzepka and S. Cherevko, Nat. Commun., 2021, 12, 2231 CrossRef PubMed.
- J. Schröder, V. A. Mints, A. Bornet, E. Berner, M. F. Tovini, J. Quinson, G. K. H. Wiberg, F. Bizzotto, H. A. E. Sayed and M. Arenz, JACS Au, 2021, 1, 247–251 CrossRef PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- Z. Morgan Chan, D. A. Kitchaev, J. Nelson Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5261–E5268 CrossRef PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- B. Zhang, H. Chen, Q. Daniel, B. Philippe, F. Yu, M. Valvo, Y. Li, R. B. Ambre, P. Zhang, F. Li, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 6311–6322 CrossRef CAS.
- K. Jin, H. Seo, T. Hayashi, M. Balamurugan, D. Jeong, Y. K. Go, J. S. Hong, K. H. Cho, H. Kakizaki, N. Bonnet-Mercier, M. G. Kim, S. H. Kim, R. Nakamura and K. T. Nam, J. Am. Chem. Soc., 2017, 139, 2277–2285 CrossRef CAS PubMed.
- L. Fan, B. Zhang, B. J. J. Timmer, N. V. R. A. Dharanipragada, X. Sheng, C.-W. Tai, F. Zhang, T. Liu, Q. Meng, A. K. Inge and L. Sun, Nano Energy, 2020, 72, 104656 CrossRef CAS.
- B. Zhang, Y. Li, M. Valvo, L. Fan, Q. Daniel, P. Zhang, L. Wang and L. Sun, ChemSusChem, 2017, 10, 4472–4478 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- Z. N. Zahran, E. A. Mohamed and Y. Naruta, ACS Catal., 2016, 6, 4470–4476 CrossRef CAS.
- G. Mattioli, I. Zaharieva, H. Dau and L. Guidoni, J. Am. Chem. Soc., 2015, 137, 10254–10267 CrossRef CAS PubMed.
- P. Chen, K. Xu, T. Zhou, Y. Tong, J. Wu, H. Cheng, X. Lu, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- V. S. Kale, U. Sim, J. Yang, K. Jin, S. I. Chae, W. J. Chang, A. K. Sinha, H. Ha, C. C. Hwang, J. An, H. K. Hong, Z. Lee, K. T. Nam and T. Hyeon, Small, 2017, 13, 1603893 CrossRef PubMed.
- H. Vrubel, T. Moehl, M. Gratzel and X. Hu, Chem. Commun., 2013, 49, 8985–8987 RSC.
- T. Takashima, K. Ishikawa and H. Irie, Chem. Commun., 2016, 52, 14015–14018 RSC.
- W. Li, F. Li, H. Yang, X. Wu, P. Zhang, Y. Shan and L. Sun, Nat. Commun., 2019, 10, 5074 CrossRef CAS PubMed.
- Y. Tanahashi, K. Takahashi, Y. Tsubonouchi, S. Nozawa, S. I. Adachi, M. Hirahara, E. A. Mohamed, Z. N. Zahran, K. Saito, T. Yui and M. Yagi, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113910118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01040f |
| This journal is © The Royal Society of Chemistry 2023 |