DOI:
10.1039/D3TA00372H
(Paper)
J. Mater. Chem. A, 2023,
11, 7498-7504
Terthiophene based low-cost fully non-fused electron acceptors for high-efficiency as-cast organic solar cells†
Received
20th January 2023
, Accepted 6th March 2023
First published on 7th March 2023
Abstract
Fully non-fused electron acceptors (FNEAs) have shown a promising prospect for commercial applications of organic solar cells (OSCs) due to their simple structures and low costs. Herein, two terthiophene-based NFEAs 3T-1 and 3T-2 were exploited from a single thiophene ring as the starting material. Octyl substituted 3T-1 exhibits strong crystallinity, poor miscibility, and forms large aggregates when blending with polymer D18, leading to an inferior power conversion efficiency (PCE). While 2-ethylhexyl substituted 3T-2 possesses good solubility and enhanced compatibility with polymer D18, the blend film exhibits an appropriate morphology, face-on molecular orientation, and efficient charge transport. As a result, the as-cast device based on 3T-2 exhibits a significantly enhanced PCE of 11.12% processed by non-halogenated o-xylene as the solvent, which is the highest value of terthiophene based FNEAs for OSCs so far. Our work provides insight into molecular design of FNEAs for high-performance OSCs.
Introduction
Organic solar cells (OSCs) as a potentially sustainable energy technology have drawn considerable attention owing to their flexibility, light weight and solution-processibility in comparison with their inorganic counterparts.1–4 Benefiting from the development of fused ring electron acceptors (FREAs), the power conversion efficiencies (PCEs) of OSCs have been significantly improved.5–9 Highly efficient FREAs generally have an acceptor–donor–acceptor (A–D–A) type molecular structure with a multi-fused ring core as the D unit and two cyanoindanone type terminal groups as the A unit.10–13 The π-extended fused ring cores play a critical role in achieving excellent photovoltaic performance. The conjugated fused ring building blocks usually possess planar and rigid configurations, which are beneficial for π-electron delocalization and intramolecular charge transport. In the meantime, the bulky side chains on the fused-ring building blocks can regulate molecular solubility and crystallinity, and provide suitable phase separation morphology to the blend film. In 2019, Zou et al. exploited a novel FREA Y6 with a N-hetero-fused-ring building block as the core.11 To date, the PCEs based on Y-series FREAs have exceeded 19% for single bulk-heterojunction OSCs.14–16 Nevertheless, these multi-fused ring cores of FREAs generally require tedious syntheses, which inevitably give rise to the low yields and expensive preparation costs. Therefore, the development of high efficiency and low-cost acceptor materials with simple molecular structures is an essential step to achieve the large-scale commercial applications for OSCs.17–21
Up to now, great efforts have been devoted to exploiting fully non-fused electron acceptors (FNEAs), which commonly consist of two or more benzene and thiophene rings linked by a C–C single bond as D units instead of a complex multi-fused ring core.22–27 On the other hand, introduction of intramolecular noncovalent interactions is an efficient method to form ladder-like structures, which can effectively prevent the low-yield ring-closure step and reduce synthetic costs. Oligothiophenes have been widely used as organic semiconductors in organic field-effect transistors and OSCs due to their good hole transport abilities and tunable optical/electrochemical properties together with their relatively simple synthesis, and high stability.28–32 Very recently, oligothiophenes as fully non-fused cores have been employed to construct high efficiency FNEAs. After comprehensive analysis of the current reports on oligothiophene cores, there are mainly two reported ways to conduct structural modifications: (i) changing the number of thiophene rings can control the length of the conjugate skeleton; such as bithiophene,33 terthiophene,24,34 and tetrathiophene;35,36 (ii) the functional side chain groups of the thiophene ring can induce the formation of noncovalent intramolecular interactions, and tune molecular solubility, packing and crystallinity.37,38 For example, Hou et al. reported a tetrathiophene based FNEA A4T-16 with bulky functional group 2,4,6-tri-isopropylphenyl as the side chain. A4T-16 can form a three-dimensional interpenetrating network and compact π–π stacking, leading to a record PCE of 15.2%.39 Subsequently, our group synthesized a tetrathiophene based FNEA 4T-3 with branched 2-ethylhexyl chains and a PCE of 12.04%, and a high figure-of-merit (FOM) value of 32.8 can be achieved.35 Compared to tetrathiophene-based FNEAs, terthiophene-based acceptors have simpler molecular structures. However, there have been limited reports on terthiophene-based FNEAs with a PCE of about 7%.24,34 Hence, designing terthiophene based FNEAs is still a challenge to realize effective photovoltaic performance for OSCs.
Herein, we report two terthiophene based FNEAs 3T-1 and 3T-2 with octyl and 2-ethylhexyl side chains, respectively. The two FNEAs are readily prepared from commercially available thiophene derivatives in high yields. Our results demonstrated that side chain engineering could regulate the molecular miscibility and crystallinity, which in turn control the morphology and molecular orientation in thin films. 3T-1 displays strong crystallinity and poor miscibility with the polymer donor D18, resulting in an apparent phase separation with an edge-on molecular orientation in the blend film. In comparison, 3T-2 has good miscibility with D18; the blend film exhibits a homogenous morphology, face-on molecular orientation and high and balanced charge mobility. As a result, the as-cast devices based on 3T-2 can achieve an excellent PCE of 11.12% processed by non-halogenated o-xylene as the solvent, which is the highest value for terthiophene based FNEAs for OSCs.
Results and discussion
Synthesis
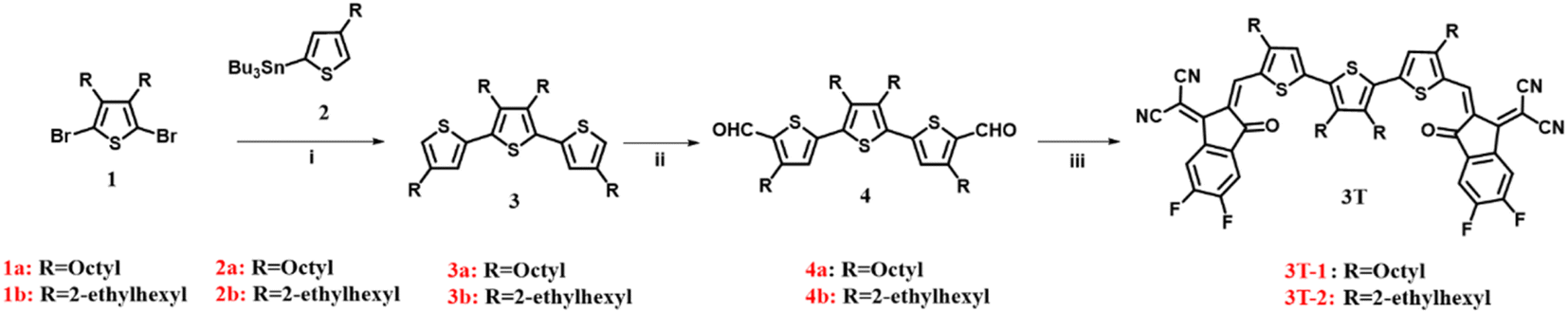
Scheme 1 shows the synthesis of 3T-1 and 3T-2. The starting materials (1 and 2) were prepared according to previously reported procedures.40,41 A Stille coupling of 2,5-dibromo-3,4-alkylthiophene (1) and compound 2 afforded 3a and 3b, respectively. Subsequently, the dialdehydes (4a and 4b) were prepared via the Vilsmeier–Haack reaction. Finally, two terthiophene based acceptors 3T-1 and 3T-2 are obtained in 87% and 91% yields by Knoevenagel condensation with dialdehyde intermediates and 3-(1,1-dicyanomethylene)-5,6-difluoro-1-indanone (2F-IC). The structures of 3T-1 and 3T-2 were confirmed by 1H and 13C NMR spectroscopy and mass spectrometry. 3T-2 are highly soluble in common halogenated solvents such as dichloromethane, chloroform, o-dichlorobenzene and non-halogenated solvents such as o-xylene at room temperature; however, 3T-1 are less soluble in the above-mentioned solvents.
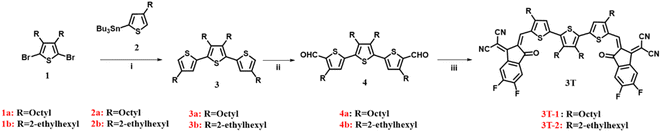 |
| | Scheme 1 The synthetic routes of 3T-1 and 3T-2. Conditions and reagents: (i) Pd(PPh3)4, toluene, 110 °C; (ii) POCl3, DMF, 1,2-dichloroethane, 85 °C; (iii) 3-(1,1-dicyanomethylene)-5,6-difluoro-1-indanone, CHCl3, pyridine, 25 °C. | |
Optical and electrochemical properties
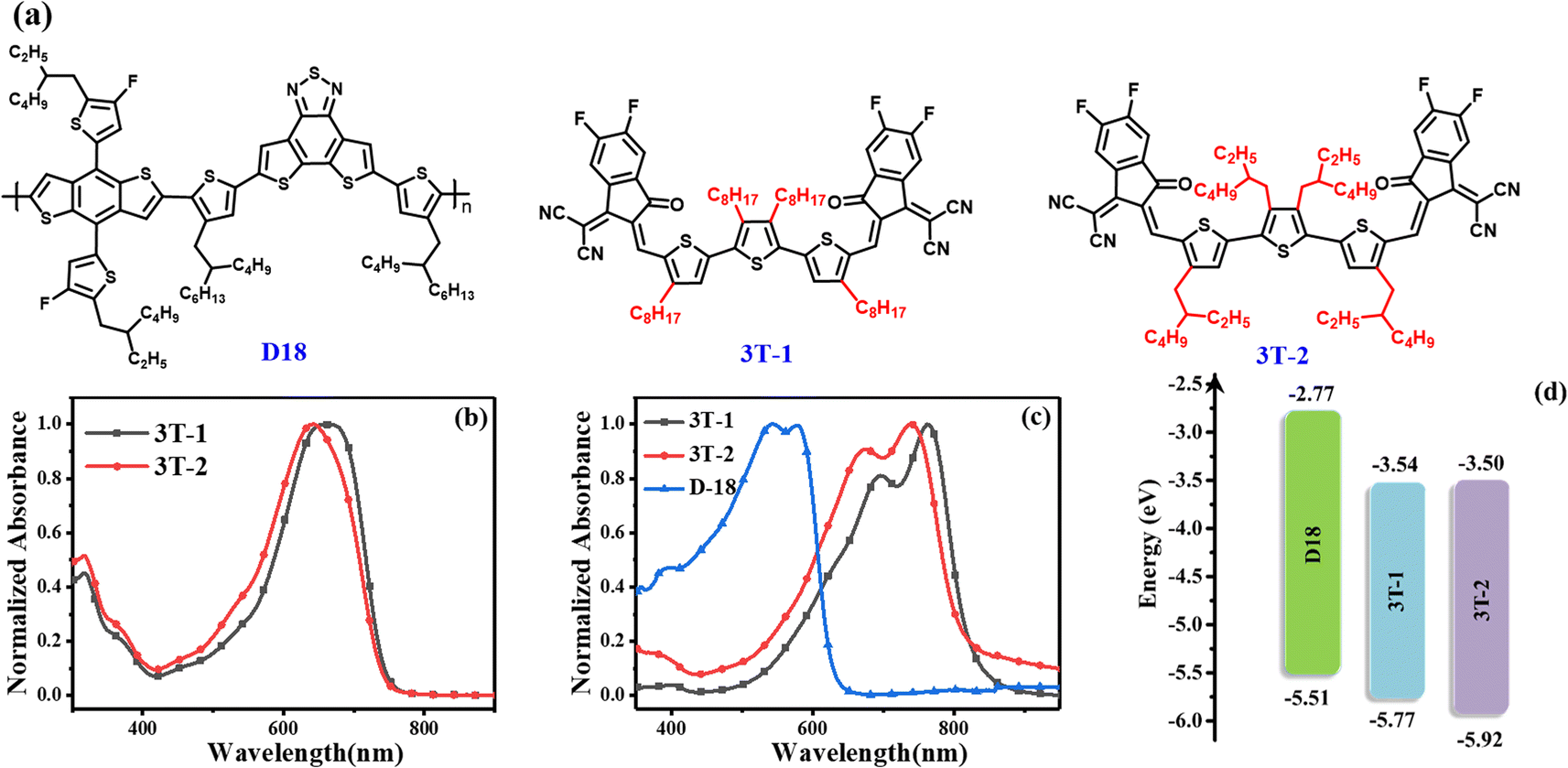
The chemical structures of the two terthiophene based FNEAs (3T-1 and 3T-2) and the wide bandgap polymer donor D18 are shown in Fig. 1a.42 The ultraviolet-visible (UV-vis) absorptions of 3T-1 and 3T-2 characterized in dilute chloroform solutions and as thin films are presented in Fig. 1 and Table 1. The main difference between 3T-1 and 3T-2 is the lateral chains on the molecular skeleton, which has a significant impact on molecular crystallization and the molecular packing in the solid state. Two acceptors 3T-1 and 3T-2 have broad and featureless absorption in solutions with the maximum absorption peaks located at 660 and 642 nm, respectively. Besides, their corresponding molar absorption coefficients are 1.87 × 105 and 1.85 × 105 M−1 cm−1 (Fig. S1†), respectively. From solutions to thin films, the absorptions of 3T-1 and 3T-2 change significantly, and both exhibit 0–0 and 0–1 absorption peaks. Compared to the absorption spectra in solutions, the absorption in the solid films covers the 500 to 850 nm region, and the main absorption peak is located at 762 and 740 nm for 3T-1 and 3T-2, respectively. The large red-shift (∼100 nm) is probably due to the formation of J-aggregation in the solid state. Meanwhile, 3T-1 possesses a more red-shifted and intensified 0–0 peak than 3T-2, which indicates a stronger intermolecular interaction and more ordered molecular packing for 3T-1. Two acceptor molecules and the polymer donor D18 display complementary absorption spectra with coverage from 300 nm to 850 nm, which offers a prerequisite for better utilization of solar radiation. According to the equation Eoptg = 1240/λonset, the optical bandgaps of 3T-1 and 3T-2 can be estimated to be 1.47 and 1.48 eV, respectively. In addition, the energy levels of 3T-1 and 3T-2 are evaluated by the electrochemical method. The cyclic voltammetry curves are exhibited in Fig. S2† and the details are shown in Fig. 1d and Table 1. According to the equation EHOMO/LUMO = −e(Eonset,ox/red − EFc/Fc+ + 4.80), their highest occupied molecular orbital (HOMO) can be determined to be −5.77 and −5.92 eV, and the lowest unoccupied molecular orbital (LUMO) energy levels are −3.54 and −3.50 eV, respectively. 3T-2 shows a slightly high-lying LUMO level, which may be beneficial to obtain a high Voc for the OSCs.
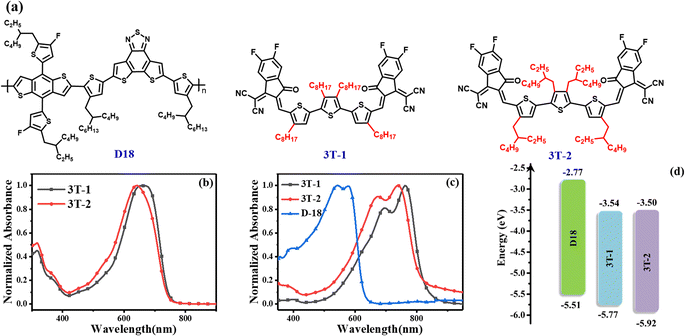 |
| | Fig. 1 (a) Molecular structures of D18, 3T-1 and 3T-2; (b) the normalized absorption spectra of 3T-1 and 3T-2 in chloroform solutions; (c) the normalized absorption spectra of the thin films for D18, 3T-1 and 3T-2; (d) energy level diagrams of the three materials D18, 3T-1 and 3T-2. | |
Table 1 Optical and electrochemical properties of 3T-1 and 3T-2
| Compound |
Solution |
Film |
HOMO (eV) |
LUMO (eV) |
E
optg (eV) |
|
λ
max (nm) |
λ
onset (nm) |
λ
max (nm) |
λ
onset (nm) |
|
3T-1
|
660 |
748 |
762 |
843 |
−5.77 |
−3.54 |
1.47 |
|
3T-2
|
642 |
751 |
740 |
836 |
−5.92 |
−3.50 |
1.48 |
Quantum calculations
Density functional theory (DFT) calculations at the B3LYP/6-31G (d,p) level are carried out to evaluate the optimal molecular geometries of 3T-1 and 3T-2. To simplify the calculations, the octyl and 2-ethylhexyl side chains are simplified to methyl and 1-methylethyl groups, respectively. As shown in Fig. S3,† the side chains of the thiophene ring would produce different influences in the dihedral angle between the center core and two cyanoindanone type terminal groups. For 3T-1, two small dihedral angles are 0.25° and 0.19°. For 3T-2, the dihedral angles are 5.73° and 0.79°, respectively. The more twisted molecular conformation of 3T-2 may be conducive to reduce the molecular crystallinity and suppress the oversized aggregation in the film.
Photovoltaic properties
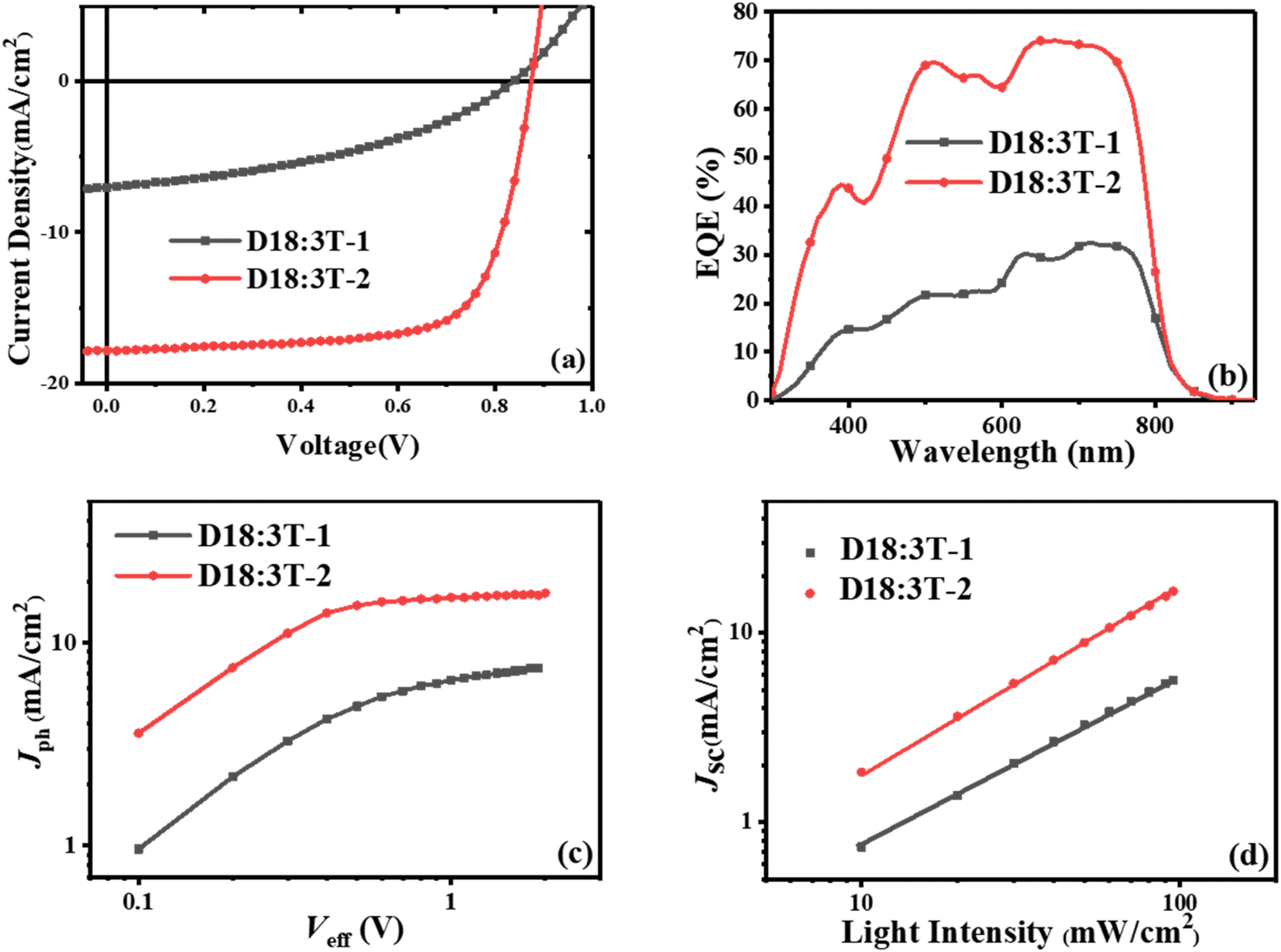
To investigated the photovoltaic properties of 3T-1 and 3T-2, OSCs were fabricated with a typical conventional device structure of ITO/PEDOT:PSS/active layer/PDINN/Ag. Here, a wide-bandgap polymer of D18 was selected as the donor polymer and aliphatic amine-functionalized perylene-diimide (PDINN) was chosen as the cathode interlayer. The device evaluation processes were systematically optimized, including the donor/acceptor ratio, additive concentration and thermal annealing temperature. The optimal photovoltaic performance can be achieved by using non-halogenated o-xylene as the processing solvent and the optimal donor/acceptor ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, without an additive and thermal annealing. The current density–voltage (J–V) curves of the optimized devices are depicted in Fig. 2, and the details are listed in Table 2. Although two terthiophene based acceptors possess the same molecular skeleton, an inferior PCE of 2.43% is obtained with a low Jsc of 7.11 mA cm−2, an FF of 40.80%, and a Voc of 0.84 V for the as-cast 3T-1-based device. When side chains were changed from the linear octyl chains to branched 2-ethylhexyl chains, the as-cast OSCs based on 3T-2 can achieve a higher Voc of 0.87 V due to the higher LUMO energy level of 3T-2 than that of 3T-1. Meanwhile an obviously enhanced Jsc of 17.85 mA cm−2 and an increased FF of 71.21% were achieved for the 3T-2-based device, contributing to a significantly enhanced PCE of 11.12%, which is among the highest values of terthiophene based fully non-fused electron acceptors. The enhancements in both the Jsc and FF values are mainly ascribed to the improved charge transport and film morphology. The external quantum efficiency (EQE) spectra of 3T-1 and 3T-2 based devices are shown in Fig. 2b. The two devices both exhibit a photoelectric response in the range of 300 nm to 850 nm and the one based on 3T-2 even possesses a maximum value of 75%, indicating that an efficient photon harvesting and conversion process takes place in the as-cast thin films. Moreover, the current density integrated by EQE curves was 6.92 and 17.58 mA cm−2 for 3T-1 and 3T-2 based devices, which are all within 5% deviation from the J–V curves.
:1, without an additive and thermal annealing. The current density–voltage (J–V) curves of the optimized devices are depicted in Fig. 2, and the details are listed in Table 2. Although two terthiophene based acceptors possess the same molecular skeleton, an inferior PCE of 2.43% is obtained with a low Jsc of 7.11 mA cm−2, an FF of 40.80%, and a Voc of 0.84 V for the as-cast 3T-1-based device. When side chains were changed from the linear octyl chains to branched 2-ethylhexyl chains, the as-cast OSCs based on 3T-2 can achieve a higher Voc of 0.87 V due to the higher LUMO energy level of 3T-2 than that of 3T-1. Meanwhile an obviously enhanced Jsc of 17.85 mA cm−2 and an increased FF of 71.21% were achieved for the 3T-2-based device, contributing to a significantly enhanced PCE of 11.12%, which is among the highest values of terthiophene based fully non-fused electron acceptors. The enhancements in both the Jsc and FF values are mainly ascribed to the improved charge transport and film morphology. The external quantum efficiency (EQE) spectra of 3T-1 and 3T-2 based devices are shown in Fig. 2b. The two devices both exhibit a photoelectric response in the range of 300 nm to 850 nm and the one based on 3T-2 even possesses a maximum value of 75%, indicating that an efficient photon harvesting and conversion process takes place in the as-cast thin films. Moreover, the current density integrated by EQE curves was 6.92 and 17.58 mA cm−2 for 3T-1 and 3T-2 based devices, which are all within 5% deviation from the J–V curves.
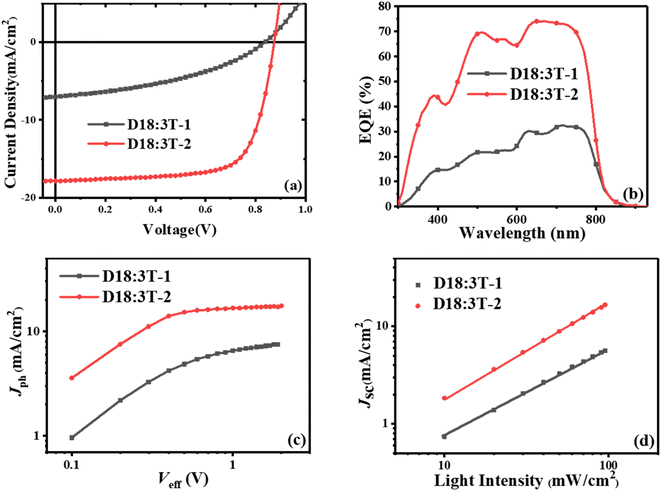 |
| | Fig. 2 (a) J–V curves and (b) EQE curves, (c) photocurrent versus effective voltage (Jph–Veff) characteristics and (d) light intensity dependence of the optimized 3T-1 and 3T-2 based devices. | |
Table 2 Photovoltaic performance of the optimized 3T-1 and 3T-2 based devices
| Active layer |
V
oc (V) |
J
sc (mA cm−2) |
FF (%) |
PCE (%) |
|
D18:3T-1 |
0.84 (0.832 ± 0.007) |
7.11 (6.78 ± 0.17) |
40.80 (41.07 ± 1.36) |
2.43 (2.32 ± 0.09) |
|
D18:3T-2 |
0.87 (0.873 ± 0.005) |
17.85 (17.73 ± 0.23) |
71.21 (69.94 ± 1.04) |
11.12 (10.85 ± 0.25) |
To further investigate the exciton dissociation and charge collection in the 3T-1 and 3T-2-based devices, the photocurrent density (Jph) versus effective voltage (Veff) was measured, and the results are shown in Fig. 2c. Under the short-circuit conditions, the abilities of exciton dissociation and charge recombination can be evaluated from the normalized photocurrent density (Pdiss = Jph/Jsat, where Jsat refers to the saturation photocurrent density). The 3T-1-based device could not achieve an effective Jsat, which indicates that charge recombination occurred in the active layer. In comparison, the Pdiss can be calculated to be 98.3% for the 3T-2-based as-cast device, indicating effective exciton dissociation, charge transport and collection in the active layer. Meanwhile, the charge recombination of 3T-1 and 3T-2-based devices was studied by measuring the current densities under various light intensities (Plight). The Jsc dependence on Plight can be expressed as Jsc ∝ Plightα, where the exponent α value reflects the bimolecular recombination information. As shown in Fig. 2d, the α values of 3T-1 and 3T-2-based devices are estimated to be 0.89 and 0.98, respectively. The α value of the device based on 3T-2 is close to unity, indicating that the bimolecular recombination can be effectively suppressed in the active layer, which is beneficial for obtaining a high FF for OSCs.
Charge transport mobilities
In order to study the influence of side chains on charge transport properties, space-charge-limited-current (SCLC) measurements were carried out to measure the hole and electron mobilities. The structures of hole-only and electron-only based devices are ITO/PEDOT:PSS/active layer/Au and ITO/ZnO/active layer/PDINN/Ag, respectively. The detailed results are summarized in Table 3 and Fig. S4.† The D18:3T-1 blend film shows inferior hole mobilities (μh) and electron mobilities (μe) of 5.77 × 10−5 and 1.29 × 10−5 cm2 V−1 s−1, respectively. In comparison, the D18:3T-2 blend film can achieve a high μh and μe of 1.99 × 10−4 and 1.49 × 10−4 cm2 V−1 s−1. And the corresponding μh/μe ratio is calculated to be 4.47 and 1.34 for 3T-1 and 3T-2 based blends, respectively. The excellent μe nearly ten times higher than that of the 3T-1-based blend and more balanced charge transport account for the higher Jsc and FF in the 3T-2-based devices, and thus, a better device performance.
Table 3 Charge transport mobilities of 3T-1 and 3T-2 based blend films
| Active layer |
μ
h [cm2 V−1 s−1] |
μ
e [cm2 V−1 s−1] |
μ
h/μe |
|
D18:3T-1 |
5.77 × 10−5 |
1.29 × 10−5 |
4.47 |
|
D18:3T-2 |
1.99 × 10−4 |
1.49 × 10−4 |
1.34 |
Film morphology
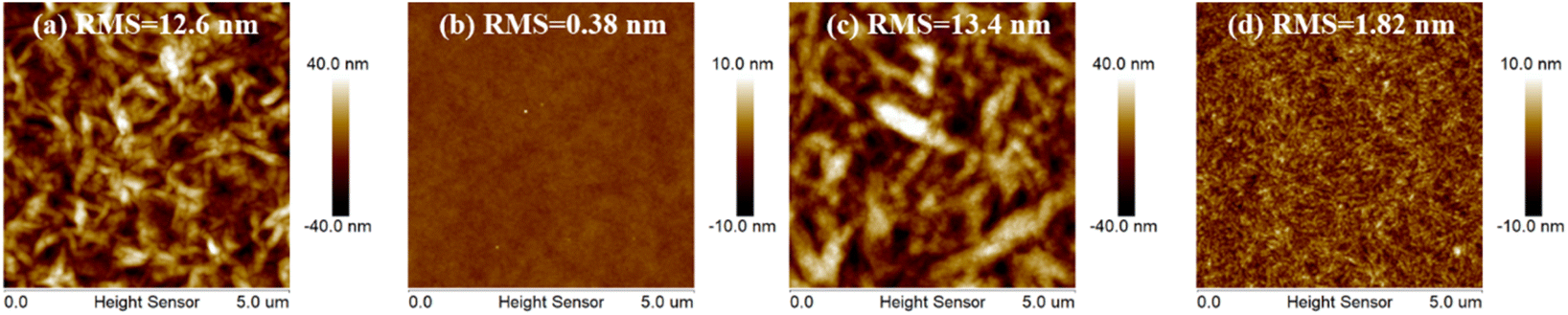
Atomic force microscope (AFM) measurements were utilized to explore the crystallinity properties and aggregation behaviors for 3T-1 and 3T-2 (Fig. 3). Due to the strong crystallinity of 3T-1, the neat film exhibits quite rough a surface with an extremely large root-mean-square (RMS) value of 12.6 nm, while a smooth surface can be observed in the pure 3T-2 film with a very small RMS value of 0.38 nm. When blending with polymer D18, the D18:3T-1 blend film shows some bright-dark areas with a large RMS value of 13.4 nm, indicating large size phase separation, which is due to the poor compatibility and strong crystallinity of 3T-1. In comparison, the D18:3T-2 blend film displays a uniform surface morphology without large size aggregates formed, which can facilitate exciton dissociation and charge transfer.
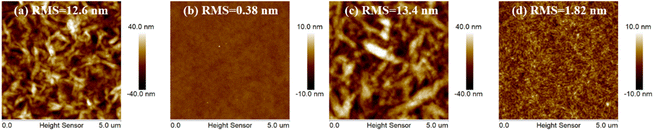 |
| | Fig. 3 AFM images of the pure and blend films: (a) the neat 3T-1 film, (b) the neat 3T-2 film, (c) the D18:3T-1 blend film, and (d) the D18:3T-2 blend film. | |
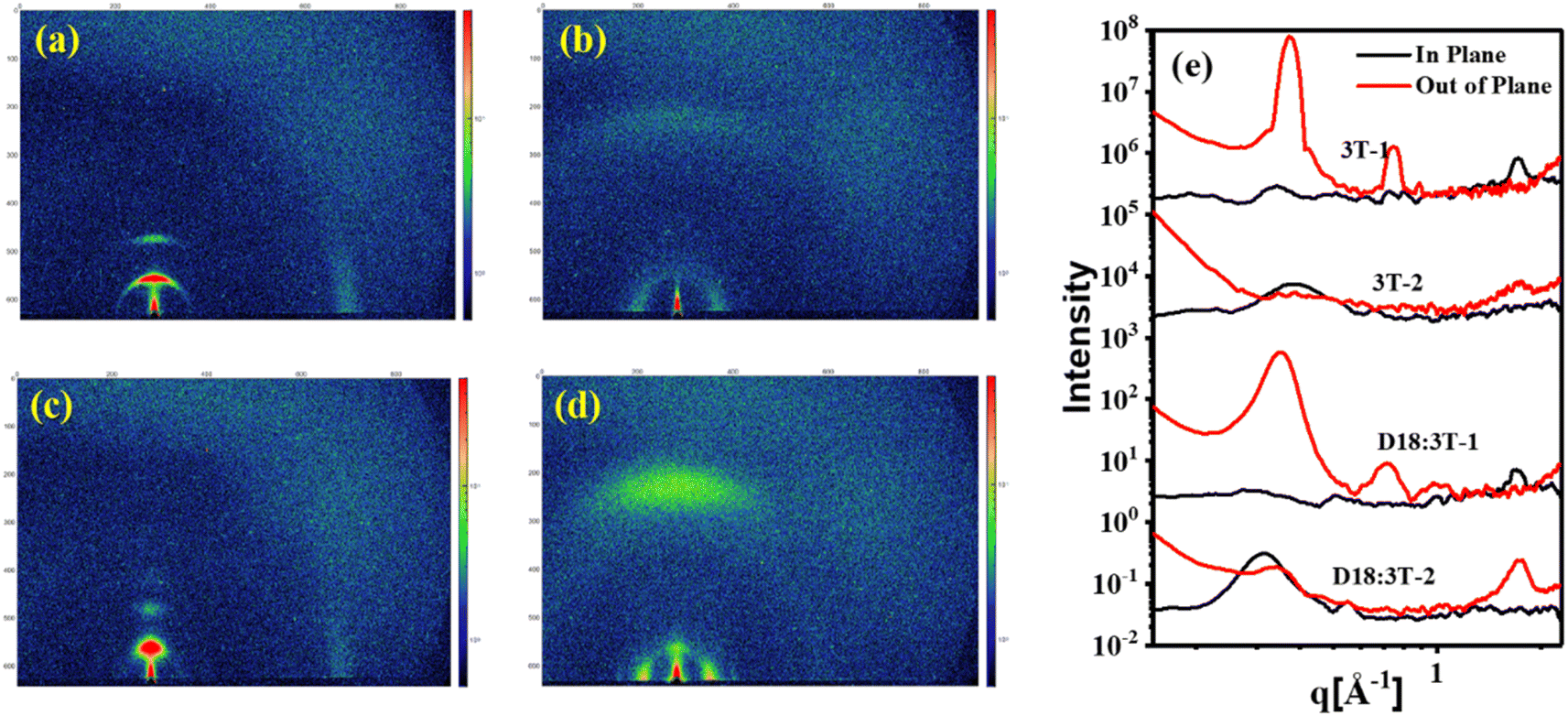
To gain insight into the impacts of side chains on the molecular orientation and crystalline properties in the pure and blend films, the grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed. As shown in Fig. 4, for the neat 3T-1 film, distinguishable (010) π–π stacking diffraction peaks at q = 1.72 Å−1 (d = 3.65 Å) in the in-plane (IP) direction can be observed, indicating the edge-on orientations. The pronounced lamellar stacking peaks (100) and (200) at q = 0.37 Å−1 and q = 0.74 Å−1 in the out-of-plane (OOP) direction suggest the highly ordered structure of 3T-1, with the corresponding lamellar stacking distances of 16.97 Å. For the neat 3T-2 film, an extremely weak (010) π–π stacking diffraction peak in the OOP direction and a (100) lamellar diffraction peak in the IP direction can be observed, indicating a face-on molecular orientation. In addition, the crystal coherence length (CCL) values of the (100) diffraction are 161.4 and 41.7 Å for the neat 3T-1 and 3T-2 films, respectively. After blending with polymer donor D18, the independent GIWAXS scattering signals originating from 3T-1 are still maintained in the D18:3T-1 blend film, demonstrating that poor miscibility and large phase separation exists in the active layer, which agrees well with the morphology study (vide supra). The D18:3T-2 blend film mainly adopted face-on orientations with an obvious (010) diffraction peak in the OOP direction, which is beneficial for improving the vertical charge transport. Additionally, the D18:3T-1 blend film shows a larger CCL value of 92.81 Å for the (100) diffraction in the OOP direction. In comparison, a markedly reduced CCL value of 69.26 Å for the D18:3T-2 blend film can be observed. The GIWAXS test results are consistent with AFM measurement. All the observations indicate that a branched side chain can efficiently adjust the molecular orientation and crystallization behavior.
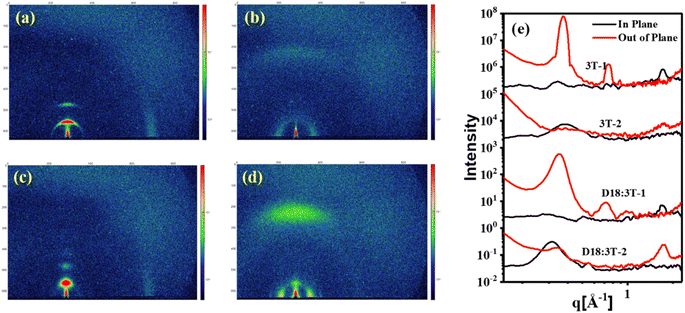 |
| | Fig. 4 GIWAXS 2D patterns of (a) the neat 3T-1 film, (b) the neat 3T-2 film, (c) the D18:3T-1 blend film, and (d) the D18:3T-2 blend film, and (e) the corresponding 1D line-cuts. | |
Femtosecond transient absorption spectra
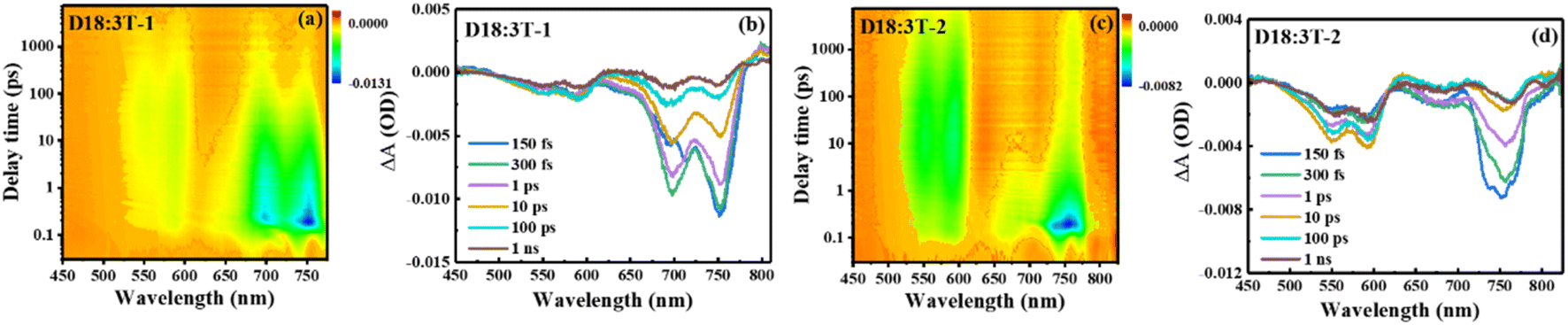
Femtosecond transient absorption (fs-TA) spectra were measured to evaluate the charge transfer dynamic process of the blend films and the results are shown in Fig. 5 and S5.† A pump wavelength of 730 nm is used to selectively excite the acceptors without the donor. For the acceptor films, two ground-state bleaching (GSB) bands mainly appear at about 750 and 697 nm for 3T-1, and a wide GSB band mainly appears at about 750 for 3T-2. In comparison with the neat acceptors, a slowly increasing negative signal can be significantly observed with a time delay covering the range of 500–630 nm for the D18:3T-2 blend film, matching well with the absorption features of D18, which indicates that an efficient hole transfer process exists at the interface of the donor and acceptor. Subsequently, the recovery signals of blend films at 592 nm are fitted with a double exponential function, and the results are shown in the ESI.† The fast signal represents the recombination of donor excitons, and the slow signal can be considered as the recovery of charge separation excitons. The D18:3T-2 blend film possesses the longest charge separated state lifetime, which can achieve efficient charge transport and photovoltaic performance for OSCs.43
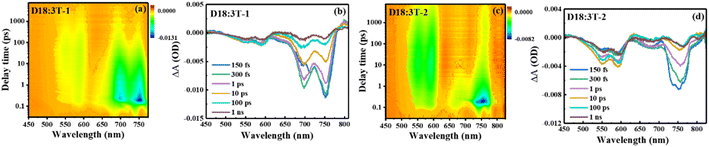 |
| | Fig. 5 . (a) and (c) Contour plots of time-resolved absorption difference spectra of the D18:3T-1 and D18:3T-2 blend film; (b) and (d) TA spectra of the D18:3T-1 and D18:3T-2 blend film recorded at selected decay times. | |
Conclusion
In summary, we have designed and synthesized two terthiophene based fully non-fused acceptors 3T-1 and 3T-2 bearing octyl and 2-ethylhexyl side chains for application in highly efficient OSCs. The two acceptors both possess a simple molecular structure and low cost compared with fused ring electron acceptors. Octyl substituted 3T-1 possesses strong crystallinity and poor miscibility, and its blend film with D18 exhibits an apparent phase separation morphology, leading to an inferior PCE of 2.43%. As for 2-ethylhexyl substituted 3T-2, it exhibits good solubility and forms an appropriate morphology with a face-on molecular orientation, which results in high and balanced charge mobility in the blend film. Finally, a markedly improved PCE of 11.12% can be achieved processed by non-halogenated o-xylene as the solvent, which is the highest value based on terthiophene based non-fused acceptors for OSCs. In all, our research can pave an effective way to design fully non-fused ring acceptors for high performance OSC application.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075069, 21871022, 22005018, 22175013, 51933001 and U1704137), the Natural Science Foundation of Henan Province (212300410002), the Program sponsored by Henan Province (No. 23ZX002 and ZYQR201912163), the Department of Education Project-Henan Province, and the Basic Research Project of Key Scientific Research Project (23A430003).
References
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Liu, L. Meng, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2022, 65, 1457–1497 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- Y. Liu, B. Liu, C.-Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z.-G. Zhang and Z. Bo, Sci. China: Chem., 2021, 65, 224–268 CrossRef.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- J. Song and Z. Bo, Chin. Chem. Lett., 2023, 108163 CrossRef.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- M. Li, Y. Zhou, J. Zhang, J. Song and Z. Bo, J. Mater. Chem. A, 2019, 7, 8889–8896 RSC.
- P. Bi, S. Zhang, Z. Chen, Y. Xu, Y. Cui, T. Zhang, J. Ren, J. Qin, L. Hong, X. Hao and J. Hou, Joule, 2021, 5, 2408–2419 CrossRef CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- M. Deng, X. Xu, Y. Duan, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 2210760 CrossRef CAS PubMed.
- X. Zhang, L. Qin, J. Yu, Y. Li, Y. Wei, X. Liu, X. Lu, F. Gao and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 12475–12481 CrossRef CAS PubMed.
- M. Li, S. Feng, S. Shen, H. Huang, W. Xue, N. Yu, Y. Zhou, W. Ma, J. Song, Z. Tang and Z. Bo, Chem. Eng. J., 2022, 438, 135384 CrossRef CAS.
- H. Huang, Q. Guo, S. Feng, C. Zhang, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu, Z. Tang, W. Ma and Z. Bo, Nat. Commun., 2019, 10, 3038 CrossRef PubMed.
- X. Wang, H. Lu, Y. Liu, A. Zhang, N. Yu, H. Wang, S. Li, Y. Zhou, X. Xu, Z. Tang and Z. Bo, Adv. Energy Mater., 2021, 11, 2102591 CrossRef CAS.
- X. Zheng, W. Liu, H. Wang, X. Man, G. Ran, X. Yu, H. Lu, Z. Bi, Y. Liu, A. Zhang, W. Ma, X. Xu, Z. Tang, W. Zhang and Z. Bo, Cell Rep. Phys. Sci., 2022, 3, 101169 CrossRef CAS.
- Y. N. Chen, M. Li, Y. Wang, J. Wang, M. Zhang, Y. Zhou, J. Yang, Y. Liu, F. Liu, Z. Tang, Q. Bao and Z. Bo, Angew. Chem., Int. Ed., 2020, 59, 22714–22720 CrossRef CAS PubMed.
- Z. P. Yu, Z. X. Liu, F. X. Chen, R. Qin, T. K. Lau, J. L. Yin, X. Kong, X. Lu, M. Shi, C. Z. Li and H. Chen, Nat. Commun., 2019, 10, 2152 CrossRef PubMed.
- Z. Yao, Y. Li, S. Li, J. Xiang, X. Xia, X. Lu, M. Shi and H. Chen, ACS Appl. Energy Mater., 2020, 4, 819–827 CrossRef.
- X. Ding, X. Chen, Y. Xu, Z. Ni, T. He, H. Qiu, C.-Z. Li and Q. Zhang, Chem. Eng. J., 2022, 429, 132298 CrossRef CAS.
- X. Zheng, W. Liu, H. Lu, N. Yu, Y. Wang, H. Huang, S. Li, X. Wang, H. Wang, Y. Liu, X. Xu, Z. Tang and Z. Bo, Chem. Eng. J., 2022, 444, 136472 CrossRef CAS.
- J. Gao, X. Zhu, H. Bao, J. Feng, X. Gao, Z. Liu and Z. Ge, Chin. Chem. Lett., 2022, 107968 CrossRef.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed.
- J. Zhou, X. Wan, Y. Liu, Y. Zuo, Z. Li, G. He, G. Long, W. Ni, C. Li, X. Su and Y. Chen, J. Am. Chem. Soc., 2012, 134, 16345–16351 CrossRef CAS PubMed.
- T. Lee, Y. Eom, C. E. Song, I. H. Jung, D. Kim, S. K. Lee, W. S. Shin and E. Lim, Adv. Energy Mater., 2019, 9, 1804021 CrossRef.
- J. Zhu, C. Yang, L. Ma, T. Zhang, S. Li, S. Zhang, H. Fan and J. Hou, Org. Electron., 2022, 105, 106512 CrossRef CAS.
- Y. Zhou, M. Li, H. Lu, H. Jin, X. Wang, Y. Zhang, S. Shen, Z. Ma, J. Song and Z. Bo, Adv. Funct. Mater., 2021, 31, 2101742 CrossRef CAS.
- L. Ma, S. Zhang, J. Ren, G. Wang, J. Li, Z. Chen, H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 2214088 Search PubMed.
- H. Lu, X. Wang, S. Li, D. Li, N. Yu, Z. Tang, Y. Liu, X. Xu and Z. Bo, Chem. Eng. J., 2022, 435, 134987 CrossRef CAS.
- C. Yang, L. Ma, Y. Xu, J. Ren, J. Hou and S. Zhang, Sci. China: Chem., 2022, 65, 2604–2612 CrossRef CAS.
- L. Ma, S. Zhang, J. Zhu, J. Wang, J. Ren, J. Zhang and J. Hou, Nat. Commun., 2021, 12, 5093 CrossRef CAS PubMed.
- S. Ko, E. Verploegen, S. Hong, R. Mondal, E. T. Hoke, M. F. Toney, M. D. McGehee and Z. Bao, J. Am. Chem. Soc., 2011, 133, 16722–16725 CrossRef CAS PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto, T. Mori and T. Michinobu, Adv. Funct. Mater., 2017, 27, 1604608 CrossRef.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- Y. Dong, H. Cha, H. L. Bristow, J. Lee, A. Kumar, P. S. Tuladhar, I. McCulloch, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2021, 143, 7599–7603 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |

 a,
XiaoDan
Tang
a,
ChaoChao
Qin
e,
Jinsheng
Song
a,
XiaoDan
Tang
a,
ChaoChao
Qin
e,
Jinsheng
Song