
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of platinum group metal nanoparticles assisted by CO2 reduction and H2 cogeneration at gas-diffusion electrodes†
Omar
Martinez-Mora
 ab,
Guillermo
Pozo‡
a,
Luis Fernando
Leon-Fernandez
a,
Jan
Fransaer
b and
Xochitl
Dominguez-Benetton
*a
ab,
Guillermo
Pozo‡
a,
Luis Fernando
Leon-Fernandez
a,
Jan
Fransaer
b and
Xochitl
Dominguez-Benetton
*a
aSeparation and Conversion Technologies, VITO, Flemish Institute for Technological Research, Boeretang 200, 2400, Mol, Belgium. E-mail: xoch@vito.be
bDepartment of Materials Engineering, Surface and Interface Engineered Materials, Katholieke Universiteit Leuven, Kasteelpark Arenberg 44 – box 2450, 3001 Leuven, Belgium
First published on 17th April 2023
Abstract
The electrochemical reduction of CO2 at gas-diffusion electrodes offers a green pathway to the synthesis of elemental nanoparticles (NPs) of the platinum group metals (PGMs) in aqueous media. The H2 and CO co-generated act on the direct reduction of the Pt, Pd and Rh chlorides. The CO2 equilibrium reactions are essential to obtain nanoparticles. The recovery and upcycling of the PGMs and the synthesis of PGM NPs are applications of the novel gas-diffusion electrocrystallization (GDEx) process portrayed.
Sustainability spotlightIndustrial transformations and recycling of the platinum group metals (PGMs) must transition towards electricity-driven alternatives (powered by renewable energy sources). Our work presents an electrochemical process, also aiming at avoiding the addition of bulk chemicals. Aside from the PGM ions to be transformed, our process only requires CO2 as a feed. All other reagents to achieve the conversions targeted are produced in situ, in small amounts. No excess chemicals need to be stored or disposed. Furthermore, it works under mild operating conditions (room temperature and atmospheric pressure). Remarkably, we achieve PGM removals of >90%, in short periods of time. Our work aligns with the UN sustainability goals: industry, innovation, and infrastructure (SDG 9) and climate action (SDG 13). |
Metal-based nanoparticles (NPs) are a lively research subject since metals hold different properties vs. their bulk materials when they are at the nanometric scale. Especially, the platinum group metals (PGMs; e.g., Pt, Pd, and Rh) display unique catalytic properties.1 PGMs, although expensive and with limited abundance in the Earth's crust, are yet the best-known catalysts for proton exchange membrane fuel cells (PEMFC) or direct alcohol fuel cells (DAFC).2,3 Pt remains the best catalyst for the oxygen reduction reaction (ORR), even in alkaline media,4 whereas Pd is best for methane oxidation,5 and Rh excels as a catalyst for hydrogenation and hydroformylation reactions.6 NPs are preferred for catalysis due to their increased surface area and, thus, high reactivity with lower PGM loading.
Amongst available methods, chemical reductions are the most extended for the synthesis of PGM NPs.7 Hydrogen (H2) gas has been used as a reducing agent since first proposed by Rampino and Nord in 1941 for the synthesis of colloidal Pt and Pd NPs.8,9 Besides, to our knowledge there are no reports in which H2 has been used for the same purpose for the synthesis of Rh NPs. Carbon monoxide (CO), as a reducing agent, has also been reported for the synthesis of shape-controlled Pd NPs.10 In addition, CO has also been used to control the size of Pt NPs during synthesis using other reducing agents.11,12 In this context, a mixture of H2 and CO has been reported for the synthesis of carbon-supported Pt NPs by Zhang and colleagues (2013). They found that in the absence of CO, pure H2 produces big and polydisperse particles, while in the presence of CO, monodisperse Pt nanocubes are formed.13
In most of these reports, H2 and/or CO are introduced to the reaction system by either bubbling the gases into the solution9 or by flowing them at different temperatures in special furnaces.13 There are disadvantages of these methods, as in some cases high temperatures (i.e. 100–180 °C) are required. Besides the inherent safety risks of working with pure H2 and CO, even at room temperature, H2 has a wide explosion range (4–75%)14 and CO is extremely toxic at levels as low as 10 ppm.15
In this work, we propose the synthesis of PGM NPs (Pt, Pd, and Rh) via the use of gas-diffusion electrodes (GDEs) in which the reduction of CO2 is conducted in a flow cell with an aqueous medium. The electrolysis of water occurs simultaneously and, thus, both H2 and CO are cogenerated in situ while a solution containing PGM chlorides passes by. The reduction of PGM ions in solution is novel and brings sustainability benefits. This process has the advantage that it is carried out at room temperature and H2 and CO are produced in small amounts eliminating the risks involved when working with these gases. There are a few reports in which H2 is electrochemically generated in situ from the water reduction reaction (WRR) and used to synthesize Pt nanoparticles with defined crystallographic orientations.16 However, to our knowledge no similar reports exist for Pd and Rh. Alternatively, the proposed method can also be used as a strategy to selectively remove PGMs dissolved in a solution and recover them as sedimented NPs.
We recently disclosed the use of GDEs for the electrosynthesis of nanoparticles of iron oxide,17 copper and zinc hydroxychlorides (spin transition materials),18 scorodite,19 and mixed metal (hydr)oxide libraries (i.e., birnessite, layered double hydroxides, and spinels),20 by a process we called gas-diffusion electrocrystallization (GDEx). In these cases, oxygen was reduced on gas-diffusion cathodes, producing hydroxyl ions (OH−) and strong oxidants (e.g., H2O2 and HO2−), acting as reactive intermediates for the formation of stable nanoparticles. Analogously, in the present work, GDEx involved the use of CO2 as the gas feedstock to be electrochemically reduced. In this case, when the GDE is polarized at an appropriate potential (e.g., circa −1.0 VAg/AgCl), the CO2 reduction reaction (CRR) generates CO and the simultaneous water reduction reaction (WRR) produces H2. H2 and CO act, thus, as reducing and size-controlling agents, respectively, for the water-soluble platinum group metal (PGM) precursors supplemented in the electrolyte, i.e., H2PtCl6, PdCl2, and RhCl3.
The materials and methods employed to conduct this research are found in the ESI (ESI-1).† Briefly, the electrochemical reactor consisted of 3 chambers: (a) one containing a VITO CoRE® multi-layered carbon-based GDE (acting as a cathode) and a Ag/AgCl reference electrode, (b) a second one containing a Pt-coated tantalum electrode (acting as an anode), and (c) the third one consisting of a gas compartment through which CO2 was fed with an overpressure of 20 mbar (g) to percolate through the hydrophobic backing of the GDE. The catholyte and anolyte compartments were separated by an anion exchange membrane (FUMASEP® FAP-4130-PK). NaCl 0.5 M adjusted to pH 3 with HCl was used as supporting electrolyte. Unless otherwise stated, the PGM NPs were synthesized using chronopotentiometry experiments at −10 mA cm−2.
The CRR on carbon-based electrodes and in aqueous electrolytes yields (primarily) either carbon monoxide (CO) or formic acid, under the conditions studied.21,22 CO was qualitatively detected to be present in the system using a portable CO detector in all experiments carried out, while traces of formic acid were detected by HPLC (at ppm levels) only in the experiments involving Pd. Thus, under the conditions studied here, CO is the major product of CO2 reduction in our process (reactions (1) and (2)). Moreover, the WRR producing H2 takes place, simultaneously (reaction (3)).23
| CO2 + 2H+ + 2e− ⇌ CO + H2O E0 = −0.106 VSHE | (1) |
| CO2 + 2H2O + 2e− ⇌ CO + 2OH− E0 = −0.934 VSHE | (2) |
| 2H2O + 2e− ⇌ H2 + 2OH− E0 = −0.828 VSHE | (3) |
Fig. 1 (gray lines) shows the evolution of pH as the CRR progressed as a function of the charge per volume, in the absence of PGMs (blank). Starting from acidic conditions (i.e., pH ∼3), an increase of the bulk pH up to ∼6 was measured when only 500 C L−1 had been consumed; after that, the pH remained buffered until ∼7.5 by the end of the experiment (104 C L−1 consumed). Both, the CRR to CO (reactions (1) and (2)) and the WRR to H2 (reaction (3)) yield OH−, resulting in a fast increase of the pH in the electrolyte.24,25 In a weakly alkaline environment, unreacted CO2 dissolves in the catholyte (reaction (4)) as HCO3−. The latter is further deprotonated under alkaline conditions to CO32− (reaction (5)).
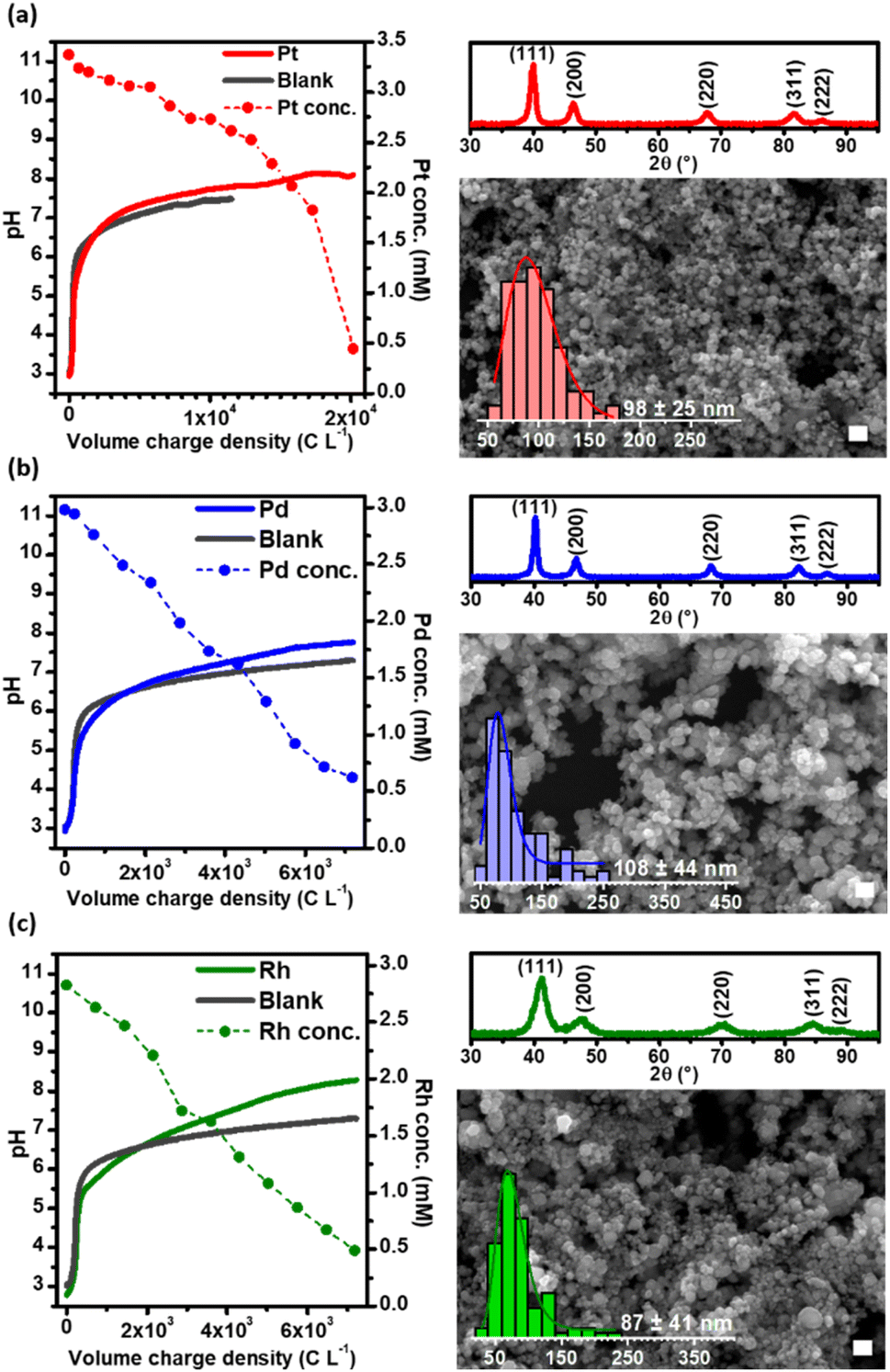 | ||
| Fig. 1 (Left) Evolution of pH and concentration of dissolved platinum group metal ions as a function of the charge consumed per unit volume of catholyte, throughout the GDEx process yielding elemental nanoparticles. (Right) X-ray diffraction patterns (up) and SEM micrographs and distribution histograms (down) of the elemental nanoparticles. The white scale bar is 250 nm. (a) Pt; (b) Pd; (c) Rh. | ||
Both equilibrium reactions consume part of the OH− anions generated at the cathode, resulting in the buffering of the electrolyte bulk.24
| CO2 + OH− ⇌ HCO3− | (4) |
| HCO3− + OH− ⇌ CO32− + H2O | (5) |
Upon addition of PGM ions to the solution (i.e., [PtCl6]2−, [PdCl4]2−, and [RhCl6]3−), the initially yellow to orange translucent solutions progressively turned into black dispersions (Fig. S2, ESI-II†). The evolution of pH and PGM concentration are shown in Fig. 1, as a function of the volumetric charge density while the chronopotentiometric curves are shown in Fig. S3, ESI-II.†
As can be seen in Fig. 1, the evolution of pH, as a function of the volumetric charge density applied in the three cases is, to some extent, comparable to that of the control experiment with the background electrolyte. A rapid increase was observed from pH 3 to 6 within the first 500–1000 C L−1 consumed, after which a buffering plateau was established, reaching a pH value of ∼8 by the end of the experiments. Fig. 1 shows that the concentration of Pd and Rh in solution decreases linearly after the electrode is polarized, while the depletion of Pt ions only starts after a delay period. The delay in the depletion of Pt suggests that the reduction of [PtCl6]2− to Pt0 is a two-step process in which [PtCl6]2− is first reduced by H2 to [PtCl4]2− and then further reduced to Pt0 while [PdCl4]2− and [RhCl6]3− are reduced directly to the zero-valence form. Cameron and Bocarsly (1985) pointed out that Pt metal formation does not occur until a ∼90% yield of [PtCl4]2− has been accumulated, as [PtCl6]2− acts as an inhibitor of Pt metal formation,26 explaining why more charge is needed to deplete Pt from the solution when compared to Pd and Rh. Thus, under GDEx conditions, the formation of PGM NPs by the electrogenerated H2 in the GDEx process can be expressed as follows, for each metal involved:
| [PtCl6]2− + H2 → [PtCl4]2+ + 2H+ + 2Cl− | (6) |
| [PtCl4]2− + H2 → Pt0 + 2H+ + 4Cl− | (7) |
| [PdCl4]2− + H2 → Pd0 +2H+ + 4Cl− | (8) |
| 2[RhCl6]3− + 3H2 → 2Rh0 + 6H+ + 12Cl− | (9) |
Our results suggest that the reduction of the metal cations to elemental nanoparticles is driven by a homogeneous chemical reduction reaction as is clear from the linear decrease of the PGM ion concentration over time, as opposed to an exponential concentration decrease which is typical for heterogeneous reactions.27 Hence, the total charge consumed is related to the simultaneous production of H2 and CO. Some electrochemical reduction of the metals could also take place at the surface of the electrode, as it is evidenced with Pd, in which the Pd deposited on the electrode can catalyse the reduction of CO2 to formic acid.28 However the electrodeposition is very limited due to the low PGM ion concentrations and the associated mass transport limitations.29
The products obtained were characterized using X-ray diffraction (XRD) and scanning electron microscopy (SEM). The diffractograms of the PGM NPs are shown in Fig. 1 (right, up). The corresponding reflections of the crystal lattice for each peak are indicated (Pt, PDF 00-001-1194; Pd, PDF 03-065-6174; Rh, PDF 03-065-2866). The broad diffraction peaks suggest that the grains are nanosized. The grain sizes calculated from XRD using Scherrer's equation,30 were 9.2 nm, 12.4 nm and 4.5 nm for Pt, Pd, and Rh, respectively. SEM was used to identify the NP morphology and to estimate the particle size in the solid-state (Fig. 1, right, bottom. The full-size images are presented in Fig. S4†). The individual nanoparticles had near-spherical shape with size diameters in the range of 98 ± 25 nm, 108 ± 44 nm and 87 ± 41 nm for Pt, Pd, and Rh, respectively. These NPs are larger when compared to NPs synthesized in previous literature, using either bubbled H2 as a reducing agent or bubbled CO as a size controller/reducing agent (for Pt and Pd NPs) (see Table S1 in ESI-III†), however, it should be noted that in our process neither surfactants, capping agents or supporting materials were used and the process was not optimized to purposefully decrease the size of the NPs.
The usability of the GDEx process for the selective recovery of PGMs was demonstrated using Rh as a model (ESI-IV†). Rh3+ solutions (1.0 to 6.0 mM) containing Al3+ 185 mM, Mg2+ 41 mM and Fe2+ 18 mM and HCl 1 M were treated with GDEx, and Rh0 was recovered as the main product (Fig S5, ESI-IV†) with more than 99% removal from the solution while the other metals remained in solution (Table S2, ESI-IV†). The composition of the synthetic solution employed is based on published work on a novel method for the leaching of PGMs from spent autocatalyst monoliths, i.e., microwave assisted leaching (MWAL).31 The potential use of MWAL followed by GDEx for PGM recovery has been suggested for providing a complete and sustainable recycling flow sheet for such kind of end-of-life materials. The present article primarily focuses on explaining the underlying mechanism of how the GDEx process works in the presence of PGM ions. Yet, it also provides preliminary evidence on the applicability of this method for the sustainable recovery of PGMs from solution.32
Additional experiments were carried out to corroborate the mechanism for the synthesis of PGM NPs here portrayed. In the first experiment, CO2 was replaced by Ar in the gas compartment, while the cathode was polarized at the same current density. Under these conditions, only the WRR takes place, and the pH increases to alkaline values, as the CO2 equilibrium does not occur (Fig. S6a, ESI-V†). The same trend was observed upon the addition of metals, in which, Pd and Rh ions precipitated as hydrated PdO, and Rh(OH)3 (Fig. S6b and c, ESI-V†), respectively. For Pt, only a change in colour of the solution, from yellow to brown-orange, was noted, when a similar charge was consumed, as in the experiments with CO2. The colour change is attributed to reduction of Pt4+ to Pt2+.33 Furthermore, it has been reported that the reduction rate of Pt2+ to Pt0 at pH 11 is reduced by at least one order of magnitude than at pH 7 (i.e., the reduction of the same amount of Pt2+ requires 25 min at pH 7 and 60 h at pH 11),34 explaining why the precipitation of metallic Pt was not observed during the experimental time. Thus, the CO2 equilibrium that buffers the pH in the GDEx process is essential to avoid the formation of Pd and Rh (hydr)oxides instead of metallic NPs. And, for Pt, whose hydroxy complexes are soluble in water, to have an optimal pH so that the reduction to Pt0 can take place.
In the second experiment, Ar was flown through the gas compartment of the reactor and CO2 was bubbled into the catholyte solution. Under the same polarization conditions, besides the WWR taking place, the CO2 equilibrium in the bulk electrolyte occurs but not the CRR to CO at the GDE. In this case the pH of the bulk electrolyte was buffered at ∼6 and the precipitates obtained were also identified as metallic nanocrystals of the three metals (Fig. S7, ESI-VI†). However, SEM micrographs evidenced that the individual particles were bigger and highly dispersed compared to their counterparts made with the GDEx process (Fig. S6, ESI-VI†). Under this condition, the average particle size was 316 ± 243 nm and 264 ± 130 for Pt and Pd respectively, while big agglomerates with no defined shape were found for Rh. CO can strongly chemisorb to the surface of all PGMs, inhibiting the growth kinetics of the nanoparticles and controlling their size.13 These results highlight the importance of the CRR to CO at the GDE for forming small and monodisperse metallic nanoparticles using the GDEx process.
Hence, based on the results from the different experimental conditions, we propose that the formation of PGM NPs in the GDEx process occurs as schematized in Fig. 2: after polarizing the GDE, the CO2 reduction and water reduction reactions lead to the formation of H2(1) and CO (2) at the vicinity of the triple phase boundary of the GDE. H2, reduces the metal ions in solution and small metal clusters are formed that are constantly recirculated (3). The interaction of CO with these clusters prevents their further growth and allows relatively small and monodisperse nanoparticles to be obtained. In contrast, if CO is not present the clusters continue to grow to a micrometric scale. Simultaneously the equilibrium of unreacted CO2(4) in the catholyte keeps the electrolyte solution at an optimal pH for the process to take place and avoids the formation of (hydr)oxide phases.
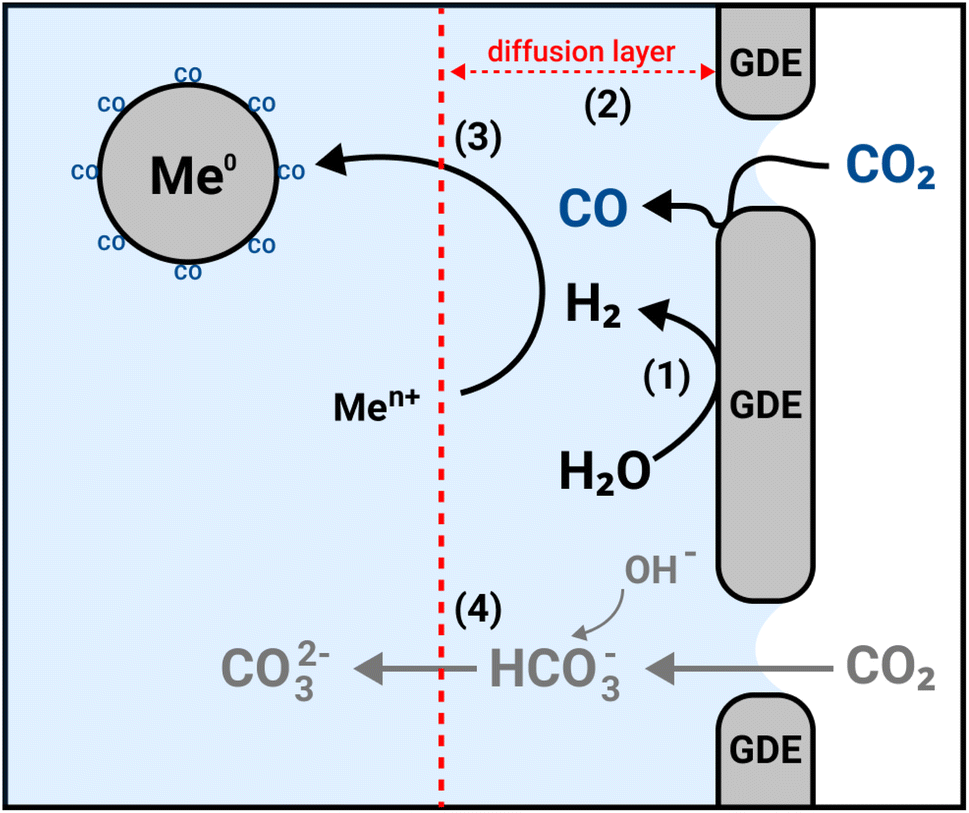 | ||
| Fig. 2 Schematic representation of the electrochemical reactor of the gas-diffusion electrocrystallization (GDEx) for the synthesis of PGM NPS. In GDEx, the H2 from the water reduction reaction (WRR) (1), acts as a reducing agent while CO from the CO2 reduction reaction (CRR) (2) acts as a size controller for the synthesis of elemental nanoparticles from metal ions in solution (3). The equilibrium of unreacted CO2 to HCO3− and CO32− (4) is crucial to consume the OH− generated in the WRR and CRR and interfere with the process. | ||
In conclusion, we present here a novel method to synthesize PGM nanoparticles in aqueous systems at room temperature and at atmospheric pressure, in which the reducing agent (i.e. H2) and the size controlling agent (i.e., CO) are produced in situ from the electrochemical reduction of CO2 and water in a gas-diffusion electrode. Besides, this process can still be modified to optimize the size of the PGM NPs synthesized (i.e., adding surfactants, stabilizers, and supporting materials). The GDEx process may also find application in producing elemental nanoparticles of other noble metals besides PGMs and in the selective recovery of PGMs from solutions containing other metals (e.g., leachates from PGMs found in catalytic converters and fuel cell electrodes), as H2 would only reduce noble metals at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the funding from the European Union's Horizon 2020 research and innovation programme, under grant agreements 730224 (PLATIRUS project) and 958302 (PEACOC project). The authors are thankful for the funding from the European Union's Horizon Europe research and innovation programme, under grant agreement 101091715 (FIREFLY project). OMM thanks CONACYT-Mexico for doctoral scholarship no. 766618.References
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- D. Banham and S. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1–12 CrossRef CAS.
- H. A. Firouzjaie and W. E. Mustain, ACS Catal., 2020, 10, 225–234 CrossRef CAS.
- P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275–286 CrossRef CAS.
- M. Guerrero, N. T. Than Chau, S. Noel, A. Denicourt-Nowicki, F. Hapiot, A. Roucoux, E. Monflier and K. Philippot, Curr. Org. Chem., 2013, 17, 364–399 CrossRef CAS.
- A. P. Reverberi, N. T. Kuznetsov, V. P. Meshalkin, M. Salerno and B. Fabiano, Theor. Found. Chem. Eng., 2016, 50, 59–66 CrossRef CAS.
- L. D. Rampino and F. F. Nord, J. Am. Chem. Soc., 1941, 63, 2745–2749 CrossRef CAS.
- J. M. Petroski, Z. L. Wang, T. C. Green and M. A. El-Sayed, J. Phys. Chem. B, 1998, 102, 3316–3320 CrossRef CAS.
- H. Zhu, Q. Chi, Y. Zhao, C. Li, H. Tang, J. Li, T. Huang and H. Liu, Mater. Res. Bull., 2012, 47, 3637–3643 CrossRef CAS.
- B. Wu, N. Zheng and G. Fu, Chem. Commun., 2011, 47, 1039–1041 RSC.
- G. Chen, Y. Tan, B. Wu, G. Fu and N. Zheng, Chem. Commun., 2012, 48, 2758–2760 RSC.
- C. Zhang, S. Y. Hwang and Z. Peng, J. Mater. Chem. A, 2013, 1, 14402–14408 RSC.
- E. Abohamzeh, F. Salehi, M. Sheikholeslami, R. Abbassi and F. Khan, J. Loss Prev. Process Ind., 2021, 72, 104569 CrossRef CAS.
- J. J. Rose, L. Wang, Q. Xu, C. F. McTiernan, S. Shiva, J. Tejero and M. T. Gladwin, Am. J. Respir. Crit. Care Med., 2017, 195, 596–606 CrossRef CAS PubMed.
- C. F. Zinola, J. Electrochem. Soc., 2017, 164, H170–H182 CrossRef CAS.
- R. A. Prato, V. Van Vught, S. Eggermont, G. Pozo, P. Marin, J. Fransaer and X. Dominguez-Benetton, Sci. Rep., 2019, 9, 15370 CrossRef PubMed.
- G. Pozo, P. de la Presa, R. Prato, I. Morales, P. Marin, J. Fransaer and X. Dominguez-Benetton, Nanoscale, 2020, 12, 5412–5421 RSC.
- G. Pozo, D. van Houtven, J. Fransaer and X. Dominguez-Benetton, React. Chem. Eng., 2020, 5, 1118–1128 RSC.
- R. A. Prato, V. Van Vught, K. Chayambuka, G. Pozo, S. Eggermont, J. Fransaer and X. Dominguez-Benetton, J. Mater. Chem. A, 2020, 8, 11674–11686 RSC.
- N. Yang, S. R. Waldvogel and X. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 28357–28371 CrossRef CAS PubMed.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161–172 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- R. E. Cameron and A. B. Bocarsly, Inorg. Chem., 1986, 25, 2910–2913 CrossRef CAS.
- K. Hoppstock, R. P. H. Garten, P. Tschöpel and G. Tölg, Fresenius. J. Anal. Chem., 1992, 343, 778–781 CrossRef CAS.
- R. Kortlever, C. Balemans, Y. Kwon and M. T. M. Koper, Catal. Today, 2015, 244, 58–62 CrossRef CAS.
- H. M. Yasin, G. Denuault and D. Pletcher, J. Electroanal. Chem., 2009, 633, 327–332 CrossRef CAS.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- T. Abo Atia and J. Spooren, Chem. Eng. Process., 2021, 164, 108378 CrossRef CAS.
- G. Nicol, E. Goosey, D. Ş. Yıldız, E. Loving, V. T. Nguyen, S. Riaño, I. Yakoumis, A. M. Martinez, A. Siriwardana, A. Unzurrunzaga, J. Jeroen, B. Michielsen, X. Dominguez-Benetton and O. Lanardi, Johnson Matthey Technol. Rev., 2021, 65, 127–147 CAS.
- H. Einaga and M. Harada, Langmuir, 2005, 21, 2578–2584 CrossRef CAS PubMed.
- A. Henglein and M. Giersig, J. Phys. Chem. B, 2000, 104, 6767–6772 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00046j |
| ‡ Current address: TECNALIA, Basque Research & Technology Alliance (BRTA), Mikeletegi Pasealekua, 2, E-20009 San Sebastian, Spain. |
| This journal is © The Royal Society of Chemistry 2023 |