
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-energy hybrid lithium-ion capacitor enabled by a mixed capacitive-battery storage LiFePO4 – AC cathode and a SnP2O7 – rGO anode†
Miguel
Granados-Moreno
 ab,
Gelines
Moreno-Fernández
*a,
Roman
Mysyk
a and
Daniel
Carriazo
*ac
ab,
Gelines
Moreno-Fernández
*a,
Roman
Mysyk
a and
Daniel
Carriazo
*ac
aCentre for Cooperative Research on Alternative Energies (CIC EnergiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, 01510 Vitoria-Gasteiz, Spain. E-mail: mamoreno@cicenergigune.com; dcarriazo@cicenergigune.com; Tel: +34 94 529 71 08
bUniversidad del País Vasco, UPV/EHU, 48080 Bilbao, Spain
cIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 17th January 2023
Abstract
In this work we present the development and optimization of a graphene-embedded Sn-based material and an activated carbon/lithium iron phosphate composite for a high-performing hybrid lithium-ion capacitor (LIC). For the negative electrode, we have synthesized and screened different tin and phosphorus-based graphene materials based on the chemical structure, morphology and particle size, selecting a composite consisting of nano-sized crystalline tin pyrophosphate (SnP2O7) particles embedded in a graphenic matrix. For the positive electrode, a composite combining a high-loading (40 wt%) faradaic material lithium iron phosphate (LFP) with a graphene-activated carbon was developed whereas other bi-material cathodes are limited to about 20 wt% of faradaic material. The homogeneous distribution of nanosized carbon-coated LFP particles along the graphene-activated carbon has enabled energy storage via faradaic, pseudocapacitive, and capacitive mechanisms. The optimized-electrode LIC delivers high energy densities and overcomes the main power limitations of LICs using high-content battery-type materials (143 Wh kg−1 at 128 W kg−1 and 45 Wh kg−1 at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W kg−1).
000 W kg−1).
1. Introduction
In recent years, lithium-ion capacitors (LICs) have emerged as promising energy storage systems filling the gap between lithium-ion batteries and supercapacitors in terms of specific energy (10–100 Wh kg−1), power density (300–5000 W kg−1) and cyclability (103–105 cycles).1–5 The excellent features of LICs are the result of the combination of a battery-type electrode along with a capacitive electrode in the same device. Within this configuration, a typical battery-type negative electrode stores energy via faradaic reactions, which enables the higher energy density of a LIC due to the higher charge storage capacity and the low redox potential, increasing the full LIC cell potential over that of non-faradaic supercapacitors. On the opposite side, a capacitor-type positive electrode stores energy electrostatically by the accumulation of charges at the electrode/electrolyte interface, which favors a fast response and long cycle life. However, due to the different charge storage mechanisms of each electrode, the multiparameter electrochemical performance of a full LIC is usually limited by the imbalance between high capacity and fast kinetics.6,7The optimization of negative electrodes would involve the use of active materials that exhibit high capacities at high rates. In this regard, tin-based compounds have attracted much attention in recent years.8 Metallic tin (Sn) has advantages of high lithium storage capacity (994 mAh g−1), low price, and earth abundance. Nevertheless, due to the huge volume changes upon lithiation, tin particles can be pulverized and aggregated, which leads to structural fracture and a loss of electrical contact.9 Consequently, the practical use of Sn poses serious challenges. To overcome them, strategies such as reduction in tin particle size10 or Sn encapsulation in a conductive matrix buffering the volume changes have been applied.11–13 Also, the use of tin oxides,14 tin-based alloys,15 tin phosphides,16 or tin phosphates17 has shown promising results. Especially, crystalline tin pyrophosphate (SnP2O7) with a reversible specific capacity of >360 mAh g−1, good capacity retention, and cycle stability has been widely studied.18–20 Several studies pointed out that the excellent electrochemical performance is a consequence of the irreversible reduction of SnP2O7 to metallic Sn and its further encapsulation into an inactive Li4P2O7 matrix that can buffer volume changes and promote fast electron transfer.21–23 This material fulfills the requirements for a negative electrode in LICs.
On the opposite side, the optimization of a positive electrode is needed to increase its specific capacity while preserving a fast response. Activated carbons (ACs) are usually the common choice due to their large surface areas and tailored pore size distribution that allow them to accumulate more charge and work fast by physical storage. Nevertheless, the physical storage capacity is still much lower than that of redox-active battery-type materials. To raise the capacity of positive electrodes, the use of composites combining capacitive and faradaic (LTO, LVNP, and LFP) materials in a single electrode has been proposed.8,24,25 Lithium iron phosphate (LFP) has been considered an attractive cathode material for lithium-ion batteries26 and, more recently, also for lithium-ion capacitors27 thanks to its low cost, low toxicity, high thermal stability, low volume change during lithiation/de-lithiation, the high specific capacity of 170 mAh g−1, a well-defined plateau at a high voltage (∼3.5 V vs. Li/Li+), and reduced reactivity toward electrolytes. Nevertheless, its poor electronic conductivity (3.7 × 10−9 S cm−1) and low Li-ion diffusion coefficient (1.8 × 10−18 m2 s−1) can limit the performance of LICs at high rates. Consequently, the extent to which LFP can be used in the positive electrode is not a trivial issue since the fast response and thus the power performance at high rates can be compromised, not to mention cycle stability.28 The reduction in LFP particle size along with carbon coating are well-known strategies to overcome these limitations.29–31 Also, smaller particle size, structural defects, and uniform distribution within the carbon matrix can induce extra pseudo-capacitive effects and improve diffusion, thus boosting the usable capacity even at high rates.32
In this study, we have developed and optimized different materials for both negative and positive electrodes for a highly performing hybrid lithium-ion capacitor. For the negative electrode, we have synthesized three different tin and phosphorus-based graphene electrodes. Using the same precursor and treating them thermally at 800, 900, and 1000 °C we obtained: (i) nano-sized crystalline tin pyrophosphate (SnP2O7) particles embedded in a graphenic matrix, (ii) an amorphous composite combining tin pyrophosphate (SnP2O7) within tin (Sn) all along graphene sheets, and (iii) micrometer-sized crystalline tin particles (Sn) embedded in a phosphorus-based graphenic matrix. The impact of different chemistries, structures, morphologies, and particle size on electrochemical performance was studied. Given the results, tin pyrophosphate (SnP2O7)-based material was chosen for the negative electrode of the final LIC thanks to its highest capacity even at high rates (420 mAh g−1 at 0.5 A g−1 and 110 mAh g−1 at 5 A g−1). For the positive electrode, a composite comprising a high loading (40% wt) of lithium iron phosphate (LFP) and 60% of our previously reported graphene-activated carbon ResFaGO-A (RG) was developed. The nanosizing of our LFP particles, further carbon coating, and the homogeneous distribution of the carbon-coated LFP throughout the final composite enabled energy storage via faradaic, pseudocapacitive, and capacitive processes. As a result, this composite material demonstrated an outstanding capacity even at high rates (102 mAh g−1 at 0.25 A g−1 and 80 mAh g−1 at 30 A g−1). Finally, the full optimization of the assembled LICs using mass balance 1:1 enabled high energy densities without compromising the power performance (143 Wh kg−1 at 128 W kg−1 and 45 Wh kg−1 at 25 000 W kg−1).
2. Experimental
2.1. Synthesis of rGO800-P-Sn and LFP/RG
The LiFePO4 (LFP)/RG composite was used as the positive electrode material. The two components of the composite were obtained as follows.The activated carbon, RG, was prepared following our previously reported method.33 Briefly, 440 mg of resorcinol (Sigma-Aldrich, 99%) were dissolved in 4.0 ml of water, 2.4 ml of ethanol, and 4.0 ml of graphene oxide (Graphenea, 4 mg ml−1). After complete dissolution of resorcinol, 600 μl of formaldehyde (Sigma-Aldrich, 37% w/w in H2O containing 10–15% methanol) and 100 μl of concentrated phosphoric acid (Sigma-Aldrich, ≥85% w/w in H2O) were added to the suspension. Then, the mixture was transferred into a closed container and thermally treated in an oven at 85 °C for 70 h. The resulting resins were pre-carbonized at 800 °C in a tubular oven for 1 h under a dynamic Ar atmosphere. The obtained carbon was grounded together with KOH (Sigma-Aldrich, ≥85%) using a C:KOH mass ratio of 1:6 and further carbonized in a tubular oven at 800 °C for 1 h under a dynamic Ar atmosphere. The resulting material was washed off once with diluted HCl, and then several times with hot deionized water to end up with the RG.
The LiFePO4 (LFP) sub-micrometric size particles were synthesized by hydrothermal synthesis as follows: 7.5 ml of 1 M H3PO4 solution (Sigma-Aldrich, ≥85% w/w in H2O) and 30 ml of ethylene glycol (EG) (FischerScientific, ≥99%) were homogenized using a magnetic stirrer to obtain the precursor solution. Then, 22.5 ml of 1 M LiOH aqueous solution was slowly introduced while stirring. Next, 15 ml of 0.5 M FeSO4 aqueous solution was dropwise added. Finally, the solution was transferred into an autoclave and then thermally treated at 180 °C for 9 h. The resulting mixture was filtered and washed 3 times with hot deionized water. The LFP particles were carbon coated to increase their electrochemical performance. For this purpose, a mixture of the synthesized LFP powder with a 10% w/w of Super P C-65 conductive carbon was ball milled in a planetary ball mill (Pulverisette 5) with a 20:1 w/w ball to sample mass ratio for 3 hours at 250 rpm, resulting in LFP/C nanoparticles.
The negative electrode materials were obtained using a facile synthesis as follows: 100 mg of SnSO4 (Sigma-Aldrich, ≥95%) were added to 50 ml of 4% w/w GO aqueous solution (Graphenea, 4 mg ml−1) that was gently stirred for 1 h. Then, 250 μl of H3PO4 (Sigma-Aldrich, ≥85% w/w in H2O) were added to the solution and, after homogenization, the solution was transferred to a closed container and thermally treated at 80 °C for 18 h. The mixture was then freeze dried for 72 h. The resulting dry powder was subsequently treated in a tubular oven at 800 °C, 900 °C, or 1000 °C for 1 h under dynamic argon flow to obtain the final materials denoted as GOPR800_Sn, GOPR900_Sn, and GOPR1000_Sn, respectively.
2.2. Physicochemical characterization
The morphology and chemical composition of the samples were characterized by scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS), respectively, using a Quanta200 FEI (3 kV, 30 kV) microscope. The microstructure of the samples was analyzed by X-ray diffraction (XRD) using a Bruker D8 X-ray diffractometer. The data were collected using CuKα radiation over 2θ within the range from 10° to 80° for the GOPRX_Sn and from 15° to 65° for the LFP samples, at steps of 0.02° and a residence time of 2 s in both cases. Nitrogen adsorption–desorption isotherms were registered at −196 °C using an ASAP 2460 instrument from Micromeritics. The samples were outgassed at 150 °C for 12 h under a vacuum prior to the analysis. Specific surface area (SBET) values were calculated using the Brunauer–Emmett–Teller (BET) equation using the Rouquerol procedure for the monolayer capacity.34 Pore size distributions were calculated with the 2D-NLDFT model applied to the data of the adsorption branches using the SAIEUS software.35 Thermogravimetric analysis was performed using a TG 209 F1 Libra between 30 and 1000 °C using a heating ramp of 10 °C min−1.2.3. Electrochemical characterization
The negative electrode was processed by mixing the active materials (GOPR800_Sn, GOPR900_Sn, or GOPR1000_Sn) with a Super P C-65 conductive carbon and a polyvinylidene fluoride (PVDF) binder in a 90:5:5 mass ratio using N-methyl-2-pyrrolidone (NMP) as the solvent. The positive electrode was processed by mixing the active materials, RG and LFP/C, with a Super P C-65 conductive carbon and a PVDF binder using a mass ratio of 54:36:5:5. The 60% w/w of the active materials corresponds to RG and the 40% w/w to the LFP/C, with the composite referred to as RG-LFP40. NMP was used as the solvent. The NMP-based inks of the negative and positive electrodes were coated on copper and aluminum foil, respectively. Laminates were dried overnight at 80 °C under a vacuum. Electrode discs of 12 mm were furtherly punched out of the laminates and dried at 120 °C overnight under a vacuum prior to cell assembly. For the sake of comparison, the RG and LFP/C electrodes were processed following the same procedure with a mass ratio of 90:5:5 and 80:10:10, respectively.
In the case of RG-LFP electrodes, the mass balance is 1.6 ± 0.2 mgAM cm−2 while in the case of GOPR800-P-Sn, GOPR900-P-Sn, GOPR1000-P-Sn the mass loading is 1.7 ± 0.2 mgAM cm−2. Exceptionally, 3.3 ± 0.3 mgAM cm−2 GOPR800-P-Sn electrodes were used for the 1:2 mass balance LIC.
The electrochemical measurements were conducted using a multichannel VMP3 generator (Biologic). Whatman D-type glass fibers discs of 13 mm in diameter and 1 M lithium hexafluorophosphate (LiPF6) in 1:1 v/v of ethylene carbonate (EC) and dimethyl carbonate (DMC) were used as a separator and an electrolyte, respectively, in all the measurements.
The electrochemical performance of the negative electrodes was first evaluated in two-electrode Swagelok-type cells using a half-cell configuration with a metallic lithium disc serving simultaneously as the counter and reference electrodes. Galvanostatic charge/discharge measurements were carried out at different current densities between 0.002 and 2.0 V vs. Li/Li+.
Similarly, the electrochemical performance of the positive electrodes was evaluated using a half-cell configuration in a three-electrode Swagelok-type cell using a metallic lithium disc as the reference electrode and an oversized Norit Activated Carbon (Kuraray) disc as the counter electrode. Galvanostatic charge/discharge measurements were carried out at different current densities with potentials between 2 and 4.2 V vs. Li/Li+.
Before the assembly of LIC cells, the negative electrodes were pre-lithiated in a two-electrode Swagelok-type cell with a lithium metal disc simultaneously used as the counter and reference electrode. The pre-lithiation procedure consists of 5 charge/discharge cycles at C/10 between 0.002 and 2.0 V vs. Li/Li+ followed by a final discharge at C/40 to 0.2 V vs. Li/Li+. After the pre-lithiation step, LICs were assembled within a three-electrode Swagelok-type cell using a pre-lithiated negative electrode, an LFP-containing positive electrode, and a lithium metal disc connected to the potentiostat as the counter, working, and reference electrodes, respectively. The negative electrode potential was set to 0.2 V vs. Li/Li+, and the positive electrode potential to 4.2 V vs. Li/Li+ to maximize the cell potential and simultaneously prevent lithium plating and electrolyte decomposition. Galvanostatic charge/discharge measurements were performed within the 1.5–4.2 V vs. Li/Li+ voltage range at different current densities.
3. Results and discussion
3.1. Characterization of the negative electrode
As explained before in the experimental section, three different materials were synthesized as negative electrodes by an easy procedure consisting of mixing SnSO4 with a graphene oxide aqueous dispersion (GO) and H3PO4, heating, freeze drying, and subsequent pyrolysis at three different temperatures of 800 °C, 900 °C, or 1000 °C.XRD patterns in Fig. 1 clearly show a huge impact of the temperature on the final structure of the materials. The material synthesized at 800 °C (GOPR800_Sn) exhibits very sharp peaks characteristic of tin pyrophosphate SnP2O7 with a crystalline cubic structure, as previously reported.17,36 Further increase in temperature to 900 °C (GOPR900_Sn) leads to crystallinity loss and amorphization. A high intensity and broad peak ascribed to the basal diffractions of graphene layers is registered in the 15–35° range.33 This structure can be ascribed to the first stage where SnP2O7 is partially reduced to metallic Sn along the randomly oriented graphene sheets. Finally, the temperature increase up to 1000 °C (GOPR1000_Sn) leads to a successful reduction of tin pyrophosphate and the formation of crystalline metallic Sn particles deposited over the P-graphene sheets.37,38 The TGA curve obtained for the precursor under the Ar atmosphere and using the same heating rate as that for the synthesis of the materials (Fig. S1†) is clearly in agreement with previous results. The first two steps of mass loss (20%) under 250 °C can be ascribed to the release of water molecules, weakly and strongly bonded, respectively. From 250 °C to 500 °C, the weight loss can be ascribed to the evolution of remaining oxygen and phosphorus-functional groups in graphene oxide. The crystallization of SnP2O7 starts at 520 °C and goes through 880 °C. Further increase in the temperature promotes the carbothermal reduction of phosphorous-tin composites leading to the formation of metallic Sn while releasing phosphorous-containing gases.20,39
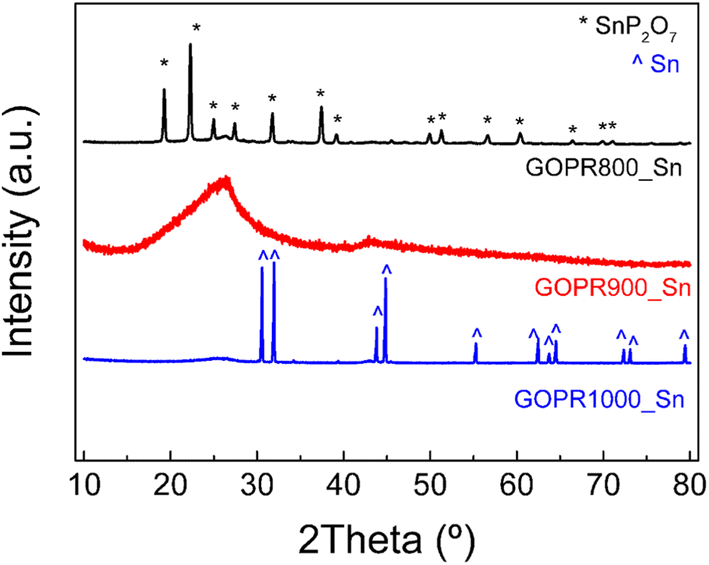 | ||
| Fig. 1 XRD patterns of the as-synthesized materials. | ||
SEM images registered for the different thermal treated samples show the morphological differences between the three materials expected from the XRD results. Fig. 2a shows that GOPR1000_Sn is composed of large Sn particles (7–14 μm) homogeneously embedded in the P-graphene matrix. Conversely, GOPR900_Sn (Fig. 2b) shows a highly open 3D macroporous network made up of an amorphous phase of Sn and SnP2O7 along the graphene sheets, confirming the beginning of the reduction of tin pyrophosphate to metallic tin. Finally, Fig. 2c and d illustrate that GOPR800_Sn is shaped by small-sized crystalline particles of SnP2O7 (∼100 nm) homogeneously embedded in the graphenic matrix. This is expected to enhance electrochemical properties since smaller particles make Li+ diffusion paths shorter, thus favoring faster kinetics. EDX results reported in Table 1 show that materials are mainly composed of carbon, tin, phosphorus, and oxygen in decreasing amounts. Also, as the temperature increases, the oxygen and phosphorus content decreases while that of tin increases, in good agreement with the further reduction of SnP2O7 to metallic Sn.
 | ||
| Fig. 2 SEM images of the as-synthesized materials: GOPR1000_Sn (a), GOPR900_Sn (b), and GOPR800_Sn (c and d). The scale is adapted to clearly show the morphological differences among the three materials. | ||
| C (wt%) | O (wt%) | P (wt%) | Sn (wt%) | |
|---|---|---|---|---|
| GOPR800_Sn | 37.7 | 12.6 | 16.5 | 33.2 |
| GOPR900_Sn | 39.2 | 9.9 | 11.6 | 39.3 |
| GOPR1000_Sn | 47.0 | 7.9 | 10.8 | 34.3 |
The three materials were electrochemically tested using the half-cell configuration in the 0.002–2.0 V vs. Li/Li+ potential range to evaluate the impact of the chemical structure and morphology on the performance as negative electrodes for lithium storage. To this end, galvanostatic charge–discharge experiments at different current densities were performed. Fig. 3a–c and S2a† show the first four cycles registered at 0.05 A g−1. Clear differences can be distinguished among the three samples, especially in the first discharge where all irreversible processes occur. During the first discharge of GOPR800_Sn (Fig. 3a), two main processes take place: (1) irreversible reduction of Sn(IV) to metallic Sn(0) and the formation of the amorphous Li4P2O7 matrix, where Sn(0) is homogeneously dispersed (see eqn (1)) and (2) the formation of reversible lithium tin alloys (see eqn (2)).11,18,40 The first irreversible process is evidenced at about 1.3 V, while the reversible alloying takes place at about 0.4 V.21,22
| SnP2O7 + 4Li+ → Sn0 + Li4P2O7 | (1) |
| Sn0 + xLi+ + xe− ↔ LixSn | (2) |
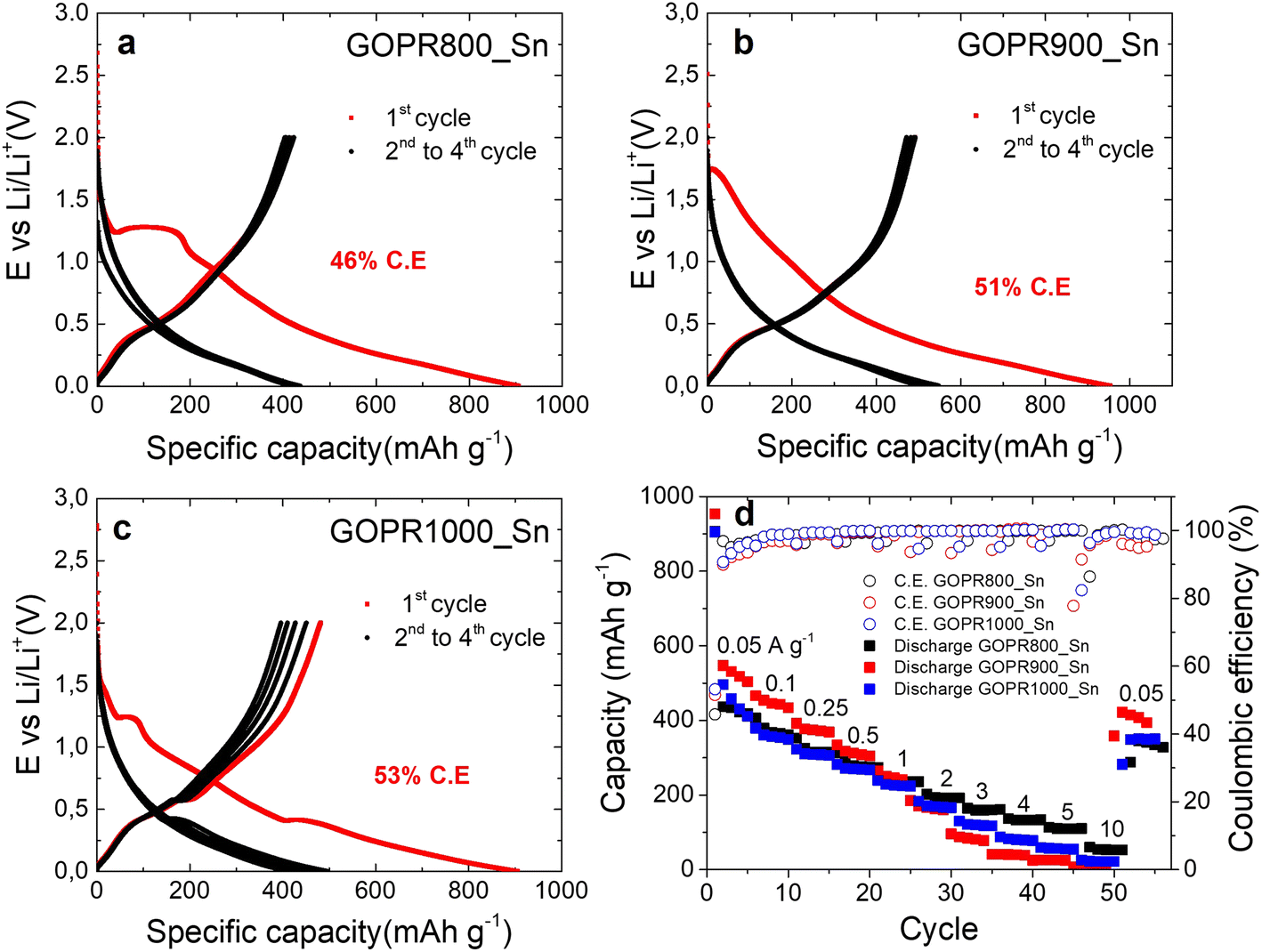 | ||
| Fig. 3 Electrochemical characterization of the negative electrodes: galvanostatic charge/discharge of the first four cycles recorded at 0.05 A g−1 for GOPR800_Sn (a), GOPR900_Sn (b), GOPR1000_Sn (c) and rate capability and their respective coulombic efficiency (d). | ||
Fig. S3a and b† show the SEM images of the cycled GOPR800-Sn obtained using Everhart–Thornley (ETD) and backscattered electron (BSED) detectors, respectively, where one can observe the SEI and the Li4P2O7 matrix covering the reduced graphene oxide sheets, and the Sn(0) particles embedded in it.
The poor coulombic efficiency of 46% recorded for the first cycle accounts for all irreversible previously explained processes as well as the solid electrolyte interface (SEI) formation. The 3 subsequent cycles show sloping profiles with no marked plateaus, characteristic of Li+ insertion into non-graphitic materials, and the coulombic efficiency is close to 100%. By contrast, no defined plateaus are recorded for the first discharge of GOPR900_Sn (Fig. 3b) due to its amorphous character where both SnP2O7 and Sn(0) coexist along the graphene sheets. Also, the coulombic efficiency is slightly improved to 51%. Finally, the first discharge of GOPR1000_Sn (Fig. 3c) shows a small plateau at 1.3 V and a prominent one at 0.4 V. The first small plateau accounts for the irreversible conversion of the remaining fraction of SnP2O7 (undetectable by XRD) to Sn(0), while the second is for the Li/Sn alloying reaction, as previously explained. The coulombic efficiency is again enhanced to 53% since less irreversible reactions take place. The subsequent 3 cycles still show marked plateaus at 0.4 V and worse coulombic efficiencies than the two previous materials due to the hindered lithiation of big Sn particles and their further breakup. Fig. S2a† clearly shows the different processes that take place during the first discharge for the three materials. Afterwards, galvanostatic charge–discharge curves at different current densities (Fig. S2b–d†) were recorded. Fig. 3d reveals that GOPR900_Sn is the material best performing at low current densities (∼500 mAh g−1), but its capacity drops drastically above 2 A g−1. GOPR1000_Sn and GOPR800_Sn show similar capacities at low current densities (∼420 mAh g−1), yet lower than that of GOPR900_Sn, but the retention at high rates is highly improved. This is especially true for GOPR800_Sn, which still retains 110 mAh g−1 at 5 A g−1. This evidences that both morphology and microstructure clearly influence the electrochemical performance upon lithiation/de-lithiation. The submicrometer size of metallic Sn after the irreversible reduction of SnP2O7 and its homogenous distribution over Li4P2O7/graphene matrix enables the faster insertion of Li+ into the GOPR800_Sn negative electrode. The better rate performance of the last material decides on its being the best choice as the negative electrode for our lithium-ion capacitor.
3.2. Characterization of the positive electrode
As stated above, a composite made of 40% of LFP/C and 60% RG was developed to be used as a positive electrode in LICs. Both components, LFP/C and RG, were synthesized and optimized separately. RG synthesis and electrochemical performance were already reported in our previous publication.33 This material was chosen as the most suitable activated carbon for the composite due to its large specific surface area, flat morphology, high specific capacitance, and excellent capacity retention.On the other hand, and as described in the experimental section, LFP was hydrothermally synthesized from the H3PO4, LiOH, and FeSO4 precursors using EG as a structure-directing agent to limit the particles' growth. Then, the LFP particles were coated with carbon to increase the conductivity and improve the contact between them. The carbon coating was performed by ball-milling a mixture of LFP with 10% w/w of Super P C65 conductive carbon using a 20:1 w/w ball-to-sample mass ratio for 3 hours at 250 rpm.
The SEM image of carbon-coated LFP (LFP/C) (Fig. 4a) shows irregular-shaped particles present on this sample and good connectivity between them. The analysis of SEM images with the ImageJ software allowed obtaining the particle size distribution (Fig. 4b) that ranges between 20 nm and 200 nm. Most of the particles (78%) are smaller than 100 nm, with 31% between 60 and 100 nm, 33% between 40 and 60 nm, and 14% between 20 and 40 nm.
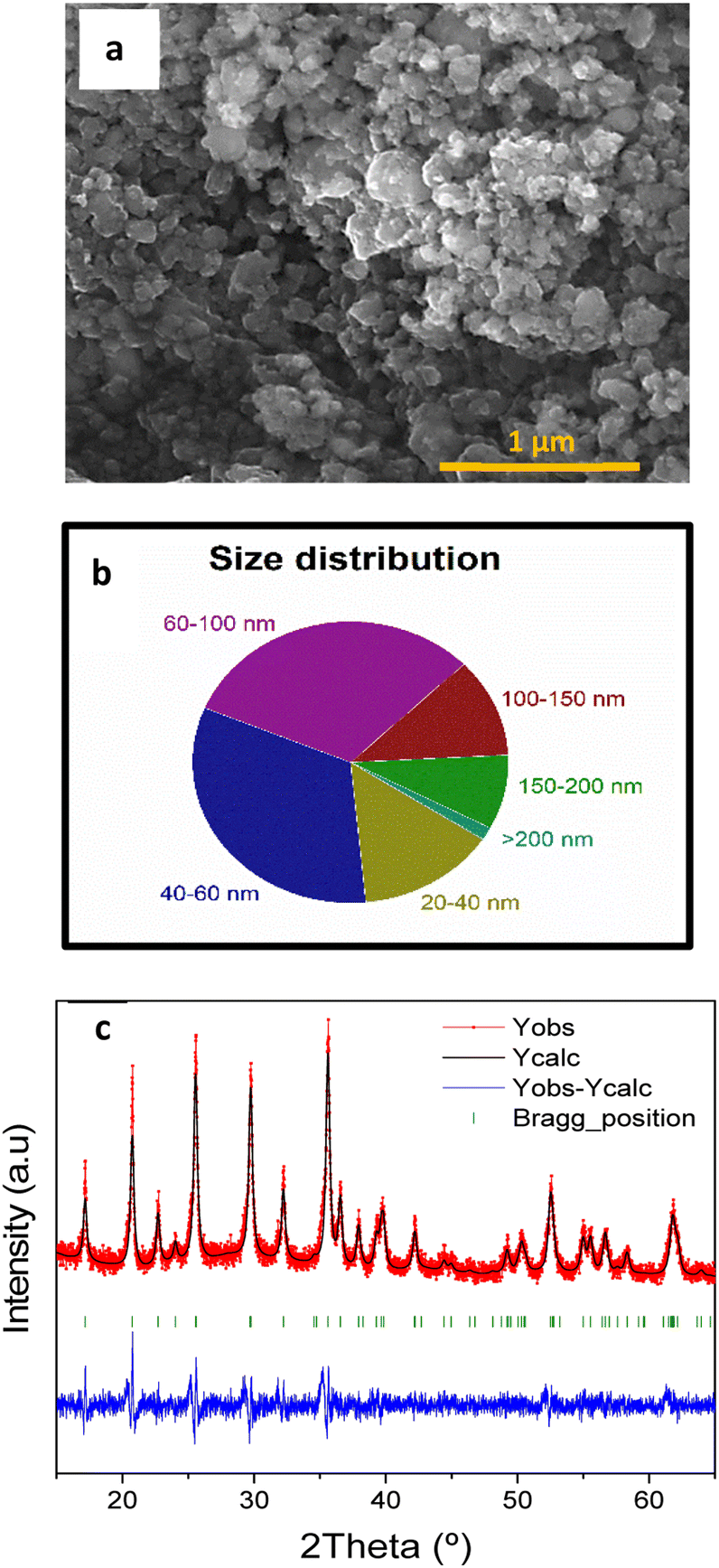 | ||
| Fig. 4 SEM image of LFP/C (a), particle size distribution of LFP/C (b), and LFP/C XRD data refinement (c). | ||
The XRD pattern in Fig. S4† shows characteristic peaks of the LFP olivine structure41 (α-LiFePO4). This confirms the successful synthesis procedure since, as previously reported, the olivine structure electrochemically outperforms other LFP structures. The XRD diffraction pattern registered for the LFP/C sample was treated using the FullProf software. After the carbon coating, the diffraction pattern still matches with the LFP olivine structure as shown in Fig. 4c, confirming that the structure is not damaged during the high-energy ball-milling. Further analysis of these patterns by FullProf software and the Scherrer equation allowed us to determine the average crystallite size of both the LFP and LFP/C samples. The average crystallite size was reduced during ball-milling from 39.5 nm for LFP to 19.7 nm for LFP/C. The crystallite size of LFP/C measured by XRD is smaller than the particle size obtained by SEM, meaning that this sample is formed by the aggregation of nanometric LFP/C crystals. Smaller particle sizes would be beneficial due to two reasons:29 (i) the Li+ diffusion pathway along LFP/C particles is shortened, allowing a faster migration of Li+ from the bulk of LiFePO4 particles, which results in an improved performance at high current densities;42 (ii) nanosized LFP/C particles can be easily and homogenously distributed along the activated carbon RG during the fabrication of the RG-LFP40 composite improving the electrical contact between the LFP/C particles and the RG matrix, and thus increasing the electrical conductivity of the electrode leading to higher capacity values and better capacity retention.
Summarizing, the carbon coating results in decreased particle size, better conductivity, and enhanced particle connectivity, which triggers improved electrochemical performance as evidenced in Fig. S5.†30
The SEM characterization results of the RG-LFP40 composite are shown in Fig. 5a and b. The composite is formed by the small-size particles of LFP/C and some agglomerates homogeneously integrated into the RG matrix. It is worth remarking that the good connectivity between the LFP/C and RG particles enhances the electronic conductivity of the composite. Also, there are some voids between particles that will enhance the electrode wettability and further improve ion adsorption.
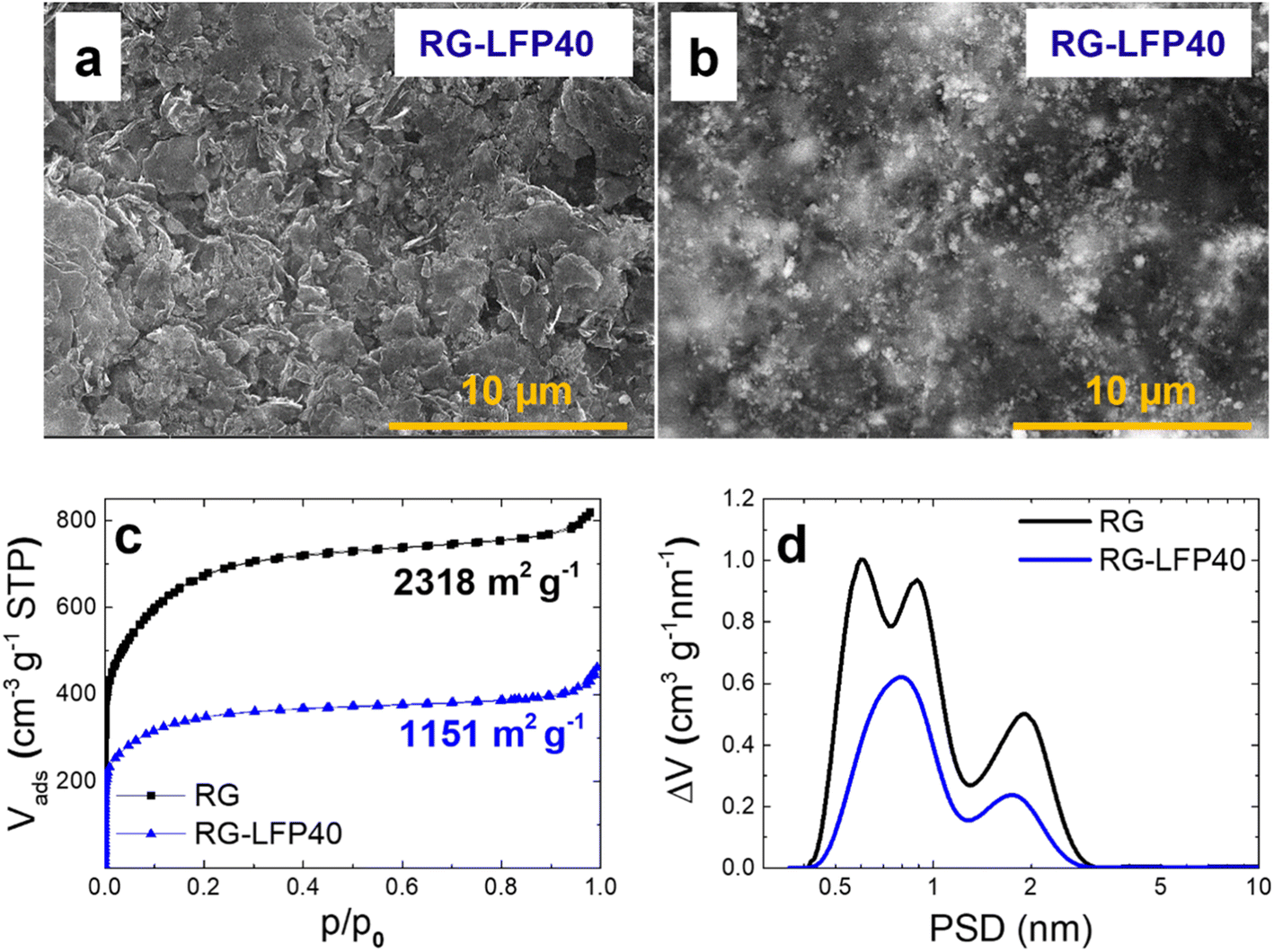 | ||
| Fig. 5 EDT and BSD SEM images of RG-LFP40 electrodes (a and b), N2 adsorption/desorption isotherms (c), and pore size distribution of RG and RG-LFP40 (d). | ||
Textural properties of RG-LFP40 composite were determined from nitrogen adsorption/desorption isotherms and included in Fig. 5c. For the sake of comparison, the textural properties of our previously reported RG are also included.33 The isotherms of both RG and RG-LFP40 can be ascribed to type I according to the IUPAC classification, which corresponds to microporous samples. RG shows a BET specific surface area (SBET) of 2318 m2 g−1. In the case of RG-LFP40, the high loading of non-porous LFP/C particles and the partial pore clogging in the RG matrix result in a decrease in SBET to only 1151 m2 g−1. Pore size distributions calculated from the isotherms are depicted in Fig. 5d. Both materials are mainly composed of micropores (<2 nm), with a high volume of small micropores (∼0.85 nm). Also, as explained before, LFP/C particles obstruct the ultramicropores (0.59 nm) of the RG matrix.
The electrochemical performance of composite RG-LFP40 was evaluated in the three-electrode cell configuration by cycling at several current densities between 2.0 and 4.2 V vs. Li/Li+. Also, to evaluate the impact of adding LFP/C to the activated carbon RG, both materials were also studied separately under the same conditions. The capacities obtained during the galvanostatic charge/discharge measurements are presented in Fig. 6a. Expectedly, at low current densities the faradaic LFP/C material (131 mAh g−1) outperforms composite RG-LFP40 (102 mAh g−1) and capacitive material RG (91 mAh g−1). Nevertheless, from 0.5 A g−1 on, the better capacity retention of RG-LFP40 must be highlighted since it exceeds 80 mAh g−1 at 30 A g−1.
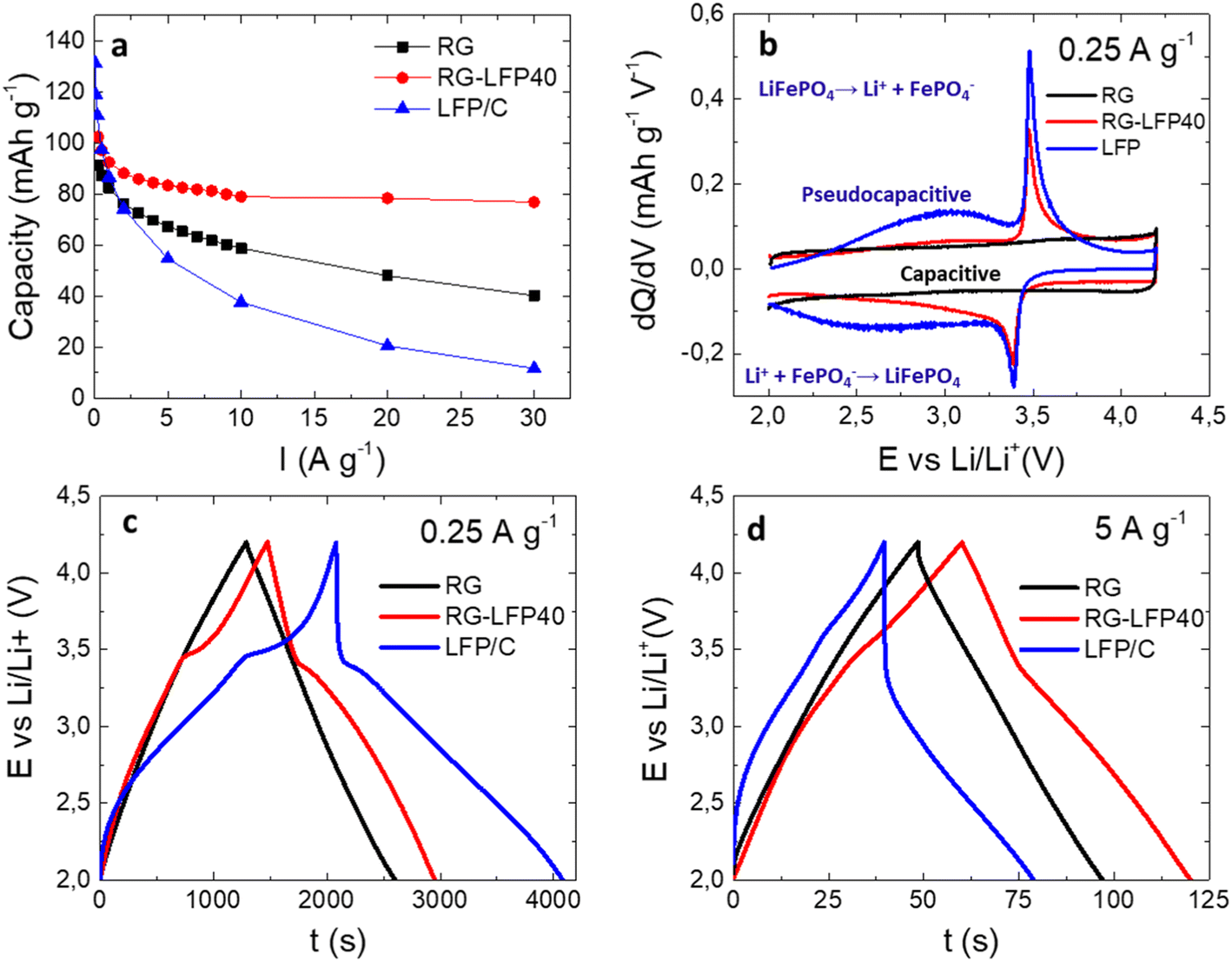 | ||
| Fig. 6 Dependence of the specific capacitance on the current density (a), differential capacity vs. voltage plot (b), galvanostatic charge/discharge profiles registered at 0.25 A g−1 (c), and 5 A g−1 (d) for the studied samples. | ||
To understand the better electrochemical performance of this material, a deeper analysis of the characteristic storage mechanisms was performed by differential capacity vs. voltage plot (Fig. 6b). On one hand, RG exhibits a typical rectangular profile ascribed to pure capacitive storage. On the other hand, two different storage mechanisms are observed for LFP/C: (i) the sharp peaks at 3.5 V and 3.4 V ascribed to the Li+ insertion/de-insertion faradaic reaction of LFP represented by eqn (3),43 and (ii) broad peaks in the 2.0–3.5 V potential window related to a pseudo-capacitive storage mechanism.
| LiFePO4 ↔ Li+ + FePO4− | (3) |
Finally, the RG-LFP40 composite shows a combination of the three mechanisms: (i) capacitive, (ii) pseudo-capacitive, and (iii) faradaic. The pseudo-capacitive mechanism is worth mentioning since it enables the good performance of the RG-LFP40 composite at high current densities.44 While the faradaic phase transition of LiFePO4 to FePO4 and vice versa is a slow process taking place in the bulk of the electrode, pseudo-capacitance is a fast mechanism caused by charge-transfer reactions at the electrode/electrolyte interphase enabling charge storage at high charge/discharge rates. The significant pseudo-capacitive feature present for the LFP/C nanoparticles is explained by two reasons: (i) nanosized particles with a high surface-to-volume ratio substantially increase the share of the surface-confined redox activity over that of the bulk redox process, boosting pseudo-capacitive contribution;45 (ii) amorphous LFP was reported to exhibit pseudo-capacitive Li+ insertion by a solid-solution reaction, the amorphization of LFP/C particles could be accomplished during the carbon coating process.32 The contact with the amorphous carbon can distort the LFP lattice and, in addition, the contact with the environment during the high-energy ball-milling can induce the formation of Fe3+ with a disordered structure.32,45 In our experiments (Fig. S6†), the amorphization of LFP is confirmed using post-electrochemistry XRD analysis where most of the characteristic peaks for LFP are not discerned upon cycling whereas SEM shows no change in the shape and size of LFP.
The integration of the optimized LFP/C nanoparticles onto the RG matrix enables a higher percentage of the faradaic material in the hybrid cathode compared with most systems recently reported in the literature, increasing the specific capacity of the electrode while keeping excellent capacity retention values at high current densities.25
The charge/discharge profiles at 0.25 A g−1 and 5 A g−1 are shown in Fig. 6c and d. At low current densities, the Li+ ions have enough time to move in and out of the bulk of the LFP/C particles. This fact is clearly shown in Fig. 6c, where LFP/C and RG-LFP40 outperform RG. As expected, high current densities have a detrimental effect on the LFP/C performance, which loses its insertion/de-insertion plateaus. In this scenario, RG-LFP40 still outperforms the bare RG. The composite successfully combines the high capacity of LFP/C and the excellent capacity retention of RG.
3.3. Characterization of the full-cell hybrid lithium-ion capacitor
As previously explained, RG-LFP40 and rGO800-P-Sn were selected as the positive and negative electrodes, respectively. Prior to LIC assembly, rGO800-P-Sn was pre-lithiated to form an SEI and supply enough lithium to counterbalance the irreversibility of the first cycles. The pre-lithiation step consists of 5 charge/discharge cycles at C/10 between 0.002 and 2 V vs. Li/Li+. Then, a cutoff potential of 0.2 V vs. Li/Li+ was set to maximize the use of the negative electrode while preventing lithium plating. Also, the positive electrode potential was set to 4.2 V vs. Li/Li+ to maximize its use at the time that electrolyte decomposition is avoided.To maximize the output capacity, a mass balancing of the electrodes was performed. The charge stored on each electrode must be equal (Q+ = Q−) and proportional to the capacitance (C+, C−), the active mass of the materials (m+, m−), and the working potential window (ΔE+, ΔE−) [eqn (4) and (5)].
| Q+ = m+C+ΔE+ | (4) |
| Q− = m−C−ΔE− | (5) |
The selection of a unique mass ratio that maximizes energy and power densities is difficult since the capacity of each electrode changes depending on the applied current density, and the working potential in the LIC differs from the one used to characterize each electrode individually. Thus, two different LICs configurations were evaluated: LIC1:1 and LIC1:2 (being LIC m+:m−). LICs were galvanostatic cycled at different current densities in the 1.5–4.2 V vs. Li/Li+ cell voltage range. The profiles of the positive and negative electrodes as well as the full LIC at the current densities of 0.1 A g−1 and 5 A g−1 are depicted in Fig. 7. At 0.1 A g−1, capacitive and faradaic storage mechanisms are clearly visible for both LIC1:1 and LIC1:2. The LFP insertion/de-insertion plateaus at 3.4/3.5 V are combined with the linear profile characteristic of capacitive materials, with a minimum ohmic drop. The positive electrode of LIC1:1 swings from 4.46 V to 2.26 V (2.2 V) while the positive electrode of LIC1:2 goes from 4.39 V to 2.02 V (2.37 V). In the case of LIC1:1, the negative electrode fluctuates between 0.26 V and 0.76 V (0.5 V) while in the case of LIC1:2, the negative electrode moves between 0.19 V and 0.52 V (0.33 V). At 5 A g−1, the effect of the LFP/C is still visible in both LIC1:1 and LIC1:2 due to its pseudo-capacitive characteristics, fast enough to store charges at high current densities. As expected, the ohmic drop increases, but is still low for both LIC1:1 and LIC1:2 at 5 A g−1. As the current density increases, there are greater differences between the voltage swing of the electrodes of the two LICs. The positive electrode of LIC1:1 fluctuates between 4.32 V and 2.43 V (1.89 V) while the positive electrode of LIC1:2 swings between 4.40 V and 2.05 V (2.35 V). The negative electrode of LIC1:1 goes from 0.12 V to 0.93 V (0.81 V) while the negative electrode of LIC1:2 moves between 0.20 V and 0.55 V (0.35 V). The overall difference between the electrode's potential ranges in the LICs is caused by the different mass ratios. This fact becomes increasingly important for the higher current densities. A heavier negative electrode is a conservative option with a shorter potential range that prevents lithium plating (the better safety option), this is easily recognizable in Fig. 7, especially at the high current density. Nevertheless, the full device will provide a lower energy density compared with a higher positive to negative electrode mass ratio.
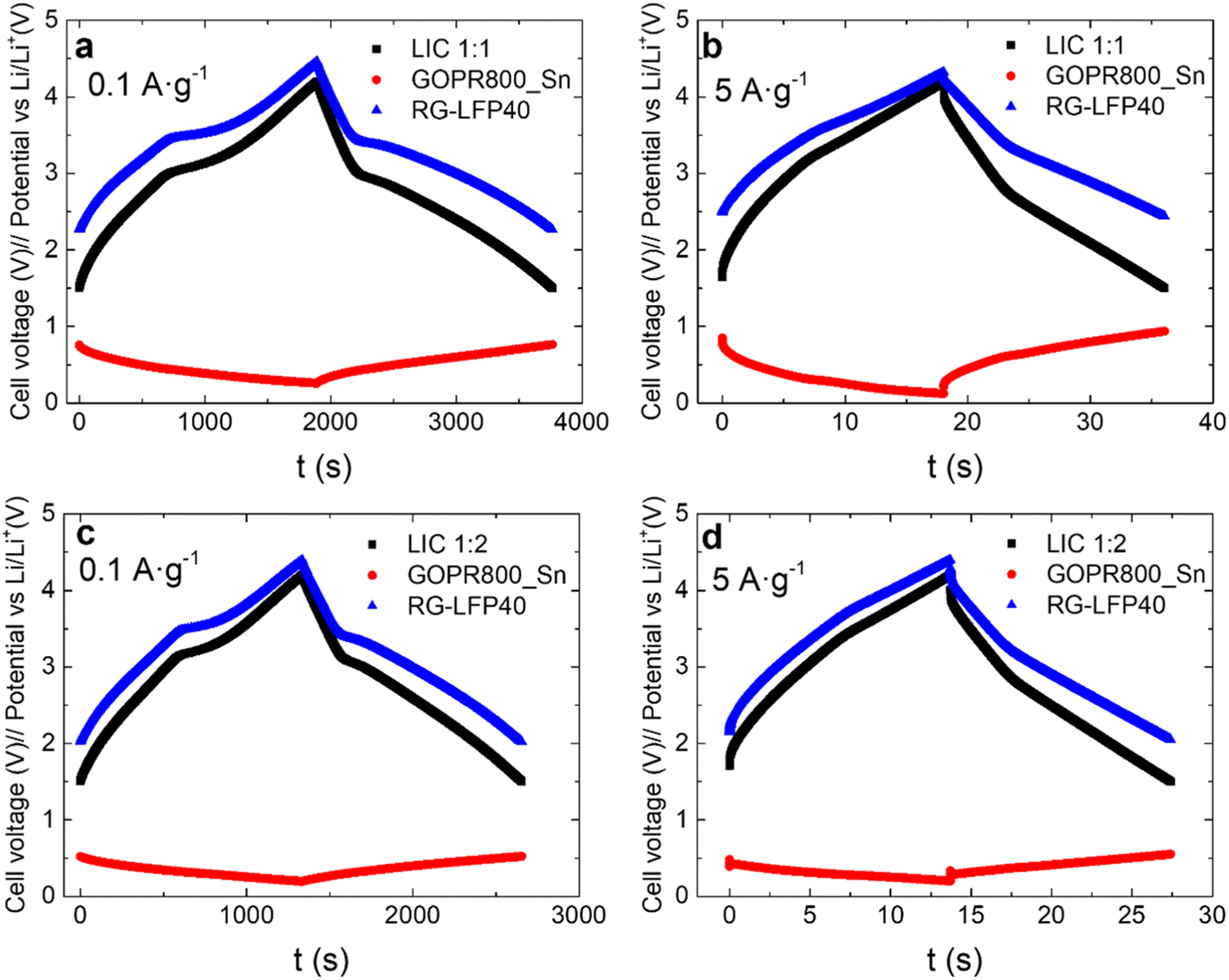 | ||
| Fig. 7 Galvanostatic charge/discharge profiles at 0.1 A g−1 and 5 A g−1 for LIC1:1 (a and b) and LIC1:2 (c and d): LIC (black), negative electrode (red), and positive electrode (blue). | ||
The gravimetric energy and power densities of the LICs, calculated from the galvanostatic curves are included in Fig. 8. LIC1:1 outperforms LIC1:2 in terms of energy and power densities. LIC1:1 shows a remarkable energy density of 143 Wh kg−1 at 128 W kg−1 and retains 100 Wh kg−1 at 2500 W kg−1 and 45 Wh kg−1 at a maximum power density of 25000 W kg−1 without any evidence of lithium plating. As far as we know, this result surpasses most of the similar devices comprising composites that contain LFP in the positive electrode (see Table 2). Even considering the high loading of LFP in our composite (40%), the power performance has been preserved, which is usually the major challenge for this type of device. This can be explained by the proper optimization of both the positive and negative electrodes. In the positive electrode, nanosized LFP/C particles homogeneously dispersed into the graphene/activated carbon RG allow energy storage via faradaic, pseudocapacitive, and capacitive mechanisms, maximizing the power performance. In the negative electrode, nanosized Sn particles homogeneously embedded into the conductive graphene/Li4P2O7 matrix can be rapidly lithiated/de-lithiated without suffering from huge volume changes and enabling an exceptional rate capability.
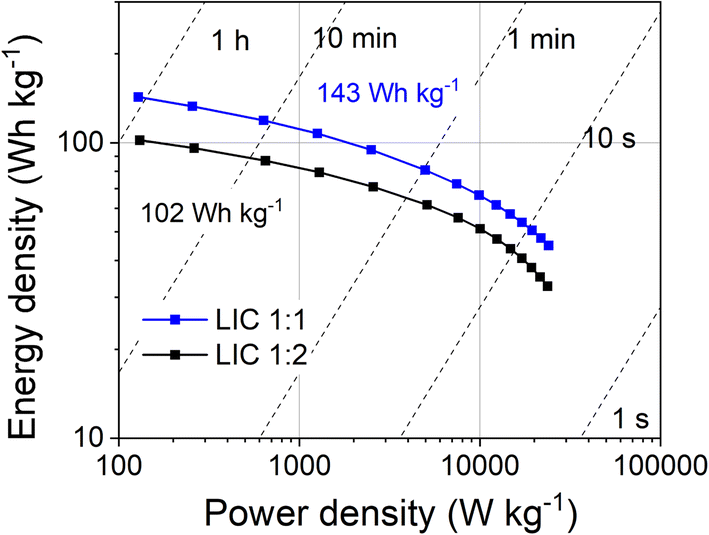 | ||
| Fig. 8 Ragone plot comparing gravimetric energy and power densities of LIC1:1 and LIC1:2. | ||
| Positive electrode | Negative electrode | LIC max. energy density (Wh kg−1) | LIC max. power density (W kg−1) | LIC power density at 100 Wh kg−1 (W kg−1) | Number of cycles (% retention) | Ref. |
|---|---|---|---|---|---|---|
| LFP/AC 40/60% (6 layers) | Hard carbon | 66 | 2000 | — | 14k (90%) 5 A g−1 | 28 |
| LFP/expanded graphite | AC/graphite 90/10 | 15,1 | 2367 | — | 10k (70%) 2 A g−1 | 29 |
| LFP/AC 20/80% multilayer | Hard carbon | 1k (94%) 1 C | 47 | |||
| LFP/AC 20/80% pouch | Hard carbon | 30 | 2000 | — | 30 k (90%) 60 C | 48 |
| LFP/AC 25/75% | Mesocarbon microbeads | 30 | 1000 | — | 1k (100%) 0.05 A g−1 | 49 |
| LFP/AC 20/80% | LTO | 90 | 15000 |
50 | ||
| LFP/AC 20/80% | Hard carbon | 5k (88.5%) | 25 | |||
| LFP/mesoporous carbon | Hard carbon | 130 | 25000 |
20000 |
3k (92%) | 32 |
| LFP/AC 40% | GOPR800_Sn | 158 | 26600 |
4000 | 2.2k (70%) 2 A g−1 | This work |
Owing to the better performance of LIC1:1, it was further subjected to a cycling stability test at 2 A g−1 (tdischarge = 1 min), showing 72% capacity retention after 2000 cycles. Although this value is somewhat lower than expected, it must be taken in the context of the high loading of faradaic materials (the full-faradaic negative electrode and 40% of LFP in the positive electrode) compared to other similar devices (Table 2 and Fig. S7†) and the limitations associated with the Swagelok-type cells used to perform the stability measurements. One approach to improve cycle life is to reduce the amount of faradaic materials, but energy delivery will be an obvious trade-off.28,46
4. Conclusions
We demonstrated a high-energy and power-performing lithium-ion capacitor comprising a tin and phosphorus-graphene-based composite as the negative electrode and a high-loading LFP/graphene-activated carbon as the positive electrode.Particularly, it has been found that the incorporation of nano-sized crystalline tin pyrophosphate (SnP2O7) particles in a graphenic matrix can promote fast kinetics for Li+ insertion/de-insertion, reaching high specific capacities even at fast rates (420 mAh g−1 at 0.5 A g−1 and 110 mAh g−1 at 5 A g−1).
The incorporation of carbon-coated LFP particles (the battery-type material) with irregular shape and nanometer size in the graphene-activated-carbon positive electrode enables energy storage via faradaic, pseudo-capacitive, and capacitive mechanisms with an outstanding capacity even at high rates (102 mAh g−1 at 0.25 A g−1 and 80 mAh g−1 at 30 A g−1).
The full optimization of the assembled LICs using mass balance 1:1 allowed high energy densities without compromising power performance (143 Wh kg−1 at 128 W kg−1 and 45 Wh kg−1 at 25000 W kg−1), overcoming the main power limitations of LFP-containing LICs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Union (Graphene Flagship, Core 3, Grant number 881603) and the Spanish Ministry of Science and Innovation (MICINN/FEDER) (PID2021-127635OB-I00) for the financial support of this work. We also want to acknowledge the company GRAPHENEA for supplying the graphene oxide used in this work. María Jauregui is acknowledged for their assistance with the XRD.References
- M. Soltani and S. Beheshti, A comprehensive review of lithium ion capacitor: development, modelling, thermal management and applications, J. Energy Storage, 2020, 34, 102019, DOI:10.1016/j.est.2020.102019.
- L. Caizán-Juanarena, M. Arnaiz, E. Gucciardi, L. Oca, E. Bekaert, I. Gandiaga and J. Ajuria, Unraveling the Technology behind the Frontrunner LIC ULTIMO to Serve as a Guideline for Optimum Lithium-Ion Capacitor Design, Assembly, and Characterization, Adv. Energy Mater., 2021, 11, 2100912, DOI:10.1002/aenm.202100912.
- S. Li, Y. Xu, W. Liu, X. Zhang, Y. Ma, Q. Peng, X. Zhang, X. Sun, K. Wang and Y. Ma, Carbon nanocages bridged with graphene enable fast kinetics for dual-carbon lithium-ion capacitors, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.10.006.
- S. Yi, L. Wang, X. Zhang, C. Li, W. Liu, K. Wang, X. Sun, Y. Xu, Z. Yang, Y. Cao, J. Sun and Y. Ma, Cationic intermediates assisted self-assembly two-dimensional Ti3C2Tx/rGO hybrid nanoflakes for advanced lithium-ion capacitors, Sci. Bull., 2021, 66, 914–924, DOI:10.1016/j.scib.2020.12.026.
- S. Cao, H. Zhang, Y. Zhao and Y. Zhao, Pillararene/Calixarene-based systems for battery and supercapacitor applications, EScience, 2021, 1, 28–43, DOI:10.1016/j.esci.2021.10.001.
- B. Sun, N. Wang, M. Li, Y. Fang, J. Gu, W. Sun, W. Zhang, D. Zhang and Q. Liu, Constructing High-Performance Lithium-Ion Hybrid Capacitors Based on the Electrode Framework Matching Strategy, ACS Appl. Energy Mater., 2022, 5, 1963–1971, DOI:10.1021/acsaem.1c03492.
- L. Li, D. Zhang, J. Deng, Y. Gou, J. Fang, H. Cui, C. Zhang and M. Cao, Application of MXene-based materials in hybrid capacitors, Sustainable Energy Fuels, 2021, 5, 3278–3291, 10.1039/D1SE00448D.
- M. L. Divya, S. Praneetha, Y.-S. Lee and V. Aravindan, Next-generation Li-ion capacitor with high energy and high power by limiting alloying-intercalation process using SnO2@Graphite composite as battery type electrode, Composites, Part B, 2022, 230, 109487, DOI:10.1016/j.compositesb.2021.109487.
- C. Botas, D. Carriazo, G. Singh and T. Rojo, Sn– and SnO2–graphene flexible foams suitable as binder-free anodes for lithium ion batteries, J. Mater. Chem. A, 2015, 3, 13402–13410, 10.1039/C5TA03265B.
- M. Winter and J. O. Besenhard, Electrochemical lithiation of tin and tin-based intermetallics and composites, Electrochim. Acta, 1999, 45, 31–50, DOI:10.1016/S0013-4686(99)00191-7.
- T. Fang, L.-Y. Hsiao, J.-G. Duh and S.-R. Sheen, A novel composite negative electrode consist of multiphase Sn compounds and mesophase graphite powders for lithium ion batteries, J. Power Sources, 2006, 160, 536–541, DOI:10.1016/j.jpowsour.2006.01.034.
- M. Granados-Moreno, G. Moreno-Fernández, R. Cid, J. L. Gómez-Urbano and D. Carriazo, Microstructured nitrogen-doped graphene-Sn composites as a negative electrode for high performance lithium-ion hybrid supercapacitors, Sustainable Energy Fuels, 2022, 6, 700–710, 10.1039/D1SE01779A.
- Z.-J. Zhao, Y.-G. Chao, F. Wang, J.-Y. Dai, Y.-F. Qin, X.-B. Bao, Y. Yang and S.-J. Guo, Intimately coupled WS2 nanosheets in hierarchical hollow carbon nanospheres as the high-performance anode material for lithium-ion storage, Rare Met., 2022, 41, 1245–1254, DOI:10.1007/s12598-021-01850-w.
- M. Arnaiz, C. Botas, D. Carriazo, R. Mysyk, F. Mijangos, T. Rojo, J. Ajuria and E. Goikolea, Reduced graphene oxide decorated with SnO2 nanoparticles as negative electrode for lithium ion capacitors, Electrochim. Acta, 2018, 284, 542–550, DOI:10.1016/j.electacta.2018.07.189.
- Z. P. Guo, Z. W. Zhao, H. K. Liu and S. X. Dou, Electrochemical lithiation and de-lithiation of MWNT–Sn/SnNi nanocomposites, Carbon, 2005, 43, 1392–1399, DOI:10.1016/j.carbon.2005.01.008.
- E. Edison, A. Chaturvedi, H. Ren, S. Sreejith, C. T. Lim and S. Madhavi, Route of Irreversible Transformation in Layered Tin Thiophosphite and Enhanced Lithium Storage Performance, ACS Appl. Energy Mater., 2018, 1, 5772–5778, DOI:10.1021/acsaem.8b01357.
- M. Behm and J. T. S. Irvine, Influence of structure and composition upon performance of tin phosphate based negative electrodes for lithium batteries, Electrochim. Acta, 2002, 47, 1727–1738, DOI:10.1016/S0013-4686(02)00017-8.
- P. S. Attidekou, F. García-Alvarado, P. A. Connor and J. T. S. Irvine, Thermodynamic Aspects of the Reaction of Lithium with SnP2O7 Based Positive Electrodes, J. Electrochem. Soc., 2007, 154, A217, DOI:10.1149/1.2424410.
- Y. Li and J. Li, Carbon-Coated Macroporous Sn2P2O7 as Anode Materials for Li-Ion Battery, J. Phys. Chem. C, 2008, 112, 14216–14219, DOI:10.1021/jp804438v.
- T. Wu, G. Dai, C. Qin, J. Cao, Y. Tang and Y. Chen, A novel method to synthesize SnP2O7 spherical particles for lithium-ion battery anode, Ionics, 2016, 22, 2315–2319, DOI:10.1007/s11581-016-1773-3.
- P. S. Attidekou, P. A. Connor, P. Wormald, D. P. Tunstall, S. M. Francis and J. T. S. Irvine, Solid state NMR studies of phosphate/tin matrix formed on electrochemical insertion into SnP2O7, Solid State Ionics, 2004, 175, 185–190, DOI:10.1016/j.ssi.2003.12.048.
- I. Bezza, M. Kaus, L. Riekehr, L. Pfaffmann, S. Doyle, S. Indris, H. Ehrenberg, A. Solhy and I. Saadoune, Electrochemical lithiation/delithiation of SnP2O7 observed by in situ XRD and ex situ7Li/31P NMR, and 119Sn Mössbauer spectroscopy, Phys. Chem. Chem. Phys., 2016, 18, 10375–10382, 10.1039/C6CP01424K.
- I. Bezza, V. Trouillet, A. Fiedler, M. Bruns, S. Indris, H. Ehrenberg and I. Saadoune, Understanding the lithiation/delithiation process in SnP2O7 anode material for lithium-ion batteries, Electrochim. Acta, 2017, 252, 446–452, DOI:10.1016/j.electacta.2017.09.023.
- M. Secchiaroli, S. Calcaterra, R. Marassi, M. Wohlfahrt-Mehrens and S. Dsoke, High Energy and High Power Lithium-Ion Hybrid Supercapacitors with Prolonged Cycle Life Based on High-Rate Capability Materials: Li4Ti5O12, Activated Carbon, Li3V1.95Ni0.05(PO4)3/C, ChemElectroChem, 2020, 7, 1631–1643, DOI:10.1002/celc.202000281.
- A. Shellikeri, S. Yturriaga, J. S. Zheng, W. Cao, M. Hagen, J. A. Read, T. R. Jow and J. P. Zheng, Hybrid lithium-ion capacitor with LiFePO4/AC composite cathode – long term cycle life study, rate effect and charge sharing analysis, J. Power Sources, 2018, 392, 285–295, DOI:10.1016/j.jpowsour.2018.05.002.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries, J. Electrochem. Soc., 1997, 144, 1188, DOI:10.1149/1.1837571.
- N. Böckenfeld, R.-S. Kühnel, S. Passerini, M. Winter and A. Balducci, Composite LiFePO4/AC high rate performance electrodes for Li-ion capacitors, J. Power Sources, 2011, 196, 4136–4142, DOI:10.1016/j.jpowsour.2010.11.042.
- J. Yan, X. J. Chen, A. Shellikeri, T. Du, M. Hagen, J. P. Zheng and W. J. Cao, Influence of Lithium Iron Phosphate Positive Electrode Material to Hybrid Lithium-Ion Battery Capacitor (H-LIBC) Energy Storage Devices, J. Electrochem. Soc., 2018, 165, A2774, DOI:10.1149/2.0911811jes.
- S. Lv, X. Zhang, P. Zhang, J. Xiang, Y. Li, S. Qiu and C. Qin, One-step fabrication of nanosized LiFePO4/expanded graphite composites with a particle growth inhibitor and enhanced electrochemical performance of aqueous Li-ion capacitors, RSC Adv., 2019, 9, 14407–14416, 10.1039/C9RA02248A.
- S. Yang, X. Zhou, J. Zhang and Z. Liu, Morphology-controlled solvothermal synthesis of LiFePO4 as a cathode material for lithium-ion batteries, J. Mater. Chem., 2010, 20, 8086–8091, 10.1039/C0JM01346C.
- S. Soylu, Electric Vehicles: the Benefits and Barriers, BoD – Books on Demand, 2011 Search PubMed.
- D. Yan, A.-H. Lu, Z.-Y. Chen, L. He and W.-C. Li, Pseudocapacitance-Dominated Li-Ion Capacitors Showing Remarkable Energy Efficiency by Introducing Amorphous LiFePO4 in the Cathode, ACS Appl. Energy Mater., 2021, 4, 1824–1832, DOI:10.1021/acsaem.0c02943.
- G. Moreno-Fernández, M. Granados-Moreno, J. L. Gómez-Urbano and D. Carriazo, Phosphorus-Functionalized Graphene for Lithium-Ion Capacitors with Improved Power and Cyclability, Batteries Supercaps, 2021, 4, 469–478, DOI:10.1002/batt.202000247.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Is the bet equation applicable to microporous adsorbents?, in, Studies in Surface Science and Catalysis, ed. P. L. Llewellyn, F. Rodriquez-Reinoso, J. Rouqerol and N. Seaton, Elsevier, 2007, pp. 49–56, DOI:10.1016/S0167-2991(07)80008-5.
- J. Jagiello and J. P. Olivier, 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation, Carbon, 2013, 55, 70–80, DOI:10.1016/j.carbon.2012.12.011.
- C.-H. Huang, O. Knop, D. A. Othen, F. W. D. Woodhams and R. A. Howie, Pyrophosphates of Tetravalent Elements and a Mössbauer Study of SnP2O7, Can. J. Chem., 1975, 53, 79–91, DOI:10.1139/v75-011.
- M. Alaf, D. Gultekin and H. Akbulut, Tin/Tinoxide (Sn/SnO2) Nanocomposites Thin Films as Negative-Electrode Materials for Li-Ion Batteries, 2013, DOI:10.12693/APHYSPOLA.123.323.
- J. Oh, J. Lee, Y. Jeon, J. M. Kim, K. Seong, T. Hwang, S. Park and Y. Piao, Ultrafine Sn Nanoparticles Anchored on Nitrogen- and Phosphorus-Doped Hollow Carbon Frameworks for Lithium-Ion Batteries, ChemElectroChem, 2018, 5, 2098–2104, DOI:10.1002/celc.201800456.
- L. Szirtes, J. Megyeri and E. Kuzmann, Thermal behaviour of tin(II/IV) phosphates prepared by various methods, J. Therm. Anal. Calorim., 2010, 99, 415–421, DOI:10.1007/s10973-009-0461-1.
- X. Guo, Z. Wan, D. Wei, X. Zeng, Z. Li, W. Jiang, H. Wang, M. Ling, H. Li and C. Liang, Dual-Carbon Confined SnP2O7 with Enhanced Pseudocapacitances for Improved Li/Na-Ion Batteries, ChemElectroChem, 2021, 8, 2708–2714, DOI:10.1002/celc.202100793.
- G. Zeng, R. Caputo, D. Carriazo, L. Luo and M. Niederberger, Tailoring Two Polymorphs of LiFePO4 by Efficient Microwave-Assisted Synthesis: A Combined Experimental and Theoretical Study, Chem. Mater., 2013, 25, 3399–3407, DOI:10.1021/cm400995g.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Particle Size Dependence of the Ionic Diffusivity, Nano Lett., 2010, 10, 4123–4127, DOI:10.1021/nl1023595.
- F. Yu, L. Zhang, Y. Li, Y. An, M. Zhu and B. Dai, Mechanism studies of LiFePO4 cathode material: lithiation/delithiation process, electrochemical modification and synthetic reaction, RSC Adv., 2014, 4, 54576–54602, 10.1039/C4RA10899J.
- L. Liu, P.-L. Taberna, B. Dunn and P. Simon, Future Directions for Electrochemical Capacitors, ACS Energy Lett., 2021, 6, 4311–4316, DOI:10.1021/acsenergylett.1c01981.
- J. Wang, J. Polleux, J. Lim and B. Dunn, Pseudocapacitive Contributions to Electrochemical Energy Storage in TiO2 (Anatase) Nanoparticles, J. Phys. Chem. C, 2007, 111, 14925–14931, DOI:10.1021/jp074464w.
- M. Hagen, J. Yan, W. J. Cao, X. J. Chen, A. Shellikeri, T. Du, J. A. Read, T. R. Jow and J. P. Zheng, Hybrid lithium-ion battery-capacitor energy storage device with hybrid composite cathode based on activated carbon/LiNi0.5Co0.2Mn0.3O2, J. Power Sources, 2019, 433, 126689, DOI:10.1016/j.jpowsour.2019.05.095.
- F. Sun, J. Gao, Y. Zhu, X. Pi, L. Wang, X. Liu and Y. Qin, A high performance lithium ion capacitor achieved by the integration of a Sn-C anode and a biomass-derived microporous activated carbon cathode, Sci. Rep., 2017, 7, 40990, DOI:10.1038/srep40990.
- L. Jin, J. Zheng, Q. Wu, A. Shellikeri, S. Yturriaga, R. Gong, J. Huang and J. P. Zheng, Exploiting a hybrid lithium ion power source with a high energy density over 30 Wh/kg, Mater. Today Energy, 2018, 7, 51–57, DOI:10.1016/j.mtener.2017.12.003.
- School of Materials Science and Engineering, Southwest University of Science and Technology, Mianyang, Sichuan 621010, China and J. Li, Pre-lithiated Mesocarbon Microbeads Anode and Bifunctional Cathode for High Performance Hybrid Lithium-Ion Capacitors, Int. J. Electrochem. Sci., 2017, 12, 3212–3220, DOI:10.20964/2017.04.59.
- S. H. Lee, C. Huang and P. S. Grant, High energy lithium ion capacitors using hybrid cathodes comprising electrical double layer and intercalation host multi-layers, Energy Storage Mater., 2020, 33, 408–415, DOI:10.1016/j.ensm.2020.08.022.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2se01459a |
| This journal is © The Royal Society of Chemistry 2023 |