
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDipyrrolonaphthyridinedione – (still) a mysterious cross-conjugated chromophore
Bartłomiej
Sadowski
 *a and
Daniel T.
Gryko
*b
*a and
Daniel T.
Gryko
*b
aCentre of New Technologies, University of Warsaw, S. Banacha 2c, 02-097 Warsaw, Poland. E-mail: b.sadowski@cent.uw.edu.pl
bInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
First published on 10th November 2023
Abstract
Dipyrrolonaphthyridinediones (DPNDs) entered the chemical world in 2016. This cross-conjugated donor–acceptor skeleton can be prepared in two steps from commercially available reagents in overall yield ≈15–20% (5 mmol scale). DPNDs can be easily and regioselectively halogenated which opens an avenue to numerous derivatives as well as to π-expansion. Although certain synthetic limitations exist, the current derivatization possibilities provided impetus for numerous explorations that use DPNDs. Structural modifications enable bathochromic shift of the emission to deep-red region and reaching the optical brightness 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. Intense absorption and strong emission of greenish-yellow light attracted the interest which eventually led to the discovery of their strong two-photon absorption, singlet fission in the crystalline phase and triplet sensitization. Dipyrrolonaphthyridinedione-based twistacenes broadened our knowledge on the influence of twisting angle on the fate of the molecule in the excited state. Collectively, these findings highlight the compatibility of DPNDs with various applications within organic optoelectronics.
000 M−1 cm−1. Intense absorption and strong emission of greenish-yellow light attracted the interest which eventually led to the discovery of their strong two-photon absorption, singlet fission in the crystalline phase and triplet sensitization. Dipyrrolonaphthyridinedione-based twistacenes broadened our knowledge on the influence of twisting angle on the fate of the molecule in the excited state. Collectively, these findings highlight the compatibility of DPNDs with various applications within organic optoelectronics.
| Bartłomiej Sadowski Bartłomiej Sadowski was educated in chemistry at the Warsaw University of Technology, Poland. He then obtained his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences in 2019 under the supervision of Prof. Daniel T. Gryko. After completing his post-doctoral work at Georg-August-Universität Göttingen in the group of Prof. Lutz Ackermann, he began his independent career at Centre of New Technologies, University of Warsaw. His research interests focus on the electrochemically-enabled synthesis of high energy intermediates as well as chemistry of functional aromatic molecules, notably dipyrrolonaphthyridinediones. |
| Daniel T. Gryko Daniel T. Gryko obtained his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences in 1997, under the supervision of Prof. J. Jurczak. After a post-doctoral stay with Prof. J. Lindsey at North Carolina State University (1998–2000), he started his independent career in Poland. He became Full Professor in 2008. The same year he received the Society of Porphyrins and Phthalocyanines Young Investigator Award and in 2017 Foundation for Polish Science Award. His current research interests are focused on the synthesis of functional dyes as well as on two-photon absorption, solvatofluorochromism, excited-state intramolecular proton transfer and fluorescence probes. |
1. Introduction
A pyrrole ring is probably the most known small aromatic heterocycle. Many of its derivatives are found in a variety of natural products1 as well as drugs.2 In addition, this small heterocyclic motif is a key part of many functional dyes such as porphyrinoids,3 corroles,4 indolizines,5 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY)6 or bis(difluoroboron)-1,2-bis((1H-pyrrol-2-yl)methylene)hydrazine (BOPHY).7Due to its intrinsic electron-rich character, a pyrrole ring seems to be an ideal candidate for an electron-donating moiety in cross-conjugated chromophores. However, there is still a huge gap in the understanding of its nature when coupled with electron-accepting units. Some of the recent reports discussed an issue of aromaticity of cross-conjugated chromophores containing a pyrrole ring in a ground and excited singlet/triplet states.8
Donor–acceptor cross-conjugated dyes are well established in the literature.9 Compounds of this type, i.e. diketopyrrolopyrroles,10 isoindigos,11 cibalakrot8a,b,12 were tested as main components in organic electronics13 or biology.14 Donor–acceptor systems of cross-conjugated nature also played formidable role in human history. Indigo and Tyrian Purple (6,6′-dibromoindigo) were used as garment dyes for millennia.15 Moving to modern times, a diketopyrrolopyrrole molecule discovered by Farnum16 in 1974 and commercialized by Ciba Geigy,17 is a key structural motif of Pigment Red 254 – better known as ‘Ferrari Red’ as it was used to paint iconic Ferrari cars. Since then, there was no discovery of new type of donor–acceptor cross-conjugated dye. The unveiling of dipyrrolonaphthyridinediones (DPNDs) in our research group in 2016 has changed this situation (Fig. 1).18 The combination of very interesting and unique photophysical properties, straightforward synthesis as well as huge potential for functionalization are responsible for their career in the literature.
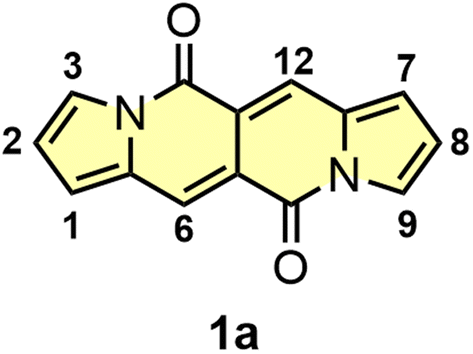 | ||
| Fig. 1 The structure and numbering of key positions of the simplest dye belonging to the DPND family. | ||
In this Perspective article, we will describe the beginnings as well as the history of DPNDs. First of all, we will briefly discuss the synthetic pathways leading to the core followed by describing already known synthetic modifications thereof. Finally, optoelectronic properties will be studied with special emphasis given to structure–property relationship followed by specific applications that has been tested until now. We believe that such structure of the Perspective will open up new avenues for this interesting and still undiscovered chromophore.
2. Synthesis of dipyrrolonaphthiridinediones
The DPND core (Fig. 1) features a 5,6,6,5-type architecture and is comprised of four linearly-fused cycles: two pyrrole rings at the periphery and two six-membered rings in the central part of a chromophore, each containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.18 From the structural point of view, the DPND core closely resembles the BOPHY fluorophore,7 however the latter bears BF2 moieties along with the bis-nitrogen bridge which results in different optoelectronic behavior.
O group.18 From the structural point of view, the DPND core closely resembles the BOPHY fluorophore,7 however the latter bears BF2 moieties along with the bis-nitrogen bridge which results in different optoelectronic behavior.
Accordingly to X-ray analysis of 1f (see Table 1 and Fig. 2),18 the core is rigidly planar and deviations from the plane are no greater than 0.018 Å. Undoubtedly, the steric clash between CO groups and alkyl chains at positions 6 and 12 has detrimental influence on the chromophore structure as bond angles between CO groups and carbon atoms adjacent to the alkyl groups deviate significantly from their ideal trigonal values of 120°. The values are closer to 120° in the case of 1a that lacks alkyl chains at positions 6 and 12, as revealed by Wang and others19 (vide infra).
|
|
|||||
|---|---|---|---|---|---|
| Carboxylic acid | Reaction time [h] | DPND | R | Yield [%] | Ref. |
| a Reagents proportions: 4 (0.5 mmol), carboxylic acid (3 mmol), TFAA (6 mmol), TFA (3 mmol). b 5 mmol scale. c Propionic anhydride was used instead of carboxylic acid. d Unpublished results. | |||||
| CH3CO2H | 5 | 1b | CH3 | 17 | 18 |
| C7H15CO2H | 3 | 1c | C7H15 | 29 | 18 |
| 23b | 18 | ||||
| Et(Me)CHCO2H | 6 | 1d | sec-Butyl | 21 | 18 |
| 4-(MeO)C6H4CH2CO2H | 6 | 1e | 4-Methoxy-benzyl | 10 | 18 |
| (C2H5CO)2Oc | 2 | 1f | C2H5 | 23 | 18 |
| C3H7COOH | 4 | 1g | C3H7 | 21 | 21 |
| C6H13COOH | 2 | 1h | C6H13 | 32 | 19 |
| 2-NO2C6H4COOH | 3 | 1i | 2-NO2C6H4 | 0 | —d |
| 2-ThienylCH2COOH | 3 | 1j | 2-ThienylCH2 | 0 | —d |
| 3,4-(MeO)2C6H4CH2CO2H | 3 | 1k | 3,4-Dimethoxy-benzyl | 0 | —d |
| (E)-Cinnamic acid | 3 | 1l | –CC–Ph |
0 | —d |
| PhCOOH | 3 | 1m | Ph | 0 | —d |
| 2-ThienylCH2COOH | 3 | 1n | 2-ThienylCH2 | 0 | —d |
| HOOC(CH2)7COOH | 3 | 1o | –(CH2)7– | 3 | 20 |
| HOOC(CH2)8COOH | 3 | 1p | –(CH2)8– | 15 | 20 |
| HOOC(CH2)9COOH | 3 | 1r | –(CH2)9– | 3 | 20 |
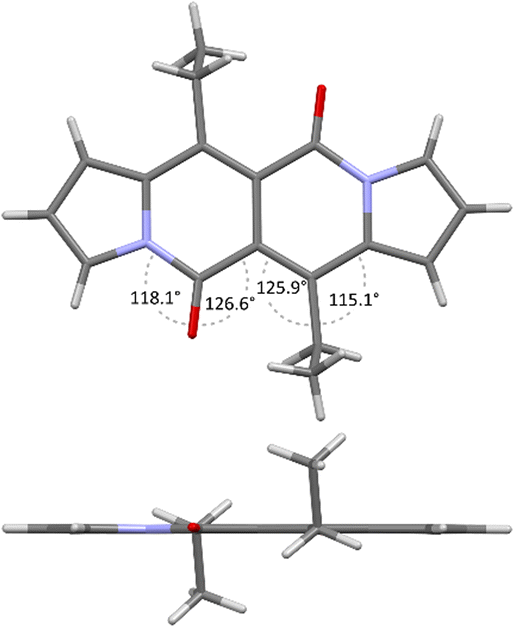 | ||
| Fig. 2 X-ray structure of compound 1f. Views perpendicular (top) and along (bottom) with respect to the chromophore plane. Adapted with permission from ref. 18. Copyright 2016 Royal Society of Chemistry. | ||
Initial attempts to assemble the DPND core were based on the use of 2-formylpyrrole (2) and succinyl chloride (3) as simple and commercially available building blocks, however 1a could be isolated in only 3.4–6.4% yield along with unreacted 2 and a large amount of black precipitate in both cases (Scheme 1).18 The presence of the black precipitate was associated with base-mediated polymerization of 3 as its formation was also noted in the absence of the aldehyde.
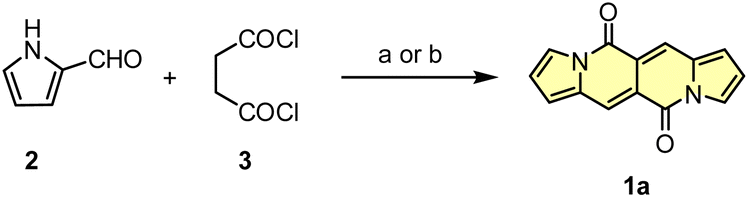 | ||
| Scheme 1 Initial attempts of the synthesis of DPND derivatives. Reaction conditions: (a) DMAP (20 mol%), Et3N, CH2Cl2, rt, 2 h. Yield: 3.4%; (b) K2CO3, DMF, 0 °C, 2 h. Yield: 6.4%. | ||
Another tested strategy towards 1a involved dipyrroyl derivative 4 as a source of pyrrole rings. Compound 4 can be easily synthesized from either 2,5-dimethoxytetrahydrofurane and succinamide18,20 or pyrrole and 3.21 Indeed, subjecting 4 to the typical Vilsmeier–Haack reaction conditions (DMF/POCl3) resulted in the formation of 1a, but still with low efficiency (Scheme 2). During the optimization process, intermediate of type 5 was not detected at all.
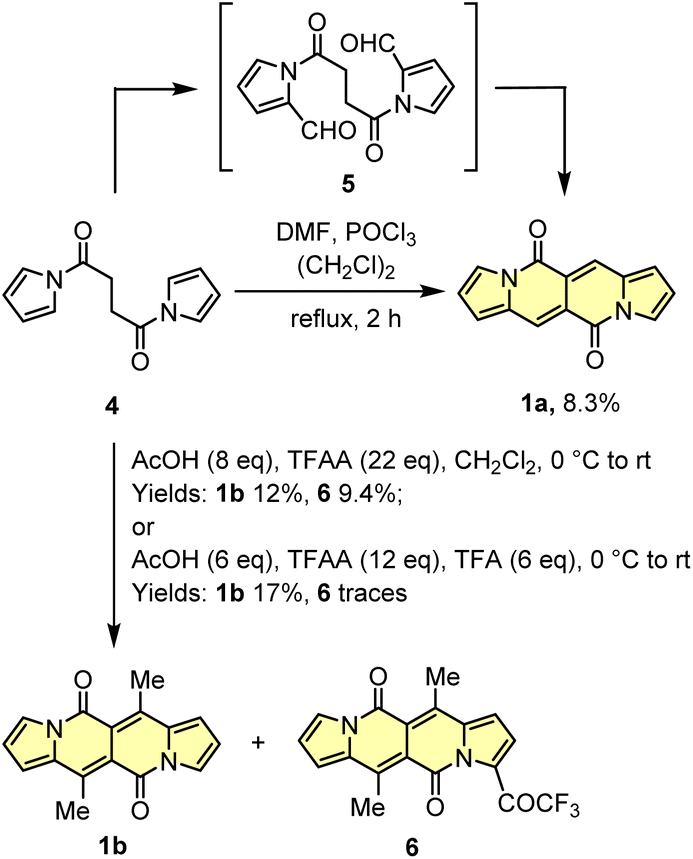 | ||
| Scheme 2 Synthesis of DPNDs via acylation of a pyrrole ring. | ||
Due to low efficiencies and relatively poor solubility of 1a in common organic solvents, we intended to introduce additional substituents that improve solubility of resulted chromophores. This idea was realized via an acylation reaction employing conditions previously developed for N-tosylpyrroles.22 Specifically, in the presence of excess of acetic acid, dipyrroyl derivative 4 undergoes double acylation followed by aldol-type condensation eventually affording 1b in 12% yield along with 9% of 6 (Scheme 2). The overall efficiency was further improved by the decrease in amounts of TFAA and TFA. In general, the developed method allows for assembling 6,12-difunctionalized DPND derivatives 1b–h as well as cyclophane analogues 1o–r (Table 1). During our ongoing adventure with these strongly fluorescent chromophores, it appeared that DPNDs are formed only when alkyl carboxylic acids are used, although with some exceptions (1j, 1k). The highest yield (32%) has been recently reported for enanthic acid (C6H13COOH)19 as a carboxylic partner. All attempts with carboxylic acids other than aliphatic ones failed.
3. Synthetic modifications of the DPND core
As the DPND core consists of two flanking pyrrole rings as well as the carbonyl-based central part, synthetic modifications of this chromophore should be a result of inherent reactivity (to a certain extent) of these two parts. It should be noticed here that all three available positions within the pyrrole ring are not equivalent due to unsymmetrical mode of fusion between 5- and 6-membered rings. As a proof of concept, we initially envisaged bromination of the pyrrole part (Scheme 3) as it is well known that aromatic halides are outstanding feedstocks in a wide range of transition-metal-catalyzed cross-coupling reactions.23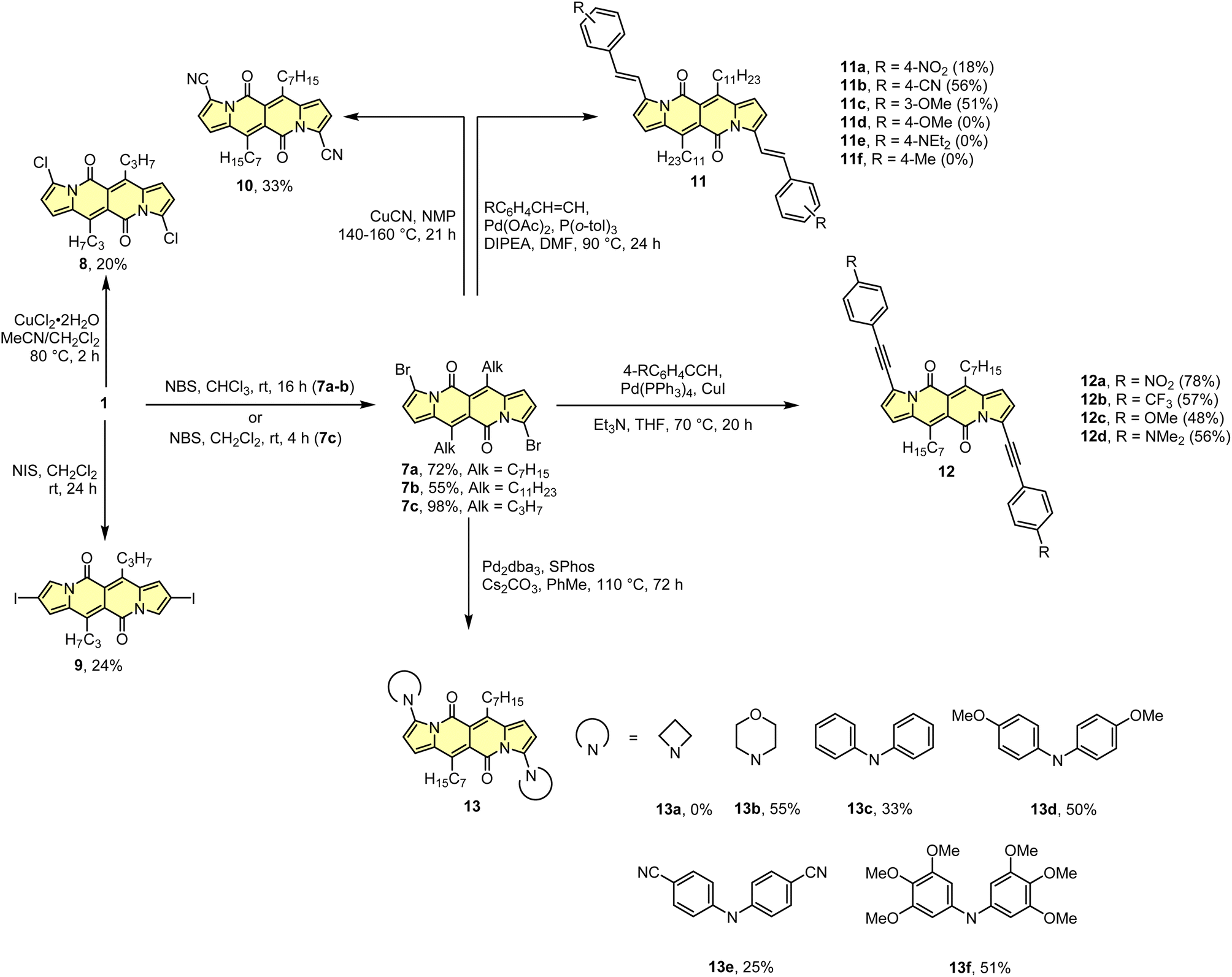 | ||
| Scheme 3 Derivatization of the DPND core based on halogenation reactions. | ||
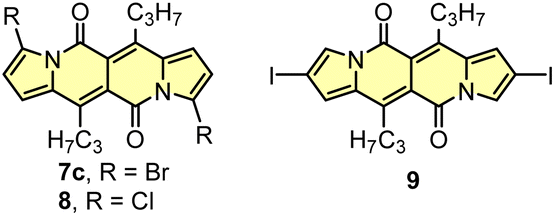
Indeed, 1 selectively undergoes double bromination reaction at the positions 3 and 9 (product 7) using NBS (N-bromosuccinimide) as a bromine atom source. Moreover, we found out that chloroform stabilized with amylene performed better in this reaction compared to that one stabilized with ethanol. Besides bromination, chlorination and iodination reactions were examined by Ayitou and others.21 While under the influence of CuCl2·2H2O double chlorination reaction occurs at the positions 3 and 9 (product 8), applying NIS (N-iodosuccinimide) as an iodination reagent results in completely different regioselectivity, giving rise to 2,8-diiodinated DPND (9). The unexpected regioselectivity of iodination was presumably ascribed to a large size of an iodine atom (potential steric clash with a carbonyl oxygen atom). However, the fact of decreased reactivity at the α position of a pyrrole ring caused by the presence of electron-withdrawing groups within this ring cannot be excluded.24
The very presence of halogen atoms at positions 3 and 9 should enable further transformations. As an example, subjecting compound 7a to the Rosenmund-von Braun reaction conditions resulted in dicyano derivative 10 in 33% yield,18 proving that further derivatization of DPND-based halides is possible.
As mentioned above, halides constitute one of the best platforms for derivatization, especially towards enlarged architectures by employing acetylenes25 or styrenes6a,26 as coupling partners. As a matter of fact, dibrominated DPNDs of type 7 were successfully transformed into a variety of π-expanded platforms employing the main types of cross-coupling reactions. The Pd(OAc)2/P(o-tol)3 system proved to be effective in the construction of quadrupolar, centrosymmetric molecules 11a–f with double bond linkages via the Heck reaction.27 Employing a typical catalytic system for the Sonogashira reaction led to molecules 12a–d bearing triple bond π-spacers.18 Finally, amine-decorated DPNDs 13a–f can be assembled via the Buchwald–Hartwig amination reaction using the Pd2dba3/SPhos system.28 It should be mentioned here that some of π-expanded derivatives, especially those bearing strongly electron-donating groups, were not sufficiently stable during isolation or it was not possible to isolate them in a pure form due to similar affinity to a stationary phase and/or lower solubility.
Compared to cross-coupling reactions of heteroaryl halides, transition-metal-catalyzed C–H activation processes are characterized by higher atom and step economy.29 In other words, employing C–H activation for the functionalization of organic chromophores helps avoiding often problematic derivatization (halogenation, borylation etc.).
The DPND core appeared to be an ideal platform for the study on direct arylation reaction (Scheme 4)30 which was widely used as a common strategy toward organic materials.31 The catalytic system involves Pd2dba3 as a catalyst, PCy3·HBF4 as a ligand, PivOH as an additive, and allows for the functionalization of the DPND core at the positions 3 and 9 with differently decorated arene rings. From the view-point of industrial research, it was proven that Pd2dba3 can be replaced with cost-efficient Pd(OAc)2.32 Variety of aryl halides are reactive towards the DPND core, nevertheless those bearing electron-withdrawing groups performed better. In a typical reaction, 2.2-3-fold excess of aryl halide was used to achieve doubly-arylated derivatives. Monoarylation is also achievable by applying 2.0 equivalents of 1cversus aryl halide, as proved for some nitroaryl-decorated DPNDs (Scheme 3).30b Interestingly, only for 1-bromo-8-nitronaphthalene we were not able to obtain doubly arylated derivative due to high steric congestion within the reaction centre. This resulted in messy reaction outcome where the expected, doubly-functionalized product was not detected at all.
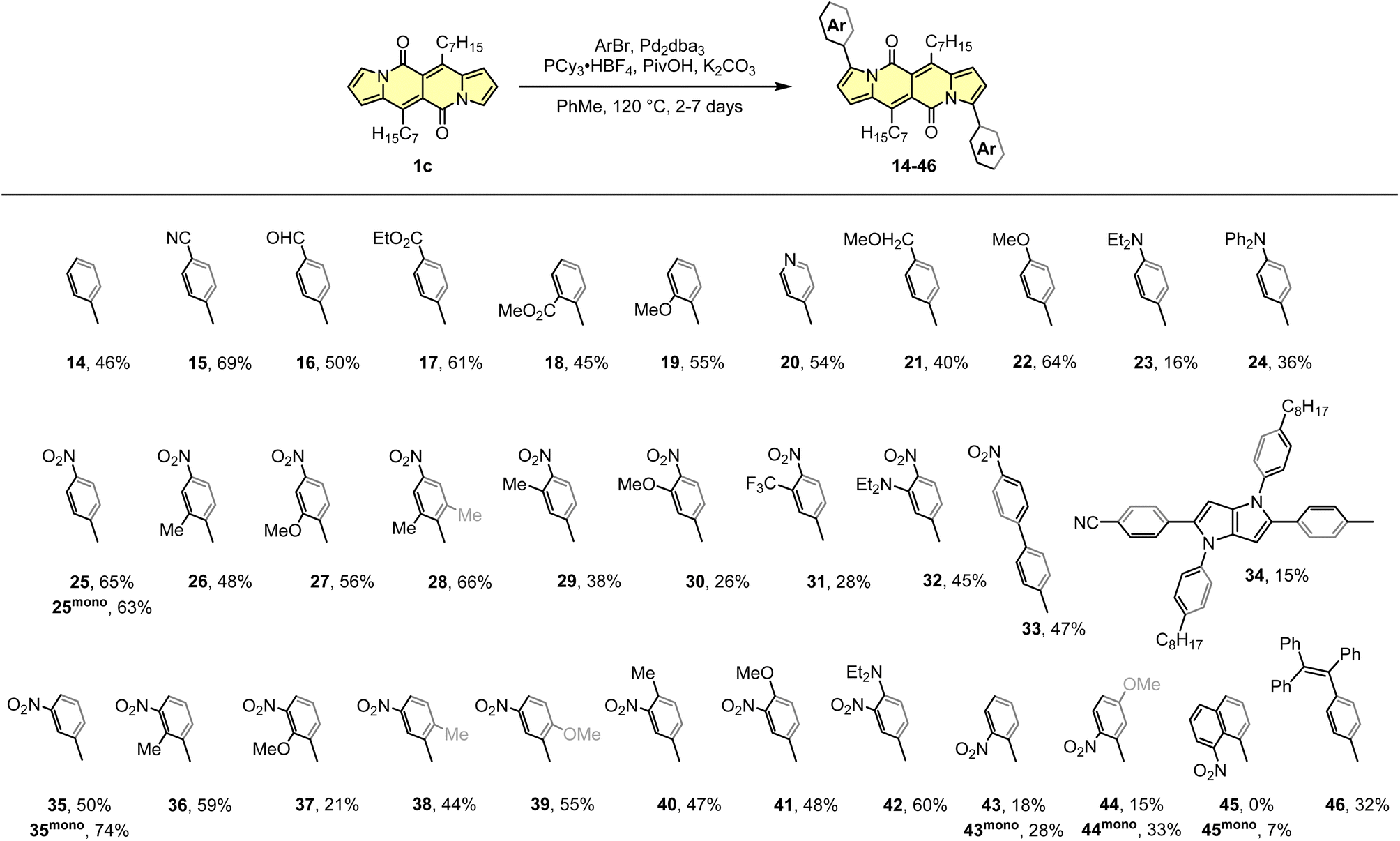 | ||
| Scheme 4 Direct arylation of the DPND core. | ||
The developed direct arylation methodology was further applied in the synthesis of N-doped analogues of polycyclic aromatic hydrocarbons (PAHs) (Scheme 5).33 Arylation of the DPND core with sterically congested aryl halides bearing acetylamino groups led to a series of dyes 47 that can be smoothly transformed into polycyclic aromatics of type 48, under the influence of strong acid(s). Here, the reactivity of carbonyl moieties within the DPND core was tested in a condensation process, similarly to the reaction described for perylene bisimides.34 Among tested derivatives, bis-arylated dye 47d failed as a precursor of 48d presumably due to low nucleophilicity of nitrogen caused by the presence of a strongly electron-withdrawing NO2 group at the para position relative to NHAc.
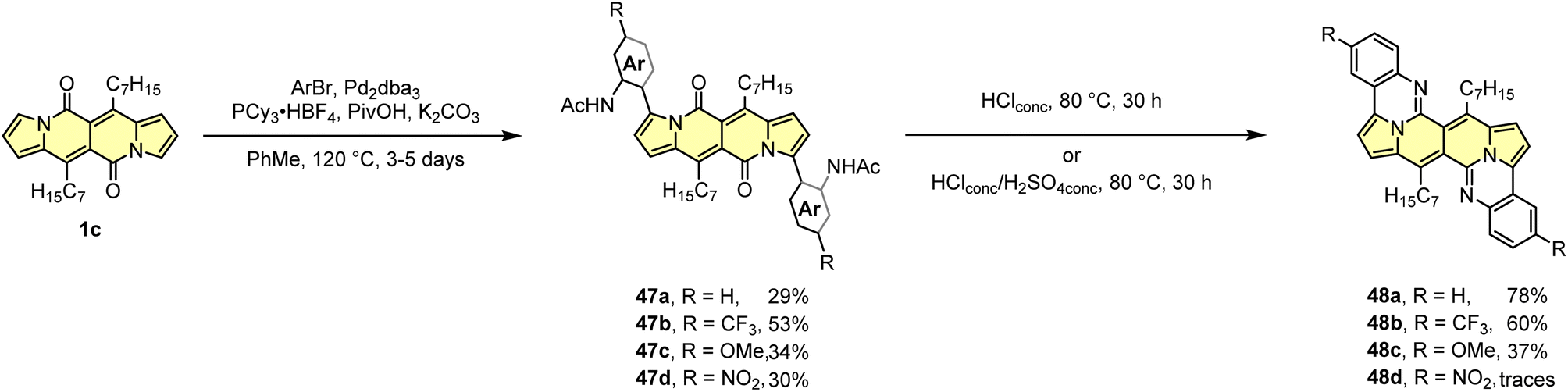 | ||
| Scheme 5 Functionalization of carbonyl moieties within the DPND core. | ||
4. The optoelectronic properties of dipyrrolonaphthyridinedione and its applications
Unsubstituted compound 1a exhibits an intense structured absorption band with the longest maxima (λmaxabs) at 509 nm in dichloromethane (CH2Cl2), and molar extinction coefficient of 26600 M−1 cm−1 (Table 2). Alkyl-substituted derivatives 1b–h shows similar structural absorption bands that are hypsochromically-shifted compared to 1a (Fig. 3 and Table 2), probably due to additional steric interactions between alkyl chains at positions 6 and 12 with carbonyl groups. The origin of these “sub-bands”, although not investigated in detail for DPNDs, one may possibly attribute to vibrational progression in the excited state, as found previously for BOPHY dyes.7 In general, DPNDs 1a–h are strongly fluorescent in CH2Cl2 (Φfl up to 0.71). Compared to BOPHYs however, DPNDs emit less intensively as values of Φfl measured for simple BOPHY-type dyes approach ≈0.9.7
| Dye | λ maxabs [nm] | ε max [M−1 cm−1] | λ maxem [nm] | Φ fl | δνa [cm−1] |
|---|---|---|---|---|---|
| a Stokes shift i.e. difference between lowest energy absorption band and highest energy emission band expressed in cm−1. b Reference: Rhodamine 6G in EtOH (Φfl = 0.94). c Reference: cresyl violet in MeOH (Φfl = 0.54). | |||||
| 1a | 509 | 26600 |
535 | 0.61b | 950 |
| 1b | 499 | 29200 |
523 | 0.66b | 900 |
| 1c | 504 | 29300 |
528 | 0.71b | 900 |
| 1d | 505 | 23100 |
543 | 0.58b | 1400 |
| 1e | 510 | 28300 |
536 | 0.26b | 950 |
| 1f | 500 | 28000 |
526 | 0.67b | 1000 |
| 6 | 503 | 24600 |
601 | 0.46b | 3240 |
| 10 | 517 | 34600 |
537 | 0.25b | 720 |
| 11a | 621 | 60200 |
699 | 0.016c | 1800 |
| 11b | 610 | 56300 |
671 | 0.022c | 1500 |
| 11c | 603 | 55100 |
662 | 0.041c | 1500 |
| 12a | 599 | 57300 |
633 | 0.51c | 900 |
| 12b | 584 | 47400 |
616 | 0.51c | 900 |
| 12c | 601 | 52400 |
643 | 0.59c | 1100 |
| 12d | 645 | 56600 |
736 | 0.17c | 1900 |
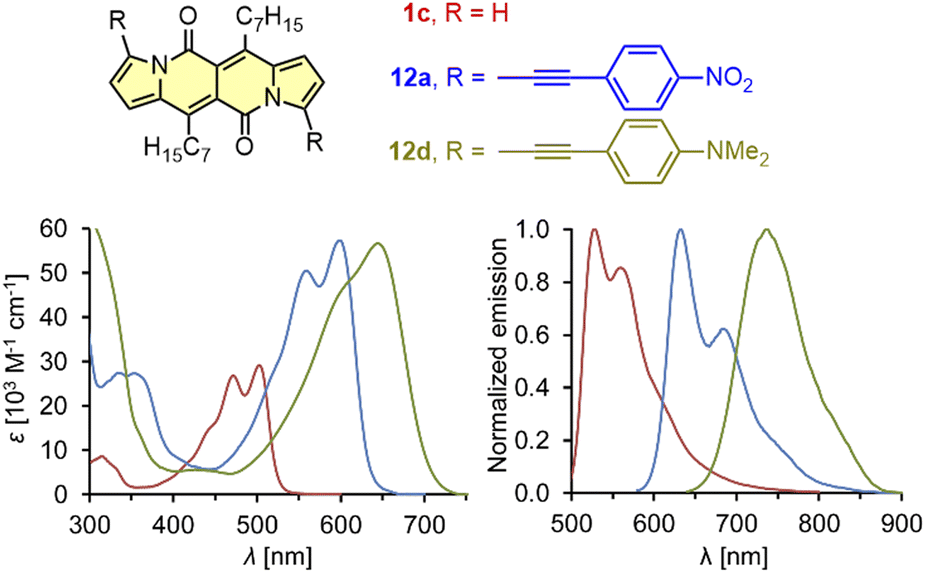 | ||
| Fig. 3 Absorption and normalized emission spectra of dye 1c (red), 12a (blue), and 12d (green). Adapted with permission from ref. 18 Copyright 2016 Royal Society of Chemistry. | ||
It should be mentioned that during the preparation of alkyl-substituted DPNDs (i.e.1c), a trifluoroacetyl derivative 6 is also obtained as a minor product (Scheme 2). Here, the emission band is red-shifted by ca. 80 nm as compared with its analog 1c (Table 2) and both absorption and emission bands become structureless.18 The most convenient way toward dyes that absorb and emit at significantly longer wavelengths is to introduce arylethynyl or arylethenyl moieties at the core's peripheries. The Sonogashira coupling of 7a with different arylacetylenes gave rise to a series of π-expanded DPNDs 12 which generally absorb at 584–601 nm and emit at 616–643 nm (Table 2 and Fig. 3).18 Importantly, the largest red-shift of both bands was observed for 12d bearing Et2N auxochromes at peripheries which suggests that the DPND core as a whole behaves as an electron acceptor. Although the value of Φfl dropped down to 0.17 (compared with 12a–c), 12d absorbs in the red region of the spectrum and emits at 736 nm.
In turn, vinylidene-linked systems 11a–c27 absorb at longer wavelengths that is around 603–621 nm (Table 2) and this is due to better electronic conjugation35 between the core and groups at the peripheries. These dyes are poorly emissive (λmaxem = 662–669 nm, Φfl < 0.1), however, probably because of an additional energy dissipation mechanism, i.e. E–Z isomerization of a CC double bond.36
An “electron-accepting” character of the DPND core was further probed by studying a variety of weakly coupled, quadrupolar dyes prepared via direct arylation methodology.30a As expected, the presence of biaryl-type connection between the DPND core and auxochromes at peripheries leads to weaker electronic conjugation between them. The dihedral angle between the DPND core and an aryl substituent was found to be ≈40°–45° or 50°–55° based on DFT methods30a and X-ray analysis,30b,30c respectively. Consequently, these dyes feature absorption and emission bands at shorter wavelengths compared to 11 and 12 (Table 3 and Fig. 4). In general, synthesized dyes are moderately fluorescent (Φfl ≈ 0.3–0.6) and the emission band is located in the red region of the spectrum (λmaxem ≈ 600–620 nm).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Dye | R | Solvent | λ maxabs [nm] | ε max [M−1 cm−1] | λ maxem [nm] | Φ fl | δν [cm−1] |
| a Solutions were sonicated for 15–30 min directly before measurement. b Not measured due to low S/N ratio. c Sulforhodamine 101 was used as a reference (Φfl = 0.95 in EtOH). d Cresyl violet was used as a reference (Φfl = 0.54 in MeOH). e 4-Pyridyl groups are attached to the DPND core instead of R-C6H4. | |||||||
| 15 | 4-CN | CH2Cl2 | 543 | 33200 |
602 | 0.36c | 1800 |
| DMSOa | 549 | 31000 |
614 | 0.38c | 1900 | ||
| 16 | 4-CHO | CH2Cl2 | 547 | 37100 |
607 | 0.36c | 1800 |
| DMSOa | 555 | 36600 |
619 | 0.36c | 1900 | ||
| 17 | 4-CO2Et | CH2Cl2 | 544 | 34000 |
600 | 0.44c | 1700 |
| DMSOa | 550 | 27200 |
614 | 0.40c | 1900 | ||
| 18 | 2-CO2Me | CH2Cl2 | 536 | 31400 |
581 | 0.63c | 1400 |
| DMSOa | 542 | 32100 |
591 | 0.57c | 1500 | ||
| 19 | 2-OMe | CH2Cl2 | 536 | 31100 |
603 | 0.60c | 2100 |
| DMSOa | 541 | 30200 |
607 | 0.54c | 2100 | ||
| 20 | 4-Pyridyle | CH2Cl2 | 535 | 31600 |
582 | 0.46c | 1500 |
| DMSOa | 540 | 31800 |
599 | 0.45c | 1900 | ||
| 21 | 4-CH2OMe | CH2Cl2 | 542 | 37100 |
601 | 0.38c | 1800 |
| DMSOa | 555 | 27100 |
620 | 0.42c | 1900 | ||
| 22 | 4-OMe | CH2Cl2 | 550 | 26600 |
624 | 0.43c | 2100 |
| DMSOa | 557 | 34800 |
643 | 0.36c | 2400 | ||
| 23 | 4-NEt2 | CH2Cl2 | 615 | 39500 |
735 | 0.10 d | 2700 |
| 24 | 4-NPh2 | CH2Cl2 | 583 | 38000 |
—b | —b | —b |
| 25 | 4-NO2 | CH2Cl2 | 548 | 29300 |
611 | 0.46c | 1900 |
| 34 | Pyrrolo[3,2-b]pyrrole core | CH2Cl2 | 399, 579 | 83600, 47300 |
—b | —b | —b |
| 43 | 2-NO2 | CH2Cl2 | 538 | 26500 |
—b | —b | —b |
| DMSOa | 545 | 26800 |
—b | —b | —b | ||
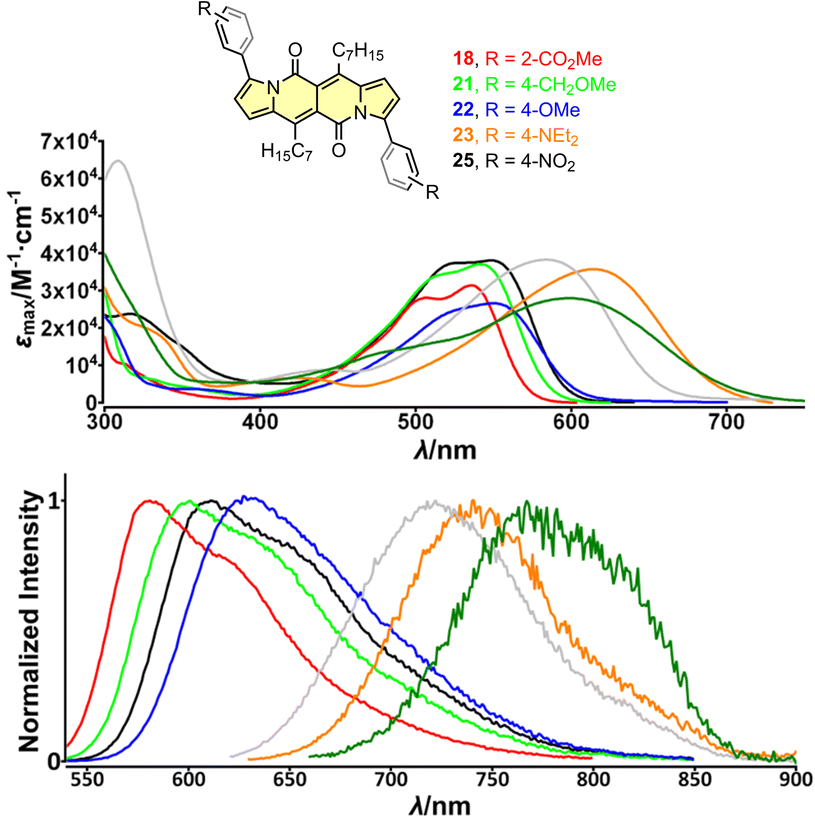 | ||
| Fig. 4 Top: absorption spectra of compounds 18 (red), 21 (green), 22 (blue), 23 (orange) and 25 (black) measured in dichloromethane. Bottom: normalized emission spectra of compounds 18 (red), 21 (green), 22 (blue), 23 (orange) and 25 (black) measured in dichloromethane. Adapted with permission from ref. 30a. Copyright 2018 Wiley. | ||
The largest change in the optical behaviour was noted for DPNDs with NR2 groups at peripheries: bathochromically-shifted both absorption and emission bands and lower value of Φfl by contrast with 15–21 (Table 3 and Fig. 4). According to theoretical calculations (Fig. 5), compounds bearing CN, H and Me groups at peripheries feature locally-excited transitions (S1-LE(π–π*)), where HOMO and LUMO wavefunctions are located mostly within the DPND core. In contrast, for NMe2 and OMe groups the electron density is shifted from outer groups toward the core upon the photoexcitation, with largest net change noted for the dye bearing NMe2 group.
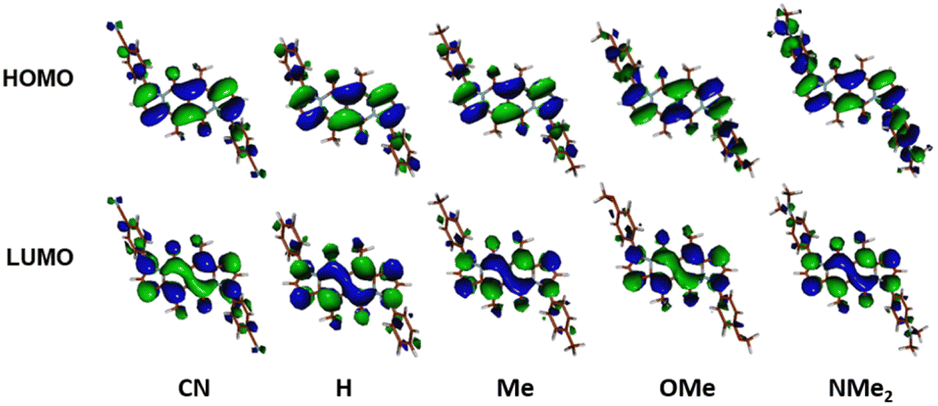 | ||
| Fig. 5 Frontier molecular orbitals of compounds from DPND(C6H4R)2 series calculated in C2 symmetry at the DFT(B3-LYP) methods using the cc-pVDZ basis set. Adapted with permission from ref. 30a. Copyright 2018 Wiley. | ||
The above-mentioned observations suggest that placing electron-donating groups at peripheries induce significant red shift of absorption and emission and the DPND core behaves like an “electron-acceptor”.
The electron-accepting character of the core was further deeply investigated28 by studying a series of dyes 13a–f decorated with an amine function connected directly with the core or by a π-spacer (12d, 23 and 24). We found that these dyes feature evident solvatofluorochromic behavior (Fig. 6) that is the emission bands shift toward lower energies (usually λmaxem > 700 nm) as well as the value of Φfl lowers as polarity of the medium increases (Table 4). Interestingly, compound 13e exhibits slightly different behavior compared to other dyes belonging to a series 13. The very presence of four CN groups withing amine moieties partially hampers electron density shift toward the DPND core and hence the efficient formation of dark charge-transfer (CT) states which results in less pronounced solvatofluorochromism. On the other hand, Φfl increases sharply in more polar environment reaching values of 0.4–0.6 (Table 4).
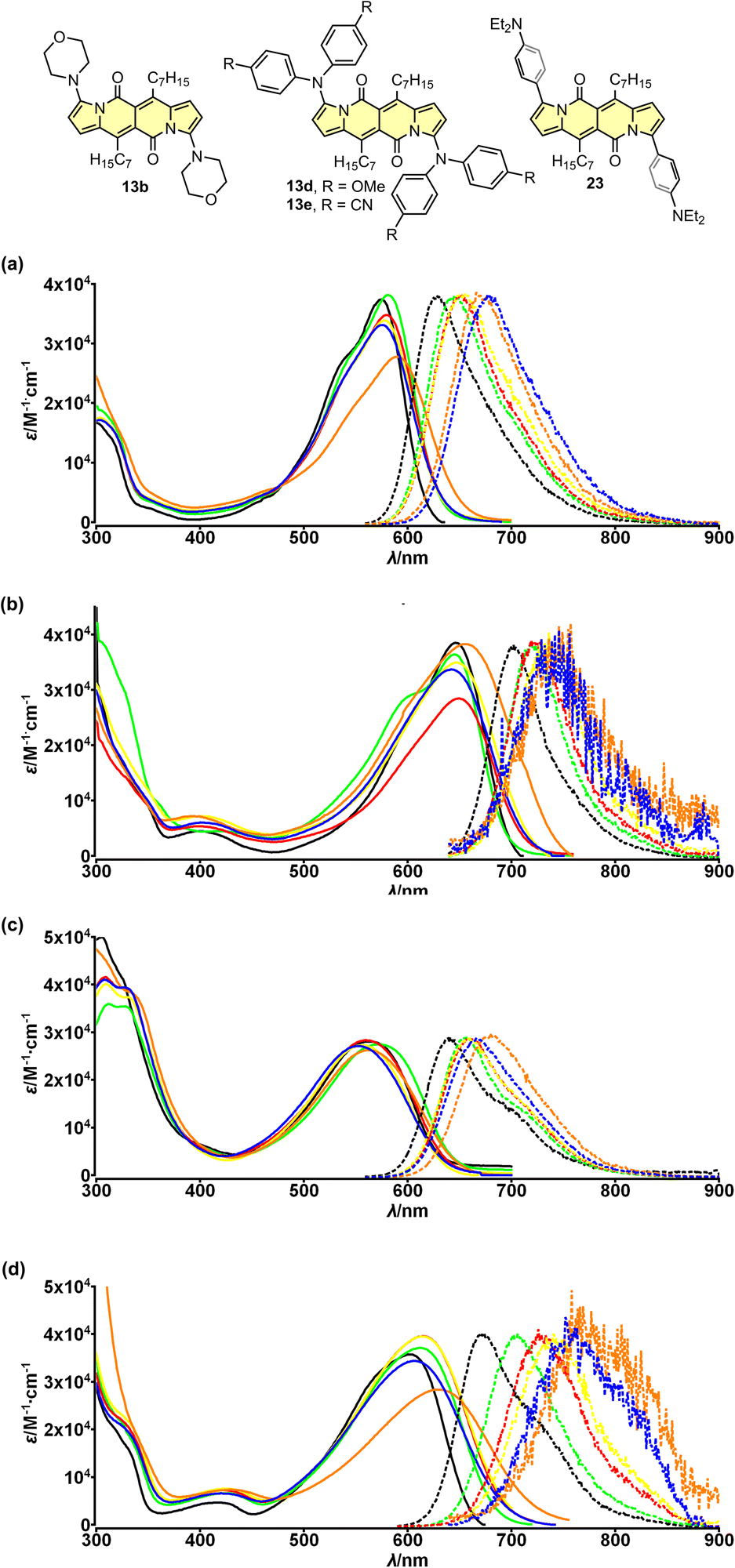 | ||
| Fig. 6 Absorption (solid) and emission (dotted) spectra of compounds 13b (a), 13d (b), 13e (c) and 23 (d) measured in cyclohexane (black), toluene (green), THF (red), CH2Cl2 (yellow), MeCN (blue) and DMSO (orange). Adapted with permission from ref. 28 Copyright 2018 American Chemical Society. | ||
| Dye | Solvent | λ maxabs [nm] | ε max [M−1 cm−1] | λ maxem [nm] | Φ fl | δνa [cm−1] |
|---|---|---|---|---|---|---|
| a Stokes shift i.e. difference between lowest energy absorption band and highest energy emission band expressed in cm−1. b Sulforhodamine 101 was used as a reference (Φfl = 0.95 in EtOH). c Cresyl violet was used as a reference (Φfl = 0.54 in MeOH). | ||||||
| 13b | Cyclohexane | 575 | 37000 |
628 | 0.35b | 1500 |
| Toluene | 582 | 38000 |
645 | 0.32b | 1700 | |
| THF | 580 | 35000 |
650 | 0.35b | 1900 | |
| CH2Cl2 | 578 | 34000 |
653 | 0.27b | 2000 | |
| MeCN | 576 | 29000 |
659 | 0.21b | 2200 | |
| DMSO | 590 | 28000 |
670 | 0.18b | 2000 | |
| 13d | Cyclohexane | 645 | 38000 |
702 | 0.39c | 1300 |
| Toluene | 654 | 35000 |
718 | 0.26c | 1400 | |
| THF | 648 | 37000 |
720 | 0.10c | 1500 | |
| CH2Cl2 | 648 | 35000 |
727 | 0.07c | 1700 | |
| MeCN | 642 | 34000 |
732 | 0.01c | 1900 | |
| DMSO | 656 | 38000 |
739 | 0.01c | 1700 | |
| 13e | Cyclohexane | 565 | nd | 639 | 0.37b | 2000 |
| Toluene | 571 | 27000 |
655 | 0.49b | 2200 | |
| THF | 560 | 28000 |
658 | 0.62b | 2700 | |
| CH2Cl2 | 558 | 27000 |
659 | 0.61b | 2700 | |
| MeCN | 554 | 27000 |
666 | 0.63b | 3000 | |
| DMSO | 564 | 26000 |
677 | 0.47b | 3000 | |
| 23 | Cyclohexane | 603 | 36000 |
670 | 0.40c | 1700 |
| Toluene | 613 | 37000 |
702 | 0.25c | 2100 | |
| THF | 615 | 40000 |
729 | 0.11c | 2500 | |
| CH2Cl2 | 615 | 39000 |
735 | 0.10c | 2700 | |
| MeCN | 607 | 34000 |
750 | 0.02c | 3100 | |
| DMSO | 631 | 28000 |
758 | 0.02c | 2700 | |
Cyclic voltammetry (CV) appeared to be an excellent methodology to probe the electronic structure of dipyrrolonaphthiridinediones (DPNDs) (Fig. 7–9). A CV trace of unsubstituted dye 1a displays irreversible oxidation/reduction events and the shape of the CV curve changes with a rising number of redox cycles (Fig. 7).18 Upon an oxidation event a black, insoluble deposit appeared on an anode which did not dissolve upon reduction. Such a deposit did not form in the case of 1c, 10 and 12b (Fig. 8). For 1c, the results show a reversible reduction with E1/2 = −1.125 V vs. SCE (ELUMO = −3.2 eV) in the accessible potential range. The appearance of reducible CN groups in the chromophore structure (compound 10, Fig. 8) results in an additional reduction wave within the CV curve (E1/2 = −0.675 V vs. SCE) that can be clearly connected with those groups, while the often reversible reduction event characteristic for the DPND core shifted toward more negative potentials (E1/2 = −1.215 V vs. SCE) and the oxidation wave is not present in the accessible potential range. This constitutes a general feature for DPND derivatives bearing electron-withdrawing groups at the peripheries.30a,b When it comes to electrochemically-driven oxidation events, most of DPND derivatives show irreversible oxidation wave (if any). The situation is changed when another, easily oxidizable groups are present in the structure (Fig. 9).28 Cyclic voltammograms of electron-rich DPND derivatives containing NR2 groups (i.e.13f, Fig. 9), beside one reversible reduction event associated with the core, frequently include two oxidation waves where the first event is connected with a reversible (often stepwise) oxidation of the –NR2 auxochrome while the second one (irreversible) comes from an oxidation process within the core.
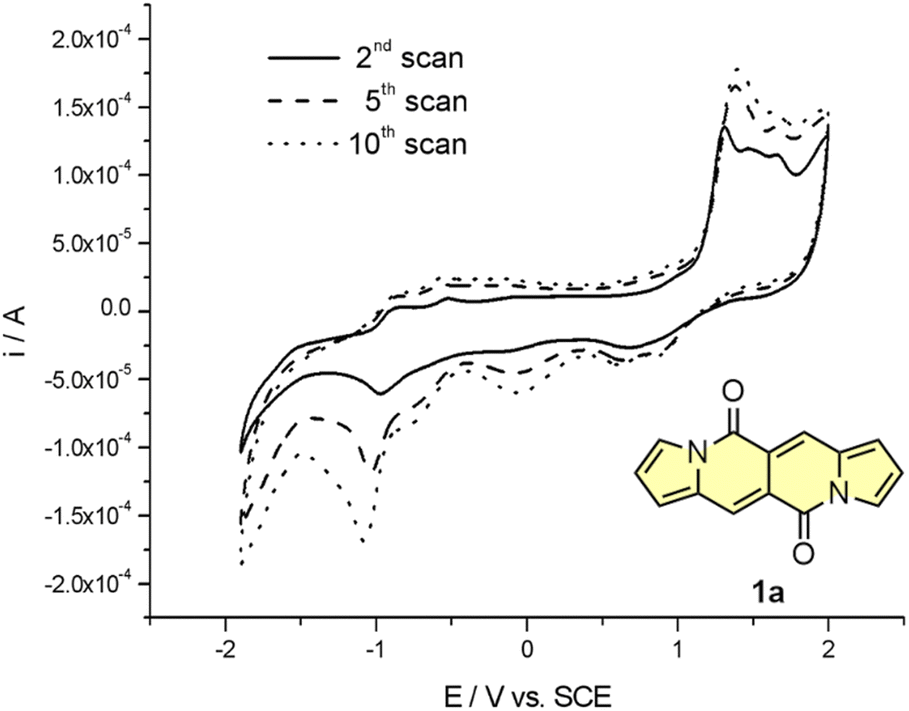 | ||
| Fig. 7 Cyclovoltammetric curve registered for 1a in dichloromethane in the entire range of examined potentials: −1800 ÷ 2100 mV, v = 100 mV s−1. Adapted with permission from ref. 18 Copyright 2016 Royal Society of Chemistry. | ||
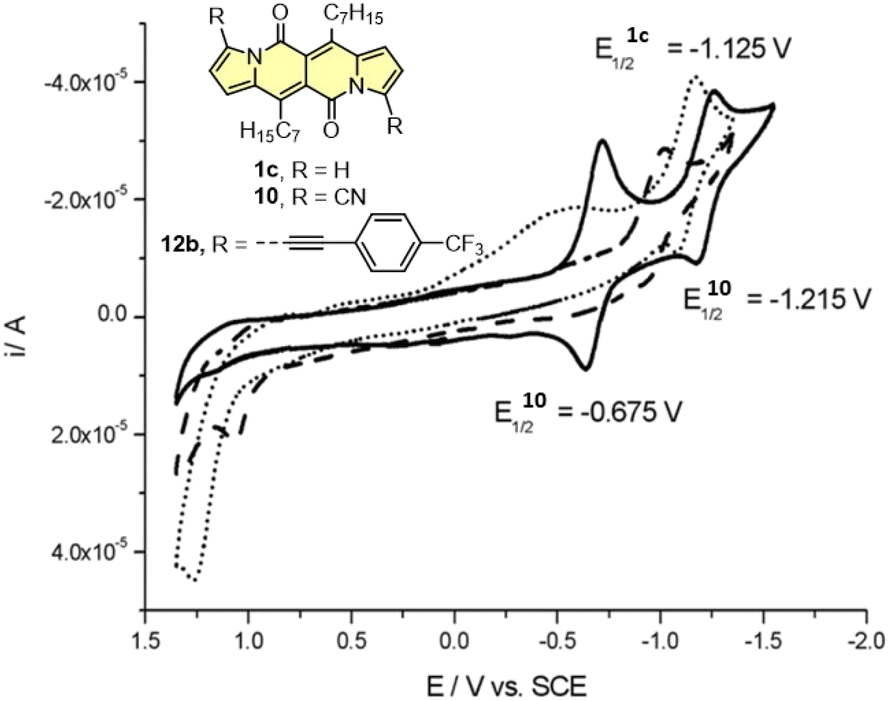 | ||
| Fig. 8 Cyclovoltammogramms of the dyes 1c (dotted line), 10 (solid line) and 12b (dashed line) in dichloromethane measured using the saturated calomel electrode (SCE) as the reference. Adapted with permission from ref. 18 Copyright 2016 Royal Society of Chemistry. | ||
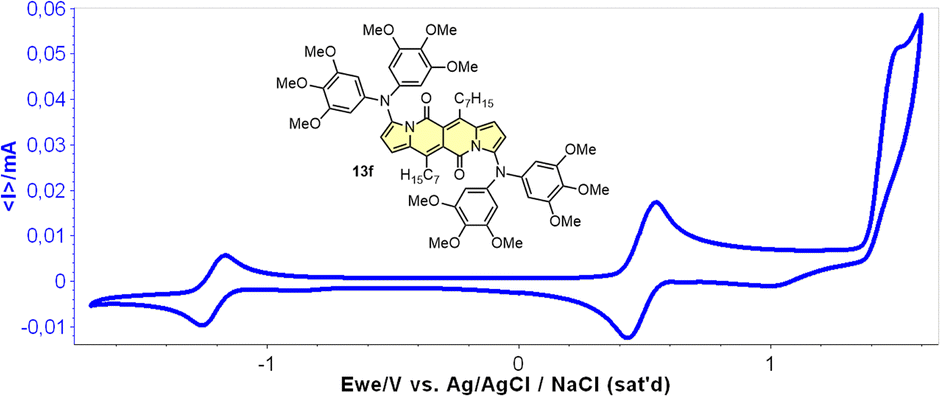 | ||
| Fig. 9 A cyclovoltammogramm of dye 13f in dichloromethane measured using Ag/AgCl/NaCl as the reference. Adapted with permission from ref. 28 Copyright 2018 American Chemical Society. | ||
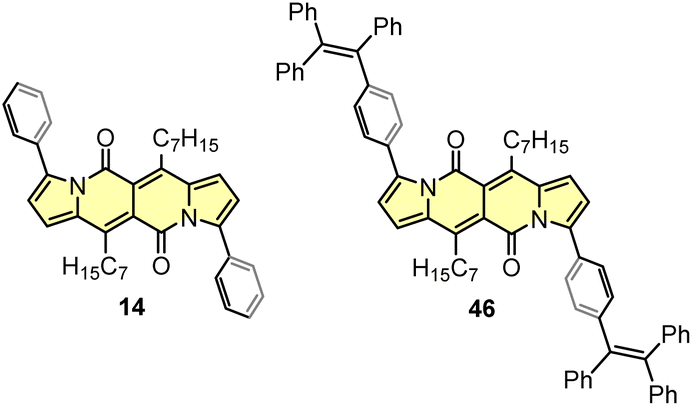
Although highly-emissive in a solution, simple DPNDs i.e.1c30c or 1h8d do exhibit weak fluorescence in the solid state, as these molecules mostly form H-aggregates in a crystalline state (Fig. 16, vide infra). The tetraphenylethylene moiety (TPE) seemed to be an ideal platform for inducing emission in the solid state,37 thus we investigated emission properties both in the solid and aggregated states for the quadrupolar dye 46 bearing two TPE units.30c Beside red-shifted absorption and emission in a CH2Cl2 solution as compared with 1c (Table 2) and 14 (Table 5), 46 emits weakly in the solid state with λmaxem = 659 nm and Φfl = 0.12 (Table 5). Fluorescence properties in the aggregated state were studied in THF/water mixtures (Fig. 10). DPND 1c showed aggregation-caused quenching effect (ACQ) as fluorescence intensity measured at 523 nm lowered at water fractions equal or higher than 80%. In turn, 46 undergoes aggregation at lower water proportions probably due to increased hydrophobicity, and then modest jump in the emission intensity was observed at water contents from 70 to 80%, possibly related to aggregation effects (aggregation-induced emission (AIE)). It means that for 46 both ACQ (dominant) and AIE (weak) effects can be observed. Direct comparison between 46 and its simple analogue 14 reveals almost no enhancement in solid state emission (Table 5). Moreover, we performed DFT calculations for some pairs of DPND molecules extracted from respective X-ray crystal structures. For most of pairs, very low or zero oscillator strengths were determined for S1 → S0 transitions.30c Additionally, some of them manifested CT character. In this work we proved somehow that the TPE moiety is not a magic group that always induces emission in the solid/aggregated state, but appearance of such feature depends only on which mode of crystal lattice arrangement is dominant (H- or J-aggregates).
|
|
||||||
|---|---|---|---|---|---|---|
| Dye | λ maxabs [nm] | ε max [M−1 cm−1] | λ maxem [nm] | Φ fl | δν [cm−1] | |
| a Determined using a spectrofluorimeter equipped with a calibrated integrating sphere. | ||||||
| 14 | CH2Cl2 | 536 | 24000 |
599 | 0.71 | 2000 |
| Solid | 562 | — | 601 | 0.15 | 1200 | |
| 46 | CH2Cl2 | 553 | 29000 |
644 | 0.42 | 2600 |
| Solid | 574 | — | 659 | 0.12 | 2200 | |
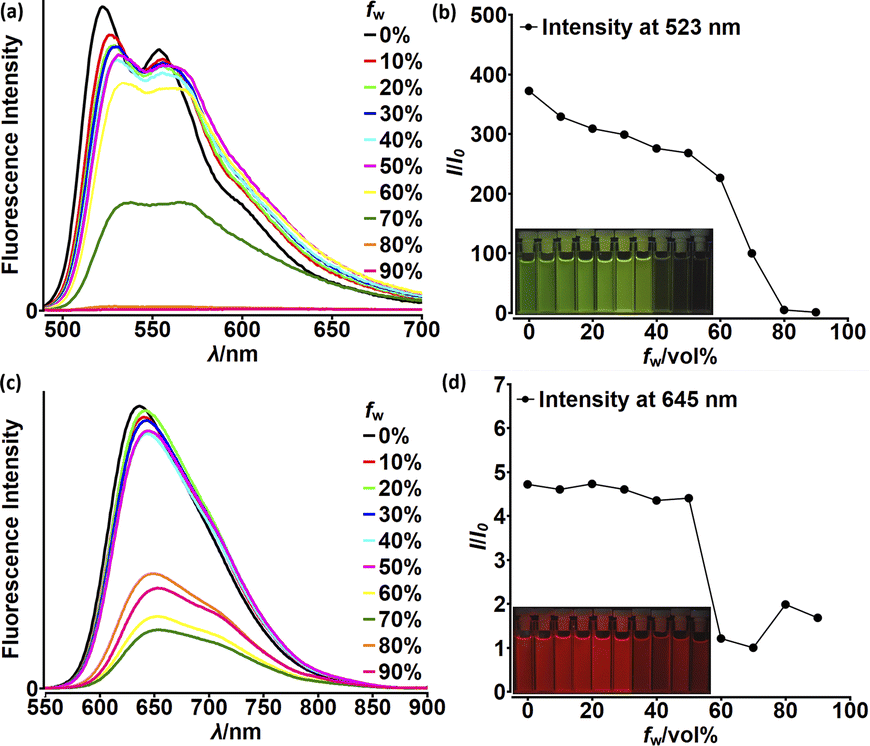 | ||
| Fig. 10 Fluorescence spectra of 1c (a) and 46 (c) in THF–water mixtures of different relative proportions. Plots of maximum intensity vs. % water fraction (fw) for dyes 1c (b) and 46 (d). Insets: photographs of 1c (b) and 46 (d) in THF–water mixtures with different water fractions under UV illumination (0% to 90% water fraction from left to right). Dye concentration: ∼10 mM. Adapted with permission from ref. 30c. Copyright 2018 Royal Society of Chemistry. | ||
Probing reactivity of CO moieties, we successfully designed and synthesized N-doped analogues of polycyclic aromatic hydrocarbons (PAHs) (48) starting from the DPND core (Fig. 11a and Table 6).33 These propeller-shaped dyes are intensively blue in a solution while having extremely weak emission (Φfl < 0.001, Table 6 and Fig. 11b). Based on transient-absorption measurements we concluded that the first singlet excited state of these dyes tend to deexcite via internal conversion rather than fluorescence or triplet state formation. A moderate red-shift of both bands can be observed when OMe group is present in the chromophore structure (48avs.48b, Table 6). Due to the presence of basic nitrogen atoms, 48a was found to be acid-responsive (Fig. 11c–e). Namely, adding an increasing amount of trifluoroacetic acid (TFA) to a solution of 48a in CH2Cl2 results in two new absorption bands centered at 467 nm and 776 nm, while the main absorption band of pure 48a vanished substantially. Broad nature of the longer-wavelength absorption band suggests that mainly monoprotonation took place and the resulted molecule features internal donor–acceptor (D–A) character. Double protonation can be achieved by employing stronger acid – methanesulphonic acid (MsOH) (Fig. 11d). First of all, significantly smaller amount of acid is needed for monoprotonation to occur as compared with TFA (10 eq. vs. 416 eq., respectively). Similarly to TFA, upon adding 10 eq. of MsOH, a new, broad absorption band appeared around 776 nm. Higher excess of MsOH (10 eq. → 250 eq.) led to complete disappearance of a broad band above 700 nm and a new absorption band emerged at 675 nm confirming stepwise double protonation of 48a. Both protonation processes are completely reversible as by adding excess of triethylamine (Et3N) the absorption spectrum as well as the color of 48a can be fully recovered (Fig. 11e).
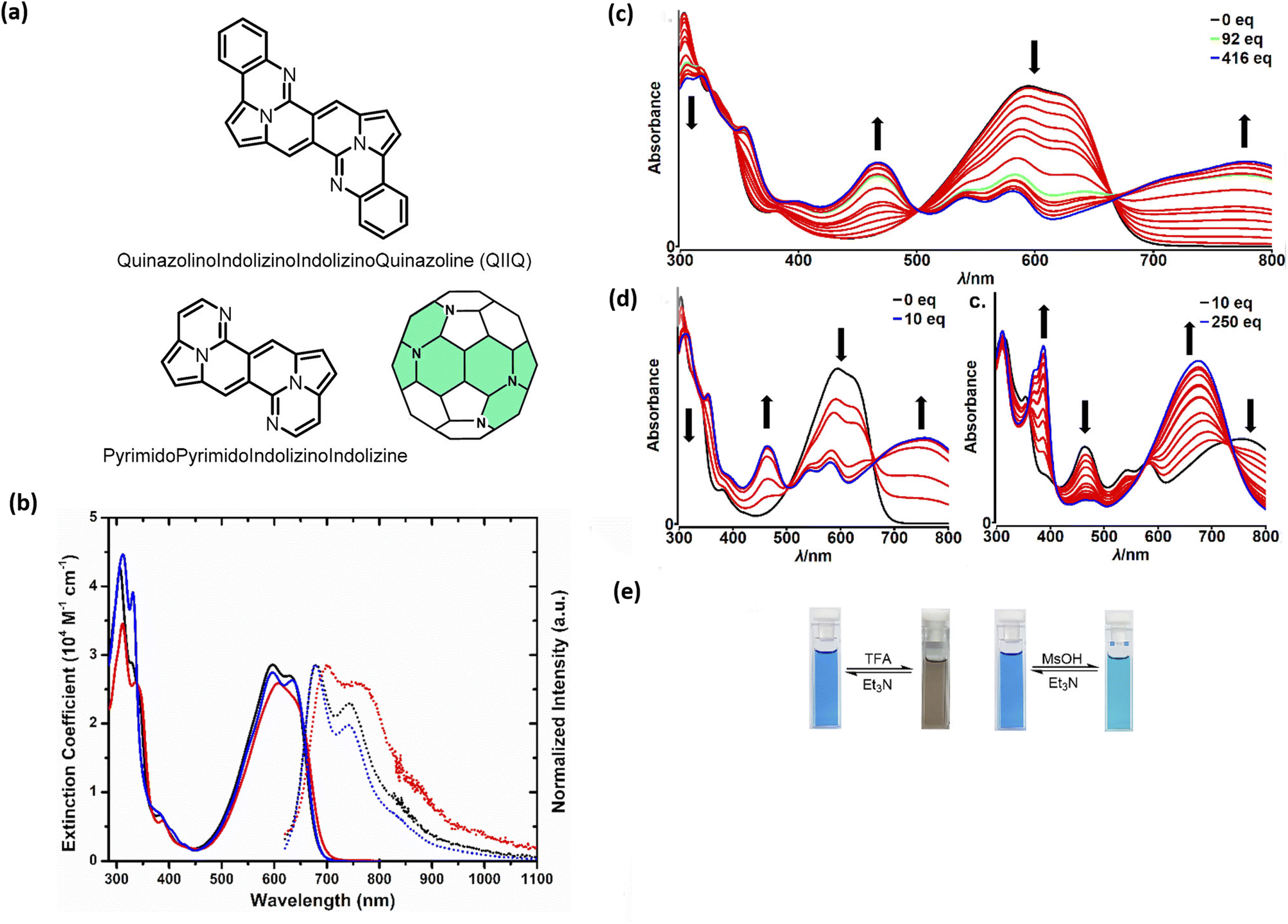 | ||
| Fig. 11 (a) General structure of the QIIQ skeleton; (b) absorption (solid) and emission (dotted) spectra of 48a (black), 48b (red), and 48c (blue) in toluene. Emission spectra from 570 nm excitation; (c) changes in the absorption spectra and color of 48a in CH2Cl2 (2.8 × 10−5 M) upon addition of MsOH (0–10 equiv.); (d) changes in the absorption spectra and color of 48a in CH2Cl2 (2.8 × 10−5 M) upon addition of MsOH (10–250 equiv.); (e) Photo of cuvettes containing 48a in CH2Cl2 solution before and after addition of a large excess of TFA (left) and MsOH (right). Adapted with permission from ref. 33. Copyright 2020 American Chemical Society. | ||
|
|
|||||
|---|---|---|---|---|---|
| Dye | λ maxabs [nm] | ε max [M−1 cm−1] | λ maxem [nm] | Φ fl | δν [cm−1] |
| a The fluorescence quantum yield (Φfl) of 48c in toluene was obtained using Nile Blue (Φfl = 0.271 in ethanol) as a standard and corrected for refractive index differences of the solvents. Compound 48c was chosen as it is the most emissive, and Φfl of 48a and 48b were then referenced to 48c. | |||||
| 48a | 596 | 29000 |
679 | 0.00083a | 2050 |
| 48b | 608 | 26000 |
704 | 0.00018a | 2240 |
| 48c | 597 | 27000 |
678 | 0.0023a | 2000 |
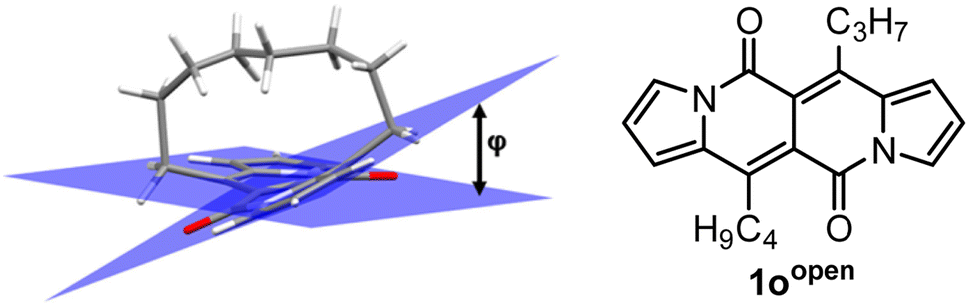
In all of the above-described chromophores, a geometry of the core was influenced only by the presence of substituents at positions 6 and 12 as well as vicinity of substituents at positions 3 and 9 (an α position within a pyrrole ring). DFT calculations for some arylated DPNDs revealed that the expected deviation from the core plane mainly caused by the presence of arene rings at position 3 and 9 should be no greater that 8°.30a In order to test how high distortion influences of the photophysical aspects, we assembled three analogues of cyclophanes based on the DPND core (1o–r)20 that differ in a length of an alkane bridge. X-ray analysis of 1p revealed that the distortion from the planarity reaches 28° (Fig. 12).
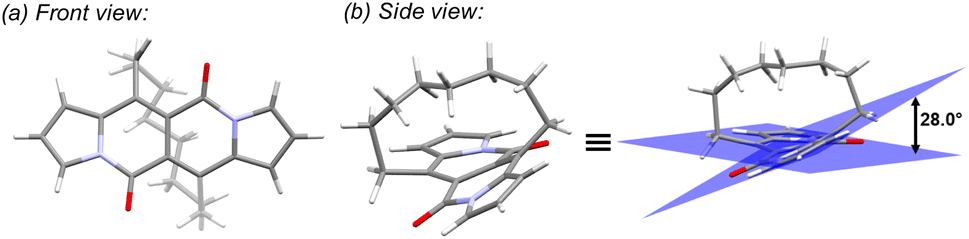 | ||
| Fig. 12 Single crystal structure of 1p (CCDC 2125168). Adapted with permission from ref. 20. Copyright 2022 Royal Society of Chemistry. | ||
DFT calculation supported the obtained degree of distortion (Table 7). Interestingly, chromophore 1o (C7 bridge) tends to increase its distortion degree upon photoexcitation while other cyclophane analogues as well as its open analogue 1oopen feature lower value of a distortion angle (Table 7). In general, 1o–r absorb at shorter wavelengths than 1c, but the most significant difference between an “open dye” and cyclophanes lies in their luminescence (Table 8). Namely, while emission bands determined for 1p–r are red-shifted by 30–40 nm as compared with 1c, in the case of 1o the emission band is located above 600 nm that is in the red region of the spectrum and particularly low values of Φfl were determined. In terms of emission intensity, 1p–r behave similarly to 1c (Φfl = 0.40–0.55), however emission bands are located at lower energies. Transient-absorption studies performed for cyclophane analogues suggested that presumably efficient formation of triplet state is responsible for distinct emission properties of 1p.
a
|
|
|||
|---|---|---|---|
| Dye | ϕ (S0)a | ϕ (S1)a | SE/kcal mol−1 |
| a ϕ – deviation from planarity. b Determined based on X-ray crystallography of 1p. c Determined using DFT/B3-LYP level of theory. SE – strain energy computed at MP2/cc-pVDZ level of theory. | |||
| 1oopen | 7.4° | 5.6° | 0 |
| 1o | 32.8° | 43.6° | 31.2 (27.8c) |
| 1p | 27.8° (28.0°b) | 25.6° | 13.6 (18.0c) |
| 1r | 15.4° | 12.8° | 8.2 (12.2c) |
|
|
|||||
|---|---|---|---|---|---|
| Dye | Solvent | λ abs [nm] | λ em [nm] | Φ fl | Δν [cm−1] |
| a Absorption maximum (bold) and shoulder. b Fluorescence maximum (bold) and shoulder. c Relative Φfl were obtained using Rhodamine 6G in ethanol (ΦPL = 0.95, λexc = 480 nm) as a reference. | |||||
| 1c | PhMe | 472, 506 | 526, 560 | 0.66 | 750 |
| THF | 470, 502 | 524, 558 | 0.57 | 800 | |
| C6H5CN | 476, 508 | 531, 564 | 0.65 | 850 | |
| MeCN | 469, 499 | 527, 557 | 0.54 | 1100 | |
| 1o | PhMe | 492 | 618 | 0.06 | 4100 |
| THF | 488 | 616 | 0.04 | 4300 | |
| C6H5CN | 493 | 625 | 0.04 | 4300 | |
| MeCN | 487 | 623 | 0.02 | 4500 | |
| 1p | PhMe | 483, 507 | 552, 577 | 0.55 | 2600 |
| THF | 482, 503 | 573, 553 | 0.41 | 3300 | |
| C6H5CN | 489, 507 | 562, 581 | 0.40 | 3200 | |
| MeCN | 481 | 577 | 0.32 | 3500 | |
| 1r | PhMe | 479, 507 | 543, 572 | 0.57 | 2500 |
| THF | 478, 505 | 542, 572 | 0.48 | 2500 | |
| C6H5CN | 483, 509 | 551, 576 | 0.52 | 2600 | |
| MeCN | 477, 500 | 572, 550 | 0.40 | 3500 | |
Dipyrrolonaphthyridinediones versus other cross-conjugated chromophores
Although cross-conjugated, the DPND core is different from well-known dyes of this type such as isoindigo (II),38 diketopyrrolopyrrole,39 or cibalakrot12 (Fig. 13) as it contains a “pure” pyrrole ring as an electron-donating moiety. This factor directly translates into particularly distinct one-photon optoelectronic behavior compared with other cross-conjugated structures (Table 9 and Fig. 13). According to the data collected in Table 9, among all dyes B-II-B is weakly- or non-fluorescent,40 features the lowest molar absorption coefficient (εmax) and absorbs at shorter wavelength (λmaxabs = 517 nm) than 14. Compound 14 emits light with Φfl = 0.71 with a maximum noted at λmaxem = 599 nm that is most red-shifted compared with other cross-conjugated chromophores, although Ph-DPP-Ph has a larger value of Stokes' shift.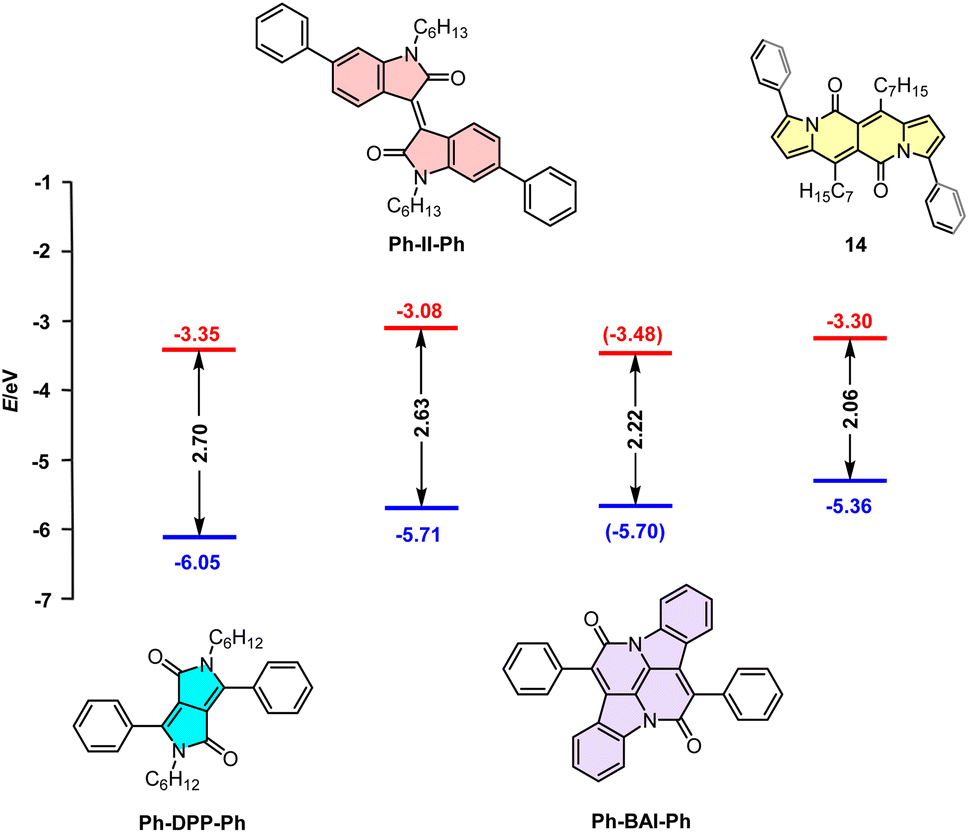 | ||
| Fig. 13 Correlation diagram of the HOMOs and LUMOs of three representative cross-conjugated units and a DPND derivative 14 determined by cyclic voltammetry (CV) measurements in CH2Cl2 (the values in parentheses were determined using DFT calculations due to lack of the experimental data). | ||
Direct comparison of electrochemically-derived energetic levels may be useful from the view-point of optoelectronics (Fig. 13). DPND 14 is the most susceptible to oxidation as its EHOMO level lies higher in energy that those noted for other cross-conjugated chromophores. When it comes to ELUMO levels, 14 features a similar value to Ph-DPP-Ph and at even lower energies than that of Ph-II-Ph which are both considered as electron-acceptors in dyes tested toward organic electronics.
DPNDs as singlet-fission (SF) materials
The singlet fission (SF) is a process in which a higher-energy singlet exciton, typically generated by the absorption of a photon, is converted into two lower-energy triplet excitons.41 This phenomenon features the potential to enhance the efficiency of solar cells42 and other optoelectronic devices as it seems to be an attractive solution to overcome Shoeckley-Queisser limit.43 Taking a lesson from cibalakrot analogues, Wang and co-workers have investigated8d the possibility of using 1h as a potential SF material. First of all, the authors found that 1h exhibits phosphorescence at 1019 and 1034 nm both in a solution and aggregated state, respectively (Fig. 14), while fluoresces weakly in the aggregated state. This result suggests that under slightly endothermic energetic conditions, a SF process can occur in polycrystalline film of 1h that outcompetes other energy dissipation channels suggesting strong intermolecular coupling between molecules in the solid state. The authors proved that the SF process takes place in 1h with up to 173% triplet yield and its film exhibits excellent stability upon exposure to air and light compared with pentalene44 or cibalakrot analogues (Fig. 15).45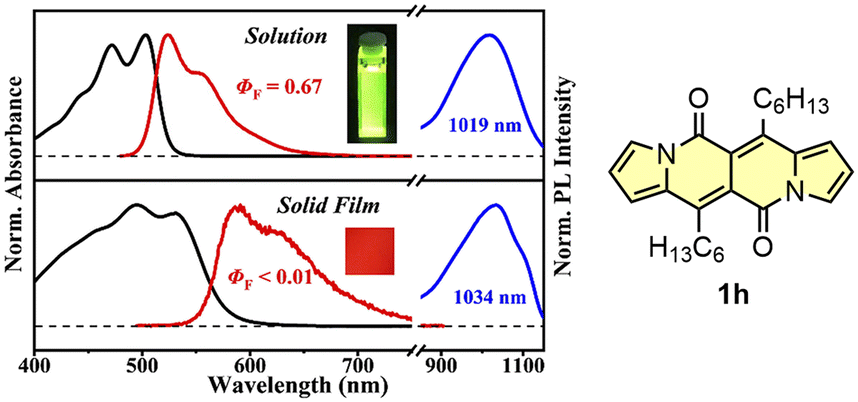 | ||
| Fig. 14 Normalized absorption (black) and fluorescence (red) spectra of DPND in CH2Cl2 solution (10−5 M) and solid thin film (100 nm), as well as sensitized phosphorescence spectra (blue) of 1h in polystyrene (PS) matrix (DPND:PtTPBP:PS = 1:5:94) and doped thin film (DPND:PtTPBP = 95:5). Inset: sample photographs of solution and thin film under 365 nm UV light. Adapted with permission from ref. 8d. Copyright 2020 American Chemical Society. | ||
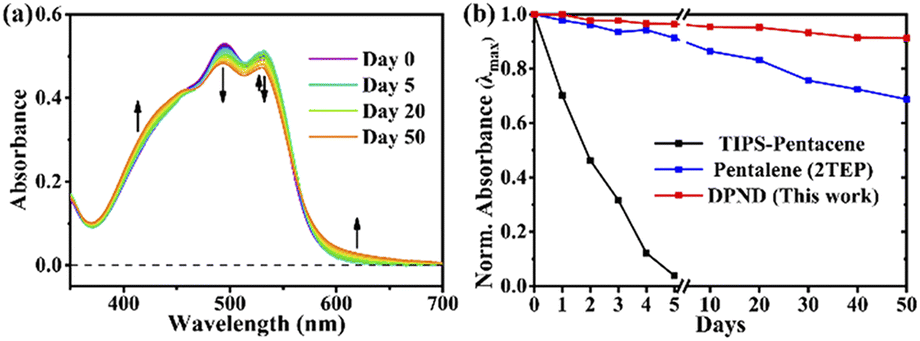 | ||
| Fig. 15 Stability testing of 1h molecule: (a) steady-state absorption spectra and (b) normalized intensity of the λmax of thin films exposed to air and light. Adapted with permission from ref. 8d. Copyright 2020 American Chemical Society. | ||
In their follow-up work, the authors investigated19 in detail effect of molecular aggregation on SF dynamics. Specifically, the DPND molecule described above contains two longer alkyl chains (1h – DPND6 in Fig. 16) which directly influences molecular arrangement in the crystal lattice with specific values of π–π distance, transverse and longitudinal offsets as well as a slipping angle (Fig. 16). In turn, compound 1a (DPND) crystallizes in a nearly cofacial pattern of packing arrangement with the smaller values of transverse and longitudinal offsets, π–π distance, and a slipping angle of 66°. Here, the formation of face-to-face dimers results in stronger coupling of molecules in the crystal lattice thus an accelerated singlet fission (SF) process was eventually observed. This result may help in the future to design more efficient SF materials.
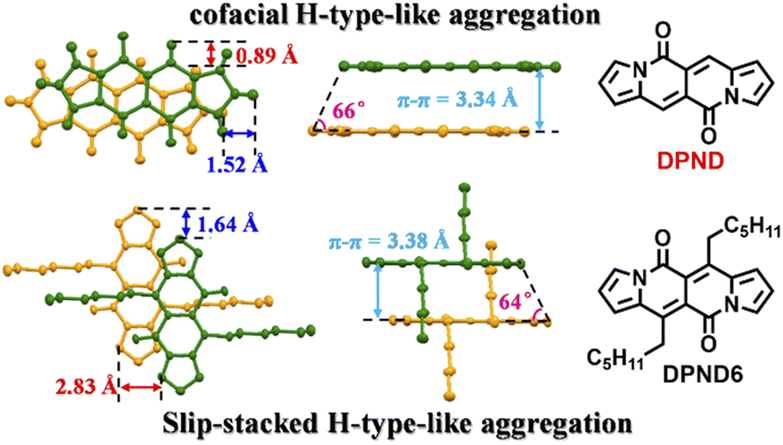 | ||
| Fig. 16 Molecular arrangement of 1a and 1h in the solid state. Adapted with permission from ref. 19. Copyright 2021 American Chemical Society. | ||
Recently, Wu and others have presented new insights into the SF mechanism by studying time-resolved spectroscopy as a function of temperature for 10.46 Here, the applied kinetic model developed based on the measurements and calculations includes initial conversion of S1 state to 1(T1T1) state followed by thermally-activated at room temperature dissociation of 1(T1T1) into two T1 states (Fig. 17). The yield of T1 states formation was determined as 154%.
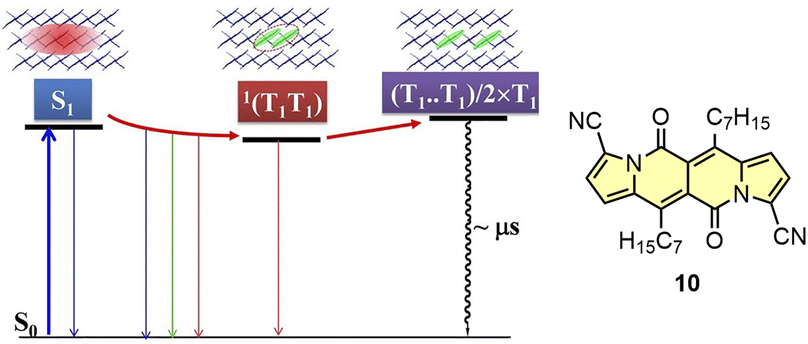 | ||
| Fig. 17 Kinetic model of the SF process determined for 10. Adapted with permission from ref. 46. Copyright 2021 American Chemical Society. | ||
Fluorescent nitroaromatics
Nitroaromatics fluoresce occasionally as numerous possible energy dissipation channels are activated after the excitation.47 The NO2 auxochrome is one of the strongest electron-withdrawing moieties which ideally should improve stability and performance of NO2-containing materials. However, the introduction of this group into the chromophore structure often results in weak emission. Although the literature outlines some strategies for enhancing emission properties of nitroaromatics,47a there are still no general rules how to make nitroaromatics glow. To gain more insight into the processes that are responsible for energy dissipation in nitroaromatics, we have studied a number of these molecules where the DPND core was chosen as a reference chromophore (Table 10, Fig. 18–20).30b,48 The introduction of the nitro groups at the peripheries undoubtedly implies emission intensity loss (from 0.71 for 14 to 0.41 for 25 in CH2Cl2), however 25 still fluoresce in the red region of the spectrum featuring red-shifted both absorption and emission bands. This initial result set the stage for the comprehensive study on fluorescence of nitroaromatics bearing the DPND core.| Dye | Solvent | λ abs [nm] | λ em [nm] | Φ fl | Δν [cm−1] |
|---|---|---|---|---|---|
| 25 | o-C6H4Cl2 | 569 | 605 | 0.45 | 1050 |
| CH2Cl2 | 562 | 601 | 0.41 | 1150 | |
| MeCN | 559 | 599 | 0.07 | 1190 | |
| 25mono | o-C6H4Cl2 | 539 | 571 | 0.76 | 1050 |
| CH2Cl2 | 534 | 568 | 0.36 | 1110 | |
| MeCN | 529 | 570 | 0.006 | 1370 | |
| 26 | o-C6H4Cl2 | 536 | 580 | 0.61 | 1410 |
| CH2Cl2 | 533 | 578 | 0.49 | 1460 | |
| MeCN | 529 | 579 | 0.013 | 1630 | |
| 27 | o-C6H4Cl2 | 547 | 612 | 0.46 | 1940 |
| CH2Cl2 | 543 | 610 | 0.21 | 2040 | |
| MeCN | 542 | 604 | 0.005 | 1910 | |
| 28 | o-C6H4Cl2 | 524 | 560 | 0.96 | 1230 |
| CH2Cl2 | 521 | 560 | 0.34 | 1340 | |
| MeCN | 518 | 564 | 0.005 | 1570 | |
| 29 | o-C6H4Cl2 | 557 | 607 | 0.58 | 1480 |
| CH2Cl2 | 549 | 605 | 0.45 | 1690 | |
| MeCN | 545 | 608 | 0.24 | 1900 | |
| 30 | o-C6H4Cl2 | 556 | 606 | 0.58 | 1480 |
| CH2Cl2 | 549 | 604 | 0.44 | 1660 | |
| MeCN | 546 | 603 | 0.31 | 1890 | |
| 31 | o-C6H4Cl2 | 558 | 606 | 0.45 | 1400 |
| CH2Cl2 | 548 | 602 | 0.31 | 1640 | |
| MeCN | 543 | 606 | 0.005 | 1940 | |
| 32 | o-C6H4Cl2 | 555 | 602 | 0.006 | 1390 |
| CH2Cl2 | 549 | 608 | 0.003 | 1760 | |
| MeCN | 542 | 584 | 0.003 | 1330 | |
| 33 | o-C6H4Cl2 | 563 | 618 | 0.38 | 1580 |
| CH2Cl2 | 555 | 615 | 0.28 | 1760 | |
| MeCN | 550 | 608 | 0.05 | 1730 | |
| 35 | o-C6H4Cl2 | 551 | 582 | 0.49 | 970 |
| CH2Cl2 | 544 | 577 | 0.28 | 1050 | |
| MeCN | 541 | 575 | 0.04 | 1090 | |
| 35mono | o-C6H4Cl2 | 529 | 555 | 0.62 | 870 |
| CH2Cl2 | 524 | 551 | 0.09 | 940 | |
| MeCN | 520 | 548 | 0.02 | 1000 | |
| 36 | o-C6H4Cl2 | 527 | 558 | 0.95 | 1050 |
| CH2Cl2 | 523 | 555 | 0.92 | 1100 | |
| MeCN | 521 | 553 | 0.43 | 1110 | |
| 37 | o-C6H4Cl2 | 532 | 583 | 0.61 | 1640 |
| CH2Cl2 | 525 | 584 | 0.52 | 1920 | |
| MeCN | 524 | 582 | 0.12 | 1900 | |
| 38 | o-C6H4Cl2 | 525 | 568 | 0.76 | 1440 |
| CH2Cl2 | 522 | 566 | 0.58 | 1490 | |
| MeCN | 520 | 569 | 0.075 | 1660 | |
| 39 | o-C6H4Cl2 | 535 | 586 | 0.63 | 1630 |
| CH2Cl2 | 531 | 582 | 0.59 | 1650 | |
| MeCN | 528 | 590 | 0.20 | 1990 | |
| 40 | o-C6H4Cl2 | 547 | 593 | 0.69 | 1420 |
| CH2Cl2 | 541 | 593 | 0.46 | 1620 | |
| MeCN | 537 | 592 | 0.17 | 1730 | |
| 41 | o-C6H4Cl2 | 553 | 604 | 0.64 | 1530 |
| CH2Cl2 | 546 | 604 | 0.43 | 1760 | |
| MeCN | 543 | 609 | 0.18 | 2000 | |
| 42 | o-C6H4Cl2 | 575 | 669 | 0.12 | 2440 |
| CH2Cl2 | 565 | 670 | 0.02 | 2770 | |
| MeCN | 557 | 688 | 0.04 | 3420 | |
| 43 | o-C6H4Cl2 | 555 | 598 | 598 | 980 |
| CH2Cl2 | 548 | 592 | 592 | 1160 | |
| MeCN | 544 | 592 | 592 | 1490 | |
| 43mono | o-C6H4Cl2 | 532 | 575 | — | 1420 |
| CH2Cl2 | 527 | 552 | — | 1020 | |
| MeCN | 522 | 561 | — | 1360 | |
| 44 | o-C6H4Cl2 | 551 | 595 | 0.06 | 1360 |
| CH2Cl2 | 545 | 594 | — | 1500 | |
| MeCN | 542 | 589 | — | 1480 | |
| 44mono | o-C6H4Cl2 | 527 | 569 | 0.02 | 1400 |
| CH2Cl2 | 524 | 551 | — | 940 | |
| MeCN | 521 | 565 | — | 1500 | |
| 45mono | o-C6H4Cl2 | 527 | 569 | — | 1400 |
| CH2Cl2 | 524 | 562 | — | 1290 | |
| MeCN | 522 | 559 | — | 1270 |
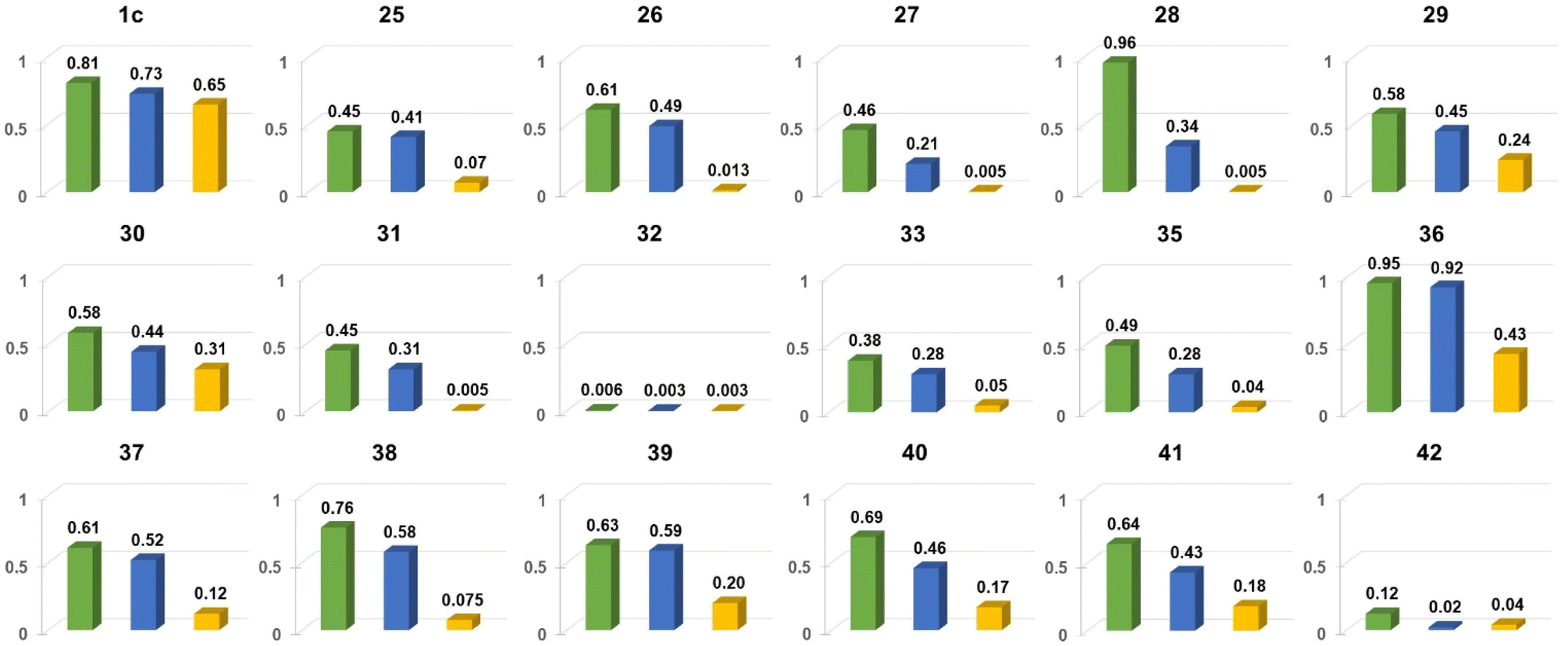 | ||
| Fig. 18 Comparison of Φfl for chosen nitroaromatics bearing the DPND core in three different solvents: 1,2-dichlorobenzene (green), CH2Cl2 (blue) and MeCN (orange). Adapted with permission from ref. 48. Copyright 2023 Royal Society of Chemistry. | ||
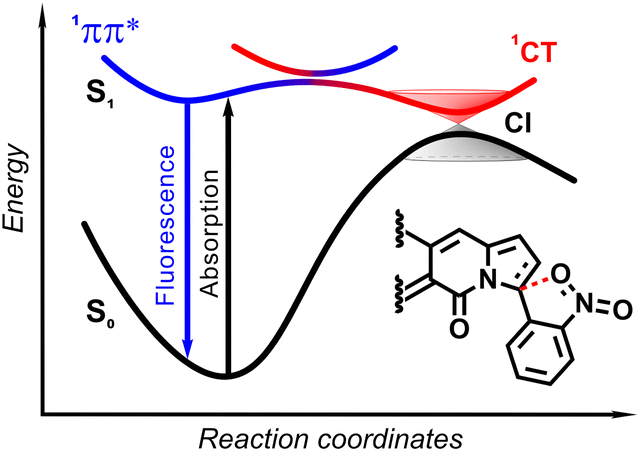 | ||
| Fig. 19 Schematic diagram showing the scenario of temporal evolution of the photoexcited ortho-nitrophenyl substituted DPND. The electrons are initially photoexcited to locally excited (LE) state (1π–π*) and is non-adiabatically transferred to the CT state along the reaction coordinate. The photoexcited system recombines radiatively from S1 to S0 or nonradiatively through 1π–π* → 1CT → S0 transitions. CI – conical intersection. Adapted with permission from ref. 30b. Copyright 2021 Royal Society of Chemistry. | ||
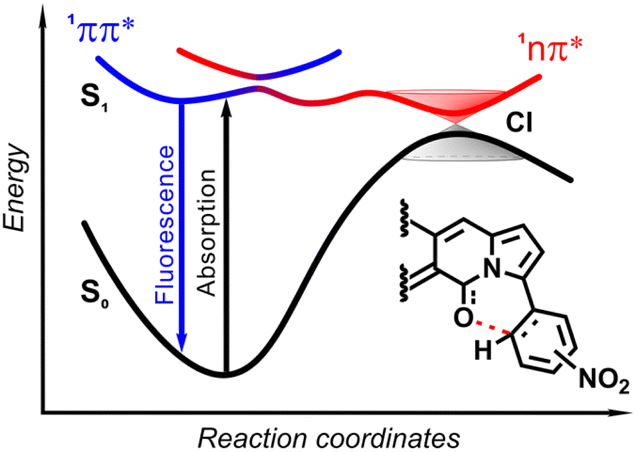 | ||
| Fig. 20 Schematic diagram showing the general scenario of temporal evolution of the photoexcited meta- and para-nitrophenyl substituted DPNDs. The system is initially photoexcited to the 1π–π* state and is non-adiabatically transferred to the 1n–π* state along the reaction coordinate. The photoexcited system recombines radiatively from S1 to S0 or nonradiatively through 1π–π* → 1n–π* → S0 transitions. CI – conical intersection. Adapted with permission from ref. 30b. Copyright 2021 Royal Society of Chemistry. | ||
In general, two structural factors play a decisive role in governing emission intensity: (1) the position of NO2 group relative to the core and (2) the substitution pattern of flanking aryl rings (Table 10, Fig. 18). Regarding the first factor, both para- and meta-NO2-subsituted derivatives are fluorescent and emission intensity lowers with the increase in solvent polarity, suggesting that a dark, charge-transfer-type (CT-type) state may be involved in energy dissipation. In contrast, close proximity of the nitro group (43–45, Scheme 4) results in weak fluorescence response (ortho- or peri-NO2-substituted molecules) in all tested solvents.
Secondly, the introduction of additional substituents (Me, OMe, NEt2) within flanking arene rings also contribute significantly to fluorescence modulation. As an example, placing either Me or OMe group at the ortho position relative to the DPND core increases emission intensity (i.e.35vs.36) by hindering rotation around Caryl–CDPND bond. Those groups may also affect electronic distribution via both inductive and mesomeric effects. As a matter of fact, careful structure optimization by taking into account all of these factors allowed us to describe nitroaromatics featuring enormous value of Φfl (up to 0.96), even in relatively polar dichloromethane (28 or 36).
The origin of the specific dependence of emission intensity on solvent polarity was investigated30b by means of theoretical calculations which revealed surprising, aborted photochemical reactivity after photoexcitation (Fig. 19 and 20). The fluorescence in the series of DPND-based nitroaromatics clearly comes from deexcitation of the lowest in energy S1 (1π–π*) state. However, the S1 state may also adiabatically relaxed via a transition to 1CT (ortho- or peri-NO2 isomers) or 1np* (para- or meta-NO2 isomers) states. Then, these states efficiently undergo non-radiative depopulation via a conical intersection (CI) with the S0 state.
Theoretical results also suggest that the nitro group twists upon photoexcitation around the CDPND–Nnitro bond with subsequent formation of a new, covalent CDPND–Onitro bond in 1CT and 1np* states. The formation of a new bond with more polar character tends to occur much easier in polar environment. Then, this bond breaks when transitioning from the CI to the more favourable ground-state configuration during relaxation. This phenomenon is called the aborted photochemistry (Fig. 19 and 20).49
Two-photon absorption (TPA) in DPNDs
Two-photon absorption (TPA) phenomenon50 have found multiple, real-life applications such as 3D imaging of materials,51 microfabrication and photopolymerization,52 photopharmacology53 or imaging of biological tissues.54 An efficient TPA absorber should feature a long system of conjugated multiple bonds and contain donor and acceptor groups that potentially would participate in the formation of strongly polarized charge-transfer states.55 As a completely new chromophore, the DPND core was investigated as a TPA absorber for the first time already in 2018.27 We have found out that dyes bearing double- or triple-bond linkages between the DPND core and flanking aryl rings exhibit superior values of two-photon absorption cross-sections (σmax2) (Table 11), where the highest value of σmax2 was noted for compound 11a bearing a double-bond linkage between the DPND core and nitroaryl flanking moieties (σmax2 = 5180 GM). Notably, while compounds from both series (11 and 12) possessed a beneficial ratio of σmax2/MW (1.6–9.8 GM g−1), dyes of type 12 show high values of two-photon brightness σmax2·Φfl, which is an important feature from the viewpoint of bioimaging applications.55b| Dye | λ max2PA/nm | σ max2/GM | σ max2 φ fl/GM | σ max2/MWc |
|---|---|---|---|---|
| a Determined using the two-photon excited fluorescence (TPEF) technique.56 b Determined using Z-scan setup.57 c The molecular weights were calculated by replacing alkyl groups at positions 6 and 12 by a methyl group (CH3). | ||||
| 1c | 750 | 44 | 32 | <1 |
| 11a | 820 | 5180 | 83 | 9.3 |
| 11b | 740 | 5100 | 112 | 9.8 |
| 11c | 740 | 1710 | 70 | 3.2 |
| 12a | 720 | 2840 | 1450 | 5.1 |
| 12b | 720 | 850 | 500 | 1.6 |
| 12c | 860 | 1990 | 340 | 3.6 |
| 25 | <690 | >3718 | >1524 | 7.3 |
| 26 | <680 | 312 | 153 | <1 |
| 27 | <690 | >835 | >175 | 1.5 |
| 28 | <680 | >197 | >67 | <1 |
| 29 | <690 | >1177 | >530 | 2.2 |
| 30 | <690 | >1326 | >583 | 2.3 |
| 31 | <690 | >1134 | >352 | 1.8 |
| 32 | <690 | >9 | 0 | 0 |
| 33 | 720 | 1384 | 388 | 2.1 |
| 35 | <680 | >233 | >65 | <1 |
| 36 | <685 | >175 | >161 | <1 |
| 37 | <685 | >167 | >87 | <1 |
| 38 | <685 | >96 | >56 | <1 |
| 39 | <685 | ≥161 | ≥95 | <1 |
| 40 | <680 | >340 | >156 | <1 |
| 41 | <700 | ≥513 | ≥221 | <1 |
| 42 | 755 | 104 | 2 | <1 |
Later on, we carefully evaluated weakly-coupled bis-arylated DPND derivatives containing nitroaryl moieties as potential TPA absorbers (Scheme 4 and Table 11).48 Weak electronic communication between the nitro group auxochrome and the DPND core resulted in weaker non-linear response compared with 11a or 12a. Among compounds tested, 25 showed a reasonable TPA response (σmax2 > 1524 GM) whereas other derivatives performed weaker (σmax2 < 600 GM) in the accessible spectral window size.48
DPNDs as triplet sensitizers
Halogenation is the most established strategy to intensify population of a triplet excited state58 due to enhanced spin–orbit coupling (SOC).59 Such halogenated derivatives have been widely studied and as emerging agents for photodynamic therapy (PDT). Along this strategy, Ayitou and others tested halogenated DPNDs (7–9) as potential (PDT) agents (Fig. 21 and Table 12).21,60 The presence of heavy halogen atoms within the DPND core was believed to enhance spin–orbit coupling (SOC) dynamics, thus leading to efficient singlet oxygen (1O2) production. They found that all halogen-substituted dyes do produce 1O2 with admirable efficiencies and the highest value of ΦΔ was determined for 9. Surprisingly, values of fluorescence quantum yields measured for 7c and 8 (75 and 76%, respectively) are comparable to the value measured for 1c, although they bear chlorine and bromine atoms within the core.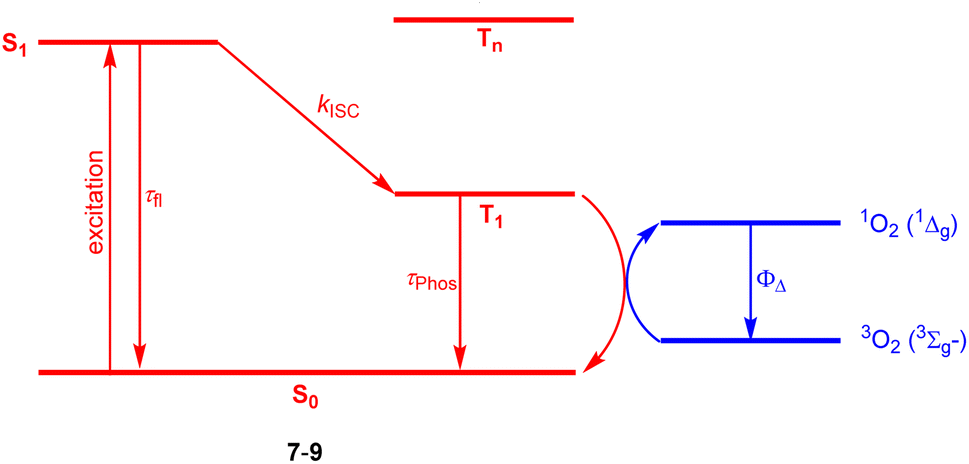 | ||
| Fig. 21 Schematic illustration of energy levels and triplet sensitization in 7–9.21,60 | ||
|
|
|||||
|---|---|---|---|---|---|
| Dye | τ fl/ns | k ISC/s−1 | τ T/μs | τ Phos/ms | Φ Δ/% |
| 7a | 5.1 | — | 5.1 | 8.2 | 31 |
| 8 | 5.7 | 1.6 × 108 | 16 | 7.9 | 28 |
| 9 | 0.34 | 2.9 × 109 | 0.31 | 9.0 | 52 |
DPND-based polymer as a potential material for OFETs
Direct arylation methodology appeared to be useful in the preparation of polymer 49 in one step from the DPND core and dibrominated derivative bearing the diketopyrrolopyrrole (DPP) unit (Fig. 22).32 The weight-average molecular weight (Mw) and polydispersity index (PD) determined by HT-GPC were 77.7 kDa and 2.64, respectively, while the UV-VIS spectrum recorded in toluene showed a strong absorption band in the NIR region with λmax = 866 nm. Polymer 49 was examined by BASF as a potential electron transport material in OFETs, nevertheless a particularly low value of electron mobility (μ = 8.85 × 10−12 cm2 V−1 m−1) was measured.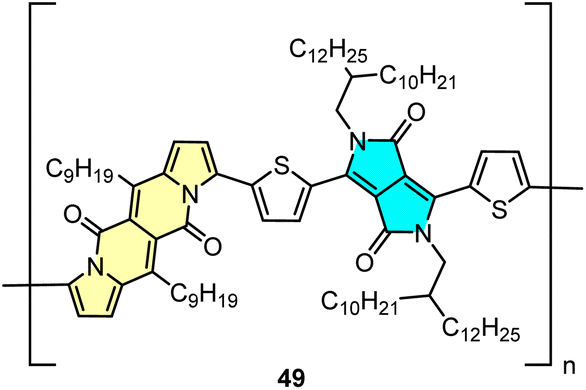 | ||
| Fig. 22 The structure of polymer 49. | ||
5. Summary and outlook
Although the history of cross-conjugated dyes dates back to the ancient times, still there are numerous possible structures that have never been either assembled or investigated. In seven years which elapsed since their discovery, dipyrrolonaphthyridinediones made a notable impact on chemistry. Their straightforward synthesis combined with functionalization possibilities are responsible for the quick initial progress in terms of structural exploitation. In the second phase the deciding factor turned out to be their intriguing photophysical characteristics and in particular: (1) strong and easily tunable emission; (2) low energy of T1 excited state which enables singlet fission in the crystalline state; (3) large two-photon absorption cross-sections after derivatization. Studies on dipyrrolonaphthyridinediones: (1) were the deciding factor enabling formulation of strategies towards strongly fluorescent nitroaromatics; (2) provided the shortest pathway to twistacenes; (3) delivered dyes with large optical brightness and exceptionally high values of two-photon absorption cross-section divided by molecular mass. Notwithstanding their success, there are still numerous synthetic limitations which have to be overcome in the nearest future to ensure continuous progress. The main envisioned areas for synthetic explorations are as follows: (1) preparation of DPNDs directly from aromatic acids; (2) extension of the DPNDs' synthesis to embrace such substrates as indole, isoindole etc.; (3) transformation of DPNDs into polar, water-soluble probes. In 2016 it would be impossible to imagine that only seven years later the chemistry of DPNDs would expand to create independent field of study within the chemistry of functional dyes. One can venture hypothesis that once these synthetic limitations are overcome these new π-expanded DPNDs could witness rapid development. The analysis of research performed within the last few years suggest that the number of known as well as possible structures of cross-conjugated nature is limited only by our imagination.Author contributions
All authors contributed to the conceptualization. B. S. performed literature search, wrote the first draft and made all schemes/figures. All authors were involved in revising, editing, and proofreading.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Polish National Science Centre (Sonata 2021/43/D/ST4/02267 and OPUS 2020/37/B/ST4/00017). This project has received funding from EU's Horizon 2020 research and innovation programme under Grant Agreement No. 860762. Authors thank Dr Marek Grzybowski for discovering DPNDs.References
- (a) N. Singh, S. Singh, S. Kohli, A. Singh, H. Asiki, G. Rathee, R. Chandra and E. A. Anderson, Org. Chem. Front., 2021, 8, 5550–5573 RSC; (b) J. T. Gupton, in Heterocyclic Antitumor Antibiotics, ed. M. Lee, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 53–92 Search PubMed.
- (a) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC; (b) G. Li Petri, V. Spanò, R. Spatola, R. Holl, M. V. Raimondi, P. Barraja and A. Montalbano, Eur. J. Med. Chem., 2020, 208, 112783 CrossRef CAS.
- (a) J. Kim, J. Oh, A. Osuka and D. Kim, Chem. Soc. Rev., 2022, 51, 268–292 RSC; (b) B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102–3137 CrossRef.
- B. Sadowski, J. Klajn and D. T. Gryko, Org. Biomol. Chem., 2016, 14, 7804–7828 RSC.
- (a) C. Bellomo, M. Chaari, J. Cabrera-González, M. Blangetti, C. Lombardi, A. Deagostino, C. Viñas, N. Gaztelumendi, C. Nogués, R. Nuñez and C. Prandi, Chem.–Eur. J., 2018, 24, 15622–15630 CrossRef CAS; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- (a) A. N. Bismillah and I. Aprahamian, Chem. Soc. Rev., 2021, 50, 5631–5649 RSC; (b) S. Boodts, E. Fron, J. Hofkens and W. Dehaen, Coord. Chem. Rev., 2018, 371, 1–10 CrossRef CAS; (c) I.-S. Tamgho, A. Hasheminasab, J. T. Engle, V. N. Nemykin and C. J. Ziegler, J. Am. Chem. Soc., 2014, 136, 5623–5626 CrossRef CAS.
- (a) W. Zeng, O. El Bakouri, D. W. Szczepanik, H. Bronstein and H. Ottosson, Chem. Sci., 2021, 12, 6159–6171 RSC; (b) W. Zeng, D. W. Szczepanik and H. Bronstein, J. Phys. Org. Chem., 2023, 36, e4441 CrossRef CAS; (c) M. Swart, Theor. Chem. Acc., 2020, 139 Search PubMed; (d) L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS.
- (a) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- J. Kaleta, M. Dudič, L. Ludvíková, A. Liška, A. Zaykov, I. Rončević, M. Mašát, L. Bednárová, P. I. Dron, S. J. Teat and J. Michl, J. Org. Chem., 2023, 88, 6573–6587 CrossRef CAS.
- (a) A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198–4207 RSC; (b) M. Vatanparast and Z. Shariatinia, Sol. Energy, 2021, 230, 260–268 CrossRef CAS; (c) B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS.
- C. Du, S. Fu, X. Ren, X. Wang, Z. Wang, J. Zhou and H. Wang, New J. Chem., 2018, 42, 3493–3502 RSC.
- (a) E. D. Głowacki, G. Voss, L. Leonat, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, Isr. J. Chem., 2012, 52, 540–551 CrossRef; (b) R. J. H. Clark, C. J. Cooksey, M. A. M. Daniels and R. Withnall, Endeavour, 1993, 17, 191–199 CrossRef CAS.
- D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 15, 2549–2552 CrossRef.
- (a) J. Pfenninger, A. Iqbal, A. C. Rochat, O. Wallquist and A. G. Ciba-Geigy, Eur. pat.Appl. 184982, 1986 Search PubMed; (b) A. Iqbal and L. Cassar, Ciba-Geigy Corporation, US Pat.4415685, 1983 Search PubMed; (c) L. Cassar, A. Iqbal, A. C. Rochat and A. G. Ciba-Geigy, Eur. Pat.Appl. 98808, 1983 Search PubMed.
- M. Grzybowski, I. Deperasińska, M. Chotkowski, M. Banasiewicz, A. Makarewicz, B. Kozankiewicz and D. T. Gryko, Chem. Commun., 2016, 52, 5108–5111 RSC.
- L. Wang, W. Cai, J. Sun, Y. Wu, B. Zhang, X. Tian, S. Guo, W. Liang, H. Fu and J. Yao, J. Phys. Chem. Lett., 2021, 12, 12276–12282 CrossRef CAS.
- B. Sadowski, D. Mierzwa, S. Kang, M. Grzybowski, Y. M. Poronik, A. L. Sobolewski, D. Kim and D. T. Gryko, Chem. Commun., 2022, 58, 3697–3700 RSC.
- J. Morgan, Y. J. Yun, A. M. Jamhawi, S. M. Islam and A. J.-L. Ayitou, Photochem. Photobiol., 2023, 99, 761–768 CrossRef CAS.
- C. Song, D. W. Knight and M. A. Whatton, Tetrahedron Lett., 2004, 45, 9573–9576 CrossRef CAS.
- (a) J. I. Ayogu and E. A. Onoabedje, Catal. Sci. Tech., 2019, 9, 5233–5255 RSC; (b) K. H. Shaughnessy, in Metal-Catalyzed Reactions in Water, Wiley, 2013, pp. 1–46 Search PubMed; (c) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS; (d) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC.
- (a) L. I. Belen’kii, T. G. Kim, I. A. Suslov and N. D. Chuvylkin, Arkivoc, 2003, 59–67 Search PubMed; (b) S. Nomiyama, T. Ogura, H. Ishida, K. Aoki and T. Tsuchimoto, J. Org. Chem., 2017, 82, 5178–5197 CrossRef CAS; (c) D. Ghorai and G. Mani, Inorg. Chem., 2014, 53, 4117–4129 CrossRef CAS; (d) T. Buchała, A. Chudoba and S. Roszak, Struct. Chem., 2016, 27, 185–189 CrossRef.
- (a) Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430–438 RSC; (b) B. Godlewski, D. Baran, M. De Robichon, A. Ferry, S. Ostrowski and M. Malinowski, Org. Chem. Front., 2022, 9, 2396–2404 RSC.
- T. Rohand, W. Qin, N. Boens and W. Dehaen, Eur. J. Org Chem., 2006, 4658–4663 CrossRef CAS.
- B. Sadowski, H. Kita, M. Grzybowski, K. Kamada and D. T. Gryko, J. Org. Chem., 2017, 82, 7254–7264 CrossRef CAS.
- B. Sadowski, M. Loebnitz, D. R. Dombrowski, D. H. Friese and D. T. Gryko, J. Org. Chem., 2018, 83, 11645–11653 CrossRef CAS.
- P. G. Chirila and C. J. Whiteoak, Dalton Trans., 2017, 46, 9721–9739 RSC.
- (a) B. Sadowski, M. F. Rode and D. T. Gryko, Chem.–Eur. J., 2018, 24, 855–864 CrossRef CAS; (b) B. Sadowski, M. Kaliszewska, Y. M. Poronik, M. Czichy, P. Janasik, M. Banasiewicz, D. Mierzwa, W. Gadomski, T. D. Lohrey, J. A. Clark, M. Lapkowski, B. Kozankiewicz, V. I. Vullev, A. L. Sobolewski, P. Piatkowski and D. T. Gryko, Chem. Sci., 2021, 12, 14039–14049 RSC; (c) B. Sadowski, S.-H. Su, T.-C. Lin, T. D. Lohrey, I. Deperasińska, P.-T. Chou and D. T. Gryko, J. Mater. Chem. C, 2018, 6, 12306–12313 RSC.
- (a) H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550–11571 RSC; (b) X. Wang, Y. Li, J. Li, Y. Zhang, J. Shao and Y. Li, Molecules, 2023, 28, 3515 CrossRef CAS; (c) I. A. Stepek and K. Itami, ACS Mater. Lett., 2020, 2, 951–974 CrossRef CAS.
- M. Grzybowski, D. T. Gryko, B. Sadowski, K. Strassel, D. Kaelblein and P. Hayoz, Polymers and compounds based on dipyrrolo[1,2-b:1’,2’-g][2,6]naphthyridine-5,11-dione, International Pat no. WO2017068009, 2018.
- B. Sadowski, D. J. Stewart, A. T. Phillips, T. A. Grusenmeyer, J. E. Haley, T. M. Cooper and D. T. Gryko, J. Org. Chem., 2020, 85, 284–290 CrossRef CAS.
- S. Ito, S. Hiroto and H. Shinokubo, Org. Lett., 2013, 15, 3110–3113 CrossRef CAS.
- J. O. Morley, Int. J. Quantum Chem., 1993, 46, 19–26 CrossRef CAS.
- P. J. McCartin, J. Chem. Phys., 2004, 42, 2980–2981 CrossRef.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- L. A. Estrada, R. Stalder, K. A. Abboud, C. Risko, J.-L. Brédas and J. R. Reynolds, Macromolecules, 2013, 46, 8832–8844 CrossRef CAS.
- J. Dhar, N. Venkatramaiah and S. Patil, J. Mater. Chem. C, 2014, 2, 3457–3466 RSC.
- (a) D. Bialas, S.-L. Suraru, R. Schmidt and F. Würthner, Org. Biomol. Chem., 2011, 9, 6127–6132 RSC; (b) S. Luňák, P. Horáková and A. Lyčka, Dyes Pigm., 2010, 85, 171–176 CrossRef.
- (a) M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS; (b) M. B. Smith and J. Michl, Annu. Rev. Physiol., 2013, 64, 361–386 CrossRef CAS.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- L. Wang, Y. Wu, Y. Liu, L. Wang, J. Yao and H. Fu, J. Chem. Phys., 2019, 151 Search PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett, C. Risko, D. O. Scanlon, C. W. M. Kay, A. Rao, R. H. Friend, A. J. Musser and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS.
- Y. Wu, L. Lu, B. Yu, S. Zhang, P. Luo, M. Chen, J. He, Y. Li, C. Zhang, J. Zhu, J. Yao and H. Fu, J. Phys. Chem. Lett., 2023, 14, 4233–4240 CrossRef CAS.
- (a) Y. M. Poronik, B. Sadowski, K. Szychta, F. H. Quina, V. I. Vullev and D. T. Gryko, J. Mater. Chem. C, 2022, 10, 2870–2904 RSC; (b) M.-C. Chen, D.-G. Chen and P.-T. Chou, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS; (c) W. Rodriguez-Cordoba, L. Gutierrez-Arzaluz, F. Cortes-Guzman and J. Peon, Chem. Commun., 2021, 57, 12218–12235 RSC.
- B. Sadowski, M. Kaliszewska, G. Clermont, Y. M. Poronik, M. Blanchard-Desce, P. Piatkowski and D. T. Gryko, Chem. Commun., 2023, 59, 11708–11711 RSC.
- (a) A. Sinicropi, W. M. Nau and M. Olivucci, Photochem. Photobiol., 2002, 1, 537–546 CAS; (b) D. Shemesh, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2009, 131, 1374–1375 CrossRef CAS; (c) D. Tuna, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2014, 16, 38–47 RSC.
- M. Göppert-Mayer, Ann. Phys., 1931, 401, 273–294 CrossRef.
- C. Dorfer, D. Hits, L. Kasmi, G. Kramberger, M. Lucchini, M. Mikuž and R. Wallny, Appl. Phys. Lett., 2019, 114 Search PubMed.
- (a) S. Maruo, O. Nakamura and S. Kawata, Opt. Lett., 1997, 22, 132–134 CrossRef CAS; (b) Z. Faraji Rad, P. D. Prewett and G. J. Davies, Microsyst. Nanoeng., 2021, 7, 71 CrossRef CAS; (c) S. O'Halloran, A. Pandit, A. Heise and A. Kellett, Adv. Sci., 2023, 10, 2204072 CrossRef.
- M. Izquierdo-Serra, M. Gascón-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, È. Ferrer, R. Alibés, F. Busqué, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS.
- (a) F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS; (b) G. Sancataldo, O. Barrera and V. Vetri, in Principles of Light Microscopy: From Basic to Advanced, ed. V. Nechyporuk-Zloy, Springer International Publishing, Cham, 2022, pp. 215–241 Search PubMed.
- (a) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS; (b) H. Myung Kim and B. R. Cho, Chem. Commun., 2009, 153–164 RSC.
- (a) M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS; (b) C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
- K. Kamada, K. Matsunaga, A. Yoshino and K. Ohta, J. Opt. Soc. Am. B, 2003, 20, 529–537 CrossRef CAS.
- (a) J. Grüne, G. Londi, A. J. Gillett, B. Stähly, S. Lulei, M. Kotova, Y. Olivier, V. Dyakonov and A. Sperlich, Adv. Funct. Mater., 2023, 33, 2212640 CrossRef; (b) W. Hu, R. Zhang, X.-F. Zhang, J. Liu and L. Luo, Part, 2022, 272, 120965 CAS.
- (a) M. Fagnoni, Angew. Chem., Int. Ed., 2010, 49, 6709–6710 CrossRef CAS; (b) C. M. Marian, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 187–203 CrossRef CAS.
- J. Morgan, Y. J. Yun and A. J.-L. Ayitou, Photochem. Photobiol., 2022, 98, 57–61 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |