
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Divergent total syntheses of ITHQ-type bis-β-carboline alkaloids by regio-selective formal aza-[4 + 2] cycloaddition and late-stage C–H functionalization†
Qixuan
Wang
a,
Fusheng
Guo
b,
Jin
Wang
b and
Xiaoguang
Lei
 *abc
*abc
aPeking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, P. R. China. E-mail: xglei@pku.edu.cn
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Department of Chemical Biology, College of Chemistry and Molecular Engineering, Synthetic and Functional Biomolecules Center, Peking University, Beijing 100871, P. R. China
cInstitute for Cancer Research, Shenzhen Bay Laboratory, Shenzhen, 518107, P. R. China
First published on 13th September 2023
Abstract
We herein report the first total syntheses of several bis-β-carboline alkaloids, picrasidines G, S, R, and T, and natural product-like derivatives in a divergent manner. Picrasidines G, S, and T feature an indolotetrahydroquinolizinium (ITHQ) skeleton, while picrasidine R possesses a 1,4-diketone linker between two β-carboline fragments. The synthesis of ITHQ-type bis-β-carboline alkaloids could be directly achieved by a late-stage regio-selective aza-[4 + 2] cycloaddition of vinyl β-carboline alkaloids, suggesting that this remarkable aza-[4 + 2] cycloaddition might be involved in the biosynthetic pathway. Computational studies revealed that such aza-[4 + 2] cycloaddition is a stepwise process and explained the unique regioselectivity (ΔΔG = 3.77 kcal mol−1). Moreover, the successful application of iridium-catalyzed C–H borylation on β-carboline substrates enabled the site-selective C-8 functionalization for efficient synthesis and structural diversification of this family of natural products. Finally, concise synthesis of picrasidine R by the thiazolium-catalyzed Stetter reaction was also accomplished.
Introduction
β-Carboline alkaloids widely exist in a variety of animals1 and plants.2 These alkaloids have been discovered to possess diverse biological activities, such as anti-cancer, anti-inflammatory, and anti-malaria.3 Since the 1980s, a large number of bis-β-carboline alkaloids have been isolated by Ohmoto,4–17 Yao,18–21 Song,22 Tian23et al. from plants of the Simaroubaceae family. Interestingly, some of these alkaloids, such as picrasidines G (1), S (2), and T (3), feature a unique indolotetrahydroquinolizinium (ITHQ) skeleton (Scheme 1a). All these ITHQ-type bis-β-carboline alkaloids bear a methoxy group at the C-4 position on each β-carboline ring, and some of them bear a hydroxy group or methoxy group at the C-8 or N-9 position. Despite the presence of a stereogenic center in ITHQ-type bis-β-carboline alkaloids, racemization can easily occur at room temperature in chloroform solution.20 Most of these ITHQ-type bis-β-carboline alkaloids show cytotoxic bioactivity. In 2015, Lin and coworkers reported the cytotoxicity and antibacterial bioactivities of picrasidines G (1), S (2), and F.24 Picrasidine F shows selective cytotoxicity against the HeLa cancer cell line (IC50 = 16.65 μM) compared to the MKN-28 (IC50 = 145.50 μM) and B-16 (IC50 = 95.48 μM) cancer cell lines, while picrasidines G (1) and S (2) show cytotoxicity against all three cancer cell lines (IC50 = 4.95–19.00 μM). These three alkaloids also show antibacterial bioactivity against MRSA P0172, MRSA H0117, MSSA P0171 and MSSA H0180 (MIC = 4–16 μg mL−1). These authors also discovered that the other two ITHQ-type bis-β-carboline alkaloids (±) quassidines I and J show cytotoxicity against the HeLa, MNK-28 and B-16 cancer cell lines (IC50 = 4.03–15.4 μM).20 In 2018, Tian and coworkers reported cytotoxicity of (±) quassidine K against the HeLa cancer cell line (IC50 = 15.8–20.1 μM).23 It is interesting to note that (+)-S configurated ITHQ-type bis-β-carboline alkaloids are generally more cytotoxic than the corresponding (−)-R configurated ones, but the difference in activity is only about 1.5 to 2-fold, probably due to the spontaneous racemization.20,23 In 2017, Kanno and coworkers reported that picrasidine G increases the caspase-dependent cell apoptosis by inhibiting the EGF-induced STAT3 phosphorylation, thus decreasing the viability of EGFR-overexpressing triple-negative breast cancer cells.25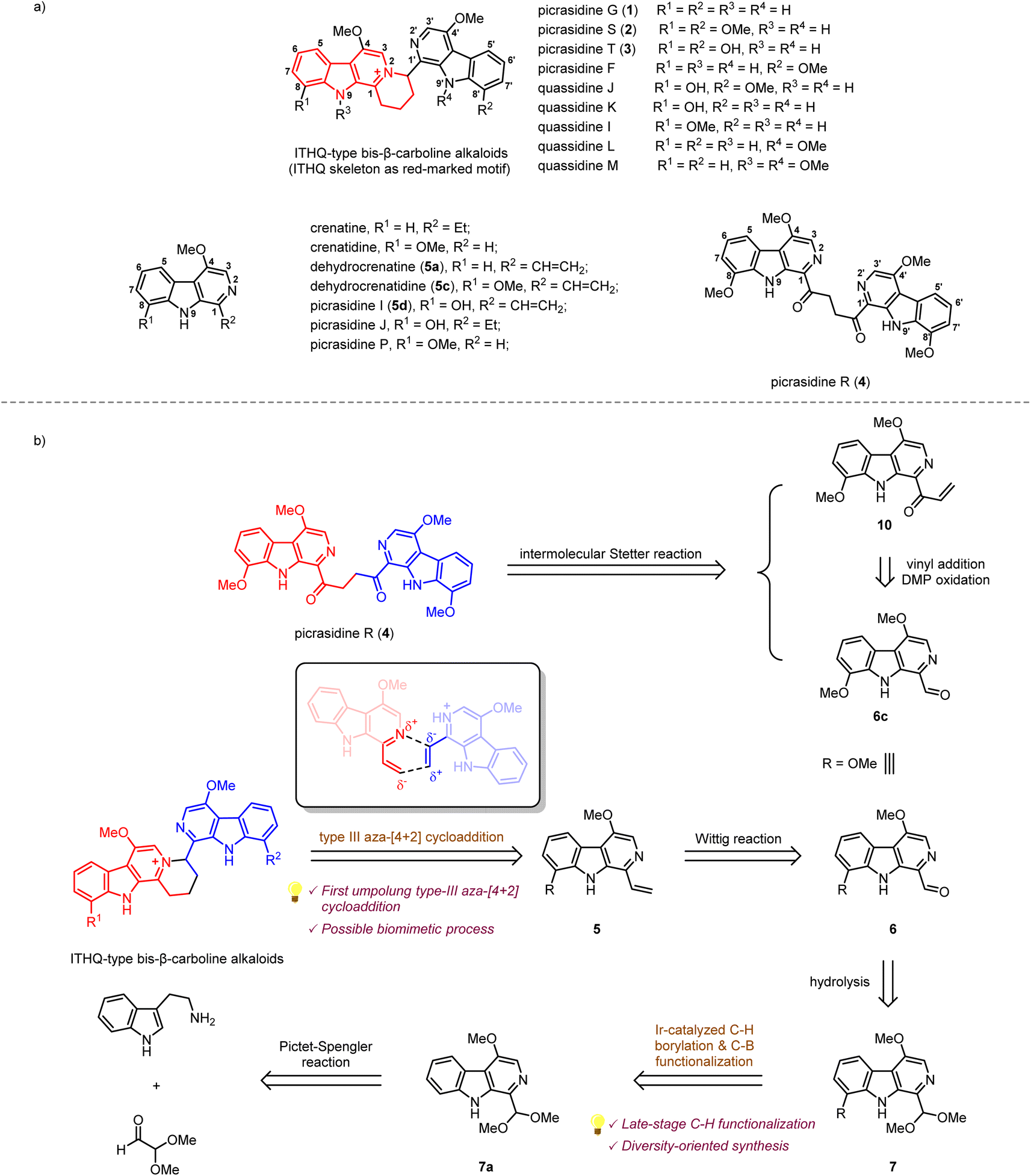 | ||
| Scheme 1 (a) Structures of several β-carboline alkaloids from Picrasma quassioides. (b) Retrosynthetic analysis of ITHQ-type bis-β-carboline alkaloids and picrasidine R. | ||
The biosynthetic pathway of ITHQ-type bis-β-carboline alkaloids is still unknown. Yao and Gao proposed a putative skeleton rearrangement mechanism. They envisaged that the ITHQ skeleton might be rearranged from a four-membered ring, which could be generated by nucleophilic substitution18 or [2 + 2] cycloaddition21,26 (Scheme S1†). However, such a hypothesis lacks corresponding experimental evidence. Here we proposed another possible synthetic route towards ITHQ-type bis-β-carboline alkaloids—aza-[4 + 2] cycloaddition. We speculated that ITHQ-type bis-β-carboline alkaloids might be generated via a late-stage regio-selective aza-[4 + 2] cycloaddition of two vinyl substituted monomeric β-carboline alkaloids, some of which were identified as natural products as well, such as dehydrocrenatine (5a), picrasidine I (5c) and dehydrocrenatidine (5d). Therefore, it would be intriguing to test such aza-[4 + 2] cycloaddition for a biomimetic synthesis.
No synthetic studies toward ITHQ-type bis-β-carboline alkaloids have been reported to date. However, several studies on the syntheses of 1-vinyl substituted monomeric β-carboline alkaloids have been reported (Scheme S2†). In 1982, Cook and coworkers reported the total syntheses of crenatine with the C-4 methoxy substituted β-carboline skeleton by a Pictet–Spengler reaction and DDQ oxidation strategy.27 In 2005, Ihara and coworkers reported the total synthesis of dehydrocrenatine (5a) via the application of Cook's strategy.28 In 1999, Murakami and coworkers reported the syntheses C-4 and C-8 disubstituted β-carboline alkaloids picrasidines I (5d), J and P, as well as crenatidine and dehydrocrenatidine (5c) by Fischer indole synthesis.29 In 2005, Murakami and coworkers reported a more concise Fischer indole synthesis strategy for the syntheses of C-4 and C-6 disubstituted β-carboline alkaloids.30
Our retrosynthetic analysis of ITHQ-type bis-β-carboline alkaloids is illustrated in Scheme 1b. As mentioned above, the ITHQ skeleton could be directly constructed by a late-stage aza-[4 + 2] cycloaddition of 1-vinyl substituted monomeric β-carboline alkaloids. The vinyl group at the C-1 position could be installed from 7 by hydrolysis of dimethyl acetal followed by a Wittig reaction. Substituent groups at the C-8 position could be installed by the iridium-catalyzed C–H bond borylation followed by C–B bond functionalization. The synthesis of precursor 7a should be achieved via a Pictet–Spengler reaction and DDQ oxidation inspired by Cook and Ihara's studies.27,28 Moreover, another bis-β-carboline alkaloid picrasidine R (4) could be synthesized via a Stetter reaction from vinyl ketone 10 and aldehyde 6c. The vinyl ketone 10 could also be obtained from aldehyde 6c by vinyl addition and a subsequent oxidation.
Results and discussion
Total syntheses of bis-β-carboline alkaloids and derivatives
Our synthesis commenced with the preparation of the monomeric C-1 vinyl substituted β-carboline dehydrocrenatine (5a) (Scheme 2a). The tetrahydro-β-carboline skeleton of compound 8 was constructed by a Pictet–Spengler reaction between tryptamine hydrochloride and 2,2-dimethoxyacetaldehyde,31 followed by tosyl protection of the secondary amine. Then benzyl oxidation of compound 8 by DDQ afforded ketone 9.27,28 Acetalmethylation of ketone 9 by the treatment with trimethyl orthoformate in acidic methanol solution led to in situ elimination of the tosyl group and aromatization to afford the key synthetic building block 7a, bearing the β-carboline structure with a methoxy group at the C-4 position. Compared with the acetyl group, usage of the tosyl group could avoid extra addition of oxidant for aromatization.28,30 After hydrolysis of the dimethyl acetal to form the aldehyde group and installation of the vinyl group by the Wittig reaction, we successfully prepared the desired C-1 vinyl substituted β-carboline dehydrocrenatine (5a).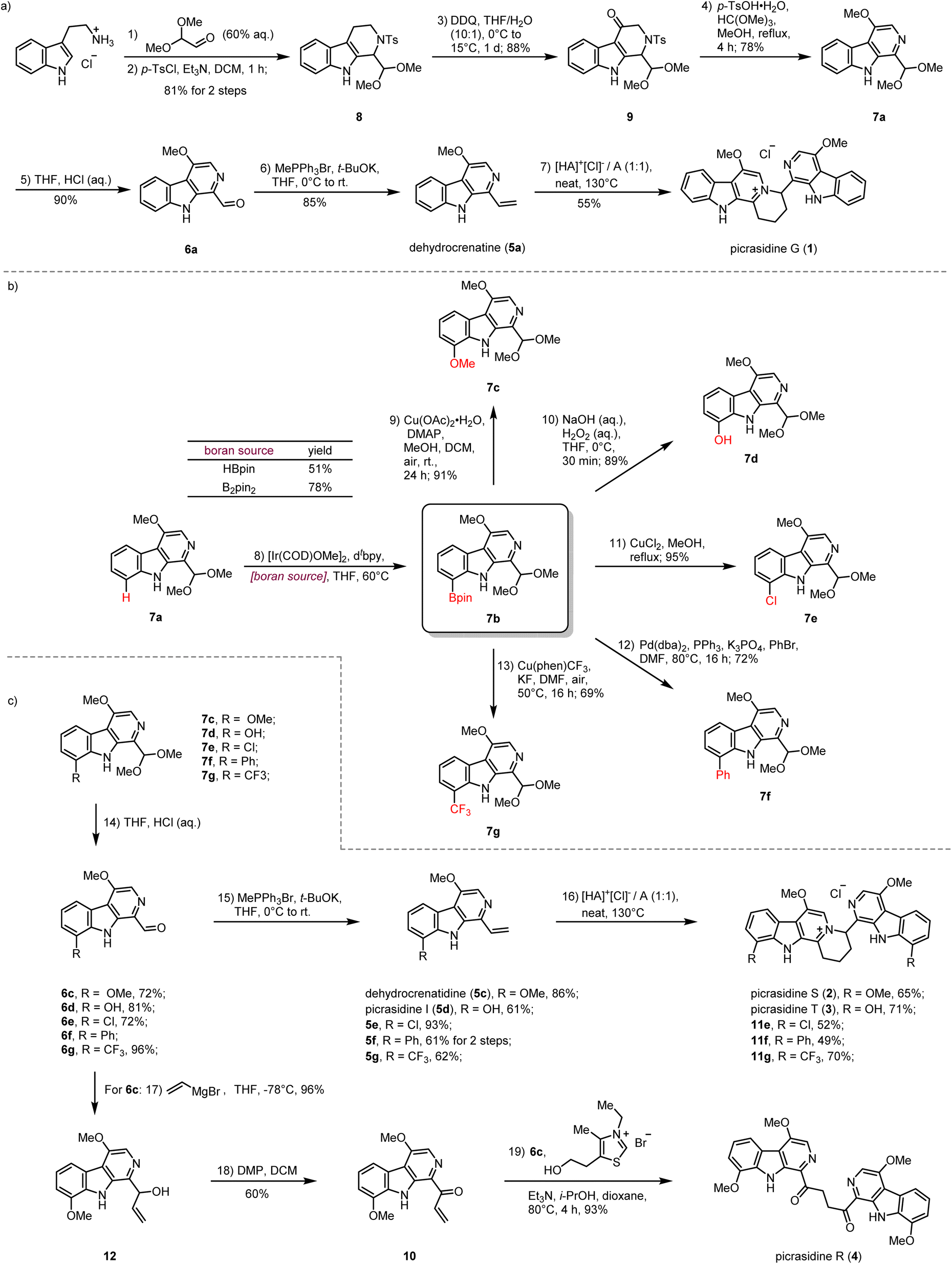 | ||
| Scheme 2 (a) Syntheses of picrasidines G (1) by the aza-[4 + 2] cycloaddition. (b) Late-stage C–H functionalization strategy for diversity-oriented syntheses of C-8 substituted β-carboline alkaloids. (c) Total syntheses of picrasidines S (2), T (3), R (4) and 11e, 11f, 11g. | ||
To explore the feasibility of the proposed regio-selective aza-[4 + 2] cycloaddition, dehydrocrenatine (5a) was chosen as a model substrate. We found that reactions performed in water solution or dioxane solution could only lead to trace formation of the dimerization product (Table 1, entries 1–5). However, upon heating an equimolar mixture of free alkaloid and its hydrochloride salt to 100 °C for 4 h under neat conditions, 40% of the reactant could be converted to the dimerization product (Table 1, entry 6). Raising the temperature to 130 °C could give almost full conversion of the reactant, with 55% isolated yield (Table 1, entry 9). The product was confirmed as the ITHQ-type bis-β-carboline alkaloid picrasidine G (1), demonstrating the feasibility of the aza-[4 + 2] cycloaddition. Although using free alkaloid as the reactant alone could also form a small amount of the dimerization product, the conversion was relatively low (Table 1, entry 11). On the other hand, using hydrochloride salt as the reactant alone could only lead to trace amounts of the dimerization product (Table 1, entry 10). This result showed that such aza-[4 + 2] cycloadditions were prone to occur between one molecule of free alkaloid and another molecule of hydrochloride salt.
|
|
||||
|---|---|---|---|---|
| Entry | Reactanta | Solventb | T/°C | Yieldg |
| a Reactions were performed using dehydrocrenatine (5a, represented by A) and its corresponding salt (represented by [HA]+X−) as reactants. b Unless otherwise stated, reactions were performed at 10 mM concentration. c In 25 mM KPi buffer (pH = 6). d In 25 mM Tris buffer (pH = 7). e In 25 mM HEPES buffer (pH = 8). f Reaction was performed in the solid state without any solvent. g Unless otherwise stated, yields were based on 1H-NMR. h Isolated yields. | ||||
| 1 | [HA]+Cl−/A (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
H2O | 100 | <5% |
| 2 | [HA]+Cl− | pH = 6 bufferc | 100 | <5% |
| 3 | [HA]+Cl− | pH = 7 bufferd | 100 | <5% |
| 4 | [HA]+Cl− | pH = 8 buffere | 100 | <5% |
| 5 | [HA]+Cl−/A (1:1) |
1,4-Dioxane | 100 | <5% |
| 6 | [HA]+Cl−/A (1:1) |
(Neat)f | 100 | 40% |
| 7 | [HA]+[AcO]− | (Neat)f | 100 | 23% |
| 8 | [HA]+[AcO]−/A (1:1) |
(Neat)f | 100 | 9% |
| 9 |
[HA]
+
Cl
−
/A (1:1) |
(Neat)f | 130 | 86% (55%h) |
| 10 | [HA]+Cl− | (Neat)f | 130 | <5% |
| 11 | A | (Neat)f | 130 | 31% |
Compared to the simplest ITHQ-type bis-β-carboline alkaloid picrasidine G (1), picrasidines S (2) and T (3) bear an additional oxidation state at the C-8 position. We envisaged a late-stage C–H borylation strategy for the functionalization of the C-8 position.32–36 Late-stage functionalization enables effective and flexible diversification at specific sites of natural products for drug discovery and chemical biology.37–40 Previously, Smith and coworkers reported the iridium-catalyzed C–H borylation at C-2 and C-7 positions of the indole ring.41 They found that if the C-2 position was blocked, the C–H borylation could only happen at the C-7 position. To our delight, such a methodology worked well for C-8 site-selective C–H borylation of 7a. Both HBpin and B2pin2 could be used as a boron source to prepare the boranate 7b, while B2pin2 showed higher yield compared to HBpin. Combined with C–B bond functionalization methodologies such as Chan–Lam coupling (7c),42 oxidation by hydrogen peroxide (7d),43 halogenation (7e),43,44 Suzuki coupling (7f)44 and trifloromethylation (7g),45 we could easily install diverse functional groups at the C-8 position (Scheme 2b). By the same route, all monomeric alkaloids 7c–g could be successfully transformed into the corresponding ITHQ-type bis-β-carboline alkaloids (Scheme 2c). Moreover, aldehyde 6c could be used for the concise synthesis of a bis-β-carboline alkaloid picrasidine R (4). Vinyl addition to the aldehyde 6c followed by Dess–Martin oxidation afforded the vinyl ketone 10, which could undergo a thiazolium-catalyzed Stetter reaction with the aldehyde 6c to afford picrasidine R (4) in 90% yield (Scheme 2c).46
Computational studies of the aza-[4 + 2] cycloaddition
It should be noticed that theoretically there may be another possible pathway of the aza-[4 + 2] cycloaddition, leading to isopicrasidine G as the product, as is shown in Fig. 1. However, isopicrasidine G was neither identified in the aza-[4 + 2] cycloaddition, nor from natural product isolation. Generally, for the type-III aza-[4 + 2] cycloaddition (Scheme S3†), the terminal alkene position of the vinyl imine structure is prone to act as an electrophile (Scheme S3,† Ihara's work as an example47–49). However, our observation of the reaction outcome was different. It is noteworthy that such a transformation is the first reported umpolung type-III aza-[4 + 2] cycloaddition, whose regioselectivity is different from those of all previously reported ones. To explain the unique regioselectivity and provide us mechanistic insights, the reaction profile was further investigated by DFT calculations (Scheme 3). Interestingly, if we select two molecules of free alkaloid (dehydrocrenatine, 5a) as the reactant, they will undergo a concerted aza-[4 + 2] cycloaddition (Scheme 3, pathways A′ and B′) with the activation Gibbs energies (ΔG) up to 40.45 kcal mol−1 for pathway A′ to form picrasidine G (1, PA) and 47.88 kcal mol−1 for pathway B′ to form isopicrasidine G (PB). However, if we select one molecule of free alkaloid and another molecule of the protonated alkaloid as reactants, they will undergo a stepwise, formal aza-[4 + 2] cycloaddition (Scheme 3, pathways A and B), and the activation Gibbs free energies decrease to 26.80 kcal mol−1 for pathway A and 30.57 kcal mol−1 for pathway B. Kinetically, the difference of activation Gibbs free energy (ΔΔG) between pathways A and B is up to 3.77 kcal mol−1, which explains such excellent regioselectivity.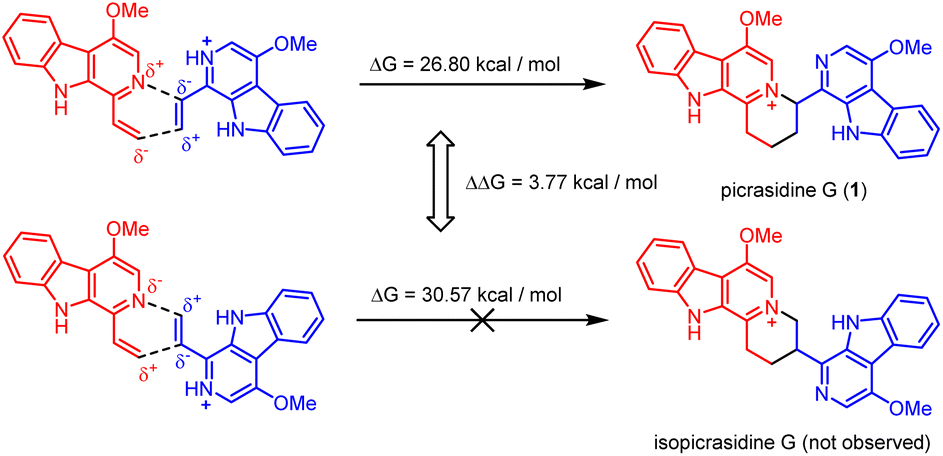 | ||
| Fig. 1 Two possible pathways of the aza-[4 + 2] cycloaddition. | ||
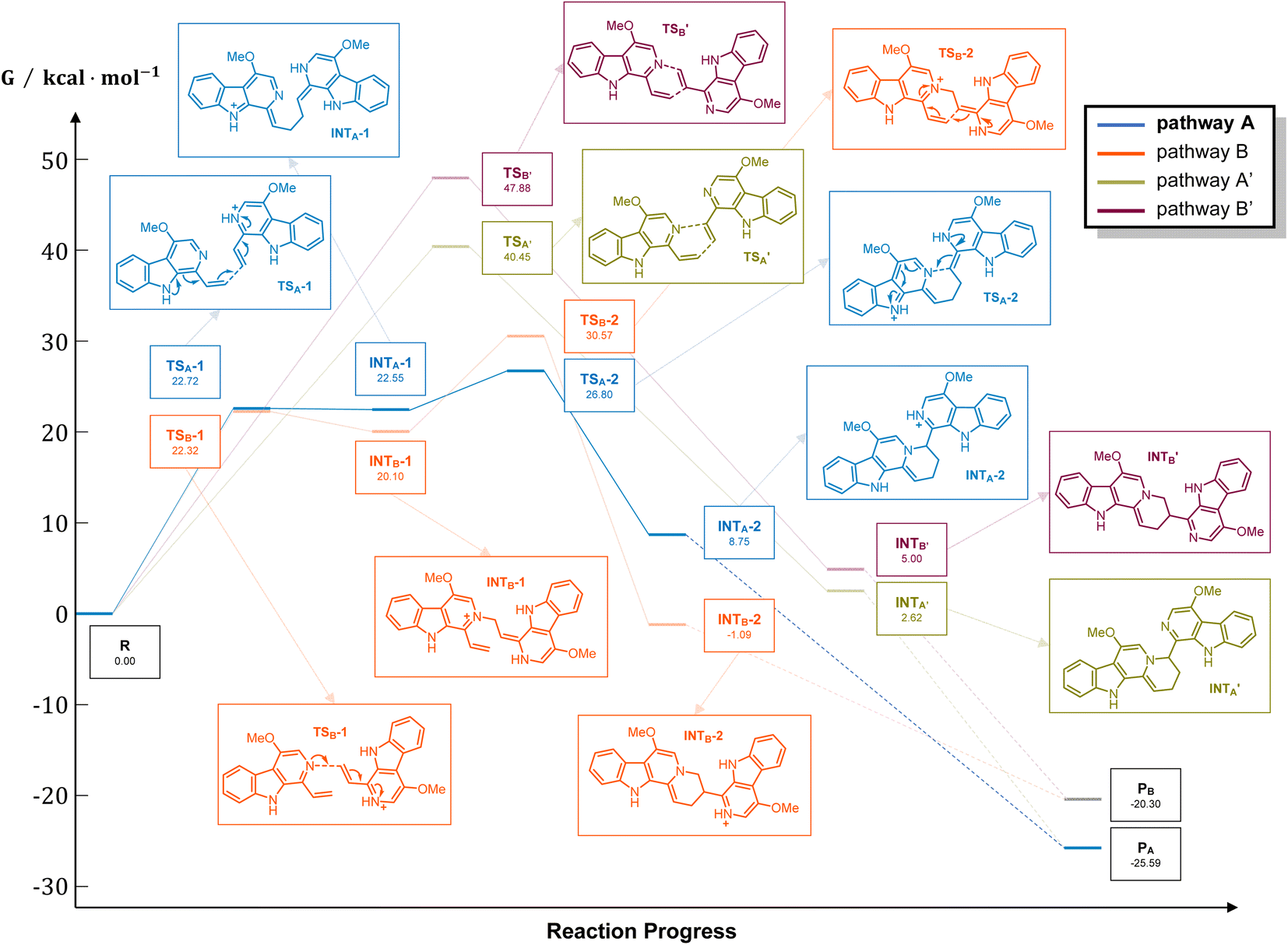 | ||
| Scheme 3 Energy profile of the aza-[4 + 2] cycloaddition. For pathways A and B, R = 1 × dehydrocrenatine (10a) and 1 × protonated dehydrocrenatine; for pathways A′ and B′, R = 2 × dehydrocrenatine. PA = picrasidine G (1), PB = isopicrasidine G. Calculation at WB97XD/6-311++G(2d,p)//WB97XD/6-31G(d) at 403.15 K. Gibbs free energies (ΔG) are in kcal mol−1. | ||
According to the computational result, the stepwise pathway A is the most favored mechanism under the experimental conditions, with just 26.80 kcal mol−1 activation Gibbs energies to overcome. For the free alkaloid, the electron-donating effect of the N-9 nitrogen atom and the C-4 methoxy group inverts the intrinsic polarity of the “pyridine ring”, making the terminal alkene nucleophilic rather than electrophilic. However for the protonated alkaloids, the “pyridine ring” maintained its intrinsic polarity due to the protonation, making the terminal alkene a Michael acceptor. First, the terminal alkene of the free alkaloid acts as a nucleophile to attack the terminal alkene of the protonated alkaloid, forming INTA-1 via TSA-1 (ΔG = 22.72 kcal mol−1). Then the succeeding annulation process of the unstable INTA-1 can easily take place via TSA-2 to form INTA-2, requiring only 4.25 kcal mol−1 energy expense. Finally, the intramolecular proton transfer decreases the total energy of the system, making the overall reaction thermodynamically favored. The computational results revealed that picrasidine G (1) is the more favorable product both kinetically and thermodynamically compared to the proposed isopicrasidine G, and the aza-[4 + 2] cycloaddition prefers a stepwise mechanism.
Biological evaluation of the synthesized bis-β-carboline compounds
We also tested the anti-cancer activities of the synthesized bis-β-carboline compounds picrasidines G (1), S (2), T (3), R (4) and 11e–g on multiple human cancer cell lines, including THP-1 (monocytic leukaemia), MCF-7 (epithelial luminal), HepG2 (hepatoma), and A375 (melanoma) (Table 2). Most of the ITHQ type bis-β-carboline alkaloids showed cytotoxicity against the above-mentioned cancer cell lines. Besides, these ITHQ type bis-β-carboline compounds showed selectivity for different cancer cell lines. For example, picraisidine G (1) showed potent cytotoxicity to THP-1 (IC50 = 3.7 μM), but relatively weak cytotoxicity to HepG2 (IC50 = 64.6 μM). Picrasidine S (2) showed stronger cytotoxicity against THP-1 and HepG2 (IC50 = 3.3–5.5 μM) compared to MCF-7 and A375 (IC50 = 15.9–20.4 μM). In contrast, 11f and 11g showed potent cytotoxicity against all four cancer cell lines (IC50 = 3.1–8.1 μM).| Compounds | IC50 (μM, at 48 h) | |||
|---|---|---|---|---|
| THP-1 | MCF-7 | HepG2 | A375 | |
| a Cell viability was assessed using the CellTiter-Glo® assay kit (Promega, USA). | ||||
| Picrasidine G (1) | 3.7 ± 1.1 | 13.2 ± 2.2 | 64.6 ± 9.6 | 16.5 ± 3.5 |
| Picrasidine S (2) | 3.3 ± 0.6 | 20.4 ± 1.6 | 5.5 ± 0.7 | 15.9 ± 3.4 |
| Picrasidine T (3) | 13.2 ± 3.4 | 52.2 ± 9.2 | 19.5 ± 2.1 | 18.9 ± 2.8 |
| Picrasidine R (4) | >100 | 81.2 ± 8.3 | >100 | >100 |
| 11e | 19.3 ± 4.2 | 22.0 ± 3.0 | 80.8 ± 8.8 | >100 |
| 11f | 3.1 ± 0.7 | 5.8 ± 1.1 | 4.9 ± 1.4 | 8.1 ± 2.2 |
| 11g | 3.1 ± 0.8 | 4.5 ± 1.4 | 3.8 ± 1.4 | 5.2 ± 1.4 |
Conclusions
We have successfully accomplished the first total syntheses of three ITHQ-type bis-β-carboline alkaloids picrasidines G, S and T and several derivatives by the regio-selective aza-[4 + 2] cycloaddition. The C-8 oxidation state was efficiently installed by the iridium catalyzed site-selective C–H borylation and C–B bond functionalization strategy, enabling the diversity-oriented syntheses of several natural product-like derivatives.50–52 Computational studies revealed a stepwise mechanism of the aza-[4 + 2] cycloaddition and explained the origin of excellent regioselectivity. Another bis-β-carboline alkaloid picrasidine R was also efficiently synthesized via the thiazolium-catalyzed Stetter reaction. This work provides synthetic evidence for the proposed biosynthetic pathway of ITHQ-type bis-β-carboline alkaloids.Data availability
Additional experimental and computational data supporting this article are included in the ESI.†Author contributions
Prof. X. Lei and Q. Wang conceived the project. Q. Wang conducted the synthetic and computational studies. F. Guo conducted the bioactivity test. All experiments were conducted under the supervision of Prof. X. Lei. J. Wang provided helpful suggestions in the project. Q. Wang wrote the manuscript with input from Prof. X. Lei, F. Guo and J. Wang. Prof. X. Lei managed the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Han Ke, Zeyuan Shen and Prof. Zhixiang Yu for assistance in computational studies. We thank Xiu Zhang, Hui Fu for NMR analysis, and Xiaoran He for HRMS analysis. This work was supported by High-performance Computing Platform and Analytical Instrumentation Center of Peking University. This work was funded by grants from the National Key Research and Development Program of China (2022YFC3401500 and 2022YFC2502500489), the National Natural Science Foundation of China (22193073 and 92253305), and the Beijing Outstanding Young Scientist Program (BJJWZYJH01201910001001).References
- G. Kleks, S. Duffy, L. Lucantoni, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2020, 83, 422–428 CrossRef CAS PubMed.
- J. Han, T. Lv, S. Song and X. Huang, Biochem. Syst. Ecol., 2023, 107, 104614 CrossRef CAS.
- Q. Chen, X. Ye, X. Liu, Y. Liang, W. Feng, C. Li and Z. Wang, Phytochemistry, 2022, 193, 112987 CrossRef CAS PubMed.
- T. Ohmoto and K. Koike, Chem. Pharm. Bull., 1982, 30, 1204–1209 CrossRef CAS.
- T. Ohmoto and K. Koike, Chem. Pharm. Bull., 1983, 31, 3198–3204 CrossRef CAS.
- T. Ohmoto and K. Koike, Chem. Pharm. Bull., 1984, 32, 3579–3583 CrossRef CAS.
- T. Ohmoto and K. Koike, Chem. Pharm. Bull., 1985, 33, 3847–3851 CrossRef CAS.
- T. Ohmoto and K. Koike, Chem. Pharm. Bull., 1985, 33, 4901–4905 CrossRef CAS.
- T. Ohmoto, K. Koike, T. Higuchi and K. Ikeda, Chem. Pharm. Bull., 1985, 33, 3356–3360 CrossRef CAS.
- K. Koike and T. Ohmoto, Chem. Pharm. Bull., 1986, 34, 2090–2093 CrossRef CAS.
- K. Koike, T. Ohmoto and K. Ogata, Chem. Pharm. Bull., 1986, 34, 3228–3236 CrossRef CAS.
- K. Koike and T. Ohmoto, Chem. Pharm. Bull., 1987, 35, 3305–3308 CrossRef CAS.
- K. Koike, T. Ohmoto and T. Higuchi, Phytochemistry, 1987, 26, 3375–3377 CrossRef CAS.
- K. Koike and T. Ohmoto, Phytochemistry, 1988, 27, 3029–3030 CrossRef CAS.
- K. Koike, T. Ohmoto and K. Ikeda, Phytochemistry, 1990, 29, 3060–3061 CrossRef CAS.
- Y. Murakami, Y. Yokoyama, C. Aoki, H. Suzuki, K. Sakurai, T. Shinohara, C. Miyagi, Y. Kimura, T. Takahashi, T. Watanabe and T. Ohmoto, Chem. Pharm. Bull., 1991, 39, 2189–2195 CrossRef CAS.
- H. Li, K. Koike and T. Ohmoto, Chem. Pharm. Bull., 1993, 41, 1807–1811 CrossRef CAS.
- W. Jiao, H. Gao, C. Li, F. Zhao, R. Jiang, Y. Wang, G. Zhou and X. Yao, J. Nat. Prod., 2010, 73, 167–171 CrossRef CAS PubMed.
- W. Jiao, H. Gao, F. Zhao, H. Lin, Y.-M. Pan, G. Zhou and X. Yao, Chem. Pharm. Bull., 2011, 59, 359–364 CrossRef CAS PubMed.
- W. Jiao, G. Chen, H. Gao, J. Li, B.-B. Gu, T. Xu, H. Yu, G. Shi, F. Yang, X. Yao and H. Lin, J. Nat. Prod., 2015, 78, 125–130 CrossRef PubMed.
- J. Zhang, S. Zhao, J. Xie, J. Yang, G. Chen, D. Hu, W. Zhang, C. Wang, X. Yao and H. Gao, Bioorg. Chem., 2020, 101, 104043 CrossRef CAS PubMed.
- W. Zhao, W. Zhou, J. Chen, G. Yao, B. Lin, X. Wang, X. Huang and S. Song, Phytochemistry, 2019, 159, 39–45 CrossRef CAS PubMed.
- X. Guo, F. Li, F. Zheng, N. Gong, Y. Li, W. Feng and L. Tian, Nat. Prod. Res., 2020, 34, 489–493 CrossRef CAS PubMed.
- G. Shi, W. Jiao, F. Yang and H. Lin, Chin. Tradit. Herb. Drugs, 2015, 46, 803–807 CAS.
- N. Yamashita, M. Kondo, S. Zhao, W. Li, K. Koike, K. Nemoto and Y. Kanno, Bioorg. Med. Chem. Lett., 2017, 27, 2608–2612 CrossRef CAS PubMed.
- L. Yi, Y. He, S. Tan, L. V. White, P. Lan, M. G. Gardiner, Z. Pei, M. L. Coote and M. G. Banwell, J. Org. Chem., 2022, 87, 12287–12296 CrossRef CAS PubMed.
- T. J. Hagen, K. Narayanan, J. Names and J. M. Cook, J. Org. Chem., 1989, 54, 2170–2178 CrossRef CAS.
- K. Takasu, T. Shimogama, C. Saiin, H.-S. Kim, Y. Wataya, R. Brun and M. Ihara, Chem. Pharm. Bull., 2005, 53, 653–661 CrossRef CAS PubMed.
- H. Suzuki, M. Unemoto, M. Hagiwara, T. Ohyama, Y. Yokoyama and Y. Murakami, J. Chem. Soc., Perkin Trans. 1, 1999, 1717–1724 RSC.
- H. Suzuki, Y. Tsukakoshi, T. Tachikawa, Y. Miura, M. Adachi and Y. Murakami, Tetrahedron Lett., 2005, 46, 3831–3834 CrossRef CAS.
- S. Gaikwad, D. Kamble and P. Lokhande, Tetrahedron Lett., 2018, 59, 2387–2392 CrossRef CAS.
- D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS PubMed.
- J. N. Newton, D. F. Fischer and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 1726–1730 CrossRef CAS PubMed.
- B. Jiang and M. Dai, J. Am. Chem. Soc., 2021, 143, 20084–20089 CrossRef CAS PubMed.
- F. Wu, J. Zhang, F. Song, S. Wang, H. Guo, Q. Wei, H. Dai, X. Chen, X. Xia, X. Liu, L. Zhang, J.-Q. Yu and X. Lei, ACS Cent. Sci., 2020, 6, 928–938 CrossRef CAS PubMed.
- B. Hong, C. Li, Z. Wang, J. Chen, H. Li and X. Lei, J. Am. Chem. Soc., 2015, 137, 11946–11949 CrossRef CAS PubMed.
- B. Hong, T. Luo and X. Lei, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS PubMed.
- S. Wang, K. Chen, F. Guo, W. Zhu, C. Liu, H. Dong, J.-Q. Yu and X. Lei, ACS Cent. Sci., 2023, 9, 1129–1139 CrossRef CAS PubMed.
- W. Liu, B. Hong, J. Wang and X. Lei, Acc. Chem. Res., 2020, 53, 2569–2586 CrossRef CAS PubMed.
- K. Chen and X. Lei, Curr. Opin. Green Sustainable Chem., 2018, 11, 9–14 CrossRef.
- S. Paul, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2006, 128, 15552–15553 CrossRef CAS PubMed.
- P. Chen, H. Yang, H. Zhang, W. Chen, Z. Zhang, J. Zhang, H. Li, X. Wang, X. Xie and X. She, Org. Lett., 2020, 22, 2022–2025 CrossRef CAS PubMed.
- R. P. Loach, O. S. Fenton, K. Amaike, D. S. Siegel, E. Ozkal and M. Movassaghi, J. Org. Chem., 2014, 79, 11254–11263 CrossRef CAS PubMed.
- D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068–4069 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed.
- P. E. Harrington and M. A. Tius, J. Am. Chem. Soc., 2001, 123, 8509–8514 CrossRef CAS PubMed.
- M. Ihara and M. Suzuki, Heterocycles, 2000, 52, 1083–1085 CrossRef.
- M. Ihara, T. Taniguchi, K. Makita, M. Takano, M. Ohnishi, N. Taniguchi, K. Fukumoto and C. Kabuto, J. Am. Chem. Soc., 2002, 115, 8107–8115 CrossRef.
- K. Takasu, N. Nishida, A. Tomimura and M. Ihara, J. Org. Chem., 2005, 70, 3957–3962 CrossRef CAS PubMed.
- J. Wang, H. Ke, J. Yang, N. Guo, K. Hu, R. Tang, Q. Ding, L. Gao and X. Lei, Chem Catal., 2023, 3, 100451 CrossRef CAS.
- K. Chen, F. Wu and X. Lei, Chin. J. Chem., 2021, 39, 838–854 CrossRef CAS.
- J. Zhang, J. Wu, B. Hong, W. Ai, X. Wang, H. Li and X. Lei, Nat. Commun., 2014, 5, 4614 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03722c |
| This journal is © The Royal Society of Chemistry 2023 |