
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-initiated 1,3-dipolar cycloaddition between dehydroalanines and tetrazoles: application to late-stage peptide and protein modifications†
Mengqian
Zhang
 a,
Peiyang
He
a and
Yanmei
Li
*abc
a,
Peiyang
He
a and
Yanmei
Li
*abc
aKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: liym@mail.tsinghua.edu.cn
bBeijing Institute for Brain Disorders, Beijing 100069, P. R. China
cCenter for Synthetic and Systems Biology, Tsinghua University, Beijing 100084, P. R. China
First published on 22nd August 2023
Abstract
As an easily introduced noncoded amino acid with unique electrophilicity distinct from the 20 natural amino acids, dehydroalanine (Dha) is not only a precise protein post-translational modification (PTM) insertion tool, but also a promising multifunctional labelling site for peptides and proteins. However, achieving a balance between the reaction rate and mild reaction conditions has been a major challenge in developing novel Dha-modified strategies. Rapid, efficient, and mild Dha modification strategies are highly desired. Additionally, catalyst-free photocontrollable reactions for Dha-containing peptide and protein modification have yet to be developed. Here, we report a photoinitiated 1,3-dipolar cycloaddition reaction between Dha and 2,5-diaryl tetrazoles. Under low-power UV lamp irradiation, this reaction is completed within minutes without catalysis, resulting in a fluorescent pyrazoline-modified peptide or protein with excellent chemoselectivity for Dha residues. Notably, this reaction exhibits complete site-specificity in the modification of thiostrepton, a natural antimicrobial peptide containing multiple Dha residues (Dha3, Dha16, and Dha17), within 20 minutes in high yields. This is currently the fastest reaction for modifying the Dha residue in thiostrepton with clear site-specificity towards Dha16. This photoinitiated reaction also provides a chemoselective strategy for precise functionalization of proteins. Additionally, the rapidity and efficiency of the reaction minimize UV light damage to the biological reaction system. Combined with fluorogenic properties, this photo-controllable methodology can be applied to live cell imaging, further broadening the application scope of the Dha modification methodology.
Introduction
Chemical modifications provide basic tools for conferring, regulating or improving peptide and protein functions in biology and medicine. As a result, this toolbox has a wide range of applications in peptide and protein science, including but not limited to post-translational modification (PTM) insertion, fluorescence tracking and targeted delivery.1–3 To date, various chemoselective modification strategies have been developed. However, strategies targeting specific canonical amino acids in peptides and proteins tend to generate multisite modification products.4,5 To achieve more precise site-selective modification, methods targeting noncoded amino acids with unique reactivity need to be further developed.1Dehydroalanine (Dha) has been an attractive option for precise late-stage modification of peptides and proteins. As a naturally occurring noncoded amino acid,6 it is formed by serine (Ser) dehydration or phosphoserine (pSer) elimination in natural peptides7–9 and proteins.10,11 In particular, employing cysteine (Cys) and serine (Ser) as precursors, Dha can be easily introduced into various peptides and proteins at the position of interest via mild chemical treatment.10,12 In addition, owing to its electrophilic α,β-unsaturated structure, Dha has a unique chemical reactivity distinct from the 20 natural amino acids, which facilitates site-selective modification of peptides and proteins. To date, various robust Dha modification strategies have been reported, such as nucleophilic additions for the construction of carbon–heteroatomic (C–X) bonds and radical additions or metal-catalysed cross-coupling reactions for the formation of carbon–carbon (C–C) bonds on the β-C of Dha residues (Fig. 1).13,14 These reactions have been applied in protein therapeutics, such as the preparation of homologous antibody–drug conjugates (ADCs),15 as well as the insertion of PTMs and their mimics, to explore the influence on protein structures and functions.16,17 In addition, the cycloaddition reaction has been another approach to Dha modification, yet it is still only in its infancy. To date, related strategies have mainly focused on the modification of small molecule Dha derivatives and peptides.18–20
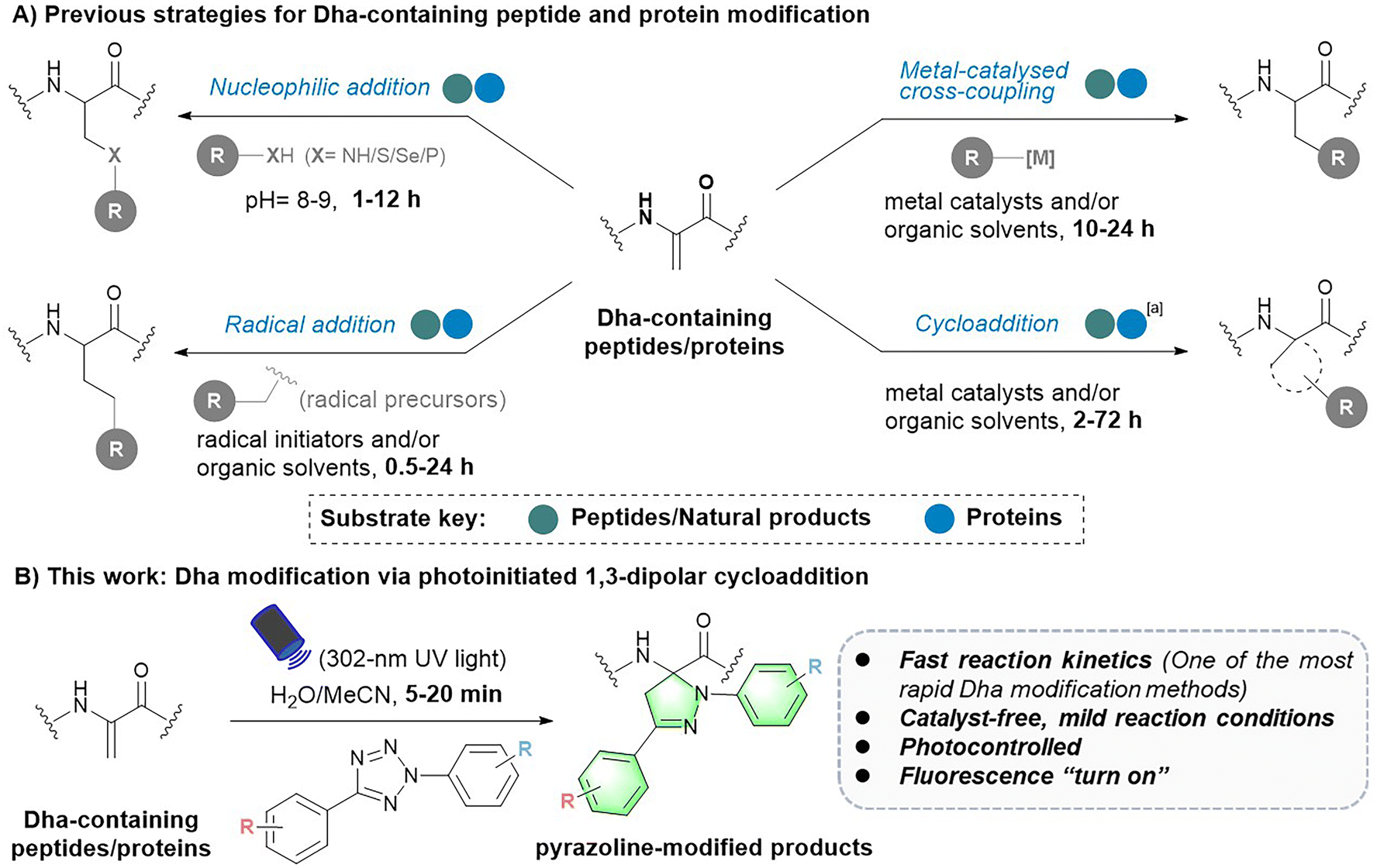 | ||
| Fig. 1 Strategies for Dha-containing peptide and protein modification. aOnly one cycloaddition strategy for protein modification has been reported so far. | ||
Despite these advances, there is still much room for the development and improvement of Dha chemistry. First, the competing electronic forces between the π-donating nitrogen substituent and electron-withdrawing carbonyl group reduce the reactivity of Dha to some extent.10,18 As a result, most peptide or protein Dha modification reactions require hours to days to complete (Fig. 1A).13 For instance, nucleophilic addition reactions typically take 1 to 12 hours15,21–25 to form C–X bonds. Free radical reactions, on the other hand, usually take 0.5 to 24 hours to complete,17,26–32 with only a small proportion of reactions17,29 finishing within an hour. Cycloaddition reactions need even longer reaction times, varying from 2 to 72 hours.18–20,33 Therefore, more rapid and efficient Dha modification strategies are highly desired. In addition, Dha modification reactions developed in recent years are generally not mild enough, requiring the use of organic solvents and/or harsh catalysts, causing difficulty in further biomedical applications. Hence, noncatalytic and mild bioorthogonal Dha modification reactions remain to be developed. Last, previously developed strategies focused more on introducing specific amino acids or PTM mimics for protein function exploration.16,29,34 However, as a powerful tool for peptide and protein multifunctionalization, more downstream applications of Dha modification need to be further explored. Reactions that can be controlled spatially and temporally are expected to expand downstream applications of the Dha modification methodology, but related reactions have yet to be developed.
To address these challenges and meet the growing needs of downstream applications, we turned our attention to the less explored 1,3-dipolar cycloaddition strategy. Although existing Dha modification strategies based on 1,3-dipolar cycloaddition have expanded the toolbox of methods available for peptide chemistry, they commonly require a long reaction time (12–72 hours) and harsh conditions (metal catalysts and/or organic solvents), thus preventing their general applicability.18,20,33 Recently, a cycloaddition strategy for protein modification using nitrile oxides as 1,3-dipoles to react with Dha has been reported, enabling rapid modification of protein substrates in two hours.35 However, nitrile oxides are prone to undergo side reactions with nucleophilic amino acids such as lysine, lacking chemical specificity for Dha. In our search for potentially more reactive and chemoselective 1,3-dipoles towards Dha residues, nitrile imines generated by photoactivation of tetrazoles attracted our attention.36,37 Considering the broad functional group tolerance of photoinduced 1,3-dipolar cycloadditions between tetrazoles and various electron-deficient alkenes, we speculated that Dha could similarly undergo a fast and catalyst-free cycloaddition with tetrazoles. Herein, we report a photoinitiated 1,3-dipolar cycloaddition between 2,5-diaryl tetrazoles and Dha residues (Fig. 1B). Tetrazoles are readily triggered by 302 nm UV light from a hand-held LED lamp to form nitrile imine dipoles (Scheme S1†), which undergo spontaneous and rapid [3 + 2] cycloadditions with the unsaturated double bond of Dha residues in peptides and proteins, resulting in the formation of fluorescent pyrazoline products under mild and catalyst-free conditions within minutes. In addition, activation by a specific wavelength of light enables precise temporal and spatial control of the reactions. Furthermore, as a fluorogenic reaction, the intense fluorescence of the pyrazoline product can be applied for cell imaging, further expanding the biomedical application of Dha chemical modification.
Results and discussion
In our initial study, protected Dha (Boc-Dha-OMe) was employed as the substrate to investigate the reactivity of Dha with 2,5-diaryl tetrazole under photoirradiation with 302 nm UV light (Table 1). A reaction mixture containing 2-(4-methoxyphenyl)-5-phenyltetrazole (1a), Boc-Dha-OMe, and the solvent was irradiated with a hand-held 302 nm UV lamp at room temperature. Notably, this reaction was completed within 10 minutes, as monitored via thin-layer chromatography (TLC), generating pyrazoline product 2a with bright green fluorescence (as shown in Fig. S1†) in high isolated yields (74–80%). The time-course analysis demonstrated that the reactions were completed within 5–7 minutes (Fig. S2†). Further experiments showed the reaction's excellent solvent tolerance and rapid, efficient performance across a range of solvents, with particularly high efficiency in protic solvents including H2O (Table 1, entries 1–6).|
|
|||||
|---|---|---|---|---|---|
| Entry | Tetrazole | Substrate concentration (mM) | Solvent | Yielda | |
| Boc-Dha-OMe | Tetrazole | ||||
| a Isolated yields are reported. | |||||
| 1 | 1a | 2.5 | 4.0 | EtOAc | 74.1% |
| 2 | 1a | 2.5 | 4.0 | CH2Cl2 | 75.8% |
| 3 | 1a | 2.5 | 3.0 | MeOH | 78.0% |
| 4 | 1a | 2.5 | 3.0 | MeCN | 79.3% |
| 5 | 1a | 2.5 | 3.0 | MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 3:1 :H2O 3:1 |
78.2% |
| 6 | 1a | 1 | 1.2 | H2O | 75.3% |
| 7 | 1b | 2.5 | 3.0 | MeCN:H2O 3:1 |
77.1% |
| 8 | 1b | 1 | 1.2 | H2O:MeCN 9:1 |
69.2% |
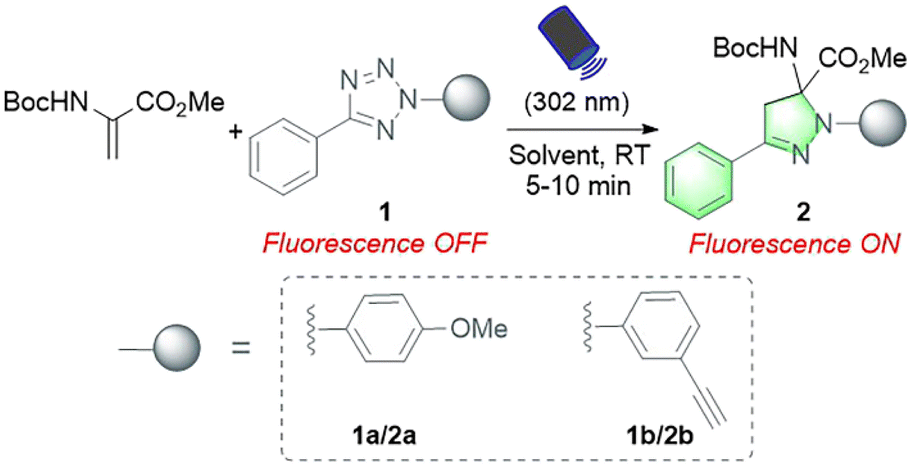
We next validated the reactivity of this reaction on Dha-containing peptides (Fig. 2). Cysteine-containing peptides 3a–e were synthesized and then treated with methyl 2,5-dibromopentanoate (MeDBP) to generate Dha-containing peptides 4a–evia bisalkylation-elimination. We initially utilized a C-terminal amide-protected peptide 4a as a model to react with tetrazole 1a in the MeCN–H2O mixed solvent. Under the same reaction conditions as those of the small-molecule substrates, the anticipated pyrazoline-modified product 5a was obtained as the only product, yet its conversion rate of 63.2% required further improvement. By optimizing the stoichiometric ratio of the reaction and performing a stepwise addition of tetrazole 1a, the conversion rate of peptide 4a was increased to 87.0% (Fig. 2A, Scheme S5†). Under similar optimized conditions, we were delighted to find out that all the modifications of peptides 4b–e with tetrazole 1a were completed within 10–15 minutes, generating pyrazoline-modified peptides with bright green fluorescence in excellent conversion rates (83–98%) and isolated yields (68–84%) without any side-product formation (Fig. 2A, Scheme S5†). These results indicated that tetrazoles could chemoselectively react with Dha moieties in both C-terminal protected (4a and 4b) and unprotected peptides (4c–e), and the reaction exhibited excellent compatibility with all natural amino acids. To further expand the scope of tetrazole reagents for downstream multifunctionalization, tetrazole 1b bearing an alkynyl group was synthesized. After confirming its reactivity with the small molecule Dha substrate (Table 1, entries 7 and 8), we further reacted 1b with peptides 4a–e to obtain modified peptides with alkyne “reaction handles” (6a–e) (Fig. 2B, Scheme S6†), which exhibited bright blue fluorescence.
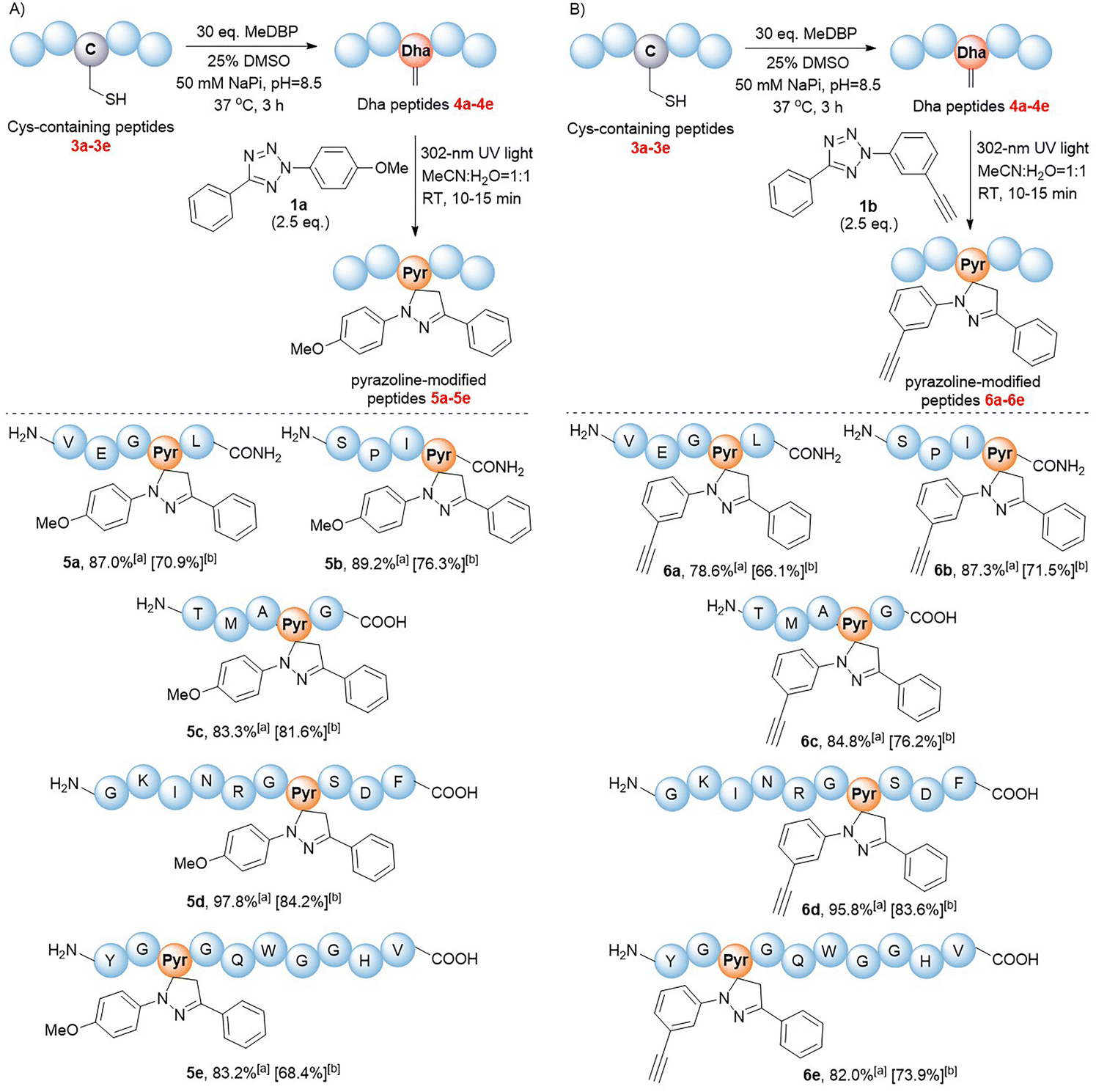 | ||
| Fig. 2 1,3-Dipolar cycloaddition reactions of Dha peptides 4a–e with tetrazoles (A) 1a and (B) 1b. All peptide substrates 4a–e were at a concentration of 1 mM. aConversions were estimated via RP-HPLC. bIsolated yields of pyrazoline-modified products. | ||
Inspired by the high yields and excellent tolerance to all natural amino acids, we aimed to extend this reaction to late-stage modifications of highly complex natural products. As a natural, complex and highly potent antibiotic against a broad range of Gram-positive bacteria,38 thiostrepton has piqued our interest. Its complexity was partly exemplified by the presence of three Dha residues (Dha3, Dha16, and Dha17), one dehydrothreonine residue (Dhb8), four thiazoles, one thiazoline, three isolated hydroxyl groups, a diol, an amine, an imine, and a highly substituted pyridine (Fig. 3A).39 Complex functional groups and multiple reaction sites bring great challenges to the site-selective Dha modification of thiostrepton. To date, only a limited number of late-stage modification strategies have been reported.19,23,39–42 Although exhibiting predominant selectivity towards Dha16 or Dha17, which are located in the solvent-exposed tail region of thiostrepton, existing reactions have been observed to yield mixtures of heterogeneous modifications, generally including Dha16-modified products, Dha17-modified products, and doubly site-labelled products. Additionally, these reactions require hours to days to complete. The development of a rapid and site-specific Dha modification strategy for thiostrepton remains an outstanding challenge.
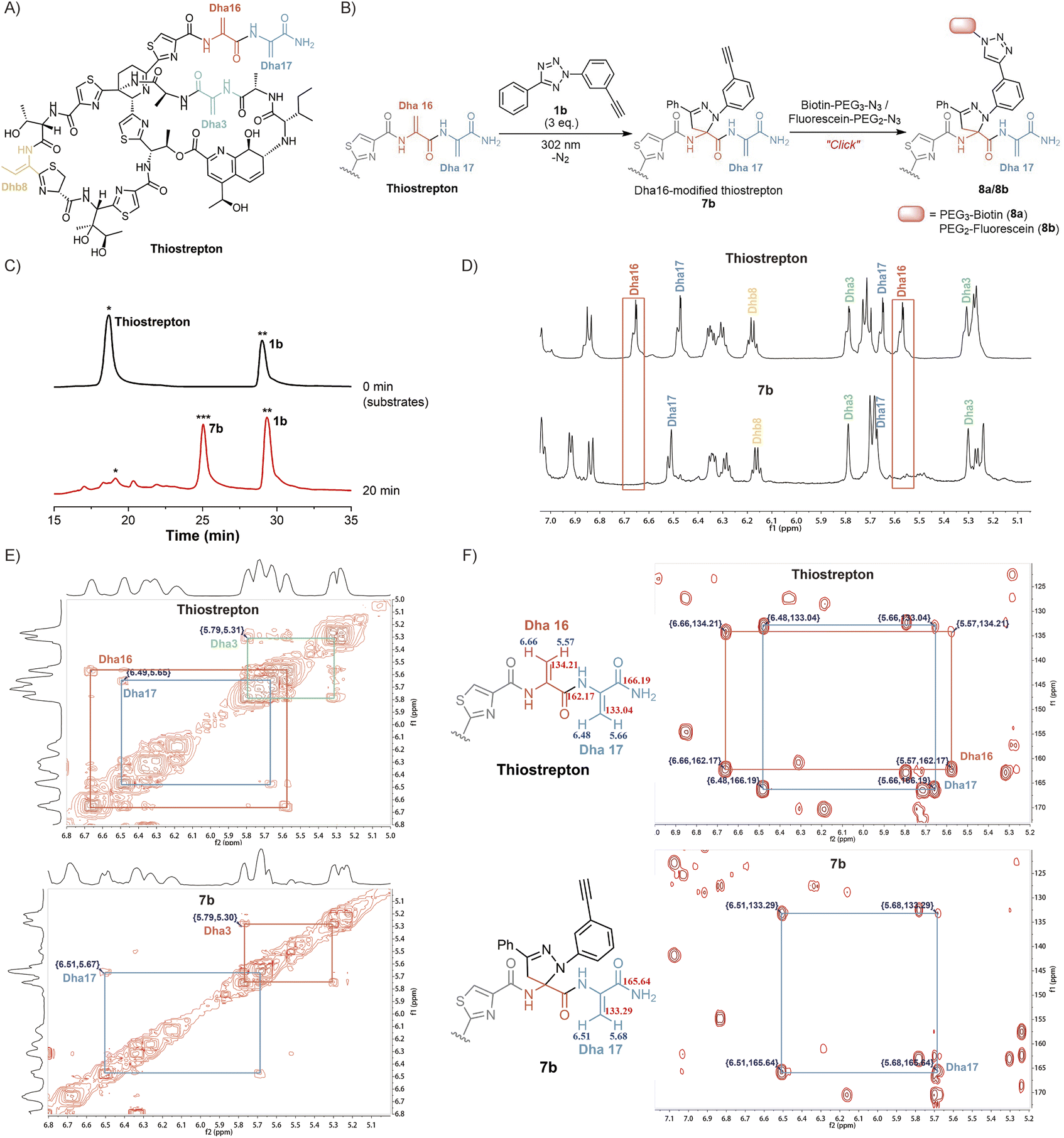 | ||
| Fig. 3 Site-selective labelling of thiostrepton via light-initiated 1,3-dipolar cycloaddition of Dha and tetrazole 1b. (A) Chemical structure of thiostrepton. (B) Two-step reaction for site-specific labelling of Dha16 on thiostrepton. (C) HPLC analysis of the reaction mixture of thiostrepton and tetrazole 1b at 0 min (top) and 20 min (bottom). (D) 1H NMR spectra of thiostrepton and Dha16-labelled product 7b. Chemical shifts of signals from the C(sp2)–H of Dha3, Dha16, Dha17, and Dhb8 are assigned. (E) 2D-NMR spectra (1H–1H COSY) of thiostrepton and 7b, which show 1H–1H couplings in Dha16 and Dha17. (F) 2D-NMR spectra (1H–13C HMBC) of thiostrepton and 7b. Long range 1H–13C couplings via a H–C–C–C path are measured and assigned. | ||
We aimed to investigate whether tetrazole reagents could tackle the aforementioned challenge. Our study started with the verification of the reactivity of tetrazole 1a with thiostrepton (Scheme S7†). After stirring the reaction under 302 nm light for 15 minutes under the optimized reaction conditions, thiostrepton derivative 7a with single-site pyrazoline modification was obtained, as characterized via reversed-phase high performance liquid chromatography (RP-HPLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) (Fig. S24, S27 and S28†). To our knowledge, this is currently the fastest Dha-based modification reaction for thiostrepton.
Next, we aspired to extend the reaction into a versatile, multifunctional derivatization strategy for thiostrepton. With this objective, we conducted the reaction between thiostrepton and tetrazole 1b to install an alkynyl handle for further multifunctionalization (Fig. 3B, Scheme S7†). During RP-HPLC analysis of the reaction mixture, the optimized reaction conditions produced a clean reaction profile, with only product 7b detected (Fig. 3C). After purification via RP-HPLC, 7b was also characterized via MS as a single-site pyrazoline-modified product (Fig. S24 and S29†). Furthermore, an absolute preference for modification at Dha16 was confirmed via the combined analyses of 1D and 2D NMR spectra of the thiostrepton substrate and modified product 7b (Fig. 3D–F). Initially, we referred to previous studies19,23,42 to confirm the chemical shifts of the 1H and 13C signals from Dha3, Dha16, Dha17, and Dhb8 in the thiostrepton substrate (as shown in Fig. 3D–F (top)). Subsequently, by comparing the 1H NMR spectra of 7b (bottom) and thiostrepton (top) in Fig. 3D, we found that there were no C(sp2)–H NMR signals of the Dha16 side chain in the remaining Dha residues of product 7b. Additionally, neither the 1H–1H correlated spectroscopy (COSY) spectra (Fig. 3E) nor the 1H–13C heteronuclear multiple bond correlation (HMBC) spectra (Fig. 3F) of 7b showed any NMR signals of Dha16, whereas the 1D and 2D NMR signals of Dha3, Dha17, and Dhb8 were all detected. These results indicated that the modification reaction occurred precisely at Dha16. We speculate that the site-selectivity for modification at Dha16 can be explained by the fact that this residue is the most electron-deficient site due to the presence of adjacent thiazole15 and Dha17, both of which are electron-withdrawing moieties.19 In addition, previous studies have indicated that the highest occupied molecular orbital (HOMO) of a nitrile imine dipole generated by the photoactivation of tetrazole undergoes electron transfer with the lowest unoccupied molecular orbital (LUMO) of the alkene dipolarophile, leading to the occurrence of a [3 + 2] cycloaddition reaction.43 The α,β-unsaturated double bond of Dha16 is adjacent to these two electron-withdrawing moieties, which lowers its LUMO energy and facilitates better alignment with the HOMO energy level of the nitrile imine. Therefore, the cycloaddition reaction exhibits a significant preference for modification at Dha16, even demonstrating explicit site-specificity.
In addition, the installation of an alkynyl group on thiostrepton enabled downstream functionalization through click reactions with various commercially available azide reagents. Herein, the reactions of biotin-PEG3-azide and fluorescein-PEG2-azide with 7b were utilized as examples to illustrate the feasibility and great potential of this reaction for the multifunctionalization of highly complex natural products (Fig. 3B). Under mild conditions, biotinylated thiostrepton derivative 8a and fluorescein-labelled product 8b were obtained with isolated yields of 61.2% and 52.7%, respectively (Scheme S8†). This reaction simplified the late-stage chemical modification of thiostrepton, allowing for more efficient and rapid site-specific modification of Dha16 under milder conditions without catalysts or additives. Compared to previous methods that needed hours or even days under metal catalysis, our method enabled the efficient synthesis of more novel analogues of homogeneous thiostrepton derivatives.
With the goal of investigating the applicability of this method to other biologically relevant molecules containing Dha motifs, we applied this reaction for Dha-containing protein modification (Fig. 4). To investigate the reactivity towards proteins containing a Dha residue, we selected alpha-synuclein (α-syn) as a model. Considering the lack of cysteine residues in the natural sequence of wild-type α-syn, we mutated Ser129 to Cys129 and further converted it to Dha to create a potential reaction site, obtaining the α-synS129Dha variant. Initially, we modified α-synS129Dha with tetrazole 1a under conditions similar to those of peptide modification. The initial stoichiometric ratio of 1a to α-synS129Dha was set at 10:1. After stirring for 30 minutes under irradiation with 302 nm UV light, the reaction mixture was analysed via RP-HPLC and MS. However, the expected product was not obtained. By optimizing the stoichiometric ratio of 1a to α-synS129Dha at 30:1 and irradiating the reaction mixture with 302 nm UV light for 15 minutes, we obtained the pyrazoline-modified protein 9a in H2O with a yield of 63.1%. Through CD spectroscopy analysis of wild-type α-syn, α-synS129Dha and pyrazoline-modified protein 9a, we have confirmed that this chemical modification did not cause any disruption to the protein's secondary and tertiary structures (Fig. S38, Table S1†). Then we proceeded to expand the scope of tetrazoles. We synthesized tetrazole 1c labelled with biotin and tetrazole 1d labelled with fluorescent dansylcadaverine for straightforward one-step protein labelling. As shown in Fig. 4, α-synS129Dha could be efficiently and rapidly functionalized with tetrazoles 1a–d, resulting in the generation of 9a–d in moderate to high yields (53–63%) in H2O or aqueous solution containing 20% MeCN (Fig. 4).
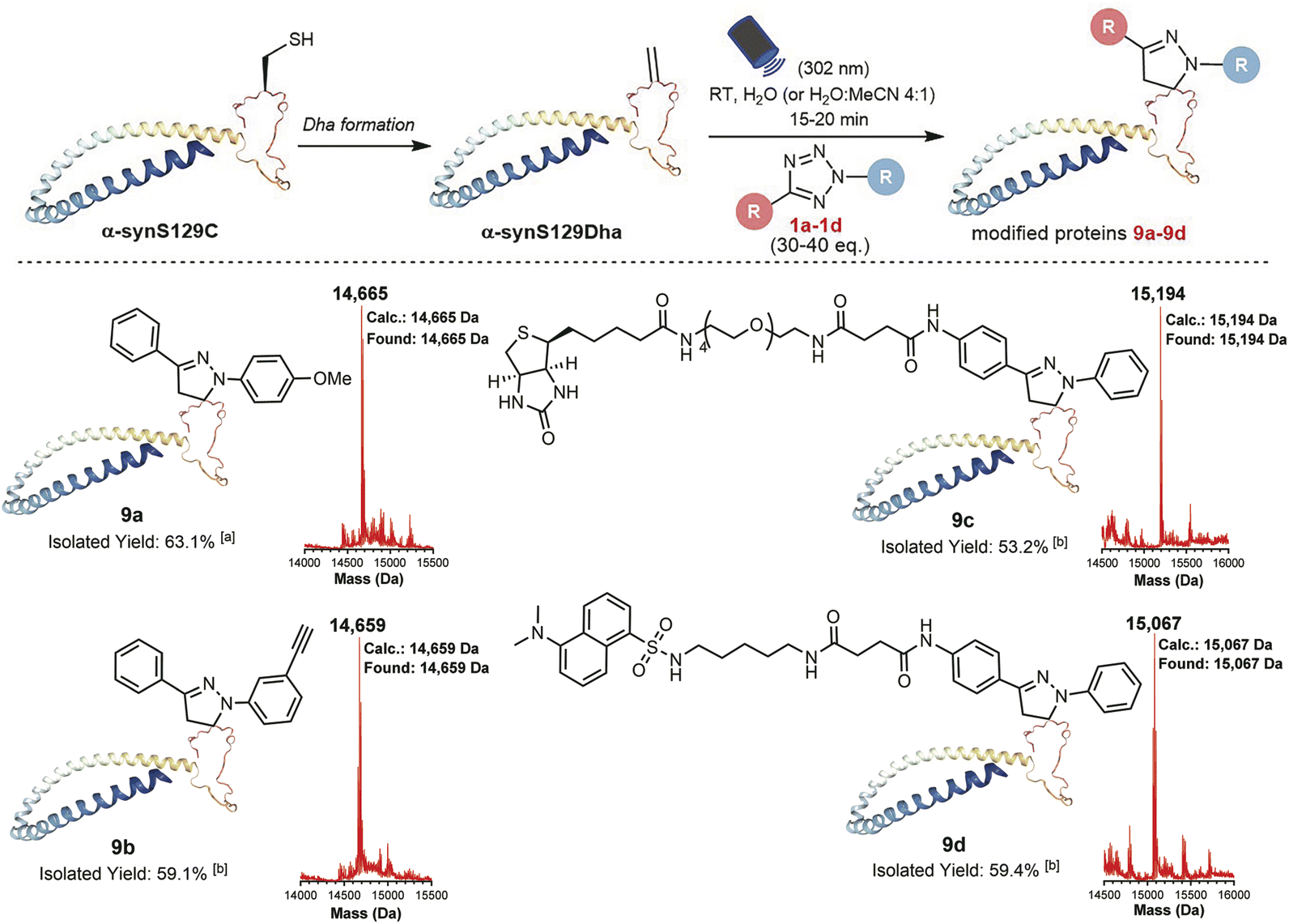 | ||
| Fig. 4 Chemoselective labelling of the α-synS129Dha protein via light-initiated 1,3-dipolar cycloaddition between Dha and tetrazoles 1a–d. The concentration of α-synS129Dha was 34.6 μM (0.5 mg mL−1) in each reaction. aThe reaction was performed in H2O. bThe reactions were conducted in the H2O–MeCN mixed solvent (4:1, v/v). | ||
The exceptional efficacy of this reaction in modifying peptides and proteins in solution prompts the broadening of its potential biological applications. The fast rate, high efficiency, mild conditions and simplicity of the reactions between Dha and tetrazoles make them highly promising for cell labeling. Furthermore, the fluorogenic properties of this reaction enables in situ imaging of cells without using additional fluorophores. Notably, the fluorescence generated in situ facilitates direct monitoring of the reaction progress to determine the effectiveness of cell labelling. Moreover, the photocontrollability of this reaction provides potential advantages in terms of time and space control. Therefore, we investigated the feasibility of in situ imaging of B16-OVA mouse melanoma cells using a light-triggered reaction between tetrazoles and cyclic RGD peptides (Fig. 5A). RGD peptides refer to a series of short peptides containing an arginine-glycine-aspartic acid sequence, which specifically bind to integrins that are overexpressed in tumour cells.44,45 Currently, there have been extensive studies on tumour cell-targeted imaging and cancer-targeted drug delivery based on the specific binding of linear RGD peptides or cyclic RGD peptides with integrin αvβ3.44–49 In our study, we chose cyclo(RGDfC) as a sensitive integrin αvβ3 receptor ligand50,51 and treated it with MeDBP to generate cyclo(RGDfDha) (10) via a bisalkylation–elimination reaction (Scheme S9†). Initially, we tested the reactions between tetrazoles (1a and 1b) and cyclic RGD peptide 10 in solution and successfully obtained modified cyclic peptides 11a with bright green fluorescence and 11b with bright blue fluorescence. The fluorescence excitation and emission spectra of both 11a and 11b were obtained via fluorescence spectral characterization (Fig. S43†).
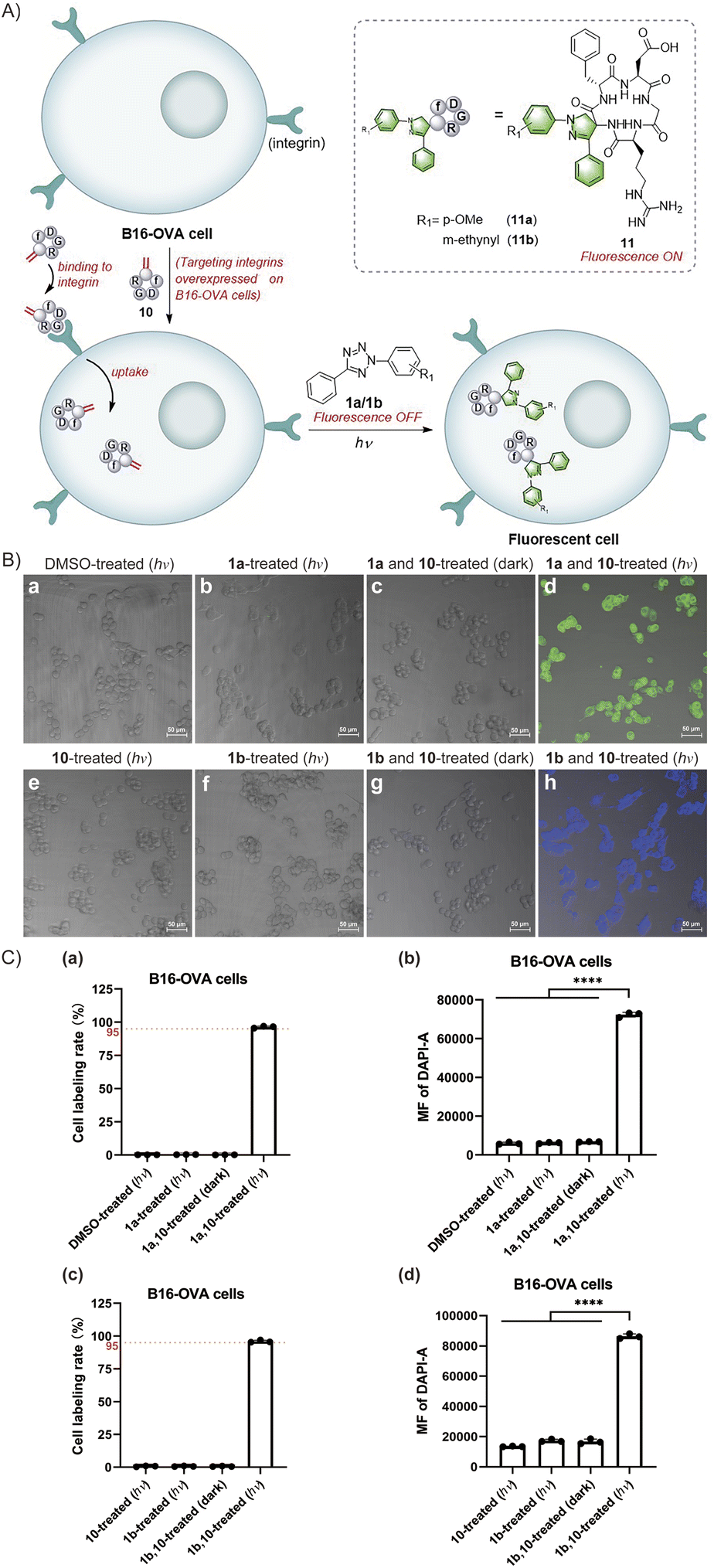 | ||
| Fig. 5 In situ imaging of tumour cells using a cyclic RGD peptide. (A) Schematic illustration. (B) Merged images of DAPI and T-PMT channels of B16-OVA cells captured using a confocal laser scanning microscope. Scale bar = 50 μm. B16-OVA cells in (a)–(h) were sequentially treated with (a) 1% DMSO (hν), (b) 160 μM tetrazole 1a (hν), (c) 160 μM 1a and 80 μM cyclic peptide 10 (dark), (d) 160 μM 1a and 80 μM 10 (hν), (e) 100 μM 10 (hν), (f) 200 μM tetrazole 1b (hν), (g) 200 μM 1b and 100 μM 10 (dark), and (h) 200 μM 1b and 100 μM 10 (hν). Among them, cells in (a), (b), (d), (e), (f), and (h) were treated with 15-min photoirradiation at 302 nm. (C) Flow cytometry analysis of B16-OVA cells. Bar graphs in (a) and (c) represent the cell labelling rate for each population. Bar graphs in (b) and (d) represent the mean fluorescence (MF) of the DAPI channel for each population. The error bars indicate the standard deviations calculated from three independent experiments. Statistical significance was determined by one-way analysis of variance (P value: ****P < 0.0001). | ||
Next, we aimed to achieve in situ imaging of B16-OVA tumour cells by targeting integrin receptors utilizing the reactions between tetrazoles and the Dha residue on cyclic peptide 10 (Fig. 5A). To begin, we evaluated the cytotoxicity of the reactants and products on B16-OVA cells using Cell Counting Kit-8 (CCK-8) and found no significant toxicity at various concentrations of tetrazoles 1a and 1b, cyclic peptide substrate 10, and modified peptides 11a and 11b (Fig. S44†). Then, we incubated the cells with cyclo(RGDfDha) (10) for 12 hours to ensure full binding of the cyclic RGD peptide to integrin receptors and subsequent cellular uptake. After washing the cells with phosphate buffered saline (PBS) to remove excess cyclic peptide 10 in the culture medium, we added tetrazole 1a or 1b to the cells and incubated them for 30 minutes, followed by a 15-minute reaction under irradiation with a 302 nm hand-held UV lamp. The cellular fluorescence was recorded using a confocal laser scanning microscope equipped with a DAPI filter. As shown in Fig. 5B(d) and (h), strong fluorescent signals were detected and effectively colocalized with the cells, and nearly all cells in the field of view were successfully labelled. In contrast, cells incubated with 10 and a tetrazole reagent (1a or 1b) without subsequent photoirradiation did not show evident fluorescence [Fig. 5B(c) and (g)]. Moreover, no significant fluorescence signals were detected in cells treated separately with tetrazole 1a and 1b and cyclic peptide 10 followed by 15 minutes of UV-light irradiation [Fig. 5B(b), (e) and (f)]. After verifying the feasibility of this reaction for cell imaging, we aimed to accurately determine the fluorescent labelling ratios of B16-OVA cells. After undergoing the same reaction process as above, the cells were analysed by flow cytometry, utilizing a 355 nm violet laser for excitation and a 450/50 bandpass filter for fluorescence detection. As shown in Fig. 5C, B16-OVA cells showed a significant increase in the population of fluorescent cells after the photoinitiated reaction compared to B16-OVA cells treated with dimethyl sulfoxide (DMSO). We demonstrated that both 1a and 1b exhibited high reaction efficiency towards cyclic peptide 10, which was able to fully bind to the integrin receptors on B16-OVA cells and undergo cellular uptake, resulting in high cellular fluorescence labelling rates of 95–100% [Fig. 5C(a) and (c)]. The bar graphs (b) and (d) in Fig. 5C indicated that the average fluorescence intensity (MF) of labelled cells was significantly enhanced compared to that of the relevant control groups. In contrast, the control groups using tetrazoles 1a and 1b and cyclic peptide 10 to incubate cells separately followed by 15-minute UV-light irradiation showed no fluorescent labelling. Similarly, cells treated with tetrazole and cyclic peptide 10 without UV-light irradiation remained unlabelled. These results were consistent with our observations via a confocal laser scanning microscope.
Conclusions
In summary, we developed a versatile method for the late-stage modification of various Dha-containing peptides and proteins using a photoinitiated 1,3-dipolar cycloaddition reaction between Dha and tetrazoles. This remarkable strategy introduces the first catalyst-free photoinitiated reaction into the existing toolbox of Dha chemical modification. During this process, we demonstrated the strategic advantages of this rapid, mild, highly selective, and photocontrollable methodology for the chemical modification of conventional peptide substrates and complex natural product peptides containing Dha moieties. The addition of an alkyne as a reaction handle on the substrates enables diverse downstream functionalizations via azide–alkyne cycloaddition. Notably, this strategy has considerable potential for expanding to the late-stage modification of other complex natural product peptides containing Dha residues. It provides a powerful tool for further structural optimization and property exploration. Moreover, we have shown the significant efficacy of this cycloaddition reaction in protein modification by utilizing diverse tetrazole reagents in aqueous solution. Additionally, by exploiting the unique fluorescence generation properties of this reaction, we have expanded the biological applications beyond those explored by previous Dha modification strategies. We utilized the reaction between a Dha-containing cyclic RGD peptide and tetrazoles to achieve in situ imaging of B16-OVA cells, providing the first application of Dha modification strategies in live cell imaging.Data availability
All experimental supporting data and procedures associated with this article have been provided in the ESI.†Author contributions
M. Z. and Y. L. conceived the idea of the research. M. Z. performed all experiments and collected the experimental data with assistance from P. H. M. Z. analyzed the results and wrote the manuscript with the guidance of Y. L. and P. H. Y. L. revised the article and supervised the project. All authors contributed to the editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0507600 and 2019YFA0904200), the National Natural Science Foundation of China (92053108 and 22237003) and the Natural Science Foundation of Shandong Province (No. ZR2022LSW014).References
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem, 2019, 3, 147–171 CrossRef CAS.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 102–112 CrossRef PubMed.
- L. H. Jones, RSC Chem. Biol., 2020, 1, 298–304 RSC.
- J.-J. Hu, P.-Y. He and Y.-M. Li, J. Pept. Sci., 2021, 27, e3286 CrossRef CAS PubMed.
- N. H. Fischer, M. T. Oliveira and F. Diness, Biomater. Sci., 2023, 11, 719–748 RSC.
- D. Siodlak, Amino Acids, 2015, 47, 1–17 CrossRef CAS PubMed.
- M. A. Ortega and W. A. van der Donk, Cell Chem. Biol., 2016, 23, 31–44 CrossRef CAS PubMed.
- H. Chen, Y. Zhang, Q.-Q. Li, Y.-F. Zhao, Y.-X. Chen and Y.-M. Li, J. Org. Chem., 2018, 83, 7528–7533 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Nagano, Y. Goto and H. Suga, J. Am. Chem. Soc., 2021, 143, 13358–13369 CrossRef CAS PubMed.
- J. Dadova, S. R. G. Galan and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef CAS PubMed.
- H. Li, H. Xu, Y. Zhou, J. Zhang, C. Long, S. Li, S. Chen, J.-M. Zhou and F. Shao, Science, 2007, 315, 1000–1003 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernandez-Gonzalez, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- J. W. Bogart and A. A. Bowers, Org. Biomol. Chem., 2019, 17, 3653–3669 RSC.
- M. Q. Zhang, P. Y. He and Y. M. Li, Chem. Res. Chin. Univ., 2021, 37, 1044–1054 CrossRef CAS.
- A. M. Freedy, M. J. Matos, O. Boutureira, F. Corzana, A. Guerreiro, P. Akkapeddi, V. J. Somovilla, T. Rodrigues, K. Nicholls, B. Xie, G. Jimenez-Oses, K. M. Brindle, A. A. Neves and G. J. L. Bernardes, J. Am. Chem. Soc., 2017, 139, 18365–18375 CrossRef CAS PubMed.
- A. Yang, S. Ha, J. Ahn, R. Kim, S. Kim, Y. Lee, J. Kim, D. Soell, H.-Y. Lee and H.-S. Park, Science, 2016, 354, 623–626 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallee, A. Phanumartwiwath, O. D. Coleman, M.-L. Thezenas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, 597 CrossRef CAS PubMed.
- G. J. Bao, P. Wang, G. F. Li, C. J. Yu, Y. P. Li, Y. Y. Liu, Z. Y. He, T. T. Zhao, J. Rao, J. Q. Xie, L. Hong, W. S. Sun and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 5331–5338 CrossRef CAS PubMed.
- R. H. de Vries, J. H. Viel, R. Oudshoorn, O. P. Kuipers and G. Roelfes, Chem. –Eur. J., 2019, 25, 12698–12702 CrossRef CAS PubMed.
- L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418–14420 CrossRef CAS PubMed.
- S. R. G. Galan, J. R. Wickens, J. Dadova, W.-L. Ng, X. Zhang, R. A. Simion, R. Quinlan, E. Pires, R. S. Paton, S. Caddick, V. Chudasama and B. G. Davis, Nat. Chem. Biol., 2018, 14, 955–963 CrossRef CAS PubMed.
- P.-Y. He, H. Chen, H.-G. Hu, J.-J. Hu, Y.-J. Lim and Y.-M. Li, Chem. Commun., 2020, 56, 12632–12635 RSC.
- R. H. de Vries, J. H. Viel, O. P. Kuipers and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 3946–3950 CrossRef CAS PubMed.
- H. K. Jiang, P. Kurkute, C. L. Li, Y. H. Wang, P. J. Chen, S. Y. Lin and Y. S. Wang, Biochemistry, 2020, 59, 3796–3801 CrossRef CAS PubMed.
- K. C. Tang, S. M. Maddox, K. M. Backus and M. Raj, Chem. Sci., 2022, 13, 763–774 RSC.
- J.-A. Shin, J. Kim, H. Lee, S. Ha and H.-Y. Lee, J. Org. Chem., 2019, 84, 4558–4565 CrossRef CAS PubMed.
- T. Rossolini, B. Ferko and D. J. Dixon, Org. Lett., 2019, 21, 6668–6673 CrossRef CAS PubMed.
- A. A. Shah, M. J. Kelly and J. J. Perkins, Org. Lett., 2020, 22, 2196–2200 CrossRef CAS PubMed.
- B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Nature, 2020, 585, 530–537 CrossRef CAS PubMed.
- R. C. W. van Lier, A. D. de Bruijn and G. Roelfes, Chem. –Eur. J., 2021, 27, 1430–1437 CrossRef CAS PubMed.
- X. X. Qi, S. Jambu, Y. N. Ji, K. M. Belyk, N. R. Panigrahi, P. S. Arora, N. A. Strotman and T. N. Diao, Angew. Chem., Int. Ed., 2022, 61, e20221331 Search PubMed.
- X. Peng, K. Xu, Q. Zhang, L. Liu and J. Tan, Trends Chem., 2022, 4, 643–657 CrossRef CAS.
- M. R. Aronoff, B. Gold and R. T. Raines, Org. Lett., 2016, 18, 1538–1541 CrossRef CAS PubMed.
- J. Dadová, K.-J. Wu, P. G. Isenegger, J. C. Errey, G. J. L. Bernardes, J. M. Chalker, L. Raich, C. Rovira and B. G. Davis, ACS Cent. Sci., 2017, 3, 1168–1173 CrossRef PubMed.
- A. Phanumartwiwath, C. Kesornpun, D. Chokchaichamnankit, A. Khongmanee, P. Diskul-Na-Ayudthaya, T. Ruangjaroon, C. Srisomsap, P. Kittakoop, J. Svasti and S. Ruchirawat, Chembiochem, 2023, e202300268 CrossRef PubMed.
- J. S. Clovis, A. Eckell, R. Huisgen and R. Sustmann, Chem. Ber., 1967, 100, 60–70 CrossRef CAS.
- Y. Z. Wang, C. I. R. Vera and Q. Lin, Org. Lett., 2007, 9, 4155–4158 CrossRef CAS PubMed.
- X. Just-Baringo, F. Albericio and M. Alvarez, Mar. Drugs, 2014, 12, 317–351 CrossRef CAS PubMed.
- H. M. Key and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 15460–15466 CrossRef CAS PubMed.
- R. J. Scamp, E. DeRamon, E. K. Paulson, S. J. Miller and J. A. Ellman, Angew. Chem., Int. Ed., 2020, 59, 890–895 CrossRef CAS PubMed.
- T. Brandhofer and O. G. Mancheno, ChemCatChem, 2019, 11, 3797–3801 CrossRef CAS.
- R. H. de Vries and G. Roelfes, Chem. Commun., 2020, 56, 11058–11061 RSC.
- R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–839 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Preat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS PubMed.
- M. Nieberler, U. Reuning, F. Reichart, J. Notni, H. J. Wester, M. Schwaiger, M. Weinmuller, A. Rader, K. Steiger and H. Kessler, Cancers, 2017, 9, 116 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- Y. J. Chen, L. X. Jia, G. L. Zhu, W. Wang, M. Geng, H. X. Lu, Y. Zhang, M. H. Zhou, F. Y. Zhang and X. Z. Cheng, Bioorganic Med. Chem. Lett., 2022, 73, 128888 CrossRef CAS PubMed.
- S. Asati, V. Pandey and V. Soni, Int. J. Pept. Res. Ther., 2019, 25, 49–65 CrossRef CAS.
- M. D. Wang, G. T. Lv, H. W. An, N. Y. Zhang and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e20211364 Search PubMed.
- R. Hennig, S. Kuespert, A. Haunberger, A. Goepferich and R. Fuchshofer, J. Drug Targeting, 2016, 24, 952–959 CrossRef CAS PubMed.
- N. Raval, H. Jogi, P. Gondaliya, K. Kalia and R. K. Tekade, Mol. Pharm., 2021, 18, 641–666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02818f |
| This journal is © The Royal Society of Chemistry 2023 |