
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective carbonylation of arenes via C(sp2)–H thianthrenation: direct access to 1,2-diarylethanones†‡
Jiajun
Zhang
ab,
Le-Cheng
Wang
ab,
Zhi-Peng
Bao
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
First published on 16th June 2023
Abstract
Herein, a new reaction for the site-selective carbonylation of arenes via C(sp2)–H thianthrenation under mild conditions has been developed. With low loadings of palladium catalysts, various desired 1,2-diarylethanones are produced in good yields. This strategy also enables the late-stage modification of complex molecules, which was previously challenging with similar carbonylative Negishi-type reactions.
Introduction
The activation and direct functionalization of inert bonds have always been a hot topic pursued by chemists,1 especially the C–H bond activation of aromatic hydrocarbons which can directly modify natural products and also macromolecules at the late stage. Among the known transformations, Friedel–Crafts acylation is a classic reaction for the C–H bond functionalization of arenes (Scheme 1a).2 However, its related low regioselectivity and over-acylation often cause synthetic problems. The halogenation of arenes to give aryl halides has been proven to be effective,3 but the synthesis of complex aromatic halides often involves site-selectivity and tolerance issues (Scheme 1a).4 On the other hand, C(sp2)–H thianthrenation has been recognized as an important means for the direct functionalization of arenes. Its mild reaction conditions and high regioselectivity are considered as the greatest advantages.5 Hence, aryl thianthrenium salts have been successfully used to construct C–C, C–B, C–N, and C–Si bonds, etc.5,6 More recently, the advantage of aryl thianthrenium salts has been applied by Cornella, Ritter and their co-workers in nickel-catalyzed synthesis of aryl halides from aryl thianthrenium salts.7 This protocol can produce aryl halides that would not readily be available by direct halogenation of arenes.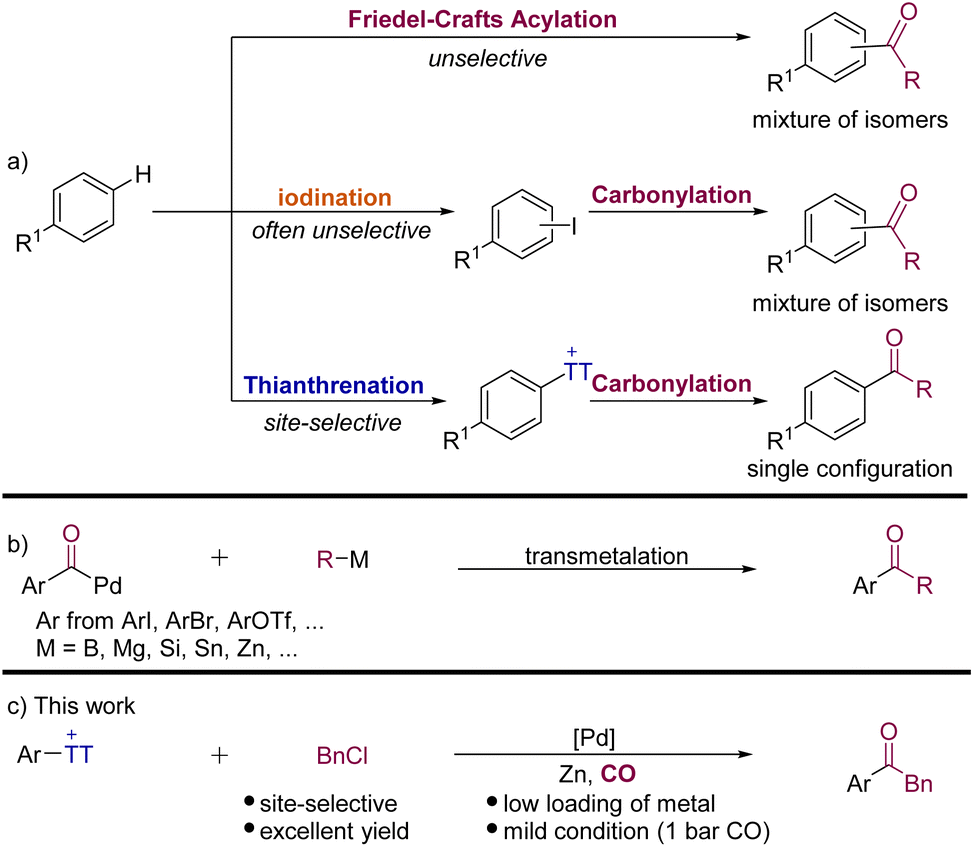 | ||
| Scheme 1 (a) Conceptual representation of various strategies for undirected C–H acylation of arenes; (b) palladium-catalyzed carbonylative coupling reactions; (c) this work: Palladium-catalyzed carbonylative Negishi-type coupling of aryl thianthrenium salts with benzyl chlorides. | ||
Transition-metal-catalyzed carbonylative cross-coupling reactions that form C–C bonds have become a powerful platform to synthesize ketones and their derivatives.8 In recent years, many palladium-catalyzed carbonylative coupling reactions for constructing C–C bonds have been established. According to different carbon nucleophilic reagents, the reactions can be divided into Suzuki (B),9 Kumada (Mg),10 Hiyama (Si),11 Stille (Sn),12 and Negishi (Zn) reactions.8,13 These reactions have been quite well developed, which provide convenient pathways for constructing ketones.
Although the carbonylative Negishi-type reaction is among the most efficient methods to obtain ketones from aryl halides, one of the long-standing challenges is competitive direct coupling reaction and also the availability and stability of substrates limited the applications of this carbonylative cross-coupling reaction. In order to overcome these limitations, alternative methods have gradually been discovered, such as the direct C–H functionalization reaction of arenes14 and alternative organometallic reagents.8 Rueping and Zhu,15 Gong,16 Hu,17 Lian,18 and others19 have achieved considerable progress in the field of carbonylative Negishi-type reactions, independently. However, the required organic halides are often not available and generally cannot be accessed in high selectivity from complex arenes. This problem can be perfectly solved by using aryl thianthrenium salts!
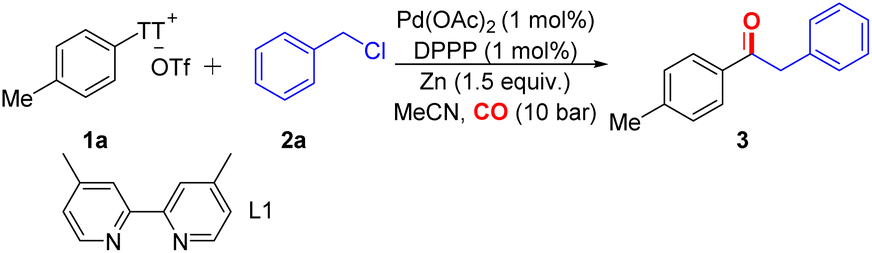
We investigated the carbonylative reaction of aryl thianthrenium salt TT-1a with benzyl chloride in the presence of a palladium catalyst and a metal agent (Table 1). Zinc was found to be crucial for the reaction, and other reagents such as manganese and iron did not produce any desired carbonylation product, which is consistent with the involvement of an organozinc intermediate (Table 1, entries 2 and 3). A preference for polar solvents such as MeCN was observed as larger amounts of unreacted initial materials remained when the reaction was conducted in less polar solvents, such as toluene (Table 1, entry 4), which could be explained by the faster rate of oxidative addition of zinc into benzyl chlorides in polar solvents.20 While nickel is the preferred transition metal for reductive aryl–alkyl bond formations,21 we identified palladium to be the catalyst of choice for this carbonylation of aryl thianthrenium salts. Various palladium catalysts have good effects on the reaction (see the ESI, Table S1,‡ entries 7–9). A series of bulky phosphine ligands were tested (see the ESI, Table S1,‡ entries 10–16). Due to the high reactivity of aryl thianthrenium salts, we attempted to use other inexpensive catalysts for the reaction, such as nickel and copper. Disappointingly, the target product was not detected (Table 1, entries 9 and 10). Surprisingly, when the pressure of carbon monoxide was decreased to 1 bar, the reaction still proceeded efficiently (Table 1, entry 11), which is still a challenge in transition metal-catalyzed carbonylation reactions.
|
|
||
|---|---|---|
| Entrya | Variation from standard conditions | Yieldb |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), CO (10 bar), Pd(OAc)2 (1 mol%), DPPP (1 mol%), Zn (0.3 mmol) in MeCN (2 mL) at 80 °C for 20 h. b Yields were determined by GC-FID analysis using n-dodecane as the internal standard. c Yield of the isolated product. n.o. = not observed. | ||
| 1 | None | 99%c (97%) |
| 2 | Mn instead of Zn | n.o. |
| 3 | Fe instead of Zn | n.o. |
| 4 | Toluene instead of MeCN | <5% |
| 5 | No catalyst | n.o. |
| 6 | No ligand | Trace |
| 7 | Pd(TFA)2 instead of Pd(OAc)2 | 94% |
| 8 | Xantphos instead of DPPP | 31% |
| 9 | Ni(acac)2 + L1 as catalyst | n.o. |
| 10 | CuI + L1 as catalyst | n.o. |
| 11 | 1 bar CO | 91% |
Different alkyl halides were tested under the optimal conditions in the presence of zinc powder (Scheme 2). Benzyl bromide can achieve an isolated yield of 91%. At the same time, some leaving groups, such as OTs and OTf, were also checked. The results showed that when the leaving group was replaced, the reaction could not proceed effectively, even in the presence of an additional amount of NaI or NaBr. Surprisingly, when using long-chain alkyl iodide, we can still detect the target product on GC-MS (see the ESI,‡ pp S8 for details). The lack of reactivity of alkyl chloride was also expected.
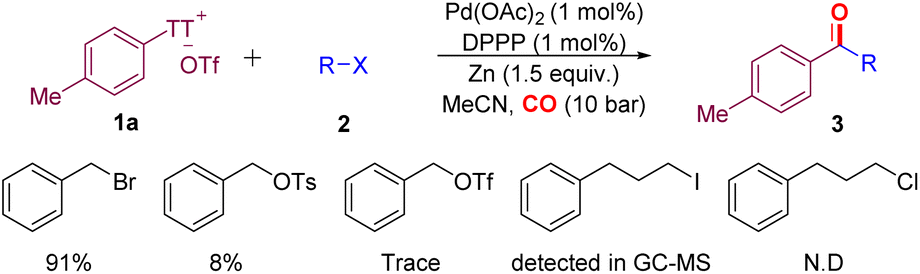 | ||
| Scheme 2 Testing of alkyl (pseudo)halides. | ||
With the optimized conditions in hand, we evaluated the scope of aryl thianthrenium salts by reacting them with benzyl chloride (Scheme 3). This method shows excellent compatibility with a broad range of aryl thianthrenium salts, and the corresponding products were isolated in moderate to excellent yields. Aryl thianthrenium salts bearing electron-neutral substituents (Scheme 3, 1-2) and monosubstituted groups including methyl, tert-butyl, ethyl, methoxy, iodo-, and chloro- (Scheme 3, 1-1, 1-3–1-7) furnished the corresponding products with moderate to good yields. Aryl thianthrenium salts bearing phenoxy or thiophenyl substituents (Scheme 3, 1-8, 1-9) reacted well and efficiently in this palladium-catalyzed carbonylation protocol. Various disubstituted substrates (Scheme 3, 1-10–1-15) also underwent the targeted carbonylative transformation successfully and gave the corresponding products in moderate to good yields. Functional groups such as cyano and acetyl groups were also compatible under the optimized conditions, and the corresponding products were produced without any problem (Scheme 3, 1-14, 1-15). The substrate based on benzofuran can gave the targeted product in excellent isolated yield as well (Scheme 3, 1-16). To further illustrate the utility of this carbonylation strategy, we sought to integrate the carbonylation approach into the diversification of complex molecules; carbonyls were subsequently installed into various pharmaceutically relevant heterocyclic substrates containing commercial natural estrogen (Scheme 3, 1-18, from estrone), commercial drug derivatives (Scheme 3, 1-17, from Gemfibrozil, Scheme 3, 1-19, from ibuprofen), natural products (Scheme 3, 1-20, from cholesterol), and others (Scheme 3, 1-21–1-23) in reasonably good to excellent yields.
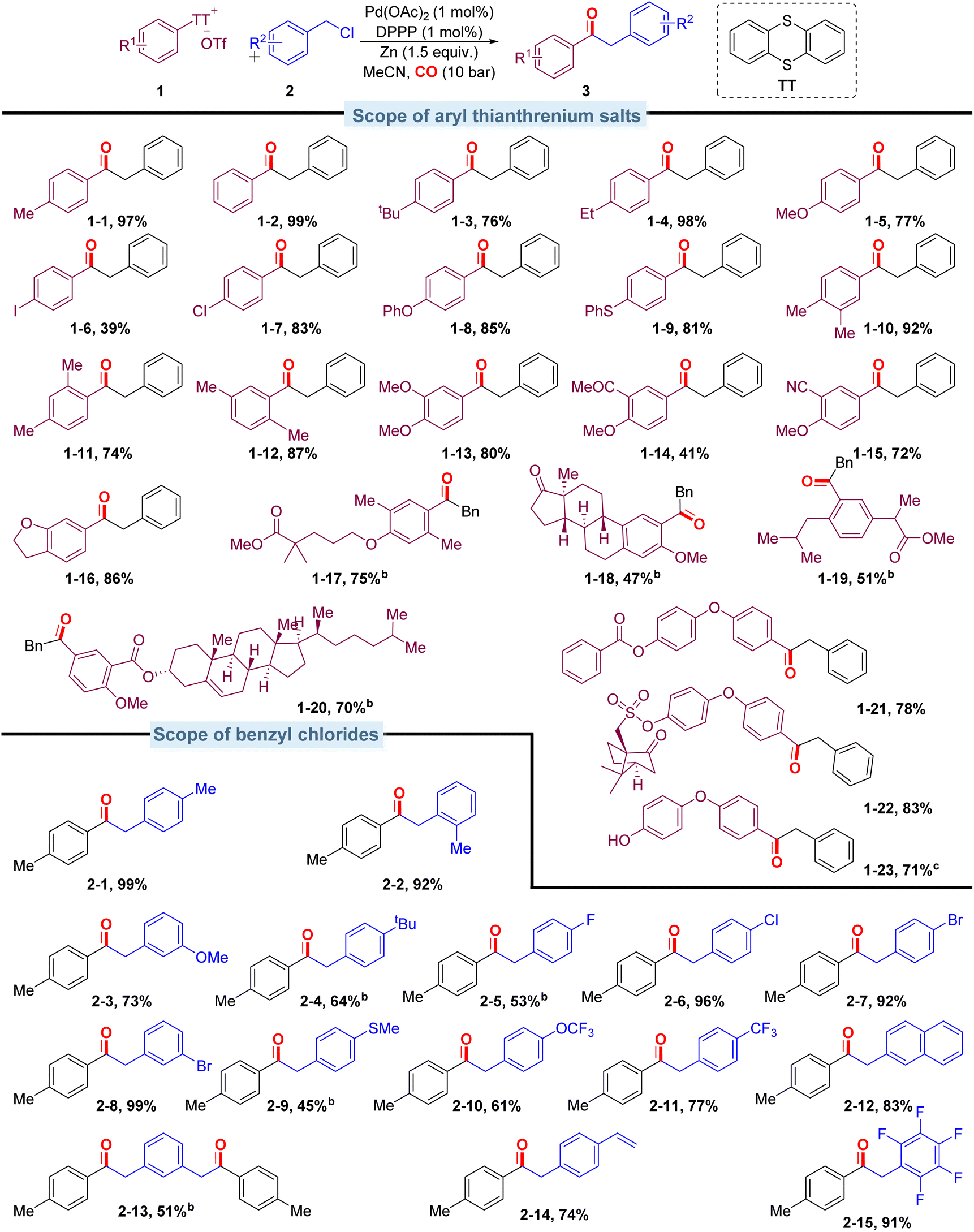 | ||
| Scheme 3 Scope of different aryl thianthrenium salts and benzyl chlorides. aReaction conditions: aryl thianthrenium salts (0.2 mmol), benzyl chlorides (0.3 mmol), Pd(OAc)2 (1 mol%), DPPP (1 mol%), Zinc powder (0.3 mmol), MeCN (2 mL), CO (10 bar), 80 °C, 20 h, isolated yields. b48 h. cWith 5-(4-(4-((6-methylpicolinoyl)oxy)phenoxy)phenyl)-5H-thianthren-5-ium as the substrate. | ||
The carbonylation protocol also displayed good compatibility across a range of benzyl chlorides. As shown in Scheme 3, when 4-methylbenzyl chloride was introduced, the product (Scheme 3, 2-1) was isolated in 99% yield. Ortho-methyl-substituted benzyl chloride gave the corresponding ketone (Scheme 3, 2-2) in good yield. Both electron-donating and electron-withdrawing substituted benzyl chlorides were successfully transformed into the desired ketones in good yields (45–99%; Scheme 3, 2-3–2-11). The reason for the lower yield of benzyl chloride substituted with methylthio- and tert-butyl is that the substrate has not been completely converted. Extending the reaction time can improve the yield, and the direct coupling products do not increase due to the extension of time. Additionally, the naphthalene motif could also be successfully incorporated into the desired products with excellent efficiency (Scheme 3, 2-12). In particular, 1,3-bis(chloroethyl)benzene modified with dual reaction sites is also compatible, and the yield was up to 51%. As the reactions proceed under mild conditions, sensitive functionalities alkene-substituent and penta-fluoro-substituent are all tolerated (Scheme 3, 2-14–2-15).
Based on these results, the following reaction mechanism is proposed in Scheme 4. We believe an oxidative addition-transmetalation-reductive elimination sequence is operative in this transformation. In principle, the active palladium (0) catalyst, as well as the zinc species, might undergo addition to the aryl thianthrenium salts and the benzyl chloride. Although the reduction potential of benzyl chloride is lower than that of aryl thianthrenium salts, that will lead to the reduction of aryl thianthrenium salts more easily. According to literature research, the oxidation addition of zinc is carried out through an inner sphere electron transfer involving a bridging ligand.22 Hence, we conclude that the desired product 1 is mainly produced via the oxidative addition transmetalation-reductive elimination sequence. After oxidative addition of palladium(0) to aryl thianthrenium salts, the insertion of carbon monoxide takes place to give the acylpalladium complex. Subsequent reaction with the in situ formed benzylic zinc reagent provides 1,2-diphenylmethane after reductive elimination. Byproduct 2, which resulted primarily from Int-I undergoes transmetalation directly with the in situ generated organic zinc reagent.
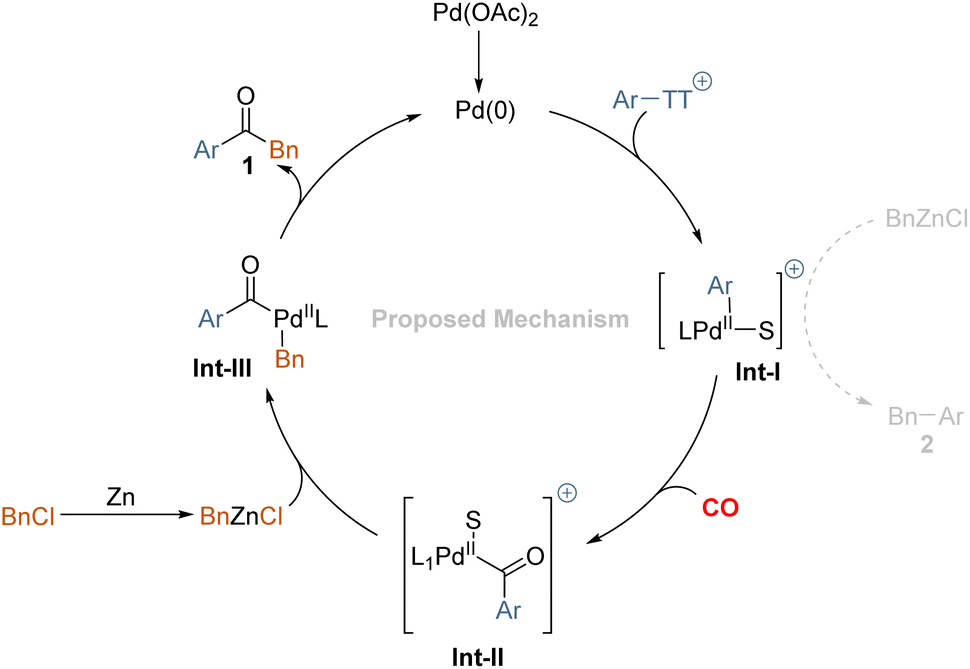 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a site-selective carbonylation method for carbonylation of aryl thianthrenium salts, which allows the production of 1,2-diarylethanones that cannot be obtained with similar efficiency by other carbonylation methods. The excellent site-selectivity and robust reactivity enable us to engage complex fragments, which could be of value in medicinal chemistry. We believe this work represents a valuable conceptual extension to existing carbonylation cross-coupling reactions with improved efficiency, reactivity, and synthetic utility.Data availability
All data supporting the findings of this study are available within the article and its ESI file.‡Author contributions
X.-F. W. conceived this project. J. Z., L.-C. W. and Z.-P. B. performed all the experiments and prepared the ESI.‡ X.-F. W. and J. Z. wrote and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from DICP and K. C. Wong Education Foundation (GJTD-2020-08).Notes and references
- (a) W. L. Pein, E. M. Wiensch and J. Montgomery, Org. Lett., 2021, 23, 4588–4592 CrossRef CAS PubMed; (b) F.-F. Tan, X.-Y. He, W. F. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS PubMed; (c) T. Shimasaki, M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2010, 49, 2929–2932 CrossRef CAS PubMed; (d) E. M. Wienschand and J. Montgomery, Angew. Chem., Int. Ed., 2018, 57, 11045–11049 CrossRef PubMed; (e) N. Ahmed, R. J. Spears, T. D. Sheppard and V. Chudasama, Chem. Sci., 2022, 13, 8626–8633 RSC; (f) C. B. Tripathi, T. Ohtani, M. T. Corbett and T. Ooi, Chem. Sci., 2017, 8, 5622–5627 RSC; (g) J. Li, S. Warratz, D. Zell, S. D. Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed; (h) C. H. Basch, J. Liao, J. Y. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS PubMed; (i) P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285 CrossRef CAS PubMed.
- C. Friedel and J. Crafts, J. Chem. Soc., 1877, 32, 725–791 RSC.
- (a) A. C. Wotal, R. D. Ribson and D. J. Weix, Organometallics, 2014, 33, 5874–5881 CrossRef CAS PubMed; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- (a) R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed; (b) H. Firouzabadi, N. Iranpoor and M. Shiri, Tetrahedron Lett., 2003, 44, 8781–8785 CrossRef CAS; (c) G. K. Surya Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770–15776 CrossRef PubMed; (d) K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Lett., 2003, 32, 932–933 CrossRef CAS.
- (a) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed; (b) M. Meng, M.-S. Liu and W. Shu, Chem. Sci., 2022, 13, 13690–13707 RSC.
- (a) X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058–5061 RSC; (b) X.-X. Nie, Y.-H. Huang and P. Wang, Org. Lett., 2020, 22, 7716–7720 CrossRef CAS PubMed; (c) Y. Wu, Y.-H. Huang, X.-Y. Chen and P. Wang, Org. Lett., 2020, 22, 6657–6661 CrossRef CAS PubMed; (d) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909–7914 CrossRef CAS PubMed; (e) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 10477–10478 CrossRef CAS PubMed; (f) Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386–2391 CrossRef CAS PubMed; (g) M. N. Wang, X. M. Zhang, M. T. Ma and B. L. Zhao, Org. Lett., 2022, 24, 031–6036 Search PubMed; (h) B. L. Zhao, Q. Z. Wang, T. X. Zhu, B. Feng and M. T. Ma, Org. Lett., 2022, 24, 5608–5613 CrossRef CAS PubMed; (i) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed; (j) J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56–62 CrossRef CAS PubMed; (k) X.-Y. Chen, Y.-H. Huang, J. Zhou and P. Wang, Chin. J. Chem., 2020, 38, 1269–1272 CrossRef CAS; (l) L. Liang, H.-Y. Niu, R.-L. Li, Y.-F. Wang, J.-K. Yan, C.-G. Li and H.-M. Guo, Org. Lett., 2020, 22, 6842–6846 CrossRef CAS PubMed; (m) E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609–13613 CrossRef CAS PubMed; (n) Y. Zhao, C. Yu, W. Liang and F. W. Patureau, Org. Lett., 2021, 23, 6232–6236 CrossRef CAS PubMed; (o) M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- S. Y. Ni, J. Y. Yan, S. Tewari, E. J. Reijerse, T. Ritter and J. Cornella, J. Am. Chem. Soc., 2023, 145, 9988–9993 CrossRef CAS PubMed.
- (a) X.-F. Wu, J. Schranck, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 40–44 CrossRef CAS PubMed; (b) X.-F. Wu, H. Neumann and M. Beller, Adv. Synth. Catal., 2011, 353, 788–792 CrossRef CAS.
- N. E. Leadbeater, Chem. Commun., 2005, 23, 2881–2902 RSC.
- (a) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed; (b) S. Schrçter, C. Stock and T. Bach, Tetrahedron, 2005, 61, 2245–2267 CrossRef; (c) A. C. Hillier, G. A. Grasa, M. S. Viciu, H. M. Lee, C. Yang and S. P. Nolan, J. Organomet. Chem., 2002, 653, 69–82 CrossRef CAS.
- (a) M. Inoue, T. Suzuki, A. Kinoshita and M. Nakada, Chem. Rec., 2008, 8, 169–181 CrossRef CAS PubMed; (b) S. A. Testero and E. G. Mata, J. Comb. Chem., 2008, 10, 487–497 CrossRef CAS PubMed.
- (a) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 116, 4808–4839 ( Angew. Chem., Int. Ed. , 2004 , 43 , 4704–4734 ) Search PubMed; (b) G. Pattenden and D. J. Sinclair, J. Organomet. Chem., 2002, 653, 261–268 CrossRef CAS; (c) M. Kosugi and K. Fugami, J. Organomet. Chem., 2002, 653, 50–53 CrossRef CAS.
- (a) V. B. Phapale and D. J. Cardenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC; (b) T. Ren, Chem. Rev., 2008, 108, 4185–4207 CrossRef CAS PubMed.
- A. Haito, M. Yamaguchi and N. Chatani, Asian J. Org. Chem., 2018, 7, 1315–1318 CrossRef CAS.
- H. F. Chen, H. F. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2022, 61, e202204144 CAS.
- X. L. Yu, T. Yang, S. L. Wang, H. L. Xu and H. G. Gong, Org. Lett., 2011, 13, 2138–2141 CrossRef CAS PubMed.
- R. Y. Shi and X. L. Hu, Angew. Chem., Int. Ed., 2019, 58, 7454–7458 CrossRef CAS PubMed.
- G. Chen, R. X. Zhou, X. M. Zhang, X. Xiao, S. Kramer, G.-J. Cheng and Z. Lian, ACS Catal., 2022, 12, 14582–14591 CrossRef CAS.
- (a) A. Rérat, C. Michon, F. Agbossou-Niedercorn and C. Gosmin, Eur. J. Org. Chem., 2016, 4554–4560 CrossRef; (b) T. Z. Lin, P. C. Qian, Y.-E. Wang, M. J. Ou, N. Cui, Y. Ye, R. Hua, D. Xiong, F. Xue, P. J. Walsh and J. Y. Mao, Asian J. Org. Chem., 2022, 11, e2022002 Search PubMed.
- (a) T. N. Majid and P. Knochel, Tetrahedron Lett., 1990, 31, 4413–4416 CrossRef CAS; (b) M. Amano, A. Saiga, R. Ikegami, T. Ogata and K. Takagi, Tetrahedron Lett., 1998, 39, 8667–8668 CrossRef CAS; (c) C. Jubert and P. Knochel, J. Org. Chem., 1992, 57, 5425–5431 CrossRef CAS.
- (a) P. Zhang, C. C. Le and D. W. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (b) H. A. Sakai, W. Liu, C. C. Le and D. W. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed; (c) J. Liu and H. Gong, Org. Lett., 2018, 20, 7991–7995 CrossRef CAS PubMed; (d) S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352–3355 CrossRef CAS PubMed; (e) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed; (f) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490–14497 CrossRef CAS PubMed.
- A. Guijarro, D. M. Rosenberg and R. D. Rieke, J. Am. Chem. Soc., 1999, 121, 4155–4167 CrossRef CAS.
Footnotes |
| † Dedicated to the 130th Anniversary of Wuhan University. |
| ‡ Electronic supplementary information (ESI) available: General comments, general procedure, analytic data, and NMR spectra. See DOI: https://doi.org/10.1039/d3sc02402d |
| This journal is © The Royal Society of Chemistry 2023 |