
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFragile intermediate identification and reactivity elucidation in electrochemical oxidative α-C(sp3)–H functionalization of tertiary amines†
Kailun
Liang‡
a,
Dongmei
Zhang‡
c,
Yanming
Su
a,
Lijun
Lu
a,
Jun
Hu
e,
Yi-Hung
Chen
*a,
Xinxing
Zhang
 *c,
Aiwen
Lei
*abd and
Hong
Yi
*a
*c,
Aiwen
Lei
*abd and
Hong
Yi
*a
aThe Institute for Advanced Studies (IAS), Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: hong.yi@whu.edu.cn; aiwenlei@whu.edu.cn; yihungchen@whu.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China
cCollege of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center (ReCAST), Tianjin Key Laboratory of Biosensing and Molecular Recognition, Shenzhen Research Institute, Frontiers Science Center for New Organic Matter, Nankai University, Tianjin, 300071, China. E-mail: zhangxx@nankai.edu.cn
dNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang, Jiangxi 330022, P. R. China
eSchool of Life Sciences and Health Engineering, Jiangnan University, Wuxi 214122, China
First published on 18th March 2023
Abstract
The direct α-C(sp3)–H functionalization of widely available tertiary amines holds promise for the rapid construction of complex amine architectures. The activation of C(sp3)–H bonds through electron transfer and proton transfer by oxidants, photoredox catalysis and electrochemical oxidation have received wide attention recently. In these reactions, the direct capture and identification of the key reactive radical intermediates are technically difficult due to their short life-time. Herein, an online electrochemical mass spectrometry (MS) methodology was utilized to probe the short-lived intermediates in the electrochemical oxidative α-C(sp3)–H functionalization of tertiary amines. The resulting electrochemical oxidation intermediates, α-amino radical cation and iminium cation were successfully detected. Further, the α-amino C(sp3) radical added to the double bond of a phenyl trans-styryl sulfone, yielding another C(sp3) radical that leads to the final vinylation. Based on the mass spectrometric elucidation of the reactivity of the α-amino radical, a scale-up electrochemical radical vinylation methodology was established, with which a large variety of allylic amines with broad functional group tolerance were synthesized.
Amine-containing compounds constitute important structural motifs of pharmaceuticals and natural products.1 Direct activation of α-C(sp3)–H bonds is an efficient way to functionalize tertiary amines. Transition metal catalysts have been used for α-C(sp3)–H functionalization of tertiary amines, in which the C(sp3)-metal species are the key intermediates for the transformations (Fig. 1A).2 Hydrogen atom transfer (HAT) is another way to functionalize tertiary amines by generating α-amino C(sp3) radicals through a direct hydrogen abstraction process.3 Halogen-,4 nitrogen-,5 and oxygen-centered radicals6 generated from HAT reagents can promote the HAT process of an inert C(sp3)–H bond on tertiary amines, which leads to further functionalization (Fig. 1B). Electrosynthesis has emerged as a broadly applicable redox tool,7 offering a mild way to drive chemical reactions without the requirement of stoichiometric oxidants. In this case, tertiary amines can be oxidized to nitrogen radical cations via a single electron transfer (SET) process, followed by deprotonation, yielding α-amino C(sp3) radicals (Fig. 1C).8 Moreover, the α-amino carbon radical can be further oxidized to an iminium cation, a reactive intermediate often involved in cross-dehydrogenative coupling reactions.9 Although the functionalization of tertiary amines has been rapidly developed, the direct capture of the intermediate radicals is scarce due to their short lifetime, and hence, the development of online methodologies that can identify short-lived intermediates is needed to deepen our understanding of these elusive intermediates.
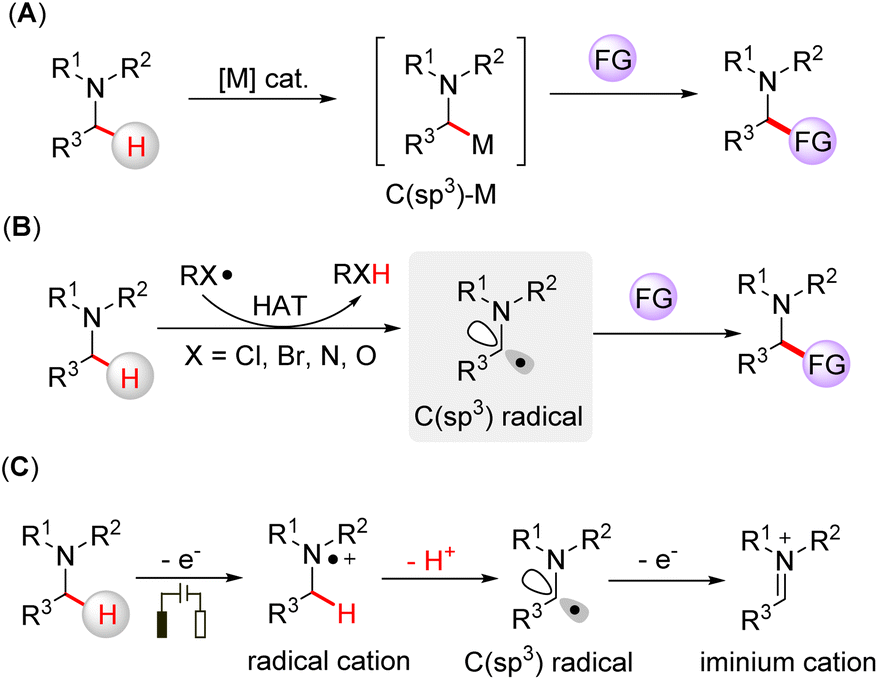 | ||
| Fig. 1 Different strategies for α-C(sp3)–H functionalization of tertiary amines. (A) Transition-metal catalyzed functionalization of tertiary amines via C(sp3)-metal intermediates; (B) functionalization of tertiary amines via a hydrogen atom transfer (HAT) process; (C) electrochemical oxidative functionalization of tertiary amines via an electrochemical SET process. FG = functional group. | ||
Among the various methods for probing the complex intermediates in electrochemical reactions, mass spectrometry stands out with remarkable advantages of fast screening speed and high sensitivity for capturing and identifying short-lived radical species. Recently, online electrochemical mass spectrometry (EC/MS) with variable ionization techniques such as differential electrochemical mass spectrometry (DEMS),10 electrospray ionization (ESI),11 desorption electrospray ionization (DESI),12 thermospray ionization,13 and nanospray desorption electrospray ionization (Nano-DESI)14 has made great progress in the mechanistic studies of electrochemical reactions. For example, Zare15 and co-workers developed a “waterwheel” working electrode, which allowed the newly electrogenerated species to be quickly desorbed with DESI and transferred and captured. Xu16 and co-workers captured several long-sought elusive intermediates by mass spectrometry using a wireless carbon bipolar electrode (BPE) deposited into the spray tip of a nanopipette.
In this study, we utilized the BPE method combined with nano-electrospray ionization mass spectrometry to investigate the short-lived reactive intermediates during the electrochemical oxidation and functionalization of the α-C(sp3)–H on tertiary amines. The α-amino radical cation and iminium cation were directly captured and identified. The neutral α-amino C(sp3) radical was also evidenced through its addition to a phenyl trans-styryl sulfone. Based on the fast screening and mechanistic elucidation of the α-vinylation reactions by mass spectrometry, scale-up synthesis of a large variety of allylic amines in undivided electrochemical cells was achieved.
Initially, we selected 1-phenylpyrrolidine as the substrate to study the reactive radical species generated by electrochemical oxidation using BPE combined with nano-electrospray ionization. Briefly, a Cu wire (0.2 mm i.d.) was inserted into a quartz capillary from its rear end and then connected to a high-voltage power supply. The BPE took advantage of the high voltage to trigger electrospray ionization and to induce potential difference and thus redox reactions at the two ends of the deposited carbon film inside of the spray tip (Fig. 2A).16 With this method, electrospray ionization and electrochemical reaction occurred simultaneously, permitting rapid transfer of the electrochemically generated short-lived intermediates into a mass spectrometer for mass and structure analysis. When a positive high voltage is applied, oxidation half reactions occur on the far end of the carbon film that is closer to the sprayer tip and mass spectrometer inlet, facilitating mass analysis of the oxidation products. Fig. 2B presents the mass spectrum of the oxidation products generated from a BPE deposited nanopipette filled with a 10 μM 1-phenylpyrrolidine solution prepared in mixed solvents (MeCN/H2O = 6/0.5). The m/z 148.2 peak is ascribed to the intact protonated 1-phenylpyrrolidine cation. Two other peaks at m/z 147.2 and 146.2 are ascribed to the 1-phenylpyrrolidine radical cation ([M]˙+) and the iminium ion ([M − H]+). Based on the observation of these two cations, the existence of the neutral α-amino C(sp3) radical ([M − H]˙) can be inferred as a deprotonation product of the 1-phenylpyrrolidine radical cation (m/z 147.2), since the latter is known to be highly acidic.17 A minor peak appeared at a half mass position, m/z 146.7, suggesting that it was a doubly charged product from the dimerization of two positively charged ions of 1-phenylpyrrolidine. Other peaks relevant to the dimerization of the reactive species were also detected in the range of m/z 291–294, providing more details for the electrooxidation process of 1-phenylpyrrolidine. The reactions involving the dimers are discussed in the ESI (Fig. S1).† Further, we used a phenyl trans-styryl sulfone as a coupling partner for the α-amino carbon radical, and the peak of the protonated addition product at m/z 391.5 was unambiguously observed, corresponding to another C(sp3) radical (Fig. 2C). This result further confirmed the existence of the α-amino C(sp3) radical.
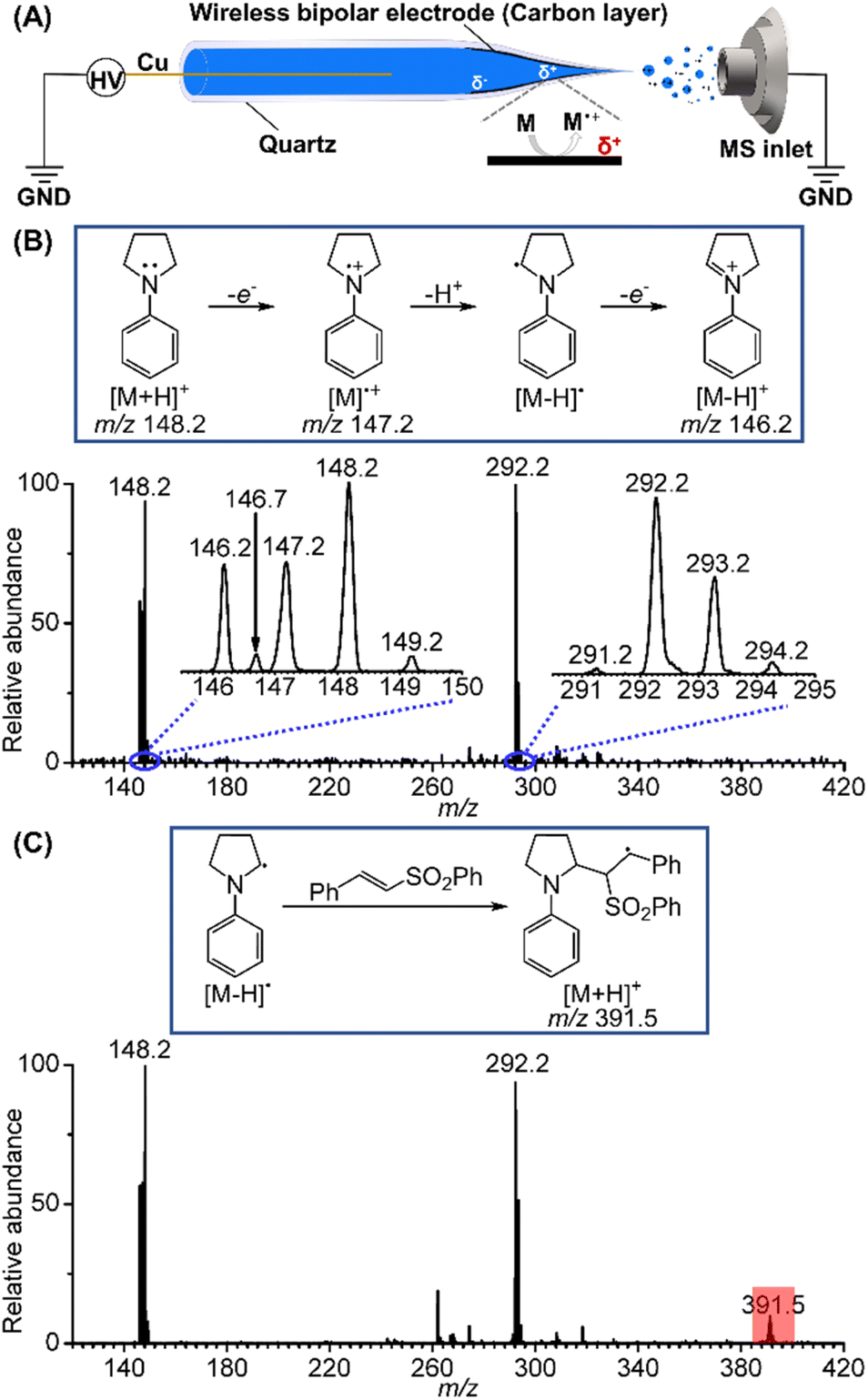 | ||
| Fig. 2 (A) Illustration of the implementation of BPE for nano-electrospray ionization mass spectrometry; (B) a typical mass spectrum showing the oxidation products of 1-phenylpyrrolidine; (C) a typical mass spectrum showing the addition product between the α-amino C(sp3) radical and a phenyl trans-styryl sulfone. | ||
Inspired by the above results, the long-sought fleeting intermediates produced by the electrooxidation of tri-n-propylamine (TPrA) were also investigated. Compared to the aromatic amines, the radical cation of TPrA, TPrA˙+, is more unstable and difficult to capture. Fig. 3A displays electrochemical mass spectra for TPrA using the same setup. Peaks at m/z 142.2, 143.2 and 144.2 are attributable to the iminium ion, TPrA˙+ and protonated TPrA, respectively. The existence of TPrA˙+ was further confirmed by comparing the intensity of the m/z 143.2 peak (18.2%) to the theoretical isotopic intensity from the neighbouring iminium ion (10.4%). Similar to the case of 1-phenylpyrrolidine, the existence of the neutral [TPrA–H]˙ radical can also be inferred. In previous studies, Xu et al.16 have successfully captured TPrA˙+ by using the BPE online electrochemical mass spectrometry method. Jiang et al.18 designed an electrochemistry-neutral reionization-mass spectrometry (EC-NR-MS) technique and detected TPrA˙+ by utilizing EC/easy ambient sonic-spray ionization (EASI). Shao et al.19 and Min et al.20 also directly detected the [Pr2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH3]+ intermediate. Further, when the mixture solution of 10 μM TPrA and 50 μM phenyl trans-styryl sulfone was sprayed, a mass peak at m/z 387.5 ascribed to the protonated product generated from the addition of [TPrA–H]˙ to a phenyl trans-styryl sulfone was also observed (Fig. 3B). Based on the fast and successful screening and mechanistic investigations of the above two systems using the electrochemical mass spectrometry methodology, we expect that this electrochemical α-vinylation strategy can be expanded to other tertiary amines using undivided cells, achieving scale-up synthesis under mild electrochemical conditions.
CHCH2CH3]+ intermediate. Further, when the mixture solution of 10 μM TPrA and 50 μM phenyl trans-styryl sulfone was sprayed, a mass peak at m/z 387.5 ascribed to the protonated product generated from the addition of [TPrA–H]˙ to a phenyl trans-styryl sulfone was also observed (Fig. 3B). Based on the fast and successful screening and mechanistic investigations of the above two systems using the electrochemical mass spectrometry methodology, we expect that this electrochemical α-vinylation strategy can be expanded to other tertiary amines using undivided cells, achieving scale-up synthesis under mild electrochemical conditions.
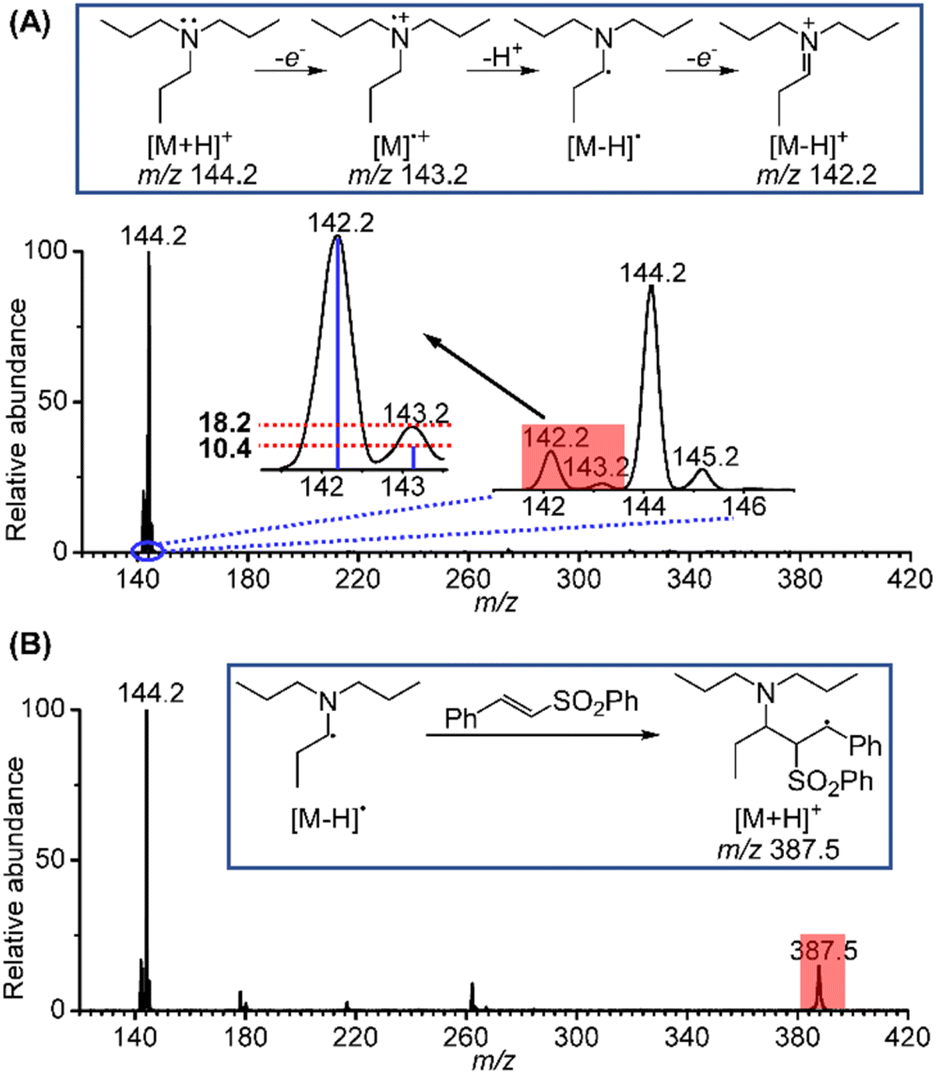 | ||
| Fig. 3 (A) A typical mass spectrum showing the oxidation products of TPrA; (B) a typical mass spectrum showing the addition product between the α-amino C(sp3) radical and a phenyl trans-styryl sulfone. | ||
The scale-up reactions were conducted with an undivided cell equipped with a carbon felt anode and a Pt cathode. The optimal conditions for the α-vinylation of tertiary amines were identified to involve constant current electrolysis in MeCN/H2O (6/0.5) (Table S1†). The substrate scope of electrochemical α-vinylation of tertiary amines was investigated and is exhibited in Fig. 4. A variety of acyclic amines bearing various alkyl substituents, such as triethylamine, tripropylamine, tributylamine and triisopentylamine afforded the corresponding products in moderate to good yields (3aa–3da). It is worth noting that an alkenyl group was selectively incorporated into the less hindered site of amines (3ea–3ga). N,N-diethylbutan-1-amine proceeded smoothly in standard conditions with 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity (3ha). N,N,N′,N′-Tetraethylethylenediamine was examined as well, and the α-vinylation occurred preferentially at N–Et (3ia). As expected, the reactivity can be smoothly extended to alcohol derivatives (3ja and 3ka). Additionally, cyclic aliphatic amines furnished the desired products with exclusive regioselectivity (3la–3oa).
:1 selectivity (3ha). N,N,N′,N′-Tetraethylethylenediamine was examined as well, and the α-vinylation occurred preferentially at N–Et (3ia). As expected, the reactivity can be smoothly extended to alcohol derivatives (3ja and 3ka). Additionally, cyclic aliphatic amines furnished the desired products with exclusive regioselectivity (3la–3oa).
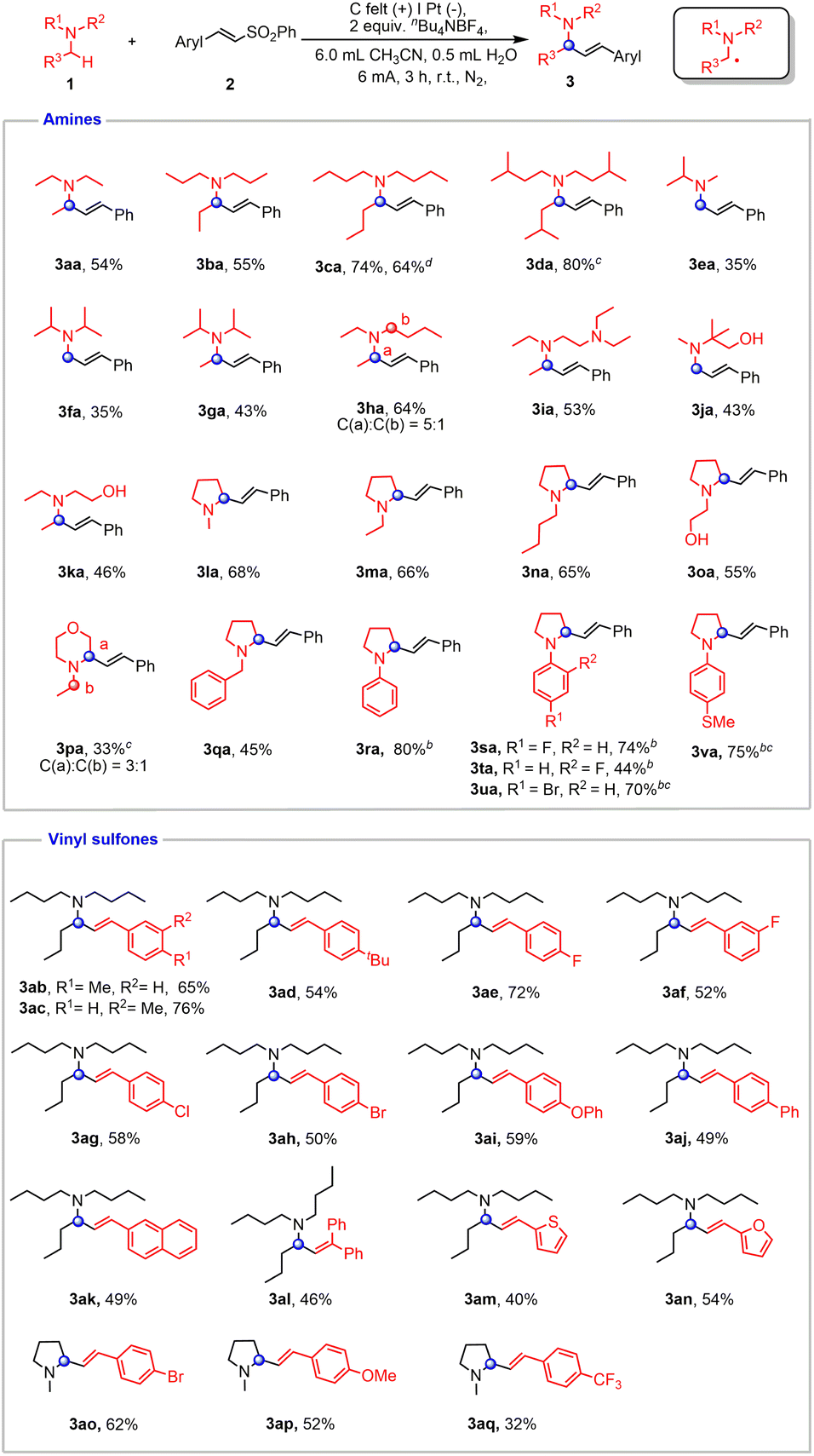 | ||
| Fig. 4 Evaluation of substrate scope. a Reaction conditions: carbon felt anode (15 mm × 10 mm × 3 mm), platinum plate cathode (15 mm × 15 mm × 0.3 mm), constant current = 6 mA, amines (1, 3.0 equiv. based on 2), vinyl sulfones (2, 0.3 mmol), nBu4NBF4 (2 equiv. based on 2), CH3CN (6.0 mL), H2O (0.5 mL), room temperature, N2 atmosphere, 3 h, undivided cell. Isolated yields were shown. b Amines (1, 2.0 equiv. based on 2), CsOAc (3.0 equiv. based on 2), DMF (6.0 mL), H2O (0.5 mL). c Yields were determined by 1H NMR, calibrated using CH2Br2 as the internal standard. d 4.5 mmol scale, CH3CN/H2O = 90 mL/7.5 mL, 45 h. | ||
N-Ethyl morpholine was also accommodated in this coupling, albeit with moderate selectivity (3pa). N-benzyl aliphatic amine gave the sole product while the benzylic methylene remained intact (3qa). In addition, aryl amines containing different functional groups all proceeded smoothly with the addition of CsOAc, which helped improve the yield and E-selectivity of products (3ra–3va). To demonstrate the feasibility of our protocol toward preparative synthesis, scale-up reaction was performed to furnish the product in 64% yield (3ca).
Then we turned our attention to the substrate scope of vinyl sulfones. Substrates containing alkyl groups on the benzene ring, such as –Me and –tBu, gave moderate to good yields (3ab–3ad). Vinyl sulfone derivatives bearing halogen groups such as F, Cl, and Br were also tolerated (3ae–3ah, 3ao). Vinyl sulfones with electron-donating groups were demonstrated to be viable coupling partners (3ai and 3ap). (E)-4-(2-(phenylsulfonyl)vinyl)-1,1′-Biphenyl and (E)-2-(2-(phenylsulfonyl)vinyl)naphthalene both gave 49% yields of desired products (3aj and 3ak). A trisubstituted alkene adduct could be obtained by using (2-(phenylsulfonyl)ethene-1,1-diyl)dibenzene (3al). Heterocyclic compounds were also compatible in this electrochemical transformation (3am and 3an). The reactivity could be also extended to a vinyl sulfone with –CF3 by using 1-methylpyrrolidine as a cross-coupling partner (3aq).
The cyclic voltammetry (CV) results of TPrA and 1-phenylpyrrolidine are provided in Fig. 5A. They both exhibit relatively low oxidation peak potentials (0.85 V for 1-phenylpyrrolidine and 0.95 V for TPrA), which can be readily oxidized at the anode. Taken together, the mechanisms deduced from the intermediates captured by MS and the products obtained from the scale-up synthesis are concluded in Fig. 5B. First, tertiary amines are oxidized at the anode to yield α-amino C(sp3) radicals (intermediate II) through single-electron oxidation and subsequent deprotonation. Then the addition of intermediate II to a vinyl sulfone can generate intermediate III, another C(sp3) radical. Finally, the allylic amines are obtained by successive elimination of a sulfinyl radical.21
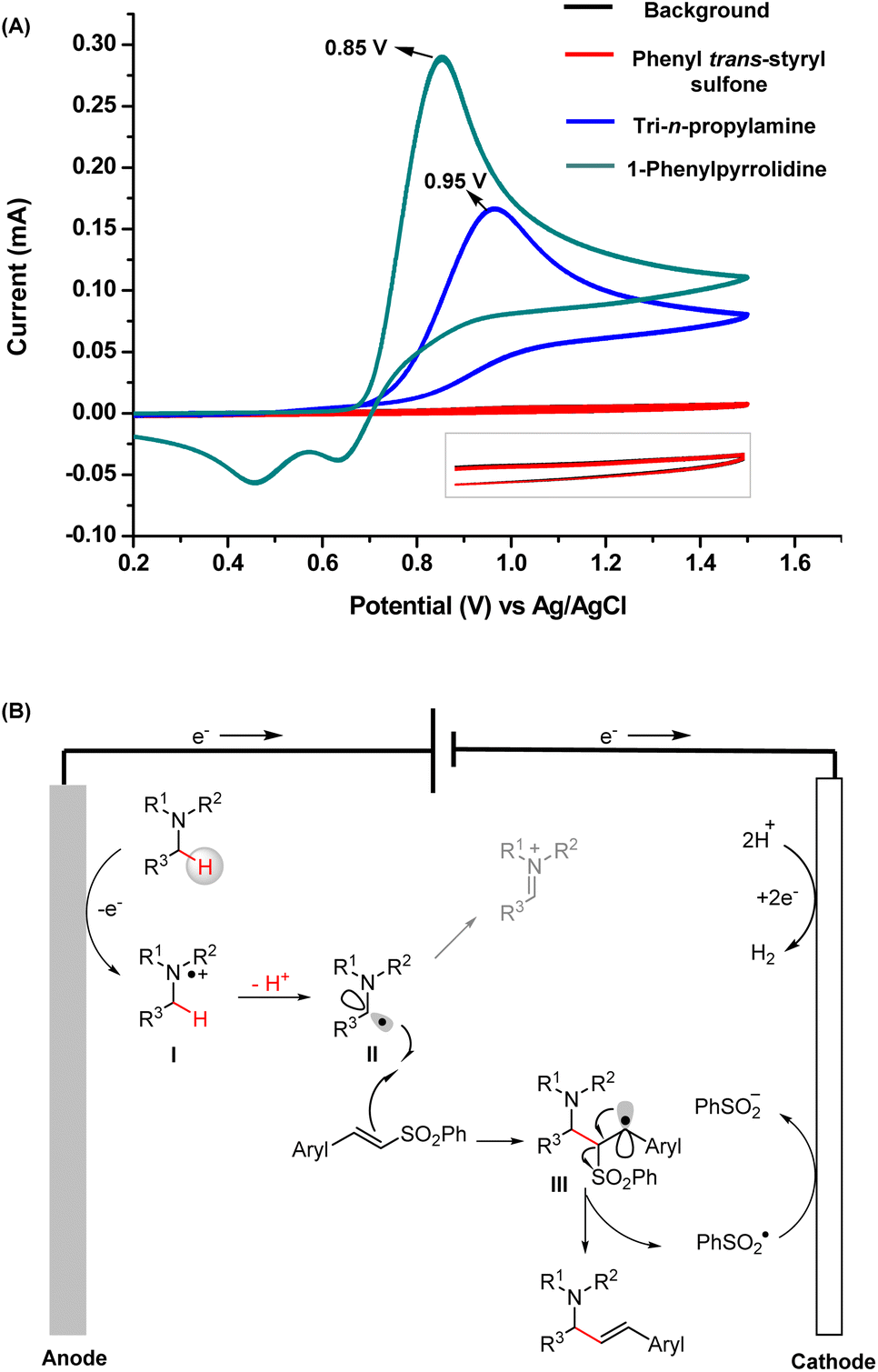 | ||
| Fig. 5 (A) CV measurements of the substrates; (B) well-elucidated mechanism of electrochemical radical vinylation of tertiary amines. | ||
Conclusions
To conclude, we captured and identified the short-lived intermediates in the electrochemical oxidative α-C(sp3)–H functionalization of tertiary amines by utilizing an online MS sampling method equipped with a BPE nano-electrospray ionization device. Direct evidence for the generation of α-amino radical cation and iminium cation was provided. Additionally, the neutral α-amino C(sp3) radical was captured and identified by its addition to a phenyl trans-styryl sulfone. Based on the fast screening and the mechanistic insights of the two systems provided by the online electrochemical mass spectrometry methodology, we successfully expanded and scaled up the synthesis to a large series of allylic amines via electrochemical α-vinylation of tertiary amines in undivided cells. From this work, we anticipate that online electrochemical mass spectrometry will be an avenue rich with opportunities for the design of new electrochemical reactions.Author contributions
H. Y., X. Z. and A. L. conceived the project. K. L., D. Z., Y. S., J. H. performed the experiments, analyzed the data, and discussed the results. K. L., D. Z., L. L., Y. C., H. Y., X. Z. and A. L. wrote the paper, supplementary methods, and related materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2022YFA1505100 and 2021YFA1500104), National Natural Science Foundation of China (22031008) Guangdong Basic and Applied Basic Research Foundation (2023A1515012260) and Science Foundation of Wuhan (2020010601012192). X. Z. acknowledges the National Natural Science Foundation of China (22174073 & 22003027), the National Key R&D Pro-gram of China (2018YFE0115000), the NSF of Tianjin City (21JCJQJC00010), Haihe Laboratory of Sustainable Chemical Transformations, Beijing National Laboratory for Molecular Sciences (BNLMS202106), and the Frontiers Science Center for New Organic Matter at Nankai University (63181206).Notes and references
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed
.
-
(a) A. J. Catino, J. M. Nichols, B. J. Nettles and M. P. Doyle, J. Am. Chem. Soc., 2006, 128, 5648–5649 CrossRef CAS PubMed
-
(a) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS
-
(a) H. P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed
-
(a) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed
-
(a) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed
-
(a) S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed
-
(a) L. Lu, R. Shi and A. Lei, Trends Chem., 2022, 4, 179–190 CrossRef CAS
-
(a) C. Y. Huang, H. Kang, J. Li and C. J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS PubMed
-
(a) H. Baltruschat, J. Am. Soc. Mass Spectrom., 2004, 15, 1693–1706 CrossRef CAS PubMed
-
(a) C. F. Bokman, C. Zettersten, P. J. Sjoberg and L. Nyholm, Anal. Chem., 2004, 76, 2017–2024 CrossRef PubMed
-
(a) P. Liu, M. Lu, Q. Zheng, Y. Zhang, H. D. Dewald and H. Chen, Analyst, 2013, 138, 5519–5539 RSC
- G. Hambitzer, J. Heitbaum and I. Stassen, Anal. Chem., 1998, 70, 838–842 CrossRef CAS PubMed
- P. Liu, I. T. Lanekoff, J. Laskin, H. D. Dewald and H. Chen, Anal. Chem., 2012, 84, 5737–5743 CrossRef CAS PubMed
-
(a) T. A. Brown, H. Chen and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 11183–11185 CrossRef CAS PubMed
- J. Hu, N. Zhang, P. K. Zhang, Y. Chen, X. H. Xia, H. Y. Chen and J. J. Xu, Angew. Chem., Int. Ed., 2020, 59, 18244–18248 CrossRef CAS PubMed
-
(a) F. G. Bordwell, J. Cheng, M. J. Bausch and J. E. Bares, J. Phys. Org. Chem., 1988, 1, 209 CrossRef CAS
- J. Liu, K. Yu, H. Zhang, J. He, J. Jiang and H. Luo, Chem. Sci., 2021, 12, 9494–9499 RSC
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC
- X. Zhang, W. Lu, C. Ma, T. Wang, J.-J. Zhu, R. N. Zare and Q. Min, Chem. Sci., 2022, 13, 6244–6253 RSC
- A. Noble and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00527e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |