
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-recovery of chiral microphase separation in an achiral diblock copolymer system†
Tengfei
Miao
*ab,
Xiaoxiao
Cheng
a,
Gong
Zhang
a,
Yuqing
Wang
a,
Zixiang
He
a,
Zhao
Wang
 a and
Wei
Zhang
*ac
a and
Wei
Zhang
*ac
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Engineering Laboratory of Novel Functional Polymeric Materials, Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, Jiangsu, China. E-mail: weizhang@suda.edu.cn
bJiangsu Key Laboratory for Chemistry of Low-Dimensional Materials, School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huaian 223300, Jiangsu, China. E-mail: miaotf@hytc.edu.cn
cSchool of Chemical and Environmental Engineering, Anhui Polytechnic University, Wuhu 241000, P. R. China
First published on 10th January 2023
Abstract
Macroscopic regulation of chiral supramolecular nanostructures in liquid-crystalline block copolymers is of great significance in photonics and nanotechnology. Although fabricating helical phase structures via chiral doping and microphase separation has been widely reported, the chiral memory and self-recovery capacity of asymmetric phase structures are the major challenge and still deeply rely on the presence of chiral additives. Herein, we demonstrate the first controllable chiral microphase separation in an achiral amphiphilic block copolymer consisting of poly(ethylene oxide) and azobenzene (Azo) groups. Chirality can be transferred to the fabricated helical nanostructures by doping with chiral additives (tartaric acid, TA). After the removal of the chiral additives and then performing cross-linking, the formed helical nanostructures will completely dispense with the chiral source. The supramolecular chirality and the micron-scale phase structure can be maintained under UV irradiation and heating-cooling treatment, enabling a reversible “on–off” chiroptical switch feature. This work is expected to avoid the tedious synthesis and expensive raw materials and shows a great application prospect in chiral separation and so on.
Introduction
The manipulation and fabrication of chiral topological structures are often considered to show a close relationship with the origin and the evolution of life.1,2 Generally speaking, hierarchical self-assembly of multiscale building units can be generated by breaking the symmetry using external chiral additives, including circularly polarized light (CPL),3–5 asymmetric liquid-crystal fields,6,7 chiral dopants,2,8,9 and chiral solvent10–14 and even vortex mixing.15 Through the design and operation of molecular or supramolecular self-assembly, the mechanisms of chiral transfer at different levels can be further understood.16–19 These research studies are of great significance in fundamental science and guide the design of novel chiral materials.20–23Self-assembly of block copolymers (BCPs), a typical bottom-up strategy for the facile fabrication of multi-morphological nano-structures,24–27 has also been applied to design and construct advanced chiral materials with regular periodicity and controlled handedness.28–34 By providing one or two BCP fragments containing chiral centers, transfer of chiral characteristics from the molecular level to the morphological level would occur during bulk microphase separation (MPS).33,35–37 Ho et al. reported the fabrication of a 3D chiral superstructure through self-assembly of polystyrene-block-poly(L/D-lactide) (PS-b-PL/DLA), demonstrating a controlled chirality evolution.38 Jinnai39 and Watkins40et al. demonstrated the construction of helical morphology from achiral triblock terpolymer polystyrene-b-polybuta-diene-b-poly(methyl methacrylate) and diblock copolymer poly-(ethylene oxide)-b-poly(tert-butyl acrylate) through the triggering of non-covalent interactions between blocks and chiral additives, respectively. Accompanying the introduction of functional liquid crystal (Azobenzene, Azo) groups in copolymer side chains, as reported by Yu29 and Lu30et al., rapid manipulation of these chiral nanostructures can be realized in a non-contact way, which provides new opportunities to construct new functional chiroptical materials.
Despite the extensive research, the evolution of chiral MPS is, however, still plagued with challenges where the chiral interactions from additives always played a vital role in the formation of helical morphologies. An ideal system is expected where this mirror symmetry breaking can occur spontaneously independent of external chiral sources, and which allows fabrication of functional chiral materials from achiral polymers and avoids tedious synthesis and expensive raw materials. Inspired by chiral self-recovery (CSR)41,42 of achiral liquid crystalline polymers, where the storage of supramolecular chirality in side chains by the cross-linking strategy can reinduce the destroyed chiral organized structures, here we set out to create a chiral covalent network in achiral liquid crystalline block copolymers (LCBCs) by cross-linking. Different from polymer networks prepared by “chiral imprinting” strategies,43–47 here cross-linking occurs after forming a well-organized chiral structure and removing the dopants or template molecules, which reflects more of the self-assembly of polymers (especially MPS of amphiphilic block copolymers) and the fixation of the formed superstructures. The expectation of our design emphasizes that the locked-in chiral information can provide a spontaneous self-recovery catalyst in the chiral microphase separation process, overcoming the absolute dependence of modulating traditional chiral MPS structures on external chiral sources.
Periodic chiral superstructures were induced in achiral LCBC, poly(ethylene oxide)-b-Azo containing poly(methyl methacrylate) (PEO-b-(Az-AzOH)), through simple doping with enantiopure tartaric acid (TA), followed by thermal annealing of the resulting film. Intermolecular hydrogen bonding between TA and PEO/carbonyl groups of the MMA (Azo) segment is widely considered the main driving force for the MPS.29,30 In order to permanently store the chirality information after removing the TA molecules, cross-linkable hydroxyl groups were randomly introduced at the terminals of the Azo segment side chains for the following acetal reaction.48 Combining with the breaking of the ordered structure triggered by trans–cis photoisomerization of Azo groups27,49 and chiral self-recovery of cross-linked covalent networks, we speculate that manipulation of chiral ordered MPS structures can be realized in the absence of chiral resources (Scheme 1).
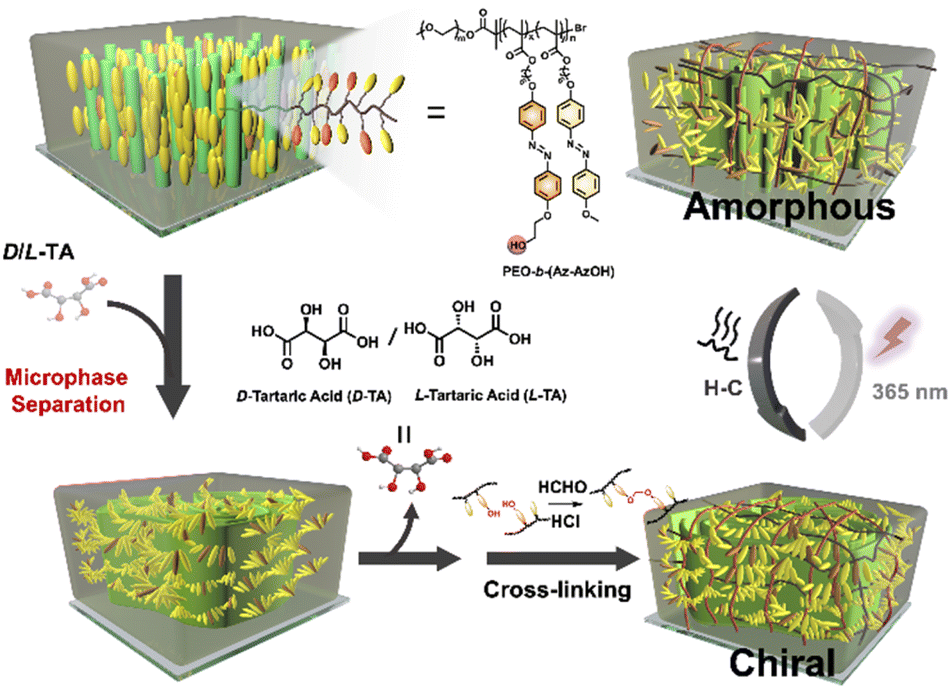 | ||
| Scheme 1 Schematic diagram showing the fabrication, fixation and reversible manipulation of chiral micron-scale structures in an achiral liquid crystalline BCP film. | ||
Results and discussion
Detailed procedures for preparing the Azo-containing monomers and the diblock copolymers are provided in the experimental section of the ESI.† The chemical structures and ratio of hydroxyl groups in diblock copolymers were confirmed using gel permeation chromatography (GPC) curves and 1H NMR spectra (Fig. S1 and S2).† The molar ratio of Az and AzOH groups in diblock copolymers calculated by 1H NMR is 1/0.53. The differential scanning calorimetry (DSC) profile of the diblock copolymer solid exhibited a sharp peak at around 40 °C and a wider phase transition at around 110 °C, which is attributed to the crystallization of the PEO segment and the clearing point temperature (Ti) of the LC segment (Fig. 1a), respectively. The polymer films were prepared by dropping a small amount of the polymer solution (with or without D/L-TA in THF; for the composite film, the molar ratio of PEO and TA was fixed at 1.2/1.0 and the relative weight ratio of TA and LC-BCP was 3.0/5.0; the polymer concentration of all samples was maintained at 2.5 mg mL−1) onto a clean quartz or silicon substrate and slowly evaporating the solvent in a THF atmosphere. After removal of the solvent by vacuum drying, the polymer films were annealed at 125 °C (beyond Ti) for 2 h for topography and structural analyses. Originally, ordered microphase structures could be formed after annealing at 125 °C (close to the clearing temperature) without adding TA. The 1D small-angle X-ray scattering (SAXS) peaks at q ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:√7 suggest a hexagonally packed cylinder phase (Fig. 1b). The d-spacing is 18.36 nm, which is consistent with the d-spacing (17.67 nm) observed in atomic force microscopy (AFM) images (Fig. 1d). Besides, no diffraction peaks were observed in the small angle region in which smectic layer diffractions are usually detected, indicating that the nematic phase of the LC segment was formed (Fig. 1b). Furthermore, the annealed copolymer films without external orientation also show that PEO nanocylinders aligned perpendicular to the film surface in AFM images, which might be attributed to the supramolecular cooperative motion of the microphase-separated PEO segment with the out-of-plane arranged Azo mesogens (Fig. 1d).50,51
:√3:√7 suggest a hexagonally packed cylinder phase (Fig. 1b). The d-spacing is 18.36 nm, which is consistent with the d-spacing (17.67 nm) observed in atomic force microscopy (AFM) images (Fig. 1d). Besides, no diffraction peaks were observed in the small angle region in which smectic layer diffractions are usually detected, indicating that the nematic phase of the LC segment was formed (Fig. 1b). Furthermore, the annealed copolymer films without external orientation also show that PEO nanocylinders aligned perpendicular to the film surface in AFM images, which might be attributed to the supramolecular cooperative motion of the microphase-separated PEO segment with the out-of-plane arranged Azo mesogens (Fig. 1d).50,51
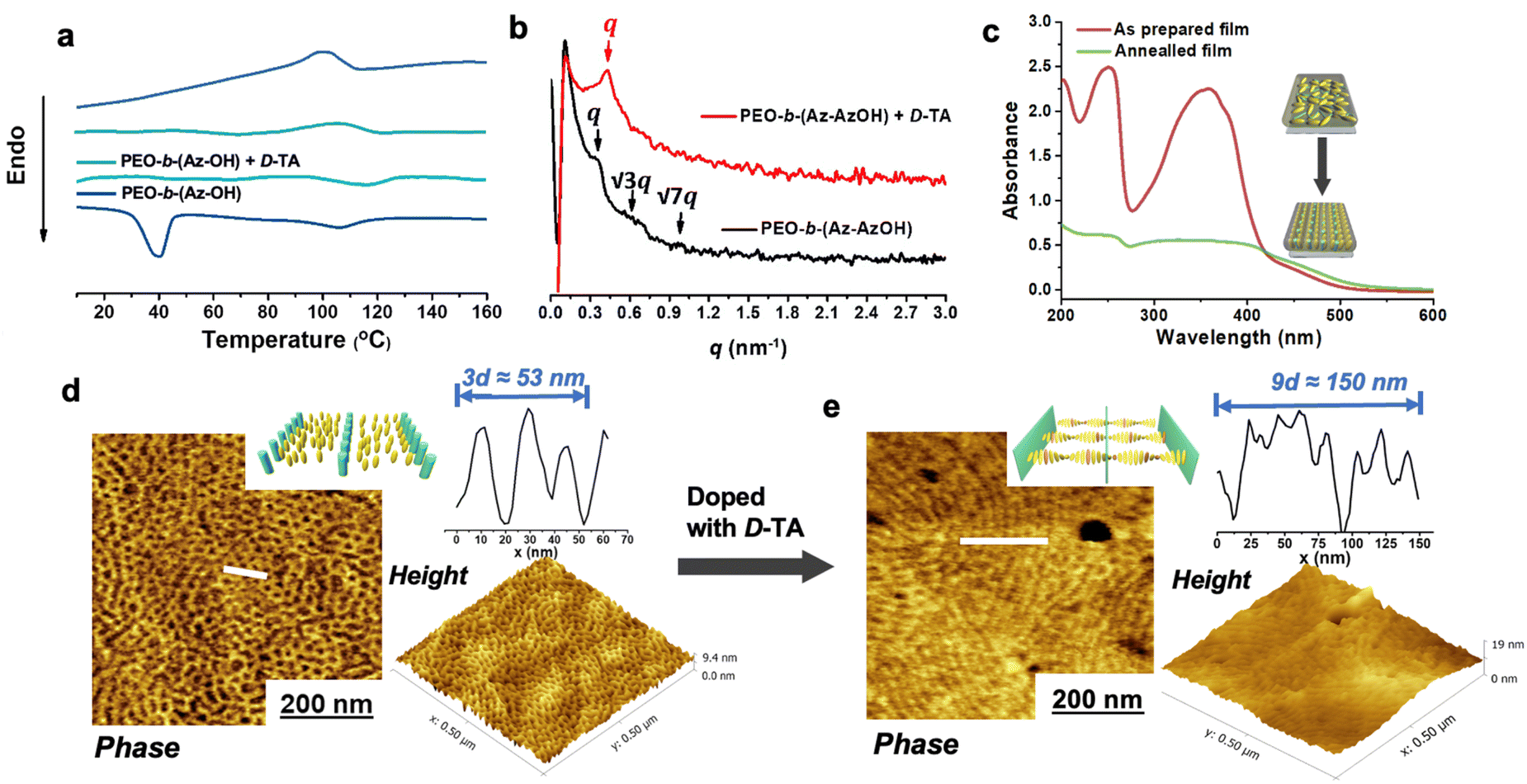 | ||
| Fig. 1 (a) DSC curves of PEO-b-(Az-AzOH) and PEO-b-(Az-AzOH) doped with D-TA. (b) UV-vis absorption spectra of the as-prepared and annealed PEO-b-(Az-AzOH) films. (c) Changes in SAXS patterns of the polymer films, recorded before and after chiral doping. (d) and (e) AFM phase structure and height image of the PEO-b-(Az-AzOH) (d) and composite film (e) measured after thermal annealing. | ||
A similar envision can be found from the change in UV-vis absorption spectrum of the LCBC film. For the as-prepared sample (in a range of 100 nm–200 nm), three peaks appeared at the band at about 270 nm, 375 nm and 435 nm, owing to the absorption of PEO, trans-Azo (π–π* transitions) and cis-Azo (n–π* transition), respectively. After annealing the polymer film at 125 °C, multiple peaks decreased greatly, indicating the out-of-plane arrangement of the Azo mesogens in the thin films (Fig. 1b).
After annealing the composite film beyond Ti, chirality transfer from the molecular level to the whole MPS structure level occurred. The PEO-b-(Az-AzOH)/D-TA solid exhibited no PEO crystallization peak due to the effect of the TA-PEO hydrogen bond. AFM images demonstrated that the cylinder morphology of the hybrid film disappeared, and instead a periodic layered phase separated structure appeared with a d-spacing of ∼16.67 nm, which is consistent with the d-spacing observed in SAXS spectra (Fig. 1e). It may have resulted from the superior intermiscibility between TA molecules and PEO segments that may slightly increase the block volume fraction (f) of the polymer, leading to further evolution of the MPS structure, according to the phase diagram of the block copolymer.52
Even though no obvious helical phase structure was observed in AFM images, helical packing of Azo units at the molecular level occurred. This is proven by the change in circular dichroism (CD) spectra and polarizing optical microscopy (POM) images of the TA hybrid thin films. As shown in Fig. 2a, the CD spectrum of the film showed only mirror-symmetric chiral signals at around 230 nm before annealing, corresponding to chirality transfer from TA molecules to the PEO phase via hydrogen bonding interaction. As expected, an intense Cotton effect with a positive maximum at around 375 nm and a negative maximum at around 315 nm was observed after annealing the D-TA doped polymer film at 125 °C for 2 hours. In the case of doping with L-TA, mirror-image Cotton effects were detected. The exciton couplet centred at 331 nm in the Cotton band could have resulted from the splitting of an H-band corresponding to the helically stacked Azo groups4,53 (Fig. 2b). This results from the formation of a chiral nematic phase, in which the orientation of the molecules is slightly twisted with respect to their neighbours and then leads to the formation of a long-range helical periodic superstructure under the command of chiral TA molecules.
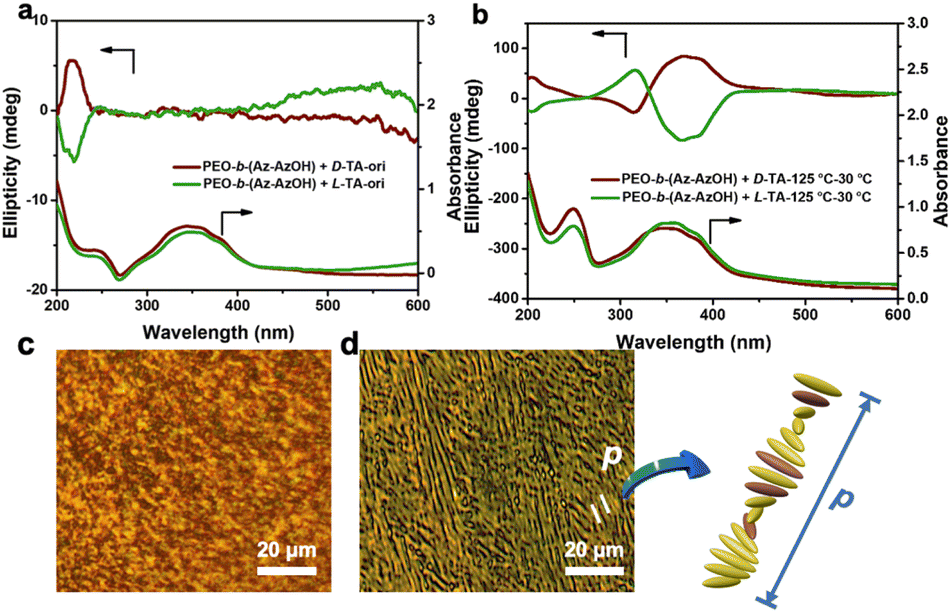 | ||
| Fig. 2 CD and UV-vis spectra of PEO-b-(Az-AzOH) doped with D/L-TA before (a) and after (b) heating-cooling treatment. POM images of PEO-b-(Az-AzOH) (c) and the composite film (d) recorded after thermal annealing. | ||
The appearance of periodic striped texture in POM corresponds to the parallelly lying helical axis of the superstructure on the substrate,54,55 which can also prove the above inference (Fig. 2c and d). Two stripe periods are approximately equal to the whole pitch length of the helical superstructure.
Remarkably, this supramolecular chirality can be memorized in the normal state even after complete removal of the chiral additives. After inducing the chiral packing by doping with D/L-TA and thermal annealing, the polymer films were then immersed in LiBr/ethanol solution to destroy the hydrogen bonds between TA molecules and PEO segments, followed by washing with pure ethanol. These procedures ensure the complete removal of the chiral dopant from the polymer films, which was confirmed using IR and 1H NMR spectra (Fig. S3†). Meanwhile, the soaking process did not affect the original chiral phase separated structure as confirmed by the layered structure in AFM images, the scattering peak in SAXS patterns and the constant CD signal (Fig. 3b–d). Besides, heating the chiral polymer films beyond their Ti could cause the erasure of the original chiral structure due to the removal of the chiral dopant (as shown in Fig. S3c†).
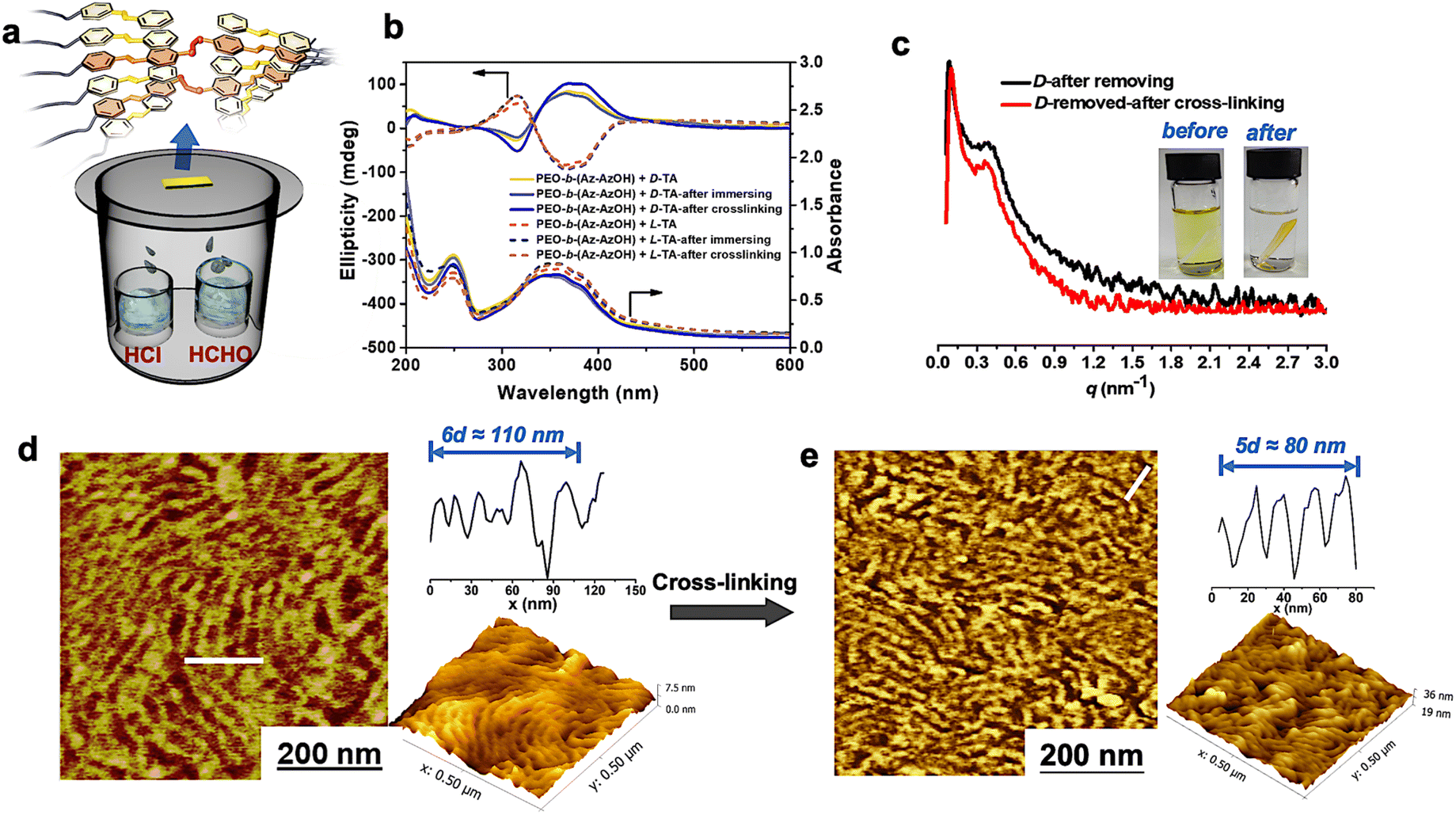 | ||
| Fig. 3 (a) Schematic diagram of the reaction method during cross-linking. (b) CD and UV-vis spectra of the chiral polymer films recorded before and after cross-linking. (c) SAXS patterns of the chiral polymer films recorded after removing the TA molecules and after cross-linking; inside: pictures of the chiral polymer films in THF solution before and after cross-linking; (d) and (e) AFM phase and height images of the immersed (d) and cross-linked (e) chiral polymer films. | ||
Then the polymer films were placed into an HCHO and HCl vapor atmosphere for covalent cross-linking via the acetal reaction (Fig. 3a). Differences in the solubility of the polymer films before and after cross-linking in THF revealed the success of cross-linking reactions (Fig. 3c).41,42,56 Besides, the shoulder peak at 286.3 eV in the C1s X-ray photoelectron spectroscopy (XPS) spectrum disappeared after cross-linking (Fig. S4†), which means that C–OH in the polymer terminal had been completely transformed into C–O–C. It is noteworthy that the separated chiral phase structure was perfectly locked in the achiral block copolymer throughout the cross-linking procedure, as confirmed by the following evidence. Firstly, almost identical ellipticity values in the CD spectra of the polymer films can be detected after cross-linking reactions (Fig. 3b). Secondly, AFM images of the cross-linked samples still displayed an ordered layered structure, with a d-spacing of about 16 nm, which is well consistent with the SAXS results (Fig. 3c–e).
To gain a deeper understanding of the storing capability of chiral superstructures, we investigated the self-recovery behaviour of supramolecular chirality and the cross-linked MPS structures upon exposure to UV light irradiation. As reported previously, trans–cis photoisomerization of Azo groups is often used to trigger manipulation of aggregation chirality, which could also assist the control of MPS helical structures. However, the presence of the chiral source (chiral TA) is indispensable during the traditional chiral recovery process. In this work, the well-organized chiral superstructure from trans-Azo is easily destroyed under UV light irradiation for 180 s, as confirmed by the disappearance of the CD signals corresponding to the contribution of Azo groups as shown in Fig. 4a. The disappearance of CD signals is attributed to a decrease in the trans-isomers and an increase in the cis-isomer percentage, and the non-coplanar structure of cis-isomers destroyed the formed ordered chiral structures (Fig. S5†). However, the chiral information still existed in the cross-linked networks contributed by the helical tendency of the polymer chains, which cannot be detected using CD spectra due to the destroyed helical packing of the chromophores.
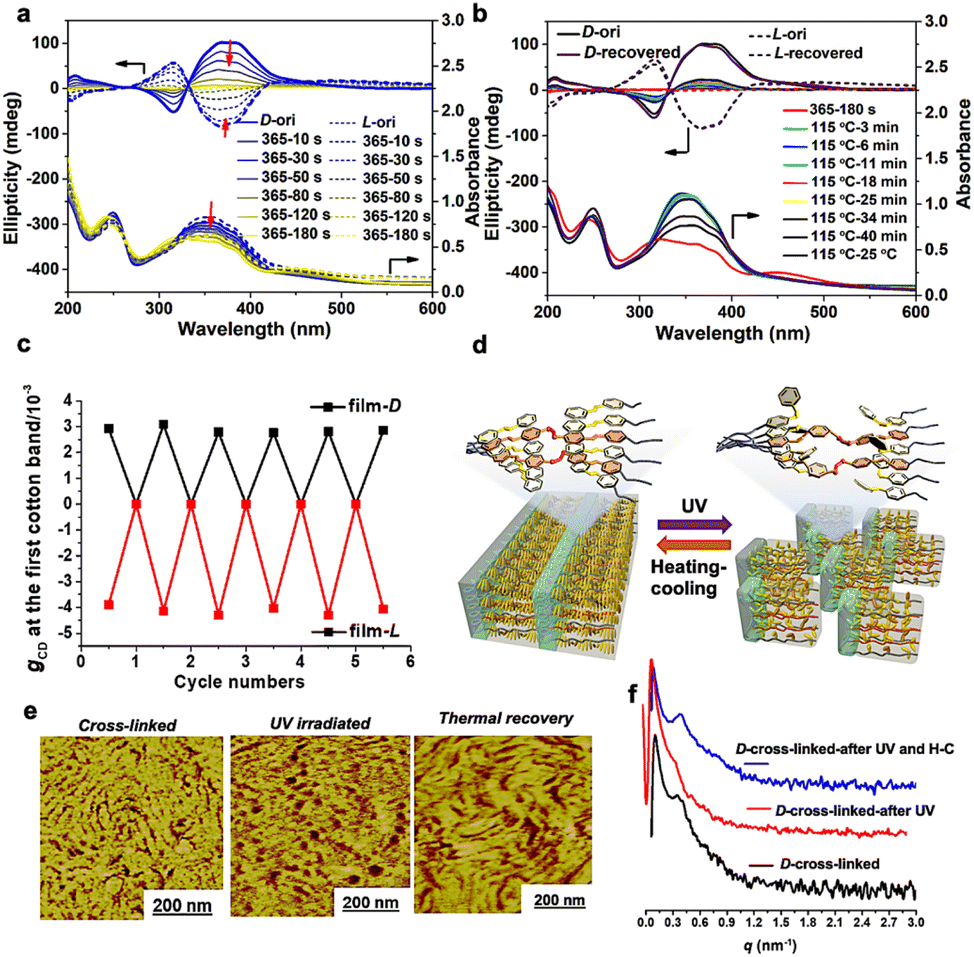 | ||
| Fig. 4 (a) Changes in the CD spectra of the cross-linked chiral polymer films (induced by D/L-TA) during 365 nm light irradiation. (b) Thermal recovery of CD intensity of the irradiated cross-linked chiral polymer films. (c) Cycle switches of the gCD values recorded in the above process (calculated from Fig. S6†). (d) Illustration for the photoinduced breaking and regeneration processes of the chiral MPS structure in absolute achiral liquid crystalline BCPs. (e) Evolution of the topological structure of the chiral polymer films (induced by D-TA) measured during the UV light irradiation and thermal recovery treatment (Fig. 4e: AFM phase images; Fig. S8:† AFM height images). (f) Changes in the SAXS patterns of the cross-linked chiral polymer films (induced by D-TA) recorded in the above process. | ||
Interestingly, the shapes and intensities of the CD spectra can return to their original states via a simple heating-cooling process (heating to 115 °C, keeping for 40 min and then cooling down to room temperature), without any help from other chiral sources (Fig. 4b). The results indicate that the organized cross-linked Azo units can provide cryptochirality to trigger the complete recovery of the whole chiral structure. This chiral “on–off” switch of achiral LCBC films could be performed at least five times without any visible loss of CD values (Fig. 4c and S6†).
Furthermore, we tracked the transformation of MPS nanostructures of the cross-linked LCBC films by AFM and SAXS. As presented in Fig. 4e, f and S7†, the periodic layered structure in AFM images disappeared after trans–cis photoisomerization of Azo units, due to the destruction of a liquid crystal field composed of regularly arranged Azo groups. At the same time, the scattering peak in the SAXS profiles of the irradiated samples also disappeared. Interestingly, both the chiral MPS morphology (AFM images) and the scattering peak in SAXS recovered after heating-cooling due to the existence of a cross-linked chiral covalent network.
Conclusions
Overall, the reversible manipulation of a chiral MPS structure in an achiral diblock system was first achieved. The interchain cross-linking structure successfully stored the chiral information in the MPS structure induced by a chiral additive (D-/L-tartaric acid). After the complete removal of chiral additives, the stored chiral information in the cross-linking structure can trigger the self-recovery of the chiral MPS structure by simple annealing treatment. The micron-scale phase structure and chiral optical properties could be manipulated by the trans–cis isomerization of Azo groups through ultraviolet irradiation and thermal annealing, where the “on–off” chiroptical switch can be achieved in an absolutely achiral system. We speculate that the novel construction of this chirality-storing diblock copolymer MPS structure and the reversible chiroptical switch will provide a new idea for developing future chiral materials.Data availability
Further details of the experimental procedure, polymer characterization and changes in chiral superstructures (1H NMR, GPC, IR AFM and CD) are available in the ESI.†Author contributions
TF. Miao and Prof. W. Zhang conceived the research idea and co-wrote the original draft. TF. Miao, XX. Cheng, YQ. Wang and ZX. He conducted experiments. The manuscript was written and proofread by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Nature Science Foundation of China (21971180 and 92056111), the China Postdoctoral Science Foundation (2022M722312), Key Laboratory of Polymeric Material Design and Synthesis for Biomedical Function, the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.Notes and references
- H. K. Bisoyi, T. J. Bunning and Q. Li, Adv. Mater., 2018, 30, 1706512 CrossRef PubMed.
- L. Wang, A. M. Urbas and Q. Li, Adv. Mater., 2020, 32, 1801335 CrossRef CAS PubMed.
- L. Wang, L. Yin, W. Zhang, X. Zhu and M. Fujiki, J. Am. Chem. Soc., 2017, 139, 13218–13226 CrossRef CAS PubMed.
- R. M. Tejedor, M. Millaruelo, L. Oriol, J. L. Serrano, R. Alcala, F. J. Rodriguez and B. Villacampa, J. Mater. Chem., 2006, 16, 1674–1680 RSC.
- Y. Wang, T. Harada, L. Q. Phuong, Y. Kanemitsu and T. Nakano, Macromolecules, 2018, 51, 6865–6877 CrossRef CAS.
- K. Akagi, Chem. Rev., 2009, 109, 5354–5401 CrossRef CAS PubMed.
- S. Yamakawa, K. Wada, M. Hidaka, T. Hanasaki and K. Akagi, Adv. Funct. Mater., 2019, 29, 1806592 CrossRef.
- Y. Kim, M. Frigoli, N. Vanthuyne and N. Tamaoki, Chem. Commun., 2017, 53, 200–203 RSC.
- H. Wang, H. K. Bisoyi, B. X. Li, M. E. McConney, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 2684–2687 CrossRef CAS PubMed.
- S. Xue, P. Xing, J. Zhang, Y. Zeng and Y. Zhao, Chem.–Eur. J., 2019, 25, 7426–7437 CrossRef CAS PubMed.
- L. Yin, Y. Zhao, M. Liu, N. C. Zhou, W. Zhang and X. L. Zhu, Polym. Chem., 2017, 8, 1906–1913 RSC.
- D. Lee, Y. J. Jin, H. Kim, N. Suzuki, M. Fujiki, T. Sakaguchi, S. K. Kim, W. E. Lee and G. Kwak, Macromolecules, 2012, 45, 5379–5386 CrossRef CAS.
- Y. Zhao, N. A. A. Rahirn, Y. J. Xia, M. Fujiki, B. Song, Z. B. Zhang, W. Zhang and X. L. Zhu, Macromolecules, 2016, 49, 3214–3221 CrossRef CAS.
- H. Nakashima, J. R. Koe, K. Torimitsu and M. Fujiki, J. Am. Chem. Soc., 2001, 123, 4847–4848 CrossRef CAS PubMed.
- Y. T. Sang, D. Yang, P. F. Duan and M. H. Liu, Chem. Sci., 2019, 10, 2718–2724 RSC.
- Y. Sang and M. Liu, Chem. Sci., 2022, 13, 633–656 RSC.
- G. Zhang, X. Cheng, Y. Wang and W. Zhang, Aggregate, 2022, e262, DOI:10.1002/agt1002.1262.
- L. Yin, M. Liu, H. Ma, X. Cheng, T. Miao, W. Zhang and X. Zhu, Sci. China: Chem., 2021, 64, 2105–2110 CrossRef CAS.
- Y. Ma, X. Cheng, H. Ma, Z. He, Z. Zhang and W. Zhang, Chem. Sci., 2022, 13, 13623–13630 RSC.
- K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429–434 CrossRef CAS PubMed.
- E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449–451 CrossRef CAS.
- T. Miyabe, H. Iida, A. Ohnishi and E. Yashima, Chem. Sci., 2012, 3, 863–867 RSC.
- M. Sun, X. Wang, X. Guo, L. Xu, H. Kuang and C. Xu, Chem. Sci., 2022, 13, 3069–3081 RSC.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- Z. Sun, Z. Chen, W. Zhang, J. Choi, C. Huang, G. Jeong, E. B. Coughlin, Y. Hsu, X. Yang, K. Y. Lee, D. S. Kuo, S. Xiao and T. P. Russell, Adv. Mater., 2015, 27, 4364–4370 CrossRef CAS PubMed.
- Y. Gan, H. Dai, Y. Ma, X. Cheng, Z. Wang and W. Zhang, Macromolecules, 2022, 55, 8556–8565 CrossRef CAS.
- X. Cheng, T. Miao, Y. Ma, X. Zhu, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24430–24436 CrossRef CAS PubMed.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed.
- S. Huang, Y. X. Chen, S. D. Ma and H. F. Yu, Angew. Chem., Int. Ed., 2018, 57, 12524–12528 CrossRef CAS PubMed.
- J. Yuan, X. Lu, Q. Li, Z. Lü and Q. Lu, Angew. Chem., Int. Ed., 2021, 60, 12308–12312 CrossRef CAS PubMed.
- G. M. Grason, ACS Macro Lett., 2015, 4, 526–532 CrossRef CAS PubMed.
- K.-C. Yang and R.-M. Ho, ACS Macro Lett., 2020, 9, 1130–1134 CrossRef CAS PubMed.
- H. Li, B. Xiong, J. Xu and J. Zhu, Aggregate, 2021, 2, e112 Search PubMed.
- F. Cai, Y.-X. Chen, W.-Z. Wang and H.-F. Yu, Chin. J. Polym. Sci., 2020, 39, 397–416 CrossRef.
- M.-C. Li, N. Ousaka, H.-F. Wang, E. Yashima and R.-M. Ho, ACS Macro Lett., 2017, 6, 980–986 CrossRef CAS PubMed.
- J. Yuan, X. Liu, Y. Y. Wang, G. J. Zeng, G. Li, X. H. Dong and T. Wen, ACS Macro Lett., 2021, 10, 1300–1305 CrossRef CAS PubMed.
- J. Yuan, X. Lu, S. Zhang, F. Zheng, Q. Deng, L. Han and Q. Lu, Macromolecules, 2022, 55, 1566–1575 CrossRef CAS.
- R. M. Ho, M. C. Li, S. C. Lin, H. F. Wang, Y. D. Lee, H. Hasegawa and E. L. Thomas, J. Am. Chem. Soc., 2012, 134, 10974–10986 CrossRef CAS PubMed.
- J. Dupont, G. Liu, K. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS PubMed.
- L. Yao, X. Lu, S. Chen and J. J. Watkins, Macromolecules, 2014, 47, 6547–6553 CrossRef CAS.
- T. F. Miao, X. X. Cheng, H. T. Ma, Z. X. He, Z. B. Zhang, N. A. C. Zhou, W. Zhang and X. L. Zhu, Angew. Chem., Int. Ed., 2021, 60, 18566–18571 CrossRef CAS PubMed.
- T. F. Miao, X. X. Cheng, H. T. Ma, W. Zhang and X. L. Zhu, Polym. Chem., 2021, 12, 5931–5936 RSC.
- J. J. BelBruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- E. Paruli, III, O. Soppera, K. Haupt and C. Gonzato, ACS Appl. Polym. Mater., 2021, 3, 4769–4790 CrossRef.
- R. A. M. Hikmet and H. Kemperman, Nature, 1998, 392, 476–479 CrossRef CAS.
- M. E. McConney, V. P. Tondiglia, J. M. Hurtubise, T. J. White and T. J. Bunning, Chem. Commun., 2011, 47, 505–507 RSC.
- A. Y. Bobrovsky and V. P. Shibaev, Adv. Funct. Mater., 2002, 12, 367–372 CrossRef CAS.
- R. Kani, Y. Nakano and S. Hayase, Macromolecules, 1995, 28, 1773–1777 CrossRef CAS.
- X. X. Cheng, T. F. Miao, L. Yin, Y. J. Ji, Y. Y. Li, Z. B. Zhang, W. Zhang and X. L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9669–9677 CrossRef CAS PubMed.
- H. Yu, T. Kobayashi and G.-H. Hu, Polymer, 2011, 52, 1554–1561 CrossRef CAS.
- H. Yu, J. Li, T. Ikeda and T. Iyoda, Adv. Mater., 2006, 18, 2213–2215 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- R. M. Tejedor, L. Oriol, J. L. Serrano, F. P. Urena and J. J. L. Gonzalez, Adv. Funct. Mater., 2007, 17, 3486–3492 CrossRef CAS.
- M. R. Wilson and D. J. Earl, J. Mater. Chem., 2001, 11, 2672–2677 RSC.
- Q. Xia, L. Meng, T. He, G. Huang, B. S. Li and B. Z. Tang, ACS Nano, 2021, 15, 4956–4966 CrossRef CAS PubMed.
- T. Miao, X. Cheng, Y. Qian, Y. Zhuang and W. Zhang, Int. J. Mol. Sci., 2021, 22, 11980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05975d |
| This journal is © The Royal Society of Chemistry 2023 |