
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity variance between stereoisomers of saturated N-heterocyclic carbenes on gold surfaces†
Gurkiran
Kaur
 a,
Nathaniel L.
Dominique
b,
Gaohe
Hu
c,
Phattananawee
Nalaoh
a,
Rebekah L.
Thimes
b,
Shelby L.
Strausser
a,
Lasse
Jensen
*c,
Jon P.
Camden
*b and
David M.
Jenkins
*a
a,
Nathaniel L.
Dominique
b,
Gaohe
Hu
c,
Phattananawee
Nalaoh
a,
Rebekah L.
Thimes
b,
Shelby L.
Strausser
a,
Lasse
Jensen
*c,
Jon P.
Camden
*b and
David M.
Jenkins
*a
aDepartment of Chemistry, The University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: jenkins@ion.chem.utk.edu
bDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: jon.camden@nd.edu
cDepartment of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: jensen@chem.psu.edu
First published on 26th September 2023
Abstract
Controlling the chirality of molecule-surface systems is essential for applications ranging from heterogeneous catalysis to biosensing. N-heterocyclic carbenes (NHCs) are quickly becoming a dominant ligand for noble metal surface passivation, but the potential of chiral NHC scaffolds remains untapped. In this work, two stereoisomers, one C2 symmetric and one Cs symmetric, of saturated N-heterocyclic carbenes (NHCs) and a structurally related unsaturated NHC (C2v symmetric) were synthesized as NHC-CO2 adducts. These CO2-protected NHCs were deposited on gold films and their presence on the films was confirmed by laser desorption ionization mass spectrometry (LDI-MS) and surface enhanced Raman spectroscopy (SERS). Surprisingly, the Cs NHC, but not the chiral C2 NHC, partially degrades to the independently synthesized unsaturated NHC upon binding to the gold. Theoretical calculations assist in explaining this phenomenon by showing that the NHCs primarily lie flat on the gold surfaces, which exposes the backbone protons on the Cs-symmetric NHC to a formal elimination of H2, while the C2-symmetric NHC remains protected from this elimination reaction. These results raise critical questions as to how the structure of NHC ligands may be tuned to influence binding and reactivity on gold surfaces.
Introduction
Self-assembled monolayers (SAMs) on gold surfaces are critical for many applications in catalysis, sensing, and medicine.1–7 Chiral thiols, in particular, are known to form SAMs on gold that deliver single enantiomer selectivity8–12 and L-cysteine-based chiral ligands are widely employed as fabricating agents for surfaces with enantiomeric recognition (Fig. 1A).13,14 Nevertheless, thiol-based SAMs degrade over time, especially when exposed to oxygen or native thiols, which limits their application in long term biomedical studies.15–17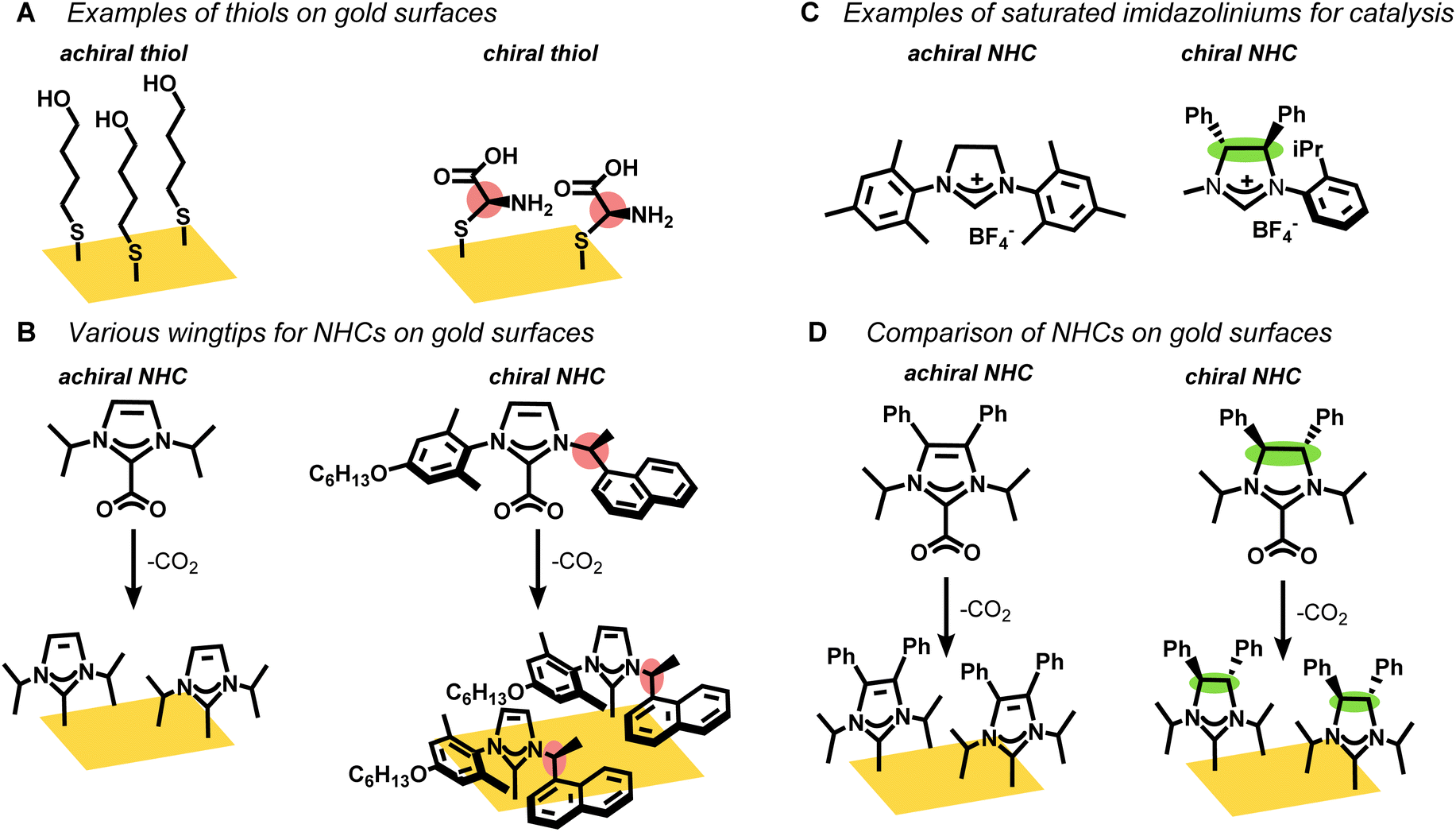 | ||
| Fig. 1 An overview of achiral and chiral monolayers on surfaces, where red and green represents chiral wingtips and chiral backbone respectively. (A) Thiol SAMs on gold surfaces. (B) NHCs with varying wingtip groups on gold surfaces. (C) Saturated imidazoliniums studied for homogeneous catalysis. (D) This work that includes saturated NHCs with chiral backbones on gold surfaces. | ||
N-heterocyclic carbenes (NHCs) are rapidly displacing thiols across myriad applications that employ SAMs due to their strong binding affinity to gold.18–20 CO2-adducts or bicarbonate salts of NHCs easily transfer the NHC ligands to a gold surface such as gold mirror or gold film-over nanospheres (AuFONs).19,21–23 However, regular NHCs, which have imidazole or benzimidazole rings, are poorly suited for developing chiral NHCs, except with chiral wingtips (Fig. 1B).24–29 Previous studies mostly focus on the organic synthesis24,26,29 of NHCs with chiral wingtips and their implementation in homogeneous catalysis.25,27,28,30 Chiral NHCs have very rarely been deposited on gold surfaces31 and even in these limited cases the chirality does not point away from the surface, which would be most valuable for sensing and medical applications.
Non-standard NHCs, specifically saturated NHCs, offer an alternative that would allow for chiral moieties at the backbone position as opposed to the wingtips (Fig. 1C). However, these saturated NHCs cannot be synthesized by simple addition of electrophilic wingtips to imidazoles or benzimidazoles.32,33 As such, this category of chiral saturated NHC has mostly been limited to a few examples for alkene metathesis catalysis.34–39
In this manuscript, we synthesized two stereoisomers, one chiral (C2) and one meso (Cs), of saturated N-heterocyclic carbenes (NHCs) and a structurally related unsaturated NHC (C2v) as their CO2 adducts (Fig. 1D). The NHC precursors were characterized by multi-nuclear NMR as well as single crystal X-ray which confirmed their absolute stereochemistry. The CO2-protected NHCs were deposited on gold films and their chemisorption was confirmed by surface enhanced Raman spectroscopy (SERS) and laser desorption ionization mass spectrometry (LDI-MS). Remarkably, we find that the Cs NHC, but not the chiral C2 NHC, partially degrades to the unsaturated NHC upon binding to the gold. This surprising result arises from the surface geometry, which exposes the backbone protons on only the Cs-symmetric NHC, but not the C2-symmetric NHC.
Results and discussion
NHC-CO2 adducts or their related bicarbonate salts are an effective method of NHC transfer to gold films.21–23,40 However, for synthesizing saturated NHCs (imidazolinium ring), the wingtips for the NHC must be appended followed by a ring closing.23,35 Thus, we followed this strategy for preparing compounds 1 and 2 as the precursors for the chiral and meso NHCs, 6 and 7 (Scheme 1).41,42 (4S,5S)-1,3-diisopropyl-4,5-diphenylethanediamine (1) was synthesized by addition of excess 2-iodopropane to (1S,2S)-(−)-1,2-diphenylethylenediamine under basic conditions in a heated pressurized glass vessel in 49% yield (see Experimental section for additional details†). meso-1,3-Diisopropyl-4,5-diphenylethanediamine (2) was prepared in a nearly identical fashion in 20% yield but required an additional purification step via column chromatography with amine-treated silica gel. | ||
| Scheme 1 Synthesis of C2, Cs, and C2v symmetric NHC-CO2 adducts, 6-CO2, 7-CO2, and 8-CO2, respectively. | ||
Refluxing 1 and ammonium chloride with excess triethyl orthoformate under acidic conditions yielded (4S,5S)-1,3-diisopropyl-4,5-diphenylimidazolinium chloride (3a) in 69% yield. meso-1,3-Diisopropyl-4,5-diphenylimidazolinium chloride (4a) was synthesized under analogous conditions in 58% yield. Since the imidazolinium salts are quite stable and the deprotonation occurs at the 2-position to form the NHC-CO2 adducts, we wanted to confirm the absolute stereochemistry for these compounds via single crystal X-ray diffraction. Despite multiple attempts, both 3a and 4a had significant disorder in their unit cells due to the chloride counter anion.
To solve the crystallographic disorder due to the counter anion, we switched to hexafluorophosphate by replacing ammonium chloride with ammonium hexafluorophosphate in the synthesis of 3 and 4. This switch led to the formation of (4S,5S)-1,3-diisopropyl-4,5-diphenylimidazolinium hexafluoro-phosphate (3b) and meso-1,3-diisopropyl-4,5-diphenylimidazolinium hexafluorophosphate (4b) in 66% and 76% yield, respectively. A comparison of the 1H NMR spectra for 3b and 4b shows mostly similarities but one key distinction due to their difference in stereochemistry (Fig. 2, left). The imidazolinium protons (labelled “a” in Fig. 2, left) are close to each other at 8.24 and 8.30 ppm for 3b and 4b, respectively, which shows that their relative acidity is similar. However, backbone protons (labelled “c” in Fig. 2, left) are at 5.11 and 5.75 ppm for 3b and 4b, respectively. The significant upfield shift for 3bversus4b is due to the proximity to the aryl ring and has been observed by Grisi for similar imidazoliniums.43
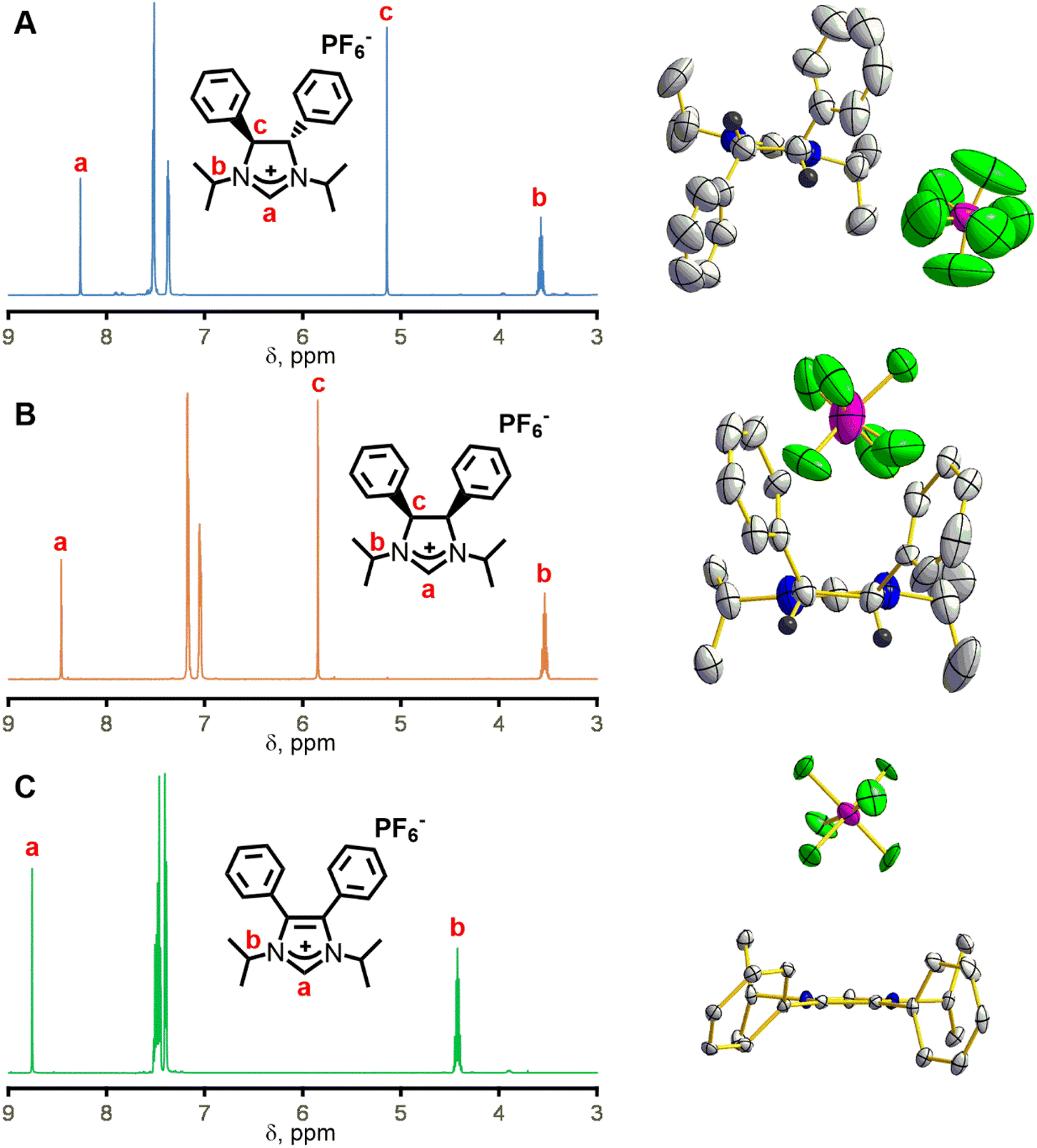 | ||
| Fig. 2 Left: 1H NMR spectra in CD3CN from 3–9 ppm of NHC hexafluorophosphate salts (A) 3b, (B) 4b, and (C) 5. Right: X-ray crystal structures of (A) 3b, (B) 4b, and (C) 5 shown with thermal ellipsoids at 50% probability. Grey, blue, black, green, and pink ellipsoids represent C, N, H, F, and P atoms, respectively. Hydrogens are omitted except for ones on stereogenic carbons. | ||
A typical reaction to append the wingtips was successfully carried out to synthesize 1,3-diisopropyl-4,5-diphenylimidazolium hexafluorophosphate (5). Excess 2-iodopropane was refluxed with 4,5-diphenylimidazole under basic conditions followed by an anion exchange with KPF6 which gave 5 in 86% yield as a pale-yellow powder (Scheme 1).44 Notably, there is no peak (labelled “c” in Fig. 2, left) between 5 and 6 ppm in 5 due to the absence of backbone protons in the 1H NMR that is observed in 3b and 4b.
Single crystal X-ray diffraction was necessary to deduce the absolute stereochemistry of 3b and 4b. Crystals for 3b cracked upon cooling, so this structure was determined at room temperature. Fig. 2A (right) shows the structure of 3b with the view from the back of the molecule, which clearly demonstrates its C2 symmetry. Likewise, 4b displays Cs symmetry as the protons are on the same side of the ring (Fig. 2B, right). 3b crystallizes in the chiral space group P21 while 4b crystallizes in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Finally, the crystal structure for compound 5 shows the expected C2v symmetry and no protons on the back of the ring (Fig. 2C, right).
. Finally, the crystal structure for compound 5 shows the expected C2v symmetry and no protons on the back of the ring (Fig. 2C, right).
The NHC-CO2 adducts for all three compounds, 3b, 4b, and 5, were prepared by deprotonation with potassium bis(trimethylsilyl)amide in THF followed by addition of excess CO2 gas (Scheme 1).45–47 After the completion of reaction, the THF was removed and the solids were extracted with pentane. The pentane was removed under vacuum to yield white solids. 6-CO2 was synthesized in 41% yield and the downfield imidazolinium proton for 3b is not observed in the 1H NMR (see ESI†). Instead, an additional resonance at 164.89 ppm is observed in the 13C NMR which is due to the bound CO2.47,48 Similar results were obtained for the synthesis of 7-CO2 and 8-CO2. The formation of these NHC-CO2 adducts was further validated by the presence of CO2 stretches in the FTIR spectra at 1672, 1672, and 1668 cm−1 for 6-CO2, 7-CO2, and 8-CO2, respectively (see ESI†).22
The deposition of the NHCs on gold mirrors followed a protocol that we previously developed for gold film-over-nanospheres (AuFONs).23 The gold mirror was placed in a vacuum oven with the solid powders placed directly on top. The vacuum oven was heated to at least 105 °C and placed under reduced pressure (<5 Torr) for 12 minutes. The gold mirror was removed from the oven and then rinsed with alcohol, water and then dried with dinitrogen gas. SERS spectra were collected for gold mirrors functionalized with 6, 7, and 8 (Fig. S51†). The resulting spectra illustrate the adsorption of each of these ligands on the gold surface and little to no spectroscopic differences were observed, which is distinct from our previous reports with benzimidazole-based NHCs.21,22 However, the relative differences in SERS intensities may arise from several different variables, such as the molecule orientation relative to the gold surface, number of molecules in the probe volume, and the Raman cross section of each NHC ligand.22,49,50
LDI-MS is a highly efficient method for detecting NHCs on gold and for tracking subsequent chemical reactions.51–53 The reason that this approach is so successful is that the NHCs do not fragment like thiols, and each gold ion binds two NHCs to form a cationic bis(NHC)gold(I) complex.51 LDI-MS analysis was carried for 6, 7, and 8 on gold mirror. LDI-MS for 6 shows the expected [(6)2Au]+ ion at 809.37 m/z (Fig. 3, bottom). However, the corresponding measurement for 7 was quite surprising. In this case, three separate ions were observed in this region at 805.35, 807.36, and 809.38 m/z (Fig. 3, middle). One possibility is that NHC 7 is losing backbone protons and becomes unsaturated, which would transform 7 into 8. A comparison to the LDI-MS of 8 supports this supposition since only one ion is observed at 805.34 m/z (Fig. 3, top). A pair of LDI-MS experiments with 7-Au at 0 ns or 110 ns delay time confirm that the dehydrogenation is not a result of the LDI-MS measurement (Fig. S52†). Since very few studies of NHCs on gold have been performed with saturated NHCs, it is difficult to say how this reaction takes place.54,55 Nevertheless, an additional piece of evidence is the small feature found at 1290 cm−1 on the SERS spectrum for 7 (Fig. S48†). This band may be due to the formation of 8 on the surface but since 8 has much weaker SERS intensity, even if most of 7 is transformed it gives a small signal.
 | ||
| Fig. 3 LDI-MS for 6 (SS, bottom), 7 (meso, middle), and 8 (Imidazolium, top) on gold mirrors. Surprisingly, NHC 7 appears to transform into 8, i.e., become unsaturated, once bound to the gold. | ||
Theoretical calculations can assist in understanding the different reactivity and binding on the gold surface between 6 and 7. Binding energies were calculated for each NHC in both vertical and flat motifs on 58-atom gold clusters.52,56 As shown in Fig. 4, the flat binding mode is significantly more energetically favorable for each NHC (with the caveat that only one NHC is modelled on the surface, which precludes NHC–NHC interactions). Of particular note, the flat binding geometry for NHC 7 with the backbone protons pointing away from the gold surface is more favorable by 17 kcal mol−1 energy than the one with protons aimed towards the gold surface. In contrast, for NHC 6, one proton must be pointed towards the gold surface in the flat position since the NHC is C2 symmetric. While future experiments are needed to unravel the mechanism of this dehydrogenation reaction, the variance in reactivity can clearly be accounted for by the differences in stereochemical relationship of the NHCs to the gold surface.
 | ||
| Fig. 4 Binding energies (BE) of NHCs in their energetically favored flat-lying geometry on gold clusters calculated with DFT. (A) represents top view of the NHCs (B) represents the side-view. Black, blue, white, and gold, spheres represent C, N, H, and Au atoms, respectively. The green spheres represent hydrogens bounds to chiral carbons. | ||
Conclusions
In conclusion, we have synthesized three NHCs which have three different symmetries (C2, Cs, and C2v) and bound them to gold mirrors via their CO2 adducts. The different stereochemistry led to key differences in the 1H NMR and their absolute configuration was determined by single crystal X-ray diffraction. This study is the first example where NHCs with a chiral backbone have been affixed to a gold surface. Once bound to the gold, all three NHCs were evaluated with SERS and LDI-MS. Notably, the Cs NHC (7), but not the chiral C2 NHC (6), partially degrades to the independently synthesized unsaturated NHC (8) upon binding to the gold. Theoretical calculations assist in explaining this phenomenon by showing that all three NHCs primarily lie flat on the gold surfaces, which exposes the backbone protons on 7 but not on 6.Experimental
Materials and general considerations
All the reactions and workups for synthesizing organic compounds were done under air unless otherwise stated. All the reactions and workups to synthesize NHC-CO2 adducts were conducted under dry N2 atmosphere using standard Schlenk techniques or a glovebox. All the glassware for air and water-sensitive reactions were dried in oven at 170 °C overnight prior to their use in glovebox or on Schlenk line. The solvents used under N2 were dried on an Innovative Technologies (Newburyport, MA) Pure Solv MD-7 solvent purification system, degassed by three freeze–pump–thaw cycles on a Schlenk line to remove dioxygen, and stored over activated 4 Å molecular sieves prior to use. Celite was dried at 240 °C under vacuum overnight and stored in the glove box. Compounds 1, 2, 3a, 3b, 4a, 4b, 5, 6-CO2, 7-CO2, and 8-CO2 were synthesized by employing modifications from previously published procedures that reported synthesis of similar compounds.41,42,44–47 (1S,2S)-(−)-1,2-Diphenylethylenediamine and meso-1,2-diphenylethylenediamine were purchased from Combi-Blocks and Ambeed, respectively, at highest available purity and used as such without further purification. All other reagents were purchased from typical commercial vendors and used without any further purification.General instrumentation
Solution 1H NMR and 13C{1H} NMR spectroscopies were performed on Varian VNMRS 500 MHz and Bruker AVANCE NEO 500 MHz. The 1H NMR spectral peaks were referenced to the residual protonated solvents. The 13C NMR spectral peaks were referenced to the solvents. Mass spectrometry analyses of NHC compounds were conducted at the Biological and Small Molecule Mass Spectrometry Center located in the Department of Chemistry at the University of Tennessee. DART analyses were performed using a JEOL AccuTOF-D time-of-flight (TOF) mass spectrometer with a DART (direct analysis in real time) ionization source from JEOL USA, Inc. (Peabody, MA). Matrix-assisted laser desorption/ionization (MALDI) mass analyses were performed using Waters Synapt G2-Si quadrupole time-of-flight mass spectrometer. Infrared spectra were collected on a Thermo Scientific Nicolet iS10 with a Smart iTR accessory for attenuated total reflectance (ATR) using pure samples of each complex.Crystal structure determination
Single crystal X-ray diffraction (SCXRD) data of all compounds were collected with a Bruker D8 Venture diffractometer with Mo Kα radiation (λ = 0.71073 Å) and PHOTON II detector. The crystal of compound 3b was mounted on the capillary glass (Hampton Research) with epoxy glue and cooled at 275 K, while the crystals of 4b and 5 were mounted on loops (MitiGen) with Paratone-N (Hampton Research) in 100 K cold stream provided by an Oxford Cryostream 800 system. The diffraction data were obtained, integrated, and calculated with Bruker APEX4 with SAINT and SADABS, SHELXL, SHELXT, and OLEX2 software. The structural figures were used by DIAMOND software for visualization.Surface-enhanced Raman spectroscopy (SERS) measurements
SERS spectra were collected on a home-made Raman microscope described previously.57–59 Briefly, one 633 nm HeNe laser (Thorlabs) was guided into an inverted microscope (Nikon Ti-U) and then focused onto the mirror-nanoparticle substrate with an objective lens (20×, N.A. = 0.5). Scattered light was collected through the same objective, filtered through a Rayleigh rejection filter (Semrock), and fed into a spectrometer (Princeton Instruments Acton SP2300, f = 0.3 mm, 1200 g mm−1). The resulting spectra were background subtracted in Matlab and plotted using Igor software.Laser desorption/ionization mass spectrometry (LDI-MS) measurements
A Bruker UltrafleXtreme MALDI-TOF-TOF mass spectrometer in positive ion, reflection mode with ultra-beam parameters and a 355 nm frequency tripled ND: YAG laser was employed for LDI-MS measurements. The laser power was set to 100% of the total available power with a global attenuator offset of 20%. Red phosphorous clusters were used to calibrate the mass spectrometer with a cubic enhanced fit.The gold substrates were mounted onto a metal target plate slide adapter (Bruker) and grounded using conductive copper tape. In general, three microliters of red phosphorous suspended in acetonitrile (1 mg mL−1) was deposited directly onto the gold mirrors and used to calibrate the mass spectrometer.60
Computational details
All calculations were carried out with Amsterdam Density Functional (ADF) engine61,62 from AMS202163 software package. The Becke-Perdew (BP86) XC functional64,65 with dispersion correction Grimme3 BJDAMP66 was used. The triple-zeta polarized slater type (TZP) basis set with large frozen cores from ADF basis set library67 was used and numerical quality is set to normal. The scalar relativistic effects were accounted for by the zeroth-order regular approximation (ZORA).68,69 Geometry optimization was performed where only carbene molecule and topmost Au atom were relaxed. The structure and normal modes diagrams were plotted using PyMOL.70Synthetic details
1H NMR (CDCl3, 500 MHz): δ 7.27 (m, 6H), 7.13 (m, 4H), 3.98 (s, 2H), 2.53 (m, J = 6.7 Hz, 2H), 0.91 (d, J = 6.3 Hz, 6H), 0.89 (d, J = 6.3 Hz, 6H). 13C NMR (CDCl3, 126 MHz): δ 128.39, 128.22, 127.42, 65.29, 45.78, 24.21, 21.84. IR: 3309, 3023, 2958, 1602, 1492, 1465, 1451, 1435, 1379, 1363, 1337, 1277, 1269, 1238, 1170, 1142, 1123, 1069, 1026, 913, 864, 835, 812, 779, 756, 724, 695 cm−1. DART HRMS (m/z): [M + H]+: (C20H29N2)+: 297.2328 (found), (C20H29N2)+: 297.2331 (calculated).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethyl acetate and hexanes. Removing the solvent via rotary evaporation yielded the pure product as an off-white powder. (0.342 g, 20.2% yield).
:1 ethyl acetate and hexanes. Removing the solvent via rotary evaporation yielded the pure product as an off-white powder. (0.342 g, 20.2% yield).
1H NMR (CD2Cl2, 500 MHz): δ 7.24 (m, 6H), 7.14 (m, 4H), 3.87 (s, 2H), 2.45 (sept, J = 6.4 Hz, 2H), 0.85 (dd, J = 7.4, 6.2 Hz, 12H). 13C NMR (CD2Cl2, 126 MHz): δ 142.49, 128.89, 128.41, 127.47, 66.00, 46.04, 24.52, 22.23. IR: 2964, 1455, 1385, 1282, 1237, 1223, 1162, 799, 758, 703 cm−1. DART HR MS (m/z): [M + H]+: (C20H29N2)+: 297.2318 (found), (C20H29N2)+: 297.2331 (calculated).
1H NMR (CD2Cl2, 500 MHz): δ 10.01 (s, 1H), 7.49 (m, 6H), 7.29 (m, 4H), 4.92 (s, 2H), 3.93 (sept, J = 6.8 Hz, 2H), 1.58 (d, J = 6.7 Hz, 6H), 1.24 (d, J = 6.6 Hz, 6H). 13C NMR (CD2Cl2, 126 MHz): δ 156.30, 137.21, 130.61, 130.47, 127.50, 73.10, 51.19, 22.03, 21.84. IR: 2975, 2876, 1627, 1495, 1455, 1391, 1370, 1343, 1269, 1228, 1211, 1177, 1118, 1080, 1045, 931, 641, 615, 755, 665 cm−1. MALDI HR MS (C21H27N2)+: 307.2178 (found), (C21H27N2)+: 307.2169 (calculated).
:Et2O.
1H NMR (CD3CN, 500 MHz): δ 8.24 (s, 1H), 7.50 (m, 6H), 7.34 (m, 4H), 5.11 (s, 2H), 3.54 (sept, J = 6.6 Hz, 2H), 1.37 (d, J = 6.8 Hz, 6H), 1.20 (d, J = 6.6 Hz, 6H). 13C NMR (CD3CN, 126 MHz): δ 154.86, 137.36, 130.90, 130.62, 128.42, 73.13, 50.93, 21.55, 21.09. 19F NMR (CD3CN, 470 MHz): δ −73.00 (d, J = 706 Hz). 31P NMR (CD3CN, 202 MHz): δ −144.62 (pent, J = 706 Hz). IR: 2985, 1725, 1632, 1497, 1457, 1397, 1379, 1270, 1222, 1179, 1120, 755.97, 700 cm−1. MALDI HR MS (C21H27N2)+: 307.2174 (found), (C21H27N2)+: 307.2169 (calculated).
1H NMR (CD2Cl2, 500 MHz): δ 9.42 (s, 1H), 7.14 (m, 6H), 7.00 (m, 4H), 5.89 (s, 2H), 3.76 (sept, J = 6.7 Hz, 2H), 1.71 (d, J = 6.8 Hz, 6H), 1.37 (d, J = 6.6 Hz, 6H). 13C NMR (CD2Cl2, 126 MHz): δ 156.58, 131.93, 129.43, 129.05, 128.95, 69.30, 50.85, 21.95, 21.85. IR: 2972, 1630, 1583, 1494, 1455, 1393, 1374, 1341, 1268, 1230, 1177, 1129, 1079, 1050, 928, 856, 772, 749, 702 cm−1. MALDI HR MS (C21H27N2)+: 307.2172 (found), (C21H27N2)+: 307.2169 (calculated).
1H NMR (CD3CN, 500 MHz): δ 8.30 (s, 1H), 7.15 (m, 6H), 6.99 (m, 4H), 5.75 (s, 2H), 3.49 (sept, J = 6.7 Hz, 2H), 1.49 (d, J = 6.7 Hz, 6H), 1.23 (d, J = 6.5 Hz, 6H). 13C NMR (CD3CN, 126 MHz): δ 156.15, 132.84, 129.74, 129.57, 129.36, 69.35, 50.68, 21.48, 20.96. 19F NMR (CD3CN, 470 MHz): δ −73.00 (d, J = 706 Hz). 31P NMR (CD3CN, 202 MHz): δ −144.63 (sept, J = 706 Hz) IR: 2972, 1632, 1584, 1495, 1455, 1375, 1266, 1178, 1129, 774, 702 cm−1. MALDI HR MS (C21H27N2)+: 307.2169 (found), (C21H27N2)+: 307.2169 (calculated).
The above yellow powder (0.80 g, 1.85 mmol, 1 eq.) was dissolved in 25 mL of dimethyl sulfoxide in a 1 L Erlenmeyer flask and a stir bar was added to it. Potassium hexafluorophosphate (5.436 g, 29.53 mmol, 16 eq.), DI water (300 mL), and a stir bar was added to another 500 mL beaker. After both the solutions had stirred for 5 min, the KPF6 solution was added to the solution in first beaker resulting in the formation of white solid. The white solid was additionally washed with 50 mL of DI water and 100 mL of diethyl ether. The product was further dried under vacuum until all the volatiles were removed to obtain pure compound 5 (0.799 g, 96% yield).
1H NMR (CD3CN, 500 MHz): δ 8.71 (s, 1H), 7.45 (m, 6H), 7.37 (m, 4H), 4.40 (sept, J = 6.7 Hz, 2H), 1.48 (d, J = 6.7 Hz, 12H). 13C NMR (CD3CN, 126 MHz): δ 132.90, 132.07, 131.34, 131.24, 129.98, 126.63, 52.14, 23.16. 19F NMR (CD3CN, 470 MHz): δ −73.00 (d, J = 706 Hz). 31P NMR (CD3CN, 202 MHz): δ −144.61 (pent, J = 706 Hz) IR: 3151, 1550, 1466, 1446, 1398, 1381, 1363, 1340, 1254, 1191, 1169, 1077, 1013, 942, 794, 761, 750, 736, 699, 664. MALDI HR MS (C21H25N2)+: 305.2011 (found), (C21H27N2)+: 305.2018 (calculated).
1H NMR (CD2Cl2, 500 MHz): δ 7.46 (m, 6H), 7.31 (m, 4H), 4.67 (s, 2H), 4.35 (sept, J = 6.8 Hz, 2H), 1.31 (d, J = 6.7 Hz, 6H), 0.89 (d, J = 6.9 Hz, 6H). 13C NMR (CD2Cl2,126 MHz): δ 164.89, 156.93, 139.91, 130.12, 129.85, 126.45, 70.14, 50.77, 22.20, 20.92. IR: 2975, 1672, 1586, 1547, 1517, 1496, 1454, 1392, 1370, 1336, 1291, 1273, 1242, 1169, 1132, 1079, 1030, 1018, 976, 932, 913, 867, 805, 788, 761, 706, 696, 664, 635, 621, 611, 595, 576, 557 cm−1.
1H NMR (CD2Cl2, 500 MHz): δ 7.14 (m, 6H), 6.95 (m, 4H), 5.33 (s, 2H), 4.10 (sept, J = 6.8 Hz, 2H), 1.41 (d, J = 6.8 Hz, 6H), 1.13 (d, J = 6.8 Hz, 6H). 13C NMR (CD2Cl2, 126 MHz): δ 167.90, 157.50, 133.37, 129.11, 128.96, 128.64, 67.99, 51.04, 21.73, 20.80. IR: 2981, 2171, 1672, 1588, 1550, 1495, 1456, 1394, 1373, 1326, 1283, 1212, 1163, 1130, 1025, 985, 927, 858, 796, 777, 759, 707, 649 cm−1.
1H NMR (CDCl3, 500 MHz): δ 7.41 (m, 6H), 7.25 (d, 4H), 4.45 (sept, J = 6.8 Hz, 2H), 1.69 (d, J = 6.8 Hz, 12H). 13C NMR (CDCl3, 126 MHz): δ 131.63, 130.84, 130.50, 129.35, 125.43, 52.11, 23.27. IR: 2979, 2937, 1668, 1623, 1548, 1463, 1443, 1370, 1298, 1262, 1203, 1120, 1075, 1010, 979, 936, 835, 796, 768, 737, 722, 704, 686, 669 cm−1.
Preparation of gold mirrors
Gold mirrors were prepared in house using a physical vapor deposition system equipped with a quartz crystal microbalance (Nano36, Kurt J. Lesker). Glass substrates were cleaned with piranha acid (caution, extremely hazardous) made by mixing concentrated sulfuric acid (VWR) and 30% hydrogen peroxide (VWR) at a 4:1 ratio. Slides were rinsed with copious amounts of water and mounted on the deposition platform. 5 nm of chromium (Kurt J. Lesker) and then 38 nm of gold (Kurt J. Lesker) were deposited via thermal evaporation at a base pressure of <10−5 Torr. Gold coated substrates were rinsed with reagent alcohol, then water, and finally blown dry with nitrogen prior to use.
Citrate-capped gold nanoparticle synthesis
60 nm quasi-spherical AuNPs were synthesized according to a modified Frens method.71,72 180 mL of 0.01% by weight tetrachloroaurate trihydrate salt (Sigma Aldrich) was brought to a boil. Then 120 μL of 1% by weight trisodium citrate (Sigma Aldrich) was added under vigorous stirring. The solution was kept at a boil for 30 minutes. The suspension was then allowed to cool to room temperature slowly. Ultrapure water (>18 MΩ) was generated using a Thermo Fisher Barnstead System in house and used for nanoparticle synthesis. All Glassware used for nanoparticle synthesis was cleaned using Aqua Regia (caution, extremely dangerous) and rinsed with ultrapure water prior to use.Deposition of NHC-CO2 adducts onto gold mirrors
NHC-CO2 adducts were deposited onto gold mirrors in a vacuum oven (National Appliance) at between 105 and 120 °C. Gold mirrors were cut into 12.5 × 12.5 mm squares using a glass cutting knife and cleaned with 0.5 M sulfuric acid prior to use. Approximately, 1 to 2 mg of the solid powder was placed directly onto the mirror and heated under vacuum (<5 Torr) for at least 12 minutes. The substrates were rinsed with reagent alcohol, then water, and finally blown dry with nitrogen. For SERS analysis, 100 μL of 60 nm, citrate-capped gold nanoparticles (AuNPs) were deposited onto the gold mirror and allowed to dry overnight. Finally, the substrates were rinsed again with reagent alcohol, then water, and dried with nitrogen. As a control, gold nanoparticles were drop-cast onto a gold mirror and allowed to dry.Author contributions
G. K. performed ligand synthesis for all NHCs along with their characterization. N. L. D. appended NHCs to gold films and performed LDI-MS measurements. G. H. performed theoretical calculations. P. N. performed single crystal X-ray diffraction analysis. R. L. T. appended NHCs to gold films and performed SERS measurements. S. L. S. performed initial synthesis for chiral compounds. L. J., J. P. C., and D. M. J. designed and supervised the project. Original draft of the manuscript was prepared by G. K. and D. M. J. Review and editing for draft of the manuscript was conducted by G. K., N. L. D., G. H., L. J., J. P. C., and D. M. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under Grants CHE-2108328 (G. K., P. N., and D. M. J.), CHE-2108330 (N. L. D., R. L. T., and J. P. C.), and CHE-1856419 and CHE-2312222 (G. H. and L. J.). Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The authors thank Dr William C. Boggess and the Mass Spectrometry and Proteomics Facility at the University of Notre Dame for use of the Bruker UltrafleXtreme instrument; Shayanta Chowdhury for collecting transmission electron microscopy (TEM) measurements; and the Notre Dame Integrated Imaging Facility for use of the Spectra 300 TEM instrument.References
- S. Rana, A. Bajaj, R. Mout and V. M. Rotello, Monolayer coated gold nanoparticles for delivery applications, Adv. Drug Delivery Rev., 2012, 64, 200–216 CrossRef CAS PubMed.
- R. B. K. C., B. Thapa and N. Bhattarai, Gold nanoparticle-based gene delivery: promises and challenges, Nanotechnol. Rev., 2014, 3, 269–280 Search PubMed.
- M. Falahati, F. Attar, M. Sharifi, A. A. Saboury, A. Salihi, F. M. Aziz, I. Kostova, C. Burda, P. Priecel, J. A. Lopez-Sanchez, S. Laurent, N. Hooshmand and M. A. El-Sayed, Gold nanomaterials as key suppliers in biological and chemical sensing, catalysis, and medicine, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129435 CrossRef CAS PubMed.
- K. Nejati, M. Dadashpour, T. Gharibi, H. Mellatyar and A. Akbarzadeh, Biomedical Applications of Functionalized Gold Nanoparticles: A Review, J. Cluster Sci., 2022, 33, 1–16 CrossRef CAS.
- P. M. Tiwari, K. Vig, V. A. Dennis and S. R. Singh, Functionalized Gold Nanoparticles and Their Biomedical Applications, Nanomaterials, 2011, 1, 31–63 CrossRef CAS PubMed.
- M. Koy, P. Bellotti, M. Das and F. Glorius, N-Heterocyclic carbenes as tunable ligands for catalytic metal surfaces, Nat. Catal., 2021, 4, 352–363 CrossRef CAS.
- G. Pieters and L. J. Prins, Catalytic self-assembled monolayers on gold nanoparticles, New J. Chem., 2012, 36, 1931–1939 RSC.
- A. Gogoi, N. Mazumder, S. Konwer, H. Ranawat, N.-T. Chen and G.-Y. Zhuo, Enantiomeric Recognition and Separation by Chiral Nanoparticles, Molecules, 2019, 24, 1007 CrossRef CAS PubMed.
- H. Li, X. Gao, C. Zhang, Y. Ji, Z. Hu and X. Wu, Gold-Nanoparticle-Based Chiral Plasmonic Nanostructures and Their Biomedical Applications, Biosensors, 2022, 12, 957 CrossRef CAS PubMed.
- C. Gautier and T. Bürgi, Chiral Gold Nanoparticles, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS PubMed.
- Y. Wang, X. Zhao, Z. Yu, Z. Xu, B. Zhao and Y. Ozaki, A Chiral-Label-Free SERS Strategy for the Synchronous Chiral Discrimination and Identification of Small Aromatic Molecules, Angew. Chem., Int. Ed., 2020, 59, 19079–19086 CrossRef CAS PubMed.
- I. I. S. Lim, D. Mott, M. H. Engelhard, Y. Pan, S. Kamodia, J. Luo, P. N. Njoki, S. Zhou, L. Wang and C. J. Zhong, Interparticle Chiral Recognition of Enantiomers: A Nanoparticle-Based Regulation Strategy, Anal. Chem., 2009, 81, 689–698 CrossRef CAS PubMed.
- I. A. Stoian, B.-C. Iacob, J. P. P. Ramalho, I. O. Marian, V. Chiş, E. Bodoki and R. Oprean, A chiral electrochemical system based on l-cysteine modified gold nanoparticles for propranolol enantiodiscrimination: Electroanalysis and computational modelling, Electrochim. Acta, 2019, 326, 134961 CrossRef CAS.
- M. Matsunaga, T. Nakanishi, T. Asahi and T. Osaka, Effect of surface coverage of gold(111) electrode with cysteine on the chiral discrimination of DOPA, Chirality, 2007, 19, 295–299 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Carbene Ligands in Surface Chemistry: From Stabilization of Discrete Elemental Allotropes to Modification of Nanoscale and Bulk Substrates, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Self-assembled monolayers of thiols and dithiols on gold: new challenges for a well-known system, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- L. Srisombat, A. C. Jamison and T. R. Lee, Stability: A key issue for self-assembled monolayers on gold as thin-film coatings and nanoparticle protectants, Colloids Surf., A, 2011, 390, 1–19 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, N-Heterocyclic Carbenes in Materials Chemistry, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- G. Kaur, R. L. Thimes, J. P. Camden and D. M. Jenkins, Fundamentals and applications of N-heterocyclic carbene functionalized gold surfaces and nanoparticles, Chem. Commun., 2022, 58, 13188–13197 RSC.
- S. Engel, E.-C. Fritz and B. J. Ravoo, New trends in the functionalization of metallic gold: from organosulfur ligands to N-heterocyclic carbenes, Chem. Soc. Rev., 2017, 46, 2057–2075 RSC.
- J. F. DeJesus, M. J. Trujillo, J. P. Camden and D. M. Jenkins, N-Heterocyclic Carbenes as a Robust Platform for Surface-Enhanced Raman Spectroscopy, J. Am. Chem. Soc., 2018, 140, 1247–1250 CrossRef CAS PubMed.
- M. J. Trujillo, S. L. Strausser, J. C. Becca, J. F. DeJesus, L. Jensen, D. M. Jenkins and J. P. Camden, Using SERS To Understand the Binding of N-Heterocyclic Carbenes to Gold Surfaces, J. Phys. Chem. Lett., 2018, 9, 6779–6785 CrossRef CAS PubMed.
- L. M. Sherman, S. L. Strausser, R. K. Borsari, D. M. Jenkins and J. P. Camden, Imidazolinium N-Heterocyclic Carbene Ligands for Enhanced Stability on Gold Surfaces, Langmuir, 2021, 37, 5864–5871 CrossRef CAS PubMed.
- B. W. Gung, L. N. Bailey, D. T. Craft, C. L. Barnes and K. Kirschbaum, Preparation and Characterization of Two New N-Heterocyclic Carbene Gold(I) Complexes and Comparison of Their Catalytic Activity to Au(IPr)Cl, Organometallics, 2010, 29, 3450–3456 CrossRef CAS.
- B. W. Gung, M. R. Holmes, C. A. Jones, R. Ma and C. L. Barnes, Structure–enantioselectivity correlation in NHC–Au(I) catalysis for 1,6-enynecyclizations, Tetrahedron Lett., 2016, 57, 3912–3915 CrossRef CAS.
- M. R. Holmes, J. F. Manganaro, C. L. Barnes and B. W. Gung, Synthesis and characterization of novel chiral [(NHC)Au(I)Cl] complexes: Featuring ortho-biphenyl substituents, J. Organomet. Chem., 2015, 795, 18–24 CrossRef CAS.
- D. Banerjee, A. K. Buzas, C. Besnard and E. P. Kündig, Chiral N-Heterocyclic Carbene Gold Complexes: Synthesis, Properties, and Application in Asymmetric Catalysis, Organometallics, 2012, 31, 8348–8354 CrossRef CAS.
- E. P. Kündig, T. M. Seidel, Y.-x. Jia and G. Bernardinelli, Bulky Chiral Carbene Ligands and Their Application in the Palladium-Catalyzed Asymmetric Intramolecular α-Arylation of Amides, Angew. Chem., Int. Ed., 2007, 46, 8484–8487 CrossRef PubMed.
- L. Benhamou, C. Besnard and E. P. Kündig, Chiral PEPPSI Complexes: Synthesis, Characterization, and Application in Asymmetric Suzuki–Miyaura Coupling Reactions, Organometallics, 2014, 33, 260–266 CrossRef CAS.
- M. Michalak and W. Kośnik, Chiral N-heterocyclic Carbene Gold Complexes: Synthesis and Applications in Catalysis, Catalysts, 2019, 9, 890 CrossRef CAS.
- J. Ren, M. Freitag, C. Schwermann, A. Bakker, S. Amirjalayer, A. Rühling, H.-Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, A Unidirectional Surface-Anchored N-Heterocyclic Carbene Rotor, Nano Lett., 2020, 20, 5922–5928 CrossRef CAS PubMed.
- P. J. Czerwiński and M. Michalak, Synthetic Approaches to Chiral Non-C2-symmetric N-Heterocyclic Carbene Precursors, Synthesis, 2019, 51, 1689–1714 CrossRef.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Synthetic Routes to N-Heterocyclic Carbene Precursors, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Chiral NHC–metal-based asymmetric catalysis, Coord. Chem. Rev., 2012, 256, 804–853 CrossRef CAS.
- V. César, S. Bellemin-Laponnaz and L. H. Gade, Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis, Chem. Soc. Rev., 2004, 33, 619–636 RSC.
- D. Janssen-Müller, C. Schlepphorst and F. Glorius, Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- V. Paradiso, V. Bertolasi, C. Costabile and F. Grisi, Ruthenium olefin metathesis catalysts featuring unsymmetrical N-heterocyclic carbenes, Dalton Trans., 2016, 45, 561–571 RSC.
- V. Paradiso, C. Costabile and F. Grisi, NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis, Molecules, 2016, 21, 117 CrossRef PubMed.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed.
- G. Lovat, E. A. Doud, D. Lu, G. Kladnik, M. S. Inkpen, M. L. Steigerwald, D. Cvetko, M. S. Hybertsen, A. Morgante and X. Roy, Determination of the structure and geometry of N-heterocyclic carbenes on Au (111) using high-resolution spectroscopy, Chem. Sci., 2019, 10, 930–935 RSC.
- J. F. DeJesus, L. M. Sherman, D. J. Yohannan, J. C. Becca, S. L. Strausser, L. F. P. Karger, L. Jensen, D. M. Jenkins and J. P. Camden, A Benchtop Method for Appending Protic Functional Groups to N-Heterocyclic Carbene Protected Gold Nanoparticles, Angew. Chem., Int. Ed., 2020, 59, 7585–7590 CrossRef CAS PubMed.
- F. Guillen, C. L. Winn and A. Alexakis, Enantioselective copper-catalyzed conjugate addition using chiral diaminocarbene ligands, Tetrahedron: Asymmetry, 2001, 12, 2083–2086 CrossRef CAS.
- V. Paradiso, V. Bertolasi and F. Grisi, Novel Olefin Metathesis Ruthenium Catalysts Bearing Backbone-Substituted Unsymmetrical NHC Ligands, Organometallics, 2014, 33, 5932–5935 CrossRef CAS.
- C. O'Beirne, H. T. Althani, O. Dada, J. Cassidy, K. Kavanagh, H. Müller-Bunz, Y. Ortin, X. Zhu and M. Tacke, Novel derivatives of the antibiotic NHC–Ag(I) drug candidate SBC3: Synthesis, biological evaluation and 109Ag NMR studies, Polyhedron, 2018, 149, 95–103 CrossRef.
- H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, CO2 Adducts of N-Heterocyclic Carbenes: Thermal Stability and Catalytic Activity toward the Coupling of CO2 with Epoxides, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS PubMed.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, A Systematic Investigation of Factors Influencing the Decarboxylation of Imidazolium Carboxylates, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- S. Naumann, F. G. Schmidt, R. Schowner, W. Frey and M. R. Buchmeiser, Polymerization of methyl methacrylate by latent pre-catalysts based on CO 2-protected N-heterocyclic carbenes, Polym. Chem., 2013, 4, 2731–2740 RSC.
- Z. Kelemen, B. Péter-Szabó, E. Székely, O. Hollóczki, D. S. Firaha, B. Kirchner, J. Nagy and L. Nyulászi, An Abnormal N-Heterocyclic Carbene–Carbon Dioxide Adduct from Imidazolium Acetate Ionic Liquids: The Importance of Basicity, Chem. – Eur. J., 2014, 20, 13002–13008 CrossRef CAS PubMed.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Surface-Enhanced Raman Spectroscopy, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- N. L. Dominique, S. L. Strausser, J. E. Olson, W. C. Boggess, D. M. Jenkins and J. P. Camden, Probing N-Heterocyclic Carbene Surfaces with Laser Desorption Ionization Mass Spectrometry, Anal. Chem., 2021, 93, 13534–13538 CrossRef CAS PubMed.
- N. L. Dominique, R. Chen, A. V. B. Santos, S. L. Strausser, T. Rauch, C. Q. Kotseos, W. C. Boggess, L. Jensen, D. M. Jenkins and J. P. Camden, Ad aurum: tunable transfer of N-heterocyclic carbene complexes to gold surfaces, Inorg. Chem. Front., 2022, 9, 6279–6287 RSC.
- N. L. Dominique, I. M. Jensen, G. Kaur, C. Q. Kotseos, W. C. Boggess, D. M. Jenkins and J. P. Camden, Giving Gold Wings: Ultrabright and Fragmentation Free Mass Spectrometry Reporters for Barcoding, Bioconjugation Monitoring, and Data Storage, Angew. Chem., Int. Ed., 2023, 62, e202219182 CrossRef CAS PubMed.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Beyond Conventional N-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- J. Li, J. Jiao, C. Zhang, M. Shi and J. Zhang, Facile Syntheses of N-Heterocyclic Carbene Precursors through I2- or NIS-Promoted Amidiniumation of N-Alkenyl Formamidines, Chem. – Asian J., 2016, 11, 1361–1365 CrossRef CAS PubMed.
- R. L. Thimes, A. V. B. Santos, R. Chen, G. Kaur, L. Jensen, D. M. Jenkins and J. P. Camden, Using Surface-Enhanced Raman Spectroscopy to Unravel the Wingtip-Dependent Orientation of N-Heterocyclic Carbenes on Gold Nanoparticles, J. Phys. Chem. Lett., 2023, 14, 4219–4224 CrossRef CAS PubMed.
- T. B. Demille, R. A. Hughes, N. Dominique, J. E. Olson, S. Rouvimov, J. P. Camden and S. Neretina, Large-area periodic arrays of gold nanostars derived from HEPES-, DMF-, and ascorbic-acid-driven syntheses, Nanoscale, 2020, 12, 16489–16500 RSC.
- A. S. Preston, R. A. Hughes, N. L. Dominique, J. P. Camden and S. Neretina, Stabilization of Plasmonic Silver Nanostructures with Ultrathin Oxide Coatings Formed Using Atomic Layer Deposition, J. Phys. Chem. C, 2021, 125, 17212–17220 CrossRef CAS.
- Z. R. Lawson, A. S. Preston, M. T. Korsa, N. L. Dominique, W. J. Tuff, E. Sutter, J. P. Camden, J. Adam, R. A. Hughes and S. Neretina, Plasmonic Gold Trimers and Dimers with Air-Filled Nanogaps, ACS Appl. Mater. Interfaces, 2022, 14, 28186–28198 CrossRef CAS PubMed.
- L. Kolářová, L. Prokeš, L. Kučera, A. Hampl, E. Peňa-Méndez, P. Vaňhara and J. Havel, Clusters of Monoisotopic Elements for Calibration in (TOF) Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2017, 28, 419–427 CrossRef PubMed.
- S. J. A. van Gisbergen, J. G. Snijders and E. J. Baerends, A density functional theory study of frequency–dependent polarizabilities and van der Waals dispersion coefficients for polyatomic molecules, J. Chem. Phys., 1995, 103, 9347–9354 CrossRef CAS.
- E. J. Baerends, T. Ziegler, A. J. Atkins, J. Autschbach, O. Baseggio, D. Bashford, et al., ADF 2021.204, SCM, Theoretical Chemistry, 2021, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- R. Rüger, M. Franchini, T. Trnka, A. Yakovlev, E. v. Lenthe, P. Philipsen, T. v. Vuren, B. Klumpers and T. Soini, AMS 2021.204, SCM, Theoretical Chemistry, 2021, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824 CrossRef PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, Optimized Slater-type basis sets for the elements 1–118, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, The zero–order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, Relativistic regular two–component Hamiltonians, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- Schrödinger, LLC, The PyMOL molecular graphics sytem, version 2.2.0 2018 Search PubMed.
- G. Frens, Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- A. S. Moody, T. D. Payne, B. A. Barth and B. Sharma, Surface-enhanced spatially-offset Raman spectroscopy (SESORS) for detection of neurochemicals through the skull at physiologically relevant concentrations, Analyst, 2020, 145, 1885–1893 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and theoretical details, including selected NMRs, IRs, MS, UV-vis, SERS, and TEM images. CCDC 2282623–2282625. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01541f |
| This journal is © the Partner Organisations 2023 |