
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic polymeric nanoparticles enable homogenous rhodium-catalysed NH insertion reactions in living cells†
Anjana
Sathyan‡
 ,
Tessa
Loman‡
,
Linlin
Deng‡
and
Anja R. A.
Palmans
*
,
Tessa
Loman‡
,
Linlin
Deng‡
and
Anja R. A.
Palmans
*
Institute for Complex Molecular Systems, Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
First published on 19th July 2023
Abstract
Rh-catalysed NH carbene insertion reactions were exported to living cells with help of amphiphilic polymeric nanoparticles. Hereto, hydrophobic dirhodium carboxylate catalysts were efficiently encapsulated in amphiphilic polymeric nanoparticles comprising dodecyl and Jeffamine as side grafts. The developed catalytic nanoparticles promoted NH carbene insertions between α-keto diazocarbenes and 2,3-diaminonaphthalene, followed by intramolecular cyclisation to form fluorescent or biologically active benzoquinoxalines. These reactions were studied in reaction media of varying complexity. The best-performing catalyst was exported to HeLa cells, where fluorescent and cytotoxic benzoquinoxalines were synthesized in situ at low catalyst loading within a short time. Most of the developed bioorthogonal transition metal catalysts reported to date are easily deactivated by the reactive biomolecules in living cells, limiting their applications. The high catalytic efficiency of the Rh-based polymeric nanoparticles reported here opens the door to expanding the repertoire of bioorthogonal reactions and is therefore promising for biomedical applications.
Introduction
The in situ synthesis of bio-active agents is paramount for the development of new innovative therapies. The development of bioorthogonal, new-to-nature reactions is a promising approach and advances in this field have caused a paradigm shift in the field of chemical biology by providing new tools for biomedical research. From studying biomolecules in their native environment to using the same chemistry to release therapeutic drugs directly near tumour sites of human cancer patients, bioorthogonal chemistry has flourished in the past decade.1–6 Even though some of these bioorthogonal reactions have unprecedented rate, stoichiometric amounts of chemical agents in vivo are still required. Introduction of a catalytic component therefore holds great potential, as this can provide signal amplification even at low concentrations. In line with this view, the field has expanded towards transition-metal-catalysed bioorthogonal reactions. These metal catalysts can be thought of as in situ chemical “factories” that generate bioactive compounds such as drugs or imaging agents repeatedly from non-toxic starting materials at controlled rates and at a place of interest.7 However, performing metal catalysis in living environments is a challenging task. The stability of the catalyst is the most important aspect, which must be maintained without compromising the catalyst's activity. Moreover, the catalyst should be biocompatible and ideally water-soluble. Finally, catalysts have to be targeted specifically to the sites of interest.Both heterogeneous and homogenous bioorthogonal catalysts have been developed in the past decade to meet the beforementioned requirements. This was achieved either by ligand modification or incorporation of the transition metal catalysts (TMCs) in synthetic or natural scaffolds, thereby enabling them to remain in a biological environment and perform reactions.8–17 Though many studies were successful in vivo, the research in this area is still in the early stage. Firstly, this is because often (super)stochiometric amounts of TMCs are required to synthesize bioactive agents in living cells or to activate pro-drugs.15,18–20 The term ‘catalysis’ has been used over the years but in the strict definition of the word many of the developed chemistries were not truly ‘catalytic’. There are only few examples where real catalytic amounts of TMCs were reported to induce biological activity in vitro.17,21–23 Additionally, so far the focus has mainly been on coupling and cleavage reactions catalysed by commonly used TMCs such as Pd, Cu, Ru and Au in biological media.8,18,23–30 In order to push the boundaries of the field for biological applications, it important to expand the bioorthogonal toolbox both in terms of developing new and efficient catalysts and thereby new bioorthogonal reactions.
In this regard, metal carbenes are an interesting class of molecules that generate novel bioorthogonal reactions,31–36 but are less explored in mammalian cell environments. Metal carbenes are formed from diazo compounds and TMCs under mild conditions, making it possible to translate them to water or complex media. Moreover, diazo compounds have been known to endure cell metabolism and were utilized for bioorthogonal reactions in cellulo.37,38 In pioneering work by Mascareñas and co-workers, Cu-catalysed NH carbene insertion reactions were exported to generate bioactive agents in situ in HeLa cells.39 Copper-catalysed reactions are important for proof-of-concept, but their feasibility in biomedical applications may be hindered due to their potential toxicity.40 Other less toxic metals such as iron, ruthenium, iridium and rhodium also can catalyse NH insertion reactions and are therefore interesting to further explore.41–46 In particular, rhodium catalysts are attractive for NH carbene insertion reactions in biological media, as they show low toxicity and high stability, where both can be tuned by proper ligand choice.47 Dirhodium carboxylate catalysts have already been explored to study biological systems exploiting carbene chemistry. Francis and co-workers showed that Rh(II)acetate can catalyse NH insertions to modify tryptophan residues, while Gillingham and co-workers showed the modification of nucleic acids in the same way.48–50 Previous work in biological environments predominantly focused on the use of water-soluble Rh(II)acetate. However, many hydrophobic dirhodium catalysts have proven to be very reactive towards a wide range of synthetic transformations such as cyclopropanation, C–H and X–H insertion, aromatic substitution and ylide formation reactions in organic solvents.51 In order to expand their use one has to accommodate for their mostly hydrophobic and thus insoluble character. The catalysts should therefore be solubilized and protected in water-soluble polymeric scaffolds to fully utilize their reactivity in aqueous environments.
An amphiphilic polymer with randomly distributed hydrophobic dodecyl grafts and hydrophilic Jeffamine M-1000 grafts can collapse into single-chain polymeric nanoparticles (SCPNs) in water.52 These nanometre-sized particles comprise hydrophobic domains in the interior that allow catalyst and substrate accumulation in aqueous solution. Such nanoparticles can be classified to the group of so-called bioorthogonal nanozymes, nanomaterials with enzyme-like properties.7,53 The nanomaterial scaffolds not only solubilize the catalysts in water, but also protect them from deactivating components in living cells and minimize overall cytotoxicity. They allow substrate accumulation near the catalyst site by the “concentrator effect”,54 enhancing the reaction rate at low concentrations. Further, the cellular uptake of these nanozymes can be modulated by changing the surface functionalities. They hold great promise for in vivo applications as they can allow increased circulation times and active targeting if functionalized.7 Rotello and co-workers have made significant contributions to this field by developing bioorthogonal polyzymes and showing the efficacy of flash nanoprecipitation strategy to increase catalyst loading and turnover frequency.55–57 Previously in our group, Liu et al. explored the potential of these nanoparticles to encapsulate Pd(II) and Cu(I) catalysts to perform bond-cleavage reactions in living cells. However, despite the promising results in vitro their catalytic activity was significantly diminished in complex environment due to the presence of deactivating agents.15 We believe the robust and remarkable activity of dirhodium carboxylates can help us overcome these issues when sequestrated inside our polymeric nanoparticles.
In this work, we investigated the possibility of using Rh carbene chemistry in living cells for the in situ synthesis of imaging or bioactive agents with the help of nanomaterial scaffolds, namely amphiphilic polymeric nanoparticles. After selection of the most active catalyst, we performed the annulation of α-keto diazocarbenes with 2,3-diaminonaphthalene in the presence of HeLa cells to form benzoquinoxaline products, a catalytic reaction initiated by an N–H carbene insertion by Rh catalysts following the work of Mascarenas.39 The results show that quinoxalines readily form using catalytic amounts of Rh embedded in amphiphilic polymeric nanoparticles.
Results and discussion
Encapsulation of dirhodium(II) carboxylate catalysts in amphiphilic polymeric nanoparticles for NH carbene insertion reactions
Dirhodium(II) carboxylates constitute a paddle-wheel-like structure formed by the bridging carboxylate ligands and a unique di-rhodium bridge that offers exceptional stability to these catalysts.58 Notably, they have two vacant axial positions that allow substrate binding, in this case for carbene formation.58,59 The hydrophobicity of these catalysts is an important factor to consider when they have to be efficiently encapsulated inside amphiphilic polymeric nanoparticles. Therefore, we chose three highly hydrophobic dirhodium carboxylate catalysts, C1–C3, with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values >10 (Fig. 1) for encapsulation. Rh2(OAC)4, C4, has low hydrophobicity (Fig. 1B) and was used as a reference as it is water soluble. Even though C1–C2 are well studied chiral catalysts, we are only interested in their hydrophobicity, stability and reactivity.
P values >10 (Fig. 1) for encapsulation. Rh2(OAC)4, C4, has low hydrophobicity (Fig. 1B) and was used as a reference as it is water soluble. Even though C1–C2 are well studied chiral catalysts, we are only interested in their hydrophobicity, stability and reactivity.
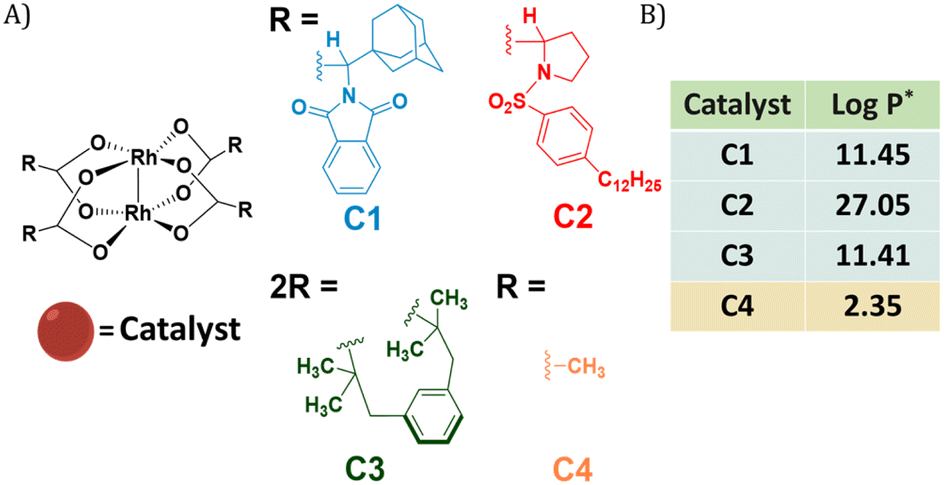 | ||
| Fig. 1 (A) Chemical structures of dirhodium(II) carboxylates-based catalysts; (B) logP values of the catalysts. *calculated using MarvinSketch 22.16. | ||
The selected dirhodium carboxylate catalysts C1–C4 were encapsulated in the hydrophobic interior of a polyacrylamide based amphiphilic polymer P1 (Fig. 2A and B), which contained 20% dodecyl grafts for hydrophobicity and 80% Jeffamine M-1000 grafts for hydrophilicity (DP = 100, Đ = 1.21). We chose the simplest design of amphiphilic polymer previously reported by our group that collapsed into SCPNs in water for loading the catalysts.52 Encapsulation of the catalysts was performed using a modification of a previously reported protocol as described in ESI.†52
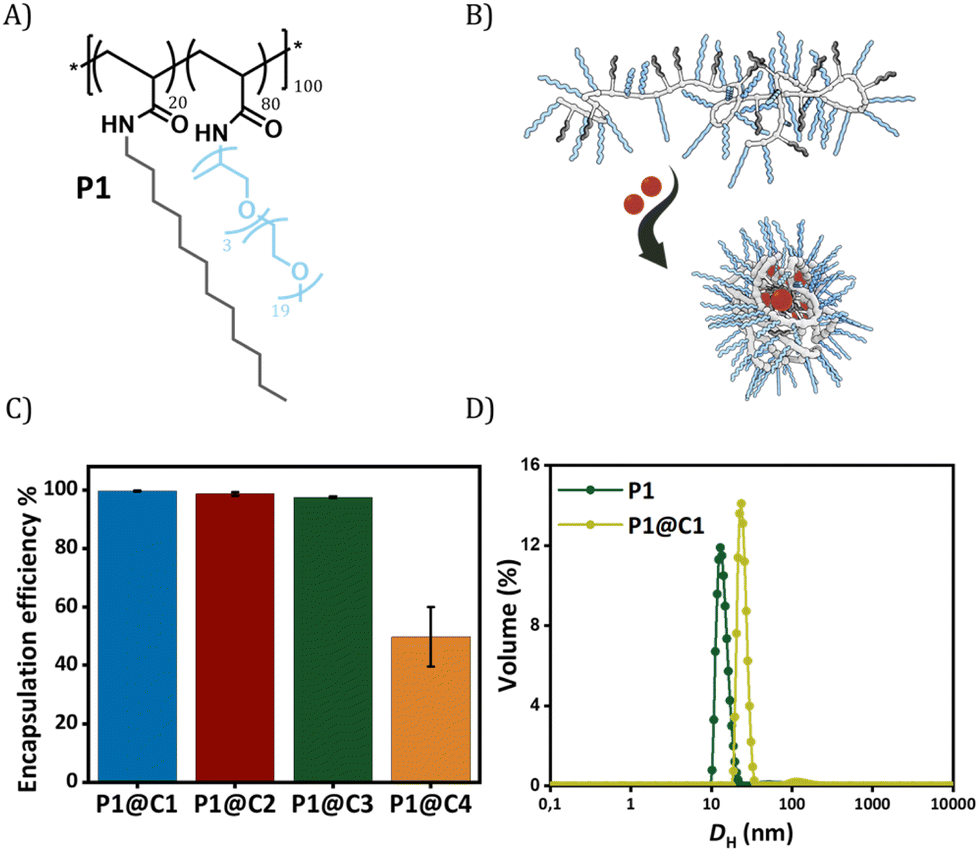 | ||
| Fig. 2 (A) Chemical structure of amphiphilic random copolymer P1, DP = 100, Đ = 1.21; (B) representation of the encapsulation of catalysts C1–C4 (red spheres) in the hydrophobic interior of polymeric nanoparticles; (C) encapsulation efficiency of the dirhodium carboxylate catalysts C1–C4 in polymeric nanoparticles calculated from the leached-out rhodium using ICP-OES. [P1] = 1 mg mL−1, P1:C = 1:5 (molar ratio), [Rh] = 10 mg L−1 before filtration. The results for the encapsulation efficiency are the average of three independent measurements; (D) volume distribution of the hydrodynamic diameter DH of P1 and P1@C1 nanoparticles ([P] = 1.0 mg mL−1, T = 20 °C, [P1]:[C1] = 1:5 (molar ratio)) monitored using DLS measurements. | ||
In order to check the efficiency of P1 to encapsulate the catalysts, the amount of rhodium that leached out from the nanoparticles was monitored using Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES). From here onwards, we represent catalyst encapsulated inside polymeric nanoparticles in solution as P1@C1–C4. The solutions of P1@C1–C4 in water with a total rhodium concentration of 10 mg L−1 were filtered using centrifugal filters with a molecular weight cut-off of 50 kDa, to ensure separation of polymeric nanoparticles (MWcal ∼ 98 kDa) from the aqueous solution. Therefore, any leached out Rh catalysts (MW = 400–1800 g mol−1) will remain in the filtered solution and the Rh concentration can be monitored using ICP-OES. ICP-OES measurements revealed that for hydrophobic catalysts C1–C3, only 0.03–0.2 mg L−1 rhodium leached out while in case of hydrophilic catalyst C4 it was 5 mg L−1. This clearly indicated that the hydrophobic catalysts C1–C3 were encapsulated with 97–99% efficiency in the hydrophobic interior of P1 in contrast to hydrophilic catalyst C4 (50% efficiency) as hypothesized (Fig. 2C).
Dynamic light scattering (DLS) measurements were performed to study the influence of the rhodium encapsulation on the particle size. The hydrodynamic diameter (DH) of polymeric nanoparticles formed from P1 without catalysts amounted to ∼14 nm (Fig. 2D). For P1@C1 with a catalyst incorporation ratio of 1:5 (P1:C1 molar ratio), DH increased to ∼20 nm, suggesting that the polymeric nanoparticles retain a small size, even after encapsulation of the catalyst. However, the size increase also indicates that there is more than 1 polymer chain present in the nanoparticles, which is why we refer to these systems as “amphiphilic polymeric nanoparticles” instead of “single-chain polymeric nanoparticles”. On varying the catalyst incorporation ratio from 1:1 to 1:10 (P1:C1 molar ratio), a size increase with increasing amount of catalyst encapsulated was observed (for more details see ESI†). We used a ratio of 1:5 (P1:C1) for catalysis studies to keep the polymer concentration low while maintaining high catalyst loading and minimizing aggregation.
Given the successful encapsulation of hydrophobic rhodium catalysts, substrates for the NH carbene insertion reactions were selected as reported earlier by Mascareñas and co-workers (Fig. 3).39 The diazo substrates 2a–c were synthesized according to previously reported protocols.39 The reaction of 2,3-diaminonaphthalene 1 with various diazo substrates 2a–c affords benzoquinoxalines 3a–c, by a process initiated by a NH carbene insertion reaction followed by an intramolecular imine condensation and oxidative aromatization (Fig. 3B).60 The products formed are fluorescent (3a) or bioactive (3c). Fluorescent benzoquinoxaline 3a allows visualization of product formation in living cells while 3c induces cytotoxic effects by promoting mitochondrial fragmentation and depolarization.39 The in situ reaction kinetics can be monitored using fluorescence spectroscopy over time.
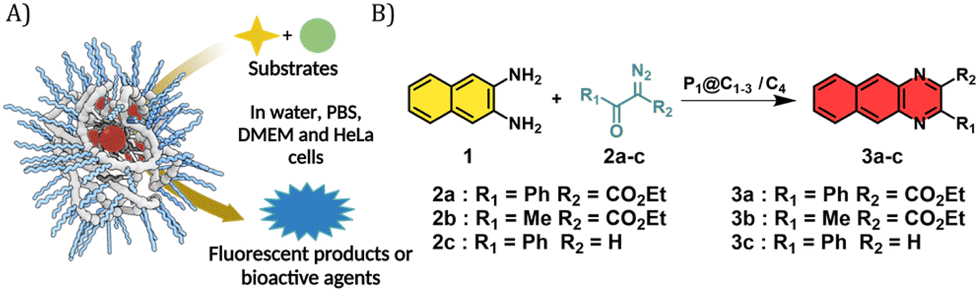 | ||
| Fig. 3 (A) Representation of reaction involving two substrates using catalytic polymeric nanoparticles; (B) formation of benzoquinoxalines 3a–c initiated by NH insertion followed by intramolecular cyclisation. | ||
Catalytic efficiency of the selected catalysts
P1@C1–C3 were further tested for their efficiency to catalyse the NH carbene insertion reactions, where 2,3-diaminonaphthalene 1 and diazo substrate 2a were used as substrates. For comparison, hydrophilic catalyst C4 was used by directly dissolving in water in the absence of polymeric nanoparticles. Reactions were performed at 500 μM concentration of substrates 1 and 2a, and 10 mol% of catalyst (C1–C4) in the appropriate medium (water, PBS and Dulbecco's modified Eagle's medium (DMEM)) at physiological temperature (37 °C). The polymer to catalyst molar ratio was fixed at 1:5 and the concentration of polymer in the reaction mixture was ∼1 mg mL−1.
The HPLC-UV chromatogram of the reaction mixture after 24 h indicated that for P1@C1 diazo substrate 2a was completely consumed, leading to the formation of product 3a (Fig. S10A, see ESI†). For P1@C2 the majority of the substrate 2a remained in the reaction mixture after 24 h in water (Fig. S10B, see ESI†). P1@C3 performed better than P1@C2, with a significant amount of 3a formed. However, it was not as efficient as P1@C1 as full consumption of diazo substrate 2a was not observed (Fig. S10C, see ESI†). With the hydrophilic catalyst C4, a large amount of diazo substrate 2a remained in the catalytic mixture, and only a negligible amount of product 3a was formed (Fig. S10D, see ESI†). The consumption of 2,3-diaminonaphthalene 1 was confirmed from the decrease in the fluorescence intensity over time as 1 was not trackable with HPLC-UV (Fig. 4A). Further, the rate of the reaction was calculated from the decrease in fluorescence intensity of 2,3-diaminonaphthalene 1 in the first 180 min (omitting the first 10 min to allow sample equilibration) for P1@C1–C3 and C4 (Fig. 4B). The catalytic efficiency followed the order P1@C1 > P1@C3 > P1@C2 > C4; the same trend as observed from product formation in the HPLC chromatograms. The hydrophobic catalysts encapsulated in polymeric nanoparticles efficiently catalysed the reaction as compared to free water-soluble catalyst C4 highlighting the advantage of this system. The hydrophobic domains in these nanoparticles allow substrate solubilisation thereby resulting in the increase in their local concentration near catalytic sites enhancing the rate of reaction. The same results and trends were observed in case of PBS or DMEM as solvent. We did not observe any side product formation such as OH-insertion product or dimerized product from the diazo substrate, which was confirmed by control experiments (for more information see ESI†).
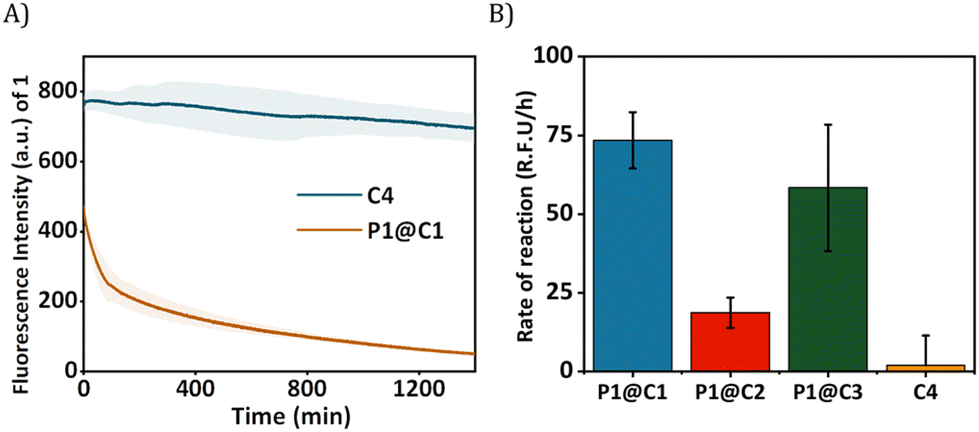 | ||
| Fig. 4 (A) Fluorescence kinetic profile of reaction progress of 1 and 2a catalysed by P1@C1 and C4 over time, wavy shadows depict the range of error in the kinetic curves, λex = 330 nm and λem = 386 nm; (B) comparison of the rate of the same reaction catalysed by P1@C1, P1@C2, P1@C3 and C4 (t0 = 10 min and tF = 180 min); reaction conditions, [1] = [2a] = 500 μM, [C] = 50 μM, [P] = 1 mg mL−1, P1:C1–C3 = 1:5 (molar ratio), T = 37 °C in deionised water. The results are the average of three independent measurements. | ||
The best performing catalyst P1@C1 was therefore used for further catalytic studies. As an additional control, we also tested the efficiency of free catalyst C1 without the presence of amphiphilic polymer but the solution was highly heterogeneous due to high hydrophobicity and conversion was poor when compared to P1@C1 (Fig. S12, see ESI†). This also highlights the importance of amphiphilic polymeric nanoparticles to achieve homogeneity when exporting these highly active hydrophobic catalysts to biological environment.
Pushing the limits of catalytic efficiency of the best performing catalyst
Reactions were performed under limiting conditions to further test the efficiency of P1@C1 before exporting it to in vitro conditions. Reactions of 1 and 2a were performed in dilute conditions by lowering the concentration of substrates to 100 μM and 50 μM, respectively, and keeping the amount of catalyst constant at 10 mol% in water. Product formation was observed within 3 h, leading to almost full conversion of 2a to 3a within 24 h as evidenced by HPLC-UV (Fig. S11, see ESI†).Further, the reaction was also performed at 5 mol% of catalyst concentration, which also resulted in significant product formation within 6 h and reaching almost full conversion within 24 h (Fig. S13, see ESI†). Given that the product 3a was formed with decreased substrate and catalyst concentrations, P1@C1 proved promising to work under limiting conditions such as dilute concentrations similar to the environment of living cells.
The next step was to further increase the medium complexity and test the efficiency of catalyst in the presence of serum proteins. For this, the same reaction was performed in cell culture medium DMEM supplemented with 10% Fetal Bovine Serum proteins (FBS) with 10 mol% catalyst at 500 μM substrate concentration. The HPLC chromatogram indicated the complete consumption of 2a and the formation of 3a. The HPLC chromatogram was compared to a control experiment where C4 was used as catalyst, which showed no peaks for the product, even after 24 h (Fig. 5A). Notably, the rate of the reaction did not diminish significantly when catalysed by P1@C1 when medium complexity was increased and the reaction did not proceed in the absence of catalyst (Fig. 5B).
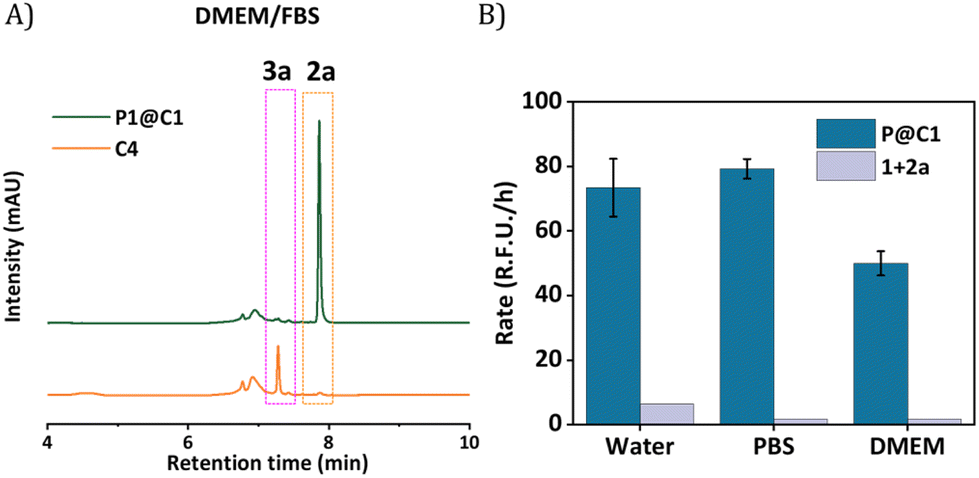 | ||
| Fig. 5 (A) HPLC-UV chromatogram of the reaction of 1 and 2a catalysed by P1@C1, monitored at 279 nm, [1] = [2a] = 500 μM in water [C1] = 50 μM in DMEM + 10%FBS (unassigned peaks arise from the presence of proteins in FBS in the reaction mixture); (B) comparison of the rate of the same reaction catalysed by P1@C1 and uncatalysed reaction in water, PBS and DMEM (t0 = 10 min and tF = 180 min) determined from fluorescence kinetic measurements. Reaction conditions, [1] = [2a] = 500 μM, [C1] = 50 μM, [P] = 1 mg mL−1, P1:C1 = 1:5 (molar ratio), T = 37 °C. The results are the average of three independent measurements. | ||
Despite numerous studies and reports on the development of bioorthogonal catalysts, most of the reported catalysts suffer deactivation from nucleophilic attack or get sequestrated by proteins present in complex biological media. This leads to the requirement of high catalyst concentration for reasonable product formation in the presence of cells. In our case, P1@C1 showed excellent catalytic activity even in the presence of cell culture media and serum proteins at only 10 mol% catalyst concentration and the reaction proceeded to full conversion even at demanding conditions.
Extending the reaction to different diazosubstrates
Given that P1@C1 proved to be a very efficient catalyst, we examined the substrate scope of these reactions in aqueous media and thereby investigated the possibility to synthesize bioactive molecules in the presence of living cells. We started investigating a NH carbene insertion reaction with very hydrophilic diazo substrate 2b (logP = −2.59) and diamine 1, which can form benzoquinoxaline 3b.
In PBS, P1@C1 showed complete consumption of 2b and formation of 3b in 24 h. In case of C4, the reaction did not go to completion even though a significant amount of product was formed (Fig. S14, see ESI†). When increasing the medium complexity to DMEM and DMEM/FBS, C4 did not catalyse the reaction; substrate 2b remained in the reaction mixture and no product formation was observed (Fig. S14B, see ESI†). Surprisingly for P1@C1, there was complete consumption of 2b but the major product peak 3b was missing. Instead, a new peak was observed which was found to be the hydrolysed product of 3b (Fig. S14A and C, see ESI†). We hypothesize that spontaneous hydrolysis of the ester may be brought forward due to the presence of charged groups.61
The next substrate investigated was diazo compound 2c, which can form bioactive cytotoxic compound 3c as previously described.39P1@C1 showed complete consumption of 2c and formation of 3c in 24 h in DMEM and DMEM/FBS (Fig. S16, see ESI†). In case of C4, the reaction did not go to completion and a significant amount of 2c remained in the reaction mixture (Fig. S16, see ESI†).
In short, P1@C1 is not only efficient, but is also a versatile catalyst that can promote NH carbene insertion reactions in different diazo substrates. The hydrophilic nature of diazo compounds did not hinder metal carbene formation in the presence of polymeric nanoparticles and also did not slow down the reaction as full conversion was obtained within 24 h. It is important to note that in all these reactions, only 10 mol% of catalyst C1 was used to achieve full conversion irrespective of the medium complexity.
Exporting reactions to living cells
The reactions in cell culture medium such as DMEM and DMEM/FBS confirmed that P1@C1 is an efficient catalyst and stays catalytically active for prolonged period of time in competitive environments. Furthermore, the fact that only a low catalyst loading is required for complete product conversion made it a promising system to be tested in the presence of living cells. The reaction between 1 and 2a was chosen as a model reaction as it forms fluorescent benzoquinoxaline 3a that allows visualization of the product in living cells.39Before performing the catalytic reaction in the presence of cells, the biocompatibility of catalysts P1@C1 and substrates 2a and 1 was evaluated. Hereto, HeLa cells were incubated with P1@C1, 2a and 1 separately for 24 h, and at varying concentrations. The CCK-8 assay was then utilized to determine the cell viability. As can be seen in Fig. S19 (see ESI),† the viability of HeLa cells remains above 80% with the increase of P1@C1 concentration (up to 15 μM), showing good biocompatibility of catalysts. Similarly, cells treated with substrate 2a in concentrations up to 200 μM exhibit viability above 80% (Fig. S19, ESI†). For substrate 1, a 24 h incubation with HeLa cells at the concentration of 100 μM caused the cell viability to decrease to around 75%, but by reducing the incubation time to 14 h, cell survival was almost completely restored (Fig. S19, ESI†).
Based on the cytotoxicity study, substrates 1 and 2a with a concentration of 100 μM were chosen for carrying out the catalytic reaction in the presence of cells. Confocal microscopy was applied to visualize the formed fluorescence product 3a in cells. To corroborate that product 3a can only be obtained by catalysing 1 and 2a using catalyst P1@C1, control experiments were performed in which HeLa cells were incubated with only substrates 1+2a, or one of the substrates (either 1 or 2a) combined with P1@C1 (10 mol%); 1+P1@C1 and 2a+P1@C1. Confocal images of control experiments show that cells incubated with 1+2a and 1+P1@C1 displayed a low fluorescence signal arising from 1 as it is weakly fluorescent when excited at 405 nm (Fig. 6A and B). Accordingly, 2a+P1@C1 did not show any fluorescence (Fig. 6C). When cells were incubated with 1+2a+P1@C1 for 1.5 h, 3 h and 5 h, the confocal images of the catalytic reactions revealed significant intracellular fluorescence, indicating that product 3a is formed, notably within even 1.5 h (Fig. 6D). This indicates that the catalytic efficiency of P1@C1 in the presence of cells is retained and also that the catalyst remains stable. Although it is not clear whether the catalytic reactions occur extracellularly or intracellularly, the most likely scenario is that catalysis occurs extracellularly and that the formed product 3a diffuses into the cells. The nanoparticles we use here are known to be taken up by endocytosis rather slowly (24 h),62 and when cells are incubated with quinoxaline 3a, they become fluorescent, indicating that 3a can cross the cell membrane (Fig. S20†).
 | ||
| Fig. 6 Confocal microscopy images of HeLa cells incubated with (A) 1+2a for 5 h; (B) 1+P1@C1 for 5 h; (C) 2a+P1@C1 for 5 h; (D) 1+2a+P1@C1 for 1.5 h; (E) 1+2a+P1@C1 for 3 h; (F) 1+2a+P1@C1 for 5 h; λex = 405 nm, λem = 579–639 nm, [1] = [2a] = 100 μM and [C1] = 10 μM, T = 37 °C, (P1:C1 = 1:5 molar ratio). | ||
Our next goal was to elicit biological effects in mammalian cells, for which we performed the reaction to form cytotoxic product 3c from substrates 1 and 2c in the presence of catalysts P1@C1. The cytotoxic product 3c belongs to a class of molecules known as Tyrphostins (Tyrosin phosphorylation inhibitors), which have distinct biological profiles due to their tyrosine kinase inhibitory activity.39 To prove that the toxicity originated from product 3c rather than substrates, product 3c and substrate 2c were incubated separately with HeLa cells for their biocompatibility study. After 24 h incubation, the CCK-8 assay demonstrated that substrate 2c is not toxic to cells; the viability of HeLa cells reaches to 90% (Fig. 7A) under various concentrations (up to 200 μM). In contrast, 3c treated cells display a viability decrease of 60% (Fig. 7B). A further viability decrease with increasing concentration of 3c from 25 μM to 100 μM was not observed, indicating the limited capability of 3c for inducing cell apoptosis.
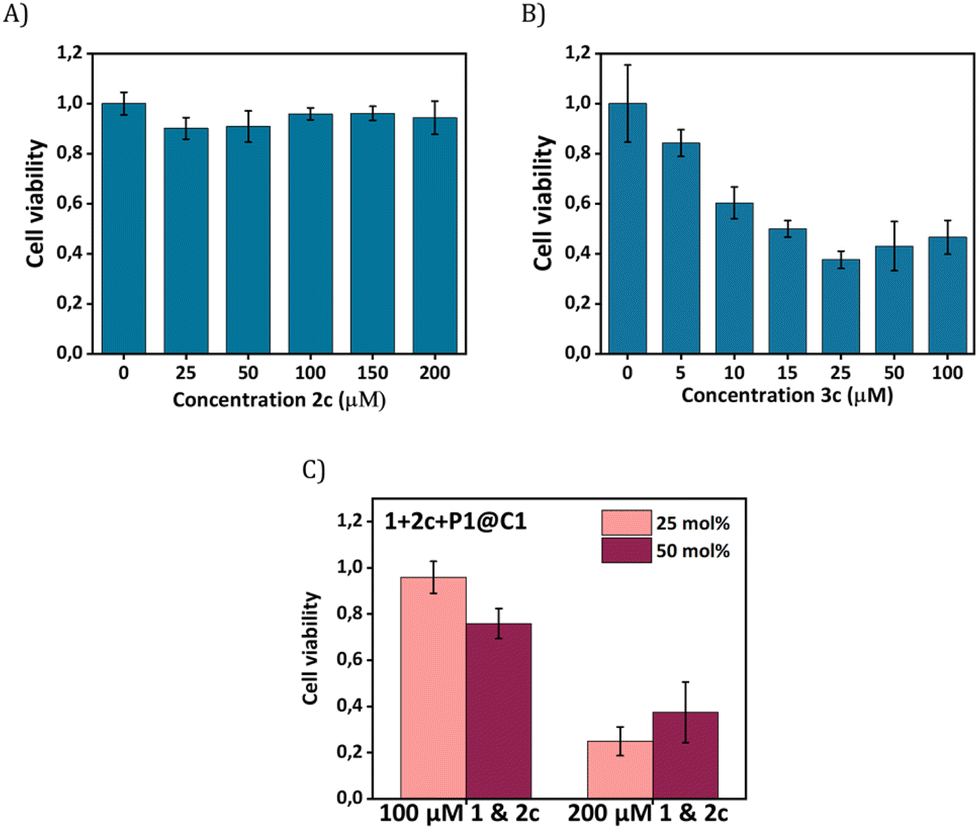 | ||
| Fig. 7 HeLa cells CCK-8 cell viability assays when incubated with (A) 3c for 24 h (25–200 μM); (B) 3c for 24 h (5–100 μM); (C) catalytic reaction 1+2c (100 & 200 μM) using P1@C1 (25 and 50 mol% C1) in HeLa cells after 14 h. | ||
To test whether 3c can be synthesized in situ in the presence of cells by P1@C1, we incubated 1+2c+P1@C1 with HeLa cells and determined the cell viability. Due to the moderate toxicity of 3c, a high substrate concentration of 1 and 2c (100 and 200 μM) with catalyst loading of 25 and 50 mol% was used, aimed to generate a sufficient amount of 3c to induce cell death. A pronounced decrease in cell viability was not observed when the reactions were performed with 100 μM substrates of 1 and 2c for 14 h (Fig. 7C). However, an increased substrate concentration of 200 μM resulted in the cell viability decreasing to less than 40% after 14 h (Fig. 7C). A low catalyst loading of 25 mol% was found to be sufficient for the efficient synthesis of cytotoxicity compound 3cin situ.
All in all, P1@C1 proved to be highly catalytically active in the presence of living mammalian cells with superior efficiency leading to the formation of fluorescent and cytotoxic compounds in situ. Therefore, it has the potential to be developed as a promising catalyst for biomedical applications as quinoxaline scaffolds are present in many biorelevant compounds.
Conclusions
In conclusion, we found that hydrophobic, stable and highly reactive dirhodium catalysts can be efficiently encapsulated in amphiphilic polymeric nanoparticles. An excellent encapsulation efficiency of 97–99% was observed for hydrophobic catalysts with logP > 10. Among the catalysts screened, P1@C1 was found to be the best performing and most efficient bioorthogonal catalyst, efficiently catalysing NH carbene insertion reactions between 2,3-diaminonaphthalene and different diazo substrates in aqueous solutions and biologically relevant media. The catalytic efficiency did not change significantly when the complexity of medium was increased from water to cell culture medium with serum proteins. In all cases, complete conversion of substrates to product was observed at 10 mol% catalyst loading within 24 h at low substrate concentrations (<500 μM). To the best of our knowledge, this is the first time highly hydrophobic rhodium catalysts C1–C2 are utilized to perform NH carbene insertion reactions in homogenous aqueous solution or in the presence of living cells. Although, C3 and C4 are already reported to catalyse the same reaction in water, results were obtained at very high substrate concentrations (100 mM) and heterogenous conditions albeit with a reasonable yield of 72% and 38%, respectively.39 Additionally, this system has advantages over the previously reported copper-catalysed NH carbene insertion reactions in vitro, as the use of copper is avoided and micromolar concentrations of substrates are used. Among the three reactions tested, the reaction between 1 and 2b forming the product 3b which spontaneously hydrolysed to 3b′ is an excellent example to highlight that polymeric nanoparticles can work in tandem with other catalytic species, likely even with enzymes.
All in all, we presented a highly active and stable catalytic system suitable for NH carbene insertion reactions in very straining conditions by exploiting encapsulation of dirhodium carboxylates in amphiphilic polymeric nanoparticles. The best-performing polymeric nanoparticle catalyst P1@C1 was exported to HeLa cells where it catalysed the NH carbene insertion reaction to produce photophysically and biologically active products. Importantly, the reactions performed in cells were ‘catalytic’, which is rarely attained in the case of TMC-catalysed bioorthogonal reactions at low substrate concentrations.
In future, the versatile nature of the dirhodium catalysts can be exploited to further expand the repertoire of efficient bioorthogonal reactions. Additionally, the easy incorporation of different hydrophobic dirhodium catalysts makes the polymeric nanoparticle platform suitable for a wide range of applications including using different TMCs. We anticipate that these studies will drive further advancements in the field, allowing to export new and stable hydrophobic catalysts and reactions to biological environments, leading to new breakthroughs.
Author contributions
This manuscript was written through the contributions of all authors. All authors have given approval to the final version of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financed by the European Union's Horizon 2020 Research and Innovation Program under the Marie Sklodowska-Curie Grant Agreement No. 765497 (THERACAT).References
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44(9), 651–653 CrossRef CAS PubMed.
- Phase 1/2a Study of SQ3370 in Patients With Advanced Solid Tumors – ClinicalTrials.gov, 5 February 2023.
- J. M. M. Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef PubMed.
- K. Wu, N. A. Yee, S. Srinivasan, A. Mahmoodi, M. Zakharian, J. M. Mejía Oneto and M. Royzen, Chem. Sci., 2021, 12, 1259–1271 RSC.
- S. Srinivasan, N. A. Yee, K. Wu, M. Zakharian, A. Mahmoodi, M. Royzen and J. M. Mejía Oneto, Adv. Ther., 2021, 4, 2000243 CrossRef CAS PubMed.
- S. Fedeli, J. Im, S. Gopalakrishnan, J. L. Elia, A. Gupta, D. Kim and V. M. Rotello, Chem. Soc. Rev., 2021, 50, 13467–13480 RSC.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814–12826 CrossRef CAS PubMed.
- A. M. Pérez–López, B. Rubio–Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd–Jones, D. Sieger, J. Santamaría and A. Unciti–Broceta, Angew. Chem., 2017, 129, 12722–12726 CrossRef.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS PubMed.
- S. Learte-Aymamí, C. Vidal, A. Gutiérrez-González and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 9149–9154 CrossRef PubMed.
- J. Konč, V. Sabatino, E. Jiménez-Moreno, E. Latocheski, L. R. Pérez, J. Day, J. B. Domingos and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2022, 61, e202113519 CrossRef PubMed.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106–114 CrossRef CAS PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- T. Völker and E. Meggers, ChemBioChem, 2017, 18, 1083–1086 CrossRef PubMed.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef PubMed.
- P. Destito, A. Sousa-Castillo, J. R. Couceiro, F. López, M. A. Correa-Duarte and J. L. Mascareñas, Chem. Sci., 2019, 10, 2598–2603 RSC.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed.
- M. C. Ortega–Liebana, N. J. Porter, C. Adam, T. Valero, L. Hamilton, D. Sieger, C. G. Becker and A. Unciti–Broceta, Angew. Chem., 2022, 134, e202111461 CrossRef.
- A. M. Pérez-López, A. Belsom, L. Fiedler, X. Xin and J. Rappsilber, J. Med. Chem., 2023, 66, 3301–3311 CrossRef PubMed.
- C. Adam, T. L. Bray, A. M. Pérez-López, E. H. Tan, B. Rubio-Ruiz, D. J. Baillache, D. R. Houston, M. J. Salji, H. Y. Leung and A. Unciti-Broceta, J. Med. Chem., 2022, 65, 552–561 CrossRef CAS PubMed.
- T. G. Rajagopalan, W. H. Stein and S. Moore, J. Biol. Chem., 1966, 241, 4295–4297 CrossRef CAS PubMed.
- D. M. Carminati and R. Fasan, ACS Catal., 2019, 9, 9683–9697 CrossRef CAS PubMed.
- M. C. M. van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 4040–4043 RSC.
- Z. J. Wang, H. Renata, N. E. Peck, C. C. Farwell, P. S. Coelho and F. H. Arnold, Angew. Chem., Int. Ed., 2014, 53, 6810–6813 CrossRef CAS PubMed.
- K. Chen, S. Q. Zhang, O. F. Brandenberg, X. Hong and F. H. Arnold, J. Am. Chem. Soc., 2018, 140, 16402–16407 CrossRef CAS PubMed.
- S. Wallace and E. P. Balskus, Angew. Chem., Int. Ed., 2015, 54, 7106–7109 CrossRef CAS PubMed.
- K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233–3244 CrossRef CAS PubMed.
- K. A. Andersen, M. R. Aronoff, N. A. McGrath and R. T. Raines, J. Am. Chem. Soc., 2015, 137, 2412–2415 CrossRef CAS PubMed.
- S. Gutiérrez, M. Tomás–Gamasa and J. L. Mascareñas, Angew. Chem., 2021, 133, 22188–22196 CrossRef.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. Ramakrishna and C. Sivasankar, Org. Biomol. Chem., 2017, 15, 2392–2396 RSC.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC.
- B. J. Anding and L. K. Woo, Organometallics, 2013, 32, 2599–2607 CrossRef CAS.
- A. Rioz-Martínez, J. Oelerich, N. Ségaud and G. Roelfes, Angew. Chem., Int. Ed., 2016, 55, 14136–14140 CrossRef PubMed.
- H. F. Srour, P. Le Maux, S. Chevance, D. Carrié, N. Le Yondre and G. Simonneaux, J. Mol. Catal. A: Chem., 2015, 407, 194–203 CrossRef CAS.
- A. M. Abu-Elfotoh, Tetrahedron Lett., 2017, 58, 4750–4754 CrossRef CAS.
- M. B. Minus, M. K. Kang, S. E. Knudsen, W. Liu, M. J. Krueger, M. L. Smith, M. S. Redell and Z. T. Ball, Chem. Commun., 2016, 52, 11685–11688 RSC.
- J. M. Antos, J. M. Mcfarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131(17), 6301–6308 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- K. Tishinov, K. Schmidt, D. Häussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 CrossRef CAS PubMed.
- J. P. Snyder, A. Padwa and T. Stengel, J. Am. Chem. Soc., 2001, 123, 11318–11319 CrossRef CAS PubMed.
- G. M. ter Huurne, L. N. J. de Windt, Y. Liu, E. W. Meijer, I. K. Voets and A. R. A. Palmans, Macromolecules, 2017, 50, 8562–8569 CrossRef CAS PubMed.
- Z. Zhang and K. Fan, Nanoscale, 2022, 15, 41–62 RSC.
- B. Helms, C. O. Liang, C. J. Hawker and J. M. J. Fréchet, Macromolecules, 2005, 38, 5411–5415 CrossRef CAS.
- R. Huang, C. M. Hirschbiegel, X. Zhang, A. Gupta, S. Fedeli, Y. Xu and V. M. Rotello, ACS Appl. Mater. Interfaces, 2022, 14, 31594–31600 CrossRef CAS PubMed.
- X. Zhang, R. F. Landis, P. Keshri, R. Cao-Milán, D. C. Luther, S. Gopalakrishnan, Y. Liu, R. Huang, G. Li, M. Malassiné, I. Uddin, B. Rondon and V. M. Rotello, Adv. Healthcare Mater., 2021, 10, 2001627 CrossRef CAS PubMed.
- C. M. Hirschbiegel, S. Fedeli, X. Zhang, R. Huang, J. Park, Y. Xu and V. M. Rotello, Materials, 2022, 15, 6487 CrossRef CAS PubMed.
- F. G. Adly, Catalysts, 2017, 7, 347 CrossRef.
- M. P. Doyle, J. Org. Chem., 2006, 71, 9253–9260 CrossRef CAS PubMed.
- R. P. Pandit, S. H. Kim and Y. R. Lee, Adv. Synth. Catal., 2016, 358, 3586–3599 CrossRef CAS.
- D. J. Blevins, R. Nazir, S. M. H. Dabiri, M. Akbari and J. E. Wulff, J. Drug Delivery Sci. Technol., 2022, 78, 103950 CrossRef CAS.
- L. Deng, L. Albertazzi and A. R. A. Palmans, Biomacromolecules, 2022, 23, 326–338 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02581k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |