
Discovering layered lead-free perovskite solar absorbers via cation transmutation†
Ming
Chen
ab,
Zhicheng
Shan
b,
Xiaofeng
Dong
b,
Shengzhong (Frank)
Liu
 *ac and
Zhuo
Xu
*a
*ac and
Zhuo
Xu
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, National Ministry of Education, Shaanxi Engineering Lab for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, Xi’an, 710119, China. E-mail: xuzh@snnu.edu.cn; szliu@dicp.ac.cn
bSchool of Electric Power, Civil Engineering and Architecture, School of Physics and Electronics Engineering, State Key Laboratory of Quantum Optics and Quantum Optics Devices, Shanxi University, Taiyuan, 030006, China
cState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
First published on 10th February 2023
Abstract
For the exploration of stable lead-free perovskites for solar cell applications, we propose a series of Dion–Jacobson (DJ) double perovskites with the formula BDA2MIMIIIX8 (BDA = 1,4-butanediamine) by substituting two Pb2+ in BDAPbI4 with an MI+ (Na+, K+, Rb+, Cu+, Ag+, and Au+) and MIII3+ (Bi3+, In3+, and Sb3+) pair. First-principles calculations demonstrated the thermal stabilities of all the proposed BDA2MIMIIIX8 perovskites. The electronic properties of BDA2MIMIIIX8 depend strongly on the choice of MI+ + MIII3+ and the structural archetype, and three out of 54 candidates with suitable solar band gaps and superior optoelectronic properties were selected for photovoltaic application. A highest theoretical maximal efficiency of over 31.6% is predicted for BDA2AuBiI8. The DJ-structure-induced interlayer interaction of apical I–I atoms is found to play a crucial role in promoting the optoelectronic performance of the selected candidates. This study provides a new concept for designing lead-free perovskites for efficient solar cells.
New conceptsDouble perovskites have the benefits of being environmentally friendly and stable. However, the majority of the synthesized bulk double perovskites have large indirect bandgaps, large carrier effective masses, and parity-forbidden transitions, which make them unsuitable for photovoltaic applications. Previous studies have demonstrated that indirect-to-direct bandgap transitions are induced by dimensionality reduction, as demonstrated in Cs2AgBiBr6 and BA4AgBiBr8, because of the avoidance of hybridization of Ag and Bi frontier orbitals. However, the large spacer cation results in suppressed carrier transportation along the interlayer direction. In this work, we construct Dion–Jacobson (DJ) double perovskites BDA2MIMIIIX8 (BDA = 1,4-butanediamine; MI+ = monovalent cation; MIII3+ = trivalent cation). Unlike the transition mechanism in BA4AgBiBr8, the indirect-to-direct bandgap transition in selected BDA2(Cu/Ag/Au)BiI8 is mainly due to interlayer apical I–I-interaction-induced band broadening, as the smaller spacer divalent cation BDA2+ is used. This band broadening can induce further-reduced bandgaps and more-dispersive band edges along the interlayer direction, which is beneficial for carrier transportation. High absorption coefficients and transition probabilities are also established in BDA2(Cu/Ag/Au)BiI8. This work indicates that the combination of a DJ archetype and double substitution provide a new approach for designing stable lead-free perovskites for high-performance solar cells. |
Hybrid lead-halide perovskites have rapidly developed as the most promising star materials for next-generation photovoltaic and optoelectronic devices because of their tunable bandgaps, balanced carrier effective masses, high absorption onset, small exciton binding energies, long carrier diffusion length, and high defect tolerance factor.1–7 However, stability issues and the toxicity of lead have hindered their commercialization.8–12 A effective way to improve the stability of metal-halide perovskites is through cation engineering, which can result in the formation of low-dimensional perovskites with intrinsically improved stability. Layered Dion–Jacobson (DJ) perovskites have attracted substantial attention because of their enhanced stability and improved carrier transportation along the interlayer direction induced by effective interlayer interaction.13–16
One effective way to solve the toxicity issue of lead-halide perovskites is through cation transmutation, in which two Pb2+ cations are replaced by one M+ cation (e.g., Na+, K+, Cu+, Ag+, Au+) and one M3+ cation (e.g., Y3+, Gd3+, Au3+, In3+, Tl3+, Sb3+, Bi3+) to form double-perovskite structures.17–24 However, the majority of the synthesized bulk double perovskites are chloride and bromide perovskites, normally with large indirect band gaps (> 1.9 eV) and large carrier effective masses because of the mismatch in angular momentum of the frontier atomic orbitals.19 Moreover, many reported double perovskites with direct band gaps have been proven to show inversion-symmetry-induced parity-forbidden transitions.17 These characteristics of most halide double perovskites make them unsuitable for photovoltaic applications. To date, Cs2AgBiBr6 has been widely investigated for solar cell application, and the record efficiency has only reached 6.37% because of the relatively large band gap of 2.19 eV.25–27
Early studies have shown that reduction of dimensionality can lead to enhanced stability of double perovskites and indirect-to-direct band gap transition because of the avoidance of frontier orbital hybridization.28–32 Additionally, the influence of interlayer coupling on the band gap feature in layered double perovskites has also been mentioned.30 Particularly, the synthesized perovskites (C6H16N2)2AgBiI8 and (C6H16N2)2CuBiI8 have measured optical band gaps of 1.93 eV and 1.68 eV, and the CuBiI system shows promising use in photovoltaic applications. Therefore, in order to obtain stable lead-free perovskites with suitable band gaps and good optical properties for solar cell applications, the use of DJ double perovskites may be an efficient strategy.
In this communication, we theoretically studied the stability and optoelectronic properties of a series of environmentally friendly DJ double perovskites proposed through the cation transmutation strategy. Potential solar cell absorber candidates with good stability, a suitable band gap, superior carrier transport properties, and good optical properties are selected for photovoltaic application, and the influences of the cation pair and structural archetype on their optoelectronic properties are thoroughly explored. Among the selected candidates, a theoretical maximum efficiency comparable with that of CH3NH3PbI3 is obtained for BDA2AuBiI8. The calculation details are given in the ESI.†
The basic concept of the proposed lead-free layered double perovskites is the replacement of two Pb2+ cations in the DJ perovskite APbX4 with a monovalent MI+ and trivalent MIII3+ cation to give the double DJ perovskite A2MIMIIIX8, as shown in Fig. 1a. Here, the A2+ cation is 1,4-butanediamine (BDA2+). The optimized structural parameters of 54 BDA2MIMIIIX8 (MI+ = Na+, K+, Rb+, Cu+, Ag+, and Au+; MIII3+ = In3+, Sb3+, and Bi3+; X− = Cl−, Br−, and I−) compounds are shown in Tables S1–S3 (ESI†), and all possess a triclinic symmetry. The doubly protonated cation BDA2+ is inserted between inorganic octahedron layers along the [001] direction, and the –NH3+ ends intrude deep into the cavity formed by the apical I atoms and can interact with the bridging I atoms through hydrogen bonds. The interlayer distance is much reduced compared to that of the RP counterpart, which could induce effective interlayer coupling and promote charge transfer between the inorganic layers.33
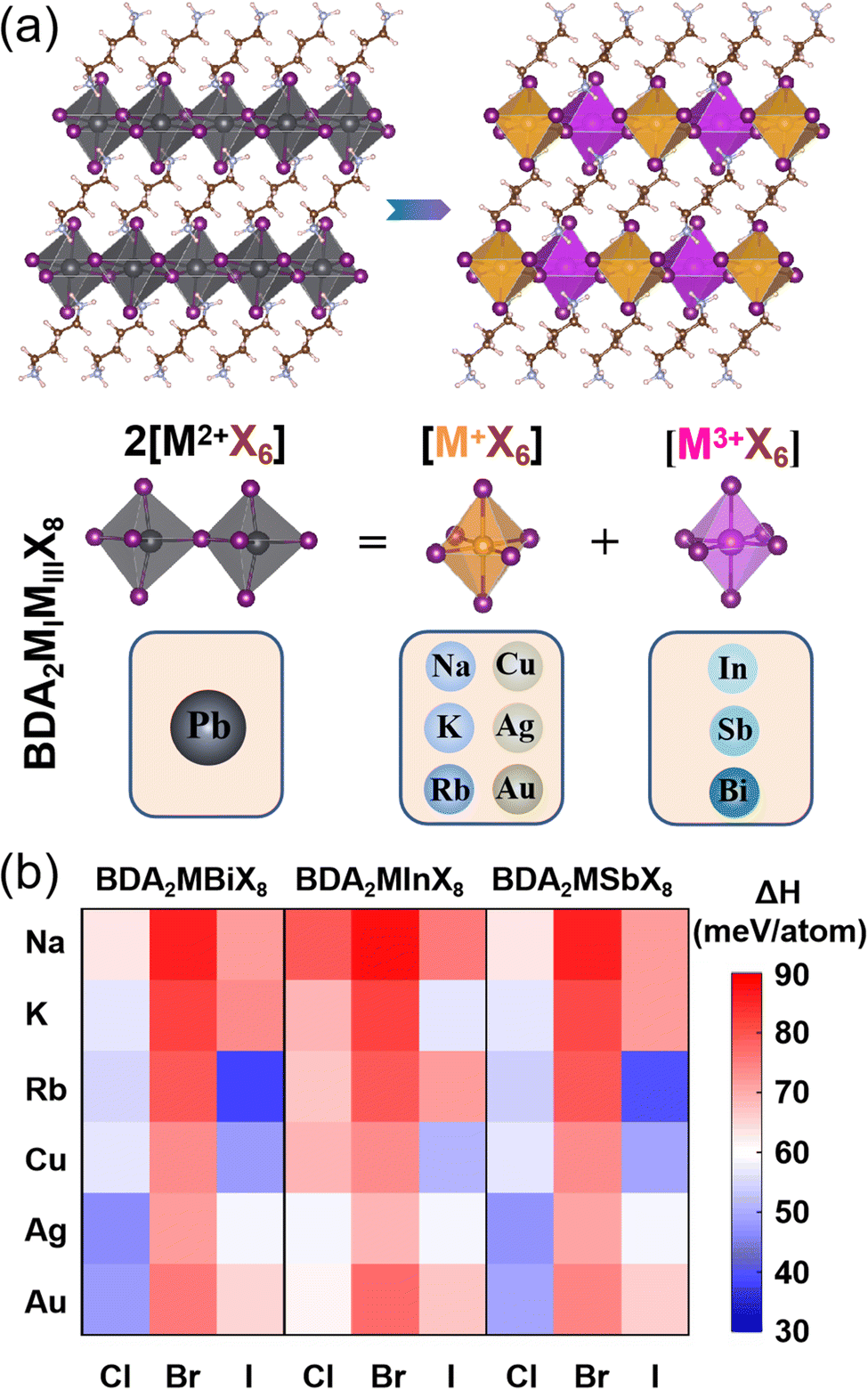 | ||
| Fig. 1 (a) Structural configurations of the 2D perovskites BDAPbI4 and 2D double perovskites BDA2MIMIIIX8, and illustration of the cation transmutation strategy to design Pb-free halide 2D double perovskites. (b) Decomposition enthalpies (ΔH) of 2D double BDA2MIMIIIX8 perovskites. | ||
To evaluate the thermodynamic stability of the BDA2MIMIIIX8 compounds, their decomposition energies with respect to possible decomposition pathways were considered. Since no ternary option is available, the most possible decomposition path of BDA2MIMIIIX8 is into the corresponding common binary compounds (BDA2MIMIIIX8 → 2BDAX2 + MIX + MIIIX3), and the perovskites can be synthesized via the inverse reaction. The decomposition enthalpy (ΔHd), which is defined via the free energy difference after and before the above decomposition path, is calculated as follows:
| ΔHd = (2E[BDAX2] + E[MIX] + E[MIIIX3] − E[BDA2MIMIIIX8])/Ntot |
The calculated values of ΔHd for all compounds are positive, as shown in Fig. 1b. Here, positive values of ΔHd mean the decomposition process is endothermic, resulting in suppressed decomposition of BDA2MIMIIIX8, demonstrating that all the BDA2MIMIIIX8 have good thermodynamic stability. This enhanced stability can be ascribed to the above-mentioned hydrogen bonds normally found in DJ perovskites.33,34 As an example, the case of BDAAgBiI8 is considered. The closest H and bridging I atoms have separation distances of 2.53 and 2.55 Å and an interaction integrated crystal orbital Hamilton population (iCOHP) value of 0.53 eV and 0.51 eV, and the nearest H and apical I atoms have a distance of 2.52 Å and an interaction iCOHP value of 0.58 eV, validating the strong hydrogen bonding between NH3+ and I atoms. These extra hydrogen bonds can promote the rigidity of the structure. Moreover, the divalent organic cation can induce fewer degrees of freedom in the structures because of the elimination of van der Waals gap.35
The band gaps of the 54 compounds calculated using the generalized gradient approximation (GGA)-Perdew–Burke–Ernzerhof (PBE) method are listed in Tables S1–S3 (ESI†). However, due to the strong relativistic effect in heavy elements, the spin–orbit coupling (SOC) effect cannot be neglected. A severe band gap reduction in Bi-based compounds is induced with the inclusion of the SOC effect because of the splitting of the Bi-6p-derived conduction bands. Detailed analysis of the band edges indicates that the locations of the valence band maximum (VBM) and conduction band minimum (CBM) of many compounds are altered when the SOC effect is considered. A comparison of band structures calculated using the PBE and PBE + SOC schemes for several representative compounds is given in Fig. S1 (ESI†) for illustration. Additionally, we also performed band structure calculations using the hybrid HSE + SOC functional, which can provide more precise band gap estimation for most semiconductors.
The variations in the band gaps for different compositions calculated using the HSE + SOC method are shown in Fig. 2. The band gaps for BDA2MIBi/In/SbX8 are found to decrease with decreasing halogen electronegativity. Furthermore, the band gaps are also closely connected with the MI cation. The band gaps of alkali compounds are larger than those of noble metal compounds with the same halide because of their strong ionic characteristics. The alkali compounds all show indirect characteristics except for BDA2RbInI8 and BDA2KSbBr8. The projected band structures in Fig. S2–S4 (ESI†) indicate that the alkali elements have negligible contributions to the VBM and CBM, which are instead dominated by the halide p-states and In 5s/Bi 6p/Sb 5p–halide p-hybridization states in BDA2MIn/Bi/SbX8, respectively. The projected density of states (PDOS) and COHP analysis of BDA2KInBr8 and BDA2KBiBr8 in Fig. S5 and S6 (ESI†), as representatives of the alkali BDA2MInX8 and BDA2MBiX8/BDA2MSbX8 compounds, indicate that the VBMs and CBMs mainly originate from the antibonding coupling of interlayer Br 4p–Br 4p states on the K sites and the antibonding coupling of In 5s/Bi 6p–Br 4p states, respectively. The decomposed charge densities of the VBMs and CBMs of BDA2KInBr8 and BDA2KBiBr8 in Fig. S7 (ESI†) indicate that the VBMs mainly originate from the antibonding coupling of interlayer Br 4p–Br 4p states on the K sites, and have negligible contribution from in-plane interactions. These features of the alkali compounds are also reasons for the less-dispersive band edges and large band gaps in these structures, which are very undesirable for solar cell applications.
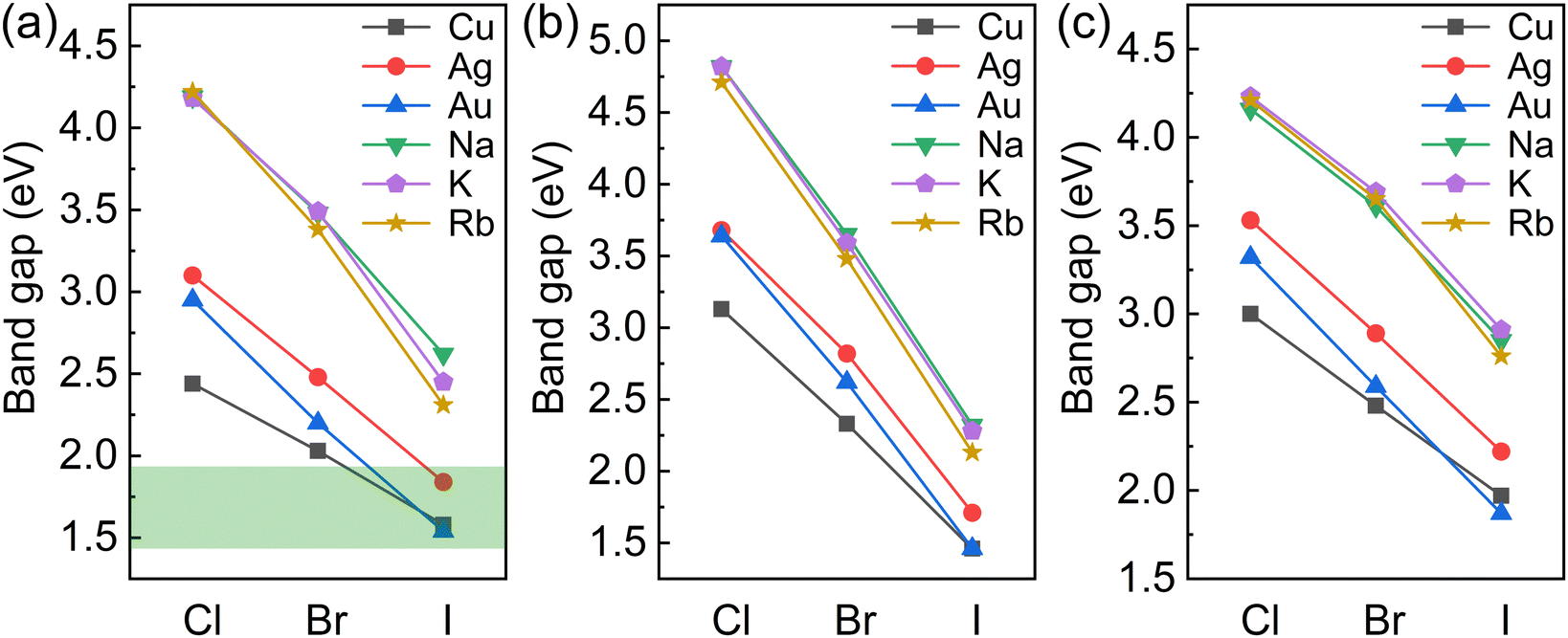 | ||
| Fig. 2 Variation in the band gaps for (a) BDA2MIBiX8, (b) BDA2MIInX8, and (c) BDA2MISbX8 (MI = Cu, Ag, Au, Na, K, and Rb; X = Cl, Br, and I) | ||
For the noble-metal BDA2MInX6 perovskites, the projected band structures of BDA2MInX8 in Fig. S8–S10 (ESI†) all show indirect band gaps. Therefore, despite the fact that BDA2(Cu/Ag/Au)InI8 have band gaps suitable for solar cells, their indirect band gap feature makes them unsuitable for thin-film solar cell application. The PDOS and CHOP analysis of BDA2AgInBr8 in Fig. S11 (ESI†), as a representative of the noble-metal BDA2MInX8 compounds, indicates that the VBM and CBM of BDA2AgInBr8 mainly originate from the antibonding coupling of Ag 4d–Br 4p states and In 5s–Br 4p states, respectively. The decomposed charge densities of the VBM and CBM are shown in Fig. S12 (ESI†) for intuitive illustration of the bonding characteristics. This bonding behavior is also the reason for the indirect band gaps of the BDA2MIInX6 compounds.
For the noble-metal perovskites BDA2MBiX8 and BDA2MSbX8, the projected band structures in Fig. S8–S10 (ESI†) all exhibit direct band gaps, except for those of BDA2AgSbI8 and BDA2AuSbI8. The projected band structures indicate that the VBMs and CBMs are dominated by the hybridization between metal d–halide p states and Bi 6p/Sb 5p–halide p hybridization states in the BDA2MBiX8/BDA2MSbX8 perovskites, respectively. Among these structures, BDA2(Cu/Ag/Au)BiI8 possess suitable band gaps for solar photovoltaic application. The PDOS and COHP analysis of BDA2(Cu/Ag/Au)BiI8 in Fig. S13 and S14–S16 (ESI†), as representatives of the noble-metal BDA2MBiX8 and BDA2MSbX8 compounds, indicates that the hybridization of Bi 6p–I 5p states and interlayer I 5p–I 5p states on Bi sites near the CBM has mainly antibonding and bonding characteristics, respectively. In contrast, the VBM is dominated by antibonding Cu 3d/Ag 4d/Au 5d–I 5p states, along with weak interlayer antibonding I 5p–I 5p states on the noble metal sites. The coupling strength between apical I–I atoms is mainly determined by their distance. It is found the apical I–I distances in BDA2AgBiI8 are smaller than those in BDA2(Cu/Au)BiI8; this is mainly because of the stronger coupling of Cu/Au–I bonds than that of Ag–I bonds, as discussed in the next paragraph. Therefore, the COHP of apical I–I near the VBM in BDA2CuBiI8 and BDA2AuBiI8 are almost negligible, while a stronger apical I–I COHP is observed in BDA2AgBiI8. The antibonding characteristics ensure relatively small and balanced in-plane carrier effective masses in BDA2(Cu/Ag/Au)BiI8. The charge density distributions of BDA2AuBiI8 at the VBM and CBM points in Fig. S17 (ESI†) show good consistency with the PDOS and COHP analysis. To further demonstrate the thermodynamic stability of BDA2(Cu/Ag/Au)BiI8, the formation energies and phonon band structures of BDA2(Cu/Ag/Au)BiI8 are calculated. Here, the formation energies of BDA2(Cu/Ag/Au)BiI8 are calculated as Ef = (E[BDA2MIMIIIX8] − NBDAμBDA − NMIμMI − NMIIIμMIII − NXμX)/Ntot, where μi is the chemical potential of the constituent element referred to its most stable phase. The calculated values for BDA2(Cu/Ag/Au)BiI8 are −0.320/−0.327/−0.317 eV/atom, respectively, indicating their thermodynamic stability. The phonon band structures of BDA2(Cu/Ag/Au)BiI8 are shown in Fig. S18 (ESI†) with no imaginary branches, indicating their dynamic stability.
Since the energy level of the Au 5d orbital is higher than that of the Ag 4d orbital because of the relativistic-effect-induced expansion of 5d states in Au, the Au 5d–I 5p antibonding coupling is stronger than the Ag 4d–I 5p antibonding coupling, resulting in a higher VBM in BDA2AuBiI8. Therefore, the band gap of BDA2AuBiI8 is smaller than that of BDA2AgBiI8. Because the energy level of Cu 3d orbital is located closer to the Au 5d orbital, BDA2CuBiI8 has a band gap almost equivalent to that of BDA2AuBiI8. The stronger couplings of the Au 5d–I 5p and Cu 3d–I 5p interactions than of Ag 4d–I 5p interactions were also verified by COHP analysis, as shown in Fig. S14–S16 (ESI†). Based on the above bonding analysis, an estimated molecular orbital bonding diagram of BDA2(Cu/Ag/Au)BiI8 is given in Fig. S19 (ESI†), which provides an intuitive way to understand the unique electronic properties of BDA2(Cu/Ag/Au)BiI8.
Notably, previous studies have demonstrated that the primitive cell of the bulk double perovskites Cs2MI(Cu, Ag, Au)MIII(Sb, Bi)X6 possess indirect band gaps.18 However, indirect-to-direct band gap transition can be induced by dimensionality reduction, as demonstrated in BA4AgBiBr8.28 The large spacer cation also results in a strong quantum confinement effect and thereby induces nearly flat band edges along the Γ-Z direction. Unlike the transition mechanism in BA4AgBiBr8, the direct band gaps in BDA2(Cu/Ag/Au)BiI8 are mainly due to the interlayer I–I-interaction-induced band-broadening as the smaller spacer divalent cation BDA2+ is used, which would induce a further-reduced band gap and more-dispersive band edges along the Γ-Z direction. This indicates that the construction of the structure archetype is also an effective way to tune the band gap type.
The effective masses of all structures were estimated by quadratic fitting of the PBE + SOC-calculated band edges, and are displayed in Tables S4–S6 (ESI†). The effective masses of BDA2(Cu/Ag/Au)BiI8 were also examined using the HSE + SOC functional, as shown in Fig. 3a. Slight differences can be noticed between the PBE + SOC and HSE + SOC functionals. Balanced electron and hole effective masses along the in-plane directions were obtained for three candidates due to the antibonding characteristic near the CBM and VBM. Low effective electron masses of 0.32–0.68 m0 and effective hole masses of 0.27–0.48 m0, which are comparable to the values for MAPbI3, ensure superior carrier transport properties in these candidates. Much heavier electron and hole effective masses are obtained along the out-of-plane direction owing to the quantum confinement effect. Despite the relatively large effective masses along the out-of-plane direction, they are still much improved compared to those of RP perovskites due to the more dispersive edges along the Γ-Z direction.34 As shown in Tables S4–S6 (ESI†), the carrier properties of the candidates are superior to those of others. Next, the Wannier–Mott exciton binding energies (Eb) were estimated using a modified hydrogen-atom-like Bohr model of Eb = μe4/2ħ2ε2, where ε, μ, and e are the high-frequency-limit dielectric constant, reduced effective mass, and charge of an electron, respectively. The results for BDA2(Cu/Ag/Au)BiI8 are displayed in Fig. 3b. Eb values as low as ∼70 meV were estimated for BDA2CuBiI8 and BDA2AuBiI8 for the in-plane directions, which are comparable with that of MAPbI3. The relatively small Eb values further establish their superior carrier transport properties. These carrier features ensure good photovoltaic performance of the selected candidates.
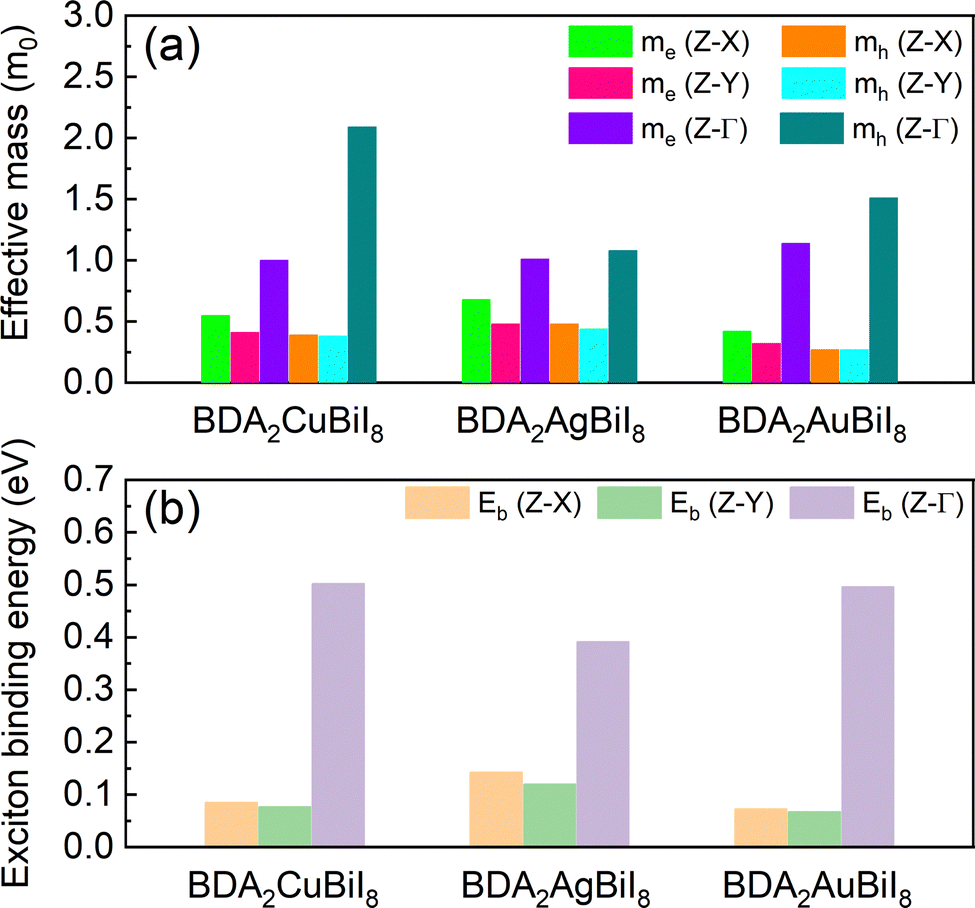 | ||
| Fig. 3 Calculated (a) effective masses of the electron (me*) and holes (mh*), and (b) exciton binding energies, Eb (meV), of BDA2Cu/Ag/AuBiI8 in different directions. | ||
In order to evaluate the photovoltaic performance of the selected absorber candidates, their absorption spectra are also calculated, along with that of MAPbI3 for comparison. Fig. 4a shows that BDA2(Cu/Ag/Au)BiI8 possess relatively high absorption edges, which are mainly contributed by the optical transition channels from the Cu 3d/Ag 4d/Au 5d–I 5p orbitals to the Bi 6p–I 5p orbitals. In addition, inversion-symmetry-induced parity-forbidden transitions in bulk double perovskites are not observed in these layered structures, as shown in Fig. S13 (ESI†). High transition probabilities are found at the Z point. Therefore, BDA2(Cu/Ag/Au)BiI8 are suitable candidates for solar cells with favorable optical absorption. Their “spectroscopic limited maximum efficiency (SLME)” was then calculated to evaluate their theoretical maximum efficiency. Fig. 4b shows the calculated SLMEs of BDA2(Cu/Ag/Au)BiI8, along with that of MAPbI3 for comparison. For the first 200 nm of film thickness, the SLME of MAPbI3 is higher than that of BDA2(Cu/Ag/Au)BiI8 because of its higher absorption intensity near the absorption edge. At a film thickness of 200 nm, the SLMEs of BDA2AuBiI8, BDA2CuBiI8, and MAPbI3 are almost equivalent and can reach a value of ∼23%. The SLME values of BDA2(Cu/Ag)BiI8 reach their theoretical limit value of about 30.8%/27.0% at a film thickness of ∼1 μm. The SLME of BDA2AuBiI8 is higher than even that of MAPbI3 from 200 nm to 1 μm, and achieved its limit value of 31.6% at ∼1 μm. Hence, it is believed that BDA2AuBiI8 and BDA2CuBiI8 may be promising as good solar absorbers that can obtain high efficiency using a low film thickness. The absorption spectra and SLMEs of BDA2(Cu/Ag/Au)InI8, which have proper indirect solar band gaps, were also calculated for comparison, as shown in Fig. S20 (ESI†). Much weaker absorption intensities are observed compared to that of BDA2(Cu/Ag/Au)BiI8. Therefore, slow increase in the SLMEs with film thickness and lower theoretical limits are obtained for BDA2(Cu/Ag/Au)InI8, indicating a much thicker film is needed for achieving suitable efficiencies.
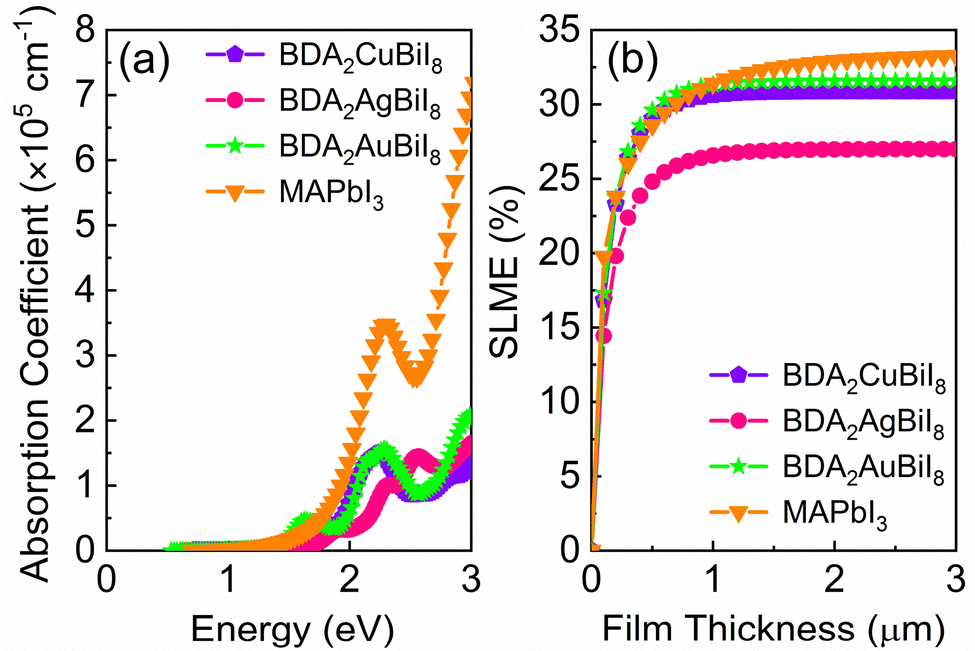 | ||
| Fig. 4 (a) Calculated absorption spectra of BDA2Cu/Ag/AuBiI8 and MAPbI3; (b) simulated spectroscopic limited maximum efficiency (SLME) of BDA2Cu/Ag/AuBiI8 and MAPbI3. | ||
In summary, DJ-type double perovskite BDA2MIMIIIX8 solar cell absorber candidates were selected and showed good stability, suitable band gaps, superior carrier transport properties, and good optical properties. Improved interlayer carrier transportation in BDA2Cu/Ag/AuBiI8 is observed because of the DJ-structure-induced interlayer I–I interaction. The crucial role of the structure archetype in their superior optoelectronic properties is revealed. Finally, theoretical maximal efficiencies of about 30.8%/27.0%/31.6% were predicted for BDA2Cu/Ag/AuBiI8. This work indicates that the combination of a DJ archetype and double substitution represents a new approach for designing stable lead-free perovskites for high-performance solar cells.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (62005148/12004235), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA17040506), the DNL Cooperation Fund CAS (DNL180311), and the 111 Project (B14041).References
- M. He, J. Liang, Z. Zhang, Y. Qiu, Z. Deng, H. Xu, J. Wang, Y. Yang, Z. Chen and C.-C. Chen, J. Mater. Chem. A, 2020, 8, 25831–25841 RSC.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V. Gustafsson, M. T. Trinh, S. Jin and X. Y. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS PubMed.
- S.-T. Ha, C. Shen, J. Zhang and Q. Xiong, Nat. Photonics, 2016, 10, 115–121 CrossRef CAS.
- S. Park, W. J. Chang, C. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- W. Chen, Y. Zhu, J. Xiu, G. Chen, H. Liang, S. Liu, H. Xue, E. Birgersson, J. W. Ho, X. Qin, J. Lin, R. Ma, T. Liu, Y. He, A. M.-C. Ng, X. Guo, Z. He, H. Yan, A. B. Djurišić and Y. Hou, Nat. Energy, 2022, 7, 229–237 CrossRef CAS.
- M. Jeong, I. W. Choi, K. Yim, S. Jeong, M. Kim, S. J. Choi, Y. Cho, J.-H. An, H.-B. Kim, Y. Jo, S.-H. Kang, J.-H. Bae, C.-W. Lee, D. S. Kim and C. Yang, Nat. Photonics, 2022, 16, 119–125 CrossRef CAS.
- B. Vargas, G. Rodríguez-López and D. Solis-Ibarra, ACS Energy Lett., 2020, 5, 3591–3608 CrossRef CAS.
- W. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 965 CrossRef PubMed.
- S. Chatterjee and A. J. Pal, J. Mater. Chem. A, 2018, 6, 3793–3823 RSC.
- D. Bryant, N. Aristidou, S. Pont, I. Sánchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- S. Chatterjee and A. J. Pal, J. Mater. Chem. A, 2018, 6, 3793–3823 RSC.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4–22 CrossRef CAS.
- X. Xiao, J. Dai, Y. Fang, J. Zhao, X. Zheng, S. Tang, P. Rudd, Z. Cheng and J. Huang, ACS Energy Lett., 2018, 3, 684–688 CrossRef CAS.
- I. Spanopoulos, I. Hadar, W. Ke, Q. Tu, M. Chen, H. Tsai, Y. He, G. Shekhawat, V. P. Dravid, M. R. Wasielewski, A. D. Mohite, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 5518–5534 CrossRef CAS PubMed.
- W. Meng, X. Wang, Z. Xiao, J. Wang, D. B. Mitzi and Y. Yan, J. Phys. Chem. Lett., 2017, 8, 2999–3007 CrossRef CAS PubMed.
- G. Volonakis, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 1254–1259 CrossRef CAS PubMed.
- Z. Deng, F. Wei, S. Sun, G. Kieslich, A. K. Cheetham and P. D. Bristowe, J. Mater. Chem. A, 2016, 4, 12025–12029 RSC.
- E. McClure, M. Ball, W. Windl and P. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- M. R. Filip, S. Hillman, A. A. Haghighirad, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 2579–2585 CrossRef CAS PubMed.
- C. Savory, A. Walsh and D. Scanlon, ACS Energy Lett., 2016, 1, 949–955 CrossRef CAS PubMed.
- X.-G. Zhao, D. Yang, Y. Sun, T. Li, L. Zhang, L. Yu and A. Zunger, J. Am. Chem. Soc., 2017, 139, 6718–6725 CrossRef CAS PubMed.
- X. Cai, Y. Zhang, Z. Shi, Y. Chen, Y. Xia, A. Yu, Y. Xu, F. Xie, H. Shao, H. Zhu, D. Fu, Y. Zhan and H. Zhang, Adv. Sci., 2022, 9, 2103648 CrossRef CAS PubMed.
- B. Wang, N. Li, L. Yang, C. Dall’Agnese, A. K. Jena, S.-I. Sasaki, T. Miyasaka, H. Tamiaki and X.-F. Wang, J. Am. Chem. Soc., 2021, 143, 2207–2211 CrossRef CAS PubMed.
- Z. Li, P. Wang, C. Ma, F. Igbari, Y. Kang, K.-L. Wang, W. Song, C. Dong, Y. Li, J. Yao, D. Meng, Z.-K. Wang and Y. Yang, J. Am. Chem. Soc., 2021, 143, 2593–2600 CrossRef CAS PubMed.
- Z. Zhang, Q. Sun, Y. Lu, F. Lu, X. Mu, S.-H. Wei and M. Sui, Nat. Commun., 2022, 13, 3397 CrossRef CAS PubMed.
- B. A. Connor, L. Leppert, M. D. Smith, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2018, 140, 5235–5240 CrossRef CAS PubMed.
- M. K. Jana, S. M. Janke, D. J. Dirkes, S. Dovletgeldi, C. Liu, X. Qin, K. Gundogdu, W. You, V. Blum and D. B. Mitzi, J. Am. Chem. Soc., 2019, 141, 7955–7964 CrossRef CAS PubMed.
- L. Mao, S. M. L. Teicher, C. C. Stoumpos, R. M. Kennard, R. A. DeCrescent, G. Wu, J. A. Schuller, M. L. Chabinyc, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2019, 141, 19099–19109 CrossRef CAS PubMed.
- L.-Y. Bi, Y.-Q. Hu, M.-Q. Li, T.-L. Hu, H.-L. Zhang, X.-T. Yin, W.-X. Que, M. S. Lassoued and Y.-Z. Zheng, J. Mater. Chem. A, 2019, 7, 19662–19667 RSC.
- L.-Y. Bi, T.-L. Hu, M.-Q. Li, B.-K. Ling, M. S. Lassoued, Y.-Q. Hu, Z. Wu, G. Zhou and Y.-Z. Zheng, J. Mater. Chem. A, 2020, 8, 7288–7296 RSC.
- Y. Zhang, Y. Liu, Z. Xu, Z. Yang and S. Liu, Small, 2020, 16, 2003145 CrossRef CAS PubMed.
- Z. Xu, M. Chen and S. Liu, J. Phys. Chem. Lett., 2019, 10, 3670–3675 CrossRef CAS PubMed.
- L. Mao, W. Ke, L. Pedesseau, Y. Wu, C. Katan, J. Even, M. R. Wasielewski, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 3775–3783 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00499b |
| This journal is © The Royal Society of Chemistry 2023 |