
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The potential of converting carbon dioxide to food compounds via asymmetric catalysis
Rui
Gao
ab,
Xinxin
Xu
*ab,
Zhimeng
Wu
 c,
Liguang
Xu
abd,
Hua
Kuang
ab and
Chuanlai
Xu
ab
c,
Liguang
Xu
abd,
Hua
Kuang
ab and
Chuanlai
Xu
ab
aInternational Joint Research Laboratory for Biointerface and Biodetection, Jiangnan University, Wuxi, Jiangsu 214122, PRC
bState Key Laboratory of Food Science and Technology, Jiangnan University, Jiangsu, PRC
cThe Key Laboratory of Carbohydrate Chemistry & Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, Jiangsu 214122, PRC
dThe Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi, Jiangsu 214122, PRC. E-mail: xxx89826@163.com
First published on 5th May 2023
Abstract
The food crisis caused by diminished arable land, extreme weather and climate change linked to increased carbon dioxide (CO2) emission, is threatening global population growth. Interestingly, CO2, the most widespread carbon source, can be converted into food ingredients. Here, we briefly discuss the progress and challenges in catalytic conversion of CO2 to food ingredients via chiral catalysis.
Biocatalytic conversion of CO2 to food
The projection that the global population will increase to 9.7 billion by 2050 poses a higher demand for food production. Specifically, agricultural production needs to increase by up to 60% in order to feed the increased population.1 However, the diminished arable land, and the increased CO2 emission amounts which lead to extreme weather and climate change make this goal more intangible.1 CO2, a well-known greenhouse gas, can serve as the most widespread and abundant carbon resource. Using CO2 as a raw material for synthesizing typical food and its ingredients such as chiral amino acids and carbohydrates can not only reduce CO2 emissions, but also alleviate the food crisis caused by rapid population growth. In this regard, nature uses photosynthesis to fix carbon dioxide and turn it into food.Recently, Zheng et al. developed a hybrid electro-biosystem by coupling spatially separated CO2 electrolysis with yeast fermentation, which efficiently converts CO2 to D-glucose or fatty acids.2D-Glucose is the predominant form of carbohydrates utilized by human beings, so the synthesis of D-glucose is very important in the field of food manufacture. In this interesting study, the researchers employed three steps to achieve the synthesis of long-chain energy substances from CO2. First, Ni-based single-atom catalysts were used to realize almost 100% CO2 electroreduction to CO which was efficiently converted to acetic acid in the second step by using a grain-boundary-rich Cu catalyst. In this step, a porous solid electrolyte reactor was developed in order to obtain high-purity acetic acid and avoid complex isolation and purification steps. Finally, high-purity acetic acid was utilized as the feedstock for genetically engineered Saccharomyces cerevisiae fermentation, which delivers an average D-glucose yield of 8.90 μmol per gram of yeast per hour. In addition to D-glucose, the production of other valuable products was also showcased by taking advantage of this platform. For example, using a similar design, the researchers engineered strain LXJ015 to electrically produce free fatty acids in vitro from pure acetic acid (Fig. 1).2
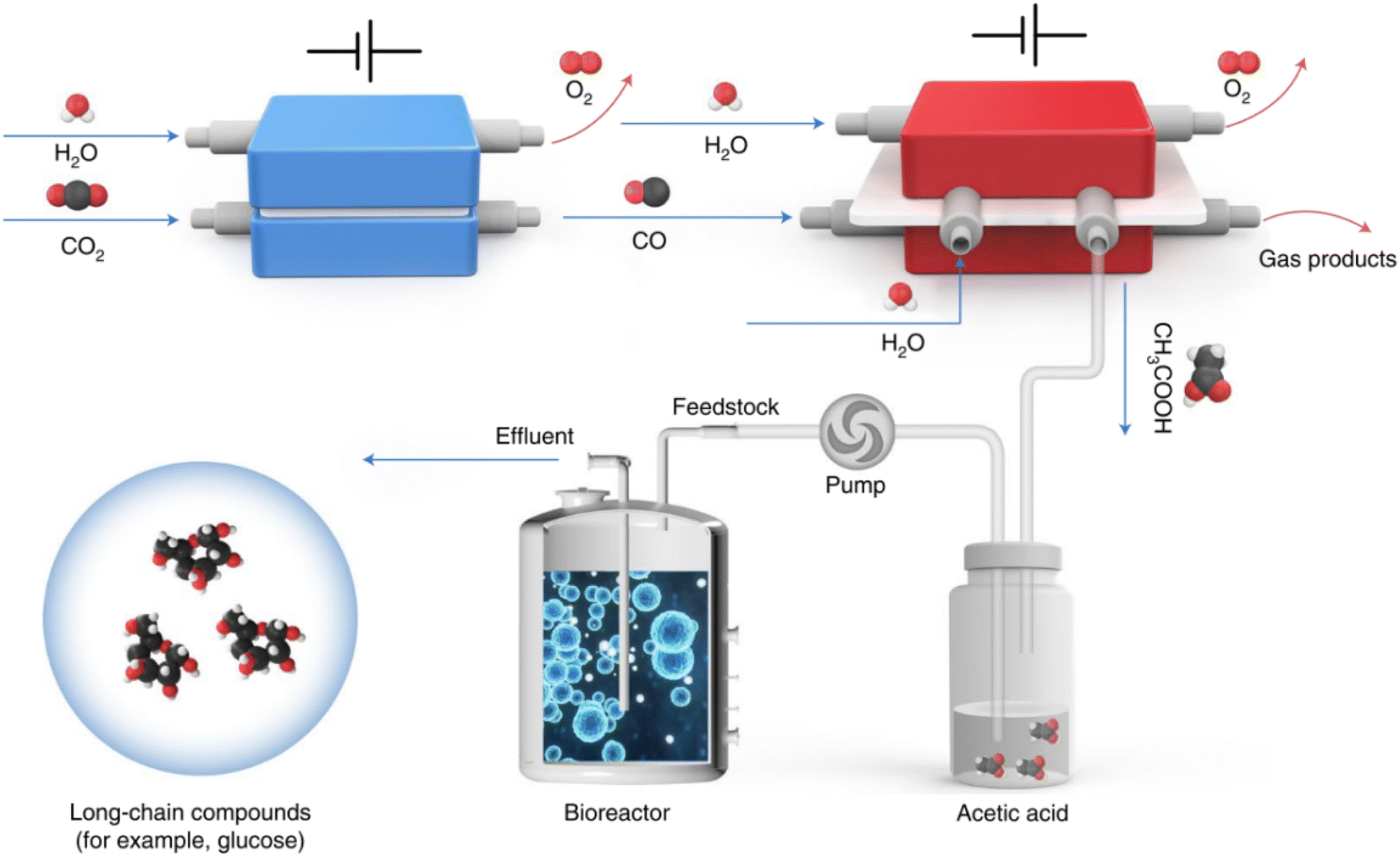 | ||
| Fig. 1 Schematic illustration of an artificial synthesis system for glucose. CO2 was first converted to acetic acid through a two-step electrochemical reaction. The generated acetic acid was then fermented in a bioreactor as a microbial substrate to produce long-chain compounds such as glucose (ref. 2). | ||
Earlier, Cai et al. reported a chemical–biochemical hybrid pathway for the synthesis of starch from CO2 and hydrogen in a cell-free system through 11 core enzyme-catalyzed reactions.3 Starch is the most important carbohydrate utilized by people. In this research, CO2 was first reduced to a C1 compound (formic acid or methanol) using a chemical catalyst. The C1 compound was then utilized as a substrate for the artificial starch anabolic pathway (ASAP) system developed by the same researchers. In the ASAP system, they divided the starch synthesis into a C1 module (for formaldehyde production), C3 module (for D-glyceraldehyde-3-phosphate production), C6 module (for D-glucose-6-phospate production), and Cn module (for starch synthesis). The enzymatic processes were delicately combined with the chemical catalysis processes, and each module of the enzymatic processes was optimized by mutating the core enzymes of catalytic pathways, namely, formolase, fructose-bisphosphatase, and ADP-glucose pyrophosphorylase. Finally, an ASAP 3.0 system, composed of C1e, C3a, C6b*, and Cnb modules, was determined with 10 enzyme-catalyzed reactions using methanol as the substrate. This system delivered a remarkably high starch productivity of ∼410 mg L−1 h−1 from CO2. This landmark work provides for the first time a way to convert waste CO2 into an economically valuable food ingredient via a combination of chemical catalysis, biosynthesis, and protein engineering (Fig. 2).3
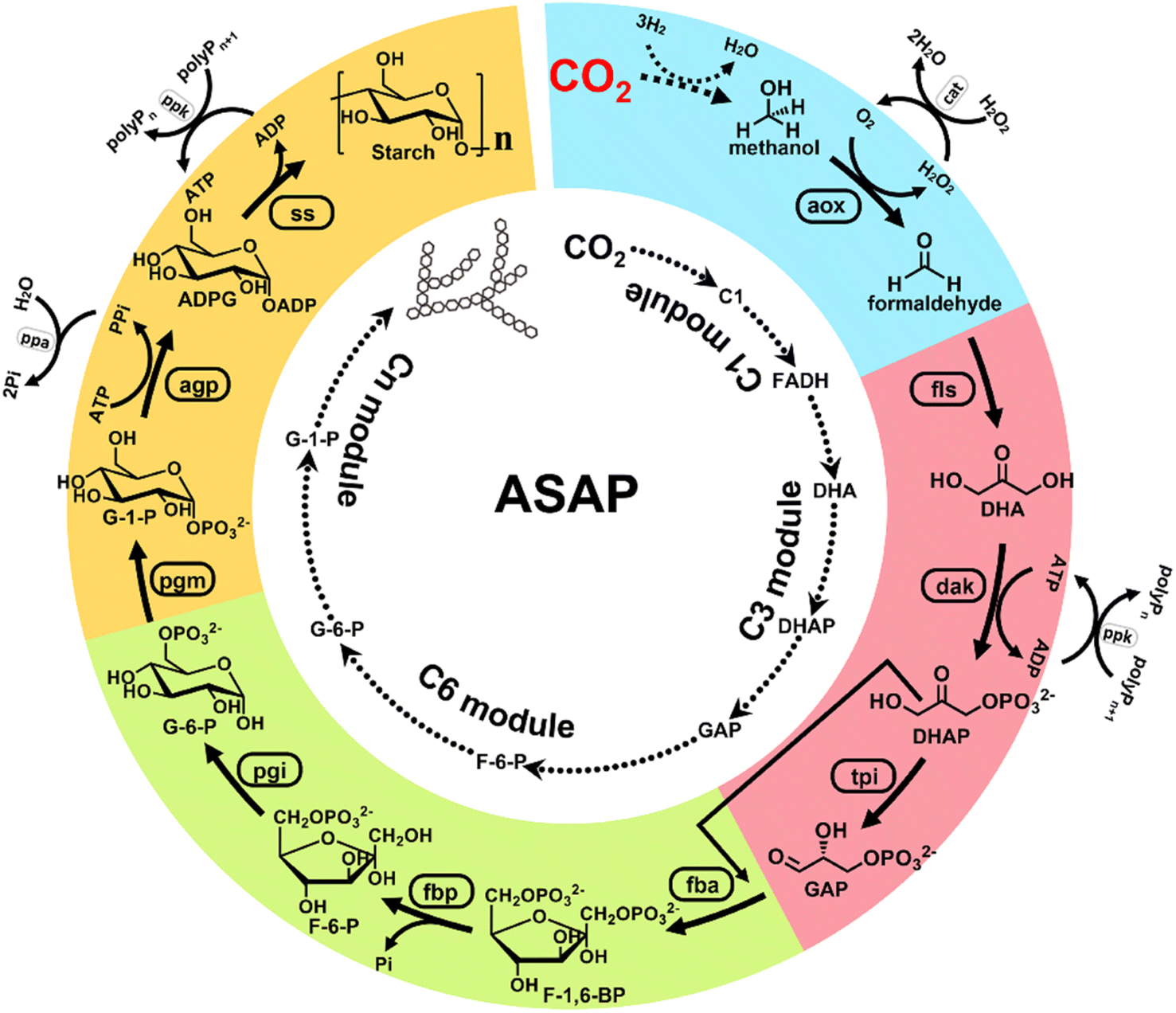 | ||
| Fig. 2 Design of an artificial starch anabolic pathway (ASAP) system that converts CO2 to starch. CO2 is first reduced to one-carbon units (C1), such as methanol. Then, methanol is oxidized to formaldehyde (FADH). FADH is treated with formolase (FLS) to produce dihydroxyacetone (DHA). Then, DHA is phosphorylated to form dihydroxyacetone phosphate (DHAP). DHAP is treated with triose phosphate isomerase (TPI) to from D-glyceraldehyde 3-phosphate (GAP). So far, the synthesis of three-carbon units (C3) has been completed. Next, GAP is treated with (FBA) to form D-fructose-1,6-bisphosphate (F-1,6-BP). Then, F-1,6-BP is treated with fructose-bisphosphatase (FBP) to form D-fructose-6-phosphate (F-6-P). F-6-P is then treated with phosphoglucose isomerase (PGI) to form D-glucose-6-phosphate (G-6-P). The presence of G-6-P implies the successful preparation of six-carbon units (C6). After that, G-6-P is treated with phosphoglucomutase (PGM) to form D-glucose-1-phosphate (G-1-P). Then, G-1-P is treated with ADP-glucose pyrophosphorylase to form ADP glucose (ADPG). ADPG then is treated with starch synthase (SS) to form starch. This is the final composite module (Cn) of the ASAP system (ref. 3). | ||
As an emerging area in CO2 utilization, the two studies mentioned above not only open new strategies for preparing food products by combining electrochemistry with biocatalysis, but also provide potential solutions to alleviate the food crisis. Nonetheless, both studies synthesize chiral food ingredients from CO2 by taking advantage of chiral biological systems. Strictly speaking, it is not a direct route to transform CO2 to chiral food ingredients, since two separate processes, chemical transformation and biological transformation, were integrated in both studies. The former harvests achiral C1 compounds or C2 compounds, while the latter delivers chiral compounds by utilizing the asymmetric catalytic transformation function of biological systems.
So far, the chemo-catalytic conversion of CO2 into chiral compounds with specific configurations, especially chiral food components (amino acids, monosaccharides, and fatty acids), has been slow to develop. This could be attributed to the following challenges that hinder the development of an operable technology for the direct asymmetric conversion of CO2. First, CO2 is a very stable and inert molecule since it is in the lowest energy state, and the carbon in CO2 is in its most stable valence (IV). Second, it is rather difficult for CO2 to approach metal catalysts because of its weak coordination ability with metal catalysts and low solubility in non-alkaline solvents. Third, direct asymmetric conversion of achiral CO2 into chiral food ingredients requires a complex and tedious organic chemical synthesis process, and the derived products from the catalytic processes are often mixed enantiomers, which require an expensive enantiomeric sieving process. Furthermore, because of the inert nature of CO2, its catalytic conversion usually requires high temperature and high pressure, which are not conducive to the preparation of chiral compounds with specific configurations.4
In conclusion, chemical methods for the catalytic conversion of CO2 into valuable chiral chemicals are feasible, but the synthesis of chiral food components still requires process optimization and cost reduction, which inspire us to search for efficient catalytic pathways and chiral catalysts for this challenging chemical transformation.
Asymmetric catalysis
Asymmetric catalysis, also known as chiral catalysis, renders catalytic reactions to proceed in the direction of the mirror isomers that match chiral catalysts, thereby enabling the controlled synthesis of chiral compounds with various specific configurations. Because of the chiral amplification effect, it is one of the most effective methods for synthesizing chiral products.5 The key to chiral catalytic reactions is the development of efficient chiral catalysts. In recent years, due to the combined amplification effect6 of chirality in asymmetric catalysis with the excellent opto-electromagnetic properties of nanomaterials,7 chiral nanomaterials have received increasing attention,8 and great progress has been made in the field of asymmetric catalysis.9 For example, Zhang et al. reported versatile and efficient asymmetric cross-coupling between carbonyl radicals (from aldehydes) and aryl radicals (from aryl halides) by utilizing porous chiral metal–organic frameworks as catalysts under light illumination.10 Wei et al. accomplished the dimerization of a specific configuration of the prochiral molecule 2-anthracenecarboxylic acid under light by controlling the chirality of metal nanohelices which were produced by glancing-angle deposition on the substrates rotated in the clockwise or counterclockwise direction.11 Chiral nanomaterials also exhibit fascinating asymmetric catalytic functions relevant to biocatalysis.12 In addition, compared with enzymes, nanomaterials cost less and have good environmental stability. Importantly, nanomaterial catalytic products often require only simple centrifugal separation to obtain high purity of the targets. Our group has demonstrated that chiral CdTe quantum dots with the function of restriction endonuclease can specifically cut DNA, and the cleavage efficiency is dependent on the configuration of the chiral CdTe quantum dots.13 Chiral Cu1.96S nanoparticles (NPs) and Cu2S NPs have been reported to display chiral-configuration-dependent cleavage capability for the tobacco mosaic virus capsid protein9 and hepatitis B core antigen,14 respectively. Chiral nanoparticles can also catalyze the polymerization of some biomolecules. Li et al. reported that chiral nanoscale inorganic superparticles can serve as photocatalysts that enantioselectively catalyze L-tyrosine and D-tyrosine into L-dityrosine and D-dityrosine, respectively.12Therefore, asymmetric catalysis based on chiral nanomaterials is thriving and could become an important direction for the direct asymmetric catalytic conversion of CO2 to chiral food ingredients. Nonetheless, catalytic conversion of CO2 into chiral food components inevitably involves complex processes including carbon chain elongation, polymerization, amination, sulfurization, etc. Access to readily available and easily convertible chiral intermediates is the key to the direct synthesis of food ingredients from CO2. Next, we briefly describe how to obtain potential chiral intermediates from CO2 since they are indispensable for the preparation of more complex chiral food components.
Progress in direct catalytic conversion of CO2 into chiral intermediates
To date, asymmetric catalytic conversion of CO2 into chiral intermediates can be mainly divided into the categories of asymmetric C–O bonds, asymmetric C–N bonds, asymmetric C–C bonds, and asymmetric C–H bonds on the basis of the products formed.The formation of asymmetric C–O bonds refers to the bonding method where the carbon atoms of CO2 become chiral carbon atoms after the addition of oxygen from reactants. The formation of asymmetric C–O bonds can be achieved by either asymmetric nucleophilic attack on CO2 by O-nucleophiles15 or kinetic resolution of epoxides with CO2.16 Strategies for constructing chiral C–O bonds directly on carbon radical centers are attractive, but the control over stereoselectivity remains a formidable challenge. Zhu et al. reported a copper-catalyzed regioselective and enantioselective carboesterification of substituted dienes using alkyl diacyl peroxides as the source of both carbon and oxygen substituents. This work represents a striking advance in the development of asymmetric radical C–O bonds and may lead to the discovery of other asymmetric radical reactions.15 Lu et al. found that chiral cyclic carbonates can be synthesized by employing cobalt(III)-based complexes as chiral catalysts and the nucleophiles as cocatalysts during the kinetic resolution of racemic propylene oxide and CO2.17 Jing et al. realized the kinetic resolution of epoxides and CO2 with an ee value of 52% by loading Ni-based chiral catalyst into a three-dimensional porous metal–organic framework and using n-Bu4NBr as a cocatalyst.18 North et al. declared that cyclic carbonates can be synthesized over a dinuclear chiral catalyst consisting of a chiral Al(III)-based catalyst and a Cr(III)-based catalyst in the presence of n-Bu4NBr as a cocatalyst.19 Johnston and coworkers developed an elegant strategy to produce chiral cyclic carbonates via the carboxylation/alkene functionalization reaction of homoallylic alcohols. They also confirmed that a properly balanced Brønsted acid/base bifunctional catalyst lowers the barrier of CO2 incorporation and facilitates the stabilization of the resulting adduct, which are essential for the nucleophilic addition of a nucleophile to CO2 (Fig. 3).20
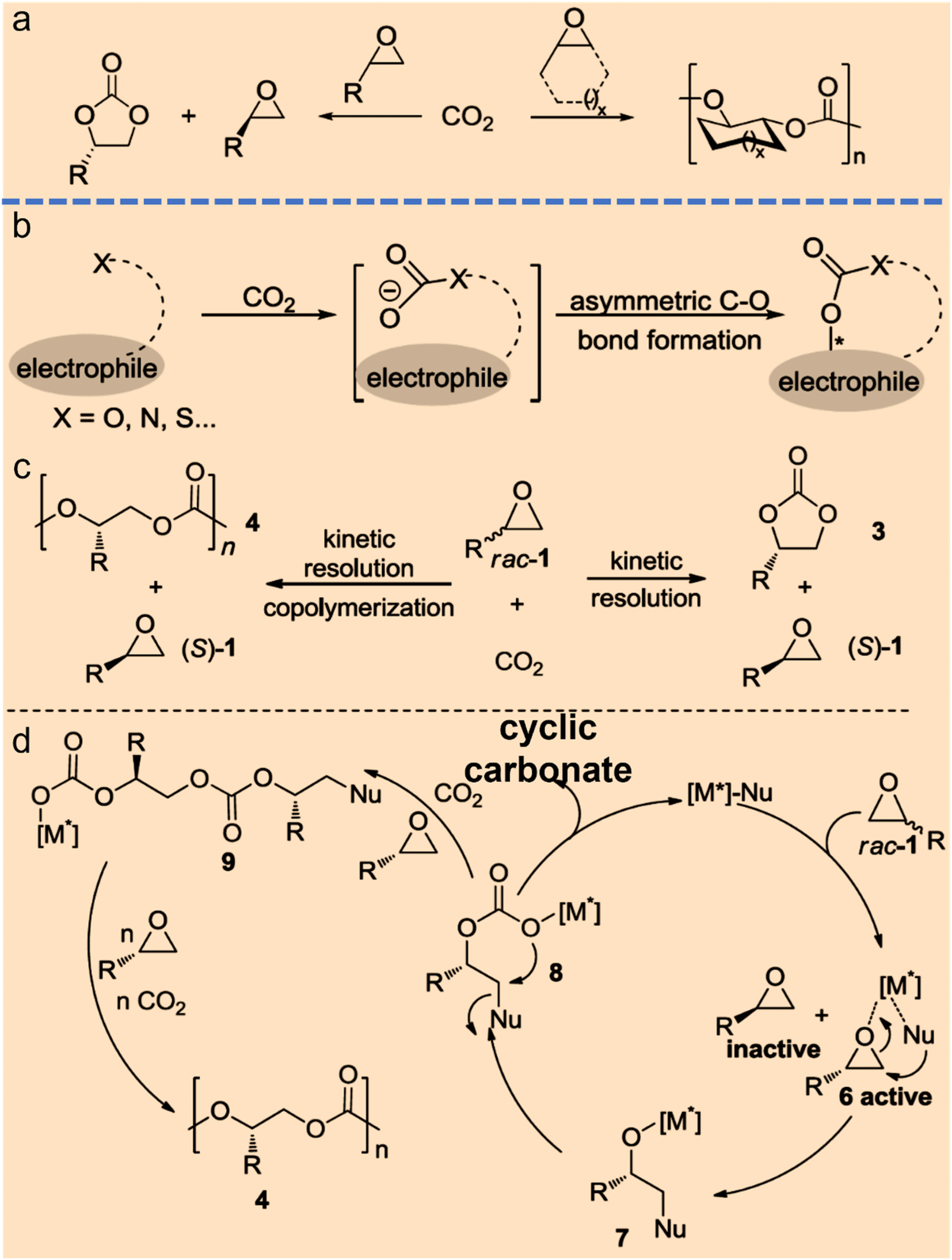 | ||
| Fig. 3 Strategies for direct catalytic conversion of CO2 into chiral intermediates. Asymmetric C–O bond formation with CO2via kinetic resolution (a) or nucleophiles (b) (ref. 4). (c and d) Chiral metal complex and nucleophile co-catalyzed kinetic resolution of racemic epoxides with CO2 to form chiral cyclic carbonates and polycarbonates (ref. 4). | ||
The formation of asymmetric C–N bonds implies that the carbon atoms of CO2 become chiral following the nitrogen addition reaction. The asymmetric C–N bond formation can be realized by either asymmetric nucleophilic attack on CO2 by N-nucleophiles21 or asymmetric carboxylation of amines and their derivatives with CO2 using chiral catalysts.22 In the former case, Zhang et al. synthesized chiral allyl carbamate from CO2 by the catalytic domino reaction for the first time with a high ee value of 94%.23 In the latter case, Zheng et al. found that the nucleophilic addition of CO2 using amine nucleophiles can enantioselectively convert allyl carbonate to branched-chain carbamate with an ee value up to 93% (Fig. 4).24
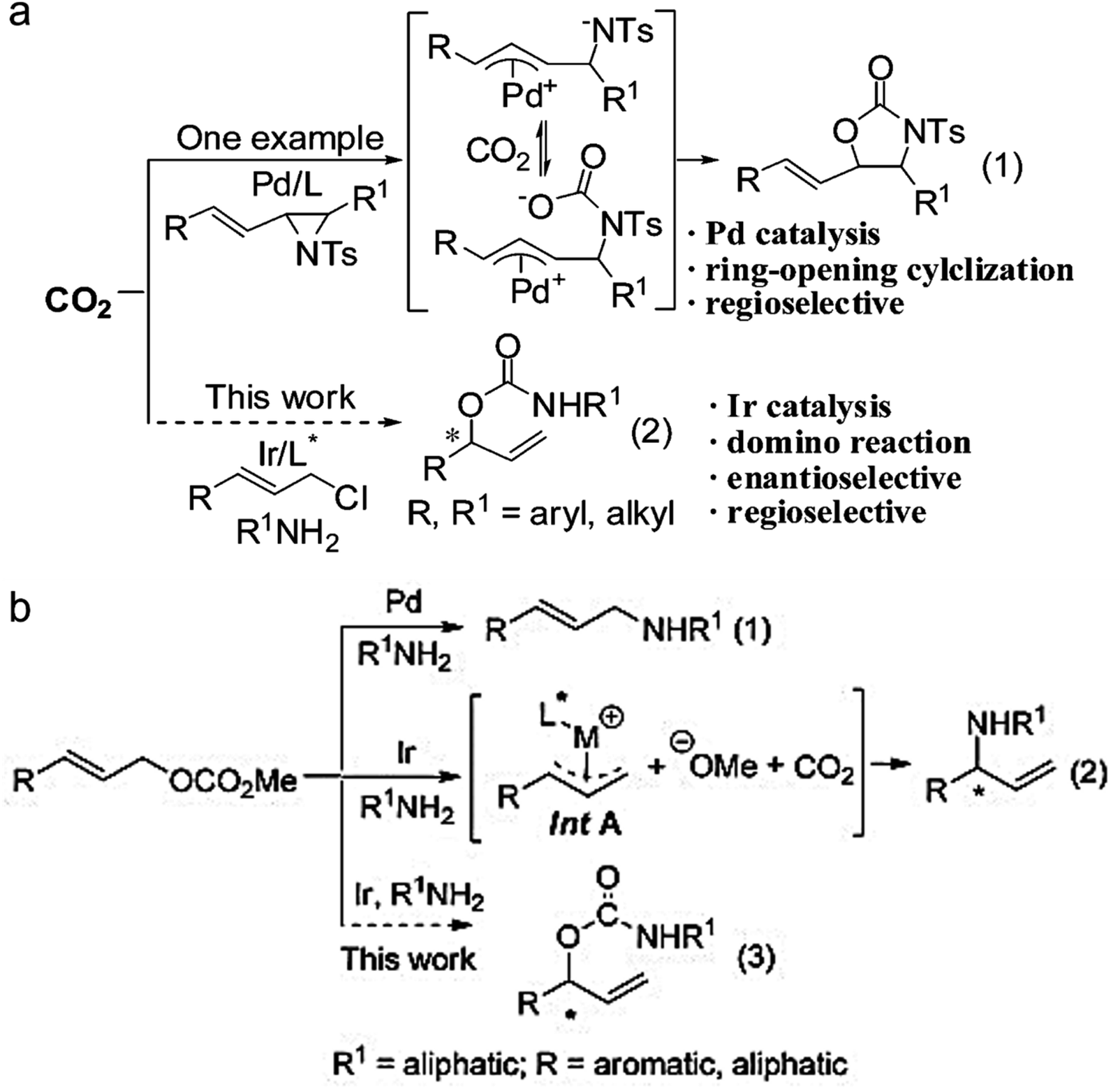 | ||
| Fig. 4 (a) Domino reactions involving Pd or Ir-catalyzed allylation reaction (ref. 23). (b) Transition-metal-catalyzed allylation of allyl carbonates with amines (ref. 24). | ||
In a similar way, the formation of asymmetric C–C bonds and asymmetric C–H bonds signifies that the carbon atoms of CO2 become chiral after reaction products are formed. Asymmetric C–C and C–H bonds25 can be formed directly via an enantioselective nucleophilic attack on CO2 by organometallic reagents. For example, directly catalyzed asymmetric binding of CO2 to unsaturated hydrocarbons is considered as an attractive CO2 chemical fixation scheme. Studies have shown that copper-based catalysts can catalyze both the reduction of two prochiral conjugated olefins and the restoration of the product. This allows for enantioselective synthesis of organoboron derivatives with multiple adjacent stereocenters in the presence of diboron compounds.26 Another study demonstrated that palladium-based catalysts can catalyze the enantioselective C–H alkylation of allyl groups to yield chiral γ,δ-unsaturated amides.27 While progress has been made in the formation of asymmetric C–C and asymmetric C–H bonds, a general method has not been fully developed. It is also worth noting that some nucleophiles possess both N-nucleophilic and O-nucleophilic addition functions. In this case, the formation of asymmetric C–O bonds, asymmetric C–N bonds, and asymmetric C–H bonds may be accompanied by the formation of asymmetric C–C bonds due to the presence of different nucleophilic adducts. The generation of these chiral compounds makes it possible to directly catalyze CO2 into chiral food components (Fig. 5).
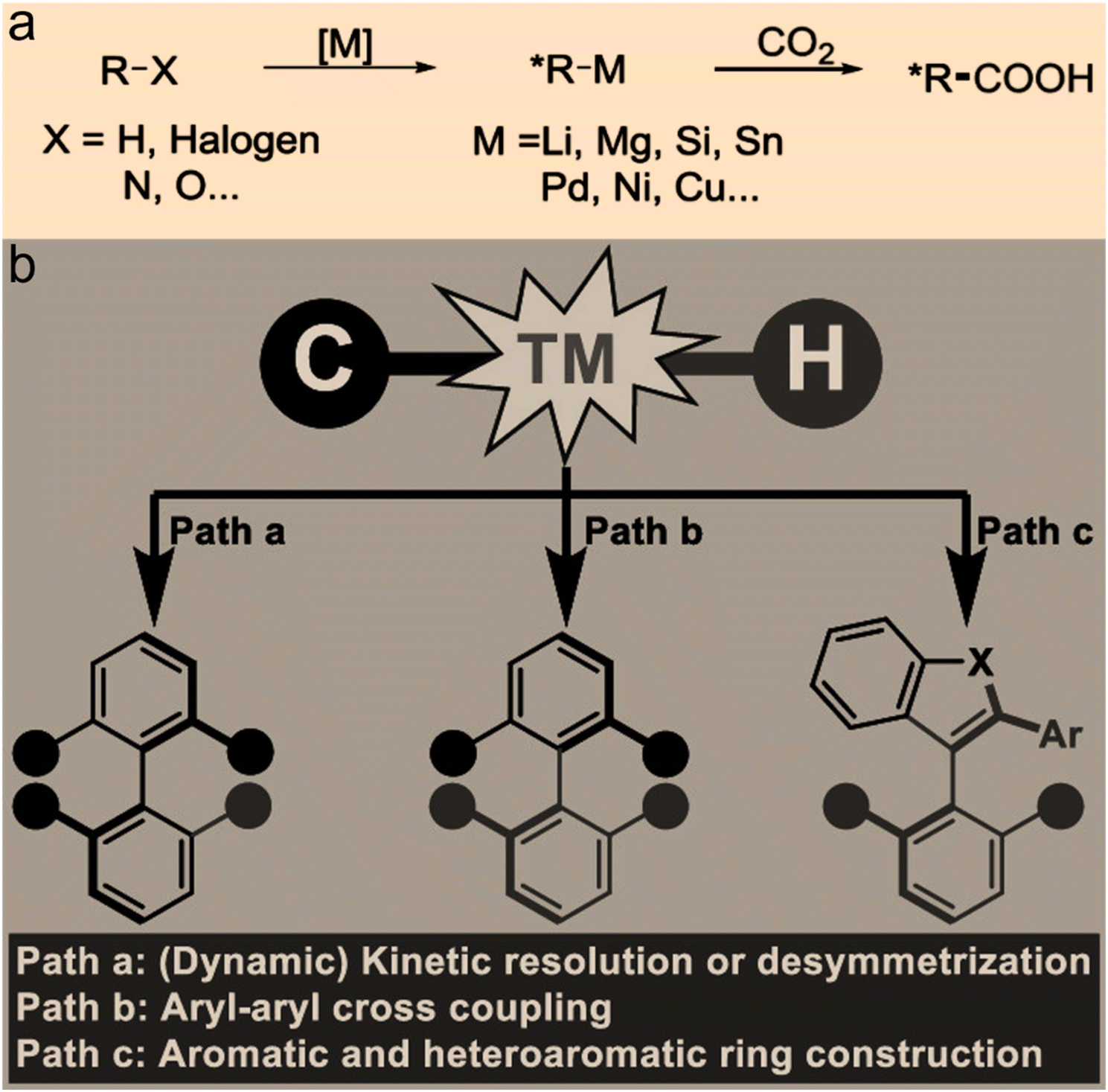 | ||
| Fig. 5 (a) Asymmetric C–C bond formation with CO2 (ref. 4). (b) Strategies for enantioselective C–H functionalization (ref. 25). | ||
The potential of directly converting CO2 to chiral food compounds
Excitingly, some reports on the synthesis of chiral amino acids from CO2 are already available.21,28 These pioneering studies confirm that the formation of asymmetric C–N bonds can facilitate the synthesis of α-amino acids with specific configurations. Significant progress has also been made in the asymmetric carboxylation of amines and their derivatives with CO2 by using chiral catalysts to synthesize α-amino acids with high ee values. On the basis of the asymmetric catalytic function of vitamin B6, Cai et al. developed a variety of chiral biomimetic catalytic systems, leading to various chiral amine compounds such as α-amino acids and peptides via biomimetic asymmetric transamination and carbonyl-catalyzed α-C–H asymmetric functionalization of primary amines.21 Moreover, the obtained α-amino acids can be further polymerized to form high-order chiral poly-amino acids by meticulously selecting specific chiral catalysts (Fig. 6).28 | ||
| Fig. 6 Strategies for direct catalytic conversion of CO2 into chiral α-amino acids (ref. 4). | ||
Although asymmetric catalytic conversion of CO2 has made tremendous progress in the past few decades, direct catalytic conversion of CO2 into chiral food components is still challenging. Currently, only a limited number of studies on the synthesis of amino acids and short peptides using CO2 exist. The scientific progress in this field is far behind to meet the needs for sufficient food, especially carbohydrates, for the world's growing population. Direct catalytic conversion of CO2 to synthesize carbohydrates, functional peptides, and proteins will help relieve the food crisis, but still faces huge obstacles. For example, D-glucose and its aggregates (starch) are the most important carbohydrates utilized by human beings; however, artificial synthesis of D-glucose suffers from complex and tedious separation and purification of chiral configurations. In addition, compared to biocatalysis, the chiral catalysis of carbon dioxide conversion can effectively separate the two enantiomer products by designing the chiral configuration of the catalyst. Thus, the rapid development of asymmetric catalysis based on chiral nanomaterials provides enormous opportunities for the direct conversion of CO2 into chiral food components.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22236002), Jiangsu Provincial Natural Science Foundation (SBK2022044122) and China Postdoctoral Science Foundation (2022M711357).References
- J. P. Giraldo, H. Wu, G. M. Newkirk and S. Kruss, Nanobiotechnology approaches for engineering smart plant sensors, Nat. Nanotechnol., 2019, 14, 541–553, DOI:10.1038/s41565-019-0470-6.
- T. Zheng, et al., Upcycling CO2 into energy-rich long-chain compounds via electrochemical and metabolic engineering, Nat. Catal., 2022, 5, 388–396, DOI:10.1038/s41929-022-00775-6.
- T. Cai, et al., Cell-free chemoenzymatic starch synthesis from carbon dioxide, Science, 2021, 373, 1523–1527, DOI:10.1126/science.abh4049.
- C.-K. Ran, et al., Recent advances in asymmetric synthesis with CO2, Sci. China Chem., 2020, 63, 1336–1351, DOI:10.1007/s11426-020-9788-2.
- N. Shukla and A. J. Gellman, Chiral metal surfaces for enantioselective processes, Nat. Mater., 2020, 19, 939–945, DOI:10.1038/s41563-020-0734-4.
- L. Xu, et al., Enantiomer-dependent immunological response to chiral nanoparticles, Nature, 2022, 601, 366–373, DOI:10.1038/s41586-021-04243-2.
- J. Cai, et al., Polarization-sensitive optoionic membranes from chiral plasmonic nanoparticles, Nat. Nanotechnol., 2022, 17, 408–416, DOI:10.1038/s41565-022-01079-3.
- A. Qu, et al., Stimulation of neural stem cell differentiation by circularly polarized light transduced by chiral nanoassemblies, Nat. Biomed. Eng., 2021, 5, 103–113, DOI:10.1038/s41551-020-00634-4.
- R. Gao, et al., Site-selective proteolytic cleavage of plant viruses by photoactive chiral nanoparticles, Nat. Catal., 2022, 5, 694–707, DOI:10.1038/s41929-022-00823-1.
- Y. Zhang, et al., Metal–organic frameworks enable regio- and stereo-selective functionalization of aldehydes and ketones, Chem, 2022, 8, 1688–1704, DOI:10.1016/j.chempr.2022.02.004.
- J. Wei, et al., Iron-Catalyzed Highly Enantioselective cis-Dihydroxylation of Trisubstituted Alkenes with Aqueous H2O2, Angew. Chem. Int. Ed., 2020, 59, 16561–16571, DOI:10.1002/anie.202002866.
- S. Li, et al., Single- and multi-component chiral supraparticles as modular enantioselective catalysts, Nat. Commun., 2019, 10, 4826, DOI:10.1038/s41467-019-12134-4.
- M. Sun, et al., Site-selective photoinduced cleavage and profiling of DNA by chiral semiconductor nanoparticles, Nat. Chem., 2018, 10, 821–830, DOI:10.1038/s41557-018-0083-y.
- X. Guo, et al., Ultrasmall Copper (I) Sulfide Nanoparticles Prevent Hepatitis B Virus Infection, Angew. Chem. Int. Ed., 2021, 60, 13073–13080, DOI:10.1002/anie.202103717.
- X. Zhu, et al., Asymmetric radical carboesterification of dienes, Nat. Commun., 2021, 12, 6670, DOI:10.1038/s41467-021-26843-2.
- R.-H. Wang, et al., Selective C(sp3)–H Cleavage of Enamides for Synthesis of 2-Pyridones via Ligand-Enabled Ni–Al Bimetallic Catalysis, ACS Catal., 2021, 11, 858–864, DOI:10.1021/acscatal.0c04585.
- X. B. Lu, et al., Asymmetric catalysis with CO2: direct synthesis of optically active propylene carbonate from racemic epoxides, J. Am. Chem. Soc., 2004, 126, 3732–3733, DOI:10.1021/ja049734s.
- T. Chang, L. Jin and H. Jing, Bifunctional Chiral Catalyst for the Synthesis of Chiral Cyclic Carbonates from Carbon Dioxide and Epoxides, ChemCatChem, 2009, 1, 379–383, DOI:10.1002/cctc.200900135.
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, Aluminum(salen) Complexes as Catalysts for the Kinetic Resolution of Terminal Epoxides via CO2 Coupling, ACS Catal., 2015, 5, 3398–3402, DOI:10.1021/acscatal.5b00235.
- B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, Enantioselective small molecule synthesis by carbon dioxide fixation using a dual Bronsted acid/base organocatalyst, J. Am. Chem. Soc., 2015, 137, 7302–7305, DOI:10.1021/jacs.5b04425.
- W. Cai, et al., Asymmetric biomimetic transamination of alpha-keto amides to peptides, Nat. Commun., 2021, 12, 5174, DOI:10.1038/s41467-021-25449-y.
- W. J. Zhou, et al., Reductive dearomative arylcarboxylation of indoles with CO2 via visible-light photoredox catalysis, Nat. Commun., 2020, 11, 3263, DOI:10.1038/s41467-020-17085-9.
- M. Zhang, X. Zhao and S. Zheng, Enantioselective domino reaction of CO2, amines and allyl chlorides under iridium catalysis: formation of allyl carbamates, Chem. Commun., 2014, 50, 4455–4458, 10.1039/c4cc00413b.
- S. C. Zheng, M. Zhang and X. M. Zhao, Enantioselective transformation of allyl carbonates into branched allyl carbamates by using amines and recycling CO2 under iridium catalysis, Chemistry, 2014, 20, 7216–7221, DOI:10.1002/chem.201402388.
- C. X. Liu, W. W. Zhang, S. Y. Yin, Q. Gu and S. L. You, Synthesis of Atropisomers by Transition-Metal-Catalyzed Asymmetric C–H Functionalization Reactions, J. Am. Chem. Soc., 2021, 143, 14025–14040, DOI:10.1021/jacs.1c07635.
- W. S. Yoon, W. J. Jang, W. Yoon, H. Yun and J. Yun, Copper-catalysed asymmetric reductive cross-coupling of prochiral alkenes, Nat. Commun., 2022, 13, 2570, DOI:10.1038/s41467-022-30286-8.
- Z.-S. Nong, et al., Palladium-catalysed branch and enantioselective allylic C–H alkylation of α-alkenes, Nat. Synth., 2022, 1, 487–496, DOI:10.1038/s44160-022-00084-7.
- Y. Wang, M. Li, S. Wang, Y. Tao and X. Wang, S-Carboxyanhydrides: Ultrafast and Selective Ring-Opening Polymerizations Towards Well-defined Functionalized Polythioesters, Angew. Chem. Int. Ed., 2021, 60, 10798–10805, DOI:10.1002/anie.202016228.
| This journal is © The Royal Society of Chemistry 2023 |