
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-density lipoprotein: a versatile nanoscale platform for targeted delivery
Luyao
Dai†
 ac,
Shuaijun
Li†
c,
Qian
Hao
ac,
Ruina
Zhou
ac,
Hui
Zhou
ac,
Wenxi
Lei
c,
Huafeng
Kang
a,
Hao
Wu
*abc,
Yuanpei
Li
*b and
Xiaobin
Ma
*a
ac,
Shuaijun
Li†
c,
Qian
Hao
ac,
Ruina
Zhou
ac,
Hui
Zhou
ac,
Wenxi
Lei
c,
Huafeng
Kang
a,
Hao
Wu
*abc,
Yuanpei
Li
*b and
Xiaobin
Ma
*a
aDepartment of Oncology, The Second Affiliated Hospital, Medical School of Xi'an Jiaotong University, Xi'an, Shaanxi 710061, China
bDepartment of Biochemistry and Molecular Medicine, UC Davis Comprehensive Cancer Center, University of California Davis, Sacramento, CA 95817, USA
cDepartment of Biophysics, School of Basic Medical Sciences, Key Laboratory of Environment and Genes Related to Diseases, Xi'an Jiaotong University Health Science Center, Xi'an, Shaanxi 710061, China
First published on 24th January 2023
Abstract
Low-density lipoprotein (LDL) is a small lipoprotein that plays a vital role in controlling lipid metabolism. LDL has a delicate nanostructure with unique physicochemical properties: superior payload capacity, long residence time in circulation, excellent biocompatibility, smaller size, and natural targeting. In recent decades, the superiority and feasibility of LDL particles as targeted delivery carriers have attracted much attention. In this review, we introduce the structure, composition, advantages, defects, and reconstruction of LDL delivery systems, summarize their research status and progress in targeted diagnosis and therapy, and finally look forward to the clinical application of LDL as an effective delivery vehicle.
1. Introduction
The development and application of nanotechnology have made remarkable progress in the medical field. Various nanoscale building blocks provided alternative delivery options for diagnosing and treating diseases.1–4 The Food and Drug Administration (FDA) has approved several nanocarriers for clinical imaging and treatment of cancer or other diseases, such as liposome and lipid-based nanoparticles, protein nanoparticles, polymeric micelles, inorganic nanoparticles, and so on.5–8 However, most nanocarriers were trapped in preclinical research for many reasons: difficulties in batch synthesis, biocompatibility problems, lack of suitable targeted selective sites, and especially potential immunotoxicity.9,10 An ideal nanocarrier should have excellent biocompatibility, loading efficiency, and targeting ability. Since natural nanoparticles based on lipoproteins can meet these requirements, it is a promising direction of nanomedicine.11So far, the research on the structure and function of lipoproteins has made remarkable progress.12 The interior of lipoprotein has abundant hydrophobic triglycerides and cholesterols, indicating that the lipoprotein has large potential to deliver various hydrophobic bioactive compounds such as amphotericin B, cyclosporine A, and halofantrine.13 The lipoprotein's outer layer comprises monolayer amphiphilic phospholipids and several apolipoproteins, and the lipoprotein is shown as a spherical nanocomplex that can be transported in the blood. This extended blood circulation property of the lipoprotein allows good systemic administration without additional modification of polyethylene glycol.14,15 Besides, lipoproteins are endogenous nanoparticles that would not be regarded as foreign substances by the body's immune system nor be absorbed by the reticuloendothelial system.16 Therefore, they have good biocompatibility and limited biological toxicity. In addition, lipoproteins target actively mainly by recognizing the specific receptor of apolipoproteins. These receptors are highly overexpressed on a variety of cancer cells. Thus, the potential ability of lipoprotein targeted delivery is generated by reducing adverse cargo interactions in normal cells and increasing cargo concentration in cancer cells.17
The density of plasma lipoproteins is different due to protein and lipid composition ratios. Lipoproteins can be divided into five categories by ultracentrifugation: chylomicrons, very low-density lipoprotein (VLDL), low-density lipoprotein (LDL), intermediate-density lipoprotein (IDL) and high-density lipoprotein (HDL).18 The general characteristics of various lipoproteins are shown in Table 1. Among them, LDL and HDL can diffuse into the interfibrillar opening of solid tumors due to their smaller size, so they are widely studied and applied as delivery carriers.19 Since the mid-1990s, HDL-based nanovesicles have been widely explored,20–22 and the coverage of the reports has significantly exceeded that of LDL.23 This review summarizes the latest content on targeted delivery using LDL. Specifically, we dissected the structure and function of LDL and the interaction of LDL receptors. Then, we illustrated the advantages and defects of using LDL as a natural nanocarrier and discussed the common strategies for LDL reconstruction. After that, we cited many examples highlighting LDL nanocarriers' diagnostic and therapeutic capabilities for different diseases. Finally, the summary and prospect of the versatile LDL are put forward.
| Diameter (nm) | Density (g cm−3) | Molecular weight (×106 Da) | Apolipoprotein | Composition (wt%) | ||||
|---|---|---|---|---|---|---|---|---|
| Protein | Cholesterol | Phospholipid | Triglyceride | |||||
| a Apo, apolipoprotein; VLDL, very low-density lipoprotein; LDL, low-density lipoprotein; IDL, intermediate-density lipoprotein; HDL, high-density lipoprotein. | ||||||||
| Chylomicron | 75–1200 | <0.95 | 400 | ApoA-I, ApoA-II, ApoA-IV, ApoB-48, ApoC-II, ApoE | 1–2 | 2–4 | 7–9 | 80–85 |
| VLDL | 30–80 | 0.95–1.006 | 10–80 | ApoB-100, ApoC, ApoE | 8–10 | 17–27 | 17–19 | 45–53 |
| LDL | 18–25 | 1.006–1.019 | 2.3 | ApoB-100 | 20–25 | 43–50 | 19–21 | 5–9 |
| IDL | 25–35 | 1.019–1.063 | 5–10 | ApoB-100, ApoC, ApoE | 19 | 23 | 20 | 23 |
| HDL | 5–12 | 1.063–1.210 | 0.17–0.36 | ApoA-I, ApoA-II, ApoC, ApoE | 50–60 | 12–25 | 17–24 | 2–3 |
2. LDL and its receptor
2.1 Structure and function of LDL
As shown in Fig. 1, LDL is spherical biological nanoparticles with an average diameter of 18–25 nm. Its interior is a hydrophobic core (cholesterol ester and triglyceride). And its exterior is an amphiphilic shell (phospholipid and free cholesterol) with B-100 apolipoprotein molecules (ApoB-100).26 Among the components contained in LDL, free and esterified cholesterol account for the most, and triglycerides account for the least. Free cholesterol is inserted between the fatty acid chains of phospholipids, which increases the rigidity of the outer layer of LDL to a certain extent.27 ApoB-100 consists of 4536 amino acid residues and covers the particle surface through complex amphiphilic α-helix protein lipid interactions to stabilize this nanostructure.28 In addition, another critical role of ApoB-100 is to specifically recognize tissue sites that express LDL receptors (LDLR), such as adrenal glands, skeletal muscles, lymphocytes, gonads, and kidneys.29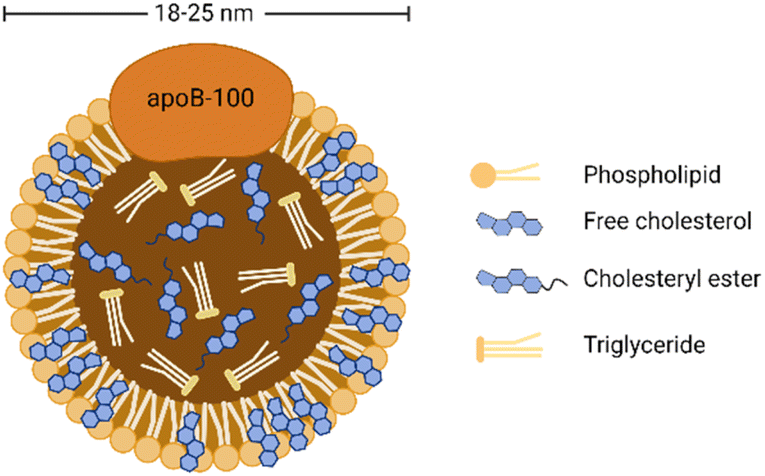 | ||
| Fig. 1 General structure diagram of LDL. | ||
LDL is the primary cholesterol carrier in blood circulation, and its most important role is to transport cholesterol to extrahepatic tissues for steroid production and membrane synthesis. Cholesterol is an essential component of cell membranes and can be obtained from dietary sources, de novo synthesis, or ingestion of circulating low-density lipoprotein.30 Although cholesterol is vital for membrane and hormone synthesis, excessive cholesterol supply in peripheral tissues triggers pathological processes such as hyperlipidemia and atherosclerotic plaque formation.29 In addition, due to the increased demand of cancer cells for lipids to synthesize new membranes for rapid proliferation, abnormal regulation in cholesterol levels is also associated with multiple types of tumorigenesis.31 Many studies have shown that the plasma cholesterol level of cancer patients is reduced and may return to normal after successful cancer treatment.32–34 Other studies have pointed out that hypocholesterolemia may be caused by increased LDLR expression in malignant tumors,35,36 especially acute myeloid leukemia, rectal cancer, adrenal cancer, lung cancer, liver cancer, brain cancer, metastatic prostate cancer cells, etc.37
2.2 Binding to LDLR
LDLR is a membrane mosaic glycoprotein containing 839 amino acids and can be divided into five independent domains. As mentioned earlier, it is distributed in many different tissues and organs of the body.38 By contrast, LDLR is relatively abundant in the liver and kidney to promote their normal uptake and utilization of LDL.39 Therefore, although the upregulation of LDLR expression in tumor tissues is conducive to the targeted delivery of LDL, the LDLR expression in normal organs affects this specific tumor-targeting effect. Therefore, reducing the side effects in normal organs is the key to improving targeting efficiency. On the one hand, researchers have identified several specific regulators to inhibit the LDLR activity in normal organs, including bile acid,40 sodium taurolaurate and hydrocortisone sodium succinate,41 saturated fats,40 cholesterol with hydrogenated coconut oil,42 modified LDL43 (acetylated LDL, methylated LDL and oxidized LDL), and angiotensin-II inhibitors.44 On the other hand, alternative receptors for ApoB-100 ligands can also reduce the binding of LDL to normal tissues.16,45 The study showed that the alkylation of the side chain of ApoB-100 lysine abolished the LDLR binding activity. Therefore, LDL can be redirected to targeted receptors other than LDLR.46The cell uptake of LDL with cargo is called LDLR-mediated endocytosis.47 This specific process is as follows (Fig. 2). Firstly, LDLR on the extracellular membrane specifically recognizes and embeds ApoB-100 of the LDL's phospholipid monolayer. Then, LDL binds to the ligand binding domain of LDLR and forms a clathrin-coated pit in the cell membrane. Subsequently, the vesicle envelopes with LDL and LDLR invaginate and form a vesicle. After continuous maturation, the vesicle undergoes depolymerization and fusion and is transformed into acidic endosomes. The low pH in the endosomes (as low as pH 5) triggers the separation of LDL from receptors. LDL is further transported into the lysosome and degraded into free cholesterol, fatty acids, and amino acids for use by cells. Finally, the receptor is recycled back to the cell surface and continues to bind and internalize with other LDL.48 The turnover time of LDLR is about 24 hours.49
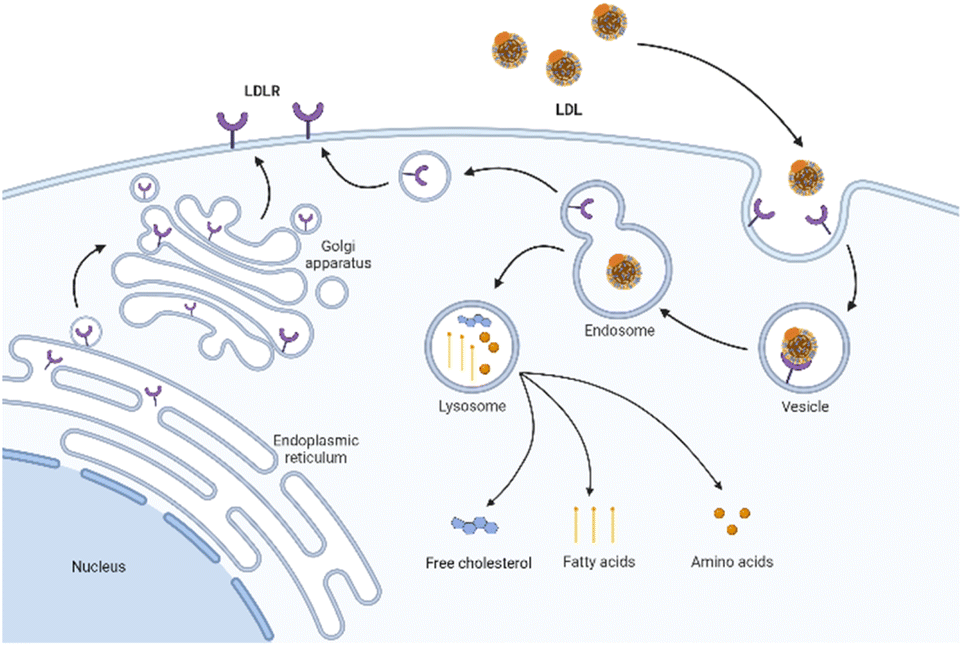 | ||
| Fig. 2 Schematic diagram of LDLR mediated endocytosis. | ||
3. LDL as a versatile nanocarrier
3.1 Methods for cargo carrying
The hydrophobic core of LDL can accommodate lipophilic payload. The amphiphilic phospholipid shell allows the loading of amphiphilic compounds. The amino acid residues exposed by ApoB-100 can covalently bind to the therapeutic or diagnostic cargo. In brief, there are three methods for cargo carrying in LDL:16 loading in the hydrophobic core (Fig. 3A), inserting into the phospholipid monolayer (Fig. 3B), and binding to the apolipoprotein (Fig. 3C).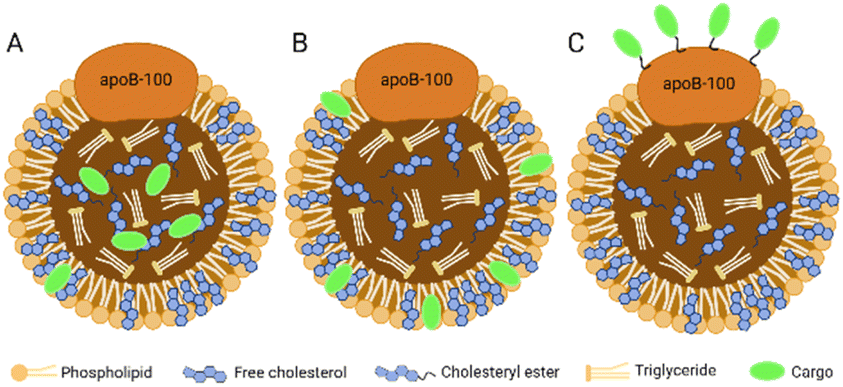 | ||
| Fig. 3 Three methods for cargo carrying in LDL. (A) Loading in the hydrophobic core; (B) inserting into the phospholipid monolayer; (C) binding to the apolipoprotein. | ||
3.2 Advantages of LDL-mediated delivery
As a versatile nanoscale building block, LDL has many unique advantages over other nanocarriers.31,58–60 First, LDL is a natural biological component with inherent biocompatibility, biodegradability, and non-immunogenicity. It is synthesized and metabolized in organisms and finally degraded into recyclable units, including cholesterol, fatty acids, and amino acids. In addition, LDL was able to evade the recognition of in vivo mononuclear phagocytes and reticuloendothelial systems, and the rapid clearance by the kidney due to the endogenous characteristics. Second, LDL has a long half-life (lasting 2–4 days). The long circulation characteristic of blood is conducive to the systemic delivery of drugs or probes. Third, the diameter of LDL is less than 30 nm in the nanometer range, making it diffuse from the blood vessel to the outside. The suitable size leads to good penetration of LDL through the interfibrillar opening (<40 nm) in solid tumors. Fourth, the primary defect of the widely used humanized targeting vectors, including monoclonal antibodies, growth factors, and hormones, is the low efficiency of cell internalization, which the LDL-mediated targeted delivery system can avoid. Furthermore, because of the LDLR over-expression in the tumor microenvironment, the selective affinity of LDL enhances the cellular uptake. Finally, LDL has a high loading capacity and can be processed by various loading strategies (specific methods will be described in the next section). The sizeable hydrophobic core allows LDL to functionally deliver hydrophobic bioactive compounds without changing the protein integrity and can effectively avoid the degradation or destruction of the load in the blood circulation. Amphiphilic shells are also well suited for loading amphiphilic compounds. In conclusion, LDL's size, structure, and various characteristics make it the preferred target vector.3.3 Defects of LDL-mediated delivery
Although LDL-based nanocarriers show a series of advantages, some defects still limit the delivery process:61 (1) LDL is challenging to obtain and extract in large quantities, and the composition and size of different batches are variable. (2) LDL particles are isolated from human blood, so there is a pathogen risk of causing infectious diseases. (3) As a lipoprotein, LDL has poor storage stability and is easily degraded by in vitro physical or chemical factors. (4) Although LDL is an effective carrier for the targeted delivery of drugs and diagnostic agents to tumor sites, its application in cancer treatment and diagnosis is limited by the expression level of LDLR. (5) Like other nanocarrier systems, LDL also has the defects of limited drug encapsulation efficiency and surface loading capacity. (6) LDL cholesterol is considered to be associated with myocardial infarction and atherosclerotic cardiovascular disease.62 Therefore, choosing LDL as a targeted delivery carrier to deliver into the body is controversial.3.4 Strategies on recombinant LDL
Because the defects of natural LDL limited the delivery applications, researchers tried to develop improved recombinant LDLs (rLDL). Recombinant lipoproteins are one of the most widely studied synthetic lipoproteins. Unlike natural lipoproteins extracted directly from human blood, recombinant lipoproteins are formed by combining isolated apolipoproteins with lipids (natural or synthetic analogs). Typically, each lipoprotein contains a single apolipoprotein type, while the lipid component type can be one or more.Based on the known functions of natural LDL, rLDL has unique advantages and performances. First, since all compounds constituting rLDL are clear, it is easier to characterize and repeat according to their respective structural components. Secondly, the synthesis of rLDL is achieved by covalent derivatization or other chemical modification of the lipid and protein components of nanoparticles.63 It means that the physicochemical properties of LDL (size, zeta potential, core, and surface loading) can be flexibly controlled by separately adjusting the stoichiometric ratio of lipid/protein, the types of lipid, apolipoprotein, or other components. Studies have shown that increasing the proportion of protein to lipids tends to reduce the diameter of lipoprotein complexes.64,65 Other factors, such as the type of apolipoprotein, also influence the final size of complexes.66
As early as the 1860 s, Scanu et al.67 separated human apolipoprotein and phospholipid from donors by a two-step method (ultra-centrifugation density centrifugation and organic solvent extraction). Then they induced apolipoprotein and phospholipid to self-assemble to recombinant lipoproteins through lyophilization, solubilization, co-incubation, and other operations. Walsh et al.68 proposed using ApoB isolated from human plasma and combining it with phospholipids to form rLDL by using sodium deoxycholate. Subsequently, Ginsburg et al.69 and Lundberg et al.70 modified the method and successfully synthesized rLDL nanoparticles.
However, there is no denying that the development of rLDL preparations is plagued by the limited availability of ApoB-100.51 Over the years, researchers have made various attempts to overcome this obstacle. Law et al.71 successfully cloned human ApoB-100 into an expression vector and obtained 560 amino acid sequences of the protein after specific screening and identification. Moreover, methods to construct lipoprotein-like nanoparticles using peptides that mimic the functional properties of apolipoproteins have been reported many times. This technology enables the mass production of lipoproteins and accelerates clinical translation for the targeted delivery of recombinant lipoproteins. In 2002, Baillie et al.72 developed a synthetic LDL (sLDL) formulation using a combination of a lipid emulsion and an amphipathic peptide containing the ApoB receptor domain. sLDLs mimic natural LDL and can specifically bind to U937 tumor cells through LDLR. In 2007, Nikanjam and colleagues27,73 synthesized an amphiphilicity α Helical peptide, composed of 29 amino acids and containing a particular sequence with nine amino acids that LDLR can recognize. This functional peptide was used to prepare a nano-LDL (nLDL) through a microemulsion with phosphatidylcholine, triolein, and cholesteryl oleate (Fig. 4). The study also demonstrated that the nLDL vector could target GBM cells with high LDLR expression. In the subsequent research, they loaded Paclitaxel Oleate (PO) into the core of nLDL to prepare drug-loaded nanocapsular-PO. The LDLR selectivity of nLDL shows the potential for targeted drug delivery as a vehicle. The advantages of peptide segments to simulate apolipoproteins are in many aspects, such as purity, quantity, processing time, and safety.74 Therefore, this method of synthesizing lipoproteins has received extensive attention from researchers.
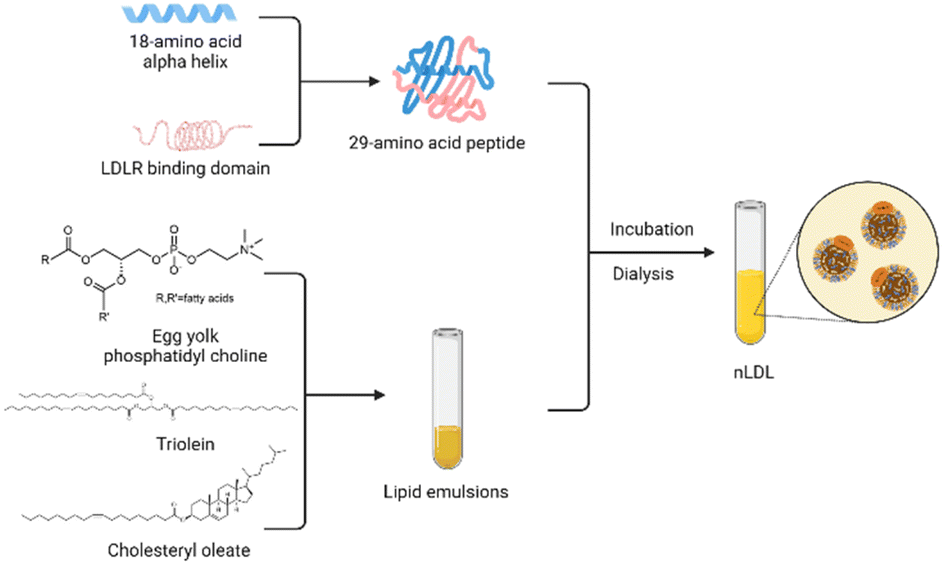 | ||
| Fig. 4 Strategy for synthesizing nLDL. | ||
4. LDL-mediated targeted delivery
4.1 Delivery for diagnosis
Combining different contrast agents allows LDL to have additional tumor imaging capabilities. We summarized the examples in Table 2 and showed the representative diagnostic cargoes in Fig. 5.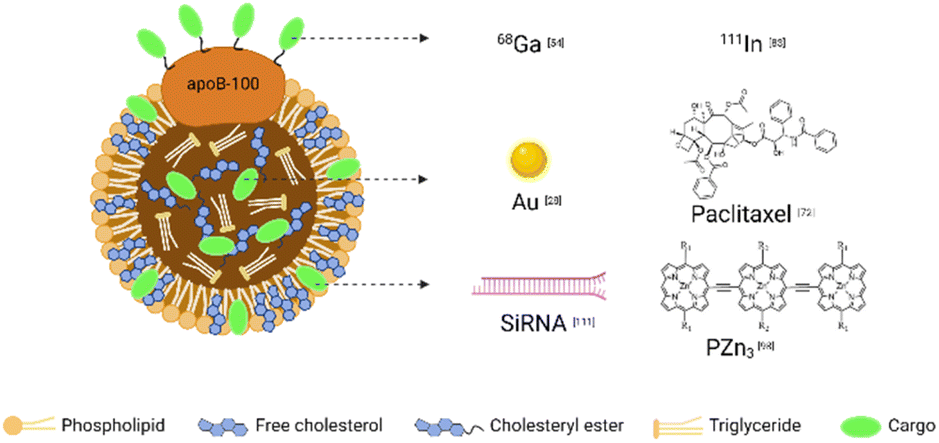 | ||
| Fig. 5 Representative cargoes in the LDL-mediated targeted delivery system for diagnosis and therapy. | ||
The experimental results of labeling LDL with iodine monochloride technology showed that the combination of l25I and LDL was stable.78 Ultrastructural autoradiography can be used to examine the effect of atherosclerotic regions of blood vessels on lipoprotein uptake and localization. Lewis and colleagues79 preferred white Carneau pigeons 0.25% cholesterol-supplemented diet to accelerate atherosclerosis in an experiment. After successful modeling, homologous 125I-LDL was injected intravenously, and the pigeons were killed one hour later. Autoradiography showed rapid absorption of 125I-LDL, which accumulated mainly in the liver, followed by the lung, kidney, spleen, and aorta. Compared with 125I, 131I has a shorter half-life but higher radiation energy. Therefore, 125I and 131I are widely used as radiolabels for various lipoproteins in vitro and in vivo.80–82 However, iodized LDL samples can cause lipid peroxidation and change the biological characteristics of LDL, and the most obvious consequence is to reduce its specific binding ability to LDLR. The immediate introduction of butylated hydroxytoluene and ascorbic acid can solve this problem. Studies have shown that these two antioxidants significantly inhibit radioiodine-induced lipid peroxidation and LDL modification.83 In addition, LDL labeled with gallium-68 (68Ga) or indium-111 (111In) has been considered as potential radioactive markers for positron emission tomography (PET) or single-photon emission computed tomography (SPECT) imaging.54,84
The method of immobilizing radionuclides directly on the surface of LDL has been reported in several studies. It has also been proposed to connect the radionuclide to apolipoprotein through the cyclic anhydride of diethylenetriaminepentaacetic acid (DTPA) to achieve chelation. However, this bifunctional agent will cause intramolecular or intermolecular crosslinking, leading to ApoB-100 deactivating.85,86 Therefore, some scholars chose to incorporate the lipophilic derivative of DTPA into the phospholipid monolayer of LDL to reduce the structural modification of ApoB-100.54 Nevertheless, these radiolabeling methods are inevitably limited by the degradable nature of lipoproteins. Once LDL is degraded in the body, radioactive markers will quickly leak and redistribute.87 So this process cannot quantitatively reflect the catabolism in tissues. The LDL-labeled residualized moiety tyramine-cellobiose can solve this difficulty. Because even when lipoproteins are metabolically degraded, tracers remain in tissue cells.84 In addition, Xiao et al.88 synthesized an (125)I-labeled hexa-iodinated diglyceride analog, named 1,3-dihydroxypropan-2-one 1,3-diiopanoate (DPIP). LDL is labeled with core reconstitution and is resistant to lysosomal degradation after cellular uptake.
Because of the polarity of conventional X-ray agents, linking these molecules to LDL is more complicated than radionuclides. Hill et al.89 explored the incorporation of poly-iodinated triglyceride (ITG) into LDL for targeted delivery of CT contrast agents. They first developed an ITG-loaded LDL ((rITG)LDL). In the cell particle CT imaging, the attenuation was weak with excessive natural LDL, which indicated that excessive natural LDL effectively inhibited (rITG)LDL from entering human hepatoblastoma G2 (HepG2) cells, further confirming that LDL modified with ITG did not damage its ability to recognize LDLR and interact with it. Allijn et al.28 established a new LDL labeling method based on sonication and ultracentrifugation purification. Specifically, the method first encapsulated various payloads (such as gold nanoparticles and lipophilic or amphiphilic fluorophores) in micelles, and then transferred them to the LDL core by sonication and ultracentrifugation. The experimental results in vitro and in vivo showed that LDL loaded with gold nanocrystals had similar characteristics in size, morphology, composition, oxidation state, ApoB-100 function, and molecular weight compared with untreated natural LDL. In conclusion, as one of the few reports on targeted CT contrast agents, Au-LDL can be used as a marker to study LDL interaction, cholesterol metabolism, atherosclerosis, tumor growth, and other fields.
Since magnetic resonance imaging (MRI) has the advantages of high spatial resolution, no radiation,90 and no depth limitation, LDL has also been explored for the delivery of MRI contrast agents. The most commonly used MRI contrast agents in clinical practice are the paramagnetic chelate of Gd3+ ions to increase the longitudinal relaxation rate of water protons distributed in tissues. Corbin et al.91 inserted amphiphilic Gd-DTPA chelate into a LDL phospholipid monolayer to achieve surface modification of LDL. The results of the structural and functional properties of Gd-labeled LDL nanoparticles showed a similar diameter and surface charge, and the ability to bind to LDLR as native LDL. In addition, LDL-containing tissues (liver and HepG2 tumors) showed significant MRI contrast enhancement after intravenous administration of Gd-labeled LDL. Thus, Gd-labeled LDL has the potential to be an MRI contrast agent for in vivo tumor detection. However, the disadvantage of this targeting system is its relatively low relaxation. On this basis, to enhance the relaxation of the complex system and improve the thermodynamic stability, Crich and colleagues37 developed a new Gd complex, namely Gd-AAZTAC17/LDL. When added to the incubation medium, the Gd-AAZTAC17/LDL adduct was absorbed by HepG2 and melanoma B16 tumor cells. In vivo MRI analysis of C57BL/6 mice transplanted with melanoma B16 cells showed that the enhancement of tumor signal intensity 8 hours after the injection of the Gd-AAZTAC17/LDL adduct was significantly higher than that of Gd-AAZTAC17 alone. Therefore, this Gd complex can be an effective and sensitive MRI probe. Several LDL-based MRI probes have been reported in recent years after the optimization of the targeting system.92–96
As early as the early 2000s, Zheng et al.98 synthesized a tricarbocyanine cholesteryl laurate (TCL) with primary amine functional groups. A stable conjugate probe TCL17-LDL was prepared by chelating TCL to the lipid region of LDL. TCL17-LDL was i.v. injected to detect HepG2 tumor tissues by using a low-temperature 3-D redox scanner. Images showed an enhanced fluorescent signal only for the tumor tissue, confirming the tumor-targeted delivery of TCL17-LDL. Wu et al.99 incorporated meso-to-meso ethyne-bridged tris [(porphinato) zinc(II)] (PZn3) into the hydrophobic core of LDL. In vitro experiments demonstrated that PZn3-containing LDL transferred core NIRFs to murine B16 melanoma cells through the LDLR pathway and achieved the imaging of B16 cells by confocal near-infrared fluorescence microscopy in a shallow dose (∼nM). Trivedi et al.100 reported the synthesis of chiral porphyrazine (pz), H2 [pz (trans-A2B2)]-247 (Pz 247). Pz 247 bound to the lipophilic core of LDL and entered the cell primarily through receptor-mediated endocytosis. In vivo studies have shown that Pz 247 exhibited preferential accumulation and retention in murine subcutaneously implanted MDA-MB-231 tumor cells, thus enabling NIRF imaging to distinguish tumors from surrounding normal tissue.
As a classic carbocyanine dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil) was used to label native or modified LDL. Li and colleagues101 synthesized and validated the possibility of noninvasive NIRF imaging of LDLR-overexpressing tumors using Dil-labeled LDL. In addition, they confirmed the selective accumulation of DiI-LDL in the tumor area using confocal microscopy and three-dimensional cryo-imaging.
Unlike the above targeting methods, Chen et al.45 introduced an LDL redirection strategy. The strategy was to transfer LDL nanoparticles from their native receptor to alternative surface receptors or epitopes by coupling tumor-directed ligands to active lysine residues exposed in ApoB-100. Following this idea, they prepared LDL nanoparticles labeled with the fluorescent dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) and modified with folic acid (FA), DIR-LDL-FA. The probe was accumulated in KB cells for the folate receptor (FR) or LDLR overexpression but only slightly absorbed in cells without FR (CHO and HT1080).
To solve the problems of long-term toxicities, low quantum yields, and poor water solubilities of some NIR-II fluorophores, Kalot et al.102 developed a water-soluble NIR-II emitting nitrogen BODIPY derivative SWIR-W AZABY-01. They used lipoprotein gel electrophoresis and ultracentrifugation to prove that the SWIR-WAZABy-01 fluorophore mainly interacted with LDL in human plasma. LDL drove the circulation of SWIR-WAZABy-01 in the blood and strongly increased its fluorescence emission. Therefore, SWIR-W AZABY-01 could be used as a new and relevant tool to label LDL for pathology effectively.
4.2 Delivery for therapy
| Load type | Examples | Ref. |
|---|---|---|
| Therapeutic agents | DOX | 103–106 |
| Paclitaxel | 72 and 107–109 | |
| siRNA | 110–114 | |
| DHA | 117–121 | |
| 5-Fluorouracil | 122 | |
| Imatinib | 123 | |
| Donepezil | 25 | |
| Oxaliplatin | 124 | |
| Thiosemicarbazone metal–ligand complexes | 125 | |
| Polysaccharide | 126 | |
| Dexamethasone palmitate | 130–132 | |
| Photosensitizers | Porphyrin compounds and derivatives | 135, 137 and 138 |
| Pyropheophorbide cholesterol oleate conjugate | 139 | |
| SiPcBOA | 140 | |
| SiNcBOA | 141 | |
| Bchl-BOA | 142 | |
| TPA DPPy | 143 |
Chu et al.104 conjugated the cytotoxic drug doxorubicin (DOX) to human LDL to form a complex (LDL-DOX). When injected into mice, LDL-DOX accumulated more in the liver than free DOX and less in the heart. Both histological and enzymatic analyses showed that LDL-DOX could reduce DOX-induced cardiotoxicity. Over the next decade, various DOX-loaded LDLs were designed and used to explore the treatment of different tumor models. For example, Pinzón-Daza and colleagues105 combined statins with DOX-loaded LDL to convert a less permeable DOX into a drug capable of crossing the blood–brain barrier (BBB), thereby achieving significant anti-brain tumor cytotoxicity. Tian and colleagues106 encapsulated DOX with self-assembled nanoparticles composed of chitosan and LDL. After the incubation of this pellet with gastric cancer SGC7901 cells for 24 h, encapsulated DOX showed higher cellular uptake than free DOX. A recent study reported a PH-sensitive ApoB-100/Oleic-DOX/NLC (AODN) nanoparticle based on a nanostructured lipid carrier. The experimental results show that AODN nanoparticles can accumulate more drugs in the tumor site, reduce systemic toxicity, and effectively inhibit in situ breast cancer.107
LDL is also reported to be used by researchers to load another common cytotoxic drug, paclitaxel. Nikanjam and coworkers73 incorporated the lipophilic prodrug paclitaxel oleate into the lipid core of LDL particles to prepare drug-loaded particles that could target and kill glioblastoma multiforme cells. Unlike this, another glioma treatment study108 developed nanoparticles loaded with dual-targeting paclitaxel by modification with peptide-22 (PNP-PTX). Su et al. designed a sLDL to encapsulate paclitaxel-alpha linolenic acid (PALA). PALA-loaded sLDL (PALA-sLDL) had higher tumor accumulation and tumor suppression efficiency than a PALA-loaded microemulsion (PALA-ME, without the LDLR binding domain). In addition, the use of paclitaxel-loaded LDL in lung cancer models has been reported. Kim et al.109 developed a solid lipid nanoparticle (SLN) with paclitaxel at its core. Qian et al.110 used lipoprotein-mimic nanoparticles modified with an amphiphilic hybrid peptide to deliver paclitaxel.
Interestingly, neither of these studies directly utilized LDL but developed biomimetic LDL nanocarriers instead. Nevertheless, both LDL-like agents showed significant antitumor activity in lung cancer models, demonstrating the potential use of these nanocarriers to improve therapeutic efficacy and reduce the side effects of antitumor drugs.
Small interfering RNA (siRNA) plays a vital role in silencing multidrug resistance genes in tumors.111 Therefore, developing effective and stable siRNA vectors is crucial for siRNA system delivery. Zhu and coworkers112 loaded cholesterol-binding siRNA onto LDL, and DOX-loaded chol-siRNA/LDL-coupled N-succinyl chitosan nanoparticles (Dox-siRNA/LDL-SCS-NPs) were prepared and characterized. In vivo tumor targeting demonstrated significant accumulation of this agent in an orthotopic liver tumor model. Jin and colleagues113 combined SLN with pegylated c-Met siRNA. This complex not only effectively down-regulated the expression level of c-Met in vitro but also reduced cell proliferation in U-87 MG. Yang et al.114 developed a new micelle loaded with paclitaxel and an LDL nanoparticle loaded with siRNA, which were coupled to form a “binary polymer”. For cancer cell targeting, this system has the potential to be used to co-deliver siRNA and antitumor drugs to address multidrug resistance in cancer. Notably, in one study,115 siRNA was also used to target connective tissue growth factors to treat liver fibrosis. This study showed that the stable nanocomplex could be precisely delivered to the liver and silence the targeted gene by cell internalization. It reduced the collagen content and pro-fibrogenic factors and significantly improved the pathophysiological symptoms of liver fibrosis model rats.
As a natural omega-3 polyunsaturated fatty acid, docosahexaenoic acid (DHA) has antitumor properties.116,117 Reynolds and colleagues118 investigated the cytotoxic effects of DHA-loaded LDL nanoparticles (LDL-DHA) on the liver. The therapeutic utility of LDL-DHA nanoparticles was evaluated in normal and malignant murine liver cell lines TIB-73 and TIB-75, respectively. After administration, therapeutic doses of LDL-DHA ultimately killed TIB-75 but did not harm TIB-73. This study demonstrated the potential of LDL-DHA nanoparticles as an anti-hepatocellular carcinoma agent. On this basis, scholars have continuously verified the effect of LDL nanoparticles loaded with DHA on liver tumors.119,120 A study also explored the molecular mechanism underlying the anticancer activity of LDL-DHA nanoparticles and indicated that LDL-DHA could induce HCC cell death through the iron-death pathway.121 Additionally, DHA also plays a role in regulating the neural function of the brain. Mulik and colleagues122 then used pulsed focused ultrasound (FUS) exposures to open up the BBB to deliver LDL-DHA locally to the brain. FUS exposures resulted in a 2-fold increase in DHA levels in target brain regions compared with nontarget regions or control treatment groups. This technique provides an alternative idea for targeting acute brain injury areas or invasive tumor cells in the brain.
In addition to these well-studied anticancer agents, other anticancer drugs such as 5-fluorouracil,123 imatinib124 donepezil,25 and oxaliplatin125 have also been explored to be loaded onto LDL for targeted delivery. In a recent study,126 the anticancer agent thiosemicarbazone metal–ligand complexes were encapsulated in LDL, targeting breast (MCF7), lung (A549), and prostate (C42) cancer cells, respectively. Cytotoxicity assessment reported effective cancer cell growth inhibition in all studied cell lines. In addition, western blot analysis showed that tubulin expression was significantly reduced when the cell line was treated with LDL encapsulated with thiosemicarbazone metal–ligand complexes. In conclusion, all the experimental results indicated the potential feasibility of LDL as an active drug delivery strategy for cancer treatment.
Interestingly, all the above studies used the targeted delivery method of intravenous administration, while Zhou et al.127 took a different approach and explored a novel oral delivery modality. They combined a polysaccharide with LDL (extracted from fresh egg yolk) to form a composite nanogel with a diameter of less than 85 nm, which was able to encapsulate curcumin. Under simulated gastrointestinal conditions, polysaccharides can achieve stable and sustained release of curcumin while protecting LDL from enzymatic degradation in the stomach. This study suggests that LDL/polysaccharides could be a potential oral delivery system for drugs or nutrients.
It is important to note that LDL can also be used as an anti-atherogenic drug carrier. When LDL in the aorta is continuously deposited and oxidized, it will be converted into foam cells after engulfing by the recruited macrophages, thus intensifying the formation of atherosclerotic plaque. Compared with natural LDL, those oxidized particles have a higher affinity for atherosclerotic plaque. Therefore, LDL can be used as an ideal carrier for the specific delivery of drugs to atherosclerotic lesions.128–130 Tauchi and colleagues131–133 focused on this exploration and reported several studies. By incorporating dexamethasone palmitate (DP) into LDL, they demonstrated the inhibitory effect of the DP-LDL complex on foam cell formation through serial in vivo and in vitro experiments. They also examined the effect of DP-LDL on cholesteryl ester accumulation in the aorta of atherosclerotic mice. The DP-LDL complex showed a 100-fold decreased cholesteryl ester accumulation in the aorta than the free dexamethasone. Although more long-term studies are needed to establish validation, the DP-LDL complex is expected to guide the treatment of atherosclerosis in the clinic.
Moreover, another advantage of using LDL during PDT is that LDL is highly oxidized after irradiation. The resulting oxide is cytotoxic to endothelial cells and can further prolong the photodynamic effect.137 The examples mentioned below can be found in Table 3.
Among all photosensitizers, porphyrin compounds and their derivatives have been widely used in cancer PDT due to their unique advantages, such as high singlet oxygen quantum yields, apparent lack of dark toxicity, and strong adsorption capacity in the near-infrared region.39 For example, haematoporphyrin,138 benzoporphyrin derivative,136 tetraphenylporphyrin,139 and other porphyrin-related PSs incorporated into LDL for PDT have been reported earlier.
Earlier studies focused on the non-covalent binding of PS to LDL or the covalent ligation of PS to ApoB-100. However, Zheng and colleagues concentrated on core-load reconstruction strategies to improve the labeling rate of PS and serum stability. In one study, they synthesized a pyropheophorbide cholesterol oleate conjugate and successfully reconstituted it into the lipid core of LDL. Laser scanning confocal microscopy showed that this rLDL could be internalized into HepG2 tumor cells via the LDLR pathway.140 Later, they141 designed SiPcBOA, a tetra-t-butyl silicon phthalocyanine compound with two oleate moieties in the axial position. The final synthesized rLDL(r-SiPcBOA-LDL) had a very high payload (SiPcBOA to LDL molar ratio > 3000 to 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001:1). Similarly, they142 synthesized a novel NIRF imaging agent tetra-t-butyl silicon naphthalocyanine bisoleate (SiNcBOA). Again, high loading (100:1 payload) was achieved while successfully reassembling it into the LDL lipid core. Besides, they143 engineered reconstituted bacteriochlorin e6 bisoleate LDL (r-Bchl-BOA-LDL) and evaluated the efficacy of PDT using a mice tumor model. Compared with the control group, the group injected with 2 μm per kg r-Bchl-BOA-LDL significantly delayed tumor regeneration.
001:1). Similarly, they142 synthesized a novel NIRF imaging agent tetra-t-butyl silicon naphthalocyanine bisoleate (SiNcBOA). Again, high loading (100:1 payload) was achieved while successfully reassembling it into the LDL lipid core. Besides, they143 engineered reconstituted bacteriochlorin e6 bisoleate LDL (r-Bchl-BOA-LDL) and evaluated the efficacy of PDT using a mice tumor model. Compared with the control group, the group injected with 2 μm per kg r-Bchl-BOA-LDL significantly delayed tumor regeneration.
Recently, Wang and coworkers144 reported for the first time that natural LDL particles were recombined with saturated fatty acids and mitochondria-targeting aggregation-induced emission (AIE) PSs. The new AIE photosensitizer (TPA DPPy) encapsulated in LDL granules was effectively absorbed by cancer cells and released to the mitochondria. Under light irradiation, reactive oxygen species produced around mitochondria lead to irreversible apoptosis of cancer cells. Unlike previous studies, this process can be monitored in real time by fluorescence, such as significantly enhanced luminescence and blue-shifted emission. This innovative method of providing real-time fluorescence feedback on treatment results has dramatically improved the quality of PDT.
5. Conclusions and prospects
Since lipoproteins are genius drug carriers and diagnostic tools, the research of lipoprotein-based nano-delivery systems has been carried out and developed continuously. Among them, LDL, as a prominent representative, has increasingly diversified applications in the field of targeted delivery. LDL-based carriers can transport various substances and reagents, including imaging agents, chemotherapeutic drugs, antiviral drugs, antibacterial agents, and siRNA. These applications provide great potential for clinical transformation. LDL-based nanoparticles show a series of advantages, such as superior payload capacity, long residence time in circulation, excellent biocompatibility, smaller size, and natural targeting. However, there are still many problems to be solved. The difficulty in obtaining and purifying natural LDL is one of the main reasons that limit its research and application. The functionalized rLDL provides a new direction for applying LDL as a carrier. Exploring the construction of novel lipoproteins or apolipoproteins can offer novel ideas and methods for designing drug delivery systems. It is affirmative that novel LDL-based nanoformulations will become a promising diagnostic and therapeutic drug delivery nano-platform. However, more systematic and comprehensive in vivo evaluation is necessary to safely use LDL preparations in humans. Admittedly, LDL and its derivatives, driven by advances in nanotechnology and bionics, will play a key role in solving more human diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 52203186), the “Young Talent Support Plan” of Xi’an Jiaotong University, China (No. YX6J014), and the Natural Science Foundation of Shaanxi Provincial Department of Education (No. 2022KW-01 and 2022JM-101). The authors thank the Library and Instrument Analysis Center of Xi'an Jiaotong University.References
- H. Lu, J. Xu, J. Yang, Z. Wang, P. Xu, Q. Hao, W. Luo, S. Li, Z. Li, X. Xue, H. Zheng, Z. Zhou, H. Wu, X. Ma and Y. Li, Biomaterials, 2022, 290, 121852 CrossRef CAS PubMed.
- S. Raj, S. Khurana, R. Choudhari, K. K. Kesari, M. A. Kamal, N. Garg, J. Ruokolainen, B. C. Das and D. Kumar, Semin. Cancer Biol., 2021, 69, 166–177 CrossRef CAS PubMed.
- H. Wu, H. Lu, W. Xiao, J. Yang, H. Du, Y. Shen, H. Qu, B. Jia, S. K. Manna, M. Ramachandran, X. Xue, Z. Ma, X. Xu, Z. Wang, Y. He, K. S. Lam, R. J. Zawadzki, Y. Li and T. Y. Lin, Adv. Mater., 2020, 32, e1903759 CrossRef PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CrossRef CAS PubMed.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS PubMed.
- A. L. Cortajarena, D. Ortega, S. M. Ocampo, A. Gonzalez-García, P. Couleaud, R. Miranda, C. Belda-Iniesta and A. Ayuso-Sacido, Nanobiomedicine, 2014, 1, 2 CrossRef PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- S. Hua, Front. Pharmacol., 2015, 6, 219 Search PubMed.
- N. Desai, AAPS J., 2012, 14, 282–295 CrossRef CAS PubMed.
- R. M. Crist, S. S. K. Dasa, C. H. Liu, J. D. Clogston, M. A. Dobrovolskaia and S. T. Stern, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1665 CAS.
- K. K. Ng, J. F. Lovell and G. Zheng, Acc. Chem. Res., 2011, 44, 1105–1113 CrossRef CAS PubMed.
- D. S. Fredrickson, Circulation, 1993, 87, III1–15 CrossRef CAS PubMed.
- N. S. Chung and K. M. Wasan, Adv. Drug Delivery Rev., 2004, 56, 1315–1334 CrossRef CAS PubMed.
- H. Huang, W. Cruz, J. Chen and G. Zheng, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 298–314 CAS.
- L. Zhang, J. Song, G. Cavigiolio, B. Y. Ishida, S. Zhang, J. P. Kane, K. H. Weisgraber, M. N. Oda, K. A. Rye, H. J. Pownall and G. Ren, J. Lipid Res., 2011, 52, 175–184 CrossRef CAS PubMed.
- G. Zheng, J. Chen, H. Li and J. D. Glickson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17757–17762 CrossRef CAS PubMed.
- C. Zhu, P. Pradhan, D. Huo, J. Xue, S. Shen, K. Roy and Y. Xia, Angew. Chem., Int. Ed., 2017, 56, 10399–10402 CrossRef CAS PubMed.
- K. M. Wasan and S. M. Cassidy, J. Pharm. Sci., 1998, 87, 411–424 CrossRef CAS PubMed.
- W. H. D. Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef PubMed.
- T. Skajaa, D. P. Cormode, E. Falk, W. J. Mulder, E. A. Fisher and Z. A. Fayad, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 169–176 CrossRef CAS PubMed.
- K. M. McMahon, L. Foit, N. L. Angeloni, F. J. Giles, L. I. Gordon and C. S. Thaxton, Cancer Treat. Res., 2015, 166, 129–150 CAS.
- R. Kuai, D. Li, Y. E. Chen, J. J. Moon and A. Schwendeman, ACS Nano, 2016, 10, 3015–3041 CrossRef CAS PubMed.
- S. Raut, J. L. Dasseux, N. A. Sabnis, L. Mooberry and A. Lacko, Ther. Delivery, 2018, 9, 257–268 CrossRef CAS PubMed.
- E. Gianazza, B. Zoanni, A. Mallia, M. Brioschi, G. I. Colombo and C. Banfi, Mass Spectrom. Rev., 2021, 1–27, DOI:10.1002/mas.21747.
- K. V. Krishna, R. N. Saha and S. K. Dubey, ACS Chem. Neurosci., 2020, 11, 4139–4151 CrossRef CAS PubMed.
- F. Alanazi, Z. F. Fu and D. R. Lu, Pharm. Res., 2004, 21, 675–682 CrossRef CAS PubMed.
- M. Nikanjam, E. A. Blakely, K. A. Bjornstad, X. Shu, T. F. Budinger and T. M. Forte, Int. J. Pharm., 2007, 328, 86–94 CrossRef CAS PubMed.
- I. E. Allijn, W. Leong, J. Tang, A. Gianella, A. J. Mieszawska, F. Fay, G. Ma, S. Russell, C. B. Callo, R. E. Gordon and E. Korkmaz, ACS Nano, 2013, 7, 9761–9770 CrossRef CAS PubMed.
- M. Teemu, V. A. Timo, H. T. Marja, M. Siewert-Jan, K. Mikko and V. Ilpo, Soft Matter, 2011, 7, 8135–8141 RSC.
- P. E. Fielding and C. J. Fielding, in New Comprehensive Biochemistry, Elsevier, 2002, vol. 36, pp. 527–552 Search PubMed.
- G. I. Harisa and F. K. Alanazi, Saudi Pharm. J., 2014, 22, 504–515 CrossRef PubMed.
- S. Muntoni, L. Atzori, R. Mereu, G. Satta, M. D. Macis, M. Congia, A. Tedde, A. Desogus and S. Muntoni, Nutr., Metab. Cardiovasc. Dis., 2009, 19, 218–225 CrossRef CAS PubMed.
- M. Moschovi, G. Trimis, F. Apostolakou, I. Papassotiriou and F. Tzortzatou-Stathopoulou, J. Pediatr. Hematol./Oncol., 2004, 26, 289–293 CrossRef PubMed.
- A. Niendorf, H. Nägele, D. Gerding, U. Meyer-Pannwitt and A. Gebhardt, Int. J. Cancer, 1995, 61, 461–464 CrossRef CAS PubMed.
- S. Vitols, G. Gahrton, M. Björkholm and C. Peterson, Lancet, 1985, 2, 1150–1154 CrossRef CAS PubMed.
- R. A. Firestone, Bioconjugate Chem., 1994, 5, 105–113 CrossRef CAS PubMed.
- S. G. Crich, S. Lanzardo, D. Alberti, S. Belfiore, A. Ciampa, G. B. Giovenzana, C. Lovazzano, R. Pagliarin and S. Aime, Neoplasia, 2007, 9, 1046–1056 CrossRef CAS PubMed.
- S. Xu, B. Z. Olenyuk, C. T. Okamoto and S. F. Hamm-Alvarez, Adv. Drug Delivery Rev., 2013, 65, 121–138 CrossRef CAS PubMed.
- J. Anupriya, J. Keerti, K. Prashant and J. K. Narendra, J. Nanopart. Res., 2013, 15, 1888 CrossRef.
- B. Angelin, C. A. Raviola, T. L. Innerarity and R. W. Mahley, J. Clin. Invest., 1983, 71, 816–831 CrossRef CAS PubMed.
- S. A. Hynds, J. Welsh, J. M. Stewart, A. Jack, M. Soukop, C. S. McArdle, K. C. Calman, C. J. Packard and J. Shepherd, Biochim. Biophys. Acta, 1984, 795, 589–595 CrossRef CAS PubMed.
- D. K. Spady and J. M. Dietschy, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4526–4530 CrossRef CAS PubMed.
- R. E. Pitas, J. Boyles, R. W. Mahley and D. M. Bissell, J. Cell Biol., 1985, 100, 103–117 CrossRef CAS PubMed.
- E. Leitersdorf, O. Stein and Y. Stein, Biochim. Biophys. Acta, 1985, 835, 183–190 CrossRef CAS PubMed.
- J. Chen, I. R. Corbin, H. Li, W. Cao, J. D. Glickson and G. Zheng, J. Am. Chem. Soc., 2007, 129, 5798–5799 CrossRef CAS PubMed.
- J. D. Glickson, S. Lund-Katz, R. Zhou, H. Choi, W. Chen, H. Li, I. Corbin, A. V. Popov, W. Cao, L. Song and C. Qi, Adv. Exp. Med. Biol., 2009, 645, 227–239 CrossRef CAS PubMed.
- A. Nykjaer and T. E. Willnow, Trends Cell Biol., 2002, 12, 273–280 CrossRef CAS PubMed.
- T. E. Willnow, A. Nykjaer and J. Herz, Nat. Cell Biol., 1999, 1, E157–E162 CrossRef CAS PubMed.
- M. S. Brown and J. L. Goldstein, Science, 1986, 232, 34–47 CrossRef CAS PubMed.
- M. Krieger, L. C. Smith, R. G. Anderson, J. L. Goldstein, Y. J. Kao, H. J. Pownall, A. M. Gotto and M. S. Brown, J. Supramol. Struct., 1979, 10, 467–478 CrossRef CAS PubMed.
- W. J. McConathy, M. P. Nair, S. Paranjape, L. Mooberry and A. G. Lacko, Anti-Cancer Drugs, 2008, 19, 183–188 CrossRef CAS PubMed.
- L. Mateu, A. Tardieu, V. Luzzati, L. Aggerbeck and A. M. Scanu, J. Mol. Biol., 1972, 70, 105–116 CrossRef CAS PubMed.
- A. A. Radwan and F. K. Alanazi, Saudi Pharm. J., 2014, 22, 3–16 CrossRef PubMed.
- F. Jasanada, P. Urizzi, J. P. Souchard, L. F. Gaillard, G. Favre and F. Nepveu, Bioconjugate Chem., 1996, 7, 72–81 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, 2013 Search PubMed.
- S. Lund-Katz, J. A. Ibdah, J. Y. Letizia, M. T. Thomas and M. C. Phillips, J. Biol. Chem., 1988, 263, 13831–13838 CrossRef CAS PubMed.
- I. R. Corbin, Methods Mol. Biol., 2013, 1049, 467–480 CrossRef CAS PubMed.
- N. Sabnis and A. G. Lacko, Ther. Delivery, 2012, 3, 599–608 CrossRef CAS PubMed.
- V. Huntosova, D. Buzova, D. Petrovajova, P. Kasak, Z. Nadova, D. Jancura, F. Sureau and P. Miskovsky, Int. J. Pharm., 2012, 436, 463–471 CrossRef CAS PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- J. M. Shaw and K. V. Shaw, Targeted Diagn. Ther., 1991, 5, 351–383 CAS.
- M. B. Mortensen and B. G. Nordestgaard, Lancet, 2020, 396, 1644–1652 CrossRef CAS PubMed.
- A. G. Lacko, M. Nair, L. Prokai and W. J. McConathy, Expert Opin. Drug Delivery, 2007, 4, 665–675 CrossRef CAS PubMed.
- M. J. Thomas, S. Bhat and M. G. Sorci-Thomas, J. Lipid Res., 2008, 49, 1875–1883 CrossRef CAS PubMed.
- I. G. Denisov, Y. V. Grinkova, A. A. Lazarides and S. G. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed.
- B. A. Chromy, E. Arroyo, C. D. Blanchette, G. Bench G, H. Benner, J. A. Cappuccio, M. A. Coleman, P. T. Henderson, A. K. Hinz, E. A. Kuhn and J. B. Pesavento, J. Am. Chem. Soc., 2007, 129, 14348–14354 CrossRef CAS PubMed.
- A. Scanu, J. Biol. Chem., 1967, 242, 711–719 CrossRef CAS PubMed.
- M. T. Walsh and D. Atkinson, Biochemistry, 1983, 22, 3170–3178 CrossRef CAS PubMed.
- G. S. Ginsburg, M. T. Walsh, D. M. Small and D. Atkinson, J. Biol. Chem., 1984, 259, 6667–6673 CrossRef CAS PubMed.
- B. Lundberg, Cancer Res., 1987, 47, 4105–4108 CAS.
- S. W. Law, K. J. Lackner, A. V. Hospattankar, J. M. Anchors, A. Y. Sakaguchi, S. L. Naylor and H. B. Brewer, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 8340–8344 CrossRef CAS PubMed.
- G. Baillie, M. D. Owens and G. W. Halbert, J. Lipid Res., 2002, 43, 69–73 CrossRef CAS.
- M. Nikanjam, A. R. Gibbs, C. A. Hunt, T. F. Budinger and T. M. Forte, J. Controlled Release, 2007, 124, 163–171 CrossRef CAS PubMed.
- C. L. Nykiforuk, Y. Shen, E. W. Murray, J. G. Boothe, D. Busseuil, E. Rhéaume, J. C. Tardif, A. Reid and M. M. Moloney, Plant Biotechnol. J., 2011, 9, 250–263 CrossRef CAS PubMed.
- R. S. Lees, H. D. Garabedian, A. M. Lees, D. J. Schumacher, A. Miller, J. L. Isaacsohn, A. Derksen and H. W. Strauss, J. Nucl. Med., 1985, 26, 1056–1062 CAS.
- A. M. Lees, R. S. Lees, F. J. Schoen, J. L. Isaacsohn, A. J. Fischman, K. A. McKusick and H. W. Strauss, Arteriosclerosis, 1988, 8, 461–470 CAS.
- J. L. Isaacsohn, A. M. Lees, R. S. Lees, H. W. Strauss, M. Barlai-Kovach and T. J. Moore, Metabolism, 1986, 35, 364–366 CrossRef CAS PubMed.
- H. Sinzinger, G. Lupattelli, H. Kritz, P. Fitscha and J. O'Grady, Prostaglandins, 1996, 52, 77–91 CrossRef CAS PubMed.
- J. C. Lewis and R. G. Taylor, Histochem. J., 1994, 26, 833–843 CrossRef CAS PubMed.
- K. Kornerup, B. G. Nordestgaard, B. Feldt-Rasmussen, K. Borch-Johnsen, K. S. Jensen and J. S. Jensen, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1168–1174 CrossRef CAS PubMed.
- L. B. Nielsen, S. Stender, K. Kjeldsen and B. G. Nordestgaard, Circ. Res., 1996, 78, 615–626 CrossRef CAS PubMed.
- R. A. Medina, L. E. Cardona-Sanclemente, G. V. Born and M. J. Brown, J. Hypertens., 1997, 15, 531–536 CrossRef CAS PubMed.
- A. S. Khouw, S. Parthasarathy and J. L. Witztum, J. Lipid Res., 1993, 34, 1483–1496 CrossRef CAS.
- S. M. Moerlein, A. Daugherty, B. E. Sobel and M. J. Welch, J. Nucl. Med., 1991, 32, 300–307 CAS.
- D. J. Hnatowich and J. McGann, Int. J. Radiat. Appl. Instrum., Part B, 1987, 14, 563–568 CrossRef CAS PubMed.
- F. Maisano, L. Gozzini and C. D. Haën, Bioconjugate Chem., 1992, 3, 212–217 CrossRef CAS PubMed.
- S. M. Moerlein, K. B. Dalal, S. N. Ebbe, Y. Yano and T. F. Budinger, Int. J. Radiat. Appl. Instrum., Part B, 1988, 15, 141–149 CrossRef CAS PubMed.
- W. Xiao, L. Wang, J. M. Ryan, A. Pater and H. Liu, Radiat. Res., 1999, 152, 250–256 CrossRef CAS PubMed.
- M. L. Hill, I. R. Corbin, R. B. Levitin, W. Cao, J. G. Mainprize, M. J. Yaffe and G. Zheng, Acad. Radiol., 2010, 17, 1359–1365 CrossRef PubMed.
- J. Wallyn, N. Anton, S. Akram and T. F. Vandamme, Pharm. Res., 2019, 36, 78 CrossRef PubMed.
- I. R. Corbin, H. Li, J. Chen, S. Lund-Katz, R. Zhou, J. D. Glickson and G. Zheng, Neoplasia, 2006, 8, 488–498 CrossRef CAS PubMed.
- S. Wen, D. F. Liu, Y. Cui, S. S. Harris, Y. C. Chen, K. C. Li, S. H. Ju and G. J. Teng, Nanomedicine, 2014, 10, 639–649 CrossRef CAS PubMed.
- D. Alberti, N. Protti, A. Toppino, A. Deagostino, S. Lanzardo, S. Bortolussi, S. Altieri, C. Voena, R. Chiarle, S. Geninatti Crich and S. Aime, Nanomedicine, 2015, 11, 741–750 CrossRef CAS PubMed.
- M. Mirzaei, M. Mohseni, N. Iranpour Anaraki, E. Mohammadi, S. Safari, B. Mehravi, A. Ghasempour and O. Pourdakan, Artif. Cells, Nanomed., Biotechnol., 2020, 48, 46–52 CrossRef CAS PubMed.
- A. Fracassi, J. Cao, N. Yoshizawa-Sugata, É. Tóth, C. Archer, O. Gröninger, E. Ricciotti, S. Y. Tang, S. Handschin, J. P. Bourgeois, A. Ray, K. Liosi, S. Oriana, W. Stark, H. Masai, R. Zhou and Y. Yamakoshi, Chem. Sci., 2020, 11, 11998–12008 RSC.
- S. Rakhshan, D. Alberti, R. Stefania, V. Bitonto and C. S. Geninatti, J. Nanobiotechnol., 2021, 19, 208 CrossRef CAS PubMed.
- Y. Ji, C. Jones, Y. Baek, G. K. Park, S. Kashiwagi and H. S. Choi, Adv. Drug Delivery Rev., 2020, 167, 121–134 CrossRef CAS PubMed.
- G. Zheng, H. Li, K. Yang, D. Blessington, K. Licha, S. Lund-Katz, B. Chance and J. D. Glickson, Bioorg. Med. Chem. Lett., 2002, 12, 1485–1488 CrossRef CAS PubMed.
- S. P. Wu, I. Lee, P. P. Ghoroghchian, P. R. Frail, G. Zheng, J. D. Glickson and M. J. Therien, Bioconjugate Chem., 2005, 16, 542–550 CrossRef CAS PubMed.
- E. R. Trivedi, A. S. Harney, M. B. Olive, I. Podgorski, K. Moin, B. F. Sloane, A. G. Barrett, T. J. Meade and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1284–1288 CrossRef CAS PubMed.
- H. Li, Z. Zhang, D. Blessington, D. S. Nelson, R. Zhou, S. Lund-Katz, B. Chance, J. D. Glickson and G. Zheng, Acad. Radiol., 2004, 11, 669–677 CrossRef PubMed.
- G. Kalot, A. Godard, B. Busser, M. Bendellaa, F. Dalonneau, C. Paul, X. Le Guevel, V. Josserand, J. L. Coll, F. Denat, E. Bodio, C. Goze, T. Gautier and L. Sancey, Biomater. Sci., 2022, 10, 6315–6325 RSC.
- L. Polo, G. Valduga, G. Jori and E. Reddi, Int. J. Biochem. Cell Biol., 2002, 34, 10–23 CrossRef CAS PubMed.
- A. C. Chu, S. Y. Tsang, E. H. Lo and K. P. Fung, Life Sci., 2001, 70, 591–601 CrossRef CAS PubMed.
- M. Pinzón-Daza, R. Garzón, P. Couraud, I. A. Romero, B. Weksler, D. Ghigo, A. Bosia and C. Riganti, Br. J. Pharmacol., 2012, 167, 1431–1447 CrossRef PubMed.
- J. Tian, S. Xu, H. Deng, X. Song, X. Li, J. Chen, F. Cao and B. Li, Int. J. Pharm., 2017, 517, 25–34 CrossRef CAS PubMed.
- W. Li, J. Fu, Y. Ding, D. Liu, N. Jia, D. Chen and H. Hu, Acta Biomater., 2019, 96, 456–467 CrossRef CAS PubMed.
- B. Zhang, X. Sun, H. Mei, Y. Wang, Z. Liao, J. Chen, Q. Zhang, Y. Hu, Z. Pang and X. Jiang, Biomaterials, 2013, 34, 9171–9182 CrossRef CAS PubMed.
- J. H. Kim, Y. Kim, K. H. Bae, T. G. Park, J. H. Lee and K. Park, Mol. Pharm., 2015, 12, 1230–1241 CrossRef CAS PubMed.
- J. Qian, N. Xu, X. Zhou, K. Shi, Q. Du, X. Yin and Z. Zhao, Int. J. Nanomed., 2019, 14, 7431–7446 CrossRef CAS PubMed.
- D. H. Kim and J. J. Rossi, Nat. Rev. Genet., 2007, 8, 173–184 CrossRef CAS PubMed.
- Q. L. Zhu, Y. Zhou, M. Guan, X. F. Zhou, S. D. Yang, Y. Liu, W. L. Chen, C. G. Zhang, Z. Q. Yuan, C. Liu, A. J. Zhu and X. N. Zhang, Biomaterials, 2014, 35, 5965–5976 CrossRef CAS PubMed.
- J. Jin, K. H. Bae, H. Yang, S. J. Le, H. Kim, Y. Kim, K. M. Joo, S. W. Seo, T. G. Park and D. H. Nam, Bioconjugate Chem., 2011, 22, 2568–2572 CrossRef CAS PubMed.
- S. D. Yang, W. J. Zhu, Q. L. Zhu, W. L. Chen, Z. X. Ren, F. Li, Z. Q. Yuan, J. Z. Li, Y. Liu, X. F. Zhou, C. Liu and X. N. Zhang, J. Biomed. Mater. Res., Part B, 2017, 105, 1114–1125 CrossRef CAS PubMed.
- W. H. Kong, K. Park, M. Y. Lee, H. Lee, D. K. Sung and S. K. Hahn, Biomaterials, 2013, 34, 542–551 CrossRef CAS PubMed.
- M. Javadian, N. Shekari, M. S. Soltani-Zangbar, A. Mohammadi, B. Mansoori, S. Maralbashi, D. Shanehbandi, B. Baradaran, M. Darabi and T. Kazemi, J. Cell. Biochem., 2020, 121, 2416–2427 CrossRef CAS PubMed.
- D. D’Eliseo and F. Velotti, J. Clin. Med., 2016, 5, 15 CrossRef PubMed.
- L. Reynolds, R. S. Mulik, X. Wen, A. Dilip and I. R. Corbin, Nanomedicine, 2014, 9, 2123–2141 CrossRef CAS PubMed.
- X. Wen, L. Reynolds, R. S. Mulik, S. Y. Kim, T. Van Treuren, L. H. Nguyen, H. Zhu and I. R. Corbin, Gastroenterology, 2016, 150, 488–498 CrossRef CAS PubMed.
- J. Yang, Y. Gong, D. P. Sontag, I. Corbin and G. Y. Minuk, Mol. Biol. Rep., 2018, 45, 1023–1036 CrossRef CAS PubMed.
- W. Ou, R. S. Mulik, A. Anwar, J. G. McDonald, X. He and I. R. Corbin, Free Radical Biol. Med., 2017, 112, 597–607 CrossRef CAS PubMed.
- R. S. Mulik, C. Bing, M. Ladouceur-Wodzak, I. Munaweera, R. Chopra and I. R. Corbin, Biomaterials, 2016, 83, 257–268 CrossRef CAS PubMed.
- A. A. Radwan and F. K. Alanazi, Molecules, 2014, 19, 13177–13187 CrossRef PubMed.
- P. Zhou, S. Hatziieremia, M. A. Elliott, L. Scobie, C. Crossan, A. M. Michie, T. L. Holyoake, G. W. Halbert and H. G. Jørgensen, J. Controlled Release, 2010, 148, 380–387 CrossRef CAS PubMed.
- X. Jiang, W. Han, J. Liu, J. Mao, M. J. Lee, M. Rodriguez, Y. Li, T. Luo, Z. Xu, K. Yang, M. Bissonnette, R. R. Weichselbaum and W. Lin, Adv. Sci., 2022, 9, e2201614 CrossRef PubMed.
- L. Jaragh-Alhadad, M. Samir, T. J. Harford and S. Karnik, Drug Delivery, 2022, 29, 2206–2216 CrossRef CAS PubMed.
- M. Zhou, Q. Hu, T. Wang, J. Xue and Y. Luo, Carbohydr. Polym., 2016, 153, 336–344 CrossRef CAS PubMed.
- R. Ross, Nature, 1993, 362, 801–809 CrossRef CAS PubMed.
- X. Wang, J. Greilberger, M. Ratschek and G. Jürgens, J. Pathol., 2001, 195, 244–250 CrossRef CAS PubMed.
- F. Alanazi, H. Li, D. S. Halpern, S. Øie and D. R. Lu, Int. J. Pharm., 2003, 255, 189–197 CrossRef CAS PubMed.
- Y. Tauchi, M. Takase, I. Zushida, S. Chono, J. Sato, K. Ito and K. Morimoto, J. Pharm. Sci., 1999, 88, 709–714 CrossRef CAS PubMed.
- Y. Tauchi, L. Zushida, M. Yokota, S. Chono, J. Sato, K. Ito and K. Morimoto, Biol. Pharm. Bull., 2000, 23, 466–471 CrossRef CAS PubMed.
- Y. Tauchi, L. Zushida, S. Chono, J. Sato, K. Ito and K. Morimoto, Biol. Pharm. Bull., 2001, 24, 925–929 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel and M. Korbelik, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- W. M. Sharman, J. E. V. Lier and C. M. Allen, Adv. Drug Delivery Rev., 2004, 56, 53–76 CrossRef CAS PubMed.
- B. A. Allison, E. Waterfield, A. M. Richter and J. G. Levy, Photochem. Photobiol., 1991, 54, 709–715 CrossRef CAS PubMed.
- J. C. Mazière, P. Morlière and R. Santus, J. Photochem. Photobiol., B, 1991, 8, 351–360 CrossRef PubMed.
- M. R. Hamblin and E. L. Newman, J. Photochem. Photobiol., B, 1994, 26, 147–157 CrossRef CAS PubMed.
- P. C. D. Smidt, A. J. Versluis and T. J. V. Berkel, Biochemistry, 1993, 32, 2916–2922 CrossRef PubMed.
- G. Zheng, H. Li, M. Zhang, S. Lund-Katz, B. Chance and J. D. Glickson, Bioconjugate Chem., 2002, 13, 392–396 CrossRef CAS PubMed.
- H. Li, D. E. Marotta, S. Kim, T. M. Busch, E. P. Wileyto and G. Zheng, J. Biomed. Opt., 2005, 10, 41203 CrossRef PubMed.
- L. Song, H. Li, U. Sunar, J. Chen, I. Corbin, A. G. Yodh and G. Zheng, Int. J. Nanomed., 2007, 2, 767–774 CAS.
- D. E. Marotta, W. Cao, E. P. Wileyto, H. Li, I. Corbin, E. Rickter, J. D. Glickson, B. Chance, G. Zheng and T. M. Busch, Nanomedicine, 2011, 6, 475–487 CrossRef CAS PubMed.
- C. Wang, X. Zhao, H. Jiang, J. Wang, W. Zhong, K. Xue and C. Zhu, Nanoscale, 2021, 13, 1195–1205 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |