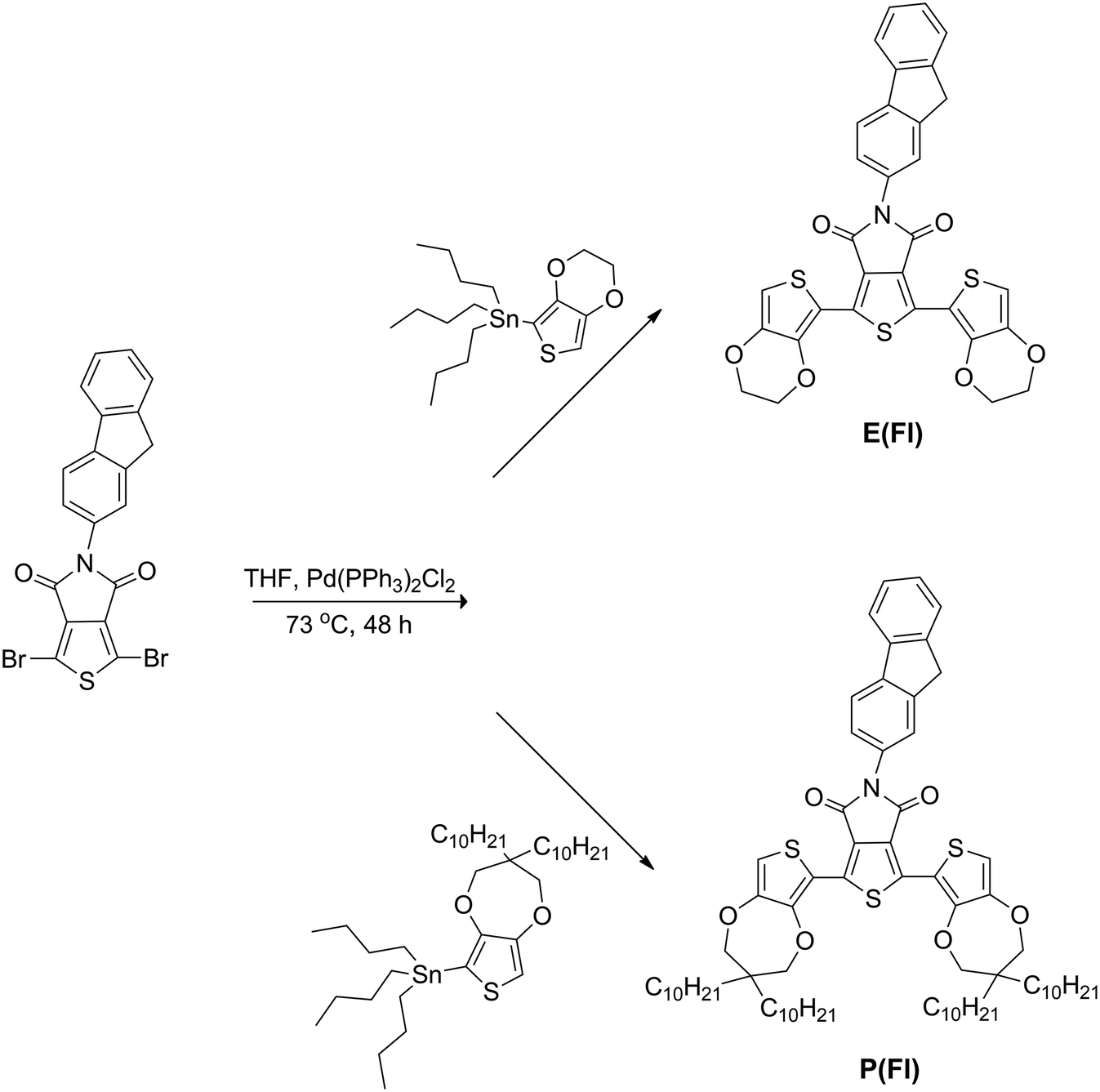
Side chain effect on the electrochemical and optical properties of thieno[3,4-c]pyrrole-4,6-dione based donor–acceptor donor type monomers and polymers†
Received
18th July 2022
, Accepted 30th September 2022
First published on 30th September 2022
Abstract
In organic π-conjugated materials, side chains play great roles that impact far beyond solubility. In this work, we mainly focused on the synthesis of new donor–acceptor–donor (D–A–D) type conjugated monomers and their corresponding polymers appending thieno[3,4-c]pyrrole-4,6-dione (TPD) acceptor with a new side chain, fluorene (Fl), to investigate the side chain effect. In this context, to reveal the precise effect of the side chains on the optical and electrochemical properties of the monomers and polymers synthesized in this work, four series of D–A–D monomers, each containing a TPD core unit with a different side chain, are compared and discussed in relation to each other. Notably, it was discovered that the TPD acceptor unit can be modified with any functional group other than common alkyl chains to impart new functionalities by maintaining their superior optoelectronic properties. New types of side chains can be used to tune the physical characteristics, such as solubility, absorption, emission, and molecular packing. In this work, Fl-appended monomers as a new class of D–A–D type π-conjugated molecules containing 3,4-ethylenedioxythiophene (EDOT (E)) and 3,4-propylenedioxythiophene (ProDOT (P)) donor units were studied and it was found that 1,3-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-5-(9H-fluoren-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (E(Fl)) and 1,3-bis(3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-5-(9H-fluoren-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (P(Fl)) exhibited reasonable quantum yields and their corresponding polymers revealed ambipolar character with slightly lower band gap as compared to the previous analogues containing other side chains. Observed experimental results were elucidated by first principle calculations. In this paper, we discussed that using side chain engineering is an effective strategy for improving next-generation organic π-conjugated materials with the desired properties.
Design, System, Application
By modifying the molecular structures, not only the band gap value can be altered but also solubility, stability and electrochromic properties can be enhanced. To have desired properties with low band gap values, different types of donor and acceptor groups have been designed and synthesized. Among the acceptor groups, thieno[3,4-c]pyrrole-4,6-dione (TPD) is a potential optoelectronic building block material since it can be easily functionalized via a bridging nitrogen center to be considered in bandgap engineering. To understand the effect of different side groups, TPD acceptor was modified with a new side chain, fluorene. The modified TPD was coupled with alkylenedioxythiophene based donor units for the synthesis of D–A–D type monomers and polymers. These fluorene appended TPD-based D–A–D architectures were compared with the previously synthesized analogs involving different side chains; ethylhexyl, POSS, azobenzene and fluoerene unveil the side chain effects on the optoelectronic properties. The investigation of these conjugated systems in donor–acceptor–donor configuration has provided a deep insight on evaluating the role of different types of functional side groups on the acceptor unit. Furthermore, optoelectronic features of these systems were investigated to elucidate the effect of aforementioned functional side groups as well as donor units.
|
1. Introduction
Organic π-conjugated materials, both polymers and small molecules, have been receiving a great deal of research interest due to their potential applications in any field of electronic applications as great alternatives to inorganic ones.1–6 Organic π-conjugated materials can provide numerous advantages over their inorganic counterparts due to their low cost, lightweight, processability, and easy modification.6–9 The great power of these materials comes from the modification of the molecular structure on the main backbone and/or side chain to endow new functionalities.9–13 With these modifications, the intrinsic properties of organic semiconducting materials can be improved by providing control over parameters, such as frontier molecular orbital energy levels, intramolecular and/or intermolecular interactions, planarity, and solubility.9–14 One of the most important modification strategies that can be applied to the main backbone is the donor–acceptor approach, which is based on an alternating arrangement of electron-rich units (donor) and electron-deficient units (acceptor).10,15–18 With this approach, it is possible to control the frontier molecular orbital energy levels to obtain low band gap π-conjugated polymer by providing intramolecular charge transfer between the donor and acceptor units.14–17 On one hand, the primary purpose of modifications on the side chains is to impart solubility to the polymer chain.11,19 As well as enabling solution processability, the intrinsic properties of π-conjugated materials can also be modulated owing to side chains as the side chains form densely packed molecular ordering, and the high molecular weight in π-conjugated polymers consequently lead to improved intrachain or interchain charge transport characteristics in the π-conjugated materials.19–21 Owing to the remarkable developments in side-chain engineering, a wide variety of side-chains having different functions can be appended to the polymer backbone and the desired property can be inserted into the molecular structure for the synthesis of novel π-conjugated materials.11,13,22–24
To date, alkyl side chains have been the most common type of side chains used for the functionalization of both donor and acceptor units.11,25–27 Although many studies have been reported for the investigation of chain length and branching effect within this class of side chains, there have been a limited number of systematic studies on how the variation of the side chain type affects the intrinsic properties of π-conjugated polymers.26–33 In this context, we have synthesized novel donor–acceptor–donor type monomer systems involving a modified acceptor with a new kind of functional side chain for the first time to investigate the intrinsic properties of both π-conjugated monomers and polymers. For this purpose, an aromatic, rigid, and planar fluorene functional group was appended to the TPD acceptor unit as a side chain to endow its highly planar and fluorescent characteristics to the π-conjugated systems.34,35 Then, we investigated the influences of this new type of side chain on the optical and electrochemical properties of π-conjugated monomers and polymers. After that, we carried out a systematic study on how the side chain variation from alkyl to the aromatic or hybrid group (ethyl hexyl (EH), POSS propyl (POSS), azobenzene (AB), and fluorene (Fl)) affect the intrinsic properties of π-conjugated monomers and polymers. In order to comprehend the side chain effect on the optical and electrochemical properties of π-conjugated systems, the previously synthesized monomers and polymers with different functional side chains EH, POSS, and AB were involved in this study for comparison sake.36–38
2. Experimental
2.1. Materials and instrumentation
The chemicals and reagents used for the synthesis of monomers and polymers were acquired from commercial sources (Merck, Sigma-Aldrich, Avocado Research Chemicals, Henan Tianfu Chemical Co.) and used as received unless noted. The solvents, hexane, ethyl acetate (EtOAc), tetrahydrofuran (THF), dichloromethane (DCM), and acetonitrile (MeCN) were freshly distilled under nitrogen purge prior to their use. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Spectrospin Avance DPX-400 Spectrometer in deuterated chloroform operating at 400 MHz and 100 MHz, respectively. The chemical shifts were recorded in ppm units by using tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) measurements were recorded by utilizing a Waters Synapt G1 mass spectrometer with the electron impact mode. Photophysical characterization was carried out by utilizing Cary 60 model UV-vis and Varian Cary Eclipse fluorescence spectrometers. Electrochemical measurements were recorded on a Gamry PCI4/300 potentiostat–galvanostat system, with a conventional three-electrode system with a Pt disk (0.02 cm2) as a working electrode, Ag/AgCl as a reference electrode, and a Pt wire as a counter electrode in an anhydrous solution of 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) dissolved in a proper solvent system. Electrochemical polymerization was carried out on both, the Pt disk electrode and indium-tin-oxide (ITO) coated glass slide electrode (Delta Tech. 8–12 Ω, standard size: 7 mm × 50 mm) in a three-electrode cell via potentiodynamic methods in a controlled way by adjusting the current passed for the Pt disc electrode and the absorbance for the ITO glass slide electrode (not to exceed 1). Electropolymerization on an ITO glass slide was carried out against an Ag wire pseudo-reference electrode and a Pt wire counter electrode. Spectroelectrochemical and kinetic measurements for the polymer films on ITO glass slides were recorded on a Specord S600 spectrometer equipped with a Gamry Reference 600 potentiostat–galvanostat system with an Ag wire pseudo-reference electrode and a Pt wire counter electrode in a UV quartz cell containing 0.1 M n-Bu4NPF6 electrolyte solution dissolved in MeCN. A ferrocene/ferrocenium (Fc/Fc+) couple with an oxidation peak potential of 0.61 V was used as an external standard for calibration of the electrochemical measurements. Cathodic measurements were performed in a deoxygenated system. The color coordinates of the polymer films were expressed as L*, a*, b* according to the International Commission of Illumination, referenced to platinum cobalt DIN ISO 621, iodine DIN EN 1557, and Gardner DIN ISO 6430 (standard illuminator D65, field of width 10° observer).
2.2. Material synthesis
Synthesis of 4,6-dibromothieno[3,4-c]furan-1,3-dione.
The synthesis of 4,6-dibromothieno[3,4-c]furan-1,3-dione was carried out in acetic anhydride according to the literature procedure in an 80% yield.39,40
Synthesis of 1,3-dibromo-5-(2-fluoerene)-thieno[3,4-c]pyrrole-4,6-dione (TPD-Fl).
358 mg of 4,6-dibromothieno[3,4-c]furan-1,3-dione (1.15 mmol) and 228 mg of 2-aminofluorene (1.26 mmol) were added to 5 mL of freshly distilled THF and then stirred at 50 °C for 4 h under an inert atmosphere. After cooling to room temperature, THF was evaporated at reduced pressure. The resulting mixture was stirred at 55 °C for 3 h after the addition of 3 mL of SOCl2 and then allowed to reach room temperature. The final mixture was poured drop by drop into a cold mixture of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (25 mL:50 mL). The resulting precipitate was recovered by filtration, washed excessively with hot ethanol, and dried under a vacuum. The beige precipitate was isolated as a final product in 54% yield. 1H NMR (400 MHz, CDCl3, δ/ppm): 7.88 (d, J = 8.1 Hz, 1H), 7.81 (d, J = 7.4 Hz, 1H), 7.56 (d, J = 7.4 Hz, 1H), 7.53 (s, 1H), 7.42–7.32 (m, 3H), 3.96 (s, 2H); 13C NMR (100 MHz, CDCl3, δ/ppm): 159.56, 144.00, 143.60, 142.17, 140.73, 134.34, 129.80, 127.28, 126.95, 125.23, 125.11, 123.26, 120.26, 120.22, 114.11, 36.99. HRMS calcd for C19H9Br2NO2S, [M + H]+: 475.8779, found: 475.8803.
:water (25 mL:50 mL). The resulting precipitate was recovered by filtration, washed excessively with hot ethanol, and dried under a vacuum. The beige precipitate was isolated as a final product in 54% yield. 1H NMR (400 MHz, CDCl3, δ/ppm): 7.88 (d, J = 8.1 Hz, 1H), 7.81 (d, J = 7.4 Hz, 1H), 7.56 (d, J = 7.4 Hz, 1H), 7.53 (s, 1H), 7.42–7.32 (m, 3H), 3.96 (s, 2H); 13C NMR (100 MHz, CDCl3, δ/ppm): 159.56, 144.00, 143.60, 142.17, 140.73, 134.34, 129.80, 127.28, 126.95, 125.23, 125.11, 123.26, 120.26, 120.22, 114.11, 36.99. HRMS calcd for C19H9Br2NO2S, [M + H]+: 475.8779, found: 475.8803.
Synthesis of 2-(tributylstannyl)-3,4-ethylenedioxythiophene (E-Sn) and tributyl(3,3-didecyl-3,4-dihydro-2H-thieno [3,4-b] [1,4]dioxepin-6-yl)stannane (P-Sn).
The donor (EDOT and ProDOT refers to the didecyl substituted form of ProDOT unless noted) (7 mmol) was dissolved in 15 mL of dry THF under an argon atmosphere and cooled to −78 °C. To this solution, n-butyllithium (2.9 mL of 2.5 M in hexane) was added dropwise and the mixture was stirred for 1.5 h while maintaining the temperature at −78 °C. Then, tributyltin chloride (1.90 mL, 7 mmol) dissolved in 3 mL of dry THF was added to this solution dropwise. The mixture was allowed to reach room temperature slowly and stirred at this temperature overnight. The solvent was removed under reduced pressure and the crude mixture was poured into DCM and washed with brine. The combined organic phases were separated and dried over MgSO4 and filtered. After filtration, the solvent was evaporated to afford a yellow oil product and used as obtained.
E-Sn (97% yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 6.61 (s, 1H), 4.22–4.16 (m, 4H), 1.67–1.50 (m, 6H), 1.41–1.30 (m, 6H), 1.18–1.08 (m, 6H), 0.99–0.85 (m, 9H).
P-Sn (93% yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 6.60 (s, 1H), 3.78–3.64 (m, 4H), 1.61–1.35 (m, 12H), 1.33–0.94 (m, 42H), 0.88–0.73 (m, 15H).
Synthesis of D–A–D monomers.
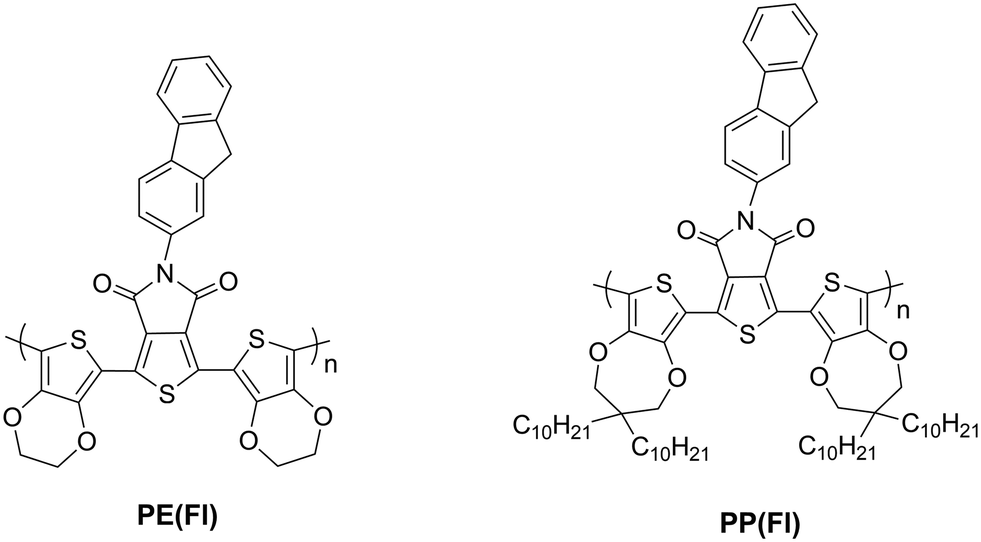
Stille cross-coupling reaction was carried out with slight modifications of the well established literature procedures for the synthesis of D–A–D monomers.36 Two novel D–A–D monomers, which are named as 1,3-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-5-(9H-fluoren-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (E(Fl)) and 1,3-bis(3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-5-(9H-fluoren-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (P(Fl)), having a new kind of modified TPD acceptor, were synthesized by using EDOT and ProDOT donor units (Scheme 1).
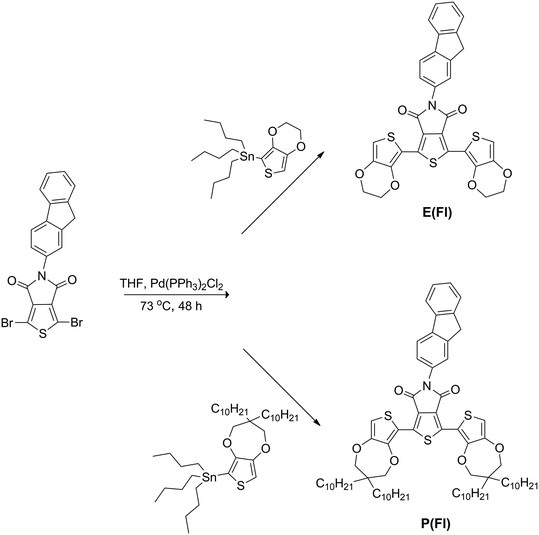 |
| | Scheme 1 Synthesis route for the monomers. | |
E(Fl)
.
The pure yellow compound was isolated by using successive silica gel columns with hexane:DCM (1:4) and hexane:EtOAc (1:4) and finally an alumina column with DCM (71% yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 7.88 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 7.3 Hz, 1H), 7.63 (s, 1H), 7.56 (d, J = 7.5 Hz, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 7.2 Hz, 1H), 7.35–7.31 (m, 1H), 6.58 (s, 2H), 4.46 (s, 4H), 4.30 (s, 4H), 3.96 (s, 2H). HRMS calcd for C31H19NO6S3, [M + H]+: 598.0453, found: 598.0477.
P(Fl)
.
The compound was isolated as a greenish-yellow solid using two successive silica gel columns with hexane:DCM (50:1) and hexane:EtOAc (25:1) and finally an alumina column with hexane:DCM (4:1) (59% yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 7.86 (d, J = 8.1 Hz, 1H), 7.80 (d, J = 7.4 Hz, 1H), 7.61 (s, 1H), 7.54 (d, J = 7.4 Hz, 1H), 7.45 (dd, J = 8.1, 1.8 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.31 (td, J = 7.4, 1.1 Hz, 1H), 6.64 (s, 2H), 4.05 (s, 4H), 3.94 (s, 2H), 3.92 (s, 4H), 1.43 (s, 8H), 1.36–1.21 (m, 64H), 0.88 (t, J = 6.8 Hz, 12H); 13C NMR (100 MHz, CDCl3, δ/ppm): 162.10, 149.02, 148.95, 143.75, 143.64, 141.39, 141.09, 136.14, 130.81, 126.93, 126.82, 126.06, 125.58, 125.05, 123.57, 120.14, 119.97, 113.78, 107.98, 77.71, 43.88, 36.98, 32.01, 31.94, 30.55, 29.74, 29.68, 29.66, 29.39, 22.95, 22.70, 14.11. HRMS calcd for C73H103NO6S3, [M + H]+: 1186.7026, found: 1186.7230.
2.3. Computational methods
Computational studies were performed for repeat units in the form of D–A–D where EDOT and ProDOT were used as a donor, and methyl, EH, POSS, AB, and Fl were used as acceptor units. ProDOT with the didecyl substitution was also constructed to determine the effect by using B3LYP hybrid functional and 6-311G(d) basis set with tight SCF convergence criteria in the Gaussian09 (Revision A.02) software package, which provided successful results in the previous studies.41–48 Geometry optimizations were started from different initial conformations by controlling the torsional angle between connected donor, bridge and acceptor units to determine the lowest energy geometry. The optimized geometries were utilized for the calculation of electrostatic potential surface (ESP), the highest occupied molecular orbitals (HOMO), and the lowest unoccupied molecular orbitals (LUMO). The band gap was calculated by using two different methods that show the direct difference between the HOMO energy and LUMO energy for the optimized ground state and the calculation of vertical excitation energy of the lowest singlet excited state. The singlet excited states of the oligomers were calculated by using time-dependent density-functional theory (TDDFT). Vertical ionization potential (VIP) and adiabatic ionization potential (AIP) were calculated by the energy difference between the neutral and cation states of the optimized ground state geometry and optimized cation geometry, respectively. Electron affinities (EA) were calculated in the same way. The effect of substitution on the density of states was studied by using Multiwfn software.49 Hole reorganization energies (λreorg) were determined depending on the method suggested by Bredas et al.50 Dipole moment (μ), isotropic polarizability (α), and static hyperpolarizability (β) were calculated for the BDBAB type optimized units. Total atomic charges (δ) on donor and acceptor groups were calculated by using the Merz-Kollman scheme fitting to elucidate charge transfer.51
3. Results and discussion
3.1. Optical and electrochemical properties of the TPD-based monomers with Fl side chains
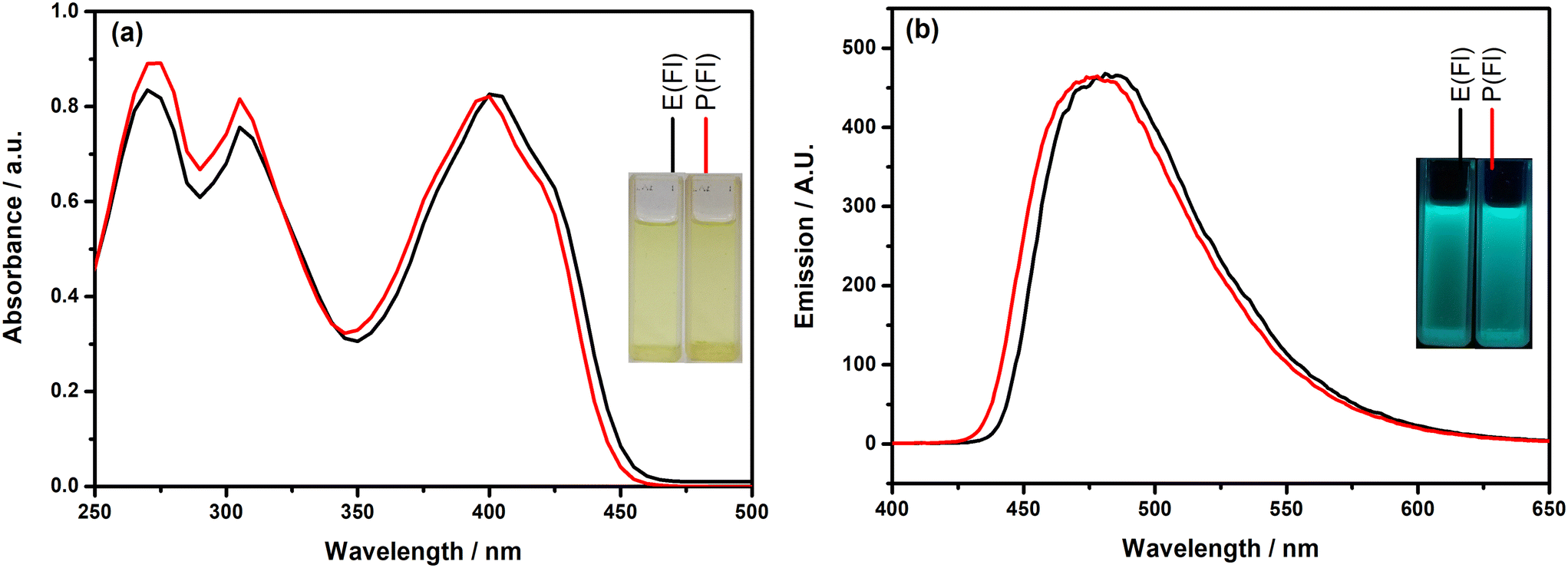
First of all, the photophysical properties of the TPD-based monomers with the Fl side chain were investigated by recording their electronic absorption and fluorescence emission spectra in DCM and the obtained spectra are given in Fig. 1. Inspection of Fig. 1(a) indicates that the monomers demonstrate similar absorption characteristics with three absorption bands. The highest energy band appearing almost at the same wavelength (271 nm for E(Fl) and 272 nm for P(Fl)) originated from the existence of the Fl side chain on the TPD core, which exhibits a characteristic absorption peak at roughly 270 nm.52 The middle band that appears at 305 nm for both monomers is most probably due to π–π* transition. The absorption bands located on the lowest energy region of spectra results from the charge transfer between donor and acceptor as a characteristic signature of push–pull type systems and the band maxima are detected at 402 nm and 398 nm for E(Fl) and P(Fl), respectively. The origin of band transitions was also supported by theoretical studies (see Table S1†). The slight bathochromic shift observed in this band when moving from ProDOT to EDOT proves the stronger effective conjugation between the acceptor and EDOT as compared to that of ProDOT. The fluorescence emission spectra of the monomers revealed strong luminescence maxima centered at 482 nm for E(Fl) and 477 nm for P(Fl) when they are excited at 340 nm and the spectra are illustrated in Fig. 1(b). Owing to the stronger donor character of EDOT, a slight red shift is observed in the emission spectrum of E(Fl) as compared to that of P(Fl). The results are tabulated in Table 1.
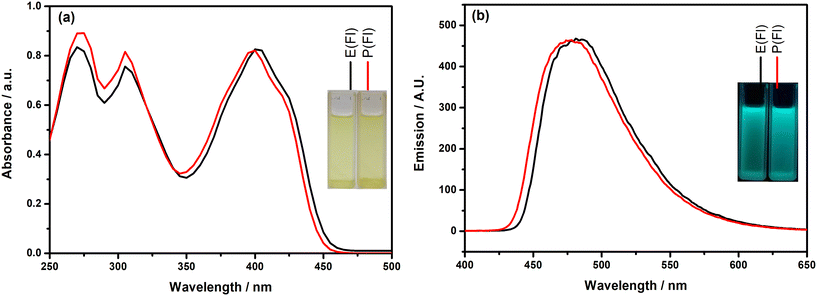 |
| | Fig. 1 (a) Absorption and (b) emission spectra of the monomers in DCM. Inset: the colors of the monomers (a) in daylight and (b) under a UV lamp at 365 nm. | |
Table 1 Optical and electrochemical properties of the monomers E(Fl) and P(Fl)
| Monomers |
E(Fl)
|
P(Fl)
|
|
HOMO and LUMO energy levels were calculated from Eox,onset and Ered,onset values of CV diagrams using eqn (1) and (2), respectively.53λmax,abs: absorption maximum wavelength, λmax,em: emission maximum wavelength, Eox,m: oxidation potential of monomer, Eox,onset,m: oxidation onset potential of monomer, Ered,m: reduction potential of monomer, Ered,onset,m: reduction onset potential of monomer, Eg,electro: electrochemical band gap, Eg,opt: optical band gap.
|
|
λ
max,abs (nm) |
270, 305, 402 |
272, 305, 398 |
|
λ
max,em (nm) |
482 |
477 |
|
E
ox,m (V) DCM (CV) |
1.10 |
1.19 |
|
E
red,m (V) DCM (CV) |
−1.70 |
−1.64 |
|
E
ox,onset,m/Ered,onset,m (V) |
0.96/−1.56 |
1.07/−1.55 |
|
E
electrog (V)/Eoptg (eV) |
2.52/2.75 |
2.62/2.78 |
| HOMOa (V) |
−5.67 |
−5.78 |
| LUMOa (V) |
−3.15 |
−3.16 |
| Quantum yield |
0.0574 |
0.0719 |
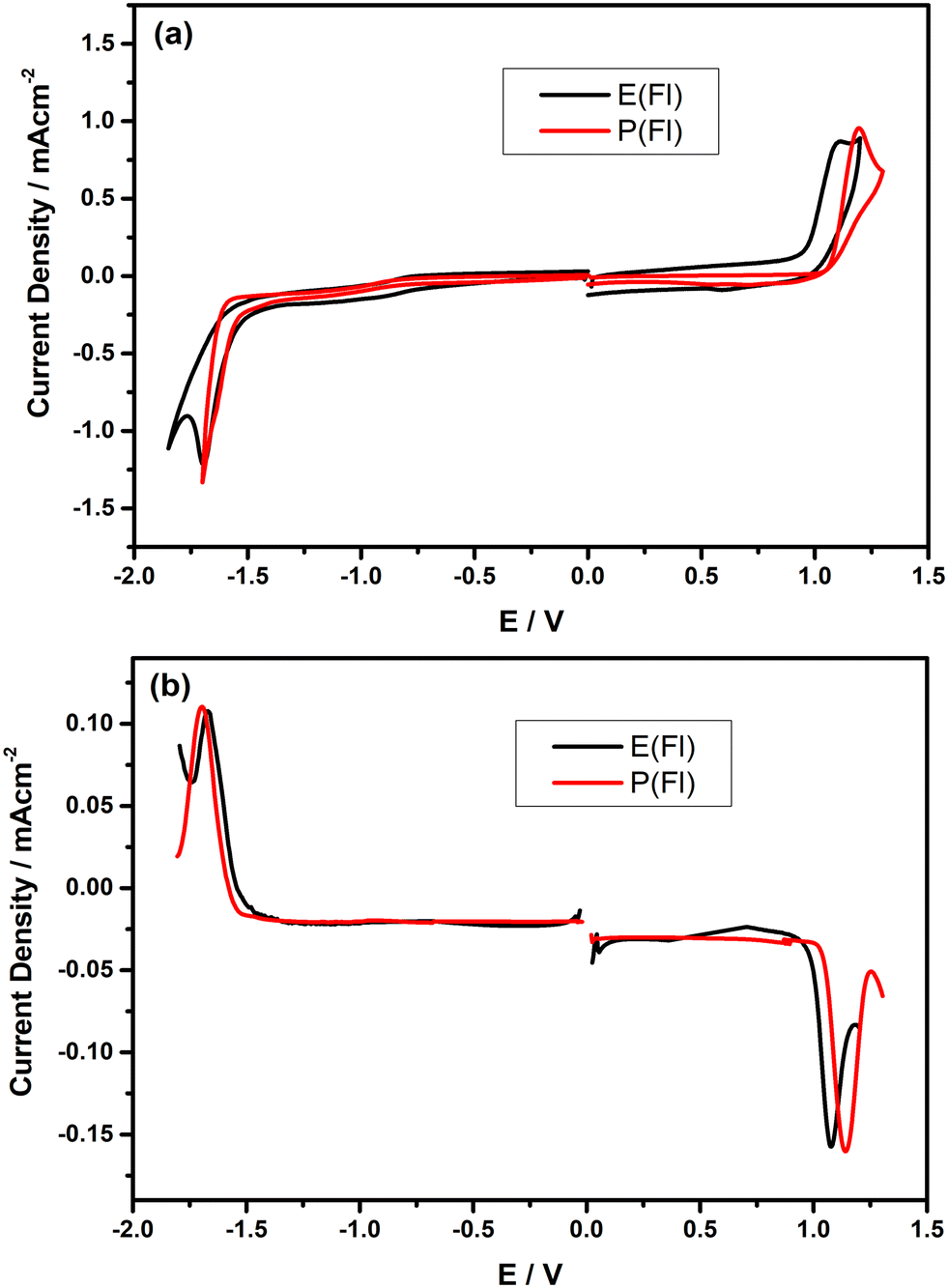
In order to comprehend the redox properties of Fl-appended TPD-based monomers, both cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements were recorded in dry DCM consisting of 0.1 M n-Bu4NPF6 as the supporting electrolyte and the resulting CVs measured on Pt disc electrode are given in Fig. 2(a). As seen from the CVs, both monomers reveal only one irreversible oxidation peak, which is responsible for electropolymerization. The oxidation peaks were observed at about 1.10 V and 1.19 V vs. Ag/AgCl for E(Fl) and P(Fl), respectively. The relatively lower oxidation potential of the E(Fl) monomer, as compared to P(Fl) demonstrates that EDOT is a stronger electron-donating unit than ProDOT, which is consistent with earlier findings.36–38E(Fl) and P(Fl) monomers exhibit pseudo-reversible reduction peaks vs. Ag/AgCl, which are centered at −1.70 V and −1.64 V, respectively. The reduction peaks are more explicitly observed in their differential pulse voltammograms as shown in Fig. 2(b). A lower shift is expected in the reduction potential when moving from EDOT to ProDOT in comparison to the change in the oxidation potential since the reduction potentials are directly associated with LUMO energy levels of the monomers that do not alter too much for the same acceptor unit as observed in this case. The results for the electrochemical measurements are tabulated in Table 1 together with HOMO/LUMO energy levels calculated by using the following equations:53
| | | EHOMO = −e(Eox,onset (vs. Ag/AgCl + 4.71) eV | (1) |
| | | ELUMO = −e(Ered,onset (vs. Ag/AgCl + 4.71) eV. | (2) |
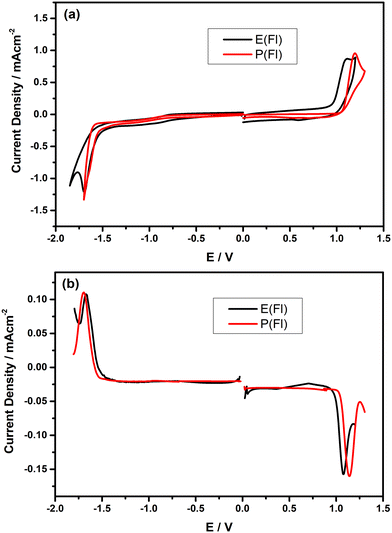 |
| | Fig. 2 (a) Full scan cyclic voltammograms of the monomers on the Pt electrode vs. Ag/AgCl in 0.1 M Bu4NPF6 dissolved in DCM at a scan rate of 100 mV s−1 and (b) differential pulse voltammograms of the monomers on the Pt electrode vs. Ag/AgCl in 0.1 M n-Bu4NPF6 dissolved in DCM. | |
3.2. Comparison of the optical and electrochemical properties of TPD-based D–A–D monomers
In order to shed light on how the side chain variation affects the photophysical characteristics of TPD-based D–A–D monomers, the electronic absorption and emission spectra of Fl-appended TPD-based monomers were compared with the previously synthesized analogues containing different side chains such as EH, POSS and AB and the related data are tabulated in Table 2 for comparison sake (see Scheme S1† for the structure of TPD based monomers with different substituents). When the absorption profile of the monomers was taken into account, all series reveal the dual-band absorption nature of conjugated donor–acceptor systems owing to a strong intramolecular charge transfer between donor and acceptor units (see Tables 1 and 2). Furthermore, it was found that alkyl-type side chains like EH or POSS, even if they have a hybrid composition, have no appreciable effect on the photophysical properties of the monomers, whereas aryl side chains like Fl and AB result in a variation in the electronic absorption characteristics of the monomers owing to the absorption of aryl side chains in the related range. In addition to this, a slight red shift was observed for aryl, Fl- and AB-appended series as compared to alkyl-appended analogues. The red shift effect of aryl functional side chains can be attributed to the different electronic nature of the Fl and AB as compared to alky-type side chains. Furthermore, the changing morphology as a consequence of side chain change may result in this red shift. When the emission characteristics of the monomers were investigated, it was observed that the emission profiles of all monomer series were almost the same except for the AB-appended series. An extremely low emission intensity that is almost invisible to the naked eye was distinctly observed in the AB-appended series. This demonstrates that the AB side chain has a quenching effect on the conjugated monomer system, unlike the other side chains. Therefore, the fluorescence quantum yields were calculated only for the other monomer systems to detect the side chain effect for comparison sake and it was found that POSS and Fl side chains have a tendency to increase quantum yields of monomers, as compared to EH chains, especially in the case of ProDOT bearing monomers. Consequently, the replacement of alkyl side chains with the aryl ones resulted in some pronounced changes in the photophysical properties of the monomers depending on the characteristics of the side chain.
Table 2 Optical and electrochemical properties of the previously synthesized monomers with different side chains36–38
| Monomers |
E(EH)
|
P(EH)
|
|
λ
max,abs (nm) |
277, 398 |
277, 393 |
|
λ
max,em (nm) |
480 |
475 |
|
E
ox,m (V) (CV) |
1.11 |
1.17 |
|
E
red,m (V) (CV) |
−1.80 |
−1.79 |
|
E
ox,onset,m/Ered,onset,m (V) |
1.02/−1.63 |
1.08/−1.64 |
|
E
electrog (V)/Eoptg (eV) |
2.65/2.79 |
2.72/2.82 |
| HOMOa (V) |
−5.73 |
−5.79 |
| LUMOa (V) |
−3.08 |
−3.07 |
| Quantum yield |
0.0650 |
0.045 |
| Monomers |
E(POSS)
|
P(POSS)
|
|
λ
max,abs (nm) |
275, 398 |
276, 393 |
|
λ
max,em (nm) |
480 |
477 |
|
E
ox,m (V) DCM (CV) |
1.15 |
1.37 |
|
E
red,m (V) DCM (CV) |
−1.89 |
−1.85 |
|
E
ox,onset,m/Ered,onset,m (V) |
0.98/−1.63 |
1.06/−1.65 |
|
E
electrog (V)/Eoptg (eV) |
2.61/2.79 |
2.71/2.81 |
| HOMOa (V) |
−5.69 |
−5.77 |
| LUMOa (V) |
−3.08 |
−3.06 |
| Quantum yield |
0.074 |
0.075 |
| Monomers |
E(AB)
|
P(AB)
|
|
HOMO and LUMO energy levels were calculated from Eox,onset and Ered,onset values of CV diagrams, respectively, using eqn (1) and (2).53
|
|
λ
max,abs (nm) |
265, 335, 405 |
265, 333, 400 |
|
λ
max,em (nm) |
475 |
479 |
|
E
ox,m (V) DCM (CV) |
1.11 |
1.20 |
|
E
red,m (V) DCM (CV) |
−1.36 |
−1.38 |
|
E
ox,onset,m/Ered,onset,m (V) |
0.99/−1.24 |
1.09/−1.25 |
|
E
electrog (V)/Eoptg (eV) |
2.23/2.72 |
2.34/2.75 |
| HOMOa (V) |
−5.70 |
−5.80 |
| LUMOa (V) |
−3.47 |
−3.46 |
The CVs of Fl-appended monomers were compared with those of the other analogues in order to acquire a deeper understanding of how side chains affect the redox behaviors of TPD-based monomers. When the effect of both alky and aryl side chains on the electrochemical behavior of the monomers was investigated, it was observed that all monomer series follow a similar pattern, with one oxidation peak and one reduction peak (see Tables 1 and 2 for the related electrochemical data). While all the oxidation peaks of the monomers show irreversible characteristics, the reduction peaks were found to exhibit irreversible, reversible, or quasi-reversible characteristics. When the peak potentials from all series of TPD-based monomers were carefully inspected, it was observed that the side chain on the acceptor unit has no appreciable effect on the oxidation potential, excluding the POSS side chain. A modest anodic shift was noted in the oxidation potentials of all POSS-appended monomers. Furthermore, when the effect of donor units on the oxidation peaks and onset potentials in all TPD-based monomers were investigated, it was observed that the oxidation peaks and onset potentials exhibit more notable variations with a decline when moving from ProDOT to EDOT donor units in each monomer series depending on the strength of the donor group. EDOT-based monomers exhibit lower oxidation and onset potentials owing to the more electron-donating property of the ethylenedioxy bridge, as compared to the propylenedioxy bridge. The stronger electron-donating nature increases the HOMO energy level and makes the removal of electrons easier. Therefore, the oxidation peaks and onset potential decreases for monomers when moving from ProDOT to EDOT. Taking all of these findings into account, it can be inferred that the oxidation potential trend is mainly determined by the strength of the donor group. A systematic examination and comparison of the reduction characteristics of these monomers revealed that the reduction potentials of TPD-based monomers differ, depending on whether the acceptor unit has aryl or alky side chains. In the alkyl side chain comparison, the reduction potentials of POSS-appended TPD-based monomers were found to be lower than those of their EH analogues since the reduction potentials are directly associated with LUMO energy levels of the donor–acceptor system that can be adjusted by structural alterations in the acceptor unit. When the effect of the aryl side chain on the reduction behavior of TPD-based monomer was inspected, the reduction potentials of all AB-appended monomers were found to be significantly less negative than those of all other alky and aryl side chain analogues. The side chain on the acceptor unit is responsible for this dramatic change in the reduction potentials. The AB side chain raises the electron deficiency of the acceptor unit owing to its electron-withdrawing nature, causing reduction potentials to shift to less negative values and providing low-lying LUMO energy levels. This was also supported by theoretical studies, as explained in the following section. Lastly, when the effect of donor units on the reduction characteristics of all TPD-based monomers was considered, due to the higher electron density of TPD-based monomers with the EDOT donor unit, as compared to the ProDOT analogues, the reduction potentials of EDOT-based monomers were found to be more negative.
3.3. Computational results
Theoretical calculations presented that both band gap calculations based on HOMO–LUMO energy differences and band gaps by time-dependent DFT calculations have slightly higher values for all ProDOT-based monomers compared with the EDOT-based monomers. This result is in agreement with the experimental results. Calculations also indicated that this difference mainly originated from deeper HOMO levels with more negative values, while LUMOs have closer values for both EDOT and ProDOT-based monomers.
The band gap decreased for both AB and Fl substitutions, supporting the experimental results. According to the theoretical calculations, the difference between the ionization potentials of EDOT and ProDOT-based monomers was not significant. Although the ProDOT-based monomer has slightly more negative electron affinities with the exception of E(Fl) and P(FI), the difference is also not significant.
While E(Fl) and P(FI) have the least positive IPs, E(AB) and P(AB) present the most negative EAs. The most negative EAs for AB-appended monomers are consistent with the experimental measurements, i.e., having less negative reduction potentials (see Table 2). Moreover, because of the least positive IPs Fl-appended monomers exhibit better hole formation, on the other hand, having the most negative EAs enhance electron accepting properties with AB substitution. These results reflect the reorganization energies, where E(Fl) and P(FI) have the lowest hole reorganization energies and E(AB) and P(AB) have the lowest electron reorganisation energies. It should be noted that Fl has close values for electron and hole reorganisation energies that indicate possible enhanced charge carrier mobilities for the doped systems.
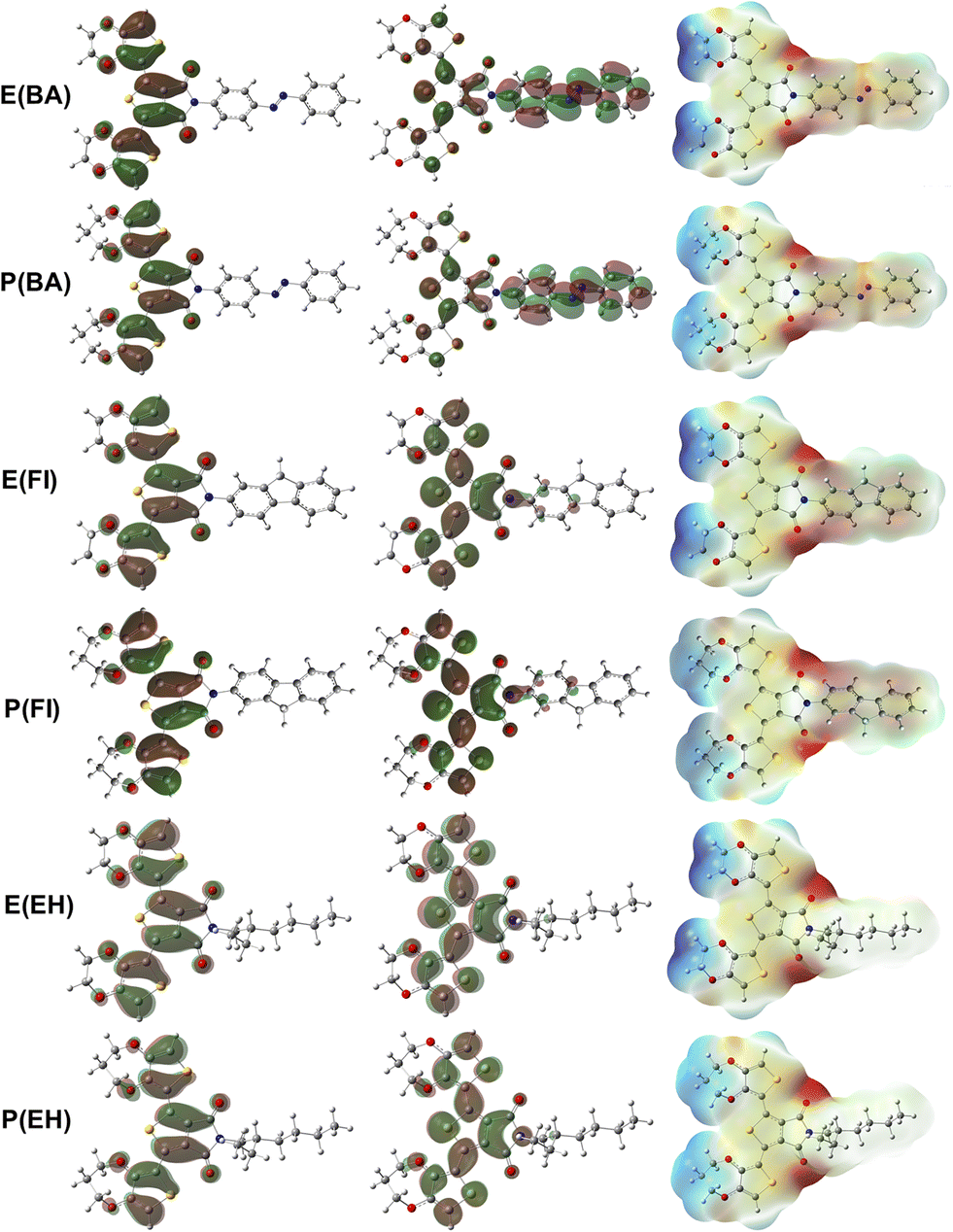
Higher polarizability values indicated the possibility of stronger dispersion and dipole–dipole intermolecular interactions with other species such as PC71BM. We determined higher polarizability values for ProDOT-based monomers compared to the EDOT-based ones. All side chains improved polarizability compared to the pristine EDOT and ProDOT with only methyl substitutions. This result is also valid for dipole moment with the exception of EH substitution that did not change dipole moment compared to the pristine monomers. Didecyl addition enhanced the polarizability values even more for both AB and FI substitutions. E(POSS) and P(POSS), which did not improve band gap or reorganization energies, provide the highest polarizability values as the POSS cages were ready to be polarized, which is an interesting result that can be exploited in optoelectronic devices. A relation between molecular hyperpolarizability, β, and the bond-length alternation in the donor–acceptor type of chain was proposed, where β increases with increasing electron delocalization and decreasing bond length alternation.54 The highest β values were calculated for E(AB) and P(AB) with a ten-fold increase, where EDOT-based monomers generally have higher hyperpolarizability values compared to the ProDOT-based ones with the P(FI) and E(FI) exceptions. There is a slight difference between the charge transfer (δ) of the TPD unit in the center with the EDOT and ProDOT groups given in Table 3 that provide higher charge transfer for EDOT-based monomers. We determined that there is a more significant charge transfer difference between the side groups and the TPD unit that can be depicted in the frontier orbitals in Fig. 3.
Table 3 Electronic and optoelectronic properties of EDOT and ProDOT-based monomers
|
|
HOMO (eV) |
LUMO (eV) |
E
directg (eV) |
E
TDg (eV) |
VIP (eV) |
AIP (eV) |
VEA (eV) |
AEA (eV) |
λ
reorg hole (eV) |
λ
reorg elec (eV) |
β (a.u) |
α (a.u) |
μ (Debye) |
δ
donor
|
|
E (methyl)
|
−5.32 |
−1.94 |
3.38 |
3.09 |
6.62 |
6.43 |
−0.66 |
−0.81 |
0.383 |
0.309 |
773.5 |
325.7 |
2.99 |
−0.14 |
|
P (methyl)
|
−5.35 |
−1.94 |
3.41 |
3.11 |
6.63 |
6.42 |
−0.67 |
−0.83 |
0.428 |
0.324 |
595.8 |
348.8 |
2.91 |
−0.10 |
|
E(AB)
|
−5.44 |
−2.33 |
3.10 |
2.53 |
6.63 |
6.46 |
−1.39 |
−1.39 |
0.385 |
0.205 |
5226.8 |
536.9 |
3.58 |
−0.11 |
|
P(AB)
|
−5.46 |
−2.33 |
3.13 |
2.54 |
6.64 |
6.45 |
−1.39 |
−1.39 |
0.425 |
0.214 |
5115.5 |
561.2 |
3.51 |
−0.08 |
|
P(AB)-Didecyl
|
−5.40 |
−2.30 |
3.10 |
2.53 |
6.54 |
6.32 |
−1.39 |
−1.39 |
0.448 |
0.215 |
6846.1 |
1050.2 |
3.91 |
−0.05 |
|
E(FI)
|
−5.37 |
−2.03 |
3.33 |
3.04 |
6.53 |
6.40 |
−1.01 |
−1.01 |
0.311 |
0.308 |
358.2 |
491.2 |
3.25 |
−0.11 |
|
P(FI)
|
−5.39 |
−2.03 |
3.36 |
3.06 |
6.54 |
6.39 |
−0.87 |
−1.03 |
0.355 |
0.322 |
638.9 |
515.2 |
3.18 |
−0.08 |
|
P(FI)-Didecyl
|
−5.33 |
−1.97 |
3.36 |
3.04 |
6.44 |
6.26 |
−0.87 |
−1.04 |
0.405 |
0.326 |
53.8 |
998.0 |
3.56 |
−0.03 |
|
E(EH)
|
−5.32 |
−1.95 |
3.37 |
3.08 |
6.59 |
6.40 |
−0.70 |
−0.85 |
0.388 |
0.335 |
1089.5 |
412.3 |
2.99 |
−0.12 |
|
P(EH)
|
−5.35 |
−1.94 |
3.40 |
3.10 |
6.60 |
6.39 |
−0.71 |
−0.87 |
0.431 |
0.325 |
898.9 |
435.7 |
2.91 |
−0.09 |
|
E(POSS)
|
−5.34 |
−1.97 |
3.37 |
3.09 |
6.59 |
6.38 |
−0.74 |
−0.93 |
0.419 |
0.430 |
1291.5 |
842.4 |
3.19 |
−0.12 |
|
P(POSS)
|
−5.37 |
−1.96 |
3.41 |
3.10 |
6.60 |
6.37 |
−0.75 |
−0.95 |
0.462 |
0.433 |
1105.0 |
866.2 |
3.11 |
−0.09 |
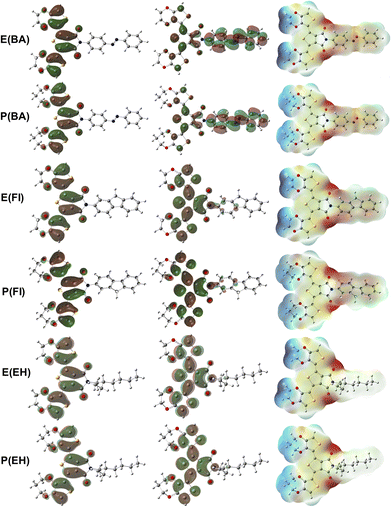 |
| | Fig. 3 HOMO (left), LUMO (middle) and ESP (right) isosurfaces for the E(AB), P(AB), E(FI), P(FI), E(EH), P(EH). | |
HOMO and LUMO orbitals have similar distributions for E(EH) and P(EH) compared to the pristine EDOT and ProDOT without an extension into the side group, as shown in Fig. S1.† This result is also valid for E(POSS) and P(POSS) given in Fig. S2.† However, E(Fl) and P(Fl) have slight extensions of unoccupied orbitals into the Fl side groups that can improve the acceptor by improving electron deficiency. Moreover, E(AB) and P(AB) have a substantial extension of unoccupied orbitals into the AB side groups. This phenomenon leads to the electron density transfer from the backbone to the side groups in the order of AB > Fl > EH ≈ POSS. This result can also be depicted by the electrostatic potential surfaces where higher electron density can be observed by the red color on the Fl and AB side groups that were not present for EH and POSS substitution. Low reorganisation energies, less positive IPs, more negative EAs, and effortless electron transfer from the backbone to the side chains in the ground state prove the possible improvement of optoelectronic properties viaAB and Fl substitutions. Theoretical calculations also showed that the effect of EDOT and ProDOT substitutions has a less significant effect on the electronic properties compared to the significant effect by the Fl side groups visualized by the density of states calculations given in Fig. S3.†
3.4. Electropolymerization of TPD-based monomers with Fl side chains and opto-electronic properties of their corresponding polymers
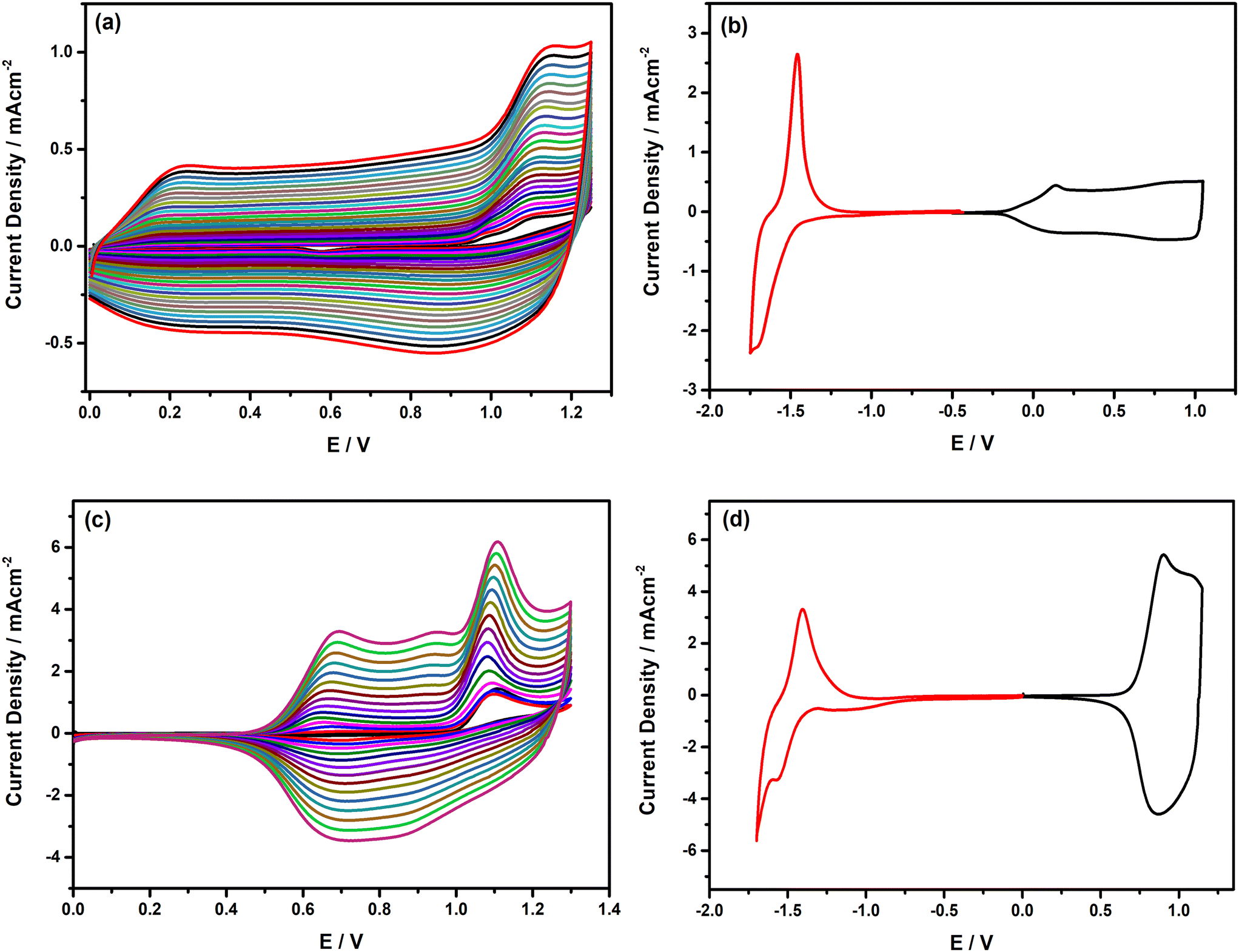
After the detailed inspection and comparison of redox features of the Fl-appended monomers with the precedent analogues, electrochemical polymerization of the monomers was performed either on Pt disc or ITO working electrodes via repetitive potential cycling from 0.0 V to +1.25 V for E(Fl) and from 0.0 V to +1.3 V for P(Fl) to obtain their corresponding polymers. The resulting voltammograms recorded during electropolymerization are depicted in Fig. 4(a) and (c). As seen from the voltammograms, during the second anodic scan, new redox couples started to appear within a broad potential range from +0.2 V to +1.0 V for PE(Fl) and at around +0.7 V for PP(Fl) and steadily intensified after each consecutive scan due to the formation of electroactive polymer films on the electrode surface. The proposed mechanism for the electrochemical polymerization of the E(Fl) monomer is depicted in Schemes S2 and S3.† The chemical structures of the polymer films are given in Scheme 2. After completing the electropolymerization, the polymer films were washed with a solvent to remove unreacted monomers and oligomeric species from the electrode surface. Then, the redox behaviours of the polymer films were inspected by recording their CVs in a monomer-free electrolyte solution. As seen from the CVs in Fig. 4(b) and (d), the polymer films exhibit reversible doping and de-doping in both anodic and cathodic regions, demonstrating the generation of charge carriers on the main backbone. A rectangle-shaped and highly broad redox couple appears between +0.2 V and +1.0 V during the anodic scan of the PE(Fl), indicating a slow doping process due to the well-ordered polymer chains. This attractive feature of PE(Fl) with a broad potential window paves its way for being used in supercapacitors. On the other hand, a narrow redox couple is observed at around +0.9 V during the p-doping of PP(Fl). Furthermore, the films reveal a well-defined redox couple in the cathodic region, with a peak potential at −1.71 V and −1.59 V for PE(Fl) and PP(Fl), respectively. All peaks and onset potential values acquired from CVs are noted in Table 4. The onset potentials from CV measurements (see Fig. 4(b) and (d)) were used to estimate the electrochemical band gap values of polymer films and it was found that the electrochemical band gap of PE(Fl) is lower than that of PP(Fl) due to different electron donating ability and steric hindrance of donor units. Moreover, during the anodic scan, the variations in peak currents with voltage scan rate were examined in order to prove the deposition of immobilized polymer films on the electrode surface. For this purpose, CVs were recorded at various scan rates ranging from 25 mV s−1 to 200 mV s−1, and the resulting CVs are presented in Fig. S4.† The redox process on the well-adhered electroactive polymer film is non-diffusion controlled, as seen from the linear relation of peak current as a function of the scan rate.
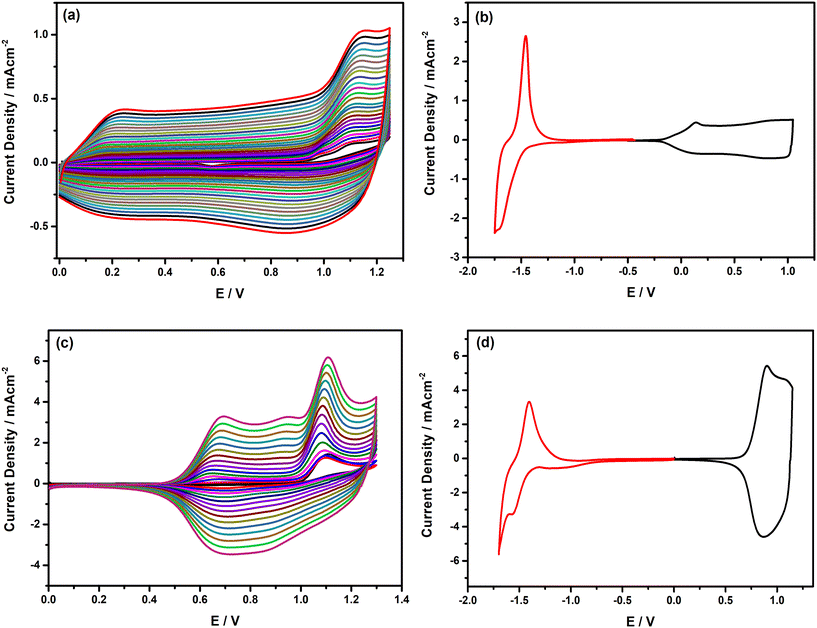 |
| | Fig. 4 Repetitive cyclic voltammograms of (a) E(Fl) and (c) P(Fl) monomer recorded in 0.1 M n-Bu4NPF6 in DCM for E(Fl), in MeCN/DCM (1/3: v/v) for P(Fl) and the cycling voltammograms of their corresponding polymer films (b) PE(Fl) and (d) PP(Fl) polymer films recorded in 0.1 M n-Bu4NPF6 in DCM for PE(Fl), in MeCN for PP(Fl) on the Pt disc electrode vs. Ag/AgCl at a scan rate of 100 mV s−1. | |
 |
| | Scheme 2 The structures of the polymer films. | |
Table 4 Electrochemical properties of the polymer films, PE(Fl) and PP(Fl)
| Polymers |
PE(Fl)
|
PP(Fl)
|
|
HOMO and LUMO energy levels were calculated from Eox,onset and Ered,onset values of CV diagrams, respectively, using eqn (1) and (2).53
|
|
E
ox,p (V) (CV) |
0.14 |
0.90 |
|
E
red,p (V) (CV) |
−1.71 |
−1.59 |
|
E
ox,onset,p (V) (CV) |
−0.11 |
0.72 |
|
E
red,onset,p (V) (CV) |
−1.49 |
−1.41 |
|
E
electrog (eV) |
1.38 |
2.13 |
|
E
optg (eV) |
1.48 |
1.59 |
| HOMOa (V) |
−4.60 |
−5.43 |
| LUMOa (V) |
−3.22 |
−3.30 |
For investigating of optoelectronic properties such as optical band gap, percent transmittance change (Δ%T), switching time (ts), and coloration efficiency (CE), in situ spectroelectrochemical measurements were carried out by using the polymer films coated on the ITO-glass electrode. Initially, in a monomer-free MeCN solution consisting of 0.1 M n-Bu4NPF6 as a supporting electrolyte, the electronic absorption spectra of the neutral state polymer films were recorded and the resulting spectra are depicted in Fig. S5.† As seen from the spectra, due to π–π* transitions, the PE(Fl) polymer film shows one broad absorption band centered at 644 nm, whereas the PP(Fl) polymer film exhibits two-well separated absorption bands centered at 609 nm and 668 nm. The presence of vibronic couplings is demonstrated by these well-separated absorption bands in the case of PP(Fl), which also shows the solid state order of polymer backbones as a consequence of π–π stacking.3,55 The optical band gaps of the Fl-appended polymer films were calculated from their fully neutral states by using the commencement in the low energy end of the π–π* transition and the obtained results were expressed as 1.48 eV for PE(Fl) and 1.59 eV for PP(Fl) and listed in Table 4. In comparison to PP(Fl), the existence of the EDOT donor group in PE(Fl) film results in a 0.11 eV decline in the band gap.
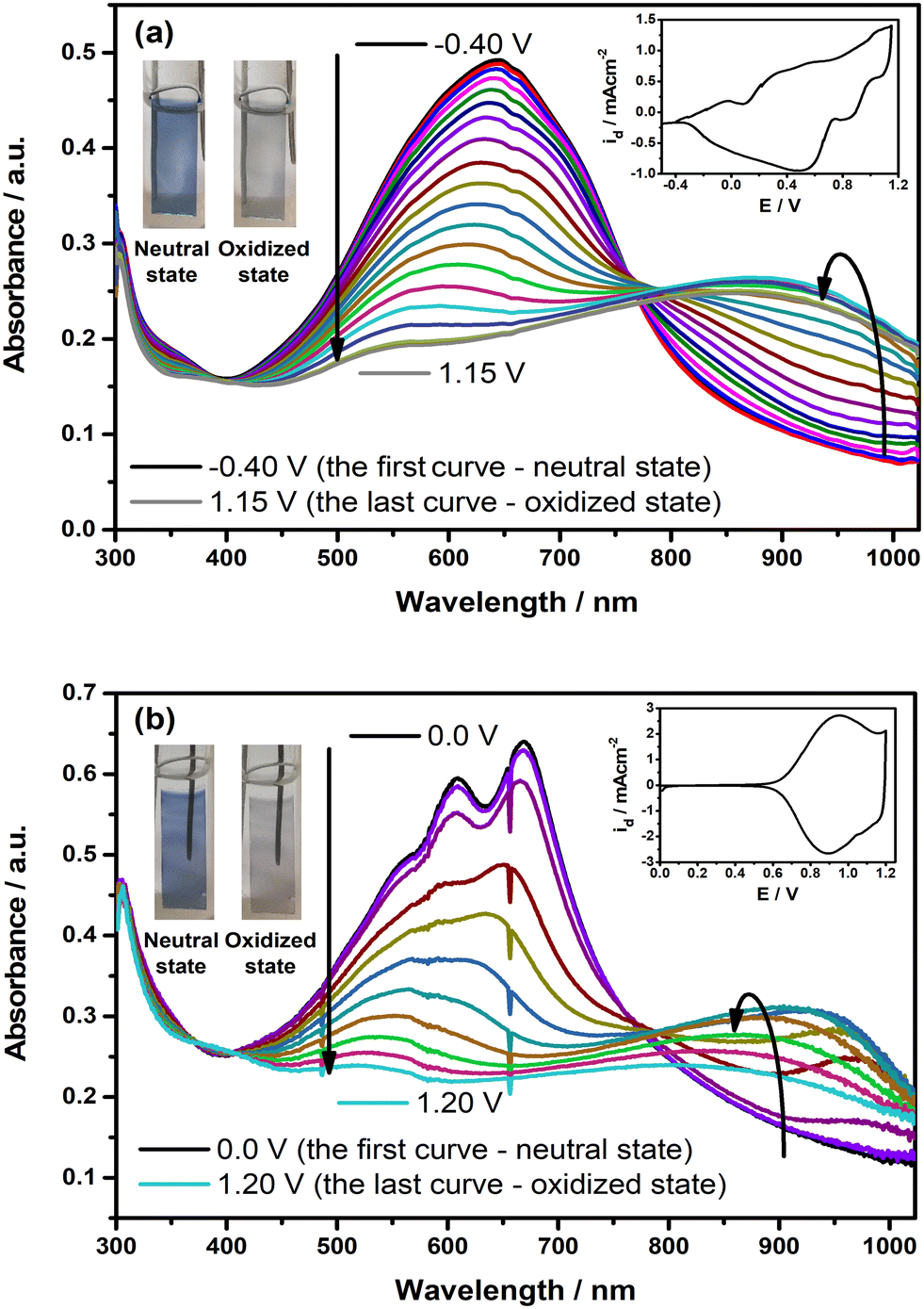
The changes in the optoelectronic features of the polymer films on ITO were observed under the applied external potential, and the resulting spectra are depicted in Fig. 5. Upon oxidation, the intensity of the π–π* transition band in each polymer film starts to decrease by a simultaneous change in the near-IR region with the formation of a new absorption band, which indicates the generation of polaron charge carriers. Upon further oxidation, this alteration is followed by a blue shift in the newly formed polaron charge carrier band. This shift is observed more explicitly in the case of PP(Fl). When the polymer films reach a fully oxidized state, the π–π* transition band completely diminishes, along with a decline in the intensity of the polaron charge carrier band. Upon oxidation, both PE(Fl) and PP(Fl) polymer films exhibit a color change from blue to gray, indicating their electrochromic characteristics (see inset photos of Fig. 5). The color coordinates of these polymer films in two extreme states, both neutral and oxidized, were expressed as L*, a*, and b* values, and are listed in Table 5.
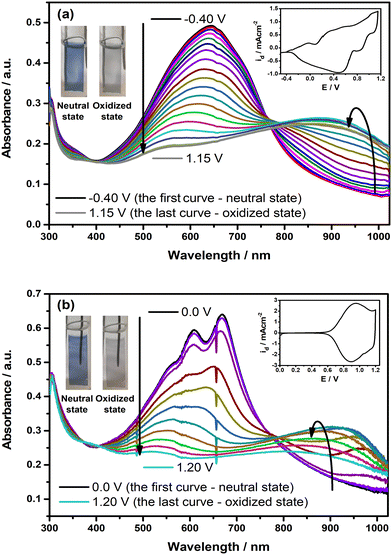 |
| | Fig. 5 The changes in electronic absorption spectra of electrochemically deposited (a) PE(Fl) and (b) PP(Fl) films on ITO recorded at various applied potentials in 0.1 M n-Bu4NPF6/MeCN. Insets: CVs of the films recorded with a scan rate of 20 mV s−1. Inset: the colors of the films at neutral and oxidized states. | |
Table 5 Optoelectronic properties of the polymer films on ITO in 0.1 M n-Bu4NPF6/MeCN
| Polymers |
PE(Fl)
|
PP(Fl)
|
|
λ
max (nm) |
644 |
609, 668 |
|
E
optg (eV) |
1.48 |
1.59 |
| Δ%T |
32 |
34, 36 |
| CE (cm2 C−1) |
407 |
501, 606 |
|
t
s,ox
(s) |
0.4 |
0.6, 0.5 |
|
t
s,neut
(s) |
0.2 |
0.2, 0.2 |
| Color at a neutral state |
71.19, −3.69, −18.14 |
68.75, −4.39, −17.24 |
|
L*, a*, b* |
| Color at an oxidized state |
84.99, −0.95, −0.73 |
81.69, 2.14, 0.19 |
|
L*, a*, b* |
| Color at a neutral state |
58.40, −2.07, −25.67 |
66.13, −2.88, −15.04 |
|
L*, a*, b* |
| Color at a reduced state |
68.56, −2.45, −9.31 |
77.22, −3.35, 0.85 |
|
L*, a*, b* |
| Polymers |
PE(EH)
|
PP(EH)
|
|
λ
max (nm) |
625, 685 |
600, 660 |
|
E
optg (eV) |
1.58 |
1.69 |
| Δ%T |
41 |
52 |
| CE (cm2 C−1) |
424 |
281 |
|
t
s,ox
(s) |
0.6 |
0.5 |
|
t
s,neut
(s) |
0.2 |
2.0 |
| Polymers |
PE(POSS)
|
PP(POSS)
|
|
λ
max (nm) |
545, 600 |
585, 640 |
|
E
optg (eV) |
1.64 |
1.78 |
| Δ%T |
46, 33 |
50, 52 |
| CE (cm2 C−1) |
472, 387 |
522, 550 |
|
t
s,ox
(s) |
0.6, 0.6 |
0.4, 0.4 |
|
t
s,neut
(s) |
0.6, 0.7 |
0.3, 0.3 |
| Polymers |
PE(AB)
|
PP(AB)
|
|
95% of the full switch from colored to bleached states.
|
|
λ
max (nm) |
630 |
612, 672 |
|
E
optg (eV) |
1.54 |
1.68 |
| Δ%T |
32 |
41, 44 |
| CE (cm2 C−1) |
454 |
750, 762 |
|
t
s,ox
(s) |
0.4 |
0.4, 0.4 |
|
t
s,neut
(s) |
0.4 |
0.4, 0.4 |
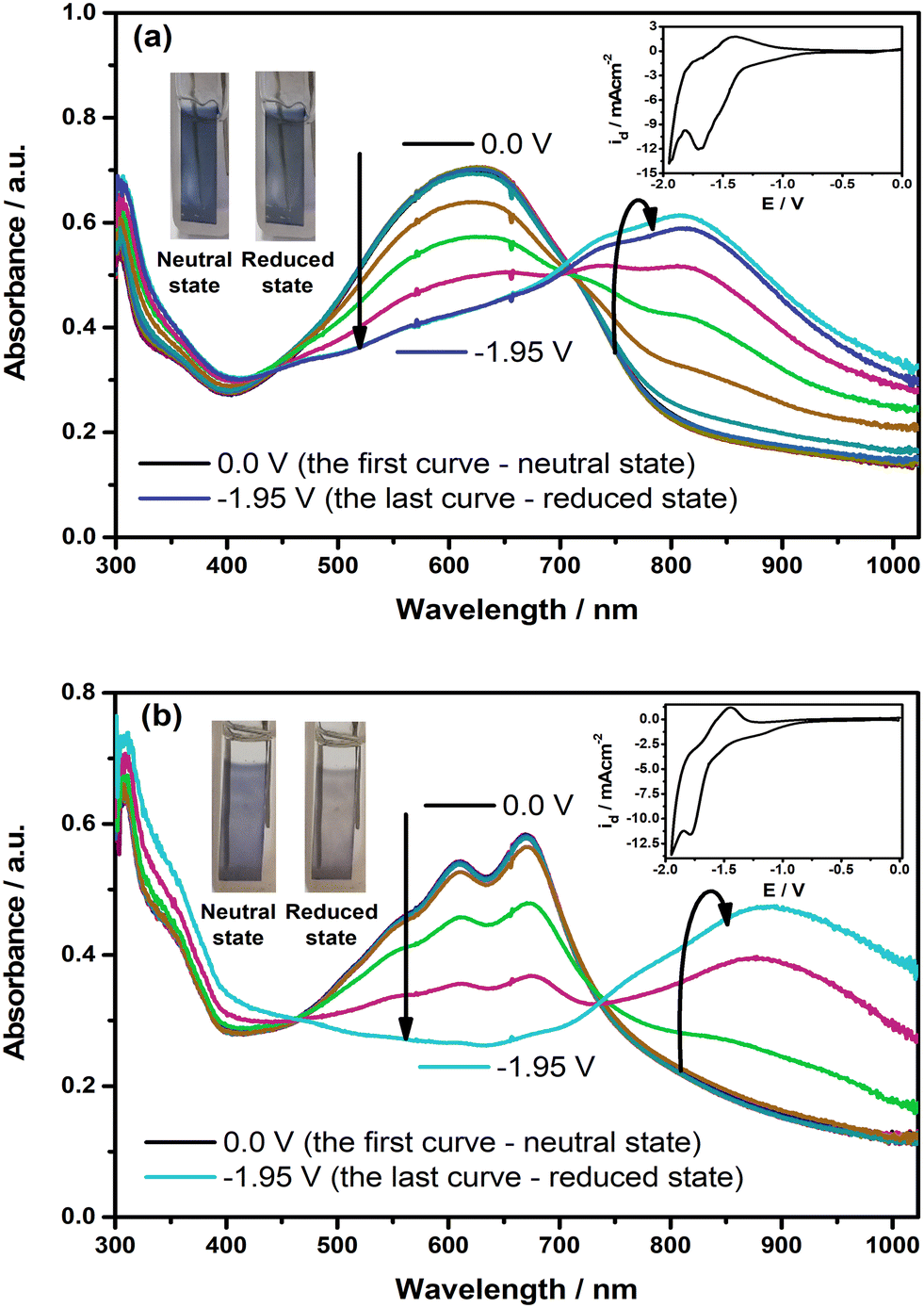
To identify whether the polymer films show spectral alterations during the reduction, the changes in their electronic absorption spectra were also recorded with the application of external potential in the cathodic region, as illustrated in Fig. 6. As shown in the figure, the intensity of π–π* transition bands starts to drop with the creation of new charge carrier bands at roughly 815 nm for PE(Fl) and 880 nm for PP(Fl) when the potential is applied, confirming the promising spectroelectrochemical characteristics of the polymer films during the reduction. L*, a*, and b* values corresponding to the colors in their neutral and reduced states are also noted in Table 5. These results demonstrated that not only electrical conductivity but also the type of doping and mechanism of the conductivity can be engineered by the molecular and electronic structures of the side chains.
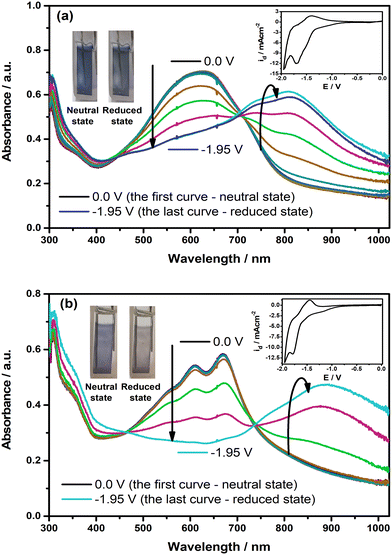 |
| | Fig. 6 The changes in electronic absorption spectra of electrochemically deposited (a) PE(Fl) and (b) PP(Fl) films on ITO recorded at various applied potentials during the reduction in 0.1 M n-Bu4NPF6/MeCN. Insets: CVs of the films recorded with a scan rate of 20 mV s−1. Inset: the colors of the films at neutral and reduced states. | |
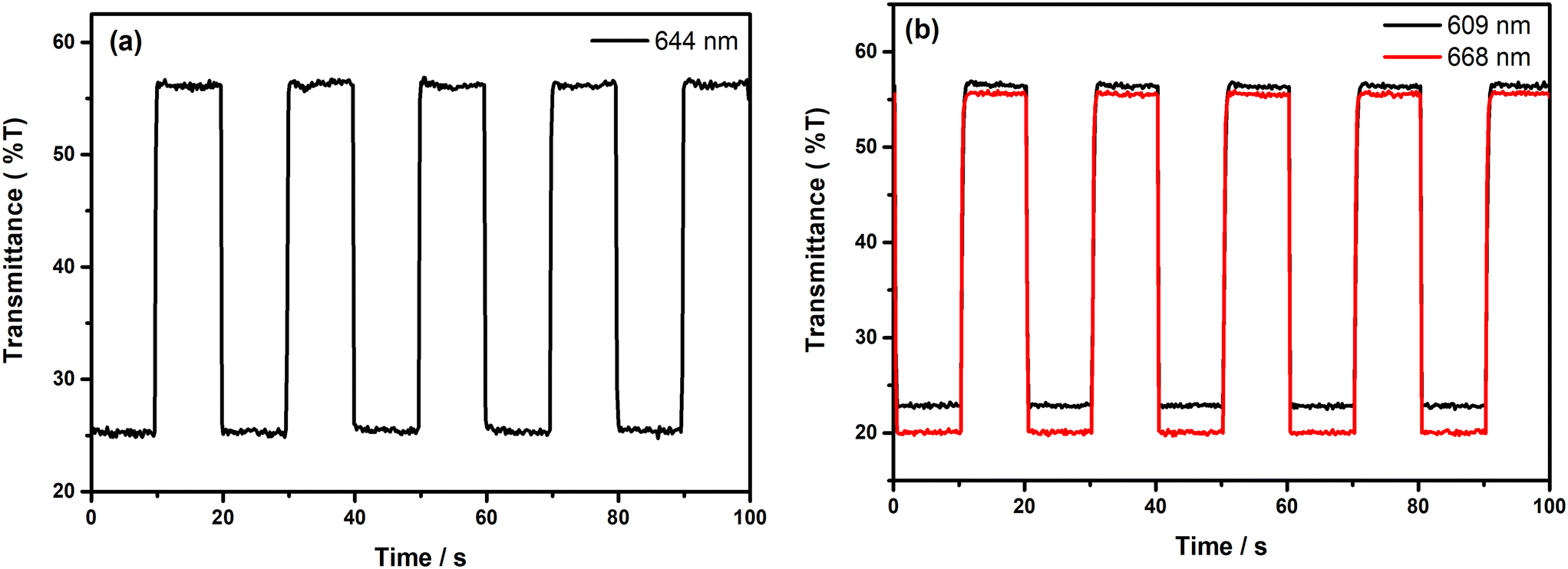
It is highly crucial for an electrochromic polymer to have good switching characteristics, such as short response time, high stability and high coloration efficiency to be utilized for display and device applications. For the investigation of the switching ability of the polymer films, chronoabsorptometry experiments were carried out. The polymer films were switched between two extreme redox states in the electrolyte solution by applying potential difference via the square wave potential method. Upon switching between redox states, the variations in the optical and electrical responses of the polymer films were recorded at λmax with a residence time of 10 s and the obtained spectra are illustrated in Fig. 7. The percent transmittance change (%T) of the films in the visible region was found to be 32% at 644 nm for PE(Fl) and 34% at 609 nm and 36% at 668 nm for PP(Fl) (see Table 5). The response time and coloration efficiency (CE) of the obtained films during a redox step were calculated at 95% of the full optical switch since the color change cannot be perceived with the naked eye beyond this point. The corresponding response times were found to be 0.4 s for PE(Fl) and 0.6 s at 609 nm and 0.5 s at 668 nm for PP(Fl) during doping and to be 0.2 s for both films during de-doping. This fast response time during the de-doping of polymer films makes them amenable for device applications. CE values were found to be 407 cm2 C−1 at 644 nm for PE(Fl) and 501 cm2 C−1 at 609 nm and 606 cm2 C−1 at 668 nm for PP(Fl). The obtained results for switching characteristics are also listed in Table 5.
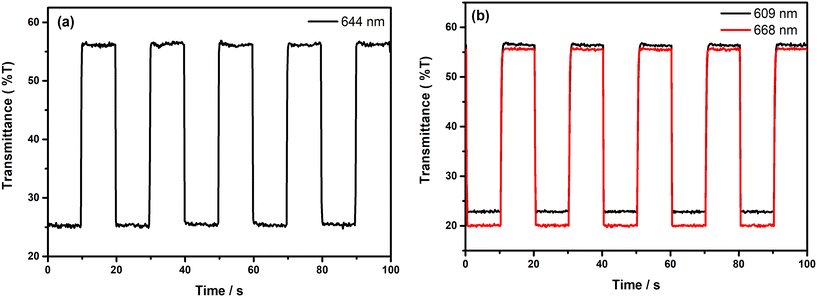 |
| | Fig. 7 Chronoabsoptometry experiments for the polymer films on ITO in 0.1 M n-Bu4NPF6/MeCN when the films were switched between redox states with an interval time of 10 s for (a) PE(Fl) (between −0.5 V and +1.2 V at 644 nm) and (b) PP(Fl) (between 0.0 V and +1.2 V at 609 nm and 668 nm). | |
3.5. Comparison of the optoelectronic properties of TPD-based D–A–D polymers
In order to shed light on the impact of side chains on the optoelectronic properties of TPD-based polymers, the optical band gap values of Fl-appended polymers recorded at the same solvent system were compared with the precedent analogues containing EH, POSS, and AB side chains and results are also tabulated in Table 5. When the optical band gap values were evaluated, it was found that the appendage of the Fl side chain instead of precedent analogues noticeably reduced the optical band gap by shifting the absorption bands to longer wavelengths. This effect might be ascribed to the difference in the electronic nature of the planar Fl side chain, which enhances the π–π interaction between polymer chains causing the red shift. The effect of the side chain on the electrochemical band gap values, on the other hand, was not evaluated because different solvent systems were required for the electrochemical measurements of polymers due to differences in their properties, such as solubility or electrochemical inactivity in the solvent system. Moreover, when the impact of the donor units on the optical band gap values of polymers was examined, the same trend was followed for each series depending on the strength of the donor unit (i.e. Eg,EDOT < Eg,ProDOT). The highest band gap alternation can be obtained by direct control of the donor and acceptor structures along the backbone, such as that reported for different donors and acceptors.56,57
In order to reveal the precise effect of the side chain on the switching properties of the polymers synthesized in this work, optical contrast, coloration efficiency, switching response time, and the color of Fl-appended polymers were compared with the previous analogues (see Table 5). It was observed that the structural modifications in the acceptor unit via side chains influence some switching characteristics more profoundly. First of all, optical contrast values of the polymers with the aryl side chains were found to be lower than those of alkyl side chains due to the more colorful oxidized state of the polymer with aromatic Fl and AB side chains. CE values for the polymers with the EDOT external unit do not exhibit significant changes depending on the side chain variations. On the contrary, the polymer films with ProDOT donor units exhibit notable variations depending on the side chain, especially for AB-appended polymer film, which indicates the highest CE among all the polymer films. When the switching time of the polymer films was investigated, it was found that the side chain variation on the acceptor unit has no systematic effect on the switching speed of the polymer films during doping and de-doping processes. Among them, the Fl-appended polymer films exhibit quite a fast switching speed, especially for the de-doping process, which showed the shortest switching time during de-doping in comparison to the other analogues containing EH, POSS, and AB side chains. When the effect of the side chain on the color of electrochromic polymer films was explored in their neutral and fully oxidized states, different cases were observed for their neutral and oxidized states. According to this, the neutral state color of polymer films does not change significantly in their neutral state depending on the side chain variation. On the contrary, the oxidized state color of the polymer films exhibits more pronounced variations depending on the type of side chain. When the alky side chains are replaced by aryl groups, the color of the oxidized state polymer films changed from blue or transparent blue to gray color.
The optical and electrochemical stabilities upon switching are important features for electrochromic polymers. The long-term redox switching stability of Fl-appended polymer films was performed by continuously stepping the voltages between their neutral and oxidized states using the square wave potential method within the potential range of −0.5 V and +1.1 V for PE(Fl) and 0.0 V and +1.1 V for PP(Fl) under ambient conditions. The absorbance and current density of the films were recorded as a function of time up to 10000 double potential steps. The obtained stability test results are given in Fig. S6† to demonstrate the variations in optical and electrochemical activities of the polymer films after thousands of cycles. As seen from Fig. S6,† it was found that PE(Fl) film reveals perfect stability by preserving 75% of its optical activity and 73% of its electroactivity under continuous switching up to 20000 cycles. The long-term stability of PE(Fl) polymer film makes it a promising candidate for electrochromic applications. In the case of PP(Fl), the polymer film also exhibited good stability by retaining approximately 89% of its optical activity with the loss of half of its electrochemical activity after 8000 deep double potential steps as depicted in Fig. S6.† When the stability test results were compared with the previous analogues, Fl-appended polymer films exhibited superior electrochemical and optical stability, especially in contrast to EH-appended ones.
4. Conclusion
In this work, an extensive study has been presented to reveal the effect of side chains on the optical, electronic, and optoelectronic features of new D–A–D type trimeric monomer systems and their corresponding polymers. With the goal of exploring such kinds of properties and different kinds of donors, the side chains were involved in this study. In order to elucidate the effect of the donor and the side chain on the acceptor, D–A–D type systems holding a new side chain, Fl has been synthesized and compared with three series of TPD-based acceptor units involving different functional side chains. Optical investigations revealed that the Fl side chain present on the TPD acceptor profoundly affects the absorption profile of monomers and also creates a change in quantum yields. In addition, the electrochemical measurements revealed that the electrochemically synthesized polymers, PE(Fl) and PP(Fl) exhibited reversible p-doping accompanied by the electrochromic response. Moreover, beyond both optical and electrochemical stabilities and robustness, Fl-appended polymers exhibit slightly lower band gaps, as compared to all previous analogues, which shows that the charge injection process with both holes and electron carriers is feasible. Besides, Fl-appended polymers exhibited quite a fast switching time, especially during the de-doping process, which makes them attractive candidates for electrochromic device applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
E. Y. gratefully acknowledges support from the 2232 International Fellowship for Outstanding Researchers Program of TÜBİTAK (118C251).
References
- F. Wolfart, B. M. Hryniewicz, M. S. Góes, C. M. Corrêa, R. Torresi, M. A. O. S. Minadeo, S. I. Córdoba de Torresi, R. D. Oliveira, L. F. Marchesi and M. Vidotti, J. Solid State Electrochem., 2017, 21, 2489–2515 CrossRef CAS.
- E. Reichmanis, H. Katz, C. Kloc and A. Maliakal, Bell Labs Tech. J., 2005, 10, 87–105 CrossRef.
- J. Roncali, Chem. Rev., 1997, 97, 173–205 CrossRef CAS PubMed.
- M. Berggren, D. Nilsson and N. D. Robinson, Nat. Mater., 2007, 6, 3–5 CrossRef CAS PubMed.
- H. Chen, Z.-G. Gu and J. Zhang, J. Am. Chem. Soc., 2022, 144, 7245–7252 CrossRef CAS.
- D.-J. Li, Q.-H. Li, Z.-G. Gu and J. Zhang, Nano Lett., 2021, 21, 10012–10018 CrossRef CAS PubMed.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
-
J. R. Reynolds, B. C. Thompson and T. A. Skotheim, Conjugated Polymers: Properties, Processing, and Applications, CRC Press, Boca Raton, FL, 4th edn, 2019 Search PubMed.
- M. C. Scharber and N. S. Sariciftci, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef CAS.
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2013, 26, 604–615 CrossRef.
- Z.-G. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC.
- N. Luo, P. Ren, Y. Feng, X. Shao, H.-L. Zhang and Z. Liu, J. Phys. Chem. Lett., 2022, 13, 1131–1146 CrossRef CAS.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- E. E. Havinga, W. Ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- Y. Li, L. Xue, H. Li, Z. Li, B. Xu, S. Wen and W. Tian, Macromolecules, 2009, 42, 4491–4499 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2019, 30, 1904545 CrossRef.
- Z. Liu, G. Zhang and D. Zhang, Acc. Chem. Res., 2018, 51, 1422–1432 CrossRef CAS PubMed.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS PubMed.
- Y. Wu, Y. Zhao and Y. Liu, Acc. Mater. Res., 2021, 2, 1047–1058 CrossRef CAS.
- B. Meng, Y. Fu, Z. Xie, J. Liu and L. Wang, Macromolecules, 2014, 47, 6246–6251 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- B. Meng, J. Liu and L. Wang, Polym. Chem., 2020, 11, 1261–1270 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- A. Welford, S. Maniam, E. Gann, X. Jiao, L. Thomsen, S. J. Langford and C. R. McNeill, Org. Electron., 2019, 75, 105378 CrossRef CAS.
- T. Lei, J. Y. Wan and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- Z. G. Zhang and Y. F. Li, Sci. China: Chem., 2015, 58, 192–209 CrossRef CAS.
- H. J. Park, H. S. Kim, J.-Y. Go, Y.-Y. Noh and D.-H. Hwang, Org. Electron., 2022, 101, 106403 CrossRef CAS.
- J. Y. Back, T. K. An, Y. R. Cheon, H. Cha, J. Jang, Y. Kim, Y. Baek, D. S. Chung, S. K. Kwon, C. E. Park and Y. H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 351–358 CrossRef CAS PubMed.
- H. B. Akkerman, S. C. Mannsfeld, A. P. Kaushik, E. Verploegen, L. Burnier, A. P. Zoombelt, J. D. Saathoff, S. Hong, S. Atahan-Evrenk, X. Liu, A. Aspuru-Guzik, M. F. Toney, P. Clancy and Z. Bao, J. Am. Chem. Soc., 2013, 135, 11006–11014 CrossRef CAS PubMed.
- L. Yang, H. Zhou and W. You, J. Phys. Chem. C, 2010, 114, 16793–16800 CrossRef CAS.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357–4363 CrossRef CAS.
- R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4215–4233 CrossRef CAS.
- S. Inaoka and R. Advincula, Macromolecules, 2002, 35, 2426–2428 CrossRef CAS.
- D. Çakal, S. Ertan, A. Cihaner and A. M. Önal, Dyes Pigm., 2018, 158, 175–182 CrossRef.
- D. Çakal, A. Cihaner and A. M. Önal, Electrochim. Acta, 2021, 377, 138064 CrossRef.
- D. Çakal, A. Cihaner and A. M. Önal, Electrochim. Acta, 2021, 398, 139325 CrossRef.
- D. Cornelis, H. Peeters, S. Zrig, B. Andrioletti, E. Rose, T. Verbiest and G. Koeckelberghs, Chem. Mater., 2008, 20, 2133–2143 CrossRef CAS.
- G. Griffini, J. D. Douglas, C. Piliego, T. W. Holcombe, S. Turri, J. M. J. Fréchet and J. L. Mynar, Adv. Mater., 2011, 23, 1660–1664 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- H. T. Turan, O. Kucur, B. Kahraman, S. Salman and V. Aviyente, Phys. Chem. Chem. Phys., 2018, 20, 3581–3591 RSC.
- E. A. Alkan, S. Göker, H. Sarigul, E. Yildirim, Y. A. Udum and L. Toppare, J. Polym. Sci., 2020, 58, 956–968 CrossRef CAS.
- S. C. Cevher, G. Hizalan, E. Alemdar Yilmaz, D. Cevher, Y. Udum Arslan, L. Toppare, E. Yıldırım and A. Cirpan, J. Polym. Sci., 2020, 58, 2792–2806 CrossRef CAS.
- M. Yasa, A. Deniz, M. Forough, E. Yildirim, O. Persil Cetinkol, Y. Arslan Udum and L. Toppare, J. Polym. Sci., 2020, 58, 3336–3348 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5003 CrossRef CAS PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- F. C. Correia, T. C. F. Santos, J. R. Garcia, L. O. Peres and S. H. Wang, J. Braz. Chem. Soc., 2015, 26, 84–91 CAS.
- Q. Sun, H. Wang, C. Yang and Y. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- S. M. Risser, D. N. Beratan and S. R. Marder, J. Am. Chem. Soc., 1993, 115, 7719–7728 CrossRef CAS.
- M. Li, A. Patra, Y. Sheynin and M. Bendikov, Adv. Mater., 2009, 21, 1707–1711 CrossRef CAS.
- M. E. Köse, J. Phys. Chem. A, 2019, 123, 5566–5573 CrossRef PubMed.
- H. T. Turan, O. Kucur, B. Kahraman, S. Salman and V. Aviyente, Phys. Chem. Chem. Phys., 2018, 20, 3581–3591 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Elif
Demir Arabacı
a,
Elif
Demir Arabacı