
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-responsive metal–organic frameworks – design strategies and emerging applications in photocatalysis and adsorption
Gabriele
Scandura
*ab,
Sana
Eid
a,
Ali A.
Alnajjar
ac,
Twinkle
Paul
abc,
Georgios N.
Karanikolos
 abcd,
Dinesh
Shetty
de,
Khalid
Omer
f,
Rami
Alqerem
f,
Alaa
Juma
f,
Huanting
Wang
g,
Hassan A.
Arafat
ac and
Ludovic F.
Dumée
*abc
abcd,
Dinesh
Shetty
de,
Khalid
Omer
f,
Rami
Alqerem
f,
Alaa
Juma
f,
Huanting
Wang
g,
Hassan A.
Arafat
ac and
Ludovic F.
Dumée
*abc
aDepartment of Chemical Engineering, Khalifa University, P.O. Box 127788, Abu Dhabi, United Arab Emirates. E-mail: gabriele.scandura@ku.ac.ae; ludovic.dumee@ku.ac.ae
bResearch and Innovation Center on CO2 and Hydrogen (RICH), Khalifa University, P.O. Box 127788, Abu Dhabi, United Arab Emirates
cCenter for Membrane and Advanced Water Technology (CMAT), Khalifa University, P.O. Box 127788, Abu Dhabi, United Arab Emirates
dCenter for Catalysis and Separations (CeCaS), Khalifa University, P.O. Box 127788, Abu Dhabi, United Arab Emirates
eDepartment of Chemistry, Khalifa University, P.O. Box 127788, Abu Dhabi, United Arab Emirates
fAbu Dhabi Agriculture and Food Safety Authority (ADAFSA), P.O Box 52150, Abu Dhabi, United Arab Emirates
gDepartment of Chemical Engineering, Monash University, VIC 3800, Clayton, Australia
First published on 24th February 2023
Abstract
Stimuli-responsive metal–organic frameworks (MOFs) are highly versatile porous materials with the ability to respond to different external stimuli, including temperature, pressure, pH, and light. The MOF properties can switch reversibly under specific light irradiation, opening the doors to various applications. This review focuses on design strategies to obtain photo-responsive MOFs, namely (i) encapsulation of photo-switchable molecules as guests in MOF porous structures, (ii) fabrication of MOF composites, (iii) post-synthesis modification, and (iv) synthesis of MOFs with photo-responsive ligands. The most recent reports from the literature are herein reviewed and analyzed in terms of material chemistry and performance. Comparisons between the different strategies are performed and future challenges are discussed. The critical aspect of the fatigue of photo-responsive MOFs applied for prolonged cycling of irradiation is also discussed.
1. Introduction
Metal–organic frameworks (MOFs) are a class of high-surface area, microporous, crystalline materials structured with inorganic nodes coordinated to organic ligands forming highly periodic structures.1,2 The inorganic nodes are typically metal ions or Secondary Building Units (SBUs), such as Cu2(COO)4, Zn4O(COO)6, and Cr3O(H2O)3(COO)6, while the organic ligands contain aromatic molecules with pyridyl, phosphonate, imidazolate, and carboxylate functional groups.3 Different framework topologies, morphologies, and properties may be achieved by careful selection of nodes and linkers,4,5 which open the door to advanced applications, primarily in sensing,6 drug delivery,7 catalysis,8–12 separation,13–19 and chemical conversion.18,20The design of MOF materials with high chemical, mechanical, and structural stability is critical to their scale-up and industrial implementation.21,22 The instability of MOF materials is largely related to solvation issues, e.g., with water and other solvents or adsorbates, which detrimentally affect the node–linker bonds within the framework, leading to hydrolysis and formation of hydroxide ligated nodes and protonated linkers.23 Thus, a thermodynamically stable MOF should have an inert node to avoid the irreversible hydrolysis reaction, but most nodes, being ionic based, are electrophilic, hence easily react with the nucleophilic O in water.23 Interestingly, a MOF may be thermodynamically unstable but kinetically stable23 if water molecules are unable to cluster close to the metal nodes either due to steric hindrance within the pore or due to the internal hydrophobicity, arising from the linker molecule pendant group chemistry.23 The efficient design of MOF hierarchical structures is therefore dictated by the conditions under which they may operate and by their response to external physical, thermal or chemical stressors.24
The design of responsive MOF materials shall therefore be based on these stressors to support reversible conformational changes.24 Breathing, swelling, linker rotation and subnetwork displacements represent the main modes of MOF flexibility.25 Breathing refers to a reversible drastic change in the unit cell volume due to a displacement of atoms within the framework, while swelling is a progressive change of the unit cell volume for a constant unit cell shape.26 The cell edge angles and rotational deformation may only change during breathing, while linker rotations lead to spatial re-alignment of the linker around a rotational axis affecting internal pore dimensions.27,28 Subnetwork displacements concern individual frameworks connected only by weak chemical bonds and where the subnets may shift, relocate, or drift towards each other.24 The unit cell volume shall change after breathing and swelling, whereas it is constant during the linker rotation and subnetwork displacement.20,29 Special functionalities may be embedded within the MOF matrix to benefit from these flexibility modes. For example, guest-selective gate opening may be due to a linker rotation upon interaction of a pendant functional group of the linker with an analyte, which chemistry may unlock the gating process at the pore opening.30
Photo-responsivity is a specific stressor, whereby external stimuli, such as light irradiation, may support conformal and non-destructive deformations within materials. Pristine MOFs are generally poor light-harvesting materials due to their density and intrinsic chemistry and yet, photo-responsive MOFs have been developed by using photo-switchable molecules or grafting specific pendant groups across their linkers. Photo-switchable molecules undergo reversible isomerization upon absorption of electromagnetic radiation at specific wavelength. Other parameters such as light irradiance, system temperature, and interactions with the surrounding environment may also affect the photo-switchable character.31 The engineering of photo-switchable MOF materials is therefore highly desired to expand the scope of MOF materials, beyond their common applications.
Reviews published on MOF-based stimuli-responsive materials include strategies to trigger light, mechanical or piezo-electric reactions upon specific stress application including thermal, pressure, chemical, or electro-magnetic stressors. An overview of 2D MOFs responding to different external stimuli was reported focusing more on their chemistry rather than their applications,32 while other reviews dealt with MOF-based stimuli-responsive systems for biomedical applications, such as nano-carriers for drug delivery.33 pH and temperature-based stimuli-responsive MOFs able to cross-react to several stimuli were also discussed, supporting the design of multi-functional meta-materials33,34 presented in detail in a feature article.35 A previous short review only addressed systems formed by a photo-responsive molecule embedded within the MOF pores,36 and this will be the first strategy to be discussed below. Light responsive MOF design strategies were however not discussed in-depth to date, and a systematic analysis of the relationship between the chemical properties and physical switchability, as well as structural characteristics and performance, is required.37–39
This review presents recent advances in the field of photo-responsive MOFs to establish relationships between design strategies, material properties, and applications towards photocatalysis and adsorption. These design strategies include (i) the formation of MOFs with a photo-switchable guest molecule physically confined inside their pores, (ii) the engineering of photo-responsive MOF/nanoparticle heterostructures, hereafter referred to as composites, (iii) post-synthetically modified photo-responsive MOFs, and (iv) the design of MOFs encompassing photo-responsive ligands within their structures. The introduction of the photo-responsive character can occur after the MOF synthesis as in the first three strategies, or during the MOF synthesis as in the fourth one. The MOF structure is chemically unaltered when following strategies (i) and (ii) since there is no covalent bond between the photo-switchable guest molecules or the nanoparticles and the MOF scaffold. In contrast, the photo-responsive functional groups are covalently bonded to the MOFs when implementing strategies (iii) and (iv). Key properties supporting enhanced photo-switchability are discussed, while material performances are analyzed and, when possible, compared and benchmarked. Challenges in the area and shortcomings of these strategies are critically discussed, therefore supporting the rationalization of advanced photo-responsive MOF-based platforms towards more scalable applications of combinatorial MOF materials.
2. MOFs engineered from photo-switchable guest molecules
The first strategy used to impart a light-responsive character to MOFs involves physically confining a photo-switchable molecule within the porous structure. The photo-isomerization of the guest molecule will affect the physicochemical properties of a guest@MOF system.40 Recent photo-switchable molecules and their derivatives that have been used as guests in MOFs include spiropyran,41–44 spirooxazine,45 azobenzene,46,47 dithienylethenes,48 and anthracene.49 Each group of photo-switchable molecules will be discussed in this section, along with their demonstrated applications.2.1 Spiropyran@MOFs
The closed-ring isomer of spiropyran (SP) is composed of indoline and 2H-1-benzopyran bound together through a spiro junction (Fig. 1). Both moieties can have substitutional groups in their structure (see for instance Table 1), which are typically bound to the N atom of indoline (R1 group) and the aromatic ring of benzopyran (R2 and R3 group).50 The open-ring isomer merocyanine (MC) is formed under UV irradiation at wavelengths between 250 and 380 nm, depending on the substitutional groups.50,51 First, the Cspiro–O bond breaks via a single-photon process resulting in a transient species (cis-MC)52; then the central C–C double bond rotates yielding trans-MC or MC.53 In addition to UV, the SP → MC isomerization may also occur in the dark or under NIR radiation (785 nm) via two-photon absorption,54,55 while visible irradiation or thermal energy can trigger the reverse MC → SP isomerization.56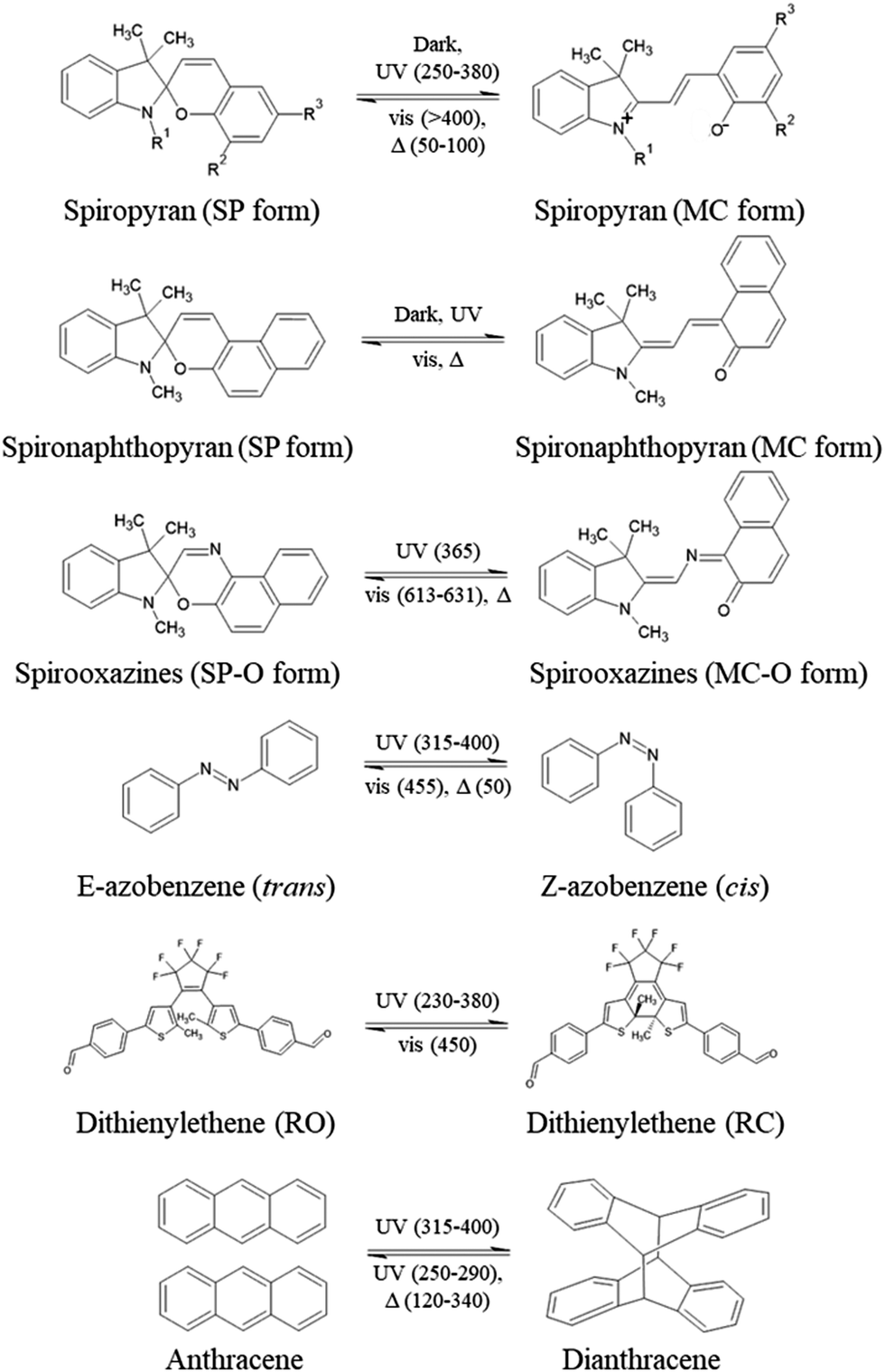 | ||
| Fig. 1 Photo-switchable molecules used in the guest@MOF systems with the isomerization wavelengths given in nm and temperature in °C. | ||
| Entry | Guest@MOF | R1 | R2 | R3 | Ref. |
|---|---|---|---|---|---|
| a SP acrylate was polymerized inside the MIL-53 pores. | |||||
| 1 | SSP@ZIF-8 | –CH3 | –H | –SO3− | 41,42 |
| 2 | NSP@UiO-67 | –CH3 | –H | –NO2 | 43 |
| 3 | PSPA@MIL-53a | –CH2CH2O-acrylate | –H | –NO2 | 44 |
| 4 | SP@MOF-5 | –CH3 | –H | –H | 57 |
| 5 | –CH3 | –OMe | –H | ||
| 6 | –CH3 | –H | –Br | ||
| 7 | –CH2CH2OH | –H | –NO2 | ||
| 8 | –CH3 | –OMe | –NO2 | ||
| 9 | –CH3 | Naphtho | |||
| 10 | SP@MIL-68(In) | –CH3 | –H | –H | |
| 11 | –CH3 | –OMe | –H | ||
| 12 | –CH3 | –H | –Br | ||
| 13 | –CH2CH2OH | –H | –NO2 | ||
| 14 | –CH3 | –OMe | –NO2 | ||
| 15 | –CH3 | Naphtho | |||
The electron-donating/withdrawing nature of the substitutional groups plays an essential role in the activation energy barrier for isomerization. An SP with an electron donor R1 group and an electron acceptor R2 or R3 group, for instance, elongated the Cspiro–O bond, decreasing the activation energy for the SP → MC isomerization and stabilizing the MC form.58 Similarly, the activation energy for the Cspiro–O bond cleavage was lowered in a polar medium, simultaneously increasing the energy barrier for the MC → SP isomerization.59
MC can aggregate because of the dipole–dipole interactions due to its negatively charged phenolate and positively charged indolium groups, and the π–π stacking. Consequently, this isomer becomes more stable hindering the reverse ring-closing reaction to SP, which undergoes significant photo-degradation with a high number of switching cycles.60 The isolation of the photo-switchable molecule, as in the case of the guest@MOF, reduces fatigue since irreversible side photo-reactions are significantly suppressed.61 Furthermore, encapsulation of spiropyran in the MOF structure prevents intermolecular interactions between the MC molecules, and, hence, their potential aggregation, as well as it improves the efficiency of the photo-isomerization in both directions.62
The optical properties of six different spiropyran molecules (Table 1, entries 4–15, also referred to as systems 4–15) loaded in polar MOF-5 and less polar MIL-68(In) were reported as a function of the SP substitutional groups.57 The guest–host interactions were investigated by comparing bulk analysis results, namely liquid-state NMR, with surface ones, namely XPS. Although the guest molecule was randomly distributed within the MOF pores with no preferential position near the metal node in systems 4, 5, 6, 11, 12, and 15 (Table 1), a weak guest–host interaction was observed in system 10 (Table 1), and a preferential location of the guest molecule close to the metal nodes was assumed in the other systems (Table 1, entries 7–9, 13, and 14). The MC form was already found in all the guest@MOF-5 systems before UV irradiation owing to the electrostatic field presented within the MOF pores. In contrast, in the MIL-68 systems, where the electrostatic field is less intense, the stable form of the guest molecule was dependent on its substitutional groups; systems 10–12 and 15 had the MC form, while 13 and 14 had the SP form before UV irradiation. The last result regarding the MIL-68 series seems to contradict previous findings.58 After UV light exposure (365 nm), systems 5 and 8 were stable, but no significant differences were recorded in the reflectance spectra before and after irradiation. Systems 13 and 14 (Table 1) instead showed a reversible photochromic response with the SP → MC and MC → SP isomerization occurring under irradiation at 365 and 630 nm, respectively, while the other systems were degraded under UV light. Thus, the significant outcome of this systematic study was that the combination of the MOF polarity and the substitutional groups of the guest molecule, in particular the nitro functionality, may lead to an efficient reversible photo-conversion between the SP and MC isomers.57
Sulfonated spiropyran (SSP, Tables 1 and 2, entry 1) was embedded into a ZIF-8 hybrid proton-conductive membrane for potential application in photo-switchable devices.41 Under dark conditions, the phenol and sulfonate groups of MC may form a hydrogen bond network inside the ZIF-8 pores, enhancing the proton transportation across the membranes by two orders of magnitude compared to the bare ZIF-8 membrane materials. Humidity dramatically affected the membrane proton conductivity since water present within the pores may also form hydrogen-bonds, hence enhancing the proton transportation. At room temperature, the conductivity increased by a factor of 180 when switching the relative humidity (RH) from 55% to 95%. The highest proton conductivity (1 ± 0.6 × 10−4 S cm−1) at 25 °C and 95% RH was achieved with an actual SSP content of 8.1%. The MC isomer switched back to the SP form upon visible light irradiation (wavelength larger than 400 nm), reducing the proton conductivity of the membrane. The on/off conductivity ratio, namely between dark conditions and visible irradiation, was ∼300 at 25 °C and 95% RH, while keeping the RH constant at 95% and increasing the temperature up to 75 °C led to a conductivity of 5 ± 1 × 10−2 S cm−1 in the dark and an on/off conductivity ratio of about 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Cycling testing, performed at 55 °C and 95% RH, showed that the SSP@ZIF-8 membrane maintains an on/off conductivity ratio of about 1200 after 100 cycles. The SP → MC reaction took 5 min, whereas the reversal process occurred 60 times faster.
000. Cycling testing, performed at 55 °C and 95% RH, showed that the SSP@ZIF-8 membrane maintains an on/off conductivity ratio of about 1200 after 100 cycles. The SP → MC reaction took 5 min, whereas the reversal process occurred 60 times faster.
| MOF | Guest@MOF | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Guest@MOF | Pore window [Å] | Pore cavities [Å] | Pore volume [cm3 g−1] | BET SSA [m2 g−1] | Molecule dimensions [Å] | Molecule loading [wt%] | Pore volume [cm3 g−1] | SSA [m2 g−1] | λ A [nm] a | λ D [nm] a | Application | Ref. |
| Spiropyran state (SP), merocyanine state (MC).a Activation and deactivation wavelengths, unit is nm unless specified.b Thermal treatment.c Average NSP molecules per MOF unit cell. | |||||||||||||
| 1 | SSP@ZIF-8 | 3.4 | 11.6 | 0.72 | 1729 | SP 10.5 × 7.1 × 11.0, MC 10.0 × 9.7 × 8.0 | 8.1 | 0.61 | 1486 | Dark | >400 | Membrane with on/off photo-switchable proton conductivity | 41 |
| Cation-selective transport membrane | 42 | ||||||||||||
| 2 | NSP@UiO-67 | SP 12.2, MC 14.0 | 7.5c | 365 | Room light | SURMOF film on interdigitated gold electrodes on quartz substrates | 43 | ||||||
| 3 | PSPA@MIL-53 | 8.5 × 8.5 2.6 × 13.6 (breathing effect) | Channels | 875 | SP 5.4 × 5.8 × 11.6, MC 3.6 × 5.6 × 12.9 | 22.5 | Dark or 254 | Vis | Ion adsorbent for water desalination | 44 | |||
| 4 | SP-O@MOF-5 | 8 | 11; 15.1 | 1467 | 15.5 × 9.5 × 8 | 48 | 237 | 365 | 625 | NA | 45 | ||
| 5 | SP-O@MIL-68(In) | 6, 16.5 | Channels | 1629 | 19 | 45 | 365 | 625 | NA | 45 | |||
| 6 | SP-O@MIL-68(Ga) | 6, 16.6 | Channels | 438 | 26 | 36 | 365 | 625 | NA | 45 | |||
| 7 | SP-O@MIL-53(Al) | 8.5 × 8.5 7.3 × 7.7 | Channels | 1140 | 10 | NA | 365 | 625 | NA | 45 | |||
| 8 | AB@UiO-67 | 8 | 12; 18 | 0.676 | 2565 | trans 9.5, cis 5.5 | 18.9 | 0.286 | 1080 | 455 | 365 | Gas permeation | 46 |
| 9 | AB@UiO-66 | 6 | 9; 12 | 1036 | 5 | 761 | 50 °Cb | 365 | Light-driven molecular gates to regulate the ion flux | 47 | |||
| 10 | AB@HKUST-1SURMOF | 25 | 365 | 455 | Photo switchable system | 63 | |||||||
| 11 | AB@ UiO 66-TEPA | 0.48 | 682 | 2.5 | 0.31 | 433 | 320 | 400 | Photo switchable CO2 capture | 64 | |||
| 12 | AB@ MIL-101-NH2- Cu+ | 1.84 | 2780 | 0.82 | 0.59 | 703 | 335 | 434 | Photo switchable CO capture | 65 | |||
| 13 | DTE@ZJU-88 | 8 × 12 | 23.38 × 8.98 × 7.75 | 300 | >450 | Reversible confidential information protection | 48 | ||||||
| 14 | ANT@ZIF-8 | 3.4 | 11.6 | 1464 | 5.6 × 9.8 | 32 | 11 | 360 | 120 °Cb | Photo-patternable and erasable surfaces | 49 | ||
This membrane also exhibited cation conductivity, which depended on the diameters of the dehydrated ions and their hydration energy.42 In fact, only the dehydrated ions can enter the channels of the membrane since the diameters of the hydrated ions were larger than the size of the ZIF-8 pore window. As a result, the SSP@ZIF-8 membrane exhibited Li+ conductivity as much as 1.6 × 10−4 S cm−1 at 25 °C, and dramatic cation selectivity, namely 77, 112, and 4913 for Li+/Na+, Li+/K+, and Li+/Mg2+, respectively. The on/off conductivity ratios were 23, 3.8, 1.7, and 1.3 for Li+, Na+, K+, and Mg2+, respectively. Only three on/off cycles were performed in the case of Li+ solutions, in which the duration of the SP → MC and MC → SP reactions were 1.5 and 15 times longer than that of a pure aqueous solution. The latter result was ascribed to the larger steric hindrance since a single Li+ ion (dehydrated ion diameter 1.2 Å) and one MC molecule (10.0 × 9.7 × 8.0 Å3) filled about 96% of the volume of the ZIF-8 pore (diameter 11.6 Å) and the SP isomer was even larger than the MC one. However, this aspect could be investigated more in depth as one SP molecule (10.5 × 7.1 × 11.0 Å3) and one Li+ ion seemed to occupy more than the available volume. In the 3rd cycle, the Li+ conductivity in the dark was about 85% of the one during the 1st cycle, likely due to the fatigue of spiropyran. Overall, SSP@ZIF-8 showed better conductivity and on/off conductivity ratio but longer response time when compared with other materials. For instance, the on/off ratio and the response time were 20 times higher and 100000 times longer than those of a Pt/graphene membrane.66 The Li+ conductivity was from 2 to 5 orders of magnitude higher, with a response time of about 20 times longer when compared to PVC/DOS/crowned spirobenzopyran membranes.67,68
Nitro-substituted spiropyran (NSP, Tables 1 and 2, entry 2) was encapsulated in an UiO-67 surface-anchored MOF (SURMOF) film, grown on an interdigitated gold electrode on a quartz substrate to study the conductance.43 This MOF structure exhibited a nonpolar pore environment without open metal sites to help stabilize the NSP within the cavity. Under 365 nm LED light irradiance, the SP → MC isomerization occurred with a yield of 70%, while the current increased from 0.35 to 3.44 nA in 4 min, which corresponded to an electrical conductivity of 4.1 × 10−11 S cm−1 and 4.1 × 10−10 S cm−1. Reverse isomerization took place upon turning the UV light off in about 24 ± 2 min, but the current was 5% lower in the following cycle, revealing that SP molecules can undergo degradation during on/off cycles. Density functional theory (DFT) simulations showed that the MC → SP isomerization of NSP results in a decrease of the molecular extension by 1.8 Å,43 instead of an increase as in the case of SSP.42 The bare MOF film was not photo-responsive, while its conductivity was found to be 3 orders of magnitude lower than that of the same films inclusive of photo-responsive molecules embedded in their cavities.43
Poly(spiropyran acrylate) (PSPA, Tables 1 and 2, entry 3) into the pores of MIL-53(Al) was used as an ion adsorbent for water desalination.44 Under UV irradiation (254 nm) or in the dark, the PSPA transformed to PMC allowing the simultaneous capture of anions and cations by electrostatic interactions with the two charged groups in the PMC structure. The maximum ion adsorption loading under UV light was 1.4 times higher than under the dark conditions, while the reverse isomerization to PSPA, carried out under solar irradiation, released at least 96% of the ions. When performing batch tests with a 1000 ppm NaCl solution, an increase in the PSPA loading from 15.6% to 22.5% enhanced the ion adsorption loading by 260%, whereas larger concentrations of the embedded polymer blocked the pores. The salt adsorption capacity varies with pH since H+ or OH− ions yield electrostatic bonds with the adsorption sites, therefore affecting adversely the adsorption performance of the material. Thus, the optimal PSPA loading of 22.5% was used to ascertain that the ideal pH was 8, while the ion adsorption loading was 3.2 times lower and practically 0 at pH of 7 and 9, respectively. Monovalent salts were adsorbed at a higher capacity than divalent salts due to their smaller hydrated ion diameters and lower requirements in terms of adsorption site occupancy.69 The ion adsorption loading was optimized at 2.5 mmol g−1 for a 10000 ppm NaCl solution after 30 min. The desorption process duration lasted from 2 h (under room light) to 4 min (under one sun or 100 mW cm−2). Finally, the ion adsorption capacity decreased by 10% after ten cycles of reversible salt adsorption with an initial concentration of 10000 ppm. The ion adsorption capacities of commercial and other reported adsorbents were in the same range of PSPA@MIL-53, with the adsorption and desorption processes lasting for 20–960 min. Compared with these materials, PSPA@MIL-53 showed a fast adsorption time of around 30 min, and an ultrafast desorption time of close to 4 min. Furthermore, the regeneration of PSPA@MIL-53 took place under sunlight irradiation, which is more cost-effective and environmentally friendly, whereas thermal energy and strong acid/base solutions were employed for the regeneration of the other materials.
2.2 Spirooxazine@MOFs
The structure and photo-isomerization of spirooxazine and spiropyran are similar (Fig. 1), with the former molecule having Cspiro bonded with a benzoxazine instead of a benzopyran. 1,3,3-Trimethyl indolino-naphthospirooxazine (SP-O) was loaded as a guest molecule in different MOFs containing the same ligand, namely, MOF-5, MIL-68(In), MIL-68(Ga), and MIL-53(Al) (Table 2, entries 4–7), in order to study the photo-switching properties and the role of the host MOF.45 The channels of MIL-53 were too small to embed the SP-O molecule, which was only adsorbed on the MOF surface, making an SP-O@MOF-5 with loading in the order of 10 wt%. Nevertheless, SP-O@MIL-53 showed good reversible SP-O/MC-O photo-conversion during three cycles of UV/vis irradiation (365 and 625 nm). The amount of SP-O was 48 wt%, 19 wt%, and 26 wt%, while the BET surface area decreased by 83.8%, 97.2%, and 92.5% in MOF-5, MIL-68(In), and MIL-68(Ga), respectively. A small amount of the MC-O form was already present in the non-irradiated SP-O@MOF-5 sample due to the highly charged [Zn4O]6+ nodes, and then SP-O completely isomerized to MC-O after 30 min of irradiation with UV light (365 nm). In the case of MOF-5, the MC → SP reverse isomerization occurred gradually at room temperature after UV irradiation, yet it was about 5–10 times faster under visible irradiation (625 nm). In another study, photodegradation of the guest molecule was observed in the SP@MOF-5 system (Table 1, entry 9),57 which is analogous to SP-O@MOF-5. However, the SP-O embedded in MOF-5 did not undergo photodegradation after 720 min under UV light, whereas it did when loaded in MIL-68(In) and MIL-68(Ga), likely because of the relatively low polarity of these MOFs.2.3 Azobenzene@MOFs
Under UV irradiation typically in the range of 315 to 400 nm, E-azobenzene (trans, planar, thermodynamically stable state) may shift to the Z-isomer (cis, non-planar), while the reverse isomerization may occur either under irradiation in the visible range or under thermal stress input (Fig. 1).70 Adding different substituents onto the aromatic rings present across the azobenzene molecule may alter their photochemical properties, including their excitation wavelength and thermal stability.70Azobenzene (AB) was confined within the cavities of the UiO-67 based membrane (Table 2, entry 8), where π-stacking adsorption complexes were formed with the aromatic rings of the MOF scaffold.46 The membrane was tested for gas separation based on a gated photo-switching mechanism to support selective and triggered gas transfer. After loading the membrane with AB (∼19 wt%), the H2 and CO2 permeances were reduced by 29% and 57%, respectively, since the trans isomer partially closed the MOF pore window (Fig. 2a), while, as expected from the pore tightening, the H2/CO2 separation factor increased from 8.9 to 14.7. During the trans → cis isomerization process, performed under UV light at 365 nm, the C4–C4′ distance of AB changed from 9 to 5.5 Å therefore broadening the gate within the MOF and reducing the H2/CO2 separation factor from 14.7 to 10.1. The duration of the trans → cis reaction, estimated by monitoring the permeance in real-time, was in the order of 120 min, while the cis → trans transformation was completed after ∼150 min under visible light irradiation at 455 nm (Fig. 2b).
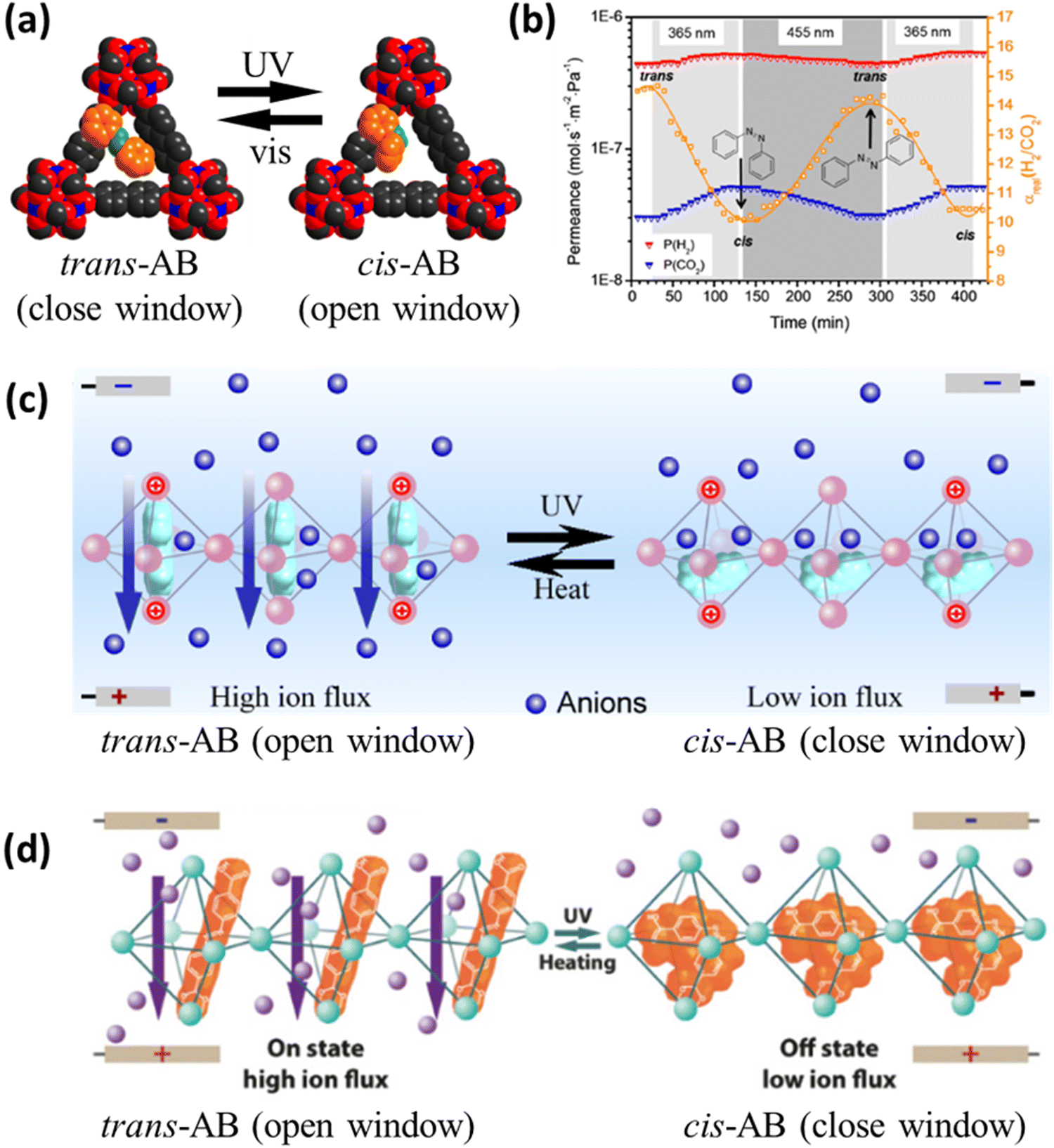 | ||
| Fig. 2 (a) Gate switching mechanism with a space-filling model of a pore window of AB@UiO-67. Oxygen atoms are colored red, nitrogen atoms are green, carbon atoms of the MOF are gray and carbon atoms of AB are orange, while zirconium atoms are blue. Hydrogen atoms are hidden to not overcrowd the structure. (b) Sinusoidal change in mixed-gas separation factor α (H2/CO2) observed in gas permeation of the equimolar H2/CO2 mixture upon in situ reversible switching of AB in UiO-67. (c) Light-gating mechanism of AB@UiO-66. Alignment of AB molecules inside the 3D UiO-66 sub-nanometer channels at an applied voltage. (d) Light-gating mechanism in the mixed-ligand UiO-66. Only the gating process of anions is depicted in panels (c and d) since cations may move in the opposite direction. Reprinted (adapted) with permission from ref. 46,47,71. | ||
3D UiO-66 sub-nanochannels were grown in an etched PET membrane and used to hold trans-AB lying in the center of the cavity based on intermolecular repulsion, leaving space for ion transport under applied voltage across the membrane immersed in a LiCl aqueous solution (Fig. 2c and Table 2, entry 9).47 When trans-AB isomerized to cis-AB upon UV irradiation (365 nm), the molecular shape switched from a rod-like planar to a bent non-planar morphology leading to a dipole moment shift from neutral to 3.1 Debye (D). Consequently, the host molecule orientation could be controlled by altering the electric field supporting partial to complete closure of the MOF pore window (Fig. 2c), leading to decreased ionic currents by a factor of 17.7 ± 0.5 and 17.8 ± 0.3, between +2 V and −2 V, respectively. The ionic current under UV and visible irradiation was similar since the cis → trans isomerization did not occur under visible light due to weak irradiation, which would not affect significantly the strong electrostatic interactions between the MOF inorganic nodes and the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– groups present within the host molecule. The ion flux could be returned to its original value after quenching the material at 60 °C for 1 h and subsequent cooling to room temperature. Only four cycles of UV irradiation and thermal treatment were performed, with the ionic current in the 3rd and 4th cycle at +2 V and the 4th cycle at −2 V being ∼5% lower than that in the first two cycles, suggesting relatively strong stability of the complexes and a repeatable mechanism. However, further cycles and testing should be performed to evaluate the kinetics of the response and assess the impact of the sweeping voltage on the response rate of the material.
N– groups present within the host molecule. The ion flux could be returned to its original value after quenching the material at 60 °C for 1 h and subsequent cooling to room temperature. Only four cycles of UV irradiation and thermal treatment were performed, with the ionic current in the 3rd and 4th cycle at +2 V and the 4th cycle at −2 V being ∼5% lower than that in the first two cycles, suggesting relatively strong stability of the complexes and a repeatable mechanism. However, further cycles and testing should be performed to evaluate the kinetics of the response and assess the impact of the sweeping voltage on the response rate of the material.
In a follow-up study, the same material was fabricated by using a mixed-ligand of benzene-1,4-dicarboxylic acid, namely the backbone of UiO-66, and azobenzene-4,4′-dicarboxylic acid (ABDC).71 This strategy was hybrid since the photo-responsive molecule had a covalent bond with the Zr node (Fig. 2d), thus it is not a guest@MOF system, yet it was neither a ligand pendant (Section 5.1) nor a ligand-backbone one (Section 5.2). The sample with a 33 mol% of ABDC in the mixed-ligand resulted in the increase of the on/off ionic current ratio from 17.7 ± 0.5 and 17.8 ± 0.3, which was the case of the AB@MOF system,47 to 40.2 ± 0.7 and 25.8 ± 0.3, at −2 V and +2 V, respectively. These values were higher than those of other AB-based light-gated nanochannel or sub-nanochannel materials for ion transport, such as proteins (on/off ratio of 1.3–2.1),72,73 pillararene (5.0),74 poly(diallyldimethylammonium chloride) (3.3),75 and diamide complex membrane (16.7).76 Higher molar percentages of ABDC led to (i) a lower on/off ratio because the ABDC works more as the backbone rather than controlling the ion flux through light-responsive gating, and (ii) a gradual loss of the reversible gating property as the MOF structure collapsed after the first UV/heating cycle when only ABDC was used as the ligand.
AB, embedded in a HKUST-1 SURMOF thin film with a concentration of 15 molecules per unit cell, showed reversible photo-isomerization when alternating irradiation at 365 and 455 nm.63 However, the amount of AB loaded in the MOF decreased by ∼40% and 5% when exposing the material to air at 60 °C for 1 week and at room temperature for 3 weeks, respectively. The causes for this depletion were not investigated and it was stated that AB likely desorbed and/or degraded. AB@HKUST-1 was tested in a guest uptake experiment using 1,4-butanediol in argon. Under visible light, the mass uptake was 7.2 μg cm−2, with this figure increasing by 8 ± 3% when switching to UV light (365 nm).
Furthermore, azobenzene@MOFs were utilized with target-specific chemical active sites to modulate adsorption by enhancing chemical interaction with active sites for selective capture. AB, embedded in UiO MOF (U-azo) with tetraethylenepentamine (TEPA) as a specific active site (Table 2, entry 11), was investigated for selective CO2 adsorption.64 TEPA efficiently boosted the binding affinity of U-azo for CO2. The CO2 adsorption capacity was changed from 43.4 to 29.8 cm3 g−1 upon UV light irradiation (320 nm) by trans/cis isomerization of TEPA-U-azo, indicating that TEPA can be successfully sheltered and exposed by photo-responsive motifs. The selective adsorption of CO2 over CH4 and N2 was further studied by evaluating the adsorption isotherm and isosteric heat value, which decreased from 69.8 to 56.6 kJmol−1 during the isomerization, confirming the distinctive interactions between TEPA and the photo-responsive motifs.
Similarly, AB decorated on MIL-101-NH2 (M-azo) along with Cu+ was investigated for the selective adsorption of CO over N2 (Table 2, entry 12).65 Cu+, as a π-complexation active site, was incorporated to trade-off between weak (physical) interactions due to steric hindrance and strong (chemical) interactions with target adsorbates. Upon UV light irradiation on Cu+@M-azo, the adsorption capacity of CO decreased from 16 to 10.5 cm3 g−1 which can be attributed to the sheltering effect of cis/azobenzene formation. During cis/azobenzene formation, the electrostatic potential of Cu+ decreased from 0.008 to −0.038 eV proving the shelter/expose effect during photoswitching.
2.4 Dithienylethene@MOFs
Dithienylethenes (Fig. 1 and Table 2, entry 11), a particular class of diarylethenes, are photo-switchable molecules having both isomers thermally stable due to the low aromatic stabilization energy of the two thiophene rings.77 In solution, the ring-opened isomer may have two conformations, antiparallel and parallel, based on the symmetry of the two aryl groups, C2 and mirror, respectively. Only the antiparallel conformation can switch to the ring-closed isomer under UV light (230–380 nm), and with dynamics in the order of picoseconds.78 The visible light induced the reverse reaction forming the ring-closed isomer, which can be photo-stable after thousands of isomerization cycles.79A photo-responsive system consisting of a dithienylethene (RO-DTE, ring-opened isomer, or RC-DTE, ring-closed isomer) encapsulated in the channels of ZJU-88, a lanthanide MOF, was designed to enable reversible information reading for anti-counterfeiting applications.48 The quantum yield of the RO-DTE@ZJU-88 system was about 7% upon exposure to UV light at 300 nm and the cyclo-reversion reaction took place under visible light with a wavelength greater than 450 nm. This photo-responsive system was designed to enable reversible information reading for anti-counterfeiting applications. Eu3+ ions within the MOF exhibited an emission band in the visible range of the light spectrum and the encrypted information was readable under visible light since the emission band of Eu3+ and the adsorption band of RO-DTE do not overlap. The adsorption band of RC-DTE and the Eu3+ emission band however do overlap producing a photochromic fluorescence resonance energy transfer between the lanthanide and the chromophore, making the information unreadable. During forty cycles of illumination with UV for 60 s followed by visible irradiation for 90 s, the Eu3+ emission band decreased by 5.8%, 4.1% and 7.4% after the 10th, 20th, and 40th cycle, respectively, relative to the 1st cycle.
2.5 Anthracene@MOFs
Anthracene (ANT) and its substituted derivatives photo-dimerize upon UV light (315–400 nm), yielding dianthracene as a result of the [4+4] cycloaddition in which a pair of new C–C bonds are formed (Fig. 1, Table 2, entry 12). The monomerization can take place either thermally (120–340 °C) or by exposure to irradiation in the range of 250–290 nm.80The presence of ANT molecules in the cavities of ZIF-8 distorted the tetrahedral angles of the Zn center, changing the MOF structure from cubic (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m) to monoclinic (C2).49 Two pairs of ANT ordered in a twisted and partially overlapping geometry were present in each ZIF-8 cage. This spatial arrangement allowed two ANT π-orbitals to overlap and give a fluorescence broad emission peak at 550 nm assigned to the formation of an excimer, namely an excited short-lived dimeric molecule. The MOF structure also affected the photo-switching performance of the system since only two ANT molecules per cage could photo-dimerize under UV light (360 nm), leaving the other two molecules as monomers because of steric hindrance. During 30 min of exposure to the UV light, the emission at 550 nm decreased while a new emission band at 415 nm emerged and increased with time. The latter emission was ascribed to the two monomers present in each cage since the dianthracene is a non-fluorescent photo-dimer. ANT@ZIF-8 could switch back to its original structure either after 2 days at 120 °C or 1 day at 140 °C, while a thermal treatment below 120 °C did not produce any effect. This reversible photo-switching mechanism with the two different emission bands was observed for an average loading of ANT per cage between 0.24 and 3.6 molecules. Based on these results, it was concluded that (i) ANT molecules were distributed in the ZIF-8 cages in pairs rather than monomers, (ii) the number of cages containing 3 molecules were deficient because no monomer emission was observed before exposure to UV light, and (iii) the 4 molecules per cage configuration was preferred to the 2 molecules per cage one since the UV-induced enhancement of the monomer emission was observed for all loadings. The enhancement factors, namely the ratio of monomer emission intensity after and before UV irradiation, were 88, 63, 74.1, 74.8, and 67.4 from the 1st to 5th cycle, respectively, indicating that irreversible reactions occurred during each cycle.
3m) to monoclinic (C2).49 Two pairs of ANT ordered in a twisted and partially overlapping geometry were present in each ZIF-8 cage. This spatial arrangement allowed two ANT π-orbitals to overlap and give a fluorescence broad emission peak at 550 nm assigned to the formation of an excimer, namely an excited short-lived dimeric molecule. The MOF structure also affected the photo-switching performance of the system since only two ANT molecules per cage could photo-dimerize under UV light (360 nm), leaving the other two molecules as monomers because of steric hindrance. During 30 min of exposure to the UV light, the emission at 550 nm decreased while a new emission band at 415 nm emerged and increased with time. The latter emission was ascribed to the two monomers present in each cage since the dianthracene is a non-fluorescent photo-dimer. ANT@ZIF-8 could switch back to its original structure either after 2 days at 120 °C or 1 day at 140 °C, while a thermal treatment below 120 °C did not produce any effect. This reversible photo-switching mechanism with the two different emission bands was observed for an average loading of ANT per cage between 0.24 and 3.6 molecules. Based on these results, it was concluded that (i) ANT molecules were distributed in the ZIF-8 cages in pairs rather than monomers, (ii) the number of cages containing 3 molecules were deficient because no monomer emission was observed before exposure to UV light, and (iii) the 4 molecules per cage configuration was preferred to the 2 molecules per cage one since the UV-induced enhancement of the monomer emission was observed for all loadings. The enhancement factors, namely the ratio of monomer emission intensity after and before UV irradiation, were 88, 63, 74.1, 74.8, and 67.4 from the 1st to 5th cycle, respectively, indicating that irreversible reactions occurred during each cycle.
2.6 Comparison of photo-isomer performance
Several classes of photo-switchable molecules, offering various photo-isomerization mechanisms and physical–chemical properties, were developed and investigated for MOF incorporation, fitted for specific applications. Spiropyrans were found to be useful as acidity switches81 and dipoles82 due to the variation in polarity and basicity between the two isomers. Azobenzenes appear as ideal candidates in the fields of photo-actuation83 and photo-pharmacology84 due to their large geometrical changes. Also diarylethenes, associated with minimal geometrical and significant electronic changes, are ideally suited to optoelectronic applications.85 The remarkable advantage of the photo-responsive MOFs is that the target properties may be modulated by turning the light on/off, opening the doors to emerging applications. These processes will, however, become functional only if the switching features occur in a relatively short time and operate for many on/off cycles, whence, photo-switchable materials are desired.The SP → MC and MC → SP isomerizations of the spiropyran and spirooxazine molecules used in the guest@MOF systems lasted from 1 to 60 min. The SP → MC reaction was usually slightly faster than the reverse one, except for the PSPA@MIL-53 system, in which the ion release process associated with the MC → SP isomerization was faster than the opposite process. The trans → cis and cis → trans isomerizations were slower, in the order of 1 to 2 h, whereas the ring-opening and -closing reaction in the dithienylethenes would take place within 1 to 2 min. In addition, although the photo-switching feature of the ANT@ZIF-8 system for potential fabrication of photo-patternable and erasable surfaces was activated within 30 min, a 1 day long thermal treatment was necessary to switch back the material to its original structure. Both direct and reverse isomerization kinetics should be fast to enable the development of practical photo-responsive materials.
The stability of the guest@MOF systems was not always investigated in-depth, and the materials were often tested only for a relatively short number of cycles. As shown in Fig. 3a, the performance of the materials seems to be decreasing for several systems after a couple of cycles only, and it is not clear at this point if this drop will quickly reach a satisfactory plateau or continue to decline adversely. This unwanted phenomenon could be related to photochemical irreversible side reactions or to the fact that the guest molecules are not covalently bound to the MOF scaffolds. In the latter case, when the guest molecules are embedded by simply soaking the MOF sample in an organic solution containing the photo-responsive molecules, the irreversible isomerization appeared to be faster, as in SSP@ZIF-8_2 and NSP@UiO-67 (Fig. 3a).42,43 The SSP@ZIF-8_1 system applied as a membrane with photo-switchable proton conductivity showed an on/off ratio 1.1% higher and 3.3% lower, after respectively 20 and 100 cycles, than in the case of cycle 1, which undoubtedly represented a highly promising result (Fig. 3a). Nevertheless, the same material (SSP@ZIF-8_2) was tested only 3 times, with the Li+ conductivity of the last cycle being 30% lower than that of the first one (Fig. 3a). Azobenzene materials (AB@UiO-66, Fig. 3a) seemed to be more suitable candidates from a stability standpoint, likely due to the simpler chemical structure than those of previously mentioned spiropyran and spirooxazine molecules. Dithienylethenes, known for their superior resistance to photodegradation,78 seemed relatively stable in the DTE@ZJU-88 system, which was tested for 40 cycles (Fig. 3a), therefore confirming the potential of the approach with proper engineering.
 | ||
| Fig. 3 Photo-switching fatigue of (a) guest@MOF systems, (b) post-synthetically modified NH2-MOFs, (c) MOF composites, and (d) MOFs containing photo-responsive ligands. Performance at cycle i divided by performance at cycle 1 (Pi/P1 ratio). The parameters used to evaluate the performance at each cycle are: proton conductivity on/off ratio (SSP@ZIF-8_1), Li+ conductivity (SSP@ZIF-8_2), electrical conductivity (NSP@UiO-67), NaCl adsorption loading (PSPA@MIL-53), ion flux (AB@UiO-66), emission intensity at 613 nm (DTE@ZJU-88), enhancement factor (ANT@ZIF-8), photo-controllable cargo release (Sty-NH-MIL-53), benzyl alcohol oxidation (QUI-N-MIL-125), benzaldehyde production (DEA-N = N-MIL-125), MB degradation (Ag-AC-NH-MIL-125), catalytic conversion (Eu-PYR-NH-MIL-125), AO7 degradation (AA-N-UiO-66), α-acyloxy amide yield (Fe-2PA-N-UiO-66), HER rate (Pt(PTA)-N-UiO-66), RhB photodegradation (NH2-UiO-66/Ag/AgCl), TC photodegradation (NH2-UiO-66/Agl), H2 generation (UCNP-Pt-NH2-UiO-66/Au), RhB photodegradation (NH2-MIL-125/CoSx-20), H2 generation (1T-MoS2/NH2-MIL-125), H2 generation (Ni2P/NH2-MIL-125), CO catalytic turnover number (Ni/Ru-UiO-67), MB conversion (PDA/UiO-66), cis-isomer fraction (Th-AB-MOF), CO2 uptake (PCN-250), and 5-aryl-2-imino-1,3-oxathiolane yield (Co-DCFB) [data adapted from ref. 41–44,47–49,86–104]. | ||
Under UV light, the trans → cis isomerization of azobenzenes opened and closed the pore window of UiO-67 and UiO-66, respectively, showing how this molecule may play an opposite topological role based on where the photo-isomer is encapsulated.
The incorporation of a photo-switchable molecule may endow a MOF with new and enhanced performance characteristics. For example, the NaCl adsorption capacity was 0 in MIL-53 and 0.96 mmol g−1 in PSPA@MIL-53, and the on/off ionic current ratio was 1 and 17.7 when using UiO-66 and AB@UiO-66, respectively. Furthermore, the guest molecule can enhance MOF properties, as in AB@UiO-67, where the H2/CO2 separation factor increased by 65% after AB encapsulation; or in the case of SSP@ZIF-8, where the presence of SSP augmented the cation selectivity by 1 to 2 orders of magnitude. Yet, SSP@ZIF-8 showed a better conductivity and on/off conductivity ratio but longer response time when compared to other materials due to the photo-isomerization reaction.
3. Photo-responsive MOF composites
When the MOF embeds a photo-switchable molecule (encapsulated as a guest) or a photo-switchable group (bound to the scaffold), its physicochemical properties, such as pore size, porosity, polarity, acidity/basicity, and wettability, might change under irradiation. Pristine MOFs may also undergo electronic structure changes, acting as semiconductors for photocatalytic applications.105–107 The empty d metal orbital of the inorganic node and the highest occupied molecular orbital (HOMO) of the organic linker fulfill the role of the conduction band (CB) and valence band (VB), respectively.108,109 Band gap energy is a fundamental parameter in photocatalytic applications because a semiconductor is active only under light with energy higher than its band gap; this fact can also be expressed by the highest wavelength above which the material is not active anymore. The band gap of a MOF depends on the metal ions and the organic linkers,110 but tuning the photocatalytic properties of pristine MOFs is complex and the excited states have too short lifetimes for practical applications.111 Therefore, it is more interesting to synthesize a MOF composite, which can enhance the photocatalytic activity.Photo-responsive MOF composites are formed from the incorporation of photo-responsive nanoparticles (NPs) within the MOF architecture to form hetero-structures, whereby MOFs are mainly used as supporting materials with limited photo-responsive ability. In Sections 3.1 and 3.2, the two most used MOFs to form photo-responsive MOF composites are reviewed and their impact on light absorption and performance enhancement for target environmental applications is reported. Other MOF composites are discussed in Section 3.3, while Section 3.4 compares the performances and highlights the potential of these materials in the field of photocatalysis.
3.1 NH2-UiO-66-based composites
UiO-66 is a Zr-based MOF with 12-connected SBUs supporting water stability and is therefore a promising platform for separation applications.112 The native photo-absorption capability of UiO-66 is however limited to wavelengths shorter than 400 nm, and routes to enhance photo-absorption have included ligand functionalization with an amino group (–NH2) to broaden light absorption from the UV to the visible region. Such a functionalization strategy supports the utilization of solar energy more effectively as a result of a band gap reduction.113 Nevertheless, the band gap reduction in the amine-functionalized MOF is still not sufficient to efficiently photo-excite electrons from the HOMO at wavelengths longer than ∼450 nm,113 which limits direct solar energy utilization. In addition, the poor separation rate of the photo-generated electron–hole pairs across the MOF affects its catalytic performance for various applications. Hence, several pathways were developed to enhance the material light absorption range including loading with metal nanoparticles or forming hetero-structures, referred to as composites.114,115Loading NH2-UiO-66 with Ag-based nanoparticles has been widely utilized to enhance the photo-responsiveness of the MOF and boost the material performance in applications such as photocatalysis, controlled adsorption/desorption, and sensing. For instance, the surface of NH2-UiO-66 was decorated with Ag/AgCl nanoparticles to enhance the visible light absorption and support photo-degradation of Rhodamine B (RhB) (Table 3, entry 1). Even though the band gap of the composite (2.82 eV) was not far below that of NH2-UiO-66 (2.85 eV) at an optimal loading of Ag/AgCl nanoparticles of 16.2 wt%, the photo-degradation rate of RhB increased by 10 times compared to pure NH2-UiO-66 and 4 times when using only Ag/AgCl (k = 0.045 min−1 for the MOF composite with 16.2 wt% of NP loading, R2 = 0.9800).86 This trend was attributed to the enhanced light absorption and electron transmission between the Ag/AgCl NPs and the MOF due to the Ag surface plasmon resonance (SPR) effect, which occurs as a result of an induced electric field that promotes charge separation in the metal atom and hence, allows electrons to flow freely.116
| Entry | MOF | Nanoparticles | Synthesis mechanism | NP optimal loading [wt%] | Light spectrum | Composite light absorption wavelength edge [nm] | Composite band gap [eV]a | Kinetics of degradation [k, min−1]a | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| NA: Not available, UNCPs: up-conversion nanoparticles.a Referring to the composite containing the optimal NP loading. | |||||||||
| 1 | NH2-UiO-66 | Ag/AgCl | Facile precipitation-photoreduction method | 16.20 | Vis | 420 | 2.82 | 0.045 | 86 |
| 2 | AgI | Facile ion exchange precipitation method | 30.00 | Vis | 420 | 2.66 | 0.036 | 87 | |
| 3 | Ag2CO3 | Modified in situ ion exchange solution method | NA | Vis | NA | NA | NA | 117 | |
| 4 | Ag3PO4 | In situ ion-exchange deposition/precipitation method | NA | Vis | NA | NA | NA | 118 | |
| 5 | Pt | de novo synthesis | 16.00 | Solar | 435 | 2.85 | NA | 119 | |
| 6 | Pt | NPs dispersed with MOFs using DMF under solvothermal conditions | 2.5 | Vis | NA | NA | NA | 120 | |
| 7 | Ni2P, Ni12P5 | In situ modification method | NA | Vis | NA | 2.93 | NA | 121 | |
| 8 | UCNPs, Pt, Au | 6 cycles of growth | 0.30 (Au loading) | UV, vis, NIR | NA | NA | NA | 88 | |
| 9 | NH2-MIL-125(Ti) | CoSx | Post-synthetic modification | 20.00 | Vis | 650 | NA | NA | 89 |
| 10 | Mo3S132−, 1T-MoS2 | Post-synthetic modification | NA | Vis | NA | NA | NA | 90 | |
| 11 | Ni2P | Post-synthetic modification | 9.20 | Vis | NA | NA | NA | 91 | |
| 12 | RuO2 | RuCl3·H2O salt was added to the MOF as post-treatment | 1.00 | Vis | 620 | NA | NA | 122 | |
| 13 | MoO3/V2O5 | Metal oxides were added to the MOF by an impregnation method | NA | Vis | NA | NA | NA | 123 | |
| 14 | Ru-UiO-67 | [NiII(bpet)(H2O)2] (bpet = 1,2-bis((pyridin-2-ylmethyl)thio)ethane) | Ship-in-a-bottle approach | 0.31 | Vis | 650 | NA | NA | 92 |
| 15 | UiO-66 | Polydopamine | One pot nucleation growth | NA | Vis | 720 | 2.95 | 0.040 | 93 |
| 16 | Ag | One pot solvothermal reaction | 0.45 | Vis | NA | NA | NA | 124 | |
| 17 | MIL-53(Fe) | TiO2 | One pot solvothermal reaction | 200 | Vis | NA | 1.98 | 0.016 | 125 |
AgI nanoparticles have also been used with NH2-UiO-66 for tetracycline (TC) degradation under visible light (Table 3, entry 2).87 The addition of AgI nanoparticles to the MOFs have resulted in a lower band gap for the composite (2.66 eV) compared to Ag/AgCl nanoparticles. This addition contributed to an enhanced light absorption capability and more efficient photocatalysis at an optimal tetracycline degradation rate of 3.8 and 2.1 times higher than that of bare NH2-UiO-66 and AgI NPs, respectively, assuming that the concentration of the MOF and AgI was the same as in the composite (k = 0.036 min−1 for the MOF composite with 30 wt% of NP loading). The enhanced performance of the composite was attributed to the presence of Ag in the initial stages of the photocatalysis reaction, which led to a more efficient charge transfer pathway between Ag, AgI, and NH2-UiO-66. These configurations are known as Z-scheme heterojunction bridges that provide the SPR functionality to the composite, thus avoiding photo-corrosion of AgI, which ultimately may enhance the separation of electron–hole pairs.87
NH2-UiO-66 MOFs decorated with Pt nanoparticles (Pt NPs) were used to enhance interfacial electron transfer and photocatalytic performance.120,126,127 To control the Pt size and morphology, polyvinylpyrrolidone (PVP), a surfactant, was used to prepare NH2-UiO-66 MOF-Pt NP composites (Table 3, entry 6), where the surfactant provides a microenvironment around the Pt NPs.120 Even though PVP can modulate the microenvironment surrounding catalytic sites, the photocatalytic H2 production rate of the PVP-capped Pt-MOF (PtPVP@UiO-66-NH2) composite was much lower than that of Pt@UiO-66-NH2, i.e., and a H2 production rate of PtPVP@UiO-66-NH2 and Pt@UiO-66-NH2 was 242.7 and 400.7 μmol g−1 h−1, respectively. The higher photocatalytic efficiency was attributed to higher electrical conductivity, leading to an enhanced electron transfer rate between the MOF and Pt sites which prevented the electron–hole recombination. The impact of the spatial location of Pt NPs on the catalytic performance, was found to be related to the electron acceptors localization, across the NH2-UiO-66 compounds.126 Remarkably, Pt NPs dispersed into the semiconductor porous NH2-UiO-66 (Pt@ NH2-UiO-66, 257.58 μmol g−1 h−1) exhibited excellent photocatalytic activity towards the HER than Pt/UiO-66-NH2 (50 μmol g−1 h−1). Thanks to the improved accessibility of protons by Pt NPs inside the MOF, which reduces the electron-transport distance and prevents electron–hole recombination. Furthermore, the absence of aggregation/leaching of Pt NPs during the reaction led to better catalytic recyclability of Pt@UiO-66-NH2 than that of Pt/UiO-66-NH2. The HER rate of Pt/Pt/UiO-66-NH2 was reduced to one-half after the first run and further on subsequent runs due to aggregation/leaching of Pt NPs during the reaction caused by high surface energy. The photocatalytic activity of the Pt@UiO-66-NH2 composite can also be modulated by engineering structural defects.128 With the generation of structural defects, Pt@UiO-66-NH2-100 and Pt@UiO-66-NH2-50 showed an enhanced activity towards the HER compared to Pt@UiO-66-NH2-0, indicating that moderate structural defects can be used to modulate photocatalytic activity by promoting the electron–hole separation.
Ag2CO3117 and Ag3PO4118 were incorporated within NH2-UiO-66 to be used for adsorption and visible light-controlled desorption of sulfonamides (SAs) (Table 3, entries 3 and 4). The addition of such Ag-based NPs enhanced the adsorption capabilities of SAs and, at the same time, utilized light-triggered and controlled desorption of these molecules from the composite. The enhanced adsorption performance was attributed to (i) the homogeneous distribution of the NPs and (ii) the electrostatic interactions and hydrogen bonding occurring between Ag+ of the composite and –NH2 of SAs. The controlled desorption occurred due to the reduction of Ag+ to Ag0 upon visible light illumination, hence releasing SAs.117 Smaller nanoparticles enhanced the light harvesting capabilities of the MOF which were achieved by increasing the content of NH2-UiO-66 within the composite. As the NH2-UiO-66 proportion increased in the composite, the adsorption rate decreased while the desorption rate increased. Thus, an optimized proportion of the composite species is essential to balance between good adsorption and desorption performance.
Owing to the expensive and limited sources of noble metals, nonprecious metal cocatalysts such as transition metal phosphides (TMPs) such as Ni2P and Ni12P5 were explored for the HER.121 The unique structural and electronic properties of Ni2P and Ni12P5 enabled catalytic activity towards the HER. Ni2P and Ni12P5 particles were assembled on UiO-66-NH2 to form the composite (Table 3, entry 7) and tested for the HER under visible light irradiation. TMP@UiO-66-NH2 exhibited exponential improvement in photocatalytic activity, in which the hydrogen production rate of Ni2P@UiO-66-NH2 was two orders of magnitude higher than that of pristine UiO-66-NH2. The enhancement in the activity was attributed to the formation of the TMP-MOF Schottky junction, where TMPs drive the swift charge transfer from the MOF to the cocatalyst, preventing electron–hole recombination and leading to efficient proton reduction. This was further confirmed by calculating the activation energy, which had a value of 69.2 and 57.7 meV for UiO-66-NH2 and Ni2P@UiO-66-NH2, respectively. The lower reaction energy barrier of Ni2P@UiO-66 supported the HER by boosting the reaction activity.
Single-atom catalysts (SAC) such as Pt, Cu and Ni have also been used with NH2-UiO-66 for photo catalytic hydrogen production under visible light.129 Here, the SnO2/MOF was used as a support to immobilize SACs and upon 380 nm light irradiation, Pt1/SnO2/NH2-UiO-66 exhibited superior photocatalytic activity compared to the SnO2/MOF and bare MOF. The hydrogen production rate of Pt1/SnO2/NH2-UiO-66 was 118.6 and 8 times higher than those of NH2-UiO-66 and SnO2/NH2-UiO-66, respectively. The enhanced performance was greatly supported by electron–hole pair formation from a photoexcited amino functionalized organic linker and transfer of these electrons to Zr–oxo clusters via the ligand-to-cluster charge transfer process.
The synthesis method of composite materials may also play a critical role in the materials characteristics and performance. For instance, instead of using post-synthetic modification to add the metal nanoparticles to the MOF, which is the most common strategy to synthesize MOF composites, de novo synthesis has also been used to successfully synthesize MOF composites where the metal and the MOF precursors are introduced together in the same reaction. For instance, Pt nanoparticles were incorporated in NH2-UiO-66 through the de novo synthesis method to enhance the photocatalytic efficiency of phenol degradation in a photocatalytic membrane reactor (Table 3, entry 5).119 The advantages of this strategy include the ability of the composite to pertain high metal content (16 wt%) with superior homogeneity in the NP dispersion, and the incorporation of very small particles of less than 2 nm in diameter is otherwise challenging to handle due to aggregation. Additionally, the high metal content within the composite showed effective immobilization, which improved the photocatalytic activity. The performance of these composites for phenol degradation (70% removal efficiency for Pt/Zr = 0.5) was related to the presence of benzoic acid, which acted as a modulator during MOF synthesis and assisted in the binding formation of Pt@NH2-UiO-66.
A composite material (Table 3, entry 6), consisting of Pt NPs deposited on lanthanide-doped up-conversion nanoparticles (UCNPs) as the core structure, and NH2-UiO-66 decorated with Au NPs as the shell, was prepared and tested for the hydrogen evolution reaction (HER) through indirect water splitting, using triethanolamine as a sacrificial agent.88 The HER took place over the Pt NPs due to the photo-generation of electrons (i) upon UV irradiation in the MOF structure, and (ii) under visible light in the Au NPs and transferred to the MOF. The role of UCNPs was to absorb NIR light, and emit UV and visible irradiation, which were absorbed by the MOF and Au NPs, respectively. Thus, the HER rate of the UNCP-Pt@MOF/Au was enhanced by approximately 3 times, compared to that of UNCP-Pt@MOF (96 μmol g−1 h−1), because the material was able to absorb in a broad range of the light spectrum, and the core–shell structure allowed improving the charge separation. Interestingly, this complex composite structure offered a constant HER for the first 2 cycles, which started to drop by only 3–5% in the 3rd and 4th cycle showing no obvious compromise of the rate when tested for four catalytic cycles of 4 h each. This indicates that the material was relatively stable and can be recycled at least within the boundaries of the performed test.
Later, bimetallic core–shell Pd@Pt NPs were also used to prepare the Pd10@Ptx/UiO-66-NH2 composite for boosting photocatalytic hydrogen production.130 The incorporation of the Pt shell on the Pd surface on MOFs to form a structure that evolves from core–shell to a single atom alloy (SAA) was precisely controlled. Therefore, the surface charge on Pd and Pt is redistributed, which regulates the electronic state of Pt active sites, leading to enhanced photocatalytic efficiency. With optimal loading of Pt in the composite (Pd10@Pt1/UiO-66-NH2-Pt-1 wt%), the highest H2 production rate of 1200.5 μmol g−1 h−1 was achieved upon visible light irradiation, which was attributed to the highest charge redistribution effect and more electron-rich Pt sites. The photocatalytic ability of Pd10@Pt1/UiO-66-NH2 was 25 and 30 times higher than those of Pd10/UiO-66-NH2 and Pt1/UiO-66-NH2, respectively.
3.2 NH2-MIL-125-based composites
NH2-MIL-125(Ti) has been widely studied in photocatalysis, being active under visible irradiation.131,132 However, the low migration capabilities of the photo-generated electron–hole pairs limit its practical applicability and need to be improved, for example, by fabricating a composite material with metals. Although noble metals, such as Pt, Au, and Ag, are usually used due to their high electrical conductivities, their high cost limits their application on a large scale. Therefore, the selection of transition metals that are less costly and able to yield at least similar performance has been the focus of several research papers.Amorphous cobalt sulfide (CoSx) nanoparticles were combined with NH2-MIL-125 (Ti) to form a composite using in situ solvothermal synthesis (Table 3, entry 7).89 These nanoparticles appear promising for photocatalytic applications due to their abundant surface sulfur-rich active sites, which are easily accessible and composed of a central Co atom surrounded and saturated by sulfur and oxygen atoms.133 The light absorption capabilities of the composite materials in the UV-visible range were improved since the light absorption wavelength edge shifted from 520 nm of the bare MOF to 800 nm. Consequently, as a synergetic effect of the composite, its RhB conversion efficiency was reported to be 95.4% in 45 min for a composite containing 20 wt% of CoSx, which was found to be the optimal composition, while the RhB degradation over the bare MOF was only 48.4%.89 However, the authors did not mention the reaction rate constant for RhB degradation for any of the tested composites.
Molybdenum sulfide co-catalysts, namely Mo3S132− centers and 1T-MoS2 NPs, in combination with NH2-MIL-125 were used to fabricate Mo3S132−/NH2-MIL-125 and 1T-MoS2/NH2-MIL-125 composites for photocatalytic hydrogen production by water splitting (Table 3, entry 8).90 The presence of the molybdenum-based nanoparticles enhanced the MOF photocatalytic performance due to the high density of sulfur atoms present around the molybdenum atom, which acts as an active site for photocatalysis. Additionally, the electrical conductivity of the MOF composite was enhanced by integrating the trigonal nanoparticle symmetry as in 1T-MoS2 (T refers to the trigonal symmetry), which enhanced the electron flow to the active sites. As a result, the H2 evolution rates of Mo3S132−/NH2-MIL-125 and 1T-NH2-MIL-125 were found to be 2094 μmol h−1 g−1 and 1454 μmol h−1 g−1, respectively. Even though the photocatalytic performance was increased, the stability of the materials remained poor, where the cycling testing of the composites showed a reduction in the H2 evolution rate, specifically for Mo3S132−/NH2-MIL-125 that lost its activity after the first run, while 1T-MoS2/NH2-MIL-125 started to lose its activity after 72 h. This lack of recyclability of the materials limits their practical applicability.
The use of nickel phosphide NPs as co-catalysts mixed with the NH2-MIL-125 catalyst towards hydrogen generation by water splitting was also investigated (Table 3, entry 9).91 The choice of NPs was justified from their simple preparation as well as low cost and overall chemical stability in harsh acidic environments. The performance of the composite materials was greatly enhanced by achieving a HER rate higher than that of the Ni2P/TiO2 composite and ∼3 times higher than that of Pt/NH2-MIL-125 (HER rate over Ni2P@NH2-MIL-125 was 894 μmol h−1 g−1). The performance enhancement was attributed to the high electrical conductivity of Ni2P and the efficient electron transfer between the catalyst and the co-catalyst achieved by the synergy within the MOF, –NH2 group, and Ti metal clusters, and between NH2-MIL-125 and Ni2P.
NH2-MIL-125 loaded with MoO3 and V2O5 metal oxides has also been utilized to improve the photocatalytic activity of the NH2-MIL-125 MOF.123 Upon visible light irradiation (> 380 nm), the photocatalytic H2 production rate of MoO3/MIL-125-NH2 and V2O5/MIL-125-NH2 composites (Table 3, entry 13) reached 399.0 and 298.6 μmol g−1h−1, respectively, which was 56 and 42 times higher than that of pristine MOFs. The improvement in the photocatalytic efficiency was attributed to band bending in MoO3/MIL-125-NH2 and V2O5/MIL-125-NH2 composites which leads to a built-in electric field and enhanced charge separation. The presence of band bending in MOF composites was confirmed by the higher photocurrent densities and lower charge transfer resistance than pristine MOFs.
The synthesis of hierarchical porous MOF composite materials was also performed to enhance ion diffusion, hence producing promising materials for various applications, including adsorption and catalysis. Owing to the hierarchical structure, the materials will have a multimode porosity allowing the diffusion of active species more easily, thus enhancing the rate of subsequent surface reactions and increasing the bubble release rate.134 To produce hierarchical MOF structures, the conventional method involves the partial degradation of microporous MOFs, as a post-treatment process, through etching/extraction of agents such as HCl.135,136 However, the disadvantage of using the post-treatment method is that it requires finding the suitable etching agent for each MOF as well as the appropriate removal process of the agent. Another method involves nanomaterials as substrates to induce hierarchy in the structure. In an attempt to test the effect of hierarchy in the MOF composite, NH2-MIL-125 in combination with ruthenium oxide (RuO2) nanoparticles has been used for photoelectrochemical water splitting (Table 3, entry 10).122 The addition of RuCl2·H2O salts during the synthesis supported both the formation of hierarchical porosity within NH2-MIL-125 and at the same time, the synthesis of RuO2 nanoparticles as a co-catalyst embedded within the composite material. The photo-electrochemical water oxidation of the composite material, when using RuCl2·H2O salt, improved by 10 times compared to NH2-MIL-125, with a photocurrent density of 0.73 mA cm−2 at an electrical input of 1.23 V. This change in performance was attributed to both the hierarchical structure of the MOF material, which enhanced ion diffusion, and the presence of RuO2 nanoparticles, which offered a more efficient electron separation.
3.3 Other MOF composites
Besides NH2-UiO-66- and NH2-MIL-125-based composites, various studies on composites have been reported utilizing other MOFs. For instance, Ru-UiO-67 combined with a Ni(II) molecular catalyst was tested for photocatalytic reduction of CO2 to CO (Table 3, entry 11).92 The Ni(II) complex, [NiII(bpet)(H2O)2] where (bpet = 1,2-bis((pyridin-2-ylmethyl)thio)ethane), was selected as it is known to have high catalytic activity, which makes it a promising molecular homogenous catalyst for the photocatalytic reduction of CO2. The photoactive MOF was primarily used as a structure to immobilize the Ni(II) complex and, hence, enhance the stability and recyclability of the material. After 20 h of irradiation (450 nm), the Ni@Ru-UiO-67 composite, with a Ni(II) complex loading of 0.31 wt% (elemental ratio Ru:Ni = 15.9:1), produced a CO evolution rate of 426 mmol g−1 h−1 with a catalytic turnover number (TON) of 581, calculated as n(CO)/n(Ni(bpet)(H2O)2, exhibiting a CO selectivity of >99% (calculated as TON (CO)/TON(CO + H2)).92 The high performance of the composite has been attributed to the efficient electron transfer within the composite, where the photo-induced electrons produced from Ru-UiO-67 are directly transferred to Ni(II) hence producing Ni0 which is able to reduce CO2 to CO.92
The core/shell template approach is another method to synthesize MOF composites with the desired properties. For instance, polydopamine (PDA) nanoparticles were used as a starting template (i.e., core) for the introduction of UiO-66 as a shell to produce a core/shell structure for the degradation of methylene blue (MB) (Table 3, entry 12).93 The effect of using a core/shell structure is to have a composite with a well-defined structure such as having spherical shape with narrow size distribution of an average diameter of ∼350 nm. Additionally, the composite had a relatively high specific surface area (90 m2 g−1) compared to PDA (14 m2 g−1), a uniform shell thickness of 10 nm, a convenient band gap of 2.95 eV, and monodispersed size distribution (Fig. 5a and b). PDA was used due to its adhesive, hydrophilic nature, and its redox-active properties, which help in the formation of core/shell composites through the chelation of Zr(IV) ions that will lead to spontaneous nucleation and growth of the Zr-based MOF as a shell on the PDA substrate. Using the PDA@MOF composite, the MB and RhB decolorization showed ∼99% and ∼80% degradation, respectively, within 80 minutes of LED light illumination (MB degradation rate k = 0.04 min−1, R2 = 0.9390) (Fig. 5c). Besides that, the photoelectrochemical water splitting performance was evaluated for the same composite and it showed a high photocurrent density (1.53 mA cm−2), compared to the bare MOF (0.083 μA cm−2), at a low potential of 0.28 V and under visible light irradiation.93
One drawback of using adsorbents for CO2 capture is their high energy required for regeneration,137,138 especially that MOFs are known for their heat insulating nature (<2 W m−1 K−1).139 Hence, in an attempt to solve this issue, silver nanocrystals (Ag NC) coupled with UiO-66 can serve as ‘nanoheaters’ within the MOF so that they absorb visible light energy and convert it to thermal energy locally, and therefore, be able to liberate CO2 in an ecofriendly process (Table 3, entry 13).124 Thereafter, the composite can operate in a light induction swing adsorption (LISA) system for regeneration. At a light intensity of 0.94 W cm−2 and under light exposure for 14 minutes, the local temperature increase of the composite, having 0.45 wt% Ag content, was 16.5 °C, while the bare MOF had a temperature increase of 5.2 °C.124 Under the same conditions and with a light intensity of 1.84 W cm−2, the composite was able to liberate 90.5% of CO2 at 0.44 bar.124
A MIL-53(Fe)/TiO2 composite was tested for the photocatalytic degradation of tetracycline (TC) antibiotic molecules (Table 3, entry 14).125 This composite showed superior light absorption capabilities since TiO2 is a semiconductor active under UV irradiation, while MIL-53(Fe) can react to visible light due to the Fe–O clusters. However, using only a bare MOF leads to electron–hole recombination, hence, coupling it with TiO2 can help to conquer the recombination issue and enhances the photocatalytic performance of the composite through forming a heterojunction between the MOF and the TiO2. Additionally, the band gap of the composite has been enhanced compared to the bare MOF or TiO2, where it was calculated to be 2.55 eV, 2.75 eV, and 1.98 eV for MIL-53(Fe), TiO2, and MIL-53(Fe)/TiO2, respectively.125 When using the sample with a MIL-53(Fe)/TiO2 molar ratio of 1:2, the adsorption capacity was found to be 287 mg g−1 after 6 h under dark conditions, which corresponded to 60% of the initial TC concentration. Then, this percentage increased to 97% after 2.5 h of visible light irradiation (k = 0.0158 min−1, R2 = 0.9592).125
3.4 Performance and potential of MOF photocatalysts
The most common applications for such heterogeneous MOF-based materials was found to be photocatalysis, water splitting reactions, as well as organic compound degradation. Photo-responsive MOF composites used for water splitting were found to offer reasonable performance compared to the benchmark catalyst P25-TiO2 and Pt/TiO2, while for the H2 evolution reaction, the rates ranged from around 500 μmol h−1 g−1140 for pure P25-TiO2 and increased when doped and mixed with metals, such as copper or nickel, up to 700–3390 μmol h−1 g−1,140 or more for Pt/TiO2 (20000 μmol h−1 g−1).140 Other MOF composites such as Mo3S132−/NH2-MIL-125 offered a HER rate in the range of 280–2094 μmol h−1 g−1,88,90,91 which represents a promising material for water splitting applications. The major drawback lies in the recyclability of the materials, which were found to loose performance steadily from the second cycle onwards.
Regarding degradation of organic compounds and dye molecules, the composites may be compared based on the kinetics of degradation using the reported reaction rate constants. Most of the kinetic data were found to fit pseudo first order models yielding R2 values close to 1. In most cases, the dyes that are used are either rhodamine B or methylene blue, otherwise, tetracycline is used to represent antibiotics. The degradation rate of dyes (MB and RhB) ranged between 0.040 and 0.045 min−1,86,93 while the TC degradation rate was between 0.016 and 0.036 min−1.87,124
The size of the NPs may also have a significant impact on the performance of the composite material. Smaller NPs have shown to enhance light harvesting capabilities of the composite and, hence, enhance the photocatalytic performance.117,118,141 It has been noticed that as the NP:MOF ratio in the composite increases, the adsorption performance increases, however, the light-triggered desorption performance drops.6 Not to mention that aggregation of the NPs increases upon increasing their loading, hence also affecting their performance negatively.117 As a result, the loading of the composite with NPs should be optimized to achieve the highest performance. MOFs containing silver-based NPs have shown to form a promising composite for photocatalytic applications, particularly in dye degradation.86,93 As a result, the photocatalytic performance has been significantly enhanced, compared to the bare MOF or bare NPs, and this was attributed to the Ag SPR effect, which played a major role in sustainable charge separation and aided the photodegradation. A one-pot synthesis method was also found to yield promising results compared to post-synthetic modification in terms of avoiding aggregation of NPs and its ability to incorporate very small particles, which in effect enhances light absorption.119
The ability of a material to absorb solar energy effectively depends on its ability to absorb a broad spectrum of light including UV, visible, and NIR, which is an important factor to determine the practical usability of such materials. Most of the composite materials studied may absorb light in the visible region with a light absorption wavelength of 400–600 nm (Table 3). Given that the solar energy at the Earth's surface contains UV light (3–5%, wavelength: <400 nm), visible light (42–43%, 400–700 nm), and near infrared radiation (52–53%, >700 nm), special attention shall be given to the absorption of near infrared radiation since it contains more than half of the radiation coming from the sun.142 Although materials able to absorb a broad spectrum of light are also of great practical importance, such structures are rarely reported in the literature, probably due to their complex synthesis procedures requiring control over NP incorporation and dispersion within the composite material. In this context, a recently noticeable work focused on synthesizing such composite materials (UNCP-Pt@MOF/Au) for water splitting and, interestingly, the composite showed great stability upon testing for up to 4 catalytic cycles.88 However, the HER rate was 280 μmol g−1 h−1, which is below the average of other reported similar materials and particularly compared to benchmark TiO2-based materials, and has to be further improved for such an application.88 Additionally, the statistical reproducibility of the data reported may be somehow arguable and some publications appear to not pursue extensive data reproduction protocols, hence, making kinetics comparisons for various photocatalysts difficult and unreliable. Stronger analysis for such complex heterogeneous catalysts involving thorough benchmarking against each constituent shall be carried out to systematically report the kinetics of the reaction.
Besides, the cost of production is critical when considering economic feasibility, especially for large scale commercial production. Recent works have shed light on this topic proposing the use of cheaper NPs, instead of noble metals such as Pt, Au, and Ag, yet giving a similar performance. Towards this effort, sulfide- and phosphide-based NPs showed promising results in terms of performance and stability.89–91
4. Post-synthetically modified photo-responsive MOFs
The MOF chemical structure can be post-synthetically modified in order to introduce photo-responsive character. In this case the MOF is early synthesized and then used in another process to introduce the photo-responsive functional group or complex, which will be covalently bonded to the MOF scaffold. Different post-synthetic modification (PSM) strategies have been reported, namely, thermal treatment, solvent-assisted ligand or metal exchange, and introduction of pendant groups in the organic ligands of MOFs.4.1 Post-synthetic modification via –NH2 functionality
Using 2-aminoterephthalic acid as the ligand instead of terephthalic acid in the MOF synthesis offered potential PSM pathways (Table 4). Subsequently, organic molecules can be grafted and metal complexes can be formed because of the amino group reactivity (Fig. 4).| Entry | MOF | Synthesis | Products | Application | λ [nm] | Ref. |
|---|---|---|---|---|---|---|
| Styrylpyrene (Sty), N,N-diisopropylethylamine (DIPEA), dichloromethane (DCM), acetonitrile (AN), 2-pyridinecarboxaldehyde (2-PA), 3-pyridinecarboxaldehyde (3-PA), 4-pyridinecarboxaldehyde (4-PA), 3-thiophenecarboxaldehyde (THI), 2-quinolinecarboxaldehyde (QUI), diethylaniline (DEA), acetylacetone (AA), ethanol (EtOH), acetyl (Ac), ethyl acetate (EtOAc), pyridine (PYR), acetic acid (Hac), pyridylimine terephthalic acid (PTA), methanol (MeOH), Passerini three component reaction (P-3CR). | ||||||
| 1 | MIL-53(Al) | Sty, DIPEA, DCM | MOF-NH-Sty | Gating/release cargo molecules | Gating: 400–500 release: 365 | 94 |
| 2 | MIL-125(Ti) | AN, 2-PA, 70 °C | MOF-N-2-PA | Benzyl alcohol oxidation | 420–780 | 95 |
| 3 | AN, 3-PA, 70 °C | MOF-N-3-PA | ||||
| 4 | AN, 4-PA, 70 °C | MOF-N-4-PA | ||||
| 5 | AN, THI, 70 °C | MOF-N-THI | ||||
| 6 | AN, QUI, 70 °C | MOF-N-QUI | ||||
| 7 | step (1) HCl, NaNO2, H2O, 0 °C step (2) DEA | MOF-NN-DEA |
Benzyl alcohol oxidation | 420–720 | 96 | |
| 8 | (1) N2, AA (2) CrCl2, EtOH | MOF-N-Cr, MOF-NH-Ac, MOF-NH2 | Methylene blue degradation | Solar | 98 | |
| 9 | (1) N2, AA (2) CH3COOAg, CH3CN, H2O | Ag np, MOF-NH-Ac, MOF-NCCH3CH3 |
||||
| 10 | (1) EtOAc, PYR (2) Eu(NO3)·6H2O, CH3OH | MOF-NH-PYR-Eu | On/off luminescence sensor | 300–450 | 99 | |
| 11 | MIL-125(Ti), MIL-101(Cr), MIL-101(Al), UiO-66 | (1) HCl, NaNO2, H2O, Hac, 0 °C (2) AA, NaAc, EtOH, H2O | MOF-N-AA | Inhibition of E. coli | 400–800 | 97 |
| 12 | UiO-66 | (1) HCl, NaNO2, H2O, Hac, 0 °C (2) AA, NaAc, EtOH, H2O | MOF-N-AA | Acid orange 7 degradation | >400, >480 | |
| 13 | (1) 2-PA, CH3CN (2) FeCl3, EtOH | MOF-N-2PA-Fe | P-3CR of 4-chlorobenzyl alcohol | UV | 100 | |
| 14 | Ar, Pt(PTA) complex, MeOH | MOF-N-Pt(PTA) | Hydrogen photo-generation | >420 | 101 | |
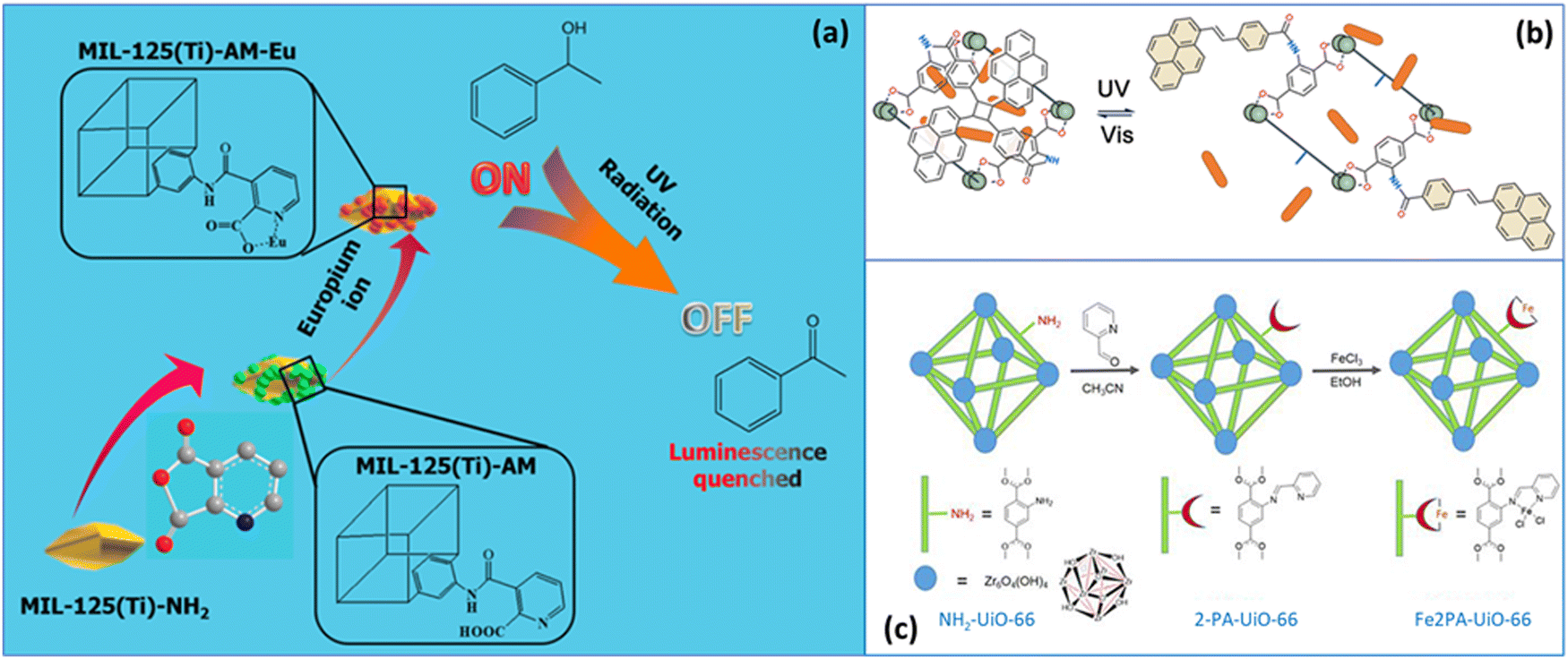 | ||
| Fig. 4 Photo-responsive MOFs prepared by the PSM strategy. (a) Acylation reaction of the amino group in NH2-MIL-125(Ti) with 2,3-pyridinedicarboxylic anhydride, to yield AM-MIL-125, which in turn was used to form an Eu complex. The lanthanide rendered the MOF structure a turn-on/turn-off luminescence sensor for small molecules (X. Lian, B. Yan, Inorganic chemistry, 55 (2016)). (b) Styrylpyrene grafted onto NH2-MIL-53(Al) via amide formation, Sty-MIL-53 was applied for photo-triggered gating and release of methyl orange (R. Ou, H. Zhang, C. Zhao, H. M. Hegab, L. Jiang, V. X. Truong, H. Wang, Chemistry of Materials, 32 (2020)). (c) 2 step-synthesis of Fe2PA-UiO-66, step 1: amine-formation reaction between NH2-UiO-66 and 2-pyridinecarboxaldehyde giving 2-PA-UiO-66, step 2: complexation of Fe(III) into the modified ligand (D. Azarifar, R. Ghorbani-Vaghei, S. Daliran, A. R. Oveisi, ChemCatChem, 9 (2017)). | ||
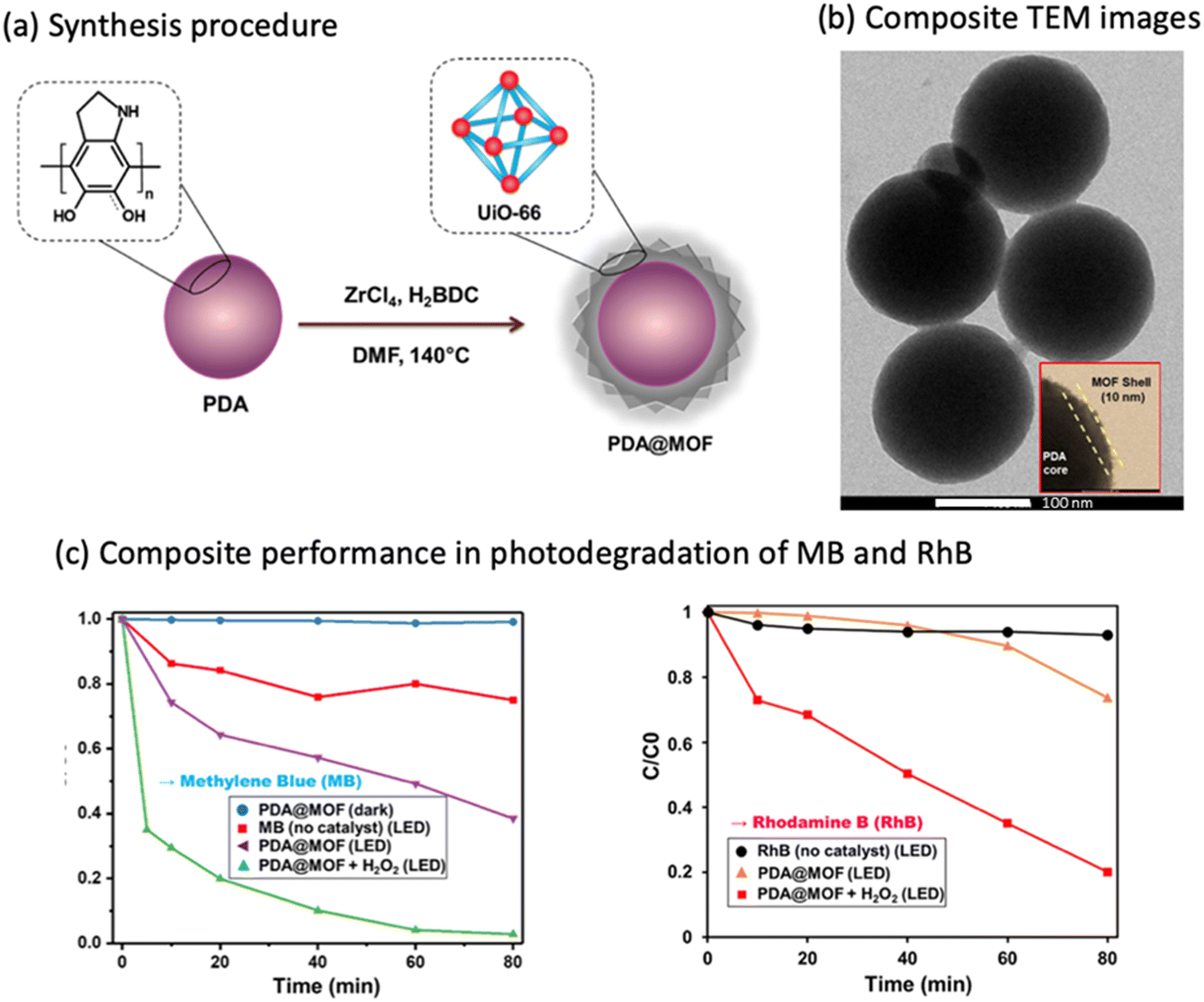 | ||
| Fig. 5 (a) A schematic representation of the synthesis procedure and reaction conditions of the formation of a PDA@MOF core/shell structure. (b) TEM image of the core/shell composite with the inset showing the MOF shell thickness. (c) Composite performance on the degradation of MB and rhodamine B. Reprinted (adapted) with permission from ref. 93 Copyright 2020 American Chemical Society. | ||
Styrylpyrene (Sty, Table 4, entry 1) was grafted onto NH2-MIL-53(Al) via amide formation, yielding Sty-MIL-53, which was applied for photo-triggered gating and release of cargo molecules.94 Firstly, methyl orange (MO) as a probe molecule was physically adsorbed on the Sty-MIL-53 porous structure. Then, under visible irradiation (400–500 nm), the Sty molecules underwent [2+2] photocycloaddition closing the MOF pore windows and entrapping the MO molecules. The latter was finally released under UV light (365 nm) since the Sty monomerization opened the pore windows. The released amount of MO was reported as a function of the Sty:NH2-MIL-53 molar ratio, the optimal value of which was 1:200, whereas the released amount halved at a ratio of 1:10 and 1:400. In fact, the pore volume might be too low at high load of Sty, while a low amount of Sty in the pores reduced the cargo functionality. The kinetics of the MO release under UV irradiation followed an exponential trend. Specifically, 50%, 90%, and 100% of the encapsulated MOs was released after 5 min, 1 h and 5 h, respectively. The release of MO was above 95% after the 5th cycle.
Aromatic heterocyclic aldehydes, namely 2-pyridine carboxaldehyde (2-PA), 3-pyridine carboxaldehyde (3-PA), 4-pyridine carboxaldehyde (4-PA), 3-thiophenecarboxaldehyde (THI), and 2-quinolinecarboxaldehyde (QUI), were grafted in NH2-MIL-125(Ti) via a Schiff base reaction (Table 4, entries 2–6).95 The presence of the aromatic heterocycles reduced the BET surface area, pore volume, and pore size of NH2-MIL-125 by 17–30%, 15–37%, and 1–4%, respectively. The band gap of NH2-MIL-125 was narrowed by 0.03 eV after the PSM loading, since conjugated Schiff bases strongly absorb light in the UV-visible region, and photocatalysis during aerobic oxidation of benzyl alcohols under broad visible irradiation (420–780 nm) was evaluated. The conversion was already significant (69%) over the pristine NH2-MIL-125, then it increased by 14%, 14%, 19%, 25%, and 27% after grafting 2-PA, 3-PA, 4-PA, THI, and QUI, respectively. The selectivity to the benzaldehyde was >99% for all the photocatalysts, including the pristine one. Only the QUI-NH2-MIL-125 sample was retested, with the benzyl alcohol conversion dropping from 88% to 85% after 5 cycles. Additionally, the photo-oxidation of various substituted benzyl alcohols over QUI-NH2-MIL-125 resulted in alcohol conversions between 46% and 99%, with the selectivity consistently above 99%. The pristine NH2-MIL-125 was however not tested for those alcohols, which means that only the impact of the substituted group for one of the photocatalysts (QUI-NH2-MIL-125) was discussed.
The amino group in NH2-MIL-125 reacted with an acidic solution of Na2NO2 forming a diazo functionality, which was then used to graft a diethylaniline molecule (Table 4, entry 7).96 Only 28 ± 2% of the linkers were successfully functionalized with the new moiety due to the unwanted denitrogenation of the diazo group and the steric effect of diethylaniline. The post-synthetically modified MOF exhibited a band gap narrowed by 0.56 eV compared to NH2-MIL-125, and consequently, the reaction rate of the selective oxidation of benzyl alcohols to benzaldehydes under visible light increased by 11%. However, when the new material was reused for a second run, the photocatalytic activity was only 2% higher than the original MOF, suggesting that the grafted molecules may have degraded upon irradiation.
NH2-UiO-66, NH2-MIL-125(Ti), NH2-MIL-101(Cr), and NH2-MIL-101(Al) were post-synthetically functionalized via the diazo coupling reaction with acetylacetone (AA) (Table 4, entries 11 and 12),97 which is a β-diketone with two tautomers used in organic synthesis as well as catalysis and photocatalysis.143–145 The presence of AA in the MOF system resulted in an intra-ligand charge transfer, which enhanced the ligand to metal cluster charge transfer and shifted the MOF light absorption edge. Under visible irradiation (400–800 nm), the new four AA-MOFs (AA-UiO-66, AA-MIL-125(Ti), AA-MIL-101(Cr), and AA-MIL-101(Al)) showed an inhibition rate of E. coli in the range of 99.990–99.996%, which was higher than those of the respective NH2-MOFs (65–99.8%) and commercial nano-TiO2 and nano-ZnO (99.9%).97 The most dramatic impact, from 65 to 99.996%, on the inactivation of E. coli by the PSM was observed in the MIL-125 system. Nevertheless, only AA-UiO-66 was further tested, specifically in the photo-degradation of acid orange 7 (AO7). The AO7 conversion increased due to the PSM from 50 to 90% and from 2 to 50%, when using a 400 nm and a 480 nm cut-off filter, respectively, since NH2-UiO-66 did not show any absorbance above 475 nm whereas a broad absorbance band was observed in the 475–550 nm region for AA-UiO-66. When AA-UiO-66 was reused, the AO7 conversion under visible irradiation was constant during the first 3 cycles, then dropped by 10 and further 5 percentage points after the 4th and 5th cycle, respectively, indicating that this photocatalyst might lose activity in a relatively short period of time.
Thermal treatment of NH2-MIL-125(Ti) in the presence of N2 and AA vapor yielded a MOF (AC-MIL-125) with modified AA-NH, acetyl, and unmodified –NH2 functionalities in the ratio 70:12:18.98 The modified AA-NH moiety was used to chelate Cr(III) (Table 4, entry 8) or to dope AC-MIL-125 with Ag nanoparticles by in situ reduction of Ag+ (Table 4, entry 9). When compared to NH2-MIL-125, the BET surface area and pore volume of the formed Cr-AC-MIL-125 and Ag-AC-MIL-125 decreased by 13.7%, 21.5%, and 15.8%, 46%, respectively. Cr-AC-MIL-125 and Ag-AC-MIL-125 had a band gap narrowed by 0.37 and 0.49 eV, resulting in the MB degradation rate constant under simulated solar light being 3.3 and 11 times higher, respectively, when compared to NH2-MIL-125. Only the stability of Ag-AC-MIL-125 was tested, showing no differences after 5 cycles of MB photodegradation.
The amino group in NH2-MIL-125(Ti) could also perform acylation with an anhydride, such as 2,3-pyridinedicarboxylic anhydride, to yield a MOF (AM-MIL-125) with a new moiety, which in turn was used to form an Eu complex (Eu-MIL-125, Table 4, entry 10).99 This material was tested in the photo-oxidation under UV light of 1-phenylethanol to acetophenone, but here the PSM did not affect the reactivity since the conversion and selectivity in the case of NH2-MIL-125 and Eu-MIL-125 were the same. Instead, the lanthanide was added to render Eu-MIL-125 a turn-on/turn-off luminescent sensor for small molecules. At the beginning of the reaction, Eu3+ emitted red light, due to the f–f transition, which gradually became weaker with reaction time until reaching a minimum. When exposed to UV light, this decrease in the lanthanide emission was due to a competition for excitation energy between AM-MIL-125 and acetophenone, both having an absorption band in the 300–450 nm range. Thus, the visible fluorescence changes of Eu-MIL-125 upon UV light exposure could be applied to detect 1-phenylethanol and acetophenone. As regards the stability, the alcohol conversion and the Eu-MIL-125 luminescence intensity did not change significantly over five cycles.
A multifunctional MOF (Fe2PA-UiO-66, Table 4, entry 13) exhibiting photocatalytic activity and active Lewis acid sites was synthesized in two steps through an amine-formation reaction between NH2-UiO-66 and 2-pyridinecarboxaldehyde giving 2-PA-UiO-66, followed by complexation of Fe(III) into the modified ligand.100 The Fe content was 1.5 wt% with a Zr:Fe ratio of 2.4, while the BET surface area was lower by 40% compared to NH2-UiO-66. The catalytic activity of Fe2PA-UiO-66 was first assessed in the synthesis of α-acyloxy amide by a tandem UV-driven oxidative Passerini three-component reaction (P-3CR) of 4-chlorobenzyl alcohol. The reaction was performed in different solvents, with acetonitrile being the best one in terms of reaction yield and time due to the longer lifetime of singlet oxygen in this solvent.146 The presence of the Fe complexes enhanced the electron transfer efficiency between the catalyst and substrates as well as added more Lewis acid strength into the material, promoting further the P-3CR. Consequently, the yield increased by 43 and 36 percentage points when compared to NH2-UiO-66 and 2-PA-UiO-66, respectively. The yield decreased by 4 percentage points after reusing the same catalyst for a second run, whereas it did not change after another run. Additional synthesis of α-acyloxy amides by the tandem photooxidative P-3CR of various substituted benzyl alcohols over Fe2PA-UiO-66 resulted in yields between 72% and 81%.
Although polypyridyl Pt(II) complexes have been previously studied as homogenous photocatalysts for the HER,147 their use in heterogeneous catalysis was suggested to enable easier recovery of the catalyst and delay potential deactivation occurring during the particulate aggregation.148 PSM of NH2-UiO-66 with 2-PA and K2PtCl4 yielded a N,N-chelating ligand and the formation of pyridylimine Pt(II) complexes on the NH2 group,101 which resulted in a new material, Pt(PTA)-NH2-UiO-66 (Table 4, entry 14), with a BET surface area 15% smaller than that of the original MOF and a Pt loading of 5 wt%. In addition, 3Pt(PTA)-NH2-UiO-66 was prepared with 3 fold amount of 2-PA, showing a BET surface area 52% smaller than that of NH2-UiO-66, and a Pt content of 15 wt%. The Pt(PTA)-NH2-UiO-66 absorption spectrum showed a new band at around 475 nm, while the intensity of this band was higher for 3Pt(PTA)-NH2-UiO-66. Pt(PTA)-NH2-UiO-66 and 3Pt(PTA)-NH2-UiO-66 were applied to the HER under visible light (>420 nm) using ethylenediaminetetraacetic acid (EDTA) as a sacrificial electron donor. The HER rate over the first sample was 178 μmol h−1 g−1, whereas it decreased by 30% for the second one. The last result was explained by the claim that the MOF windows were obstructed by the excessive amount of Pt(PTA), and consequently, the access of EDTA into the pores was hindered. No significant reduction of the photocatalytic activity was observed after 24 h. In the same study, PtCl2-UiO-67-bpy was synthesized by Pt complexation on the nitrogen atoms of the pyridine rings in UiO-67-bpy. The HER rate over PtCl2-UiO-67-bpy was about 7% higher than that over Pt(PTA)-NH2-UiO-66.
4.2 Other post-synthetic modification strategies
An early attempt included thermal treatment of MOF-5 in the presence of Cr(CO)6, a photoactive compound, to yield the piano-stool complex (Zn4O[(η6-1,4-benzenedicarboxylate)Cr(CO)3]3).149 However, this product was not soluble in common organic solvents and decomposed in the presence of water, base, or acid. Photolysis under visible light (450 nm) and N2 or H2 flow resulted in the substitution of a single CO ligand per metal by N2 or H2. Both of the resulting compounds showed improved stability, compared to molecular (η6-arene)Cr(CO)2(N2) complexes, owing to the Cr atoms bonded to the MOF structure.The SP molecule can be covalently bonded to a MOF via post-synthetic solvent-assisted ligand exchange. The indolium group of SP was first bonded to the Zr6 nodes of MOF-808, and then functionalized to yield nitro-SP-MOF-808, which was tested for CO2 capture.150 Under UV irradiation (254 nm), at room temperature and 1.2 bar, the CO2 uptake increased by 17% due to the SP → MC isomerization, showing the strong affinity between CO2 and the charged group in the MC form. After the PSM, the BET surface area decreased by ca. 30%, and the total pore volume reduced by 72%, albeit the average pore size did not change. Then, the BET surface area reduced further by 23% after UV irradiation. Remarkably, the MC → SP reaction did not occur when increasing the temperature up to 100 °C, but, instead, a blue-shift in the UV-vis spectrum of MC-MOF-808 was observed, suggesting that the heat promoted the ring opening reaction, or the polar MC form was more stable in a confining pore environment.151 An SP molecule was grafted in the SURMOF made of Cu2(e-BPDC)2(dabco) via ethynyl-azide click reactions,152,153 yielding the Cu2(SP-BPDC)2(dabco) structure.154 The PSM consisted of a first step in which the SURMOF was immersed in a solution of SP in toluene at room temperature, followed by thermal treatment at 80 °C for 9 days and a washing step. The ionic conduction properties of the Cu2(SP-BPDC)2(dabco) SURMOF were tested by impedance spectroscopy with the sample exposed to nitrogen gas and the probe molecule (water, methanol, or ethanol). When switching from visible to UV light, the sample proton conductivity was reduced by 82, 14, and 21 times in the case of water, methanol, and ethanol, respectively, due to the SP → MC isomerization. The MC form has a dipole moment 3 times higher than the SP form, and, consequently, the probe molecules resulted in stronger bonds with the film surface, decreasing their mobility.
A post-synthetic metal ion exchange method155 was applied to enhance the photocatalytic performance of NH2-UiO-66(Zr) for CO2 reduction, under visible light (UV-cut filter at 420 nm) and by using triethanolamine as an electron donor (sacrificial agent).156 When exposing the original MOF to a N,N-dimethylformamide solution containing TiCl4(THF)2, a certain number of the Zr ions, depending on the exposure temperature and time, is substituted by the Ti ions. The sample used in the reactivity tests was prepared by exposing the original MOF at 120 °C for 16 days and had roughly 50% of the Zr center substituted by Ti atoms, without changing the MOF morphology and with a BET surface area reduced by only 1.1%. The only detected product during the photocatalytic CO2 reduction was formate in an amount 1.7 times higher than the unsubstituted MOF.
5. MOFs containing photo-responsive ligands
This section discusses the potential of photo-switchable MOFs, whereby functionality is achieved through photo-responsive ligand selection. The photo-responsive functional groups are introduced during the MOF synthesis, and eventually they will be present as pendant groups of the existing ligands (Section 5.1) or form effectively the backbone ligands. Such strategies enable photo-isomers to undergo structural and reversible conformational changes upon light irradiation at specific wavelength and irradiance, thus supporting a high level of mechanical and chemical flexibility.157 The performance of such materials for CO2 capture, water splitting, dye guest adsorption and drug release is discussed.5.1 MOFs with photo-switchable pendant groups
MOFs with photo-switchable pendant groups, most-commonly azobenzene side groups, have been reported. As opposed to MOFs with a photo-switchable backbone or ligand, the light-responsive moiety protrudes into the MOF's pore to induce a light-responsive functionality such as CO2 adsorption/desorption. AB-DMOF-1,158 whose parent structure is Zn(bdc)(dabco)0.5 (Dabco MOF or DMOF-1), possesses Zn as the metal source and both 2-phenyldiazenyl terephthalic acid and 1,4-diazabicyclo[2.2.2]octane (DABCO) as ligands. AB-DMOF-1 applicability for CO2 capture was investigated by measuring its CO2 uptake capacity as well as investigating its photo-switching ability for low-energy CO2 capture. The CO2 uptake capacity for AB-DMOF-1 at 1.5 bar was found to be 73 and 41 cm3 g−1 at 0 and 25 °C, respectively. The CO2 uptake of AB-DMOF-1 was lower under UV light irradiation (static conditions) at 1.5 bar, and it was found to be 36 and 29 cm3 g−1 at 0 and 25 °C, respectively. Under dynamic conditions where UV light was continuously switched on and off, the CO2 adsorption behaved similarly to static conditions of UV light irradiation. The CO2 uptake capacity was however 35% lower than that obtained under no UV light irradiation, while the CO2 uptake capacity was found to be restored to its initial value upon switching the UV light off. AB-DMOF-1 also maintained its structure and dynamic photo-switching ability even after being stored under ambient conditions for one month. Compared to PCN-123,159 which was also studied for CO2 capture (22.9 cm3 g−1 at 22 °C and 1 bar), AB-DMOF-1 showed superior CO2 adsorption capacity.An azobenzene-modified photo-responsive thorium–organic framework [Th6O4(OH)4(H2O)6L6] was built using H2L: (E)-2′-p-tolyldiazenyl-1,1′:4′,4′-terphenyl-4,4′′-dicarboxylic acid as the photo-responsive linker and thorium, an actinide element with a large ionic radius (0.97 Å), as its metal node, resulting in one of the largest pore-size reported azobenzene-containing MOFs.102 The larger pore sizes in thorium organic frameworks compared to their Zr-based analogs aided in reducing the steric hindrance surrounding azobenzene, providing sufficient volume for the trans–cis isomerization, hence allowing for improved photo-response. Six Th(IV) atoms, six coordination water molecules, and six L2− make up the asymmetric unit of Th-AB-MOF. Six thorium metal ions combine to form a [Th6] octahedron, Th6O4(OH)4, with μ3-O(H) groups capped to each face that coordinate to three thorium metal centers. The presence of the μ3-O(H) groups fulfills the requirement for the electroneutrality of the framework. The thorium oxide wheel clusters Th6O4(OH)4(COO−)12 are formed through bridging each octahedron edge by the bidentate carboxyl groups from the azobenzene-functionalized L linker. Each thorium atom has a nine-coordinated environment in this hexanuclear cluster, which contains four carboxyl oxygen atoms coming from four different azobenzene ligands, four oxygen atoms from the μ3-O(H) group, and one atom from a coordinated water molecule. The Th-AB-MOF is made up of 12-connected hexanuclear units, Th6O4(OH)4(COO)12, that are propagated by organic linkers to produce a three-dimensional framework with the fcu structure, providing the MOF with small tetrahedral and big octahedral pores with diameters of 29.6 and 44.8 Å, respectively, connected by a triangular plane (Fig. 6a). The Th-AB-MOF can respond promptly to the light stimulus, even when the power of UV light is as low as 6 W. Only 3 min of UV irradiation (365 nm) was sufficient to support the cis isomer of Th-AB-MOF formation and the proportion of the cisTh-AB-MOF rapidly increased within the first 30 min. After 30 min of UV irradiation, the isomerization ratio reached 14.5% compared to 17.1% after 60 min. The conversion rate of the trans-Th-AB-MOF to the cis-Th-AB-MOF gradually reduced after 60 min, eventually reaching a plateau at ∼120 min, with a maximum ratio of the cisTh-AB-MOF of 19.7%. Reverse cis → trans isomerization may be induced by blue light (460 nm) or thermal treatment in the dark. In general, cis → trans isomerization was more effective through heating compared to blue light irradiation. Complete cis → trans isomerization of the Th-AB-MOF was achieved when heating at 60 °C for 180 min under dark conditions, while the yield of the transTh-AB-MOF after 180 min of blue light irradiation at 460 nm was ∼0.83%. The limited penetration depth of blue light within solid materials could be used to explain the incomplete cis → trans isomerization of the Th-AB-MOF.
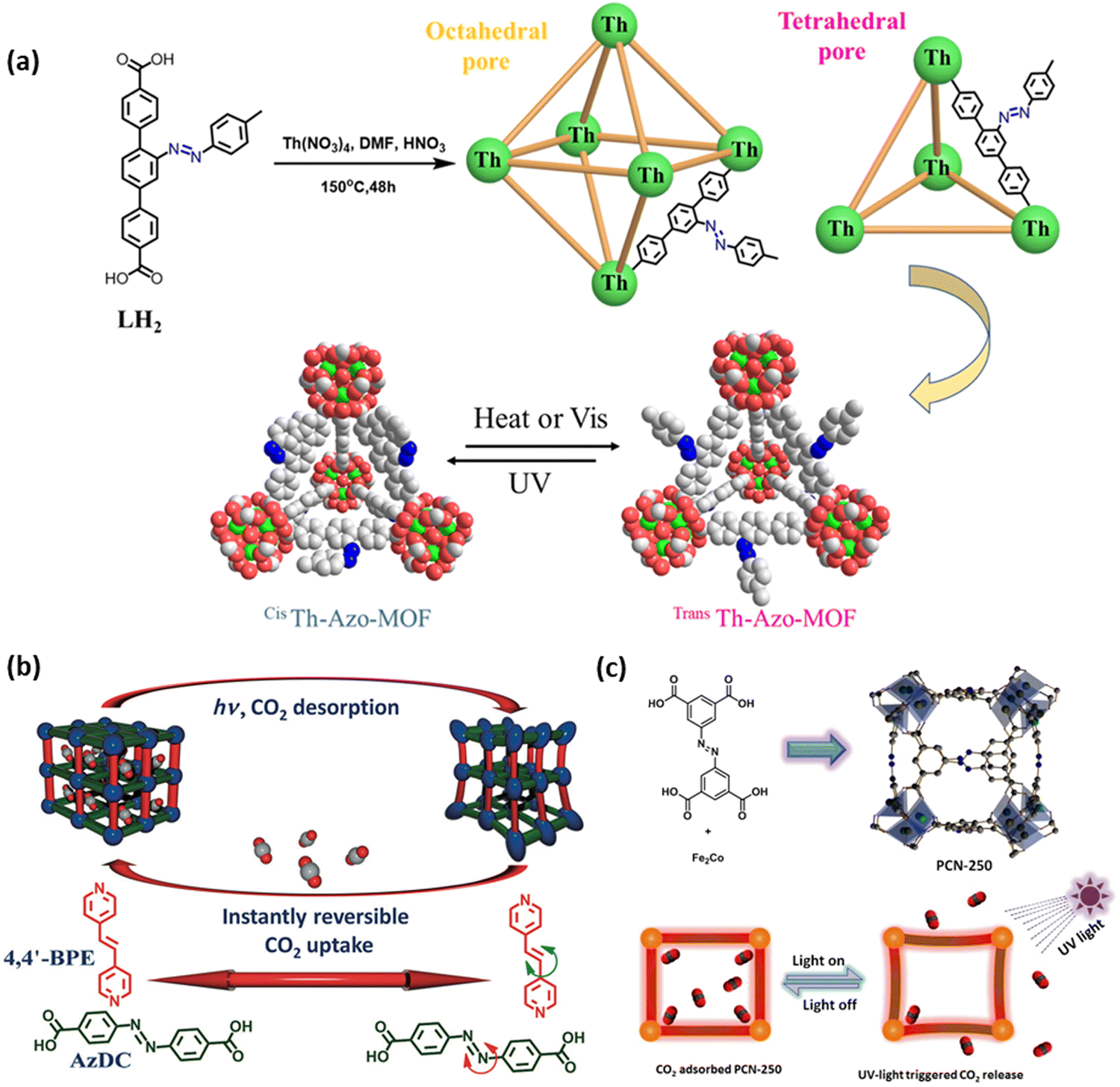 | ||
| Fig. 6 (a) Schematic for the synthesis of a Th-Azo-MOF and its photo-triggered cis–trans isomerization.102 (b) Schematic of Zn-(AzDC)(4,4′-BPE)0.5's dynamic photo-switching leading to reversible CO2 uptake.160 (c) Schematic of the crystalline structure of PCN-250 and its light-induced CO2 uptake and release.103 Reprinted (adapted) with permission. | ||
The Th-AB-MOF may be used to adsorb dye guests, as demonstrated for rhodamine B in dimethylformamide (DMF). The adsorption mechanism may not however be attributed to the electrostatic forces involved, since the Th-AB-MOF only slightly adsorbed cationic dyes, such as methylene blue, crystal violet, and methyl green. The molecular size of rhodamine B matches well the pores of the Th-AB-MOF, and the size of the dye guest played a significant role in the adsorption performance of the Th-AB-MOF. For example, compared to the adsorption capacity of rhodamine B (479.02 g mol−1), the adsorption capacity values for dyes with larger molecular size like Congo red (696.67 g mol−1) or dyes with smaller molecular size like methyl orange (327.33 g mol−1) were significantly lower by ∼97% and ∼81%, respectively. The Th-AB-MOF, on the other hand, showed a slightly better adsorption performance towards thymol blue (466.59 g mol−1), which was lower by ∼75% than that of Rhodamine B, owing to the similar molecular size of the two dyes. Both thymol blue and rhodamine B, which include pendant oxygen-rich groups with lone pair electrons, may coordinate with unsaturated Th sites within the skeleton of the framework after dehydration, which offered extra driving force for guest adsorption and retention.
Furthermore, the effect of cis → trans isomerization on the adsorption performance of Rhodamine B was also investigated. The adsorption kinetics of both 17.1% cisTh-AB-MOF (UV, 1 h) and transTh-AB-MOF (UV, 0 h) fit well the pseudo-second-order kinetic model with rate constants of 0.0168 and 0.0171 g mg−1 min−1, respectively, meaning that the adsorption performance of the transTh-AB-MOF MOF was slightly faster. After 1 h of UV irradiation, the adsorption capacity decreased from 110.01 mg g−1 for the transTh-AB-MOF to 97.9 mg g−1 for the 17.1% cisTh-AB-MOF. This drop in capacity cannot be attributed to the collapse of the crystal structure, as the latter remained intact after 1 h of UV irradiation. However, it was speculated that this phenomenon was caused by the change in polarity of the trans- and cis-azobenzene, as the cis-isomer has higher molecular polarity than the trans-isomer.161 Due to this higher polarity, the cis-isomer attracts more water molecules from the solvent compared to the trans-isomer, thus more water molecules were adsorbed in the MOF pores repelling the dye guest molecules and reducing their adsorption. Furthermore, the polarity of the solvent also affects the adsorption performance. For example, when ethanol, which has a lower polarity than water, was used instead of water, the difference in the adsorption capacity between the two Th-AB-MOF isomers was reduced significantly, i.e. 54 mg g−1 for the transTh-AB-MOF compared to 51.4 mg g−1 for the 17.1% cisTh-AB-MOF.
These recent developments call for further optimization of photoactive thorium-based MOFs in photo-regulated cargo adsorption and release, since the adsorption rate constant of the Th-AB-MOF towards Rhodamine B is considered low compared to other MOFs like MIL-68(Al), with an adsorption rate constant of 4.01 g mg−1 min−1,162 or covalent organic frameworks (COFs) like the Ttba-TPDA-COF, with an adsorption rate constant of 0.136 g mg−1 min−1.163 The adsorption rate constant of the Th-AB-MOF towards Rhodamine B was also lower than that of the Fe-MOF, which had a value of 0.99 g mg−1 min−1.164
The Cu2(DCam)2(AB-BiPyB) SURMOF is a pillared-layer MOF thin film, which possesses both photo-switchable and chiral properties due to its linker with photo-switchable azobenzene side groups AB-BiPyB (=(E)-2-(phenyldiazenyl)-1,4-bis(4-pyridyl)benzene), and its homochiral layer linker D-camphoric acid (DCam).165 The enantioselective adsorption capacity of this nanoporous homochiral azobenzene-MOF thin film was tested by performing uptake experiments with enantiopure probe molecules (S)- or (R)-phenylethanol. It was shown that the Cu2(DCam)2(AB-BiPyB) SURMOF can be photo-switched between enantioselective and unselective forms. The maximum yields of the trans → cis and cis → trans isomerizations were 60% and 100%, under UV irradiation (320 nm) and thermal or blue light (455 nm), respectively. It is worth noting that the chiral properties of the SURMOF remained unchanged regardless of the state of the azobenzene groups, namely cis or trans. When the enantiopure uptakes of (S)- or (R)-phenylethanol were investigated for both the trans- and cis-states, it was found that in the trans-state, the adsorption capacity for (S)-phenylethanol was approximately 2.9 times higher than that for (R)-phenylethanol, showing strong enantioselective behavior. Upon UV irradiation, the enantioselective behavior dramatically changed, and the difference between the adsorption capacities of (S)- and (R)-phenylethanol dropped in the cis-state, where the adsorption capacity for (S)-phenylethanol was approximately 1.2 times higher than that for (R)-phenylethanol, indicating minute enantio-selectivity. The theoretical enantiomeric excess (ee) was found to be 7% in the cis-state compared to 49% in the trans-state. Thus, this work showed the potential of using homo-chiral SURMOF membranes in enantioselective separation; for example, the homo-chiral MOF [(ZnLBr)·H2O]n was investigated for the enantio-separation of racemic drugs, showing great potential in that field.166 It is also possible that chirality in photo responsive MOFs can be induced by polarized light.167 The chirality and enantioselective enrichment of the Cu2(F2AzoBDC)2(dabco) MOF was investigated using circularly polarized light (CPL) and CPL induced chiral photoresolution, resulting in an optically active material.
Cu2(F2AzoBDC)2(dabco), a nanoporous photo-switchable MOF thin film functionalized with o-fluoroAB moieties, was synthesized, where the fluorinated AB side groups can undergo reversible isomerization with visible light irradiation only.168 The fluorinated AB side groups switched from the trans to cis configuration by irradiation with green light (530 nm) and from cis to trans by irradiation with violet light (400 nm), whereas the MOF scaffold remained unchanged. Without applying UV light, simulating the photo-switching ability of this MOF with visible light is only an advantage, as UV light is known to destroy organic materials and typically cannot be used in life sciences and biomaterials applications. Based on this, thin films of MOFs with fluorinated AB side groups were fabricated in a layer-by-layer fashion resulting in surface-mounted MOFs (SURMOFs). Upon green and violet light irradiation, the amount of trans-AB was switched from 14 to 87%, which showed a higher switching yield than Cu2(AzoBPDC)2(AzoBiPyB) SURMOFs having plain photo-responsive AB groups switching upon UV irradiation (∼63%).169
In order to investigate the potential application of visible-light-responsive SURMOFs, a thin Cu2(F2AzoBDC)2(dabco) SURMOF film was grown on a mesoporous Al2O3 substrate to fabricate a membrane, and hydrogen-separation experiments were performed. The feed consisted of a mixture of hydrogen and either ethylene or propene. It was found that the trans/cis switching had no impact on hydrogen permeance; on the other hand, the permeance of ethylene and propene increased by 25% and 30%, respectively, upon irradiation with green light. When irradiating with violet light, the initial permeance was recovered indicating a reversible remote-controlled switching of the membrane selectivity. For the mixture with ethylene, the separation factor can be switched between 6.6 and 8.2, and for the mixture with propene, it can be switched between 8.8 and 12.6. Tuning of the separation factor further can be done by adjusting the irradiation duration or mixing green and violet light together.
Five photoswitchable SURMOFs composed of copper(II)-acetate–paddle-wheel nodes with ditopic carboxylic acid linkers and ditopic nitrogen-terminated linkers with AB side groups, namely Cu2(AzoBPDC)2(dabco), Cu2(AzoTPDC)2(dabco), Cu2(AzoBPDC)2(AzoBiPyB), Cu2(BDC)2(AzoBiPyB), and Cu2(DMTPDC)2(Azo-BiPyB), were investigated.170 These SURMOFs have similar structures but different pore sizes and amounts of AB per pore. The maximum amount of cis-AB obtained upon UV irradiation at 365 nm was 64.5% for Cu2(BDC)2(AzoBiPyB), followed by 63.8% for Cu2(AzoBPDC)2(dabco), and 63% for Cu2(AzoBPDC)2(AzoBiPyB). The uptake amount of all AB containing SURMOFs increased upon trans-to-cis isomerization, which can be explained by the attractive interaction between the polar cis-AB and the polar OH group in the guest molecule, butanol.171
Even though the SURMOFs were similar in structure as mentioned above, they displayed different uptakes of the butanol guest molecule. Upon trans-to-cis isomerization of AB side groups, the butanol uptake increased by only 5% in Cu2(BDC)2(AzoBiPyB), compared to 27% in Cu2(AzoBPDC)2(dabco). A clear general trend was observed between the butanol uptake and density of AB groups per volume, as the density of AB groups per volume increased butanol uptake. A high AB density can be achieved by incorporating multiple AB-containing linkers within the SURMOF such as in Cu2(AzoBPDC)2(AzoBiPyB) with three AB groups per unit cell or by having relatively small pores such as in Cu2(AzoBPDC)2(dabco) with two AB groups per unit cell. This trend between the uptake and density of the photo-switchable AB groups per volume could be used to evaluate and optimize the performance of the already reported AB-containing MOFs.
The photo-switchability of a hybrid SURMOF, consisting of an initial layer of Cu2(BPDC)2(BiPy) (BPDC: biphenyl-4,4′-dicarboxylic acid, BiPy:4,4′-bipyridine) was evaluated. The SURMOF was grown on a functionalized gold surface and top layer (second layer) of Cu2-(AB-BPDC)2(BiPy) (AB-BPDC: 2-azobenzene 4,4′-biphenyldicarboxylic acid). It was found that butanediol uptake was significantly influenced by the photoswitchable top layer and the trans state favoured the uptake, which was 15 times faster than in the cis state and the release of molecule was optically triggered upon irradiation of visible light.172
A mesoporous MOF, PCN-222, possesses Zr as the metal source and tetrakis(4-carboxyphenyl)-porphyrin (H2TCPP) as a ligand.173 The feasibility of PCN-222 towards CO2 capture was investigated by measuring CO2 uptake at various temperatures and 1 atm. The highest CO2 uptake of 58 g cm−3 was measured at 273 K, which was 4.14 times higher than that at a temperature of 308 K. Furthermore, the photocatalytic reduction of CO2 was performed upon visible light irradiation to evaluate the photocatalytic efficiency of PCN-222, and it was found that PCN-222 significantly reduced CO2 to HCOO− in 10 h. The photocatalytic efficiency of PCN-222 was 12.5 times higher than that of the H2TCPP ligand in the presence of triethanolamine (TEOA), where TEOA is a sacrificial agent (electron donor). The photocatalytic behavior of PCN-222 could be attributed to the presence of a deep electron trap state which effectively hinders the unfavourable, radiative electron–hole recombination during the reaction. It was also found that there was no significant change in the yield of the HCOO− anion during the three continuous reaction runs, confirming the stability of PCN-222 during the photocatalytic reaction. PCN-222 was also used for aerobic oxidative coupling of amines under visible light and ambient conditions.174
In order to investigate the potential of photosensitive MOFs incorporated with single atom cocatalysts, Pt metalation was employed in the porphyrin centers of the (AlOH)2 4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate (H2TCPP) MOF (Al-TCPP-Pt) and photocatalytic hydrogen production by water splitting experiments was performed.175 Hydrogen production experiments were performed using triethanolamine as a sacrificial agent and CH3CN as a solvent by visible-light irradiation. The hydrogen production activity of Al-TCPP-Pt (0.1) was significantly higher than that of Al-TCPP-PtNPs as well as that of TCPP-0.1Pt, which had a value of 129 μmol g−1 h−1 with a turnover frequency (TOF) of 35 h−1. The TOF was 30 times higher than that of Al-TCPP-PtNPs, confirming that the photocatalytic activity of Al-TCPP-Pt was enhanced by improving charge separation during the reaction. An indium-based porphyrinic MOF, USTC-8(In), also showed enhanced photocatalytic activity for hydrogen production by improving electron–hole separation, and here, indium ions are located in indium-oxo chains as well as above the porphyrin plane by affording an unusual out-of-plane (OOP) porphyrin.176 The hydrogen production rate was 341 μmol g−1 h−1 for USTC-8(In), which was about 18–37 times higher than the activities of other USTC-8(M), where M is Co, Cu, and Ni.
5.2 MOFs with photo-switchable backbone ligands
MOFs with photo-switchable backbone ligands in their scaffold have been intensively investigated for applications like CO2 capture, photocatalysis and drug release. Zn–(AzDC)(4,4′-BPE)0.5 MOFs (Fig. 6b),160 which combine two photo-responsive ligands, namely 4,4′-dicarboxylate (AzDC) and trans-1,2-bis(4-pyridyl) ethylene (4,4′-BPE), were designed for CO2 capture due to their high capacity and selective CO2 adsorption.160 Upon UV irradiation at 370 nm, AzDC converted from the trans to cis form acting as a CO2 trap, which may be reversed by relaxation of the matrix upon exposure to visible light at 460 nm, to desorb CO2.177 The 4,4′-BPE ligand in the framework similarly underwent trans–cis transition, however to a less extent than AzDC.160 The CO2 capture performance was higher during dynamic measurements, with a 64% desorption capacity, compared to static irradiation conditions (42%).160 Zn(AzDC)(4,4′-BPE)0.5 however is built upon weak electrostatic interactions between soft Lewis-acidic species (Zn2+) and hard basic carboxylate ligands, which was found to detrimentally affect the structural stability of the MOF.178PCN-250 MOFs (Fig. 6c) were demonstrated to offer better moisture-resistance and chemical-tolerance properties, owing to the unique structure generated by rectangular tetratopic 3,3′,5,5′-azobenzene tetracarboxylic acid (ABTC) ligands and 6-connected Fe2Co(μ3-O)(CH3COO)6 metal clusters.103 Hard Lewis acidic metal nodes with a greater connectivity with carboxylate ligands are provided by the Fe2Co(μ3-O)(CH3COO)6 metal clusters, endowing PCN-250 with good structural stability in both aqueous and non-aqueous environments. When exposed to UV light (365 nm) at room temperature and 120 mbar, the ABTC ligand in PCN-250 bent, causing the adsorbed CO2 to be liberated and PCN-250 to be regenerated.103 The CO2 desorption capacity of PCN-250 increased with the light intensities, as it was 57.5% at 1050 mW cm−2 compared to 36.2% and 23.2% at 710 and 350 mW cm−2, respectively.103
JUC-62 (Cu2(ABTC)(H2O)2·(DMF)2(H2O), which is based on copper and 3,3′,5,5′-azobenzene tetracarboxylic acid, was evaluated for CO2 capture because of its dynamic photo-switching properties.179 The rigid framework offered by JUC-62 is advantageous as it prevents the azobenzene linker from undergoing complete photo-isomerization under UV irradiation compared to its free molecule state. Hence, due to restricted bending, JUC-62 can be instantly restored to its original state upon UV light being switched off, leading to complete restoration of the CO2 adsorption capacity during cycling testing. Under UV irradiation (365 nm) and at 0 and 25 °C and 1 bar, JUC-62 showed 51% and 34% lower CO2 uptake, respectively, than when UV light was off, proving the viability of JUC-62 for CO2 capture. The maximum CO2 quantity adsorbed at 0 and 25 °C and 1 bar was 102 and 46 cm3 g−1, respectively, indicating that the CO2 adsorption capacity decreased as the temperature increased. The dynamic CO2 adsorption of a freshly activated JUC-62 was 46 cm3 g−1 compared to 42 cm3 g−1 for a two-week old JUC-62, proving that the photo-switching ability of JUC-62 was maintained after 2-weeks storage at room temperature and without any special treatment.
Photocatalytic MOFs which exhibit efficient performance for water splitting are yet rare.180 While there are many reported MOF photocatalysts that can promote only the HER or only the oxygen evolution reaction (OER), the ones dealing with overall water splitting (both the HER and OER) are rare. A Cd-TBAPy MOF,181 containing 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy) as the organic linker, was demonstrated for solar light-induced water splitting. A particularity of this MOF is the 2D eclipsed structure generated by the coordination of each Cd metal center with 8 H4TBAPy linkers. Generally, to promote overall water splitting, it is theoretically mandatory for the photocatalyst to have a bandgap of more than 1.23 eV.180 For visible light photocatalytic water splitting applications, on the other hand, where the photocatalyst absorbs the photon energy in the visible-light region, the bandgap energy should be less than 2.75 eV (corresponding to a wavelength of 450 nm).180 Along with that, the energy of the valence band (VB) must be more positive than the potential of water oxidation (1.23 V vs. NHE), and the energy of the conduction band (CB) must be more negative than the reduction of a proton (0 V vs. NHE).180 Although common semiconductors used in photochemistry include TiO2, SrTiO3, and KTaO3, with bandgaps of 3.2, 3.3, and 3.4 eV, respectively, which can only be active under UV irradiation due to their large bandgap energy, Cd-TBAPy has a bandgap of ∼2.15 eV with the conduction band and valence band estimated as −0.05 and 2.10 eV, respectively, meeting the thermodynamic requirement for visible-light water splitting.181
In the presence of a cocatalyst, such as Pt or CoPi, and hole and electron scavengers, such as TEOA or AgNO3, respectively, the kinetic requirement for visible-light water splitting can be met as Cd-TBAPy presented a dual and active function supporting both water reduction and oxidation under visible-light irradiation, and passivation in the dark. Therefore, Cd-TBAPy may act as a photocatalyst towards a one-step total water splitting, demonstrating the intriguing future of MOF materials in solar-to-chemical energy conversion.181 Among the noble metals loaded into the MOFs such as rhodium (Rh), platinum (Pt), iridium (Ir), palladium (Pd), ruthenium (Ru), and nickel (Ni), Pt promoted the most efficient HER with an optimal rate of 4.3 μmol h−1 for a 3.5 wt% Pt loading. Furthermore, the loading of Cd-TBAPy with CoPi promoted the OER and an optimal performance of 81.7 μmol h−1 was achieved at a 0.4 wt% CoPi loading. Under 420 nm irradiation, the apparent quantum efficiency (AQE) of water oxidation reached 5.6% compared to 0.32% at 400 nm for Pt-RuOx-NH2-MIL-125 that can also function as a photocatalyst for both water reduction and oxidation under natural sunlight.182 The production rate of neither H2 nor O2 was calculated for Cd-TBAPy making it hard to compare its performance to other MOF photocatalysts.
Co-DCFB, a visible-light responsive MOF, was applied as a catalyst for the di-functionalization of styrenes to form 1,3-oxathiolane-2-iminos under visible light, ambient air, and at room temperature.104 Co-DCFB was synthesized from the reaction of the ligands 2′,7′-dichlorofluorescein and 4,4′-bipyridine (bby) with Co(NO3)2·6H2O under solvo-thermal conditions. The resulting MOF photocatalyst was proven to be recyclable without sacrificing the catalytic efficiency. Co-DCFB showed excellent absorption for visible light by displaying a broad absorption band, i.e. 340 to 550 nm. A catalyst amount of Co-DCFB (2.5 μmol) in acetonitrile (CH3CN) was added to the reaction of styrene with ammonium thiocyanate (NH4SCN) in open air and under green LED irradiation (535 nm) for 6 h. 5-Aryl-2-imino-1,3-oxathiolanes were formed as the product without any byproducts. To demonstrate that Co-DCFB, visible light, and O2 are essential for the reaction to take place, several control experiments were conducted, where one of the three mentioned conditions/reagents above was missing. As a result, the product was either not detected or produced only in trace amounts. Furthermore, Co-DCFB's recyclability was tested for the reaction of styrene with NH4SCN. The yield of 5-aryl-2-imino-1,3-oxathiolanes was 100%, 99%, 98%, 96%, and 96% for runs 1 to 5, respectively. The yield decreased with each run but remained stable for runs 4 and 5. More experimental runs will be needed in order to claim whether Co-DCFB would show a good recyclability performance for this particular application. The synthesis of 5-aryl-2-imino-1,3-oxathiolanes was scaled up to gram amounts to further illustrate Co-DCFB's synthetic utility. With an eight-fold increase in reactant concentration and a 48 h reaction time instead of 6 h, the total turnover number for Co–DCFB was 3136, producing a total product amount of 1.41 g, noting that no other reaction parameters were changed.
Eosin Y was used as a catalyst for the reaction of styrene and NH4SCN under green LED irradiation (535 nm) in open air.183 The yield of 5-aryl-2-imino-1,3-oxathiolanes was 92% when Eosin Y was used compared to 100% when Co-DCFB was used. Furthermore, the use of rose bengal, another organic photocatalyst, gave a relatively low product yield (72%).183 There is therefore great potential for MOFs to be used as alternatives to organic photocatalysts to produce 5-aryl-2-imino-1,3-oxathiolanes from styrenes in ambient air and under visible light due to their ease of modification and mild synthesis conditions.
The MOF architecture was also utilized for niche applications. A photo responsive two-component core–shell MOF structure based on UiO-68 type MOF crystals with p-terphenyldicarboxylic acid containing two tetra-ortho-fluorinated azobenze as a linker molecule was investigated for guest uptake and release as a container upon light irradiation.184 The photo switch between E and Z isomers was achieved by irradiation of green light (565 nm) and blue light (420 nm). Uptake experiments demonstrated a substantial difference between both switching, that is, E-rich and Z-rich, states, confirming the large steric modulation upon photoisomerization.
An azobenzene-based photo-switchable zirconium framework UiO-AZB-F, which contains 4,4′-(diazene-1,2-diyl)bis(3,5-difluorobenzoic acid) (AZB-F) as the linker, was investigated for drug delivery vehicle (DDV) applications by measuring its photo-responsive ability using greenlight.185 5-Fluorouracil (5FU) was used as a cargo substrate due to its broad chemotherapeutic spectrum and small molecular size. The substrate was incorporated into the UiO-AZB-F MOF by an in situ encapsulation method and further functionalized with amine treated polyethylene glycol (PEGNH2) to enhance aqueous stability and impart stealth properties. Upon irradiation at 515 nm, the encapsulated cargo release rate increased to 77% within 120 min in comparison with control experiments (heating at 37 °C in the dark). The average particle size was decreased by 50% after the test, indicating that particles photo-exfoliate upon isomerization of the incorporated ligand.
6. Comparison of the photo-responsive MOF design strategies
Photo-responsive MOFs have been tested for a wide range of applications, including adsorption/desorption of target species (CO2, dyes, and salts), photocatalysis (aromatic compound oxidation, HER, OER, CO2 reduction, and dye degradation), membranes (gas permeation and ion selective transport), and sensors. Most of these applications are based on two light-induced mechanisms, which are (i) photocatalysis, where specific light irradiation triggers the generation of electron/hole pairs and, in turn, other active species; and (ii) molecular photo-switchability.Post-synthetic modification and composite fabrication are the two most used strategies in MOF photocatalysis since techniques including molecule grafting, metal complex formation, metal ion exchange, and decoration with metal nanoparticles can narrow the MOF band gap, enhance the separation of photo-generated electron/hole pairs, and hence, significantly improve the photocatalytic properties. The band gap, an important parameter to evaluate the light absorption capabilities, was not always reported in the case of composite materials. The other photo-responsive MOF design strategies are based on molecular photo-switches, either as guests, pendants, or ligands in the MOF structure. These molecules switch between their two isomers under irradiation in a specific light spectrum range, yielding MOFs with on/off properties.
The guest@MOF strategy would not be recommended for photocatalysis because the MOF/guest molecule interface might not allow for an efficient electron/hole separation, which is a very crucial step in a photocatalytic process. None of the guest@MOF systems discussed in this review was applied in photocatalysis, but instead, photo-switchable guest molecules were used to endow a MOF with new photo-responsive properties. The loading of the photo-switchable molecule is a crucial design parameter because when the guest molecules are densely packed within the MOF structure, they will not photo-isomerize.186 In the majority of those guest@MOF applications, the MOF acts as the host structure, whereas selecting a MOF with the breathing effect, e.g., ZIF-8 and MIL-53, could promote the molecule structural change. Leaching of the guest molecules might be observed in long-term application, and therefore grafting the photo-switchable molecules to the MOF scaffold appears a more efficient strategy, which can be achieved through MOF post-synthetic modification or by using a photo-responsive ligand during the MOF synthesis.
The molecule conformation change will interfere with the MOF structure changing the pore dimensions of the latter and resulting in a gating effect. For example, the spiropyran molecules, usually encapsulated as a guest rather than being grafted, are ideal for ion transportation across a membrane or reversible salt adsorption due to the electrostatic charges of their MC structure. The ion selectivity and the kinetics of the process depend on the molecule and pore sizes, ion charge, and size. The photo-switchable azo compounds, embedded or grafted, were used to modify the MOF pore window (gate) and thus, control the ion flux or separate H2/CO2 mixtures. Additionally, MOFs with the azobenzene moiety as the structural ligand showed noteworthy results in reversible CO2 capture, with the scaffolds deforming under irradiation and releasing the CO2 adsorbed. Potential adverse effects when grafting a photo-switchable molecule might be (i) steric hindrance, for instance the azo-moiety could not rotate in MOF Cu2(NDC)2(AzoBiPy) (NDC: 2,6-naphthalenedicarboxylic acid, AzoBiPy: 3-azobenzene-4,40-bipyridine),187 and (ii) lattice stabilization of the molecule metastable state, which lowers the reverse isomerization yield.188 The photo-responsive backbone ligand strategy should be cautiously applied since the photo-isomerization could lead to structural collapse of the material after one or several light on/off cycles due to irreversible reactions. Furthermore, the azobenzene isomerization yield could be lower than the case with the photo-isomer as a pendant group because the molecule has fewer degrees of conformational changes.
Over 40 different photo-responsive MOFs have been analyzed in this review, and approximately 40% of them have not been tested with respect to the recyclability, which is a crucial feature in order to scale up these materials. Fig. 3 reports the fatigue of 26 different samples under light irradiation in terms of performance, with only 61.5% of them tested in at least 5 cycles. This percentage drops to 37.5% in the case of MOF composites (Fig. 3c). Several samples showed a reduction in photochemical activity after a couple of cycles, which was rarely discussed in-depth. On the one hand, photocatalysts, including MOFs, could deactivate due to strong by-product deposition on the surface.189,190 Furthermore, photochemical side reactions could occur during the isomerization of photo-switchable molecules yielding by-products that are not photo-responsive. In both cases, the performance decreases, hindering the materials’ long-term usage and preventing their applicability at the industrial level.
7. Conclusions and prospects
Photo-responsive MOFs are an emerging new class of light-controlled porous materials, which have been explored for use in various applications, including CO2 capture, catalysis, water splitting, dye adsorption, gas separation, sensing, and data storage. These materials can be fabricated by (i) using photo-switchable molecules as guests, pendant groups or building blocks in the MOF structure, and (ii) incorporating other photo-active compounds in the MOF architecture to form composites. Notwithstanding the numerous studies, this field is still relatively undeveloped, and key challenges should be addressed in future works.Currently, the composites with better performance, particularly in photocatalysis, contain costly noble metals, albeit in low loading percentage. Thus, more effort should be directed towards producing composites formed by cheaper alternatives to nanoparticles, yet with stability and activity close to or exceeding those of the benchmark catalysts.
Light penetration depth across the material, namely the depth at which the intensity of the radiation decreases to 1/e (or 37%) of its original value at the surface, is another fundamental aspect to be investigated scrupulously. Bulk photochemical reactions are hindered by the high photo-switchable molecule molar attenuation coefficient (usually in the order of 104 M−1 cm−1),191 light scattering, and high material density. It was reported that the penetration depth at 365 nm in a 150 nm thick Cu2(BDC)2(AzoBiPyB) SURMOF film was 110 nm.192 This result would suggest that designing a core–shell structure or SURMOF could help to minimize the light absorption pathways, but a more challenging and interesting strategy consists of using donor–acceptor Stenhouse adducts (DASAs).193 DASA is a new class of molecular photoswitches with negative photochromism, namely the colored stable isomer undergoes photo-bleaching when absorbing visible light, switching to a colorless, visible-transparent, state. Thus, the light absorption at greater depth of the material becomes easier as the photoisomerization proceeds. Light penetration depth across the material is another fundamental aspect to be investigated scrupulously. Bulk photochemical reactions are hindered by the high photo-switchable molecule molar attenuation coefficient (usually in the order of 104 M−1 cm−1),191 light scattering, and high material density. Designing a core–shell structure or surface-anchored MOF (SURMOF) could help to minimize the light absorption pathways, but a more challenging and interesting strategy involves using donor–acceptor Stenhouse adducts (DASAs).193
Further studies are required to investigate the stability of photo-responsive MOFs by testing them for a high number of cycles. Since this task may be time demanding, new recyclability testing protocols should be developed, for instance, by operating under harsh conditions in order to accelerate the aging process and assess how the material behaves. Molecular simulations and machine learning algorithms are also suggested for the design of optimum MOF materials with targeted characteristics and performance.
Finally, real-time applications of photo-responsive MOFs at scale further require studies including (i) viability of large-scale equipment set-up and its efficiency due to low light penetration depth across the materials and requirement of UV/visible light transparent equipment, (ii) techno-economic studies to probe the overall cost and energy requirements of the process, and (iii) life cycle analysis to analyze the sustainability of the process from the synthesis of photo-responsive MOFs to application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Partial financial support from Khalifa University through project RC2-2019-007 is gratefully acknowledged. Funding from ADAFSA is also gratefully acknowledged.References
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat.Rev. Mater., 2016, 1, 1–15 Search PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- T. Ma, H. Li, J.-G. Ma and P. Cheng, Dalton Trans., 2020, 49, 17121–17129 RSC.
- J. W. M. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2020, 2000005 Search PubMed.
- Y. Zhang, Y. Zhou, Y. Zhao and C.-j Liu, Catal. Today, 2016, 263, 61–68 CrossRef CAS.
- M. Ali, E. Pervaiz, T. Noor, O. Rabi, R. Zahra and M. Yang, Int. J. Energy Res., 2021, 45(2), 1190–1226 CrossRef CAS.
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. Prechtl, F.-K. Shieh and K. C.-W. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS.
- S. Gautam, H. Agrawal, M. Thakur, A. Akbari, H. Sharda, R. Kaur and M. Amini, J. Environ. Chem. Eng., 2020, 8, 103726 CrossRef CAS.
- L. He, L. F. Dumée, D. Liu, L. Velleman, F. She, C. Banos, J. B. Davies and L. Kong, RSC Adv., 2015, 5, 10707–10715 RSC.
- B.-M. Jun, Y. A. Al-Hamadani, A. Son, C. M. Park, M. Jang, A. Jang, N. C. Kim and Y. Yoon, Sep. Purif. Technol., 2020, 116947 CrossRef CAS.
- C. Petit, Curr. Opin. Chem. Eng., 2018, 20, 132–142 CrossRef.
- T. A. Agbaje, S. Singh, K. S. K. Reddy, K. Polychronopoulou, L. F. Vega, M. Khaleel, K. Wang and G. N. Karanikolos, Microporous Mesoporous Mater., 2021, 324, 111265 CrossRef CAS.
- N. Bhoria, G. Basina, J. Pokhrel, K. S. Kumar Reddy, S. Anastasiou, V. V. Balasubramanian, Y. F. AlWahedi and G. N. Karanikolos, J. Hazard. Mater., 2020, 394, 122565 CrossRef CAS PubMed.
- A. Gholidoust, J. W. Maina, A. Merenda, J. A. Schütz, L. Kong, Z. Hashisho and L. F. Dumée, Sep. Purif. Technol., 2019, 209, 571–579 CrossRef CAS.
- J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345–361 RSC.
- J. Kujawa, S. Al-Gharabli, T. M. Muzioł, K. Knozowska, G. Li, L. F. Dumée and W. Kujawski, Coord. Chem. Rev., 2021, 440, 213969 CrossRef CAS.
- J. W. Maina, J. A. Schütz, L. Grundy, E. Des Ligneris, Z. Yi, L. Kong, C. Pozo-Gonzalo, M. Ionescu and L. F. Dumée, ACS Appl. Mater. Interfaces, 2017, 9, 35010–35017 CrossRef CAS PubMed.
- H. Wu, T. Yildirim and W. Zhou, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS PubMed.
- A. M. Varghese, K. S. K. Reddy, N. Bhoria, S. Singh, J. Pokhrel and G. N. Karanikolos, Chem. Eng. J., 2021, 420, 129677 CrossRef CAS.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- D. Bousquet, F.-X. Coudert, A. G. Fossati, A. V. Neimark, A. H. Fuchs and A. Boutin, J. Chem. Phys., 2013, 138, 174706 CrossRef PubMed.
- K. A. S. Usman, J. W. Maina, S. Seyedin, M. T. Conato, L. M. Payawan, L. F. Dumée and J. M. Razal, NPG Asia Mater., 2020, 12, 58 CrossRef CAS.
- J. W. Maina, C. P. Gonzalo, A. Merenda, L. Kong, J. A. Schütz and L. F. Dumée, Appl. Surf. Sci., 2018, 427, 401–408 CrossRef CAS.
- J. W. Maina, C. Pozo-Gonzalo, J. A. Schütz, J. Wang and L. F. Dumée, Carbon, 2019, 148, 80–90 CrossRef CAS.
- L. F. Dumée, J.-B. Lemoine, A. Ancel, N. Hameed, L. He and L. Kong, Nanomaterials, 2015, 5, 1766–1781 CrossRef PubMed.
- J. Seo, R. Matsuda, H. Sakamoto, C. Bonneau and S. Kitagawa, J. Am. Chem. Soc., 2009, 131, 12792–12800 CrossRef CAS PubMed.
- M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC.
- C. Li, K. Wang, J. Li and Q. Zhang, ACS Mater. Lett., 2020, 2, 779–797 CrossRef CAS.
- W. Cai, J. Wang, C. Chu, W. Chen, C. Wu and G. Liu, Adv. Sci., 2019, 6, 1801526 CrossRef PubMed.
- S. Karimzadeh, S. Javanbakht, B. Baradaran, M.-A. Shahbazi, M. Hashemzaei, A. Mokhtarzadeh and H. A. Santos, Chem. Eng. J., 2020, 127233 Search PubMed.
- Z. Liu, L. Zhang and D. Sun, Chem. Commun., 2020, 56, 9416–9432 RSC.
- H. Schwartz, U. Ruschewitz and L. Heinke, Photochem. Photobiol. Sci., 2018, 17, 864–873 CrossRef CAS PubMed.
- C. Jones, A. Tansell and T. Easun, J. Mater. Chem. A, 2016, 4, 6714–6723 RSC.
- W. Danowski, T. van Leeuwen, W. R. Browne and B. L. Feringa, Nanoscale Adv., 2021,(1) RSC.
- N. D. Shepherd and D. M. D'Alessandro, Chem. Phys. Rev., 2021, 2, 011301 CrossRef.
- M. D. Allendorf, M. E. Foster, F. Leonard, V. Stavila, P. L. Feng, F. P. Doty, K. Leong, E. Y. Ma, S. R. Johnston and A. A. Talin, J. Phys. Chem. Lett., 2015, 6, 1182–1195 CrossRef CAS PubMed.
- H. Q. Liang, Y. Guo, Y. Shi, X. Peng, B. Liang and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 7732–7737 CrossRef CAS PubMed.
- H.-Q. Liang, Y. Guo, X. Peng and B. Chen, J. Mater. Chem. A, 2020, 8, 11399–11405 RSC.
- S. Garg, H. Schwartz, M. Kozlowska, A. B. Kanj, K. Müller, W. Wenzel, U. Ruschewitz and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 1193–1197 CrossRef CAS PubMed.
- R. Ou, H. Zhang, V. X. Truong, L. Zhang, H. M. Hegab, L. Han, J. Hou, X. Zhang, A. Deletic and L. Jiang, Nat. Sustainability, 2020, 3, 1052–1058 CrossRef.
- H. Schwartz, M. Werker, C. Tobeck, R. Christoffels, D. Schaniel, S. Olthof, K. Meerholz, H. Kopacka, H. Huppertz and U. Ruschewitz, ChemPhotoChem, 2020, 4(3), 195–206 CrossRef CAS.
- A. Knebel, L. Sundermann, A. Mohmeyer, I. Strauß, S. Friebe, P. Behrens and Jr Caro, Chem. Mater., 2017, 29, 3111–3117 CrossRef CAS.
- T. Qian, H. Zhang, X. Li, J. Hou, C. Zhao, Q. Gu and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13051–13056 CrossRef CAS PubMed.
- Z. Li, G. Wang, Y. Ye, B. Li, H. Li and B. Chen, Angew. Chem., 2019, 131, 18193–18199 CrossRef.
- M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens and I. F. Vankelecom, Angew. Chem., 2019, 131, 2445–2449 Search PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- N. W. Tyer Jr and R. S. Becker, J. Am. Chem. Soc., 1970, 92, 1289–1294 CrossRef.
- J. T. Wojtyk, A. Wasey, N.-N. Xiao, P. M. Kazmaier, S. Hoz, C. Yu, R. P. Lemieux and E. Buncel, J. Phys. Chem. A, 2007, 111, 2511–2516 CrossRef CAS PubMed.
- C. Lenoble and R. S. Becker, J. Phys. Chem., 1986, 90, 62–65 CrossRef CAS.
- O. Ivashenko, J. T. van Herpt, B. L. Feringa, P. Rudolf and W. R. Browne, Langmuir, 2013, 29, 4290–4297 CrossRef CAS PubMed.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CrossRef CAS PubMed.
- J. Whelan, D. Abdallah, J. Wojtyk and E. Buncel, J. Mater. Chem., 2010, 20, 5727–5735 RSC.
- S. Kremer, I. Ober, V. Greussing, H. Kopacka, H. G. Gallmetzer, B. Trübenbacher, D. Demmel, S. Olthof, H. Huppertz and H. A. Schwartz, Langmuir, 2022, 38(14), 4295–4309 CrossRef PubMed.
- Y. Sheng, J. Leszczynski, A. A. Garcia, R. Rosario, D. Gust and J. Springer, J. Phys. Chem. B, 2004, 108, 16233–16243 CrossRef CAS.
- J. B. Flannery Jr, J. Am. Chem. Soc., 1968, 90, 5660–5671 CrossRef.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- J. Whelan, J. T. Wojtyk and E. Buncel, Chem. Mater., 2008, 20, 3797–3799 CrossRef CAS.
- M. Irie, T. Iwayanagi and Y. Taniguchi, Macromolecules, 1985, 18, 2418–2422 CrossRef CAS.
- K. Müller, J. Wadhwa, J. S. Malhi, L. Schöttner, A. Welle, H. Schwartz, D. Hermann, U. Ruschewitz and L. Heinke, Chem. Commun., 2017, 53, 8070–8073 RSC.
- Y. Jiang, P. Tan, S. C. Qi, X. Q. Liu, J. H. Yan, F. Fan and L. B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS PubMed.
- Y. X. Li, W. Zhong, J. J. Zhou, S. C. Qi, X. Q. Liu and L. B. Sun, Angew. Chem., Int. Ed., 2022, 61, e202212732 CAS.
- M. Lozada-Hidalgo, S. Zhang, S. Hu, V. G. Kravets, F. J. Rodriguez, A. Berdyugin, A. Grigorenko and A. K. Geim, Nat. Nanotechnol., 2018, 13, 300–303 CrossRef CAS PubMed.
- K. Keiichi, Y. Takashi and Y. Masaaki, Chem. Lett., 1991, 965–968 Search PubMed.
- K. Kimura, T. Yamashita and M. Yokoyama, J. Phys. Chem., 1992, 96, 5614–5617 CrossRef CAS.
- S. Adapa and A. Malani, Sci. Rep., 2018, 8, 1–12 CAS.
- H. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- T. Qian, C. Zhao, R. Wang, X. Chen, J. Hou, H. Wang and H. Zhang, Nanoscale, 2021, 13, 17396–17403 RSC.
- A. Mourot, M. A. Kienzler, M. R. Banghart, T. Fehrentz, F. M. Huber, M. Stein, R. H. Kramer and D. Trauner, ACS Chem. Neurosci., 2011, 2, 536–543 CrossRef CAS PubMed.
- A. Rullo, A. Reiner, A. Reiter, D. Trauner, E. Isacoff and G. Woolley, Chem. Commun., 2014, 50, 14613–14615 RSC.
- Y. Sun, J. Ma, F. Zhang, F. Zhu, Y. Mei, L. Liu, D. Tian and H. Li, Nat. Commun., 2017, 8, 1–6 CrossRef PubMed.
- K. Y. Chun, Y. J. Son, S. Jo and C. S. Han, Small, 2018, 14, 1703618 CrossRef PubMed.
- M. Ahmad, S. Metya, A. Das and P. Talukdar, Chem. – Eur. J., 2020, 26, 8703–8708 CrossRef CAS PubMed.
- S. Takami, S. Kobatake, T. Kawai and M. Irie, Chem. Lett., 2003, 32, 892–893 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Hanazawa, R. Sumiya, Y. Horikawa and M. Irie, J. Chem. Soc., Chem. Commun., 1992, 206–207 RSC.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- Q. Shen, Y. Cao, S. Liu, M. L. Steigerwald and X. Guo, J. Phys. Chem. C, 2009, 113, 10807–10812 CrossRef CAS.
- Y. Zhao and T. Ikeda, Smart light-responsive materials: azobenzene-containing polymers and liquid crystals, John Wiley & Sons, 2009 Search PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- X. Zhang, L. Hou and P. Samorì, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- W. Zhao, T. Ding, Y. Wang, M. Wu, W. Jin, Y. Tian and X. Li, Chin. J. Catal., 2019, 40, 1187–1197 CrossRef CAS.
- Y. Pan, X. Yuan, L. Jiang, H. Wang, H. Yu and J. Zhang, Chem. Eng. J., 2020, 384, 123310 CrossRef CAS.
- D. Li, S. H. Yu and H. L. Jiang, Adv. Mater., 2018, 30, 1707377 CrossRef PubMed.
- S. Liu, Q. Zou, Y. Ma, W. Sun, Y. Li, J. Zhang, C. Zhang, L. He, Y. Sun and Q. Chen, J. Mater. Sci., 2020, 55, 16171–16183 CrossRef CAS.
- T. N. Nguyen, S. Kampouri, B. Valizadeh, W. Luo, D. Ongari, Ol. M. Planes, A. Züttel, B. Smit and K. C. Stylianou, ACS Appl. Mater. Interfaces, 2018, 10, 30035–30039 CrossRef CAS PubMed.
- S. Kampouri, T. N. Nguyen, C. P. Ireland, B. Valizadeh, F. M. Ebrahim, G. Capano, D. Ongari, A. Mace, N. Guijarro and K. Sivula, J. Mater. Chem. A, 2018, 6, 2476–2481 RSC.
- Z.-H. Yan, B. Ma, S.-R. Li, J. Liu, R. Chen, M.-H. Du, S. Jin, G.-L. Zhuang, L.-S. Long and X.-J. Kong, Sci. Bull., 2019, 64, 976–985 CrossRef CAS PubMed.
- N. Celebi, M. Y. Aydin, F. Soysal, N. Yıldız and K. Salimi, ACS Appl. Nano Mater., 2020, 3, 11543–11554 CrossRef CAS.
- R. Ou, H. Zhang, C. Zhao, H. M. Hegab, L. Jiang, V. X. Truong and H. Wang, Chem. Mater., 2020, 32, 10621–10627 CrossRef CAS.
- Z. Wu, X. Huang, H. Zheng, P. Wang, G. Hai, W. Dong and G. Wang, Appl. Catal., B, 2018, 224, 479–487 CrossRef CAS.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- W. Huang, X. Wang, W. Zhang, S. Zhang, Y. Tian, Z. Chen, W. Fang and J. Ma, Appl. Catal., B, 2020, 273, 119087 CrossRef CAS.
- R. M. Abdelhameed, M. M. Simões, A. M. Silva and J. Rocha, Chem. – Eur. J., 2015, 21, 11072–11081 CrossRef CAS PubMed.
- X. Lian and B. Yan, Inorg. Chem., 2016, 55, 11831–11838 CrossRef CAS PubMed.
- D. Azarifar, R. Ghorbani-Vaghei, S. Daliran and A. R. Oveisi, ChemCatChem, 2017, 9, 1992–2000 CrossRef CAS.
- Z. Lionet, T. H. Kim, Y. Horiuchi, S. W. Lee and M. Matsuoka, ChemNanoMat, 2019, 5, 1467–1470 CrossRef CAS.
- J.-s Geng, K. Liu, Y.-y Liang, J.-p Yu, K.-q Hu, L.-H. Yuan, W. Feng, Z.-f Chai, L. Mei and W.-q Shi, Inorg. Chem., 2021, 60, 8519–8529 CrossRef CAS PubMed.
- H. Li, M. R. Martinez, Z. Perry, H.-C. Zhou, P. Falcaro, C. Doblin, S. Lim, A. J. Hill, B. Halstead and M. R. Hill, Chem. – Eur. J., 2016, 22, 11176–11179 CrossRef CAS PubMed.
- Y. Liu, C. Lin, B. Li, J. Wang, M. Wang, N. Zhang, Y. Feng and P. Wu, Inorg. Chem. Front., 2020, 7, 3541–3547 RSC.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, J. Mater. Chem. A, 2013, 1, 11473–11482 RSC.
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC.
- F. X. Llabrés i Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2007, 111, 80–85 CrossRef.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- J.-J. Du, Y.-P. Yuan, J.-X. Sun, F.-M. Peng, X. Jiang, L.-G. Qiu, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 190, 945–951 CrossRef CAS PubMed.
- J. Gascon, M. D. Hernández-Alonso, A. R. Almeida, G. P. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- J. Pokhrel, N. Bhoria, C. Wu, K. S. K. Reddy, H. Margetis, S. Anastasiou, G. George, V. Mittal, G. Romanos, D. Karonis and G. N. Karanikolos, J. Solid State Chem., 2018, 266, 233–243 CrossRef CAS.
- T. Musho, J. Li and N. Wu, Phys. Chem. Chem. Phys., 2014, 16, 23646–23653 RSC.
- P. Salcedo-Abraira, S. M. F. Vilela, A. A. Babaryk, M. Cabrero-Antonino, P. Gregorio, F. Salles, S. Navalón, H. García and P. Horcajada, Nano Research, 2021, 14, 450–457 CrossRef CAS.
- A. Melillo, S. Navalón, B. Ferrer and H. García, in Photocatalysis by metal-organic frameworks, ed. E. I. García-López, L. Palmisano, Materials Science in Photocatalysis, Elsevier, 2021, ch 32, pp. 543–559 Search PubMed.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS PubMed.
- X.-Y. Xu, J. Zhang, X. Zhao, H. Fu, C. Chu, P. Wang and C.-C. Wang, ACS Appl. Nano Mater., 2018, 2, 418–428 CrossRef.
- X.-Y. Xu, C. Chu, H. Fu, X.-D. Du, P. Wang, W. Zheng and C.-C. Wang, Chem. Eng. J., 2018, 350, 436–444 CrossRef CAS.
- S. S. Chen, C. Hu, C.-H. Liu, Y.-H. Chen, T. Ahamad, S. M. Alshehri, P.-H. Huang and K. C.-W. Wu, J. Hazard. Mater., 2020, 397, 122431 CrossRef CAS PubMed.
- M. Xu, D. Li, K. Sun, L. Jiao, C. Xie, C. Ding and H. L. Jiang, Angew. Chem., Int. Ed., 2021, 133, 16508–16512 CrossRef.
- K. Sun, M. Liu, J. Pei, D. Li, C. Ding, K. Wu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 132, 22937–22943 CrossRef.
- N. Afzali, S. Tangestaninejad, R. Keshavarzi, V. Mirkhani, J. Nematollahi, M. Moghadam, I. Mohammadpoor-Baltork, M. Reimer, S. Olthof and A. Klein, ACS Sustainable Chem. Eng., 2020, 8, 18366–18376 CrossRef CAS.
- C. Zhang, C. Xie, Y. Gao, X. Tao, C. Ding, F. Fan and H. L. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202204108 CAS.
- H. Li, M. R. Hill, C. Doblin, S. Lim, A. J. Hill and P. Falcaro, Adv. Funct. Mater., 2016, 26, 4815–4821 CrossRef CAS.
- X. Zhang, N. Yuan, Y. Li, L. Han and Q. Wang, Chem. Eng. J., 2022, 428, 131077 CrossRef CAS.
- J. D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- C. Zhang, D. Lei, C. Xie, X. Hang, C. He and H. L. Jiang, Adv. Mater., 2021, 33, 2106308 CrossRef CAS PubMed.
- X. Ma, L. Wang, Q. Zhang and H. L. Jiang, Angew. Chem., Int. Ed., 2019, 131, 12303–12307 CrossRef.
- J. Sui, H. Liu, S. Hu, K. Sun, G. Wan, H. Zhou, X. Zheng and H. L. Jiang, Adv. Mater., 2022, 34, 2109203 CrossRef CAS PubMed.
- Y. Pan, Y. Qian, X. Zheng, S.-Q. Chu, Y. Yang, C. Ding, X. Wang, S.-H. Yu and H.-L. Jiang, Natl. Sci. Rev., 2021, 8, nwaa224 CrossRef CAS PubMed.
- J. D. Xiao, L. Han, J. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS PubMed.
- Y. Pan, J. Wang, S. Chen, W. Yang, C. Ding, A. Waseem and H.-L. Jiang, Chem. Sci., 2022, 13, 6696–6703 RSC.
- N. Kornienko, J. Resasco, N. Becknell, C.-M. Jiang, Y.-S. Liu, K. Nie, X. Sun, J. Guo, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2015, 137, 7448–7455 CrossRef CAS PubMed.
- S. Yuan, L. Zou, J.-S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme and T. Cagin, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed.
- L. Peng, J. Zhang, Z. Xue, B. Han, X. Sang, C. Liu and G. Yang, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- G. Cai and H. L. Jiang, Angew. Chem., Int. Ed., 2017, 56, 563–567 CrossRef CAS PubMed.
- A. M. Varghese and G. N. Karanikolos, Int. J. Greenhouse Gas Control, 2020, 96, 103005 CrossRef CAS.
- G. N. Karanikolos, G. E. Romanos and L. F. Vega, Front. Chem., 2021, 9 Search PubMed.
- H. Babaei, M. E. DeCoster, M. Jeong, Z. M. Hassan, T. Islamoglu, H. Baumgart, A. J. McGaughey, E. Redel, O. K. Farha and P. E. Hopkins, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- A. T. Montoya and E. G. Gillan, ACS Omega, 2018, 3, 2947–2955 CrossRef CAS PubMed.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- E. Austin, A. N. Geisler, J. Nguyen, I. Kohli, I. Hamzavi, H. W. Lim and J. Jagdeo, J. Am. Acad. Dermatol., 2021, 84(5), 1219–1231 CrossRef CAS PubMed.
- D. Gibson, Coord. Chem. Rev., 1969, 4, 225–240 CrossRef CAS.
- S. R. Tamang, A. Singh, D. Bedi, A. R. Bazkiaei, A. A. Warner, K. Glogau, C. McDonald, D. K. Unruh and M. Findlater, Nat. Catal., 2020, 3, 154–162 CrossRef CAS.
- S. Zhang, X. Liu, M. Wang, B. Wu, B. Pan, H. Yang and H.-Q. Yu, Environ. Sci. Technol. Lett., 2014, 1, 167–171 CrossRef CAS.
- M. Hajimohammadi, N. Safari, H. Mofakham and F. Deyhimi, Green Chem., 2011, 13, 991–997 RSC.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2008, 130, 806–807 CrossRef CAS PubMed.
- K. Healey, W. Liang, P. D. Southon, T. L. Church and D. M. D'Alessandro, J. Mater. Chem. A, 2016, 4, 10816–10819 RSC.
- F. Zhang, X. Zou, W. Feng, X. Zhao, X. Jing, F. Sun, H. Ren and G. Zhu, J. Mater. Chem., 2012, 22, 25019–25026 RSC.
- Z. Wang, J. Liu, S. Grosjean, D. Wagner, W. Guo, Z. Gu, L. Heinke, H. Gliemann, S. Bräse and C. Wöll, ChemNanoMat, 2015, 1, 338–345 CrossRef CAS.
- M. A. Gotthardt, S. Grosjean, T. S. Brunner, J. Kotzel, A. M. Gänzler, S. Wolf, S. Bräse and W. Kleist, Dalton Trans., 2015, 44, 16802–16809 RSC.
- A. B. Kanj, A. Chandresh, A. Gerwien, S. Grosjean, S. Bräse, Y. Wang, H. Dube and L. Heinke, Chem. Sci., 2020, 11, 1404–1410 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- D. Sun, W. Liu, M. Qiu, Y. Zhang and Z. Li, Chem. Commun., 2015, 51, 2056–2059 RSC.
- C. L. Jones, A. J. Tansell and T. L. Easun, J. Mater. Chem. A, 2016, 4, 6714–6723 RSC.
- N. Prasetya and B. P. Ladewig, ACS Appl. Mater. Interfaces, 2018, 10, 34291–34301 CrossRef CAS PubMed.
- J. Park, D. Yuan, K. T. Pham, J.-R. Li, A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 99–102 CrossRef CAS PubMed.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS PubMed.
- J. Park, L. B. Sun, Y. P. Chen, Z. Perry and H. C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 5842–5846 CrossRef CAS PubMed.
- M. Saghanejhad Tehrani and R. Zare-Dorabei, RSC Adv., 2016, 6, 27416–27425 RSC.
- T. Xu, S. An, C. Peng, J. Hu and H. Liu, Ind. Eng. Chem. Res., 2020, 59, 8315–8322 CrossRef CAS.
- F. Wei, D. Chen, Z. Liang and S. Zhao, Nanomaterials, 2018, 8(4) CrossRef PubMed.
- A. B. Kanj, J. Bürck, S. Grosjean, S. Bräse and L. Heinke, Chem. Commun., 2019, 55, 8776–8779 RSC.
- X. Kuang, Y. Ma, H. Su, J. Zhang, Y.-B. Dong and B. Tang, Anal. Chem., 2014, 86, 1277–1281 CrossRef CAS PubMed.
- A. B. Kanj, J. Bürck, S. Grosjean, S. Bräse and L. Heinke, Chem. Commun., 2019, 55, 8776–8779 RSC.
- K. Müller, A. Knebel, F. Zhao, D. Bléger, J. Caro and L. Heinke, Chem. – Eur. J., 2017, 23(23), 5434–5438 CrossRef PubMed.
- Z. Wang, A. Knebel, S. Grosjean, D. Wagner, S. Bräse, C. Wöll, J. Caro and L. Heinke, Nat. Commun., 2016, 7, 13872 CrossRef CAS PubMed.
- Z. Wang, K. Müller, M. Valášek, S. Grosjean, S. Bräse, C. Wöll, M. Mayor and L. Heinke, J. Phys. Chem. C, 2018, 122, 19044–19050 CrossRef CAS.
- Z. Wang, S. Grosjean, S. Bräse and L. Heinke, ChemPhysChem, 2015, 16, 3779–3783 CrossRef CAS PubMed.
- L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges, S. Bräse and C. Wöll, ACS Nano, 2014, 8, 1463–1467 CrossRef CAS PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- C. Xu, H. Liu, D. Li, J.-H. Su and H.-L. Jiang, Chem. Sci., 2018, 9, 3152–3158 RSC.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- F. Leng, H. Liu, M. Ding, Q.-P. Lin and H.-L. Jiang, ACS Catalysis, 2018, 8, 4583–4590 CrossRef CAS.
- H. A. Nair, G. S. Rajawat and M. S. Nagarsenker, Stimuli-responsive micelles A nanoplatform for therapeutic and diagnostic applications, in: Drug Targeting and Stimuli Sensitive Drug Delivery Systems, 2018, Ch. 8, pp. 303–342 Search PubMed.
- D. Feng, K. Wang, Z. Wei, Y. P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T. F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H. C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed.
- N. Prasetya and B. P. Ladewig, Sci. Rep., 2017, 7, 13355 CrossRef PubMed.
- H. L. Nguyen, Sol. RRL, 2021, 5, 2100198 CrossRef CAS.
- Y. Xiao, Y. Qi, X. Wang, X. Wang, F. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- S. Remiro-Buenamañana, M. Cabrero-Antonino, M. Martínez-Guanter, M. Álvaro, S. Navalón and H. García, Appl. Catal., B, 2019, 254, 677–684 CrossRef.
- A. K. Yadav and L. D. S. Yadav, Green Chem., 2015, 17, 3515–3520 RSC.
- D. Mutruc, A. Goulet-Hanssens, S. Fairman, S. Wahl, A. Zimathies, C. Knie and S. Hecht, Angew. Chem., Int. Ed., 2019, 58, 12862–12867 CrossRef CAS PubMed.
- H. D. Cornell, Y. Zhu, S. Ilic, N. E. Lidman, X. Yang, J. B. Matson and A. J. Morris, Chem. Commun., 2022, 58, 5225–5228 RSC.
- D. Hermann, H. Emerich, R. Lepski, D. Schaniel and U. Ruschewitz, Inorg. Chem., 2013, 52, 2744–2749 CrossRef CAS PubMed.
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges, S. Bräse, K. Reuter and C. Wöll, Phys. Chem. Chem. Phys., 2015, 17, 14582–14587 RSC.
- D. G. D. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Chem. Commun., 2014, 50, 2653–2656 RSC.
- S. Weon, F. He and W. Choi, Environ. Sci.: Nano, 2019, 6, 3185–3214 RSC.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- Y. Jiang and L. Heinke, Langmuir, 2020, 37, 2–15 CrossRef PubMed.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
| This journal is © The Royal Society of Chemistry 2023 |